 Shalabh Srivastava1,2
Shalabh Srivastava1,2 Elisa Molinari
Elisa Molinari John A. Sayer
John A. Sayer- 1Institute of Genetic Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
- 2Renal Unit, City Hospitals Sunderland and South Tyneside NHS Foundation Trust, Sunderland, United Kingdom
- 3Department of Histopathology, Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle upon Tyne, United Kingdom
- 4Renal Services, Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle upon Tyne, United Kingdom
Nephronophthisis (NPHP) is a renal ciliopathy and an autosomal recessive cause of cystic kidney disease, renal fibrosis, and end-stage renal failure, affecting children and young adults. Molecular genetic studies have identified more than 20 genes underlying this disorder, whose protein products are all related to cilia, centrosome, or mitotic spindle function. In around 15% of cases, there are additional features of a ciliopathy syndrome, including retinal defects, liver fibrosis, skeletal abnormalities, and brain developmental disorders. Alongside, gene identification has arisen molecular mechanistic insights into the disease pathogenesis. The genetic causes of NPHP are discussed in terms of how they help us to define treatable disease pathways including the cyclic adenosine monophosphate pathway, the mTOR pathway, Hedgehog signaling pathways, and DNA damage response pathways. While the underlying pathology of the many types of NPHP remains similar, the defined disease mechanisms are diverse, and a personalized medicine approach for therapy in NPHP patients is likely to be required.
Introduction
Nephronophthisis (NPHP) is an autosomal recessive inherited kidney disease, which leads to end-stage renal disease (ESRD) typically within the first three decades of life (1). Traditionally, this disease was diagnosed using clinical and histological features. However, over recent years, many of the genetic causes underlying NPHP have been identified allowing both a precise molecular diagnosis to be made and some mechanistic insights into the underlying disease process. The known NPHP genes encode proteins that are almost all expressed in centrosomes and primary cilia. NPHP is therefore considered to be a ciliopathy disease (2), consistent with the fact that extrarenal manifestations, consistent will a ciliopathy syndrome, occur in around 20% of cases. Here we will review the clinical and histological features of the disease and its conventional classification before reviewing the underlying genetic causes and the ciliopathy syndromes associated with NPHP.
Based on the original histological descriptions, which included corticomedullary cysts, atrophy, and interstitial fibrosis, NPHP literally means disappearance or disintegration of nephrons (3). The clinical symptoms of NPHP, which reflect reduction in GFR and loss of distal tubular function (4), include polyuria, polydipsia, secondary enuresis, and growth retardation. Unfortunately, NPHP is associated with a progressive loss of kidney function and ESRD typically occurs before 30 years of age. Cases have historically been classified based on the age of onset of ESRD as infantile, juvenile, adolescent, and late onset. These are worth reviewing, although it is worth noting that a single genotype may present at a wide range of ages.
Juvenile NPHP is the classical form of NPHP and is characterized by polyuria and polydipsia symptoms and often anemia in patients within the first decade of life. Progressive loss of kidney function leads to ESRD at a median age of 13 years (5).
Kidneys affected by NPHP are grossly normal or have a shrunken appearance, typical of ESRD. There may be corticomedullary cysts that are up to 1.5 cm in size. Cysts often develop in later stages of the disease. The renal ultrasound scan appearances may often display a loss of corticomedullary differentiation.
Where renal biopsies have been performed in NPHP patients, distinct histological features have been reported. The histological changes can be divided into early or late stages of disease. In the early stages of the NPHP, there is interstitial fibrosis with sparse inflammation and lack of infiltration with neutrophils or monocytes. The tubules are tortuous and atrophic with segmented tubular basement membrane thickening (6). The distal tubules have focal diverticulum like protrusions. The glomeruli are usually normal but there may be periglomerular fibrosis that can extend into the glomerular tuft leading to focal or global collapse of the tuft and obsolescence of the glomeruli (7, 8). In later stages of the disease, the tubules may demonstrate basement membrane abnormalities with both atrophy and thickening. There often is cystic dilatation of the distal tubules, and the glomeruli may show collapse and severe periglomerular fibrosis (8, 9) (Figure 1). NPHP is not an immune mediated disease, and consequently there is no immune or complement deposition (6, 8). Electron microscopy may reveal tubular basement membrane duplication, thickening, and folding (6, 8). When examining clinical, pathological, and histological features of NPHP, it must be remembered that a separate disorder, known as medullary cystic kidney disease may share similar features. Medullary cystic kidney disease is an autosomal dominant condition, which is now classified under the term autosomal dominant tubulointerstitial kidney disease (ADTKD). Typical extrarenal manifestations include gout and anemia. A comparison of NPHP and ADTKD, alongside is given in Table 1 and has been discussed elsewhere (10).
