 Federica Camela1
Federica Camela1 Marcella Gallucci1
Marcella Gallucci1 Emanuela di Palmo1Salvatore Cazzato2
Emanuela di Palmo1Salvatore Cazzato2 Mario Lima3
Mario Lima3 Giampaolo Ricci1*
Giampaolo Ricci1* Andrea Pession1
Andrea Pession1
- 1Department of Pediatrics, S. Orsola-Malpighi Hospital, University of Bologna, Bologna, Italy
- 2Department of Mother and Child Health, Salesi Children’s Hospital, Ancona, Italy
- 3Department of Pediatric Surgery, S. Orsola-Malpighi Hospital, University of Bologna, Bologna, Italy
The inflammatory myofibroblastic tumor (IMT) is a rare lesion of unclear etiology and variable clinical course, consisting of a proliferation of fibroblasts and myofibroblasts, mixed with inflammatory cells. Synonyms of IMT are inflammatory pseudotumor and plasma cell granuloma reflecting the alleged inflammatory nature attributed to this lesion, even though this heterogeneity in the disease denomination is probably involved in a dispersion of the literature data. Among primary pulmonary neoplasms, it represents the most frequent endobronchial tumor of childhood and beyond the lung it has been described mainly in the bladder, mediastinum and mesentery. Despite having a tendency for local recurrence, the risk of distant metastasis is low. Clinical presentation depends on localization therefore lung peripheral lesions are often asymptomatic resulting in a delayed diagnosis. Radiological findings can suggest the diagnosis that must be confirmed by histopathology assessment. The tumor has been characterized by the application of immunohistochemical techniques, molecular biology and cytogenetics, which are very precious for the diagnosis. The therapeutic approach consists in the complete surgical excision of the lesion that normally ensures excellent survival. Due to the potential risk of recurrence, close clinical trial is indicated. To date only 24 cases of pulmonary IMT have been described, although the prevalence is probably higher. We present a case report of a 3-year-old girl with pulmonary IMT and a brief review of known literature cases in order to highlight the most common clinical presentations, the most useful diagnostic tools and therapeutic approach.
Introduction
Primary pulmonary neoplasms in pediatric patients are uncommon diseases. Among these disorders, the inflammatory myofibroblastic tumor (IMT) represents 20% of all primary lung tumors and more of 50% of all benign masses (1, 2).
The World Health Organization (WHO) defines IMT as “a lesion composed of a proliferation of myofibroblastic spindle and stellate cells with abundant eosinophilic cytoplasm mixed with infiltrative plasma, inflammatory cells, lymphocytes and eosinophils” (3, 4).
The same entity was called in the past with other names; the most recognized ones were inflammatory pseudotumor (IPT), plasma cell granuloma, and postinflammatory tumor.
Inflammatory pseudotumor was considered as a reaction to an inflammatory insult (1, 5). It involve more often liver and biliary tract (31.8% of cases), head and neck (20.6%), lung (18.2%), abdomen (15.5%), and urogenitary system (7.4%) (6). Although it was considered in the past a non-neoplastic reactive inflammation, in 2006 WHO defined it an intermediary lesion with clinical recurrence and malignant potential (4).
These generic appellations reflect the alleged inflammatory and non-neoplastic nature attributed to these lesions, even though confusion regarding the distinction of these tumors from other entities such as the “inflammatory fibrosarcoma” (considered indistinguishable from the IMT), still remains (5, 7). This ambiguity in the denomination may be a factor favoring the dispersion of the literature data and then an underreporting.
Case Report
We describe the case of a 3-year-old girl, born at 33 weeks of gestational age with a good neonatal adaptation. Starting from the age of 18 months, she presented with recurrent infections of lower and upper airways, which were often treated with antibiotic therapy. She came to our attention from another medical center.
In the previous 2 months, the child showed intermittent fever, loss of appetite and dry cough.
The chest X-ray (CXR) showed a rounded 2 cm diameter opacity in the left parahilar side, therefore she consecutively was treated with oral therapy with penicillin, cephalosporin of third generation and macrolide. Despite therapy, at first evaluation to our center the child was still having evening fever and poor general condition.
