 Paula Giménez-Mascarell
Paula Giménez-Mascarell Carlotta Else Schirrmacher
Carlotta Else Schirrmacher Luis Alfonso Martínez-Cruz
Luis Alfonso Martínez-Cruz Dominik Müller
Dominik Müller
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr. , 09 April 2018
Sec. Pediatric Nephrology
Volume 6 - 2018 | https://doi.org/10.3389/fped.2018.00077
This article is part of the Research Topic Genetic Kidney Diseases of Childhood View all 27 articles
Magnesium (Mg2+) is indispensable for several vital functions, such as neurotransmission, cardiac conductance, blood glucose, blood pressure regulation, and proper function of more than 300 enzymes. Thus, Mg2+ homeostasis is subject to tight regulation. Besides the fast and immediate regulation of plasma Mg2+, a major part of Mg2+ homeostasis is realized by a concerted action of epithelial molecular structures that tightly control intestinal uptake and renal absorption. This mechanism is provided by a combination of para- and transcellular pathways. Whereas the first pathway provides the organism with a maximal amount of vital substances by a minimal energy expenditure, the latter enables controlling and fine-tuning by means of local and regional regulatory systems and also, hormonal control. The paracellular pathway is driven by an electrochemical gradient and realized in principal by the tight junction (TJ), a supramolecular organization of membrane-bound proteins and their adaptor and scaffolding proteins. TJ determinants are claudins (CLDN), a family of membrane spanning proteins that generate a barrier or a pore between two adjacent epithelial cells. Many insights into molecular mechanisms of Mg2+ handling have been achieved by the identification of alterations and mutations in human genes which cause disorders of paracellular Mg2+ pathways (CLDN10, CLDN14, CLDN16, CLDN19). Also, in the distal convoluted tubule, a basolateral protein, CNNM2, causes if mutated, familial dominant and also recessive renal Mg2+ wasting, albeit its true function has not been clarified yet, but is assumed to play a key role in the transcellular pathway. Moreover, mutations in human genes that are involved in regulating these proteins directly or indirectly cause, if mutated human diseases, mostly in combination with comorbidities as diabetes, cystic renal disease, or metabolic abnormalities. Generation and characterization of animal models harboring the corresponding mutations have further contributed to the elucidation of physiology and pathophysiology of Mg2+ disorders. Finally, high-end crystallization techniques allow understanding of Mg2+ handling in more detail. As this field is rapidly growing, we describe here the principles of physiology and pathophysiology of epithelial transport of renal Mg2+ homeostasis with emphasis on recently identified mechanisms involved.
Magnesium (in its ionized and biologically active form: Mg2+) belongs to the group of alkaline earth metals and is the second most abundant intracellular divalent cation. It is the eleventh most abundant element by mass in the human body. Mg2+ is indispensable for several vital functions, such as neurotransmission, cardiac conductance, blood glucose control, and blood pressure regulation. In muscle, Mg2+ regulates contraction by antagonizing calcium (1–4). Mg2+ has also been identified as a second messenger, e.g., in T-Cells, where mutations in the gene MAGT1 have been linked to human immunodeficiency. There, mutations disable transient Mg2+ influx induced by the activation of the T-cell receptor (5).
Adenosine triphosphosphate (ATP) is the major source of cell energy, and must bind Mg2+ in order to be biologically active. The resulting complex, Mg2+-ATP is vital for the stability of all polyphosphate compounds in cells, including those associated with synthesis of DNA and RNA. More than 300 enzymes are dependent on Mg2+ for their biocatalytic function, including those that utilize or synthesize ATP, or those that use other nucleotides to synthesize DNA and RNA (6). In plants, Mg2+ is the central ion of chlorophyll and, therefore, vital for photosynthesis. In higher organisms, hemoglobin, the essential O2 carrier, has high structural similarities with chlorophyll but here, Fe2+ replaced Mg2+ as the central ion. Magnesium is an essential mineral nutrient (i.e., element) and is present in every cell type and in every organism. In the blood and serum, Mg2+ is mostly bound to serum albumin (like the most abundant divalent cation, Ca2+) and stored in muscle fibers and in bone. The biologically active form is the ionized form and dietary sources rich of magnesium are plants [Almonds, Cashews, Cocoa, Pumpkin Seeds, Spinach, and Fish (Halibut, Mackeral)]. Clinically, deficiency of Mg2+ causes nausea, appetite loss, fatigue, and general weakness. At a later stage, patients present with numbness, tingling, muscle cramps, cerebral seizures, and cardiac arrhythmias. Moreover, Mg2+ deficiency can lead to hypocalcemia and hypokalemia (7). On the other hand, little is known about the consequences of hypermagnesemia as can be encountered, e.g., in patients with end stage renal disease (8). However, Mg2+ complexes are involved in the development of vascular calcifications, a major cause of morbidity and mortality of patients with chronic kidney disease (9).
Compared to Ca2+ storage (around 1000 g in adults), the whole body content of Mg2+ only sums up to 20 g. In addition, Mg2+ stores are not as readily accessible as Ca2+ stores by, e.g., PTH action. Therefore, the organisms’ intra- and extracellular Mg2+ levels are kept tightly within narrow limits. Likewise, the available, ionized, biologically active and relevant form (Mg2+) is dependent on a more or less a continuous nutritional supply. Mg2+ serum concentrations in adults range from 0.7 to 1.1 mmol/l, while newborns and toddlers can have lower limit serum concentrations (e.g., from 0.45 mmol/l on). The daily need of Mg2+ in adults is around 300–350 mg/day and fractional intestinal uptake varies depending on the amount of intake. Fine et al. found that intestinal Mg2+ absorption increases with intake but also that fractional Mg2+ absorption fells progressively (from 65% at low to 11% at high intake) (10). The authors described the intestinal absorption by an equation containing a hyperbolic function plus a linear function. They concluded that Mg2+ absorption must, therefore, be realized by a twin-mechanism that simultaneously reaches an absorptive maximum, and in addition a mechanism that endlessly absorbs a defined fraction (7%, as the authors concluded) (10). Although the molecular mechanisms have not been identified at that time, their and the findings of other groups later, predicted the coexistence of two distinct mechanisms of Mg2+ absorption, the trans- and paracellular transport in the intestine. Moreover, as we know nowadays, the same holds true for the kidney.
In both organs, the intestine and the kidney, Mg2+ is absorbed via paracellular as well as by transcellular routes. Whereas the first pathway is in principal driven by an existing electrochemical gradient, the latter requires energy-consuming mechanisms in order to conduct apical uptake, buffering, transport, and basolateral extrusion (Figure 1). The paracellular route guarantees the organism to retrieve a considerable amount of vital substances with a minimal energetic expenditure. On the other side, the energy-consuming transcellular route provides a fine-tuning at the several steps of transport and is, therefore, subject to precise local, regional, or global regulation. This can be realized by hormones such as 1,25(OH)D2 or parathyroid hormone according to the basic but also the actual needs of the organism (e.g., growth).
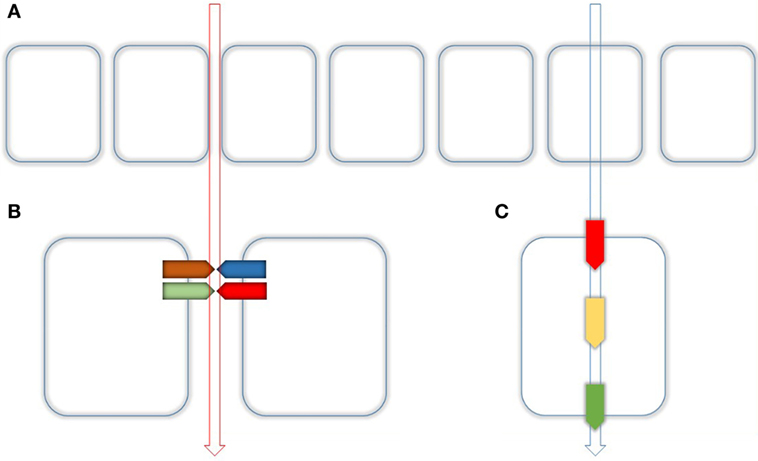
Figure 1. Routes of epithelial transport. (A) Epithelial transport is performed by two different routes. Paracellular (left) and transcellular (right) as indicated by the vertical arrows. (B) The paracellular transport is realized through the tight Junction (TJ) which is composed of several Proteins mainly by Claudins. Together with their adaptor and scaffolding proteins, they make up as hetero- or homodimers dimers the main part of the TJ. (C) Paracellular transport is realized by apical uptake (red), intracellular buffering and transport (yellow), and basolateral extrusion (green).
In the kidney, the driving force of transepithelial Mg2+ absorption is provided by a continuously maintained electrochemical gradient between the apical (tubular fluid) and the basolateral (blood) compartment. This process is realized by a battery of transcellular (and apical to basolateral) and paracellular transport mechanisms. As a principle, paracellular transport mainly takes place where the transepithelial concentration gradient is high, i.e., the proximal jejunum in the gut and in the proximal tubule (PT) as well the thick ascending loop of Henle in the kidney. By contrast, transcellular transport takes place in the late ileum, colon, and the distal part of the renal tubule (11, 12).
After glomerular filtration, the non-protein-bound fraction, Mg2+ is absorbed along the nephron before it is finally lost irretrievable for the organism within the urine. Under normal conditions (Glomerular filtration rate >90 ml/min/1.73 m2), more than 95% of filtered Mg2+ in the pro-urine is reabsorbed along the tubular system by several coordinated transport processes (13). Besides their anatomical localization, regulatory mechanisms of renal Mg2+ homeostasis can also be classified by their involvement in the hierarchy of transport. Therefore, we classify here the mechanisms of Mg2+ transport and homeostasis according to their involvement in the network of Mg2+ handling (Table 1). The first category comprises proteins or elements that transport Mg2+ by itself, e.g., a direct Mg2+ transport mechanism. The second category includes proteins and mechanisms that regulate elements of category 1. The third category comprises members that influence Mg2+ handling in a more remote way, e.g., by regulating ions, other than Mg2+ but thereby influence Mg2+ handling. To the latter category belong also proteins that influence Mg2+ homeostasis, e.g., by causing polyuria and thereby a “wash out” of Mg2+ by reducing the time of contact of absorbing mechanisms with the corresponding substance (as can be seen by the treatment with furosemide). With this newly proposed classification, we aim to focus on the current knowledge on Mg2+ handling toward a more interactive model, the magnesiome, and the Mg2+ interactome.
