
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pediatr., 12 December 2018
Sec. Pediatric Neurology
Volume 6 - 2018 | https://doi.org/10.3389/fped.2018.00394
This article is part of the Research TopicModeling Neurogenetic and Inflammatory Neurodevelopmental Disorders with Stem Cell PlatformsView all 7 articles
 Beatriz C. Freitas1
Beatriz C. Freitas1 Arianna Mei2
Arianna Mei2 Ana Paula D. Mendes2
Ana Paula D. Mendes2 Patricia C. B. Beltrão-Braga1,3
Patricia C. B. Beltrão-Braga1,3 Maria Carolina Marchetto2*
Maria Carolina Marchetto2*Recent reports show an increase in the incidence of Autism Spectrum Disorders (ASD) to 1 in every 59 children up to 8 years old in 11 states in North America. Induced pluripotent stem cell (iPSC) technology offers a groundbreaking platform for the study of polygenic neurodevelopmental disorders in live cells. Robust inflammation states and immune system dysfunctions are associated with ASD and several cell types participate on triggering and sustaining these processes. In this review, we will examine the contribution of neuroinflammation to the development of autistic features and discuss potential therapeutic approaches. We will review the available tools, emphasizing stem cell modeling as a technology to investigate the various molecular pathways and different cell types involved in the process of neuroinflammation in ASD.
Autism Spectrum Disorder (ASD) is a lifelong neurodevelopmental disorder characterized by the impairment of social abilities and cognitive functions that can manifest cortical disorganization or neuroinflammation states (1). As a complex polygenic disorder with several degrees of severity, the causes of ASD are not completely elucidated. Autism can be of unknown gene etiology (idiopathic) (2) or a specific syndromic disorder, such as Rett Syndrome or Fragile X where single gene mutations can manifest ASD features. Elucidating the etiology of ASD could help understanding the increase in incidence (3) and drive research efforts toward novel therapeutic approaches.
Earlier recognition of ASD symptoms results in better patient prognosis after behavioral therapy (4). Cortical changes, such as increased head circumference and brain volume reported for a subset of ASD toddlers, allow for potential clinical diagnosis before 1 year of age, especially when aligned with a genetic investigation of known genes that play a role in neural development (4). Sequencing of whole genomes of families where the proband was affected with ASD uncovered rare de novo mutations on genes that segregate with ASD (5, 6). These mutations are heterogeneous and only account for < 1% of ASD cases (7), highlighting the polygenic nature of ASD. A subset of the genes identified in ASD (FMR1, CHD8, DYRK1, NLGN3, PTEN) and 16p11 deletion, are likely involved in brain volume changes and neural connectivity, and its particular function can be studied in animal models and genetically engineered human cells (8). The majority of idiopathic ASD cases lack a known culprit gene(s), with phenotypes ranging across a wide pathological spectrum. In those cases, differential scientific approaches, such as the use of stem cell modeling and comprehensive in silico data analysis are required. Furthermore, twin studies suggest that hereditary mechanisms and environment also play a role in ASD during mid-fetal brain development (9).
Several studies report immune system misbalances in ASD (10–12). Particularly, immune system involvement has been associated with three major circumstances: 1-specific immune-related genes, 2-maternal risk factor (ex. gestational diabetes and/or obesity), and 3-maternal immune activation (MIA) during pregnancy (ex. viral infection) (13). A common feature associated with these three scenarios is the altered number of synapses in the fetus leading to deficits during neurodevelopment.
Global expression differences in genes related to inflammatory response in ASD individuals were reported by several studies (14–16). Transcriptome analysis of ASD post-mortem brains revealed a close association between ASD and the genes related to glial cell activation and genes belonging to immune and inflammatory categories (17). The MET gene (proto-oncogene receptor, tyrosine kinase) known to regulate cell immune functions amongst other roles (18) is the immune gene most closely associated with ASD (19), coding for a receptor that binds to hepatocyte growth factor (HGF). Single nucleotide polymorphisms (SNPs) were described at high incidence in ASD and reduced expression of aberrant MET was shown in post-mortem brains of ASD individuals (20). SNPs generating deficiencies of the complement component 4 (C4) gene function, located in the MHC region, have also been associated with ASD (21, 22). Additional components of the complement cascade (C3, Masp1, Masp2, C3aR, and C5aR) participate in microglial function and neuronal migration and are potentially implicated in altered cortical development in ASD (23, 24).
Maternal history of increased inflammation, such as development of eczema, psoriasis, and asthma was correlated with a 20–40% increased risk for both ASD and other developmental disorders (25). Other risk factors apart from the immune deficiencies mentioned are gestational diabetes, obesity, and aging that were linked to an increased chance of a child to developing ASD (26, 27). Lastly, acquiring MIA due to a viral infection during pregnancy shows evidence of an additional increased risk for developing ASD (28).
Anomalies in immune signaling pathways are reported for ASD (29). Specifically, altered cytokine profiles and immunoglobulin G (IgG) levels were associated with intensified cellular immunity and general amplified neuroinflammation. Detection of misbalanced expression of inflammatory cytokines (IL-6, TNFα, TGFβ, IL-17, and IL-2) was reported in ASD patients' blood (30). Unfortunately, due to the complexity of this disorder, none of these alterations resulted in a biomarker panel for predicting ASD prognosis. In this review we will highlight the role of inflammation in ASD focusing on the data obtained from stem cell modeling.
