 Pui Y. Lee
Pui Y. Lee
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pediatr. , 18 October 2018
Sec. Pediatric Immunology
Volume 6 - 2018 | https://doi.org/10.3389/fped.2018.00282
This article is part of the Research Topic Childhood Vasculitis View all 19 articles
Deficiency of adenosine deaminase 2 (DADA2) is a monogenic form of systemic vasculopathy that often presents during early childhood. Linked to biallelic mutations in ADA2 (previously CECR1), DADA2 was initially described as a syndrome of recurrent fever, livedo racemosa, early-onset strokes, and peripheral vasculopathy that resembles polyarteritis nodosum. However, the wide spectrum of clinical findings and heterogeneity of disease, even among family members with identical mutations, is increasingly recognized. Evidence of systemic inflammation and vasculopathy is not uniformly present in DADA2 patients and some can remain asymptomatic through adulthood. Humoral immunodeficiency characterized by low immunoglobulin levels and increased risk of infection is another common feature of DADA2. Variable cytopenias including pure red cell aplasia that mimics Diamond-Blackfan anemia can also be primary manifestations of DADA2. How defects in a single gene translate into these heterogeneous presentations remains to be answered. In this review, we will summarize lessons learned from the pleiotropic clinical manifestations of DADA2.
Vasculitis is group of disorders characterized by inflammation of blood vessels leading to organ dysfunction. Given the ubiquitous presence of blood vessels, vasculitis can result in a wide array of clinical manifestations from the pathologic consequences of inflammation, hemorrhage, and/or ischemia (1). Vasculitides are traditionally classified based on the size of affected vessels, involvement of specific organs, findings on microscopic examination, and presence of other associated etiologies (2, 3).
Most adult vasculitides can be seen in the pediatric setting; the annual incidence of childhood vasculitis is estimated to be 20 per 100,000 individuals under 17 years of age (4). Despite the advances in the diagnosis and treatment, the etiology of most vasculitides remains to be determined. These conditions share features of autoimmunity caused by aberrant activation of adaptive immunity, as illustrated by IgA vasculitis and anti-neutrophil cytoplasmic antigen (ANCA)-associated vasculitis. Similar to other autoimmune diseases, associations with human leukocyte antigen (HLA) variants have been reported in small, medium, and large vessel vasculitis (5). On the other hand, vessel wall inflammation also exhibits features of autoinflammation triggered by components of the innate immune system such as neutrophils and complement (6). Supporting this view, manifestations of vasculitis have been reported in a number of monogenic autoinflammatory syndromes (7, 8).
Deficiency of adenosine deaminase 2 (DADA2) is a unique monogenic autoinflammatory disease that often presents as childhood-onset small and medium vessel vasculitis. Initially described in 2014, DADA2 is now recognized as a mimic of polyarteritis nodosum (PAN) with primary features of early-onset strokes, peripheral vasculopathy and systemic inflammation (9, 10). Subsequent studies in the field have greatly expanded the spectrum of pathology caused by DADA2 (11). Beyond its early description as a monogenic vasculitis, DADA2 is also associated with immunodeficiency and hematologic defects. The heterogeneity of DADA2 manifestations is striking as some individuals with biallelic pathogenic mutations experience rapid deterioration and fatal disease during childhood while other can remain asymptomatic through adulthood (12, 13). To date, more than 160 cases of DADA2 have been described in the literature. In this article, we will discuss the biology of ADA2 and review the major clinical manifestations of DADA2.
Adenosine is a purine nucleoside generated from the hydrolysis of adenine triphosphate (ATP) and degradation of endogenous nucleic acids. Signaling through several receptors, extracellular adenosine has numerous biological roles including regulation of immune functions (14). Adenosine deaminases (ADA) are enzymes that irreversibly convert adenosine to inosine and 2′-deoxyadenosine to deoxyinosine as a key step in purine metabolism. ADA1, a 40 kD monomeric protein encoded by ADA on chromosome 20, is an intracellular enzyme present in most cell types. The absence of ADA1 results in cytotoxic levels of 2′-deoxyadenosine and deoxyATP, leading to severe combined immunodeficiency (SCID) due to death of developing lymphocytes (15).
ADA2 was discovered later on the basis of residual enzymatic activity in patients with ADA1 deficiency (16). The gene that encodes ADA2 in humans was not identified until two decades later by a group of geneticists studying a development disorder called cat eye syndrome. This condition is associated with duplication of a 1.1 megabase region on chromosome 22q11.2, termed cat eye syndrome critical region (CECR) (17). McDermid and colleagues identified a number of candidate genes within this cluster, including a homolog of adenosine deaminase-related growth factor (ADGF) described in insects, which they named CECR1 (18).
In 2005, Zavialov and Engstrom purified the protein responsible for ADA2 activity in the plasma (19). Mass spectrometry of this protein matched the product of CECR1 and showed extensive peptide homology to the chicken homolog of ADA2. Unlike the ubiquitous expression of ADA1, ADA2 is expressed primarily by monocytes and macrophages (20, 21). Recently, the HUGO Gene nomenclature committee (HGNC) officially changed the gene symbol to ADA2, replacing the former nomenclature of CECR1, ADGF, and IDGFL (insect-derived growth factor like) used in earlier studies (Table 1).