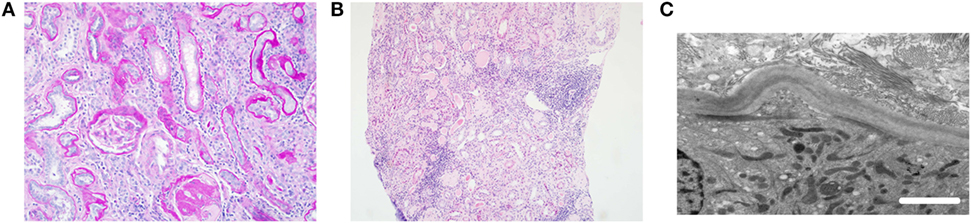
Figure 1. Typical histological features of nephronophthisis. (A) Light microscopy image. PAS stain demonstrates a globally sclerosed glomerulus and some periglomerular fibrosis. There is moderate interstitial fibrosis with chronic inflammation and thickening of tubular basement membranes. (B) Light microscopy image. H&E stain shows tubular atrophy with hyaline casts, moderate interstitial fibrosis, and patchy mononuclear inflammation. (C) Electron microscopy image. Tubular basement membrane demonstrates thickening and multilayering. Scale bar 2 µm.
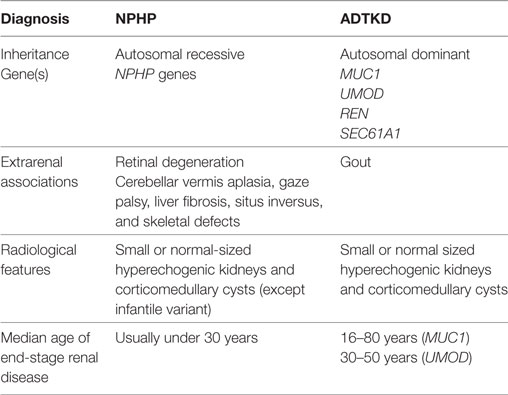
Table 1. Comparison of nephronophthisis (NPHP) with autosomal dominant tubulointerstitial kidney disease (ADTKD).
Infantile NPHP is rare, but is noteworthy, due to its severe phenotype with ESRD typically occurring during the first year of life (7). There may be antenatal presentations with oligohydramnios and bilateral enlarged cystic kidneys. Infantile NPHP is usually caused by mutations in INVS (11) and NPHP3 (12) but has been reported for other genetic forms of NPHP such as NEK8 (13) and CEP83 (14). The macroscopic and histological kidney phenotype is markedly different from other varieties of NPHP, with enlarged cystic kidneys, as opposed to micro and small corticomedullary cysts. Histologically, infantile NPHP lacks the tubular basement membrane changes seen in other NPHP phenotypes and may resemble autosomal recessive polycystic kidney disease. There may also be severe cardiac anomalies including situs inversus and ventricular septal defects (15).
The adolescent form of NPHP was originally described in a large Venezuelan pedigree (16). Biallelic mutations in NPHP3 were found in this family, resulting in ESRD at a median age of 19 years (16). It is now known that NPHP3 mutations may lead to a broad range of phenotypes including perinatal lethal Meckel–Gruber syndrome and infantile presentations. The term “adolescent NPHP” is thus somewhat arbitrary and merely extends the phenotypic spectrum from juvenile NPHP.
A number of case reports have highlighted the fact that NPHP may first present later in life. Georges et al. reported three (genetically unsolved) families with retinal dystrophy, NPHP on renal biopsy and slowly progressive renal failure and ESRD between the ages of 42 and 56 years (17). In another family with a homozygous NPHP1 deletion (18) ESRD was reported in three patients between 27 and 43 years of age. These cases of NPHP extend the age of ESRD from birth to up to the sixth decade of life.