Neurodevelopmental polygenic disorders, such as ASD are notably attractive for the application of stem cell technology as early hallmarks of neuronal development can be dissected in vitro and patients' unique genetic background can be recapitulated (31). Embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) have the ability to generate functional human neuronal and glial cells in vitro, and can self-organize into 3D brain organoid cultures (32) (Figure 1). iPSCs recapitulate aspects of neuronal development process while preserving the genetic background from ASD patients and thus are more frequently used to model idiopathic conditions. Additionally, isogenic lines can be developed to understand the impact of a specific mutation in different genetic backgrounds. While gene-editing technology is ideal for understanding the function of a single gene, the results are not always generalizable to the autistic condition. In that case, having iPSC lines from a cohort of patients with a specific endophenotype (ex. macrocephaly) can be informative for understanding ASD in vitro phenotypes. Unfortunately for idiopathic polygenic ASD, isogenic lines are not always a viable option.
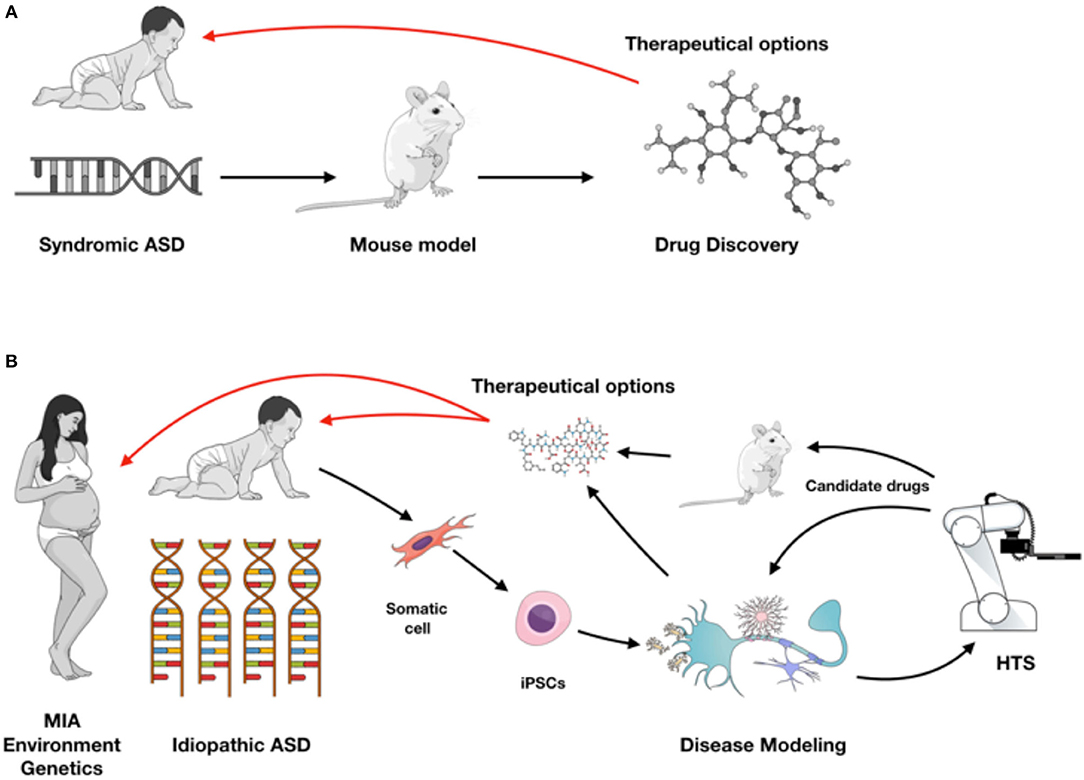
Figure 1. (A) Syndromic ASD historically has been studied using rodent models leading to drug discovery for therapeutic approaches (red arrow). (B) With iPSC modeling both syndromic and idiopathic ASD forms can be investigated in different cell types from the nervous system, or in a complex co-culture model system and used for high-throughput screening (HTS). Validation of target compounds can be performed on iPSC-derived and rodent models, providing therapeutic possibilities as early as during brain development (red arrows). Figure created in the Mind the Graph platform with subscription purchased by the authors.
Despite being a polygenic disorder, there appears to be converging phenotypes in ASD. A common finding from ASD studies reporting the generation of iPSC-derived neurons from syndromic or idiopathic ASD is altered synaptic activity (33–41). iPSC technology allows for the interrogation of the specific neurobiological foundations underlying common synaptic defects and synaptopathy in the context of neurodevelopmental disorders such ASD (42).
Aiming to recapitulate clinical post-mortem findings in neurons, one group using gene expression analyses on three-dimensional cultures derived from iPSCs showed up-regulation of genes involved in cell proliferation, neuronal differentiation, and synaptic formation (34). In this study, Mariani et al. (34) suggested that overexpression of the gene FOXG1 can generate an overproduction of GABAergic neurons, which can cause increase of brain volume and imbalance of excitation and inhibition systems in the developing cortex in ASD.
Increased proliferation of iPSC-derived neural progenitors from ASD individuals with enlarged brains was also reported by Marchetto et al. (35). Importantly, the authors observed a significant correlation between ASD patients' brain volumes in vivo and cell proliferation rates in vitro, indicating that iPSC-derived neural cells can be used as a proxy to infer specific patients' metrics (i.e., brain volume). Marchetto et al. (35) also showed a significant reduction in the density of excitatory synapses in ASD iPSC-derived neurons and decreased neuronal bursting. To date, these findings suggest that despite the overgrowth of the brain size in these young children, their neurons are immature or unhealthy.