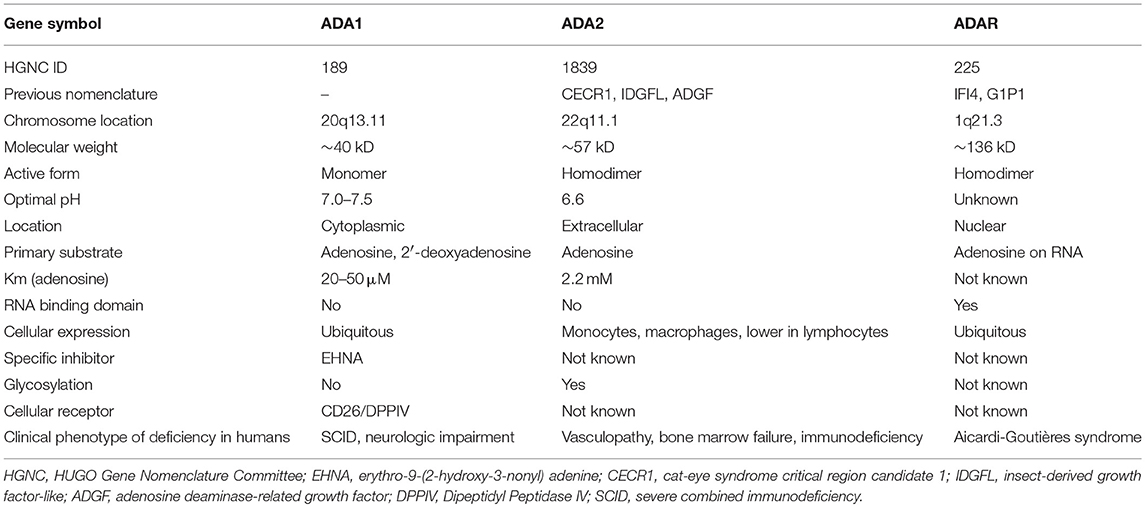
Table 1. Comparison of human adenosine deaminase enzymes.
The structure of ADA2 was solved by Zavialov et al. (22). Consistent with its function as an extracellular protein, ADA2 possesses an N-terminal signal peptide for entry into the endoplasmic reticulum. The protein has a dimerization domain, a putative receptor binding (PRB) domain and a catalytic domain that shares about 20% amino acid identity with the same domain of ADA1. The formation of homodimers with a molecular weight of ~110 kD is required for the enzymatic function of ADA2 in vitro (22). Unlike ADA1, ADA2 is heavily glycosylated and N-linked glycosylation is necessary for proper trafficking to the extracellular space (23). ADA2 has also been shown to bind to the surface of various immune cells, possibly through the PRB domain (24). The structural differences between ADA1 and ADA2 are highlighted in Table 1.
ADA2 possesses much lower affinity for adenosine compared to ADA1 (16), which raises the question of whether it truly has ADA activity at physiologic concentrations of adenosine (25). Notably, ADA2 is insensitive to the ADA1 inhibitor EHNA, a property that allows discrimination of ADA isoenzyme activity in biological samples. Although both enzymes are conserved through evolution, differences in their cell type of origin, cellular localization, and substrate-binding affinity suggest that ADA1 and ADA2 are not functionally redundant.
Elevated plasma ADA2 concentrations are associated with infection, autoimmune diseases, and malignancy, but whether ADA2 plays a role in these inflammatory processes is not clear (20, 26, 27). In vitro studies with human cells have suggested a potential roles of ADA2 in macrophage differentiation and angiogenesis (28, 29). ADA2 orthologs in insects also have growth factor activity that promotes cell proliferation dependent on the ability to metabolize adenosine (30). The precise physiologic function(s) of ADA2 in humans remains a subject of active investigation.
Plasma ADA2 activity can be quantified by determining the release of ammonium during the adenosine to inosine conversion (31). Direct measurement of reaction product inosine (in combination with the downstream product hypoxanthine) is also possible using high performance liquid chromatography (9). In both methods, the specific activity of ADA2 is distinguished from ADA1 activity using the ADA1 inhibitor EHNA. While it is possible to quantify ADA2 protein by enzyme-linked immunosorbent assay or western blotting, these methods are not commonly used clinically.
It is worth noting that human cells express additional ADA enzymes with nucleic acid binding properties (32). ADAR (ADA acting on RNA), the best-studied member of this family, resides in the nucleus and catalyzes the conversion of adenosine to inosine on RNA (Table 1). Among its many biological roles, ADAR edits endogenous double-stranded RNA to prevent activation of the innate immune system (33). Deficiency of ADAR causes Aicardi-Goutières syndrome characterized by childhood-onset encephalopathy and overproduction of type-I interferons (34). Despite the similarities in enzymatic function, ADAR bears minimal structural resemblance to ADA1 or ADA2.