An Approach to the Clinical Diagnosis of NPHP
Clinical recognition of NPHP is important, and the renal and extrarenal features of a ciliopathy syndrome (discussed below) may allow a clinical diagnosis to be made. NPHP occurs in isolation in around 80% of cases and is associated with a variety of other ciliopathy phenotypes in 20% of cases. A detailed review with specific emphasis on the family history and extrarenal features known to be associated with NPHP is therefore an essential prerequisite to an exact diagnosis. NPHP is characterized by a urinary concentrating defect early on in life that leads to polyuria and polydipsia. The onset of the disease may be easily missed, as there is typically no severe hypertension, minimal or no proteinuria, and a bland urine sediment. Clinical spectrums of disease are wide and widening. Besides extensive investigations of renal function, clinical phenotyping should also encompass a full neurological screening to assess for cerebellar signs and fundoscopy to assess for retinal degeneration. A formal ophthalmological examination is advised. The role of renal biopsy in diagnosing NPHP is contentious and should be limited to cases where a tissue diagnosis will serve to distinguish it from other differential diagnoses. In most cases, a histopathological diagnosis should be superseded by a molecular genetic diagnostic approach, because genetic screening allows for early diagnosis and prevents complications of renal biopsy. NPHP1 mutations and deletions are the most frequent genetic cause of NPHP and may be screened for using standard PCR assays (19). Given the large numbers of other NPHP genes involved multiplex PCR (20), targeted exon capture or whole-exome sequencing approaches are recommended (21).
Extrarenal Manifestations of NPHP
There are several important additional phenotypes that may be associated with NPHP (Table 2). These multisystem features are consistent with the fact that NPHP is a ciliopathy and may affect retina, brain, liver, and other tissues either by prenatal-onset dysplasia or by postnatal organ degeneration and fibrosis. Extrarenal manifestations are seen in ~20% of cases (22). In a recent study where 89 patients with NPHP mutations were analyzed, NPHP1 mutations were the most common genetic cause and gave rise to typical renal presentations. These included increased echogenicity of the kidney and loss of corticomedullary differentiation, with cystic kidney disease presenting later in the disease course (median age 12.3 years) and ESRD at a median age of 12.8 years (23). Extrarenal manifestations of NPHP1 mutations were seen more frequently than expected, with 8% presenting with liver symptoms, 19% having developmental delay and 7% epilepsy and seizures (23). The important syndromes associated with NPHP are briefly described below.
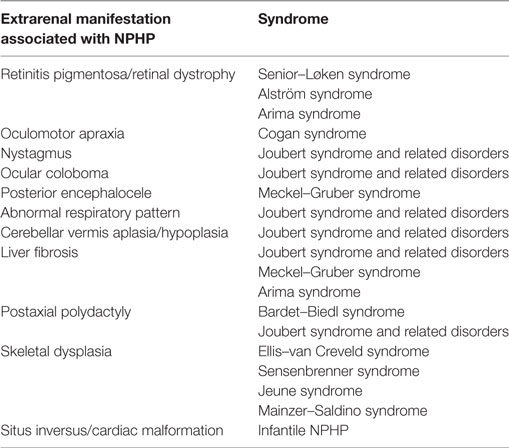
Table 2. Extrarenal manifestations of nephronophthisis (NPHP) and their associated syndromes.
NPHP with Retinitis Pigmentosa (Senior–Løken Syndrome)
Retinal dysplasia and degeneration is seen in 10–15% of patients with NPHP and may lead to an early and severe visual loss resembling Leber congenital amaurosis (LCA) (24, 25). Later onset forms present initially with night blindness, which then progresses to visual loss.