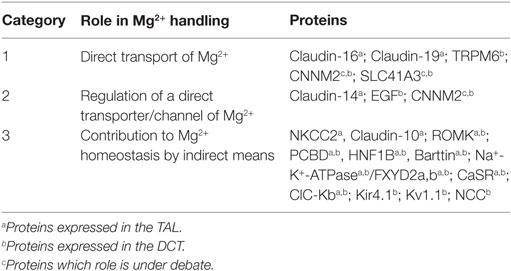
Table 1. Protein classification based on the role played by each molecule in renal magnesium handling.
Anatomically, the major sites of renal Mg2+ reabsorption are, besides the PT (10–20%), the thick ascending loop of Henle (TAL, 65–70%) and the distal convoluted tubule (DCT, 10%) (Figure 2). Beyond the DCT, no significant mechanisms of Mg2+ absorption have been described so far (11, 12).
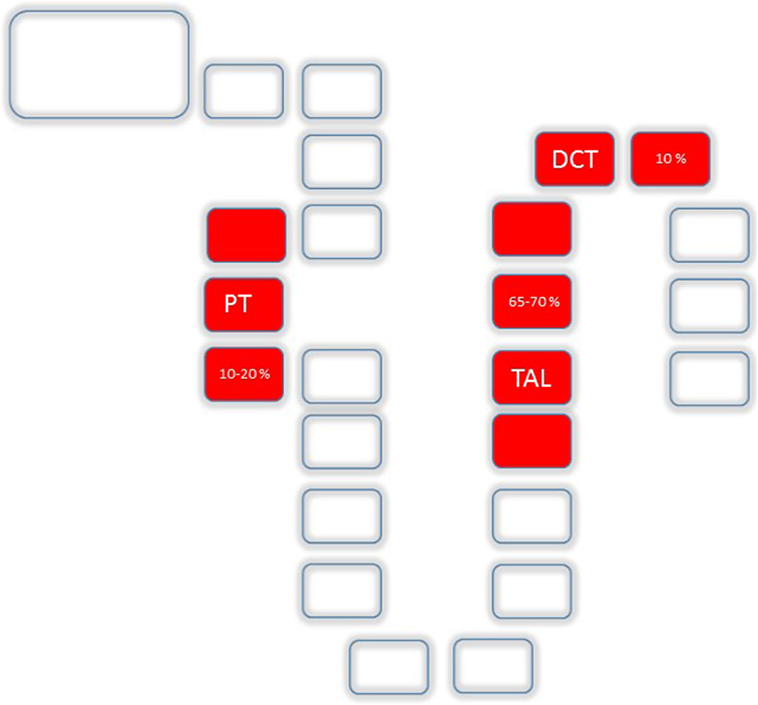
Figure 2. Mg2+ absorption along the nephron: Uptake along the nephron is realized by the proximal tubule (PT) (10–20%), the TAL (65–70%) and the distal convoluted tubule (DCT, 10%).
Mg2+ absorption in the PT and TAL occurs mainly via paracellular route and in the DCT the transcellular route (13). The paracellular route is mainly determined by the Tight Junction (TJ), a supramolecular organization of membrane-bound proteins and their intracellular adaptor- and scaffolding proteins. The major proteins of the TJ comprise Claudins (from lat. claudere: to seal), a protein family consisting of at least 24 members in Eukaryotes enabling the TJ to function as either (a) barrier, (b) fence, or (c) channel (Figure 3). The Barrier function of the TJ enables the organism to increase transepithelial resistance (TER; Ω × cm2) where needed. As such, the PT has a low resistance (6–10 Ω × cm2) whereas downstream, toward TAL (11–34 Ω × cm2) and the Collecting Duct (60 Ω × cm2; MDCK cells) TER is constantly increasing (14, 15). The bladder, in order to fulfill its function of a tight reservoir, has the highest epithelial resistance [>300 kΩ × cm2 (16); Figure 3A]. The Fence function of the TJ is a key element that contributes to the apical-to-basolateral orientation of epithelial cells. In order to guarantee a coordinated, regulated transcellular transport, membrane-bound proteins must be oriented stable at either apical or the basolateral side. This principal requirement of all epithelial tissues is mainly realized by the TJ (17). Likewise, the loss of the apical-to-basolateral orientation, i.e., the loss of polarity and even cell-to-cell contact is an early event in tumor development. Several claudins have been shown to be potential markers of gastrointestinal tumors and their progression (18). Moreover, other Claudins (e.g., Claudin-3) have been demonstrated to be involved, in men and animals in tumor invasiveness and in autoimmune disorders (18, 19). Also, several Claudins are vital for teeth development in mice and humans (20). A great step toward the determination of the role of Claudins has been achieved by the resolution of the crystal structure of the Claudin protein Claudin-15 (21, 22) (Figure 3B). Although their contribution to human disease has been demonstrated, proteins that directly interfere with Claudins have only been preliminarily characterized (23, 24). The Channel function of the TJ is crucial for ions (like Mg2+ and Ca2+) and H20 absorption and is dependent on an existing electro-chemical gradient (17). The Channel function is realized by a surplus of one charge on one side of the epithelial layer than on the other side (e.g., apical vs. basolateral or vice versa) or on the surplus of a molecule or substance over the epithelial layer. Although the TJ does not possess pumps or antiporters, transport can nevertheless be selective by the variation and regulation of TJ composition and protein expression (25). Thus, the expression of different Claudins along epithelial tissues and their regulation and expression can determine Mg2+ spatial and temporal reabsorption (26) (Figure 4).
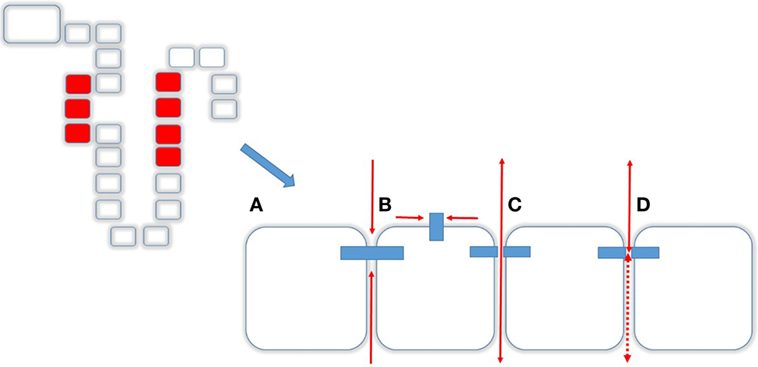
Figure 3. Transcellular transport. Tight junctions (TJs) and Caudins are responsible for different functions (A) Barrier, i.e., no transit in any direction, (B) Fence, enabling a cell apical-to-basolateral orientation, (C) Channel function, allowing molecules to pass from apical to basolateral and vice versa, and (D) depending on the composition of the TJ.
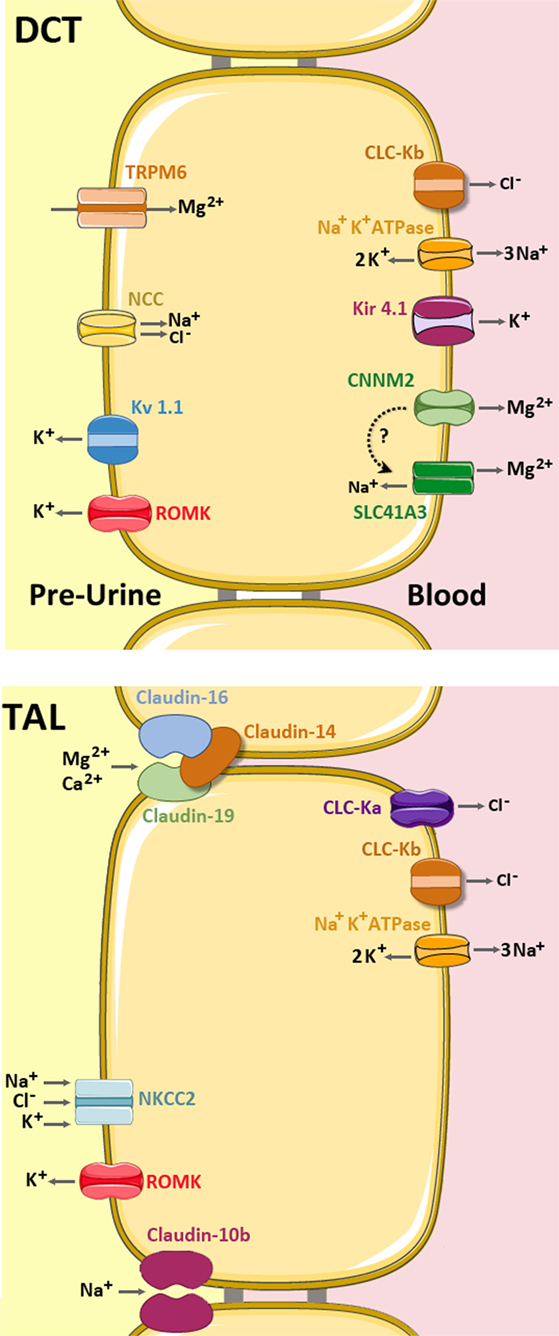
Figure 4. Mg2+ handling network along the TAL and distal convoluted tubule (DCT). In the TAL (bottom), Mg2+ and Ca2+ are absorbed via the permeable channel formed by Claudin-16 and Claudin-19, which is inhibited by Claudin-14 (light brown). Claudin-10b is a cation-selective channel with high preference for Na+. The apical force needed for Mg2+ reabsorption is created by the cooperative activities of the basolateral Na+ K+ ATPase pump, the Chloride channels ClC-Ka and ClC-Kb, and the apical Na+ K+ 2Cl− cotransporter (NKCC2) and the K+ extruder, ROMK. (up) In the DCT, Mg2+ is mainly absorbed by transcellular routes. The Mg2+ ions enter into the cell through the apical TRPM6 channel and are then extruded by either CNNM2 and/or SLC41A3 (still under debate). The apical force needed for Mg2+ transport is created by the cooperative action of the basolateral Na+ K+ ATPase, the Cl- channel ClC-Kb, the K+ transporter Kir 4.1 and the apical Na+ Cl− cotransporter (NCC) and the K+ extruders, ROMK and Kv 1.1 (5, 27).