The clinical relevance of human ADA2 was not appreciated until the discovery of individuals with deleterious mutations in this gene. In 2014, two groups independently described a novel syndrome of systemic inflammation and vasculitis caused by mutations in ADA2 (9, 10). Zhou and colleagues studied several young children with recurrent fevers, early-onset strokes, livedo racemosa, hepatosplenomegaly, and systemic vasculopathy. Using whole exome sequencing (WES), they determined biallelic loss-of-function ADA2 mutations in 9 patients and named the condition DADA2. The authors also described the first experimental model of the disease using zebrafish, which showed intracranial hemorrhage and neutropenia after knockdown of the ADA2 homolog (9).
In parallel, Navon Elkan and colleagues studied cases of a medium-vessel vasculitis that closely resembles polyarteritis nodosum (PAN) in the Georgian Jewish population as well as additional patients from Germany and Turkey (10). These patients had recurrent fever, livedo, skin ulceration, neuropathy and aneurysms, among the most prevalent findings. WES identified loss-of-function mutations in ADA2 and biallelic mutations were identified in 21 patients, with the majority under 10 years of age. The mortality of this condition was highlighted by two deaths in early childhood.
These two landmark studies established DADA2 as a monogenic cause of childhood-onset vasculitis. Over the past few years, our understanding of the clinical spectrum of DADA2 has advanced significantly, with more than 30 publications describing over 160 patients with confirmed mutations world-wide. The collective experience from these studies has been instrumental in guiding treatment approaches. Although the pathogenesis of DADA2 is still under investigation, anti-TNF (tumor necrosis factor) agents seem to be most effective to manage the inflammatory vasculopathy, while hematopoietic stem cell transplantation (HSCT) provides an option for definitive cure (35). Therapeutic options for DADA2 were recently reviewed (11) and will not be extensively discussed here.
The clinical manifestations of DADA2 are summarized in Figure 1. Consistent with the initial studies, vascular involvement is the hallmark of DADA2 and features of vasculitis or vasculopathy have been described in more than 75% of cases. Organs affected include skin, brain, GI tract, and kidneys. The distribution of medium and small vessel involvement mimics the pattern seen in PAN (10). Not surprisingly, DADA2 patients are often diagnosed with PAN before the diagnosis is confirmed by gene sequencing or plasma activity assay. Caorsi and colleagues reviewed 48 cases of childhood PAN in Italy and found 15 patients with biallelic ADA2 mutations (36). Furthermore, DADA2 has also been found in cases previously diagnosed as Sneddon syndrome, an adult-onset vasculopathy characterized by livedo and strokes (37).
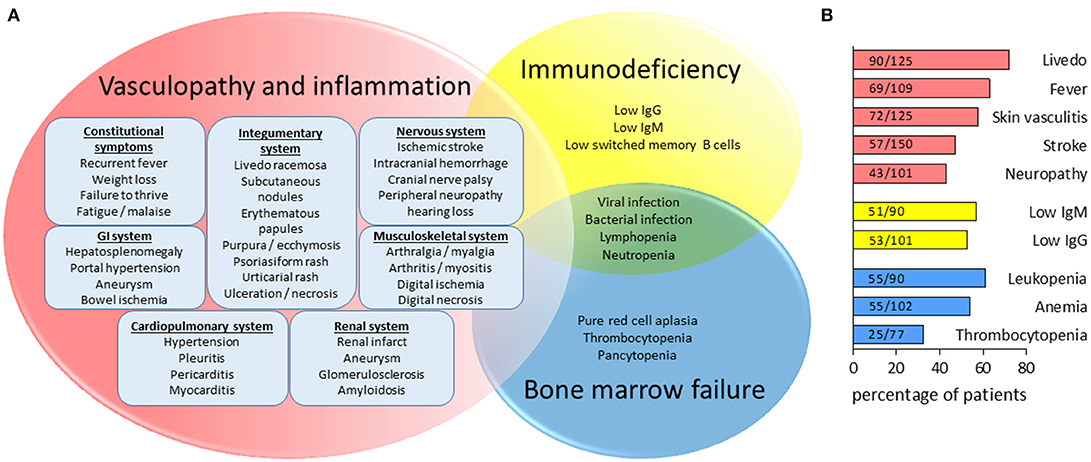
Figure 1. Pleiotropic manifestations of DADA2. (A) Depiction of overlapping features of inflammatory vasculopathy, immunodeficiency and hematologic defects in DADA2. (B) Bar graph illustrating the estimated percentage of patients with the common clinical features seen in DADA2. As displayed by the Fractions in the graph, estimates are based on the number of patients reported to have the specific clinical features in the literature (160 cases published prior to July 1, 2018) divided by the total number of patients included in the studies in which the parameters were assessed. The category “stroke” include both ischemic strokes and hemorrhagic strokes.