Cerebellar Vermis Aplasia/Hypoplasia with NPHP (Joubert Syndrome)
Joubert syndrome is a developmental disorder characterized by cerebellar vermis hypoplasia (26). Brain imaging (MRI) reveals the typical sign known as the “molar tooth sign.” Clinical features include hypotonia, cerebellar ataxia, neonatal tachypnea, and developmental delay. There may also be ocular coloboma, polydactyly, and hepatic fibrosis. NPHP is found in up to 30% of Joubert syndrome patients (27–29). Large cohorts of Joubert syndrome patients have been described, allowing some genotype/phenotype correlations to be made in the more frequent genetic causes. Mutations in TMEM67 in Joubert syndrome are the most frequently associated with kidney disease, whereas mutations in CEP290 were most likely to give retinal, renal, and brain phenotypes (28). In a recent cohort analysis of 97 patients with Joubert syndrome, renal phenotypes were detect in 30% of cases and was commonly associated with NPHP genes including CEP290, TMEM67, and AHI1 (29). In this study, renal phenotypes in Joubert syndrome extended beyond classical NPHP and included an overlapping phenotype resembling autosomal recessive polycystic kidney disease and NPHP (mimicking infantile NPHP), unilateral multicystic dysplastic kidney and indeterminate cystic kidney disease phenotypes (29).
Oculomotor Apraxia (OMA) Type Cogan
Oculomotor apraxia type Cogan is an eye movement disorder. It is characterized by abnormal horizontal eye movements that include nystagmus and difficulty with saccades (smooth visual pursuits) and has been associated with NPHP (2, 30). OMA may be a mild form of Joubert syndrome, as cerebellar vermis aplasia has been described in this condition (31).
Perinatal Lethality (Meckel–Gruber Syndrome)
Meckel–Gruber syndrome is characterized by occipital encephalocele, polydactyly, bile ductal proliferation, and cystic kidney dysplasia. Typically, the condition is perinatally lethal. The syndrome is associated with severe biallelic mutations in NPHP genes that include NPHP3, CEP290, and RPGRIP1L (1, 32–35).
Skeletal Defects [Jeune Syndrome (JS), Sensenbrenner Syndrome, and Saldino-Mainzer Syndrome]
Various skeletal defects have been reported in association with NPHP. These include cone-shaped epiphyses (16, 36), shortening of limbs, and ribs, scoliosis, polydactyly, brachydactyly, and craniosynostosis. Mutations are in genes encoding intraflagellar transport (IFT) proteins including TTC21B and WDR19 (37–41).
Episodic Hyperpnea (Joubert Syndrome)
The original report of Joubert syndrome (42) described episodes of fast breathing followed by a period of apnea. This feature is demonstrable only when the patient is awake. Abnormal respiratory pattern is not a consistent feature of Joubert syndrome and the reported incidence varies (44–71%) (43).
Anosmia As an Extrarenal Manifestation of Renal Ciliopathies
Several renal ciliopathy syndromes have been associated with anosmia, secondary to olfactory cilia defects. This has been studied in most detail in Bardet–Biedl syndrome (BBS) (44, 45) but has been reported in patients with LCA secondary to mutations in CEP290 and in a murine model of Cep290 (46). Such data also suggest a link between ciliary defects in the olfactory neurons and kidney disease. Indeed, proteins that mediate olfactory-like chemosensory signaling pathways were found expressed in the renal tissue (47), including adenylate cyclase III, which is localized to the primary cilium (48). These pathways may be vital in tubuloglomerular feedback and blood pressure control. There is a real need now to assess patients with renal ciliopathies/NPHP (and indeed corresponding murine models) for defects in smell and to determine a role for olfactory-like signaling within the kidney.
Known Genetic Causes of NPHP
There are now more than 20 genes that if mutated may lead to NPHP (Table 3). It is worth reviewing these genetic causes as they all point toward some mechanistic insights into the pathogenesis of NPHP. Finding commonality among the genetic causes relies on a connection to the centrosome/basal body/primary cilium, although this may not be true for every genetic cause.