Though neuronal phenotypes have been explored in more depth in ASD; glial cells play an important role in the disease as well. A recent study using iPSC-derived neurons and iPSC-derived astrocytes from idiopathic ASD individuals demonstrated that ASD astrocytes influence neuronal morphology and function, evidenced by co-culture experiments (43). Remarkably, when astrocytes from neurotypical individuals were co-cultured with ASD neurons they improved morphological and functional features that were not observed when ASD neurons were co-cultured with ASD astrocytes. The researchers identified interleukin-6 (IL-6) as a key cytokine responsible for the effect on neuronal activity by showing synaptogenesis recovery in ASD neurons after blocking IL-6 release in ASD astrocytes. The researchers also hypothesized that an increased expression of IL-6 in ASD astrocytes might be one of the triggers of the observed neuronal defects (43). Interestingly, elevated levels of IL-6, along with other inflammatory cytokines, were reported in the brains and blood of autistic subjects and IL-6 has been found to be a key mediator in MIA models (30, 44, 45).
RTT is a monogenetic progressive neurologic disorder that shares proposed mechanistic and core symptoms with ASD, with mutations in the X-linked gene MeCP2 occurring in ~90% of patients with RTT (46–48). RTT patients are predominantly female, as affected male patients are rare, and those who survive usually die at early ages (49–51). RTT patients have progressive neurological abnormalities starting at 6–18 months after birth. Following a period of progressive neurological damage there is deceleration of head growth, severe motor abnormalities, and loss of language skills. Along with the RTT onset of development stagnation comes impaired motor function, stereotypic hand wringing, hypotonia, seizures, autistic behavior, encephalopathy, and respiratory dysfunction (46, 49). Amir et al. identified mutations in methyl CpG binding protein 2 (MeCP2) as the primary genetic cause of RTT (47) and Chahrour et al. showed that MeCP2 binding can both repress and activate transcription (52).
Reduced MeCP2 expression is also found in forebrain post-mortem tissue from idiopathic ASD individuals, suggesting similar epigenetic dysregulation in some cases of idiopathic ASD and RTT (48). Human iPSC-derived neural cells from RTT patients show fewer synapses, reduced soma size and dendritic spine density, altered calcium signaling and decreased spontaneous excitatory post-synaptic currents (33, 46). Marchetto et al. showed rescue in synapses phenotype of RTT after treatment with IGF-1 (Insulin Growth Factor Like-1) compound, which is now in clinical trials for RTT (33). Increased dosage of MeCP2 protein also results in severe neurodevelopmental delay with epilepsy and autistic behavior. Interestingly, iPSC-derived neurons from patients with the MECP2 duplication syndrome show increased synaptogenesis and dendritic complexity resulting on abnormal network synchronization compared to controls (37). These results indicate that MECP2 gene dosage is critical for proper human neurodevelopment.
Neurons were thought to be the most relevant cell type for RTT pathology due to the high expression levels of MeCP2 protein in these cells. Based on previous data showing the non-cell autonomous effect in rodents (53), a group in 2014 was able to confirm the negative effect of MeCP2-mutant astrocytes in neuronal morphology using iPSCs. Furthermore, IGF-1 and GPE (IGF-1 cleaved peptide) was used to partially rescue the neuronal deficits caused by mutant astrocytes (54). To date, the direct role of RTT-derived astrocytes in inflammation has not been extensively explored.
Several studies demonstrated that the immune system is involved in RTT in early life. Microglia activation and/or proliferation and defective BDNF signaling is described in RTT (55). Maezawa et al. proposed that RTT resting microglia are sensitive to both immunological stimuli and neuronal/astrocytic signals causing neuroinflammation and, consequently, affecting brain development (56, 57).
CDKL-5 is a kinase protein highly expressed in neurons, but its specific function inside the cell is poorly known (58). Mutations in X-linked CDKL5 gene have been identified in the early onset seizures variant of RTT and male X-linked epileptic encephalopathy. CDKL5 expression is present in neurons suggesting an important role in neuronal development. In certain brain regions, MeCP2 protein can regulate the expression of CDKL5 gene and in turn CDKL5 can phosphorylate MeCP2. Downregulation of CDKL5 expression inhibits neurite outgrowth and maturation of the neuronal network in rodents (59). iPSC-derived neurons from patients with CDKL5 mutations exhibit aberrant dendritic spines, suggesting a common function of CDKL5 in mice and humans (58, 60).
The immune system misbalance in Rett syndrome (RTT) can be explained in part by impaired regulation of the inflammatory system. In rodent models MeCP2 regulates gene expression in microglia and macrophage (61). Leoncini et al. (62) showed that cytokine levels of RTT patients are misregulated in the blood and that there are differences in cytokine profiling depending on the RTT mutation (MECP2 vs. CDKL5). The researchers also pointed out that omega-3 polyunsaturated fatty acids (PUFAs) partially counterbalanced the cytokine changes and the aberrant redox homeostasis and ameliorated the inflammatory status (62).
Patients with FXS show intellectual disability, hyperactivity and a range of autistic-like behaviors. FXS is characterized by the absence of expression of the fragile X mental retardation 1 (FMR1) gene due to CGG trinucleotide repeat expansion in the 5′ untranslated region of FMR1. This aberrant expression leads to hypermethylation and gene silencing, leading to the decrease or absence of FMRP (fragile X mental retardation protein) (46, 63).
FMRP is a selective RNA-binding protein that regulates the translation of many genes at synaptic sites. It was shown that the lack of FMRP leads to the excess and dysregulation of mRNAs translation, which leads to alterations in the synapse function, increased numbers of spines and neuronal overexcitability (64). A recent study using a Drosophila Fragile-X model showed phagocytosis defects in innate immune cells (65). Loss of FMR1 also causes aberrant differentiation in human iPSC-derived neural progenitor cells, particularly a marked induction of the astrocyte marker glial fibrillary acidic protein (GFAP) (66). In parallel, research also shows increased astrocyte activation in FMR1 knockout mouse cerebellum (67), indicating a possible increase in inflammation on both human and mouse models. Although cytokine imbalance was seen with increased IL-6 levels in mouse models (68), no iPSC-derived study has recapitulated these glial findings in vitro. Using ASD and Fragile-X iPSC-derived neurons, a study compared the molecular pathways between these two disorders by global gene expression and DNA methylation profiling, and identified several cytokine molecules, BMP7, and HLA-related abnormalities with aberrant FMRP1 expression (69).