When compared to other patients with PAN, DADA2 patients had earlier disease onset and greater prevalence of skin and neurologic manifestations (36). Overall, skin involvement is the most common manifestation of DADA2, reported in nearly 90% of cases. Based on studies with adequate description of skin findings, livedo racemosa and livedo reticularis are the most common observations (72%), followed descriptions of cutaneous vasculitis (57%) including subcutaneous nodules, petechiae/purpura, ulcerations, and necrosis. Non-specific erythematous maculopapular rash, urticaria, and psoriatic lesions are less common. Biopsy of inflammatory skin lesions typically demonstrates evidence of vasculitis, with variable descriptions including non-granulomatous necrotizing arteritis, leukocytoclastic vasculitis, and panniculitis (9, 10, 38).
Neurologic complications of DADA2 include ischemic strokes, intracranial hemorrhage, and a wide range of neuropathy. Ischemic strokes have been reported in more than 50 patients (~38%) and many have experienced recurrent episodes. In some cases, stroke is the initial feature that leads to the diagnosis (12, 39). The severity of strokes varies from small lacunar infarcts with minimal sequelae to larger infarcts leading to permanent neurologic impairments and even death (40). Transient ischemic attacks without radiographic findings have also been reported (41). As brain imaging is not routinely performed in the absence of neurologic findings, subclinical infarcts may escape detection and the prevalence of strokes in this disease may be underestimated.
Intracranial hemorrhage or hemorrhagic strokes have been reported in at least 20 cases. The severity again ranges from subclinical to life-threatening hemorrhage that requires urgent evacuation. Structural abnormalities, such as aneurysms of the cerebral vasculature and basal ganglia calcification seen in interferonopathies are typically absent (42). Whether intracranial hemorrhage is a spontaneous phenomenon in DADA2 or secondary to ischemic stroke conversion remains a question. While coagulopathy is uncommon in this disease, complicating this determination is the use of anticoagulants and/or anti-platelet agents in some patients after an ischemic stroke. Interestingly, radiographic evidence did not suggest cerebral vasculitis in 5 patients with intracranial hemorrhage, and biopsy from 2 of these patients revealed extravasation of red blood cells from small vessels without findings of inflammation (9). In the absence of inflammation, vasculopathy rather than vasculitis may be a more accurate description of the pathology in some cases. A disconnect between brain hemorrhage and inflammatory features of DADA2 is also evident in a case where recurrent spontaneous hemorrhage occurred after effective management of systemic inflammation (23).
Diverse manifestations of central and peripheral neuropathy have been reported in patients with DADA2. In fact, neuropathy is another distinguishing feature of DADA2 compared to other cases of childhood PAN, but its connection with vasculopathy is not entirely clear (36). Cranial nerve (CN) palsies can involve CN III, IV, VI, and VII, and several patients experienced neurosensory hearing loss. Ophthalmologic complications including vision loss, central retinal artery occlusion, optic nerve atrophy, uveitis, diplopia, nystagmus, and strabismus have all been described (9, 40). Peripheral neuropathy is noted in half of patients in some cohorts and spastic paraplegia has been reported as a presenting symptom of DADA2 (36, 43).
Consistent with a systemic inflammatory process, most patients experience recurrent fever. Myalgia and arthralgia are also frequent complaints, but arthritis and myositis are less common. Inflammatory markers such as erythrocyte sedimentation rate and C-reactive protein are often elevated in parallel to the clinical features of inflammation. Early studies established hepatosplenomegaly as a common finding of DADA2 and portal hypertension has been noted in several patients (9).
In keeping with the resemblance to PAN, DADA2 can affect small to medium-size vessels in different organs. Inflammation of arteries supplying the extremities results in digital infarct and necrosis. Vasculitis and aneurysm of the gastrointestinal vasculature leads to bowel perforation and intestinal hemorrhage. Renal aneurysm and infarction have been reported in a handful of cases (10). Hypertension can be seen independent of other renal pathology and may be a risk factor for the development of stroke and intracranial hemorrhage (13, 36, 51). Lastly, rare inflammatory manifestations including pleuritis, pericarditis, myocarditis, meningitis, and amyloidosis have been reported in patients with DADA2 (9, 10).
In the initial description of DADA2, Zhou et al. noted mild immunodeficiency with low IgM levels as a clinical feature in 5 patients (9). Schepp and colleagues subsequently found compound heterozygous ADA2 mutations with absent plasma ADA2 activity in an adult with hypogammaglobinemia and recurrent respiratory infection (44). Expanding on this work, they screened 181 patients with antibody deficiencies and found 9 additional patients with DADA2. Recurrent infection, rather than the classic features of inflammation and vasculopathy, was the primary presentation for more than half of this cohort (45). These patients generally present later in life and several have no clinical evidence of vasculitis. Importantly, low IgG and IgM levels are common features of DADA2, with or without concurrent findings of vasculopathy (Figure 1). Across studies that quantified immunoglobulins, low IgG and/or IgM levels are reported in ~67% of DADA2 patients. The prevalence of IgG deficiency, IgM deficiency, and combined antibody deficiency are ~52, 57, and 35%, respectively.