Chest auscultation revealed reduced air entry and fine crackles on the left hemithorax. The laboratory tests showed a stable increase of inflammatory markers: C-reactive protein 9.5 mg/dL (normal range <0.08 mg/dl), erythrocyte sedimentation rate 102 mm, leukocytes 11 × 106/L (59% neutrophils, 32% lymphocytes, 8% monocytes, 0.4% eosinophils, and 0.7% basophils), hemoglobin 12 mg/dL, and platelet 809 × 103/μL.
The nasal-pharyngeal samples culture and blood culture were both negative, including research for multidrug-resistant staphylococcus.
Nevertheless, we started with empiric therapy with Piperacillin/Tazobactam and Linezolid. A few days later, because of persistent fever with altered laboratory and radiological findings, the child underwent a chest high-resolution computer tomography (HRCT) that showed a large lung lesion with thickened wall and fluid content at the lingula level (Figure 1). The mass diameter was about 35 mm. Suspecting a necrotizing pneumonia, surgical removal of the lesion was performed by atypical resection. The subsequent histological assessment showed an IMT positive for actin, anaplastic lymphoma kinase 1 (ALK1), and desmin (Figure 2). Subsequently, to achieve a complete removal of the lesion margins, the left upper lobectomy by thoracotomy was necessary.
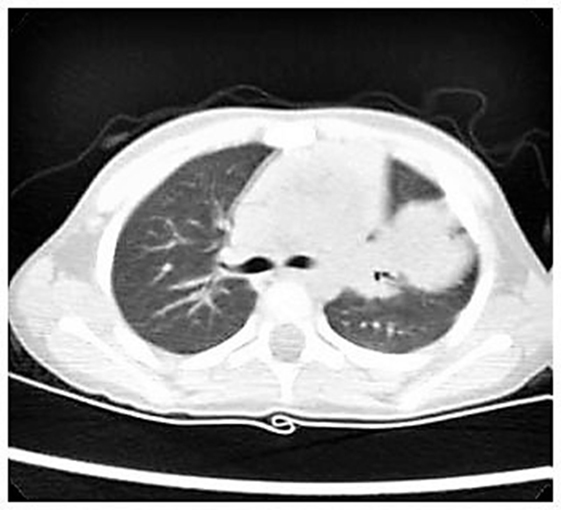
Figure 1. High-resolution computer tomography imaging showing a large lung lesion in the left parahilar side.
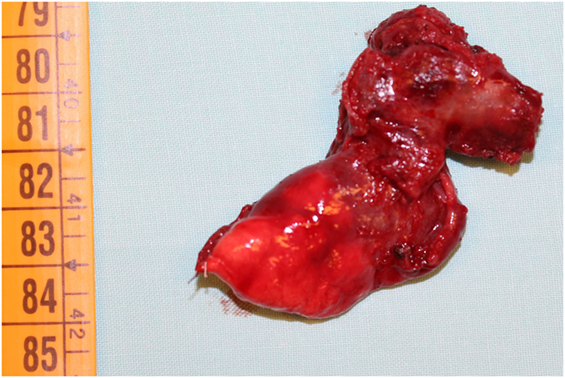
Figure 2. Surgical sample of lingula inflammatory myofibroblastic tumor in 3-year-old child.
The postoperative course was uncomplicated. After surgery, the clinical picture of the child gradually improved with remission of fever. At 6, 12, and 18 months of follow-up, she presented good clinical conditions and radiological evidence of atelectasis to the left lower lobe, compatible with recent surgery.
Discussion
Although the spectrum of primary lung tumors in the childhood is the same as in adults, the lesions have different prevalence (8). IMT shows a high incidence in children and young people, representing 50% of primary benign lung tumor in infancy (1, 2).
Pulmonary IMT typically presents itself as a parenchymal nodule and occasionally as an endobronchial lesion (2). It has a tendency to grow slowly but can invade local structures (1).
The clinical picture depends primarily on the location of the tumor but most patients have a few respiratory symptoms so the diagnosis may be delayed.
Children with IMT may exhibit symptoms of chronic inflammation as a low-grade fever, weight loss, anemia, thrombocytosis, polyclonal hypergammaglobulinemia, and elevated sedimentation rate. Several cases are asymptomatic and are detected only incidentally on imaging studies.