Mouse modeling is a crucial tool for autism spectrum studies with the generation of genetically modified strains focusing on understanding the basic features of abnormal neurodevelopment in ASD.
A relevant environmental risk factor known to recapitulate aspects of ASD pathology was extensively studied by Patterson and his collaborators (70). His group induced an acute inflammatory response on expectant pregnant rodents, causing maternal immune activation (MIA) leading to autistic phenotypes in the offspring (70). The mechanism for the development of autistic traits was later correlated with the release of IL-6, amongst other cytokine misbalances especially in CD4+ T cells. The offspring from MIA models displayed general alteration of their immune profile and function, and interestingly, bone marrow transplantation of MIA-derived mice into irradiated control mice did not recapitulate the MIA phenotypes illustrating the importance of the peripheral system context in the immune system activation (71). Using CRE-induced models for local depletion of IL-6 receptors, Patterson's group showed that the placental IL-6 signaling is directly involved in fetal brain development and behavior and when IL-6 receptors were depleted in the placenta the MIA model had no effect on the offspring (72). Recent results from Cho et al. (73) also implicate the maternal interleukin-17a pathway in mice promoting autism-like phenotypes in the offspring. Their data suggest that therapeutic targeting of Th17 cells in susceptible pregnant mothers could potentially reduce the likelihood of bearing children with inflammation-induced ASD-like phenotypes (73).
Another relevant study proposed the hypothesis that blood marrow transplantations could improve the phenotype of mice with syndromic forms of ASD, such as RTT (74). The concept was based on the idea that healthy microglia would rescue RTT phenotypes in mouse models. While this is an appealing concept, several groups attempted to repeat the experiment using similar methodologies and the results have not been confirmed (75). Although microglia was not necessarily the culprit in the rescue of MeCP2-deficient phenotype, it is anticipated that this cell type plays an important role in the disease, particularly given its ability for synaptic pruning (76). Future studies will be crucial to consider the potential use of microglia replacement as a therapeutic strategy for ASD.
Mouse models lacking microglial fracktalkine receptor gene (CX3CR1) displayed increased microglial IL-1β resulting in an enhanced tauopathy, immature synapses throughout the cortex and connectivity imbalance with ASD-like behaviors (77). Blocking IL-1β/p38 MAPK pathways significantly improved the mouse phenotype and could be a promising therapeutic option (78). Interestingly, mouse models for RTT using MeCP2 KO mice crossed with mice with CX3CR1 mutations recovered the MeCP2 deficient phenotype, suggesting that blocking the fracktalkine receptor is a possible therapeutic approach for RTT (79).
In 2011, a group led by Gail Mandel demonstrated the role for glia in the progression of RTT (53). Using MeCP2 KO animals and astrocyte-specific GFAP promoter controlling MeCP2 expression, they showed that astrocyte depletion of MeCP2 was detrimental to neurons in a non-cell autonomous fashion. Interestingly, the re-expression of MeCP2 in GFAP-driven cells was sufficient to rescue the RTT phenotype, and systemic delivery of MeCP2 was used as a therapeutic approach for female mouse models (80). Recent work demonstrated that MeCP2 expression in astrocytes affects glutamatergic synapses, highlighting the importance of astrocytes to ensure proper synaptic connectivity (81). Using primary rodent cells another group found that MeCP2-mutant astrocytes had an exaggerated inflammatory and oxidative stress response to LPS (Lipopolysaccharide) stimulation when compared to control cells, suggesting anti-inflammatory approaches for disease treatment (82).
All current in vivo and in vitro models have increased our overall mechanistic knowledge of ASD, however the complexity of idiopathic autism and the influence of epigenetic and environmental factors poses a challenge to modeling non-syndromic forms using rodent models. Furthermore, when attempting to study aspects of neuroinflammation, where specific cytokines can play a crucial role in the disorder and are species-specific (83), humanized models can be instrumental to obtain therapeutically relevant information. As iPSCs emerge as an important in vitro model for translational research, current limitations include the restricted complexity for cytokine signaling and the absence of body systemic interaction. Future improvements to iPSC technology will incorporate circulating blood and mature cell types to the current model systems.
Increasing evidence supports the idea of immune system activation in ASD. iPSCs are crucial for a better understanding of complex polygenic diseases with heritable and sporadic forms, such as ASD in order to develop novel biomarkers and identify targets for future therapeutics (35). Deriving brain cells from iPSCs can in fact provide a unique opportunity to explore and identify early phenotypes in autism in opposition to secondary phenotypes that are a downstream consequence of the pathology. Moreover, iPSC technology is particularly valuable over the two most used research approaches to study and understand brain biology and pathology, namely post-mortem samples and mouse models. Post-mortem tissue has inherent limitations for studying a neurodevelopmental disease, mainly because it cannot be manipulated to test experimental hypotheses and provides only a brief snapshot in disease progression (84). Animal models can be genetically and pharmacologically manipulated, however, when it comes to behavioral, affective and cognitive changes, animal models may not always recapitulate the human phenotype (84, 85). Ideally for a complex disorder, such as ASD one would take advantage of multiple models, using them as a base to build the background knowledge of the mechanism of the disorder, studying each individual player (i.e., neuron, glia) and as a complex niche (in vivo, in co-culture or brain organoid systems). The next step would be developing a high-throughput screening (HTS) system capable of identifying disease biomarkers and potentially detecting compounds that rescue a phenotype. Since ASD is a complex developmental disorder, studying iPSC-derived fetal-like cells is ideal for establishing a therapeutic approach as early as during gestation time (Figure 1). Recent research indicates a relevant role for the gut microbiota in autism (86), specially regarding inflammatory components (87–89). It would be interesting to see upcoming technologies, such as “organs-on-a-chip” that can integrate brain development with gut signaling to an in vitro platform.