Both B cell intrinsic and extrinsic factors may contribute to the low immunoglobulin levels. Switched memory B cells are markedly decreased in DADA2 patients and in vitro stimulation demonstrates reduced antibody production (9, 45). Defective class-switching, however, cannot account for deficient IgM production. Reduced number of antibody producing cells may be a contributing factor, as DADA2 is associated with lymphopenia (further discussed in the next section). Schepp et al. demonstrated an inverse correlation between CRP and immunoglobulin levels in one patient, suggesting that the inflammatory milieu of DADA2 may directly compromise the B cell compartment (45). This intriguing observation contradicts the observation in pediatric autoimmune conditions, where hypergammaglobulinemia is recognized as a feature of active disease (46).
On the other hand, immunologic studies in DADA2 have not revealed significant impairments in the T cell compartment. Naïve and memory T cell activation, mitogen-induced proliferation, and ex-vivo cytokine production by CD4+ and CD8+ T cells were comparable in patients with DADA2 and control subjects (9). Since specific defects in T cell function are not apparent, infection related to suboptimal T cell function in patients with DADA2 may be better explained by a quantitative defect due to generalized lymphopenia.
Viral and bacterial respiratory tract infections and herpes infection are common in DADA2 patients with antibody deficiencies (45). Infection of the brain and meninges, gastrointestinal tract, urinary tract have also been reported. The risk of infection is escalated further by immunosuppressive agents used to treat the inflammatory features of the disease, although the risk may be partially mitigated if the humoral defects are driven by inflammation. Fungal and mycobacterial infections are unusual in DADA2 but have been noted in cases with severe neutropenia or pancytopenia (47, 48). Fungal infection was also reported in two sibling with a large genomic deletion that spans ADA2 and the adjacent gene IL-17RA, in line with the role of IL-17 in fungal immunity (49).
The spectrum of hematologic involvement adds another intriguing dimension to our understanding of DADA2 (Figure 1). Severe reduction in erythrocytes, leukocytes, and platelets have all been reported and hematologic defects can be the presenting finding, even without features of inflammation or vasculopathy. Reduced number of immune cells further fuels the infection risk posed by low immunoglobulin levels. Notably, it is often the severity of anemia or pancytopenia that drive the necessity of curative treatment with HSCT (35).
Anemia is present in >50% of cases, often with a severity extending beyond what is expected for anemia of chronic inflammation. Several patients with DADA2 presented with pure red cell aplasia with transfusion dependence that mimics Diamond-Blackfan anemia (47, 50). Genetic evaluation, typically by WES show biallelic ADA2 mutations while DBA associated mutations in ribosomal proteins and GATA1 are absent.
Leukopenia occurs in about 60% of cases and variably affect myeloid and lymphoid cells. Thrombocytopenia is less common, described in ~32% of cases. These hematologic defects often do not occur alone as multiple cases of pancytopenia have been reported. Bone marrow biopsy typically demonstrates hypoplasia of the affected lineage(s), suggesting a defect in cell production. Lymphoid cell aggregates in the bone marrow have been documented, although their significance is unclear (48, 50). Evidence of cell consumption is generally absent, but two cases of macrophage activation syndrome has been described (9, 51).
How a plasma protein produced mainly by monocytes and macrophages affects multi-lineage hematopoiesis is unclear. Knockdown of ADA2 in the Zebrafish model was shown to cause neutropenia, supporting an intrinsic role of ADA2 in normal hematopoiesis (9). Whether human ADA2 functions as a cellular growth factor similar to the insect orthologs remains to be seen. In vitro, human ADA2 can promote T cell-dependent monocyte maturation (28), though a broader mechanism is necessary given the multi-lineage involvement.
The clinical spectrum of DADA2 spans beyond the three major phenotypes discussed thus far. Blurring the line the between autoinflammation and autoimmunity, several patients display prominent features of systemic lupus erythematous including the presence of specific autoantibodies and a type I interferon signature (52, 53). Such presentation bears some resemblance to Aicardi-Goutières syndrome associated with mutations in ADAR (43). More recently, cases of lymphoproliferative disease with resemblance to large granular lymphocyte leukemia have been described in a Finnish cohort of DADA2 patients (54).
Missense mutations, frameshift mutations, splicing defects and deletions have been described in pathogenic ADA2 mutations. Mutations in all structural domains of ADA2 have also been reported. Neither the type of mutation or location of the mutation seems to correlate with the diverse manifestations of DADA2. Homozygosity of most common pathogenic variants G47R or R169Q explains more than 60 cases of DADA2, but the clinical findings are highly variable among family members that share the same homozygous mutations (10, 12, 51). The amounts of residual ADA2 activity, rather than genotype, may be more predictive of phenotype (12).
It is also important to recognize that individuals (including adults) with confirmed biallelic mutations and absent plasma ADA2 activity can remain entirely asymptomatic (13). These individuals are often found on the basis of having affected family members. On the other hand, some features of the disease including lacunar strokes has been described in heterozygous individuals (9, 55). Additional studies are needed to determine the clinical significance of ADA2 haploinsufficiency.
Advances in genomic analysis have significantly improved our ability to diagnose monogenic disorders with variable presentations. Less than 5 years after the discovery of DADA2, the clinical spectrum of the disease has drastically expanded from simply a monogenic form of childhood-onset PAN. The pleiotropic presentations of the disease span many sub-specialties, including rheumatology, immunology, neurology, dermatology and hematology in pediatrics as well as in adult medicine. Given the multi-organ involvement of DADA2, recognizing the diverse manifestations is a crucial step toward timely diagnosis and management of this potentially fatal but often treatable syndrome.