Among patients with endobronchial lesions, symptoms of bronchial irritation such as cough and hemoptysis may be accompanied by chest pain (3).
In the current case, the previous clinical history, CXR and the presence of intermittent fever suggested at first an infectious disease. Nevertheless, the failure to respond to broad-spectrum antibiotics brought us to investigate the presence of a non-infectious lesion. The chest HRCT was decisive even though the diagnosis could not have been possible without histological assessment.
According to literature data, there are only 24 published cases of pediatric pulmonary IMT (the age thereof ranges from 3 to 13 years), even though the real incidence is presumed to be higher. Peripheral lung lesions appear to be more frequent than central and endobronchial tumors that may be present about in 10% of the cases resulting in bronchial obstruction and atelectasis (2).
Among the 24 described cases, the tumor grew in trachea in 5 patients (3 in carina; 20% of total) and was peripheral in 19 cases it (80% of cases). These peripheral masses were located in 11 cases (46%) in the left lobes and in 8 cases (33%) in the right lobes. At the onset, the mass was average on 4.5 cm of the diameter (1.4–12 cm); four subjects (17%) presented metastasis and secondary masses were discovered in one case after 11 months of follow-up. At the time of diagnosis, the IMT involved adjacent structures in five cases (21%): chest wall, main bronchus, mediastinum, hilum, or pulmonary artery (Table 1). In some of these cases, the patients came to clinicians’ attention with symptoms related to the local effects of the mass such as stridor or a history of wheezing (5).
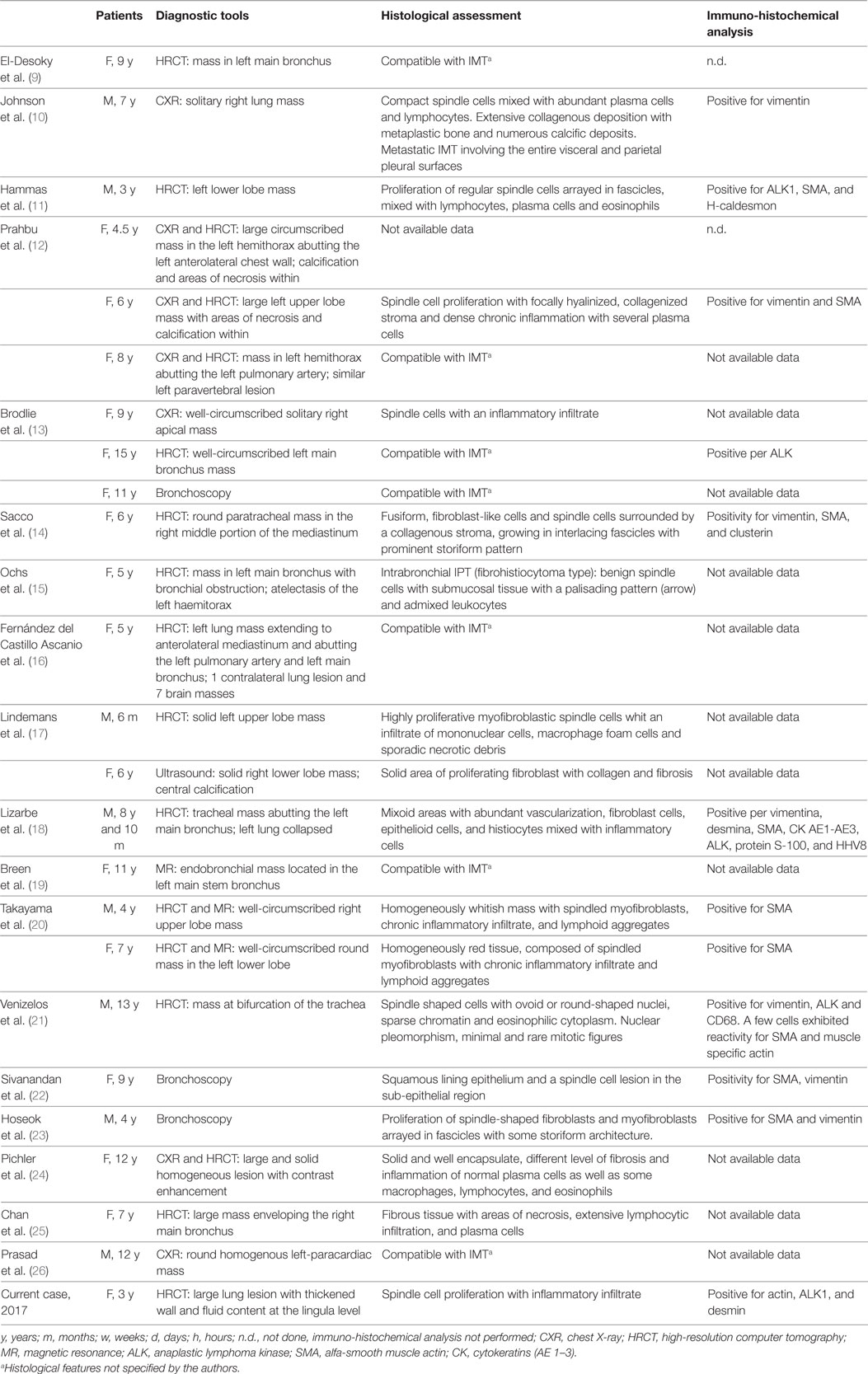
Table 1. Review of 25 known cases of pediatric pulmonary IMT, including our case: diagnostic tools, histological assessment, and immune-histochemical analysis of lung bioptic samples.