Although our knowledge on Mg2+ handling in the more distal nephron has increased significantly, little is known about Mg2+ transport in the PT, where it is believed to be transported by the paracellular way. There, terminus “bulk reabsorption” is used widely although this phenomenon has not been elucidated in detailed molecular or physiological context.
Bartter’s Syndrome, first described by the endocrinologist Frederick Bartter, is characterized by renal wasting of Na+, K+, and polyuria. Clinically, hyperaldosteronism in preterm borns, polyhydramnion and prematurity became a hallmark. The seminal work of the Lifton and Hildebrandt groups showed that mutations in the genes that code for the apical transport in the TAL of Na+, K+ and Cl− (NKCC2, ROMK2) as well as the basolateral extrusion mechanisms (ClC-Ka, Barttin) are essential for Na+ K+ and Cl− handling in the TAL (Bartter’s Syndrome types 1–4) (28–33) Here, Mg2+ wasting is not caused by a direct mechanisms, and likewise Mg2+ wasting is a clinical hint, but not a mandatory feature of Bartter’s syndrome. Mg2+ wasting in Bartter’s Syndrome is also believed to be secondary to polyurias (and polydipsia) present in these patients. Thus, all the proteins are classified into Category 3 (Table 1) supported by the fact that also mice with a targeted deletion of NKCC2 show a Bartter-like phenotype but no hypomagnesemia (34). Interestingly, a transient form of Bartter’s syndrome has been described in several patients recently. Patients displayed the prenatal (Polyhydramnion, Prematurity) and postnatal (Hyponatrimia, Hypokalemia) clinical hallmarks of Bartter’s syndrome. The authors have shown that these disorders are caused by mutations in the gene encoding melanoma-associated antigen D2, explaining that fact that only males were affected (35, 36). Although the authors have not reported on Mg2+ levels, it is intriguing that this disorder is a more common cause of neonatal hypomagnesemia than anticipated that far.
The TAL is separated anatomically but also by its functional elements in the medullary and the cortical part (mTAL and cTAL). An important driving force in epithelial tissues and, therefore, also in the TAL, in order to maintain a transepithelial gradient is generated by the basolateral Na+ K+ ATPase. Mutations in the gene (FYXD2) coding for the γ-subunit of this protein have been shown to cause dominant familial Hypomagnesemia. Thus, it has been shown that this protein is a “conditio-sine-qua-non” for generating the necessary transepithelial gradient for transport systems and, thus, also for renal Mg2+ handling (11, 12, 37), this protein can be classified into the Category 2.
During the last years, several proteins have been shown to be involved in renal paracellular ion transport. However, a clear phenotype–genotype correlation has established in some but not in all of the genes and proteins involved. An example is provided by CLDN14. Mutations in CLDN14 cause non-syndromic deafness in men and mice (38, 39). Affected human individuals do not display overt renal abnormalities and the same holds true for mice with targeted deletions in CLDN14 (38). On the other hand, genome-wide association studies identified CLDN14 variants as a major risk gene associated with hypercalciuric stone disease (4, 40, 41). Furthermore, in the TAL, mutations in human genes (CLDN10, CLDN14, CLDN16, and CLDN19), that define the paracellular pathway (Figure 4) of Mg2+ absorption have been shown to cause rare human disorders. In the TAL, where significant Mg2+ and Ca2+ transcellular transport is absent, paracellular transport is of vital importance and is driven by a lumen-positive potential. Mutations in CLDN16 cause an autosomal-recessive disorder called “Familial Hypomagnesemia with Hypercalciuria and Nephrocalcinosis” (FHHNC) (42). Patients affected display renal Mg2+ and Ca2+ wasting accompanied by nephrocalcinosis. This disorder causes in most of the cases end stage renal disease, leading in many, but not all cases to the need for renal transplantation (43). A clinical significant problem is given by the fact that there currently is no general screening procedure at the neonatal stage or later for toddles and schoolchildren on hypercalciuria or hypermagnesemia. Thus, if such patients are referred to a secondary or tertiary center, respectively, chronic renal insufficiency and calcifications have often already progressed to a severe and often, irreversible state. In contrast to the human situation, mice with targeted deletion of CLDN16 recapitulate human renal Mg2+ and Ca2+ wasting, but do not show any signs of overt renal calcifications and, moreover, no signs of apparent or progressing renal insufficiency (44). There is currently no explanation for this significant difference; however, it is intriguing that solving this striking difference, a possible route for treatment of patients may be opened. A novel recent finding was that the absence of CLDN16 in ameloblasts explains the clinical finding of amelogenesis imperfecta in FHHNC patients and mice with CLDN16 deficiency (45). A similar finding was reported for patients with mutations in the genes coding for CLDN19 and similar for mice (CLDN3) (20, 46).
In 2006, Konrad et al. reported that mutations in human CLDN19 also lead to renal Mg2+ and Ca2+ wasting, clinically an almost phenocopy of patients with CLDN16 mutations (47). Although still a matter of debate, it is believed that CLDN16 and CLDN19 interact and form a heteromeric paracellular channel, with a cation selectivity including Ca2+ and Mg2+ (48) (Figure 4). However, they could also show that patients harboring CLDN19 mutations suffer from severe ocular involvement, as Claudin-19, but not Claudin-16 is expressed in the retinal pigment epithelium TJs, leading besides the renal Mg2+ and Ca2+ wasting to major vision problems (47, 49).
Another Claudin (Claudin-10) that contributes to Mg2+ handling exists at least in two principal forms. Claudin-10a and -10b, both confer different electrophysiological properties (anion-selective channel vs. cation-selective channel with a high preference for Na+) and their tissue distribution (Kidney and Uterus for Claudin-10a and ubiquitously for Claudin-10b) (50–52) (Figure 4). The expression of the Claudin-10b in almost every epithelial tissue might explain that mice with a targeted deletion die soon after birth. In sharp contrast, the generation of a kidney specific CLDN10-KO mouse (by the use of a ksp-Cre deleter strain) led to a vital mouse model. However, these mice displayed hypermagnesemia, hypocalciuria, nephrocalcinosis, and polyuria (53). Isolated mouse tubules of the TAL demonstrated a decreased paracellular Na+ permeability as well as higher expression of Claudin-16. Interestingly, recently four independent groups have reported mutations in human CLDN10 (54–57). Bongers et al. reported on two non-related patients presenting with alkalosis, hypokalemia, hypocalciuria, and hypercalcemia and a serum Mg2+ in the upper range of normal. They identified heterozygous mutations (P149R, Glu157_Tyr192del, and D73N) in two unrelated families. Hadj-Rabia et al. reported mutations in six patients from two unrelated families (S131L, M1T), resulting in an absence of CLDN10 at the plasma membrane. Affected members had high serum Mg2+ levels and renal loss of K+, Na+ and Cl−. Of interest is that patients also suffered from a variety of skin and teeth disorders (hypolacrymia, ichthyosis, xerostomia, and severe enamel wear). A similar renal phenotype was reported by Klar and colleagues (55). They identified a CLDN10 Mutation (N48K) in two distantly related families with 13 affected individuals presenting with anhidrosis and the inability to produce tears. Although serum levels of Na+ and K+ were in the normal range, all patients present with high Mg2+ serum levels. All groups identified homozygous or compound heterozygous mutations in CLDN10 and demonstrated thereby unequivocally the importance of Claudin-10 for human Mg2+ homeostasis. However, the recently described homozygote mutation (G163A) in a patient by Terliesner was reported to have normomagnesemia (57). So far, several different mutations in human CLDN10 have been shown to cause a renal tubular disorder that is characterized by hypokalemia, alkalosis, and hypermagnesemia. Moreover, as Claudin-10 is expressed in skin tissues, and several different symptoms of disordered dermal Na+ homeostasis could, therefore, be attributed to this defect.
Interestingly, the full CLDN10 knock-out mouse dies a few hours after birth, indicating that one or more organs different than the kidney and skin must be, if deficient for Claudin-10, vital for survival. Thus, the fact that human mutation does not lead to a lethal phenotype are intriguing in terms of compensatory mechanisms. However, the fact that Claudin-10 is expressed in the lung leads to spectate that the primary cause of postnatal death is caused by the absence of Claudin-10 in the lung (58, 59).
A corresponding mouse model has been generated by Breiderhoff and colleagues, by crossbreeding Claudin-16-deficient mice with a kidney-specific Claudin-10-deficient mouse strain (44, 53). Combining a hypomagnesemic model (CLDN16−/−) with a hypermagnesemic model (kidney specific CLDN10−/−) resulted in a normomagnesemic mouse, thus a “restored” normal phenotype (60). These findings point at the high compensatory, and more than so far anticipated capacity of the DCT. Put in perspective, the development of a selective renal Claudin-10b blocking agent could be a therapeutic option for Claudin-16 patients, since obviously the block of a Na+ pore restores the capacity of Mg2+ and Ca2+ recovery more distantly, i.e., the DCT.
In the DCT, Mg2+ reabsorption takes place mainly by transcellular route. Here, apical uptake, intracellular buffering, transport, and the extrusion at the basolateral site is concerted by a highly defined and regulated (e.g., hormonal) molecular machinery that has been recently reviewed in Ref. (5, 27). Among the eight different types of Mg2+ transport factors identified in eukaryotes (TRPM6/M7, Mrs2, MMgT, MagT1, SLC41 family, NIPA, HIP14, and CNNMs) (61–67), only three are expressed at the DCT. The selected list includes (1) the transient receptor potential channel melastatin member 6 (TRPM6) (68), (2) the third member of the solute carrier SLC41A family (SLC41A3) (69), and (3) the “Cyclin and CBS Domain Divalent Metal Cation Transport Mediator-2” (CNNM2), also referred to as ACDP2 (ancient conserved domain protein-2) (63, 65) (Figure 4).
These three proteins are classified in the first category of Table 1 as they are directly involved in Mg2+ transport or have been related to direct Mg2+ handling in the DCT.