The author confirms being the sole contributor of this work and has approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author would like to thank Dr. Peter Nigrovic for helpful discussions and critical reading of this manuscript. This work was supported by the Rheumatology Research Foundation Investigator Award and Boston Children's Hospital Faculty Career Development Fellowship.
1. Petty RE, and Cabral DA. Vasculitis and its classification. In: Petty RE, Laxer RM, Lindsley C, Wedderburn LR, editors. Textbook of Pediatric Rheumatology. Philadelphia, PA: Elsevier (2016). p. 448–532.
2. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum. (2013) 65:1–11. doi: 10.1002/art.37715
3. Ozen S, Ruperto N, Dillon MJ, Bagga A, Barron K, Davin JC, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. (2006) 65:936–41. doi: 10.1136/ard.2005.046300
4. Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schonlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet (2002) 360: 1197–202. doi: 10.1016/S0140-6736(02)11279-7
5. Monach PA, Merkel PA. Genetics of vasculitis. Curr Opin Rheumatol. (2010) 22:157–63. doi: 10.1097/BOR.0b013e32833654a8
6. Lamprecht P, Gross WL. Antineutrophil cytoplasmic antibody-associated vasculitis: autoinflammation, autodestruction and autoimmunity–key to new therapies. Trends Immunol. (2008) 29:587–8. doi: 10.1016/j.it.2008.09.003
7. Peleg H, Ben-Chetrit E. Vasculitis in the autoinflammatory diseases. Curr Opin Rheumatol. (2017) 29:4–11. doi: 10.1097/BOR.0000000000000347
8. Jain A, Misra DP, Sharma A, Wakhlu A, Agarwal V, Negi VS. Vasculitis and vasculitis-like manifestations in monogenic autoinflammatory syndromes. Rheumatol Int. (2018) 38:13–24. doi: 10.1007/s00296-017-3839-6
9. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Stone DL, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. (2014) 370:911–20. doi: 10.1056/NEJMoa1307361
10. Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. (2014) 370:921–31. doi: 10.1056/NEJMoa1307362
11. Caorsi R, Penco F, Schena F, Gattorno M. Monogenic polyarteritis: the lesson of ADA2 deficiency. Pediatr Rheumatol Online J. (2016) 14:51. doi: 10.1186/s12969-016-0111-7
12. Van Montfrans JM, Hartman EA, Braun KP, Hennekam EA, Hak EA, Nederkoorn PJ, et al. Phenotypic variability in patients with ADA2 deficiency due to identical homozygous R169Q mutations. Rheumatology (2016) 55:902–10. doi: 10.1093/rheumatology/kev439
13. Nanthapisal S, Murphy C, Omoyinmi E, Hong Y, Standing A, Berg S, et al. Deficiency of adenosine deaminase type 2: a description of phenotype and genotype in fifteen cases. Arthritis Rheumatol. (2016) 68:2314–22. doi: 10.1002/art.39699
14. Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. (2008) 7:759–70. doi: 10.1038/nrd2638
15. Giblett ER, Anderson JE, Cohen F, Pollara B, Meuwissen HJ. Adenosine-deaminase deficiency in two patients with severely impaired cellular immunity. Lancet (1972) 2:1067–9.
16. Schrader WP, Pollara B, Meuwissen HJ. Characterization of the residual adenosine deaminating activity in the spleen of a patient with combined immunodeficiency disease and adenosine deaminase deficiency. Proc Natl Acad Sci USA. (1978) 75:446–50.
17. Footz TK, Brinkman-Mills P, Banting GS, Maier SA, Riazi MA, Bridgland L, et al. Analysis of the cat eye syndrome critical region in humans and the region of conserved synteny in mice: a search for candidate genes at or near the human chromosome 22 pericentromere. Genome Res. (2001) 11:1053–70. doi: 10.1101/gr.154901
18. Riazi MA, Brinkman-Mills P, Nguyen T, Pan H, Phan S, Ying F, et al. The human homolog of insect-derived growth factor, CECR1, is a candidate gene for features of cat eye syndrome. Genomics (2000) 64:277–85. doi: 10.1006/geno.1999.6099
19. Zavialov AV, Engstrom A. Human ADA2 belongs to a new family of growth factors with adenosine deaminase activity. Biochem J. (2005) 391:51–7. doi: 10.1042/BJ20050683
20. Ratech H, Martiniuk F, Borer WZ, Rappaport H. Differential expression of adenosine deaminase isozymes in acute leukemia. Blood (1988) 72:1627–32.