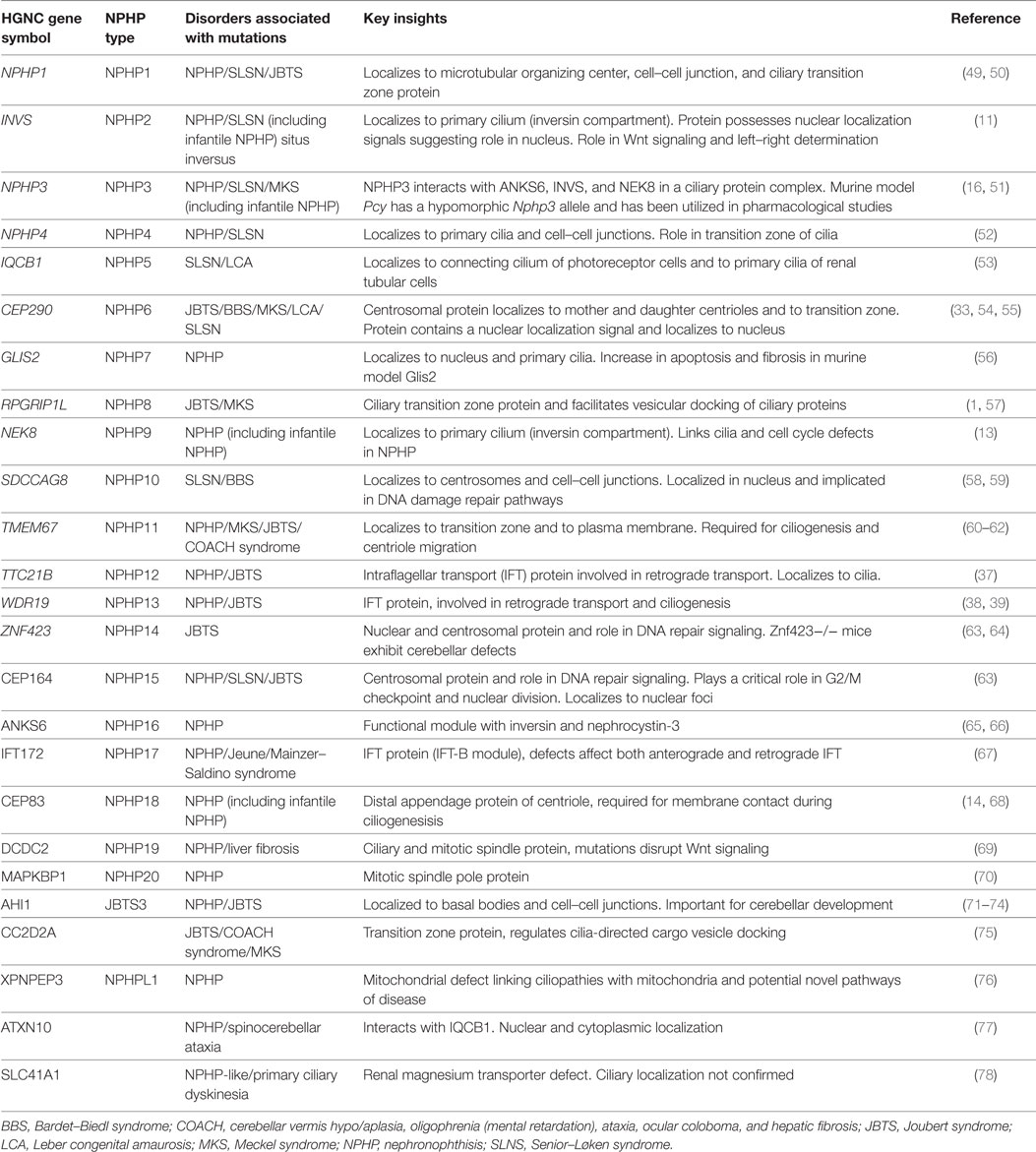
Table 3. Genetic classification of NPHP, its related disorders and the key insights obtained.
The most common genetic cause of NPHP is mutations in NPHP1, which account for around 20% of cases. The most common NPHP1 gene defect is a large homozygous deletion affecting the whole gene (49, 79). Each of the remaining NPHP genes probably accounts for 1% or less of all cases of NPHP, meaning that around two-thirds of cases remain genetically unsolved (2). It is noteworthy that mutations in a single NPHP gene may give an extremely wide spectrum of clinical phenotypes that may include isolated NPHP, NPHP with additional features, such as Senior–Løken syndrome and Joubert syndrome and severe neonatal lethal forms, such as of Meckel–Gruber syndrome. Linkage studies and painstaking mapping approaches led to the identification of NPHP1 in 1997 (49). Similar approaches for the next decade (sometimes combined with candidate gene screens) allowed the discovery of eight genes (at a rate of around one new gene per year). Since 2010, next-generation sequencing approaches have been utilized (80) allowing the detection of NPHP genes at a much faster rate.