TRPM6 was the first molecularly identified protein involved in active Mg2+ reabsorption (68). This channel, which associates in homotetramers, and may also interact with its closest homolog TRPM7 to form heterotetrameric species (70), is five times more permeable to Mg2+ than to Ca2+, and permits the reabsorption of these cations through the apical membrane of the epithelial cells (68). The three-dimensional structure of TRPM6 still remains unsolved, but its domain distribution is known and includes a cytosolic N-terminus followed by a transmembrane region of six α-helices and a long intracellular C-terminus that contains a serine-threonine active kinase domain similar to that present in α-kinases. The ion pore is putatively located between the fifth and sixth α-helices of the transmembrane section (71). Interestingly, mutations that impair the phosphorylation of threonine at position 1851 (72) decrease the protein transport activity. These findings have led to propose that autophosphorylation is a key step in the regulatory mechanism of Mg2+ transport through this channel (73). Clinical or genetical disturbances in TRPM6 are linked to different diseases. For example, mutations in its amino acid sequence cause the rare autosomal-recessive familial hypomagnesemia with secondary hypocalcemia (74–76). Other variants have been linked to hypoparathyroidism (77) and breast cancer (78). Of note, genetic ablation of the TRPM6 gene in mice results in early embryonic lethality (79). Interestingly, TRPM6± mice showed reduced expression of the channel in kidney and colon, resulting in mild hypomagnesemia with no hypocalcemia (80).
A set of other mutations in genes that are not primarily associated with Mg2+, but relevant for the establishment of the apical membrane potential to drive Mg2+ entry through TRPM6 (and, therefore, classified in categories 2 and 3 in Table 1), have been identified causing secondary Mg2+ wasting. The corresponding genes code for transcriptional factors (HNF1B and PCBD1), growth factors (EGF), (co)-transporters (NCC, encoded by gene SLC12A3), or even ion-channels (Kir4.1 encoded by KCNJ10) (66, 81–84) (Figure 4). According to the gene mutated, the resulting phenotype comprises cystic kidney disease, diabetes, electrolyte disturbances other than Mg2+, or seizures. Clinically, hypomagnesemia is, compared to coexisting diabetes (MODY5), the chronic kidney disease and the hyperphenylinaemia a subordinated problem to the patient. However, identifying hypomagnesemia might be of value to identify the comorbidities at an early stage (85). Furthermore, elucidating the mechanisms that contribute to the disordered handling of Mg2+ in these patients may also enable a better understanding and treatment of diabetes, cyctic kidney disease, and hyperphenylinemia.
Inactivating mutations in SLC12A3 cause Gitelman syndrome, the most frequent cause of hereditary hypomagnesemia and characterized by hypokalemic metabolic alkalosis with hypomagnesemia and hypocalciuria. It has been proposed that a decrease activity of the NCC protein affects the membrane potential necessary for Mg2+ reabsorption in the apical membrane of DCT by TRPM6 (86–89).
SLC41A3 was originally described by Quamme as part of the solute carrier family 41 (65), which encompasses three integral cytoplasmic membrane putative Mg2+ transporters (SLC41A1, -A2, and -A3) (90). Mutations in this family are linked to Parkinson’s disease (91), diabetes (92), and nephrolithiasis (93). As in the case of TRPM6, the three-dimensional structure of these transporters remains unsolved, but is known to be built up of 10 or 11 transmembrane α-helices (94). SLC41A3, whose molecular function and interaction partners remain also elusive, is the highest enriched member in the DCT. Recently, de Baaij et al. found that, a Slc41a3−/− knockout mice suffer from hypomagnesemia and normomagnesiuria, accompanied by upregulation of TRPM6 and SLC41A (69). These results underlined the relevant role played by SLC41A3 in Mg2+ reabsorption.
The third identified Mg2+ transport mediator expressed in the DCT is CNNM2, which belongs to the Cyclin M family. This family encompasses four different members (CNNM1-4). Mutations in CNNM2 cause dominant familial hypomagnesemia (67), and have been linked to brain development anomalies (95), hypertension, diabetes, and obesity (96, 97). Moreover, the CNNM2 locus has been linked by GWAS to neuro-psychiatric disorders (e.g., Schizophrenia) (98, 99). In 2014, Arjona et al. found that knockdown of CNNM2 orthologs in zebrafish results in brain abnormalities, increase of spontaneous contractions, and Mg2+ waste (95). These authors also identified five new families with mutations in CNNM2 that suffered hypomagnesemia with mental retardation and seizures. These findings suggested an essential role of CNNM2 in Mg2+ homeostasis and brain development. The relevant role of CNNM2 is underlined by the fact that mice lacking CNNM2 are embryonic lethal (100). Heterozygous (Cnnm2+/−) mice show lower Mg2+ levels in serum, thus suggesting defects in Mg2+ reabsorption in kidney. In addition, these animals showed lower blood pressure than compared to control mice. These results highlighted the importance of Mg2+ and its reabsorption in the kidney to maintain blood pressure (100).
The CNNMs represent the least-studied members across the mammalian transporters and share with MgtE and with the CLC family of chloride channels the presence of a cystathionine β-synthase (CBS) domain pair in their amino acid sequence (101–103). The four CNNM family members were first identified in 2003 by Wang et al. (61, 104) and show very strong homology to the bacterial CorC protein [which is involved in Mg2+ and cobalt (Co2+) efflux (63)], and with the Mam3p proteins (67, 105). It was initially suggested that CNNMs might be involved in cell-cycle regulation (61), as they contain a cyclin box-like motif and are located in the plasma membrane. However, the cyclin M function has that far not been proved in vivo.
The second member of the Cnnm family, CNNM2, is abundant in brain and kidney (64, 65), and shows a complex modular architecture composed by four structural domains (Figure 5) (106). The N-terminal section (likely to be an extracellular compoment) consists of a β-stranded enriched region (residues 1–250) and precedes a DUF21 domain (residues 251–400, Pfam code PF01595) with three or four transmembrane α-helices (107). The following intracellular region includes a CBS domain pair (so called “Bateman” module; Pfam code PF00571) (106) and a C-terminal cyclic nucleotide monophosphate (cNMP) like binding domain (Pfam code PF00027) (106) (Figure 5). Although the concrete function of each domain remains unknown, recent biophysical and structural data supports a regulatory role for the Bateman module.
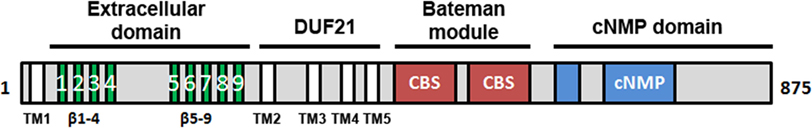
Figure 5. Domain distribution of CNNM2. CNNM2 is a 97 kDa protein of 875 amino acid residues, formed by four different domains: (i) the N-terminal extracellular region is enriched in β-strands and presents a large cleavage signal peptide of approximately 65 residues. This zone is preceded by a transmembrane α-helix (TM1); (ii) the following DUF21 domain includes 3 or 4 transmembrane helices (TM1-4) that are putatively responsible for Mg2+ transport across the membrane; the intracellular part includes two domains: (iii) a Bateman module consisting of two consecutive CBS domains and (iv) a cyclic nucleotide monophosphate (cNMP)-like domain. CNNM2 shows two isoforms (Iso1 and Iso2). Iso2_CNNM2 lacks residues 721–742.
Despite the universally recognized relevance of magnesium in maintaining key life processes as mentioned above, current knowledge about the CNNMs and their role in the DCT, as well as the molecular mechanisms involved in its transport across the cellular membranes remain still to be explored. This is largely due to the scarce structural information available on Mg2+ transporters and channels, that so far was limited to the crystal structures of two prokaryotic proteins: (i) CorA from Thermotoga maritima (108–111) and (ii) MgtE from Thermus thermophilus (112–115). These proteins are homologs of two eukaryotic Mg2+ transport mediators: the mitochondrial Mg2+ channel Mrs2 and the solute carrier (SLC) family 41 members (homologs of CorA and MgtE, respectively). CorA represents the major transport machinery responsible for Mg2+ uptake in bacteria and it translocates Mg2+ by using an inwardly biased electrochemical gradient that serves as the driving force for Mg2+ permeation (108, 109, 111, 116).
MgtE is a dimeric Mg2+ selective channel (117) that permeates Mg2+ ions and maintains the intracellular Mg2+ homeostasis in bacteria. MgtE shares with CNNM2 (but not with SLC41) the presence of an intracellular CBS domain pair (101, 102). An ATP/Mg-dependent open-to-close gating process that involves binding of the nucleotide at this region defines the threshold of intracellular Mg2+ for the channel inactivation and provides sensory capacity to this protein (113, 114, 118).
The recent elucidation of the crystal structure of the Bateman module of CNNM2 (119, 120) has shed new light on the molecular mechanisms underlying Mg2+ transport through the basolateral membrane of the DCT (Figure 6). We and others recently confirmed that this region may host ATP in a Mg2+-dependent manner, as well as independent Mg2+ atoms that interact with some acidic clusters located in the CBS1 motifs (106, 119). The fact that the Mg2+ independent sites are far away from the nucleotide suggest that, as observed in MgtE (114), binding of Mg2+ at concrete positions may not be directly coupled with ATP binding, although it may complement its effect in conformational transformations suffered by the whole module (106, 121).