21. Iwaki-Egawa S, Yamamoto T, Watanabe Y. Human plasma adenosine deaminase 2 is secreted by activated monocytes. Biol Chem. (2006) 387:319–21. doi: 10.1515/BC.2006.042
22. Zavialov AV, Yu X, Spillmann D, Lauvau G. Structural basis for the growth factor activity of human adenosine deaminase ADA2. J Biol Chem. (2010) 285:12367–77. doi: 10.1074/jbc.M109.083527
23. Lee PY, Huang Y, Zhou Q, Schnappauf O, Hershfield MS, Li Y, et al. Disrupted N-linked glycosylation as a disease mechanism in deficiency of ADA2. J Allergy Clin Immunol. (2018). doi: 10.1016/j.jaci.2018.05.038. [Epub ahead of print].
24. Kaljas Y, Liu C, Skaldin M, Wu C, Zhou Q, Lu Y, et al. Human adenosine deaminases ADA1 and ADA2 bind to different subsets of immune cells. Cell Mol Life Sci. (2017) 74:555–70. doi: 10.1007/s00018-016-2357-0
25. Hashem H, Kelly SJ, Ganson NJ, Hershfield MS. Deficiency of adenosine deaminase 2 (DADA2), an inherited cause of polyarteritis nodosa and a mimic of other systemic rheumatologic disorders. Curr Rheumatol Rep. (2017) 19:70. doi: 10.1007/s11926-017-0699-8
26. Gakis C, Calia G, Naitana A, Pirino D, Serru G. Serum adenosine deaminase activity in HIV positive subjects. A hypothesis on the significance of ADA2. Panminerva Med. (1989) 31:107–13.
27. Sari RA, Taysi S, Yilmaz O, Bakan N. Correlation of serum levels of adenosine deaminase activity and its isoenzymes with disease activity in rheumatoid arthritis. Clin Exp Rheumatol. (2003) 21:87–90.
28. Zavialov AV, Gracia E, Glaichenhaus N, Franco R, Zavialov AV, Lauvau G. Human adenosine deaminase 2 induces differentiation of monocytes into macrophages and stimulates proliferation of T helper cells and macrophages. J Leukoc Biol. (2010) 88:279–90. doi: 10.1189/jlb.1109764
29. Zhu C, Chrifi I, Mustafa D, van der Weiden M, Leenen JM, Duncker DJ, et al. CECR1-mediated cross talk between macrophages and vascular mural cells promotes neovascularization in malignant glioma. Oncogene (2017) 36:5356–68. doi: 10.1038/onc.2017.145
30. Zurovec M, Dolezal T, Gazi M, Pavlova E, Bryant PJ. Adenosine deaminase-related growth factors stimulate cell proliferation in Drosophila by depleting extracellular adenosine. Proc Natl Acad Sci USA. (2002) 99:4403–8. doi: 10.1073/pnas.062059699
31. Muraoka T, Katsuramaki T, Shiraishi H, Yokoyama MM. Automated enzymatic measurement of adenosine deaminase isoenzyme activities in serum. Anal Biochem. (1990) 187:268–72.
32. Song C, Sakurai M, Shiromoto Y, Nishikura K. Functions of the RNA editing enzyme ADAR1 and their relevance to human diseases. Genes (2016) 7:129. doi: 10.3390/genes7120129
33. Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science (2015) 349:1115–20. doi: 10.1126/science.aac7049
34. Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. (2012) 44:1243–8. doi: 10.1038/ng.2414
35. Hashem H, Kumar AR, Muller I, Babor F, Bredius R, Dalal J, et al. Hematopoietic stem cell transplantation rescues the hematological, immunological, vascular phenotype in DADA2. Blood (2017) 130:2682–8. doi: 10.1182/blood-2017-07-798660
36. Caorsi R, Penco F, Grossi A, Insalaco A, Omenetti A, Alessio M, et al. ADA2 deficiency (DADA2) as an unrecognized cause of early onset polyarteritis nodosa and stroke: a multicentre national study. Ann Rheum Dis. (2017) 76:1648–56. doi: 10.1136/annrheumdis-2016-210802
37. Bras J, Guerreiro R, Santo GC. Mutant ADA2 in vasculopathies. N Engl J Med. (2014) 371:478–80. doi: 10.1056/NEJMc1405506
38. Gonzalez Santiago TM, Zavialov A, Saarela J, Seppanen M, Reed AM, Abraham RS, et al. Dermatologic features of ADA2 deficiency in cutaneous polyarteritis nodosa. JAMA Dermatol. (2015) 151:1230–4. doi: 10.1001/jamadermatol.2015.1635
39. Elbracht M, Mull M, Wagner N, Kuhl C, Abicht A, Kurth I, et al. Stroke as initial manifestation of adenosine deaminase 2 deficiency. Neuropediatrics (2017) 48:111–4. doi: 10.1055/s-0036-1597611
40. Sahin S, Adrovic A, Barut K, Ugurlu S, Turanli ET, Ozdogan H, et al. Clinical, imaging and genotypical features of three deceased and five surviving cases with ADA2 deficiency. Rheumatol Int. (2018) 38:129–36. doi: 10.1007/s00296-017-3740-3
41. Van Eyck L Jr, Hershfield MS, Pombal D, Kelly SJ, Ganson NJ, Moens L, et al. Hematopoietic stem cell transplantation rescues the immunologic phenotype and prevents vasculopathy in patients with adenosine deaminase 2 deficiency. J Allergy Clin Immunol. (2015) 135:283–7.e5. doi: 10.1016/j.jaci.2014.10.010
42. Bulut E, Erden A, Karadag O, Oguz KK, Ozen S. Deficiency of adenosine deaminase 2; special focus on central nervous system imaging. J Neuroradiol. (2018). doi: 10.1016/j.neurad.2018.05.002. [Epub ahead of print].