Inflammatory myofibroblastic tumor can be sometimes detected as incidental finding on a routine CXR (27). In all cases, at presentation patients had fever, respiratory distress, arthralgia, clubbing, night sweat, vomiting, and hemoptysis. At the onset, fever and cough were the commonest symptoms (Table 2).
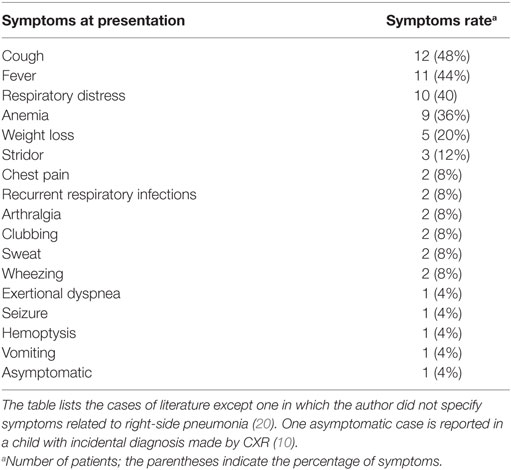
Table 2. Frequency of symptoms in pediatric pulmonary IMT, including our case.
Data collected from the literature show that central IMT are recognized earlier, probably due to the resulting symptoms (acute obstruction of the trachea, carena, or main bronchus). On the contrary, peripheral parenchymal tumors cause more misleading and nonspecific symptoms leading to delayed diagnosis.
Including our patient, in nine cases the children presented with variable degree of anemia. They also showed laboratory findings of chronic inflammation such as leukocytosis, hypergammaglobulinemia, elevated platelet count, increase of erythrocyte sedimentation rate, and C-reactive protein (12, 14, 15, 17, 18, 24).
The anatomopathological differential diagnosis is wide and must be conducted on the clinical picture, morphological alterations, and the immune phenotype of spindle cells.
Differential diagnosis must include foreign body aspiration, infectious disease (Aspergillum and Mycobacterium) but also necrotizing pneumonia and spindle cell neoplasms (1).
The histological assessment is essential to rule out pseudo-lymphomas, malignant lymphomas, lymphoid hyperplasia and sarcomas such as leiomyosarcoma, fibrosarcoma, rhabdomyosarcoma, myxoid sarcoma, malignant fibrous histiocytoma, and gastrointestinal stromal tumor (12, 14, 15, 17, 18, 24, 27, 28).
Chest X-ray usually reveal a peripheral lesion or nodule of variable size (diameter usually ranges from 1.2 to 15 cm,) more often localized in the lower lobes (29). Rarely, patients may have multiple lesions (30).
Among the literature cases, 16/24 subjects underwent HRCT often showing solid masses with homogeneous or heterogeneous enhancement (Table 1). These lesions appear as single, well-defined, lobulated mass and frequently contain punctate calcifications that mimics malignant tumors (20).