iPSC technology is also a remarkable scientific tool to test compounds that could be used in the clinic for ASD individuals. The grouth factor IGF1 was first tested in RTT mouse models (90) and then iPSC from ASD patients (33, 35) and it is now in clinical trials for syndromic and idiopathic forms of ASD1, 2, 3, 4, 5 (91, 92). The compound Luteolin (93) attenuates IL-6-mediated astrogliosis in human iPSC-derived neural aggregates and was proposed as a candidate preventive substance for maternal immune activation-induced abnormalities (94). Remarkably, Luteolin dietary intake improves ASD symptoms and reduces serum levels of TNF and IL-6 (95).
In summary, iPSC technology is an invaluable tool for asking questions about human-specific neuroinflammation mechanisms, such as the ones observed in syndromic and non-syndromic forms of ASD. Importantly, iPSCs can generate a platform for the discovery of new anti-inflammatory compounds that are tailored to the ASD-specific genetic background and neuropathological phenotypes and offer potential therapeutic options for both onset and progression of the disorder.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to acknowledge the support from the National Institutes of Health (NIH) grant number R03 MH115426-01A1 (MM).
1. ^A Pilot Treatment Study of Insulin-Like Growth Factor-1 (IGF-1) in Autism Spectrum Disorder. Available online at: https://ClinicalTrials.gov/show/NCT01970345
2. ^A Safety Study of NNZ-2566 in Patients With Fragile X Syndrome. Available online at: https://ClinicalTrials.gov/show/NCT01894958
3. ^Treatment of Rett Syndrome With Recombinant Human IGF-1. Available online at: https://ClinicalTrials.gov/show/NCT01777542
4. ^Clinical Trial in 22q13 Deletion Syndrome(Phelan-McDermid Syndrome). Available online at: https://ClinicalTrials.gov/show/NCT01525901
5. ^Treatment of Rett Syndrome With rhIGF-1 (Mecasermin rDNA Injection). Available online at: https://ClinicalTrials.gov/show/NCT01253317
1. Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S, et al. Patches of disorganization in the neocortex of children with autism. N Engl J Med. (2014) 370:1209–19. doi: 10.1056/NEJMoa1307491
2. Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron (2012) 74:285–99. doi: 10.1016/j.neuron.2012.04.009
3. Baio J, Wiggins L, Christensen DL, Maenner MJ, Daniels J, Warren Z, et al. Prevalence of autism spectrum disorder among children aged 8 years–autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveill Summ. (2018) 67:1–23. doi: 10.15585/mmwr.ss6706a1
4. Piven J, Elison JT, Zylka MJ. Toward a conceptual framework for early brain and behavior development in autism. Mol Psychiatry (2018) 23:165. doi: 10.1038/mp.2017.212
5. Geschwind DH, State MW. Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol. (2015) 14:1109–20. doi: 10.1016/S1474-4422(15)00044-7
6. Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science (2007) 316:445–9. doi: 10.1126/science.1138659
7. Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Curr Opin Genet Dev. (2012) 22:229–37. doi: 10.1016/j.gde.2012.03.002
8. de la Torre-Ubieta L, Won H, Stein JL, Geschwind DH. Advancing the understanding of autism disease mechanisms through genetics. Nat Med. (2016) 22:345–61. doi: 10.1038/nm.4071
9. Volk HE, Kerin T, Lurmann F, Hertz-Picciotto I, McConnell R, Campbell DB. Autism spectrum disorder: interaction of air pollution with the MET receptor tyrosine kinase gene. Epidemiology (2014) 25:44–7. doi: 10.1097/EDE.0000000000000030
10. Onore C, Careaga M, Ashwood P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav Immun. (2012) 26:383–92. doi: 10.1016/j.bbi.2011.08.007
11. Scott O, Shi D, Andriashek D, Clark B, Goez HR. Clinical clues for autoimmunity and neuroinflammation in patients with autistic regression. Dev Med Child Neurol. (2017) 59:947–51. doi: 10.1111/dmcn.13432
12. Mazur-Kolecka B, Cohen IL, Gonzalez M, Jenkins EC, Kaczmarski W, Brown WT, et al. Autoantibodies against neuronal progenitors in sera from children with autism. Brain Dev. (2014) 36:322–9. doi: 10.1016/j.braindev.2013.04.015
13. Estes ML, McAllister AK. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat Rev Neurosci. (2015) 16:469–86. doi: 10.1038/nrn3978
14. Lanz TA, Guilmette E, Gosink MM, Fischer JE, Fitzgerald LW, Stephenson DT, et al. Transcriptomic analysis of genetically defined autism candidate genes reveals common mechanisms of action. Mol Autism (2013) 4:45. doi: 10.1186/2040-2392-4-45
15. Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J, et al. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun. (2014) 5:5748. doi: 10.1038/ncomms6748
16. Ansel A, Rosenzweig JP, Zisman PD, Melamed M, Gesundheit B. Variation in gene expression in autism spectrum disorders: an extensive review of transcriptomic studies. Front Neurosci. (2016) 10:601. doi: 10.3389/fnins.2016.00601
17. Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature (2011) 474:380–4. doi: 10.1038/nature10110