43. Poswar Fde O, da Fonseca RM, de Albuquerque LC, Zhou Q, Jardim LB, Monte TL, et al. Adenosine deaminase 2 deficiency presenting as spastic paraplegia and systemic vasculitis. J Neurol. (2016) 263:818–20. doi: 10.1007/s00415-016-8070-y
44. Schepp J, Bulashevska A, Mannhardt-Laakmann W, Cao H, Yang F, Seidl M, et al. Deficiency of adenosine deaminase 2 causes antibody deficiency. J Clin Immunol. (2016) 36:179–86. doi: 10.1007/s10875-016-0245-x
45. Schepp J, Proietti M, Frede N, Buchta M, Hubscher K, Rojas Restrepo J, et al. Screening of 181 patients with antibody deficiency for deficiency of adenosine deaminase 2 sheds new light on the disease in adulthood. Arthritis Rheumatol. (2017) 69:1689–700. doi: 10.1002/art.40147
46. Lo MS, Zurakowski D, Son MB, Sundel RP. Hypergammaglobulinemia in the pediatric population as a marker for underlying autoimmune disease: a retrospective cohort study. Pediatr Rheumatol Online J. (2013) 11:42. doi: 10.1186/1546-0096-11-42
47. Ben-Ami T, Revel-Vilk S, Brooks R, Shaag A, Hershfield MS, Kelly SJ, et al. Extending the clinical phenotype of adenosine deaminase 2 deficiency. J Pediatr. (2016) 177:316–20. doi: 10.1016/j.jpeds.2016.06.058
48. Hsu AP, West RR, Calvo KR, Cuellar-Rodriguez J, Parta M, Kelly SJ, et al. Adenosine deaminase type 2 deficiency masquerading as GATA2 deficiency: successful hematopoietic stem cell transplantation. J Allergy Clin Immunol. (2016) 138:628–30.e2. doi: 10.1016/j.jaci.2016.03.016
49. Fellmann F, Angelini F, Wassenberg J, Perreau M, Arenas Ramirez N, Simon G, et al. IL-17 receptor A and adenosine deaminase 2 deficiency in siblings with recurrent infections and chronic inflammation. J Allergy Clin Immunol. (2016) 137:1189–96.e2. doi: 10.1016/j.jaci.2015.07.053
50. Sasa G, Elghetany MT, Bergstrom K, Nicholas S, Himes R, Krance R, et al. Adenosine deaminase 2 deficiency as a cause of pure red cell aplasia mimicking diamond blackfan anemia. Blood (2015) 126:3615.
51. Batu ED, Karadag O, Taskiran EZ, Kalyoncu U, Aksentijevich I, Alikasifoglu M, et al. A case series of adenosine deaminase 2-deficient patients emphasizing treatment and genotype-phenotype correlations. J Rheumatol. (2015) 42:1532–4. doi: 10.3899/jrheum.150024
52. Skrabl-Baumgartner A, Plecko B, Schmidt WM, Konig N, Hershfield M, Gruber-Sedlmayr U, et al. Autoimmune phenotype with type I interferon signature in two brothers with ADA2 deficiency carrying a novel CECR1 mutation. Pediatr Rheumatol Online J. (2017) 15:67. doi: 10.1186/s12969-017-0193-x
53. Uettwiller F, Sarrabay G, Rodero MP, Rice GI, Lagrue E, Marot Y, et al. ADA2 deficiency: case report of a new phenotype and novel mutation in two sisters. RMD Open (2016) 2:e000236. doi: 10.1136/rmdopen-2015-000236
54. Trotta L, Martelius T, Siitonen T, Hautala T, Hamalainen S, Juntti H, et al. ADA2 deficiency: clonal lymphoproliferation in a subset of patients. J Allergy Clin Immunol. (2018) 141:1534–7.e8. doi: 10.1016/j.jaci.2018.01.012
Keywords: DADA2, vasculitis, vasculopathy, immunodeficiency, bone marrow failure, cytopenias
Citation: Lee PY (2018) Vasculopathy, Immunodeficiency, and Bone Marrow Failure: The Intriguing Syndrome Caused by Deficiency of Adenosine Deaminase 2. Front. Pediatr. 6:282. doi: 10.3389/fped.2018.00282
Received: 02 August 2018; Accepted: 17 September 2018;
Published: 18 October 2018.
Edited by:
Christian Michael Hedrich, University of Liverpool, United KingdomReviewed by:
Marina Cavazzana, Necker-Enfants Malades Hospital, FranceCopyright © 2018 Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pui Y. Lee, cHVpLmxlZUBjaGlsZHJlbnMuaGFydmFyZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.