Positron emission tomography/computed tomography is highly sensitive but has low specificity for IMT. It could be useful to evaluate the response to treatment in patients not eligible for surgery (31).
Bronchoscopy is usually performed when there exists a suspect of endoluminal lesion. It is a useful diagnostic and therapeutic tool since it has both a function of removing tumoral obstruction and providing biopsy samples (13, 19).
Moreover, due to local recurrence risk, bronchoscopy may have a potential role in the follow-up even though future evidences are necessary to define the timing of endoscopic revaluation.
Among all pediatric known cases, only three subjects underwent magnetic resonance imaging (MRI): in two cases the chest MRI confirmed the HRCT findings and MRI was performed only in one case in place of HRCT (19, 20).
Takayama et al. suggest that radiological presentation and contrast-enhancement in pediatric IMT are variable and nonspecific. Moreover, calcifications within the tumor are present more often in children than in adults (these were reported in seven children; 29% of cases) (20).
In association with lesions, there could be pneumothorax, pleural effusion, atelectasis, and rarely cavitations and lymphadenopathies (Table 1).
Molecular rearrangements on chromosome 2p23 were detected in several cases of IMTs (in about 34–56% of both pulmonary and extrapulmonary IMTs). This locus is the site of human ALK gene, which codes for a tyrosine kinase receptor called ALK. ALK rearrangements are identified by immunohistochemical analysis (fluorescence in situ hybridization) using monoclonal ALK-1 antibody and seems to be highly specific for these lesions, even though it does not seem to be a sensitive marker in children (32, 33). Furthermore, ALK-positivity does not seems to be related to recurrence (1, 5, 33, 34).
Abnormal karyotypes of tumor cells, like aneuploidy, were found in 16% of cases (32, 33). RET gene rearrangement, EML4-ALK inversion, and fusions of other kinases gene have also been implicated (35).
Alaggio et al. in a recent report found that cytologic atypia and positive ALK status are more frequent in aggressive tumors, whereas metastatic tumors are negative for ALK.
Among previous pediatric cases described, positivity to vimentin (29%), alpha-smooth muscle actin (alpha-SMA) (21.6%), ALK (16.6%), and other markers like desmin, cytokeratins (AE 1–3), CD34, protein S-100, and HHV8 was reported (32–34).
Treatment is primarily a complete but conservative surgical excision (3). This approach is necessary to prevent recurrence (1, 28). An appropriate histologic assessment should be obtained before the surgery (needle biopsy by bronchoscopy), in order to avoid an unnecessarily procedure (28).
Among all cases, 15 patients underwent open lung surgery, 2 resections by laser, 1 surgery by thoracoscopy, and 1 child was treated with bronchoscopic excision. In one case treated with open surgery, coexistent cerebral masses were detected and they required chemotherapy (16).
The most appropriate surgical approach (such as use of the wedge resection, lobectomy, and pneumonectomy) depends on the specific dimension and tumor localization, its relationship with the surrounding structures and the surgical equipment experience (36).
Partial resection may be necessary in cases where it is not possible to remove the lesion because of invasion of the vital structures. Among these cases, chemotherapy may be an alternative option for patients who have microscopic or macroscopic residual disease, although the results are controversial (32).
Described therapy for expanded masses were vinblastine plus methotrexate (14).
Although systemic steroids showed some beneficial effects in IMT, Sacco et al. found that the use of Prednisone may favorite the progression of lesion probably due to immunosuppression, therefore the authors suggest caution in the use of this drug in IMT (15).
In two cases without metastasis or local invasion, chemotherapy (methotrexate or cyclophosphamide associated with prednisone) was started as primary treatment (13).
In others two cases, the clinicians decided to undertake a therapy with only corticosteroids without evidence of recurrence at follow-up (15). Another case was treated only with COX-2 inhibitor after evidence of unresectable mass, with complete resolution at 8 months of follow-up (25).
Among all patients, only three children had tumor recurrence (10, 12, 17).
Although recurrence is described as a rare phenomenon (14% of pulmonary IMT) a close follow-up should be made for the early recognition of tumor relapse. In addition, the local invasion at diagnosis was highly correlated with local relapse (9, 11, 37).