18. Imamura R, Matsumoto K. Hepatocyte growth factor in physiology and infectious diseases. Cytokine (2017) 98:97–106. doi: 10.1016/j.cyto.2016.12.025
19. Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C, Trillo S, et al. A genetic variant that disrupts MET transcription is associated with autism. Proc Natl Acad Sci USA. (2006) 103:16834–9. doi: 10.1073/pnas.0605296103
20. Peng Y, Huentelman M, Smith C, Qiu S. MET receptor tyrosine kinase as an autism genetic risk factor. Int Rev Neurobiol. (2013) 113:135–65. doi: 10.1016/B978-0-12-418700-9.00005-8
21. Warren RP, Burger RA, Odell D, Torres AR, Warren WL. Decreased plasma concentrations of the C4B complement protein in autism. Arch Pediatr Adolesc Med. (1994) 148:180–3. doi: 10.1001/archpedi.1994.02170020066011
22. Odell D, Maciulis A, Cutler A, Warren L, McMahon WM, Coon H, et al. Confirmation of the association of the C4B null allelle in autism. Hum Immunol. (2005) 66:140–5. doi: 10.1016/j.humimm.2004.11.002
23. Gorelik A, Sapir T, Haffner-Krausz R, Olender T, Woodruff TM, Reiner O. Developmental activities of the complement pathway in migrating neurons. Nat Commun. (2017) 8:15096. doi: 10.1038/ncomms15096
24. Campbell DB, D'Oronzio R, Garbett K, Ebert PJ, Mirnics K, Levitt P, et al. Disruption of cerebral cortex MET signaling in autism spectrum disorder. Ann Neurol. (2007) 62:243–50. doi: 10.1002/ana.21180
25. Croen LA, Qian Y, Ashwood P, Daniels JL, Fallin D, Schendel D, et al. Family history of immune conditions and autism spectrum and developmental disorders: findings from the study to explore early development. Autism Res. (2018). doi: 10.1002/aur.1979
26. Keil A, Daniels JL, Forssen U, Hultman C, Cnattingius S, Söderberg KC, et al. Parental autoimmune diseases associated with autism spectrum disorders in offspring. Epidemiology (2010) 21:805–8. doi: 10.1097/EDE.0b013e3181f26e3f
27. Atladóttir HO, Pedersen MG, Thorsen P, Mortensen PB, Deleuran B, Eaton WW, et al. Association of family history of autoimmune diseases and autism spectrum disorders. Pediatrics (2009) 124:687–94. doi: 10.1542/peds.2008-2445
28. Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA, et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol. (2014) 10:643–60. doi: 10.1038/nrneurol.2014.187
29. Goines P, Van de Water J. The immune system's role in the biology of autism. Curr Opin Neurol. (2010) 23:111–7. doi: 10.1097/WCO.0b013e3283373514
30. Eftekharian MM, Ghafouri-Fard S, Noroozi R, Omrani MD, Arsang-Jang S, Ganji M, et al. Cytokine profile in autistic patients. Cytokine (2018) 108:120–6. doi: 10.1016/j.cyto.2018.03.034
31. Ross CA, Akimov SS. Human-induced pluripotent stem cells: potential for neurodegenerative diseases. Hum Mol Genet. (2014) 23:R17–26. doi: 10.1093/hmg/ddu204
32. Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature (2013) 501:373–9. doi: 10.1038/nature12517
33. Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell (2010) 143:527–39. doi: 10.1016/j.cell.2010.10.016
34. Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell (2015) 162:375–90. doi: 10.1016/j.cell.2015.06.034
35. Marchetto MC, Belinson H, Tian Y, Freitas BC, Fu C, Vadodaria K, et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry (2017) 22:820–35. doi: 10.1038/mp.2016.95
36. Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature (2013) 503:267–71. doi: 10.1038/nature12618
37. Nageshappa S, Carromeu C, Trujillo CA, Mesci P, Espuny-Camacho I, Pasciuto E, et al. Altered neuronal network and rescue in a human MECP2 duplication model. Mol Psychiatry (2016) 21:178–88. doi: 10.1038/mp.2015.128
38. Sheridan SD, Theriault KM, Reis SA, Zhou F, Madison JM, Daheron L, et al. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS ONE (2011) 6:e26203. doi: 10.1371/journal.pone.0026203
39. Cheung AY, Horvath LM, Grafodatskaya D, Pasceri P, Weksberg R, Hotta A, et al. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum Mol Genet. (2011) 20:2103–15. doi: 10.1093/hmg/ddr093
40. Paşca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Paşca AM, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. (2011) 17:1657–62. doi: 10.1038/nm.2576
41. Griesi-Oliveira K, Acab A, Gupta AR, Sunaga DY, Chailangkarn T, Nicol X, et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry (2015) 20:1350–65. doi: 10.1038/mp.2014.141
42. Shen X, Yeung HT, Lai KO. Application of human-induced pluripotent stem cells (hiPSCs) to study synaptopathy of neurodevelopmental disorders. Dev Neurobiol. (2018). doi: 10.1002/dneu.22644
43. Russo FB, Freitas BC, Pignatari GC, Fernandes IR, Sebat J, Muotri AR, et al. Modeling the interplay between neurons and astrocytes in autism using human induced pluripotent stem cells. Biol Psychiatry (2018) 83:569–78. doi: 10.1016/j.biopsych.2017.09.021
44. Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li XM, et al. Elevated immune response in the brain of autistic patients. J Neuroimmunol. (2009) 207:111–6. doi: 10.1016/j.jneuroim.2008.12.002