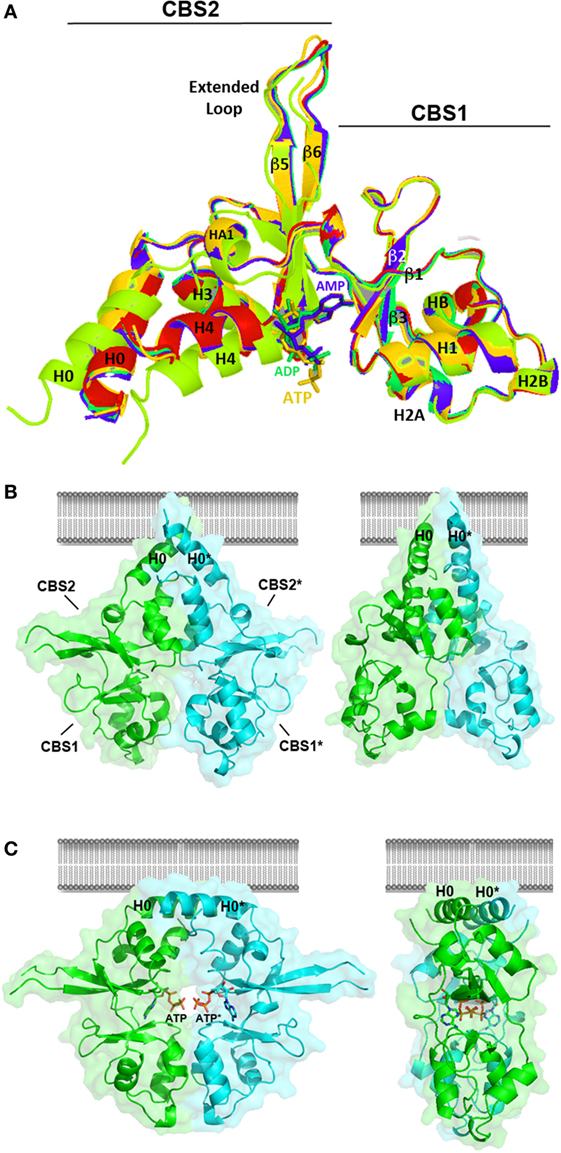
Figure 6. Crystal structure of the Bateman module of CNNM2. (A) The Bateman module of CNNM2 consists of two consecutive cystathionine β-synthase (CBS) motifs (CBS1, residues 445–508; CBS2, residues 509–578). A long extended loop links strands β5 and β6 in the CBS2 motif. The H0 helix connects CBS1 with the DUF21 transmembrane domain in the full-length protein. The H4 helix connects CBS2 with the cyclic nucleotide monophosphate domain. Nucleotides, ca. AMP (blue), ADP (green), or adenosine triphosphosphate (ATP) (orange) bind independently at the S2 site, thus disrupting the interactions formerly existing in the cavity between residues of the CBS1 and CBS2 motifs. This induces a displacement of helices H0, H1, and H4 in each Bateman subunit. The apo- and the nucleotide-bound Bateman module are represented in green and marine, respectively. The crystal structure of the T568I protein variant is in red. As shown, the T568I mutation mimics the structural effect of ATP binding; although in the first case, the structural change is irreversible, thus locking the protein in the nucleotide-bound like conformation. (B,C) Conformational changes induced by ATP. The Bateman module of CNNM2 associates in disk-like dimers known as “CBS modules,” which adopt a twisted (B) or a flat (C) state depending on whether the site S2 of each subunit is empty or hosts an ATP (or MgATP) molecule, respectively. Note that the H0 helices connecting the CBS2 motif with the DUF21 domain are differently oriented in each case, thus likely transmitting the transformation suffered by the Bateman module to the transmembrane region.
The Bateman module of CNNM2 features two major cavities (named as S1 and S2) that are located at opposite ends of the central β-sheets of the CBS motifs. In contrast with S1, which is partially occluded and full of bulky residues, the site S2 is fully accessible and can accommodate phospho-nucleotides, such as AMP, ADP, or ATP (106, 121) (Figure 6). Site S2 is built by three different structural blocks: (i) the central residues from the linker preceding the first β-strand (β1) of the CBS1 domain, (ii) the C-terminal residues from the last β-strand (β6) of the CBS2 motif, and (iii) the first two turns of helix H4 of CBS2 (Figure 6). The upper and right walls of the cavity are mainly hydrophobic and help accommodating the bulky adenine ring of ATP (106, 121). By contrast, the left wall of the cleft is hydrophilic and is built from the last β-strand and the following α-helix (H4) of the CBS2 motif. A conserved threonine (T568) and an aspartate residue (D571) from this helix are key in help orienting the ribose ring of the nucleosides inside the cavity, and if mutated, impede the allocation of ATP inside (106). Interestingly, the repulsive effect otherwise exerted by the acidic cluster formed by residues E570, D571, and E574 (at the first turn of α-helix H4 of CBS2) over the polyphosphate chain of ATP (106) is neutralized by the Mg atom that accompanies the ATP molecule. The positive dipole end of helix H4 and the nearby arginine residue, R480, complement the neutralizing effect (106). The Bateman module of CNNMs associates in head-to-head oriented disk-like dimers known as CBS modules (Figures 6B,C) (106, 121).
At low concentrations of Mg2+ and in the absence of MgATP, the CBS module adopts a “twisted” shape (Figure 6B), in which the CBS2 domains from complementary subunits remain in contact while the CBS1 motifs are separated and retain only scarce hydrophobic interactions. Binding of MgATP at site S2 disrupts a network of H-bonds centered on the conserved threonine at position 568 (T568) as well as a salt bridge between R480 and E570 and causes the displacement the C-terminal helix of the CBS2 domain (helix H4) as well as of the long α-helix (helix H0) that connects the Bateman module with the DUF21 domain (Figure 5). These structural changes, which occur concomitantly in the two complementary subunits of the dimer, trigger an overall rearrangement of the CBS module that makes it to evolve from a “twisted” (Figure 6B) toward a “flat” disk structure (Figure 6C). The conformational effect of ATP is likely transferred to the DUF21 transmembrane domain through helix (H0) that connects it with the CBS2 motif. This sequence of events has been postulated as the mechanism by which CNNM2 might regulate the gating of Mg2+ ions through the basolateral cell membrane (106, 121) (Figure 7).
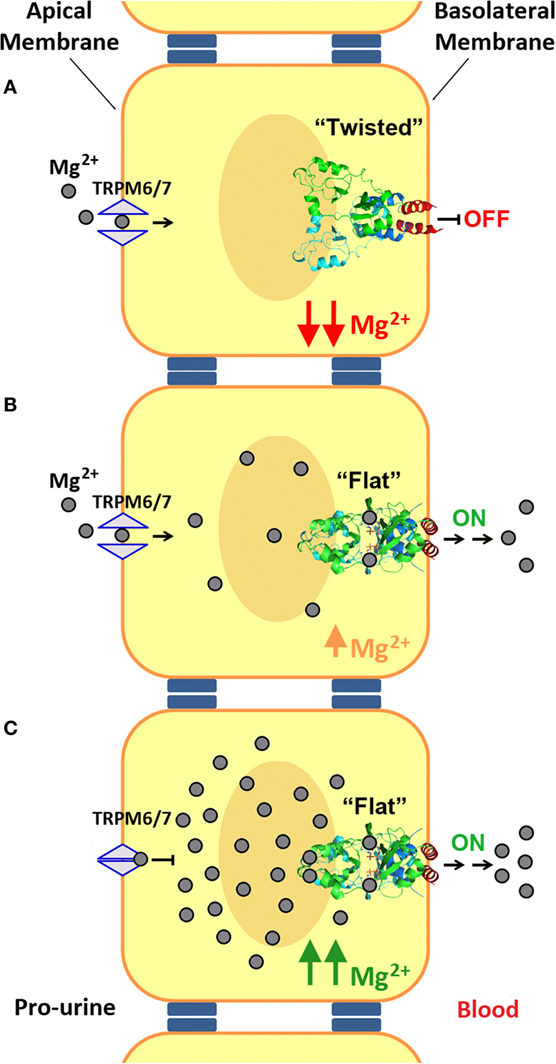
Figure 7. Mechanism of Mg2+ transport at the distal convoluted tubule (DCT). (A) Mg2+ enters into the DCT epithelial cells through the apical membrane with the help of TRPM6/7 channels. At low Mg2+ concentrations, the cystathionine β-synthase (CBS) module of CNNM2, located at the basolateral membrane, remains in its twisted conformation. (B,C) Upon increasing the intracellular concentration of Mg2+, binding of these cations and of MgATP to CNNM2, triggers the progression of the CBS module toward its flat state, and the transport of Mg2+ through the basolateral membrane toward the blood torrent. In addition, an increased intracellular Mg2+ concentration inhibits apical transport by TRPM6.
Interestingly, an ATP/Mg-mediated gating process ruled out by CBS domains has also been postulated as the mechanism by which the MgtE transporter senses and regulate the Mg2+ homeostasis in bacteria (114). In contrast with the twisted-to-flat transformation observed in CNNM, the rearrangement of the Bateman modules of MgtE responds to an open-to-close mechanism (113–115). In the absence of Mg2+ ions, the complementary CBS2 motifs of MgtE subunits remain apart in the dimer due to the repulsion exerted by acidic clusters located at the interfacial helices of the CBS2 domains. In this state, the CBS module impairs the transport of Mg through the membrane and maintains the protein in an “open” state. While the intracellular concentration of Mg2+ is low, the CBS module remains open and allows the influx of Mg2+ ions toward the interior of the cell. Upon increasing the intracellular concentration of Mg2+, the pre-existing repulsive acidic clusters become sequentially neutralized by newly bound Mg2+ ions, thus allowing the approximation of the CBS2 motifs. The sequential binding of Mg atoms progressively causes a closure of the CBS module that, when completed, adopts a “flat” disk-like arrangement as that observed in the MgATP/Mg2+ bound form of CNNM2 (106). The new state is transferred to the transmembrane region and results in the closure of the membrane pore (114).
The identification of mutations in human genes has led to a deeper understanding of Mg2+ handling in health and disease. On that basis, technologies, such as mouse genetic engineering as well as crystallography, have contributed in this field of physiology and pathophysiology. However, our current knowledge of the molecular mechanisms underlying magnesium transport through the cell membranes is very scarce, and represents an incipient field of research that will mature, as we are able to identify new molecular partners involved in this process. Proteins involved in trans- and paracellular pathways have the potential of being key players in Magnesium Homeostasis and also other disorders, such as diabetes, hypertension, and schizophrenia, thus being a potential target for pharmaceutical interventions.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part by Departamento de Educación, Universidades e Investigación del Gobierno Vasco Grant PI2010-17, Departamento de Industria, Innovación, Comercio y Turismo del Gobierno Vasco Grants ETORTEK IE05-14 and IE07-202, Diputación Foral de Bizkaia Grants 7/13/08/2006/11 and 7/13/08/2005/14, Spanish Ministerio de Ciencia e Innovación (MICINN), Grant BFU2010-17857, Spanish Ministry of Economy and Competitiveness Grant BFU2013-47531-R, BFU2016-77408-R from Spanish Ministry of Economy and Competitiveness (MINECO) and Ministerio de Ciencia e Innovación CONSOLIDER-INGENIO 2010 Program Grant CSD2008-00005 (to LM-C). We also thank MINECO for the Severo Ochoa Excellence Accreditation (SEV-2016-0644) and by a PhD fellowship from MINECO (REF BES-2014-068464) awarded to PG-M. DM was supported by the German Research Foundation (DFG, Graduate School 2318) and by The Berlin Institute of Health (BIH) G(CRG 12.01.134).
1. Rude RK. Magnesium. 11th ed. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, editors. Modern Nutrition in Health and Disease. Baltimore, MA: Lippincott Williams & Wilkins (2012). p. 159–75.