In our case, we decided to perform a complete surgical excision of the lesion followed by left upper lobectomy; nevertheless, to date we do not know the rate of survival after surgery versus others managements. The most recent evidence in young adult (mean age 33 years) suggests a 5- and 10-year disease-free survival of 89% after complete resection (23, 26, 37).
Take Home Message
• Pulmonary IMT is an uncommon disease but with significant morbidity among the pediatric population.
• Children may show non-specific and variable symptoms therefore they often underwent several antibiotic treatments before diagnosis.
• Radiological techniques are useful in suspected lesions but the diagnosis must be confirmed by histopathological assessment.
• Surgical complete resection is the treatment of choice.
• Because of the potential malignant behavior of these tumors, a close follow-up should be done to recognize the recurrences earlier.
• Due to the rarity of the disease among the pediatric population, it may be useful for the future to include all known cases in a national registry in order to improve knowledge of the disease and encode the best diagnostic and therapeutic approach.
Ethics Statement
The authors state that “written informed consent was obtained from the parents of the patient for the publication of this case report.”
Author Contributions
GR coordinated the writing group. FC and MG performed the literature review. All authors critically reviewed the manuscript, read, and approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors state that no funding has been received.
Abbreviations
IMT, inflammatory myofibroblastic tumor; IPT, inflammatory pseudotumor; HRCT, high-resolution computer tomography; ALK1, anaplastic lymphoma kinase 1; MRI, magnetic resonance imaging; CXR, chest X-ray.
References
1. Weldon CB, Shamberger RC. Pediatric pulmonary tumors: primary and metastatic. Semin Pediatr Surg (2008) 17:17–29. doi:10.1053/j.sempedsurg.2007.10.004
2. Al-Qahtani AR, Di Lorenzo M, Yazbeck S. Endobronchial tumors in children: institutional experience and literature review. J Pediatr Surg (2003) 38:733–6. doi:10.1016/jpsu.2003.50195
3. Dishop MK, Kuruvilla S. Primary and metastatic lung tumors in the pediatric population. A review and 25-year experience at a large children’s hospital. Arch Pathol Lab Med (2008) 132:1079–103. doi:10.1043/1543-2165(2008)132[1079:PAMLTI]2.0.CO;2
4. Fletcher CD. The evolving classification of soft tissue tumours: an update based on the new WHO classification. Histopathology (2006) 48:3–12. doi:10.1111/j.1365-2559.2005.02284.x
5. Dehner LP. Inflammatory myofibroblastic tumor. The continued definition of one type of so-called inflammatory pseudotumor. Am J Surg Pathol (2004) 28:1652–4. doi:10.1097/00000478-200412000-00016
6. Höhne S, Milzsch M, Adams J, Kunze C, Finke R. Inflammatory pseudotumor (IPT) and inflammatory myofibroblastic tumor (IMT): a representative literature review occasioned by a rare IMT of the transverse colon in a 9-years-old child. Tumori (2015) 101:249–56. doi:10.5301/tj.5000353
7. Gleason BC, Hornick JL. Inflammatory myofibroblastic tumours: where are we now? J Clin Pathol (2008) 61(4):428–37. doi:10.1136/jcp.2007.049387
8. Yu DC, Grabowski MJ, Kozakewich HP, Perez-Atayde AR, Voss SD, Shamberger RC, et al. Primary lung tumors in children and adolescents: a 90-year experience. J Pediatr Surg (2010) 45:1090–5. doi:10.1016/j.jpedsurg.2010.02.070