45. Deverman BE, Patterson PH. Cytokines and CNS development. Neuron (2009) 64:61–78. doi: 10.1016/j.neuron.2009.09.002
46. Acab A, Muotri AR. The use of induced pluripotent stem cell technology to advance autism research and treatment. Neurotherapeutics (2015) 12:534–45. doi: 10.1007/s13311-015-0354-x
47. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. (1999) 23:185–8. doi: 10.1038/13810
48. Liao W, Gandal MJ, Ehrlichman RS, Siegel SJ, Carlson GC. MeCP2+/− mouse model of RTT reproduces auditory phenotypes associated with Rett syndrome and replicate select EEG endophenotypes of autism spectrum disorder. Neurobiol Dis. (2012) 46:88–92. doi: 10.1016/j.nbd.2011.12.048
49. Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron (2007) 56:422–37. doi: 10.1016/j.neuron.2007.10.001
50. Schanen C, Francke U. A severely affected male born into a Rett syndrome kindred supports X-linked inheritance and allows extension of the exclusion map. Am J Hum Genet. (1998) 63:267–9. doi: 10.1086/301932
51. Villard L, Kpebe A, Cardoso C, Chelly PJ, Tardieu PM, Fontes M. Two affected boys in a Rett syndrome family: clinical and molecular findings. Neurology (2000) 55:1188–93. doi: 10.1212/WNL.55.8.1188
52. Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science (2008) 320:1224–9. doi: 10.1126/science.1153252
53. Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, et al. A role for glia in the progression of Rett's syndrome. Nature (2011) 475:497–500. doi: 10.1038/nature10214
54. Williams EC, Zhong X, Mohamed A, Li R, Liu Y, Dong Q, et al. Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wild-type neurons. Hum Mol Genet. (2014) 23:2968–80. doi: 10.1093/hmg/ddu008
55. Theoharides TC, Athanassiou M, Panagiotidou S, Doyle R. Dysregulated brain immunity and neurotrophin signaling in Rett syndrome and autism spectrum disorders. J Neuroimmunol. (2015) 279:33–8. doi: 10.1016/j.jneuroim.2014.12.003
56. Maezawa I, Calafiore M, Wulff H, Jin LW. Does microglial dysfunction play a role in autism and Rett syndrome? Neuron Glia Biol. (2011) 7:85–97. doi: 10.1017/S1740925X1200004X
57. Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. (2010) 30:5346–56. doi: 10.1523/JNEUROSCI.5966-09.2010
58. Amenduni M, De Filippis R, Cheung AY, Disciglio V, Epistolato MC, Ariani F, et al. iPS cells to model CDKL5-related disorders. Eur J Hum Genet. (2011) 19:1246–55. doi: 10.1038/ejhg.2011.131
59. Chen Q, Zhu YC, Yu J, Miao S, Zheng J, Xu L, et al. CDKL5, a protein associated with rett syndrome, regulates neuronal morphogenesis via Rac1 signaling. J Neurosci. (2010) 30:12777–86. doi: 10.1523/JNEUROSCI.1102-10.2010
60. Ricciardi S, Ungaro F, Hambrock M, Rademacher N, Stefanelli G, Brambilla D, et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat Cell Biol. (2012) 14:911–23. doi: 10.1038/ncb2566
61. Cronk JC, Derecki NC, Ji E, Xu Y, Lampano AE, Smirnov I, et al. Methyl-CpG binding protein 2 regulates microglia and macrophage gene expression in response to inflammatory stimuli. Immunity (2015) 42:679–91. doi: 10.1016/j.immuni.2015.03.013
62. Leoncini S, De Felice C, Signorini C, Zollo G, Cortelazzo A, Durand T, et al. Cytokine dysregulation in MECP2- and CDKL5-related rett syndrome: relationships with aberrant redox homeostasis, inflammation, and omega-3 PUFAs. Oxid Med Cell Longev. (2015) 2015:421624. doi: 10.1155/2015/421624
63. Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell (2010) 6:407–11. doi: 10.1016/j.stem.2010.04.005
64. Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron (2008) 60:201–14. doi: 10.1016/j.neuron.2008.10.004
65. O'Connor RM, Stone EF, Wayne CR, Marcinkevicius EV, Ulgherait M, Delventhal R, et al. A Drosophila model of Fragile X syndrome exhibits defects in phagocytosis by innate immune cells. J Cell Biol. (2017) 216:595–605. doi: 10.1083/jcb.201607093
66. Sunamura N, Iwashita S, Enomoto K, Kadoshima T, Isono F. Loss of the fragile X mental retardation protein causes aberrant differentiation in human neural progenitor cells. Sci Rep. (2018) 8:11585. doi: 10.1038/s41598-018-30025-4
67. Pacey LK, Guan S, Tharmalingam S, Thomsen C, Hampson DR. Persistent astrocyte activation in the fragile X mouse cerebellum. Brain Behav. (2015) 5:e00400. doi: 10.1002/brb3.400
68. Krasovska V, Doering LC. Regulation of IL-6 secretion by astrocytes via TLR4 in the Fragile X mouse model. Front Mol Neurosci. (2018) 11:272. doi: 10.3389/fnmol.2018.00272
69. Boland MJ, Nazor KL, Tran HT, Szücs A, Lynch CL, Paredes R, et al. Molecular analyses of neurogenic defects in a human pluripotent stem cell model of fragile X syndrome. Brain (2017) 140:582–98. doi: 10.1093/brain/aww357
70. Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun. (2012) 26:607–16. doi: 10.1016/j.bbi.2012.01.011
71. Hsiao EY, Patterson PH. Placental regulation of maternal-fetal interactions and brain development. Dev Neurobiol. (2012) 72:1317–26. doi: 10.1002/dneu.22045