2. Romani AM. Chapter 3. Magnesium in health and disease. In: Sigel A, Sigel H, Sigel RKO, editors. Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences. (Vol. 13), Dordrecht: Springer (2013). p. 49–79.
3. Kass L, Weekes J, Carpenter L. Effect of magnesium supplementation on blood pressure: a meta-analysis. Eur J Clin Nutr (2012) 66(4):411–8. doi:10.1038/ejcn.2012.4
4. Corre T, Arjona FJ, Hayward C, Youhanna S, de Baaij JHF, Belge H, et al. Genome-wide meta-analysis unravels interactions between magnesium homeostasis and metabolic phenotypes. J Am Soc Nephrol (2018) 29(1):335–48. doi:10.1681/ASN.2017030267
5. Li H, Sun S, Chen J, Xu G, Wang H, Qian Q. Genetics of Magnesium Disorders. Kidney Dis (Basel) (2017) 3(3):85–97. doi:10.1159/000477730
6. Cowan JA. Introduction to the Biological Chemistry of Magnesium. The Biological Chemistry of Magnesium. New York, NY: VCH (1995).
7. Rude RK. Magnesium. 2nd ed. In: Coates PM, Betz JM, Blackman MR, Cragg GM, Levine M, Moss J, et al., editors. Encyclopedia of Dietary Supplements. New York, NY: Informa Healthcare (2010). p. 527–37.
8. Kanbay M, Goldsmith D, Uyar ME, Turgut F, Covic A. Magnesium in chronic kidney disease: challenges and opportunities. Blood Purif (2010) 29(3):280–92. doi:10.1159/000276665
9. Verberckmoes SC, Persy V, Behets GJ, Neven E, Hufkens A, Zebger-Gong H, et al. Uremia-related vascular calcification: more than apatite deposition. Kidney Int (2007) 71(4):298–303. doi:10.1038/sj.ki.5002028
10. Fine KD, Santa Ana CA, Porter JL, Fordtran JS. Intestinal absorption of magnesium from food and supplements. J Clin Invest (1991) 88(2):396–402. doi:10.1172/JCI115317
11. de Baaij JH, Hoenderop JG, Bindels RJ. Magnesium in man: implications for health and disease. Physiol Rev (2015) 95(1):1–46. doi:10.1152/physrev.00012.2014
12. de Baaij JH, Dorresteijn EM, Hennekam EA, Kamsteeg EJ, Meijer R, Dahan K, et al. Recurrent FXYD2 p.Gly41Arg mutation in patients with isolated dominant hypomagnesaemia. Nephrol Dial Transplant (2015) 30(6):952–7. doi:10.1093/ndt/gfv014
13. Blaine J, Chonchol M, Levi M. Renal control of calcium, phosphate, and magnesium homeostasis. Clin J Am Soc Nephrol (2015) 10(7):1257–72. doi:10.2215/CJN.09750913
14. Seely JF. Variation in electrical resistance along length of rat proximal convoluted tubule. Am J Physiol (1973) 225(1):48–57.
15. Burg M, Good D. Sodium chloride coupled transport in mammalian nephrons. Annu Rev Physiol (1983) 45:533–47. doi:10.1146/annurev.ph.45.030183.002533
16. Reuss L, Finn AL. Passive electrical properties of toad urinary bladder epithelium. Intercellular electrical coupling and transepithelial cellular and shunt conductances. J Gen Physiol (1974) 64(1):1–25. doi:10.1085/jgp.64.1.1
17. Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol (2016) 17(9):564–80. doi:10.1038/nrm.2016.80
18. Ahmad R, Kumar B, Chen Z, Chen X, Müller D, Lele SM, et al. Loss of claudin-3 expression induces IL6/gp130/Stat3 signaling to promote colon cancer malignancy by hyperactivating Wnt/β-catenin signaling. Oncogene (2017) 36(47):6592–604. doi:10.1038/onc.2017.259
19. Kooij G, Kopplin K, Blasig R, Stuiver M, Koning N, Goverse G, et al. Disturbed function of the blood-cerebrospinal fluid barrier aggravates neuro-inflammation. Acta Neuropathol (2014) 128(2):267–77. doi:10.1007/s00401-013-1227-1
20. Bardet C, Ribes S, Wu Y, Diallo MT, Salmon B, Breiderhoff T, et al. Loss-of-function disrupts tight junctions and impairs amelogenesis. Front Physiol (2017) 8:326. doi:10.3389/fphys.2017.00326
21. Suzuki H, Nishizawa T, Tani K, Yamazaki Y, Tamura A, Ishitani R, et al. Crystal structure of a claudin provides insight into the architecture of tight junctions. Science (2014) 344(6181):304–7. doi:10.1126/science.1248571
22. Suzuki H, Tani K, Fujiyoshi Y. Crystal structures of claudins: insights into their intermolecular interactions. Ann N Y Acad Sci (2017) 1397(1):25–34. doi:10.1111/nyas.13371
23. Müller D, Kausalya PJ, Claverie-Martin F, Meij IC, Eggert P, Garcia-Nieto V, et al. A novel claudin 16 mutation associated with childhood hypercalciuria abolishes binding to ZO-1 and results in lysosomal mistargeting. Am J Hum Genet (2003) 73(6):1293–301. doi:10.1086/380418
24. Liu F, Koval M, Ranganathan S, Fanayan S, Hancock WS, Lundberg EK, et al. Systems proteomics view of the endogenous human claudin protein family. J Proteome Res (2016) 15(2):339–59. doi:10.1021/acs.jproteome.5b00769
25. Milatz S, Himmerkus N, Wulfmeyer VC, Drewell H, Mutig K, Hou J, et al. Mosaic expression of claudins in thick ascending limbs of Henle results in spatial separation of paracellular Na+ and Mg2+ transport. Proc Natl Acad Sci U S A (2017) 114(2):E219–27. doi:10.1073/pnas.1611684114
26. Gong Y, Hou J. Claudins in barrier and transport function-the kidney. Pflugers Arch (2017) 469(1):105–13. doi:10.1007/s00424-016-1906-6
27. Viering DHHM, de Baaij JHF, Walsh SB, Kleta R, Bockenhauer D. Genetic causes of hypomagnesemia, a clinical overview. Pediatr Nephrol (2017) 32(7):1123–35. doi:10.1007/s00467-016-3416-3
28. Bartter FC, Pronove P, Gill JR Jr, MacCardle RC. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. Am J Med (1962) 33:811–28. doi:10.1016/0002-9343(62)90214-0
29. Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet (1996) 13(2):183–8. doi:10.1038/ng0696-183
30. Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, et al. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet (1996) 14(2):152–6. doi:10.1038/ng1096-152
31. Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet (1997) 17(2):171–8. doi:10.1038/ng1097-171
32. Birkenhäger R, Otto E, Schürmann MJ, Vollmer M, Ruf EM, Maier-Lutz I, et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet (2001) 29(3):310–4. doi:10.1038/ng752
33. Estévez R, Boettger T, Stein V, Birkenhäger R, Otto E, Hildebrandt F, et al. Barttin is a Cl- channel beta-subunit crucial for renal Cl- reabsorption and inner ear K+ secretion. Nature (2001) 414(6863):558–61. doi:10.1038/35107099
34. Kemter E, Rathkolb B, Bankir L, Schrewe A, Hans W, Landbrecht C, et al. Mutation of the Na(+)-K(+)-2Cl(-) cotransporter NKCC2 in mice is associated with severe polyuria and a urea-selective concentrating defect without hyperreninemia. Am J Physiol Renal Physiol (2010) 298(6):F1405–15. doi:10.1152/ajprenal.00522.2009
35. Laghmani K, Beck BB, Yang SS, Seaayfan E, Wenzel A, Reusch B, et al. Polyhydramnios, transient antenatal Bartter’s syndrome, and MAGED2 mutations. N Engl J Med (2016) 374(19):1853–63. doi:10.1056/NEJMoa1507629
36. Kömhoff M, Laghmani K. Pathophysiology of antenatal Bartter’s syndrome. Curr Opin Nephrol Hypertens (2017) 26(5):419–25. doi:10.1097/MNH.0000000000000346
37. Meij IC, Koenderink JB, van Bokhoven H, Assink KF, Groenestege WT, de Pont JJ, et al. Dominant isolated renal magnesium loss is caused by misrouting of the Na(+), K(+)-ATPase gamma-subunit. Nat Genet (2000) 26(3):265–6. doi:10.1038/81543
38. Ben-Yosef T, Belyantseva IA, Saunders TL, Hughes ED, Kawamoto K, Van Itallie CM, et al. Claudin 14 knockout mice, a model for autosomal recessive deafness DFNB29, are deaf due to cochlear hair cell degeneration. Hum Mol Genet (2003) 12:2049–61. doi:10.1093/hmg/ddg210
39. Wilcox ER, Burton QL, Naz B, Riazuddin S, Smith TN, Ploplis B, et al. Mutations in the gene encoding tight junction claudin-14 cause autosomal recessive deafness DFNB29. Cell (2001) 104:165–72. doi:10.1016/S0092-8674(01)00200-8
40. Corre T, Olinger E, Harris SE, Traglia M, Ulivi S, Lenarduzzi S, et al. Common variants in CLDN14 are associated with differential excretion of magnesium over calcium in urine. Pflugers Arch (2017) 469(1):91–103. doi:10.1007/s00424-016-1913-7
41. Thorleifsson G, Holm H, Edvardsson V, Walters GB, Styrkarsdottir U, Gudbjartsson DF, et al. Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density. Nat Genet (2009) 41(8):926–30. doi:10.1038/ng.404
42. Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science (1999) 285(5424):103–6. doi:10.1126/science.285.5424.103
43. Konrad M, Hou J, Weber S, Dötsch J, Kari JA, Seeman T, et al. CLDN16 genotype predicts renal decline in familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol (2008) 19(1):171–81. doi:10.1681/ASN.2007060709
44. Will C, Breiderhoff T, Thumfart J, Stuiver M, Kopplin K, Sommer K, et al. Targeted deletion of murine Cldn16 identifies extra- and intrarenal compensatory mechanisms of Ca2+ and Mg2+ wasting. Am J Physiol Renal Physiol (2010) 298(5):F1152–61. doi:10.1152/ajprenal.00499.2009