9. El-Desoky T, Nasef N, Osman E, Osman A, Zaki A, Zalata K. Endobronchial inflammatory pseudotumor: a rare cause of a pneumothorax in children. J Bronchology Interv Pulmonol (2013) 20:256–60. doi:10.1097/LBR.0b013e31829bccba
10. Johnson K, Notrica DM, Carpentieri D, Jaroszewski D, Henry MM. Successful treatment of recurrent pediatric inflammatory myofibroblastic tumor in a single patient with a novel chemotherapeutic regimen containing celecoxib. J Pediatr Hematol Oncol (2013) 35:414–6. doi:10.1097/MPH.0b013e3182915cef
11. Hammas N, Chbani L, Rami M, Boubbou M, Benmiloud S, Bouabdellah Y, et al. A rare tumor of the lung: inflammatory myofibroblastic tumor. Diagn Pathol (2012) 7:83. doi:10.1186/1746-1596-7-83
12. Prabhu SM, Choudhury SR, Solanki RS, Shetty GS. Multifocal intrathoracic inflammatory myofibroblastic tumour in children. Jpn J Radiol (2012) 30:453–7. doi:10.1007/s11604-012-0065-8
13. Brodlie M, Barwick SC, Wood KM, McKean MC, Welch A. Inflammatory myofibroblastic tumours of the respiratory tract: paediatric case series with varying clinical presentations. J Laryngol Otol (2011) 125:865–8. doi:10.1017/S0022215111000648
14. Sacco O, Gambini C, Gardella C, Tomà P, Rossi UG, Jassoni V, et al. “Atypical steroid response” in a pulmonary: inflammatory myofibroblastic tumor. Pediatr Pulmonol (2010) 45:721–6. doi:10.1002/ppul.21237
15. Ochs K, Hoksch B, Frey U, Schmid RA. Inflammatory myofibroblastic tumour of the lung in a five-year-old girl. Interact Cardiovasc Thorac Surg (2010) 10:805–6. doi:10.1510/icvts.2009.219089
16. Fernández del Castillo Ascanio M, González CG, Pérez SP, Delgado LE. Inflammatory pseudotumor in a five-year-old girl. Radiologia (2013) 55:82–5. doi:10.1016/j.rx.2010.06.014
17. Lindemans CA, Bruinsma RA, Nikkels PG, Bröker FH, Arets HG. Pulmonary consolidation and inflammation: inflammatory pseudotumour in young children. Monaldi Arch Chest Dis (2009) 71:71–5.
18. Lizarbe MO, Olascoaga JH, García ER, Castiella TM, Sagaseta de Ilúrdoz Uranga María M, Garicano JM. Paediatric myofibroblastic tumours. A presentation of three cases. An Pediatr (Barc) (2009) 71:331–5. doi:10.1016/j.anpedi.2009.06.010
19. Breen DP, Dubus JC, Chetaille B, Payan MJ, Dutau H. A rare cause of an endobronchial tumour in children: the role of interventional bronchoscopy in the diagnosis and treatment of tumours while preserving anatomy and lung function. Respiration (2008) 76:444–8. doi:10.1159/000101718
20. Takayama Y, Yabuuchi H, Matsuo Y, Soeda H, Okafuji T, Kamitani T, et al. Computed tomographic and magnetic resonance features of inflammatory myofibroblastic tumor of the lung in children. Radiat Med (2008) 26:613–7. doi:10.1007/s11604-008-0284-1
21. Venizelos I, Papathomas T, Anagnostou E, Tsanakas J, Kirvassilis F, Kontzoglou G. Pediatric inflammatory myofibroblastic tumor of the trachea: a case report and review of the literature. Pediatr Pulmonol (2008) 43:831–5. doi:10.1002/ppul.20869
22. Sivanandan S, Lodha R, Agarwala S, Sharma M, Kabra SK. Inflammatory myofibroblastic tumor of the trachea. Pediatr Pulmonol (2007) 42:847–50. doi:10.1002/ppul.20651
23. Hoseok I, Joungho H, Ahn KM, Lee SI, Jhingook K. Complete surgical resection of inflammatory myofibroblastic tumor with carinal reconstruction in a 4-year-old boy. J Pediatr Surg (2005) 40:e23–5. doi:10.1016/j.jpedsurg.2005.08.043
24. Pichler G, Eber E, Thalhammer G, Muntean W, Zach MS. Arthralgia and digital clubbing in a child: hypertrophic osteoarthropathy with inflammatory pseudotumour of the lung. Scand J Rheumatol (2004) 33:189–91. doi:10.1080/03009740310004702