72. Wu WL, Hsiao EY, Yan Z, Mazmanian SK, Patterson PH. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav Immun. (2017) 62:11–23. doi: 10.1016/j.bbi.2016.11.007
73. Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science (2016) 351:933–9. doi: 10.1126/science.aad0314
74. Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG, et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature (2012) 484:105–9. doi: 10.1038/nature10907
75. Wang J, et al. Wild-type microglia do not reverse pathology in mouse models of Rett syndrome. Nature (2015) 521:E1–4. doi: 10.1038/nature14444
76. Schafer DP, Heller CT, Gunner G, Heller M, Gordon C, Hammond T, et al. Microglia contribute to circuit defects in Mecp2 null mice independent of microglia-specific loss of Mecp2 expression. Elife (2016) 5:e15224. doi: 10.7554/eLife.15224
77. Limatola C, Ransohoff RM. Modulating neurotoxicity through CX3CL1/CX3CR1 signaling. Front Cell Neurosci. (2014) 8:229. doi: 10.3389/fncel.2014.00229
78. Lee S, Xu G, Jay TR, Bhatta S, Kim KW, Jung S, et al. Opposing effects of membrane-anchored CX3CL1 on amyloid and tau pathologies via the p38 MAPK pathway. J Neurosci. (2014) 34:12538–46. doi: 10.1523/JNEUROSCI.0853-14.2014
79. Horiuchi M, Smith L, Maezawa I, Jin LW. CX3CR1 ablation ameliorates motor and respiratory dysfunctions and improves survival of a Rett syndrome mouse model. Brain Behav Immun. (2017) 60:106–16. doi: 10.1016/j.bbi.2016.02.014
80. Garg SK, Lioy DT, Cheval H, McGann JC, Bissonnette JM, Murtha MJ, et al. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci. (2013) 33:13612–20. doi: 10.1523/JNEUROSCI.1854-13.2013
81. Rakela B, Brehm P, Mandel G. Astrocytic modulation of excitatory synaptic signaling in a mouse model of Rett syndrome. Elife (2018) 7:e31629. doi: 10.7554/eLife.31629
82. Nance E, Kambhampati SP, Smith ES, Zhang Z, Zhang F, Singh S, et al. Dendrimer-mediated delivery of N-acetyl cysteine to microglia in a mouse model of Rett syndrome. J Neuroinflammation (2017) 14:252. doi: 10.1186/s12974-017-1004-5
83. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
84. Habela CW, Song H, Ming GL. Modeling synaptogenesis in schizophrenia and autism using human iPSC derived neurons. Mol Cell Neurosci. (2016) 73:52–62. doi: 10.1016/j.mcn.2015.12.002
85. Jones CA, Watson DJ, Fone KC. Animal models of schizophrenia. Br J Pharmacol. (2011) 164:1162–94. doi: 10.1111/j.1476-5381.2011.01386.x
86. Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell (2013) 155:1451–63. doi: 10.1016/j.cell.2013.11.024
87. Sharon G, Sampson TR, Geschwind DH, Mazmanian SK. The central nervous system and the gut microbiome. Cell (2016) 167:915–32. doi: 10.1016/j.cell.2016.10.027
88. Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. (2009) 9:313–23. doi: 10.1038/nri2515
89. Doenyas C. Gut microbiota, inflammation, and probiotics on neural development in autism spectrum disorder. Neuroscience (2018) 374:271–86. doi: 10.1016/j.neuroscience.2018.01.060
90. Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, et al. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci USA. (2009) 106:2029–34. doi: 10.1073/pnas.0812394106
91. Khwaja OS, Ho E, Barnes KV, O'Leary HM, Pereira LM, Finkelstein Y, et al. Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc Natl Acad Sci USA. (2014) 111:4596–601. doi: 10.1073/pnas.1311141111
92. Kolevzon A, Bush L, Wang AT, Halpern D, Frank Y, Grodberg D, et al. A pilot controlled trial of insulin-like growth factor-1 in children with Phelan-McDermid syndrome. Mol Autism (2014) 5:54. doi: 10.1186/2040-2392-5-54
93. Jeon IH, Kim HS, Kang HJ, Lee HS, Jeong SI, Kim SJ, et al. Anti-inflammatory and antipruritic effects of luteolin from Perilla (P. frutescens L.) leaves. Molecules (2014) 19:6941–51. doi: 10.3390/molecules19066941
94. Zuiki M, Chiyonobu T, Yoshida M, Maeda H, Yamashita S, Kidowaki S, et al. Luteolin attenuates interleukin-6-mediated astrogliosis in human iPSC-derived neural aggregates: a candidate preventive substance for maternal immune activation-induced abnormalities. Neurosci Lett. (2017) 653:296–301. doi: 10.1016/j.neulet.2017.06.004
Keywords: autism spectrum disorder (ASD), disease modeling, neuroinflammation, neurodevelopmental disorders, induced pluripotent stem (iPS) cells
Citation: Freitas BC, Mei A, Mendes APD, Beltrão-Braga PCB and Marchetto MC (2018) Modeling Inflammation in Autism Spectrum Disorders Using Stem Cells. Front. Pediatr. 6:394. doi: 10.3389/fped.2018.00394
Received: 25 September 2018; Accepted: 29 November 2018;
Published: 12 December 2018.
Edited by:
Sofia Beatriz Lizarraga, University of South Carolina, United StatesReviewed by:
Steven Sloan, Emory University, United StatesCopyright © 2018 Freitas, Mei, Mendes, Beltrão-Braga and Marchetto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Carolina Marchetto, bWFyY2hldHRvQHNhbGsuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.