45. Bardet C, Courson F, Wu Y, Khaddam M, Salmon B, Ribes S, et al. Claudin-16 deficiency impairs tight junction function in ameloblasts, leading to abnormal enamel formation. J Bone Miner Res (2016) 31(3):498–513. doi:10.1002/jbmr.2726
46. Yamaguti PM, Neves FA, Hotton D, Bardet C, de La Dure-Molla M, Castro LC, et al. Amelogenesis imperfecta in familial hypomagnesaemia and hypercalciuria with nephrocalcinosis caused by CLDN19 gene mutations. J Med Genet (2017) 54(1):26–37. Erratum in: J Med Genet (2017) 54(11):786. doi:10.1136/jmedgenet-2016-103956
47. Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet (2006) 79:949–57. doi:10.1086/508617
48. Hou J, Renigunta A, Gomes AS, Hou M, Paul DL, Waldegger S, et al. Claudin-16 and claudin-19 interaction is required for their assembly into tight junctions and for renal reabsorption of magnesium. Proc Natl Acad Sci U S A (2009) 106:15350–5. doi:10.1073/pnas.0907724106
49. Claverie-Martín F, García-Nieto V, Loris C, Ariceta G, Nadal I, Espinosa L, et al. Claudin-19 mutations and clinical phenotype in Spanish patients with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. PLoS One (2013) 8(1):e53151. doi:10.1371/journal.pone.0053151
50. Van Itallie CM, Rogan S, Yu A, Vidal LS, Holmes J, Anderson JM. Two splice variants of claudin-10 in the kidney create paracellular pores with different ion selectivities. Am J Physiol Renal Physiol (2006) 291(6):F1288–99. doi:10.1152/ajprenal.00138.2006
51. Günzel D, Stuiver M, Kausalya PJ, Haisch L, Krug SM, Rosenthal R, et al. Claudin-10 exists in six alternatively spliced isoforms that exhibit distinct localization and function. J Cell Sci (2009) 122(Pt 10):1507–17. doi:10.1242/jcs.040113
52. Milatz S, Breiderhoff T. One gene, two paracellular ion channels-claudin-10 in the kidney. Pflugers Arch (2017) 469(1):115–21. doi:10.1007/s00424-016-1921-7
53. Breiderhoff T, Himmerkus N, Stuiver M, Mutig K, Will C, Meij IC, et al. Deletion of claudin-10 (Cldn10) in the thick ascending limb impairs paracellular sodium permeability and leads to hypermagnesemia and nephrocalcinosis. Proc Natl Acad Sci U S A (2012) 109(35):14241–6. doi:10.1073/pnas.1203834109
54. Bongers EMHF, Shelton LM, Milatz S, Verkaart S, Bech AP, Schoots J, et al. A novel hypokalemic-alkalotic salt-losing tubulopathy in patients with CLDN10 mutations. J Am Soc Nephrol (2017) 28(10):3118–28. doi:10.1681/ASN.2016080881
55. Klar J, Piontek J, Milatz S, Tariq M, Jameel M, Breiderhoff T, et al. Altered paracellular cation permeability due to a rare CLDN10B variant causes anhidrosis and kidney damage. PLoS Genet (2017) 13(7):e1006897. doi:10.1371/journal.pgen.1006897
56. Hadj-Rabia S, Brideau G, Al-Sarraj Y, Maroun RC, Figueres ML, Leclerc-Mercier S, et al. Multiplex epithelium dysfunction due to CLDN10 mutation: the HELIX syndrome. Genet Med (2018) 20(2):190–201. doi:10.1038/gim.2017.71
57. Terliesner N, Halbritter J, Bergmann C, Schitke J, Dittrich K. Phänotyp einer neuartigen CLDN10-Mutation. Nieren- und Hochdruckkrankheiten, Jahrgang 47, Nr. 2/2018, S. 1–61. (2018).
58. Merikallio H, Pääkkö P, Harju T, Soini Y. Claudins 10 and 18 are predominantly expressed in lung adenocarcinomas and in tumors of nonsmokers. Int J Clin Exp Pathol (2011) 4(7):667–73.
59. Ohta H, Chiba S, Ebina M, Furuse M, Nukiwa T. Altered expression of tight junction molecules in alveolar septa in lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol (2012) 302(2):L193–205. doi:10.1152/ajplung.00349.2010
60. Breiderhoff T, Himmerkus N, Drewell H, Plain A, Günzel D, Mutig K, et al. Deletion of claudin-10 rescues claudin-16-deficient mice from hypomagnesemia and hypercalciuria. Kidney Int (2018) 93(3):580–588. doi:10.1016/j.kint.2017.08.029
61. Wang C-Y, Shi J-D, Yang P, Kumar PG, Li Q-Z, Run Q-G, et al. Molecular cloning and characterization of a novel gene family of four ancient conserved domain proteins (ACDP). Gene (2003) 306:37–44. doi:10.1016/S0378-1119(02)01210-6
62. Rainier S, Chai JH, Tokarz D, Nicholls RD, Fink JK. NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6). Am J Hum Genet (2003) 73(4):967–71. doi:10.1086/378817
63. Wang C-Y, Yang P, Shi J-D, Purohit S, Guo D, An H, et al. Molecular cloning and characterization of the mouse Acdp gene family. BMC Genomics (2004) 5:7. doi:10.1186/1471-2164-5-7
64. Goytain A, Quamme GA. Functional characterization of ACDP2 (ancient conserved domain protein), a divalent metal transporter. Physiol Genomics (2005) 22:82–9. doi:10.1152/physiolgenomics.00058.2005
65. Quamme GA. Molecular identification of ancient and modern mammalian magnesium transporters. Am J Physiol Cell Physiol (2010) 298:C407–29. doi:10.1152/ajpcell.00124.2009
66. Ferre S, Hoenderop JG, Bindels RJ. Insight into renal Mg2+ transporters. Curr Opin Nephrol Hypertens (2011) 20:169–76. doi:10.1097/MNH.0b013e3283435ee4
67. Stuiver M, Lainez S, Will C, Terryn S, Günzel D, Debaix H, et al. CNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemia. Am J Hum Genet (2011) 88(3):333–43. doi:10.1016/j.ajhg.2011.02.005
68. Voets T, Nilius B, Hoefs S, van der Kemp AW, Droogmans G, Bindels RJ, et al. TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J Biol Chem (2004) 279(1):19–25. doi:10.1074/jbc.M311201200
69. de Baaij JH, Arjona FJ, van den Brand M, Lavrijsen M, Lameris AL, Bindels RJ, et al. Identification of SLC41A3 as a novel player in magnesium homeostasis. Sci Rep (2016) 6:28565. doi:10.1038/srep28565
70. Schmitz C, Dorovkov MV, Zhao X, Davenport BJ, Ryazanov AG, Perraud AL. The channel kinases TRPM6 and TRPM7 are functionally nonredundant. J Biol Chem (2005) 280:37763–71. doi:10.1074/jbc.M509175200
71. Schlingmann KP, Waldegger S, Konrad M, Chubanov V, Gudermann T. TRPM6 and TRPM7 – Gatekeepers of human magnesium metabolism. Biochim Biophys Acta (2007) 1772(8):813–21. doi:10.1016/j.bbadis.2007.03.009
72. Cao G, Thébault S, van der Wijst J, van der Kemp A, Lasonder E, Bindels RJ, et al. RACK1 inhibits TRPM6 activity via phosphorylation of the fused alpha-kinase domain. Curr Biol (2008) 18:168–76. doi:10.1016/j.cub.2007.12.058
73. Zhang Z, Yu H, Huang J, Faouzi M, Schmitz C, Penner R, et al. The TRPM6 kinase domain determines the Mg·ATP sensitivity of TRPM7/M6 heteromeric ion channels. J Biol Chem (2014) 289(8):5217–27. doi:10.1074/jbc.M113.512285
74. Schlingmann KP, Weber S, Peters M, Niemann Nejsum L, Vitzthum H, Klingel K, et al. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet (2002) 31(2):166–70. doi:10.1038/ng889
75. Walder RY, Landau D, Meyer P, Shalev H, Tsolia M, Borochowitz Z, et al. Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat Genet (2002) 31(2):171–4. doi:10.1038/ng901
76. Coulter M, Colvin C, Korf B, Messiaen L, Tuanama B, Crowley M, et al. Hypomagnesemia due to two novel TRPM6 mutations. J Pediatr Endocrinol Metab (2015) 28(11–12):1373–8. doi:10.1515/jpem-2014-0394
77. Astor MC, Løvås K, Wolff AS, Nedrebø B, Bratland E, Steen-Johnsen J, et al. Hypomagnesemia and functional hypoparathyroidism due to novel mutations in the Mg-channel TRPM6. Endocr Connect (2015) 4(4):215–22. doi:10.1530/EC-15-0066
78. Zhang Y, Cai Q, Shu XO, Gao YT, Li C, Zheng W, et al. Whole-exome sequencing identifies novel somatic mutations in chinese breast cancer patients. J Mol Genet Med (2015) 9(4):183. doi:10.4172/1747-0862.1000183
79. Komiya Y, Runnels LW. TRPM channels and magnesium in early embryonic development. Int J Dev Biol (2015) 59(7–9):281–8. doi:10.1387/ijdb.150196lr
80. Woudenberg-Vrenken TE, Sukinta A, van der Kemp AW, Bindels RJ, Hoenderop JG. Transient receptor potential melastatin 6 knockout mice are lethal whereas heterozygous deletion results in mild hypomagnesemia. Nephron Physiol (2011) 117(2):11–9. doi:10.1159/000320580
81. Samite D, Kofent J, Gong M, Rüschendorf F, Jia S, Arn P, et al. Recessive mutations in PCBD1 cause a new type of early-onset diabetes. Diabetes (2014) 63(10):3557–64. doi:10.2337/db13-1784
82. Kompatscher A, de Baaij JHF, Aboudehen K, Hoefnagels APWM, Igarashi P, Bindels RJM, et al. Loss of transcriptional activation of the potassium channel Kir5.1 by HNF1β drives autosomal dominant tubulointerstitial kidney disease. Kidney Int (2017) 92(5):1145–56. doi:10.1016/j.kint.2017.03.034
83. Glaudemans B, van der Wijst J, Scola RH, Lorenzoni PJ, Heister A, van der Kemp AW, et al. A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J Clin Invest (2009) 119(4):936–42. doi:10.1172/JCI36948