25. Chan PW, Omar KZ, Ramanujam TM. Successful treatment of unresectable inflammatory pseudotumor of the lung with COX-2 inhibitor. Pediatr Pulmonol (2003) 36:167–9. doi:10.1002/ppul.10308
26. Prasad M, Thankachen R, Parihar B, Shukla V. Inflammatory pseudotumour of the lung. Interact Cardiovasc Thorac Surg (2004) 3:323–5. doi:10.1016/j.icvts.2003.11.018
27. Panagiotopoulos N, Patrini D, Gvinianidze L, Woo WL, Borg E, Lawrence D. Inflammatory myofibroblastic tumour of the lung: a reactive lesion or a true neoplasm? J Thorac Dis (2015) 7:908–11. doi:10.3978/j.issn.2072-1439.2015.04.60
28. Karnak I, Senocak ME, Ciftci AO, Cağlar M, Bingöl-Koloğlu M, Tanyel FC, et al. Inflammatory myofibroblastic tumor in children: diagnosis and treatment. J Pediatr Surg (2001) 36:908–12. doi:10.1053/jpsu.2001.23970
29. Choi BY, Kim WS, Cheon JE, Kim IO, Kim CJ, Yeon KM. Inflammatory myofibroblastic tumour of the liver in a child: CT and MR findings. Pediatr Radiol (2003) 33:30–3. doi:10.1007/s00247-002-0786-4
30. Athanassiadi K, Laenger F, Dickgreber N, Haverich A. Multiple inflammatory myofibroblastic tumors involving lung and mediastinum: a rare clinical entity. Thorac Cardiovasc Surg (2009) 57:343–6. doi:10.1055/s-0029-1185574
31. Oguz B, Ozcan HN, Omay B, Ozgen B, Haliloglu M. Imaging of childhood inflammatory myofibroblastic tumor. Pediatr Radiol (2015) 45:1672–81. doi:10.1007/s00247-015-3377-x
32. Alaggio R, Cecchetto G, Bisogno G, Gambini C, Calabrò ML, Inserra A, et al. Inflammatory myofibroblastic tumors in childhood: a report from the Italian Cooperative Group studies. Cancer (2010) 116:216–26. doi:10.1002/cncr.24684
33. Coffin CM, Patel A, Perkins S, Elenitoba-Johnson KS, Perlman E, Griffin CA. ALK1 and p80 expression and chromosomal rearrangements involving 2p23 in inflammatory myofibroblastic tumor. Mod Pathol (2001) 14:569–76. doi:10.1038/modpathol.3880352
34. Siminovich M, Galluzzo L, López J, Lubieniecki F, de Dávila MT. Inflammatory myofibroblastic tumor of the lung in children: anaplastic lymphoma kinase (ALK) expression and clinico-pathological correlation. Pediatr Dev Pathol (2012) 15:179–86. doi:10.2350/11-10-1105-OA.1
35. Antonescu CR, Suurmeijer AJ, Zhang L, Sung YS, Jungbluth AA, Travis WD, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 gene fusions and rare novel RET rearrangement. Am J Surg Pathol (2015) 39:957–67. doi:10.1097/PAS.0000000000000404
36. Messineo A, Mognato G, D’Amore ES, Antoniello L, Guglielmi M, Cecchetto G. Inflammatory pseudotumors of the lung in children: conservative or aggressive approach? Med Pediatr Oncol (1998) 31:100–4. doi:10.1002/(SICI)1096-911X(199808)31:2<100::AID-MPO10>3.0.CO;2-R
Keywords: inflammatory myofibroblastic tumor, inflammatory pseudotumor, high-resolution computer tomography, histopathology assessment, anaplastic lymphoma kinase rearrangement
Citation: Camela F, Gallucci M, di Palmo E, Cazzato S, Lima M, Ricci G and Pession A (2018) Pulmonary Inflammatory Myofibroblastic Tumor in Children: A Case Report and Brief Review of Literature. Front. Pediatr. 6:35. doi: 10.3389/fped.2018.00035
Received: 06 September 2017; Accepted: 07 February 2018;
Published: 27 February 2018
Edited by:
Elif Dagli, Emeritus Marmara University, TurkeyReviewed by:
Andrew A. Colin, University of Miami, United StatesYusei Ohshima, University of Fukui, Japan
Copyright: © 2018 Camela, Gallucci, di Palmo, Cazzato, Lima, Ricci and Pession. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giampaolo Ricci, Z2lhbXBhb2xvLnJpY2NpQHVuaWJvLml0