84. Groenestege WM, Thébault S, van der Wijst J, van den Berg D, Janssen R, Tejpar S, et al. Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. J Clin Invest (2007) 117(8):2260–7. doi:10.1172/JCI31680
85. van der Made CI, Hoorn EJ, de la Faille R, Karaaslan H, Knoers NV, Hoenderop JG, et al. Hypomagnesemia as first clinical manifestation of ADTKD-HNF1B: a case series and literature review. Am J Nephrol (2015) 42(1):85–90. doi:10.1159/000439286
86. Knoers NV, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis (2008) 3:22. doi:10.1186/1750-1172-3-22
87. Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest (2005) 115(6):1651–8. doi:10.1172/JCI24134
88. Yogi A, Callera GE, O’Connor SE, He Y, Correa JW, Tostes RC, et al. Dysregulation of renal transient receptor potential melastatin 6/7 but not paracellin-1 in aldosterone-induced hypertension and kidney damage in a model of hereditary hypomagnesemia. J Hypertens (2011) 29(7):1400–10. doi:10.1097/HJH.0b013e32834786d6
89. Pham PC, Pham PA, Pham SV, Pham PT, Pham PM, Pham PT. Hypomagnesemia: a clinical perspective. Int J Nephrol Renovasc Dis (2014) 7:219–30. doi:10.2147/IJNRD.S42054
90. Schweigel-Röntgen M, Kolisek M. SLC41 transporters – molecular identification and functional role. Curr Top Membr (2014) 73:383–410. doi:10.1016/B978-0-12-800223-0.00011-6
91. Kolisek M, Sponder G, Mastrototaro L, Smorodchenko A, Launay P, Vormann J, et al. Substitution p.A350V in Na+/Mg2+ exchanger SLC41A1, potentially associated with Parkinson’s disease, is a gain-of-function mutation. PLoS One (2013) 8(8):e71096. doi:10.1371/journal.pone.0071096
92. Chan KH, Chacko SA, Song Y, Cho M, Eaton CB, Wu WC, et al. Genetic variations in magnesium-related ion channels may affect diabetes risk among African American and Hispanic American women. J Nutr (2015) 145:418–24. doi:10.3945/jn.114.203489
93. Hurd TW, Otto EA, Mishima E, Gee HY, Inoue H, Inazu M, et al. Mutation of the Mg transporter SLC41A1 results in a nephronophthisis-like phenotype. J Am Soc Nephrol (2013) 24:967–77. doi:10.1681/ASN.2012101034
94. Fleig A, Schweigel-Röntgen M, Kolisek M. Solute carrier family SLC41, what do we really know about it? Wiley Interdiscip Rev Membr Transp Signal (2013) 2(6). doi:10.1002/wmts.95
95. Arjona FJ, de Baaij JHF, Schlingmann KP, Lameris ALL, van Wijk E, Flik G, et al. CNNM2 mutations cause impaired brain development and seizures in patients with hypomagnesemia. PLoS Genet (2014) 10(4):e1004267. doi:10.1371/journal.pgen.1004267
96. Kieboom BC, Ligthart S, Dehghan A, Kurstjens S, de Baaij JH, Franco OH, et al. Serum magnesium and the risk of prediabetes: a population-based cohort study. Diabetologia (2017) 60(5):843–53. doi:10.1007/s00125-017-4224-4
97. Lv WQ, Zhang X, Zhang Q, He JY, Liu HM, Xia X, et al. Novel common variants associated with body mass index and coronary artery disease detected using a pleiotropic cFDR method. J Mol Cell Cardiol (2017) 112:1–7. doi:10.1016/j.yjmcc.2017.08.011
98. Ohi K. Influences of schizophrenia risk variant rs7914558 at CNNM2 on brain structure. Br J Psychiatry (2015) 206(4):343–4. doi:10.1192/bjp.206.4.343b
99. Duarte RRR, Troakes C, Nolan M, Srivastava DP, Murray RM, Bray NJ. Genome-wide significant schizophrenia risk variation on chromosome 10q24 is associated with altered cis-regulation of BORCS7, AS3MT, and NT5C2 in the human brain. Am J Med Genet B Neuropsychiatr Genet. (2016) 171(6):806–14. doi:10.1002/ajmg.b.32445
100. Funato Y, Yamazaki D, Miki H. Renal function of cyclin M2 Mg2+ transporter maintains blood pressure. J Hypertens (2017) 35(3):585–92. doi:10.1097/HJH.0000000000001211
101. Ignoul S, Eggermont J. CBS domains: structure, function, and pathology in human proteins. Am J Physiol Cell Physiol (2005) 289(6):C1369–78.
102. Biemans-Oldehinkel E, Mahmood NA, Poolman B. A sensor for intracellular ionic strength. Proc Natl Acad Sci U S A (2006) 103(28):10624–9.
103. Ereño-Orbea J, Oyenarte I, Martínez-Cruz LA. CBS domains: ligand binding sites and conformational variability. Arch Biochem Biophys (2013) 540(1–2):70–81. doi:10.1016/j.abb.2013.10.008
104. Ochoa B. Can a congenital dysfunctional bladder be diagnosed from a smile? The Ochoa syndrome updated. Pediatr Nephrol (2004) 19:6–12. doi:10.1007/s00467-003-1291-1
105. Yang M, Jensen LT, Gardner A, Culotta VC. Manganese toxicity and Saccharomyces cerevisiae Mam3p, a member of the ACDP (ancient conserved domain protein) family of proteins. Biochem J (2005) 383:479–87. doi:10.1042/BJ20041582
106. Corral-Rodríguez MA, Stuiver M, Abascal-Palacios G, Diercks T, Oyenarte I, Ereño-Orbea J, et al. Nucleotide binding triggers a conformational change of the CBS module of the magnesium transporter CNNM2 from a twisted towards a flat structure. Biochem J (2014) 464(1):23–34. doi:10.1042/BJ20140409
107. de Baaij JH, Stuiver M, Meij IC, Lainez S, Kopplin K, Venselaar H, et al. Membrane topology and intracellular processing of cyclin M2 (CNNM2). J Biol Chem (2012) 287(17):13644–55. doi:10.1074/jbc.M112.342204
108. Eshaghi S, Niegowski D, Kohl A, Martinez Molina D, Lesley SA, Nordlund P. Crystal structure of a divalent metal ion transporter CorA at 2.9 angstrom resolution. Science (2006) 313:354–7. doi:10.1126/science.1127121
109. Lunin VV, Dobrovetsky E, Khutoreskaya G, Zhang R, Joachimiak A, Doyle DA, et al. Crystal structure of the CorA Mg2+ transporter. Nature (2006) 440:833–7. doi:10.1038/nature04642
110. Payandeh J, Pai EF. A structural basis for Mg2+ homeostasis and the CorA translocation cycle. EMBO J (2006) 25:3762–73. doi:10.1038/sj.emboj.7601269
111. Matthies D, Dalmas O, Borgnia MJ, Dominik PK, Merk A, Rao P, et al. Cryo-EM structures of the magnesium channel CorA reveal symmetry break upon gating. Cell (2016) 164(4):747–56. doi:10.1016/j.cell.2015.12.055
112. Smith RL, Thompson LJ, Maguire ME. Cloning and characterization of MgtE, a putative new class of Mg2+ transporter from Bacillus firmus OF4. J Bacteriol (1995) 177(5):1233–8. doi:10.1128/jb.177.5.1233-1238.1995
113. Hattori M, Tanaka Y, Fukai S, Ishitani R, Nureki O. Crystal structure of magnesium transporter MgtE. Nature (2007) 448:1072–5. doi:10.1038/nature06093
114. Tomita A, Zhang M, Jin F, Zhuang W, Takeda H, Maruyama T, et al. Crystal structure of the Mg2+ channel MgtE in complex with ATP. Nat Commun (2017) 8:148–148. doi:10.1038/s41467-017-00082-w
115. Takeda H, Hattori M, Nishizawa T, Yamashita K, Shah ST, Caffrey M, et al. Structural basis for ion selectivity revealed by high-resolution crystal structure of Mg2+ channel MgtE. Nat Commun (2014) 5:5374. doi:10.1038/ncomms6374
116. Dalmas O, Sompornpisut P, Bezanilla F, Perozo E. Molecular mechanism of Mg2+-dependent gating in CorA. Nat Commun (2014) 5:3590. doi:10.1038/ncomms4590
117. Moomaw AS, Maguire ME. The unique nature of Mg2+ channels. Physiology (Bethesda) (2008) 23:275–85. doi:10.1152/physiol.00019.2008
118. Ishitani R, Sugita Y, Dohmae N, Furuya N, Hattori M, Nureki O. Mg2+-sensing mechanism of Mg2+ transporter MgtE probed by molecular dynamics study. Proc Natl Acad Sci U S A (2008) 105(40):15393–8. doi:10.1073/pnas.0802991105
119. Hirata Y, Funato Y, Takano Y, Miki H. Mg2+-dependent interactions of ATP with the cystathionine-β-synthase (CBS) domains of a magnesium transporter. J Biol Chem (2014) 289(21):14731–9. doi:10.1074/jbc.M114.551176
120. Gómez-García I, Stuiver M, Ereño J, Oyenarte I, Corral-Rodríguez MA, Müller D, et al. Purification, crystallization and preliminary crystallographic analysis of the CBS-domain pair of cyclin M2 (CNNM2). Acta Crystallogr Sect F Struct Biol Cryst Commun (2012) 68(Pt 10):1198–203. doi:10.1107/S1744309112035348
Keywords: magnesium, crystallography, CNNM2, kidney, genetics
Citation: Giménez-Mascarell P, Schirrmacher CE, Martínez-Cruz LA and Müller D (2018) Novel Aspects of Renal Magnesium Homeostasis. Front. Pediatr. 6:77. doi: 10.3389/fped.2018.00077
Received: 31 October 2017; Accepted: 13 March 2018;
Published: 09 April 2018
Edited by:
Max Christoph Liebau, Universitätsklinikum Köln, GermanyReviewed by:
Tom Nijenhuis, Radboud University Nijmegen, NetherlandsCopyright: © 2018 Giménez-Mascarell, Schirrmacher, Martínez-Cruz and Müller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis Alfonso Martínez-Cruz, YW1hcnRpbmV6QGNpY2Jpb2d1bmUuZXM=;
Dominik Müller, ZG9taW5pay5tdWVsbGVyQGNoYXJpdGUuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.