NPHP1
NPHP1 encodes nephrocystin-1 (alias nephrocystin). It was shown to interact with p130cas, tensin, filamin, and focal adhesion kinase 2, all molecules involved in cell–cell adhesion and cell signaling (81–83). In the primary cilium, nephrocystin-1 interacts with nephrocystin-4 and RPGRIP1L at the transition zone and links it to inversin (77).
INVS
INVS causes ESRD in the first 2 years of life and presents typically as an infantile form of NPHP as described earlier. The frequency of INVS mutations has been reported to be as high as 78% in the group of patients reaching ESRD before 2 years of age (12). The kidney size in INVS is often enlarged unlike most other forms of NPHP in which the kidneys are normal in size or shrunken (12). The distribution of cysts is corticomedullary and is more reminiscent of autosomal recessive polycystic kidney disease, given the kidneys can be massively enlarged.
Inversin, the gene product of INVS interacts with nephrocystin-1 and nephrocystin-3 and plays a vital role in intercellular adhesion (84). It localizes to the cilium and serves as a switch between the canonical and non-canonical Wnt pathway (85). Otto et al. established a link between cystogenesis and the primary cilia in humans disease during the study of this disease in 2003 (11) establishing this as a landmark paper in the study of NPHP and ciliopathies. Inversin is also plays a role in planar cell polarity (PCP) processes, discussed below. Loss of inversin leads to abnormal mitotic spindle orientation (86), which may drive cystogenesis.
NPHP3
Omran et al. first described mutations in NPHP3 in a large Venezuelan family in 2000 (87). It is characterized by NPHP, situs inversus, and structural heart defects. Hoff et al. uncovered a link between nephrocystin-3, inversin, and NEK8 (65) in a report on the role of ANKS6, linking the above proteins at the proximal part of the primary cilium known as the inversin compartment. This may explain the overlap seen in the phenotype of patients with mutation in INVS, NPHP3, and NEK8.
NPHP4
NPHP4 was identified by homozygosity mapping and genome wide linkage analysis by Mollet et al. in patients with NPHP who did not have mutations in the NPHP1, 2, and 3 genes. Nephrocystin-4 localizes to the primary cilia and cortical actin cytoskeleton in the polarized cells. In dividing cells, it localizes to the centrosomes. It has been shown to interact with p130 (Cas), tensin, and filamin (88).
IQCB1
Patients with IQCB1 mutations are characterized by the presence of retinitis pigmentosa with NPHP (renal–retinal or Senior–Løken syndrome). In a study investigating the association of retinitis pigmentosa with NPHP, Otto et al. found a novel gene IQCB1 that associates with retinitis pigmentosa GTPase regulator (RPGR) and calmodulin in the retinal connecting cilia, an analogous structure of the ciliary transition zone (53).
CEP290
Mutations in the CEP290 gene underlie NPHP6 and are the leading cause of Joubert syndrome and related diseases, a cerebello–retinal–renal syndrome. The association of CEP290 with NPHP was established in 2006 in a cohort of families with Joubert syndrome, Senior–Løken syndrome, and NPHP (54, 55). CEP290 was found to interact with the transcription factor ATF4, which is involved in cyclic adenosine monophosphate (cAMP) mediated cyst formation (54). CEP290 mutations are the most common inherited cause of retinal degeneration (LCA). Mutations in CEP290 may also cause BBS phenotypes (89).
TMEM67 (33, 90) and CC2D2A (91, 92) are both interacting partners of CEP290 and can cause severe ciliopathy phenotypes including Meckel–Gruber syndrome and Joubert syndrome. There is emerging evidence of the role of CEP290 in ciliogenesis (93), cell signaling (94, 95), DNA damage response (DDR) (96), and consequently renal cystogenesis (97).
GLIS2
In 2007, Attanasio et al. reported a mutation in GLIS2 as a novel cause for NPHP. The loss of this transcription factor leads to increased fibrosis and apoptosis (56). In a recent paper, GLIS2 loss has been found to increase cell senescence. Kif3a null mice show increased cyst formation due to unrestrained proliferation, destabilization of p53 and increased DNA damage. This is partially rescued by ablation of GLIS2 and pharmacological stabilization of p53 (98).
RPGRIP1L
Arts et al. identified mutations in RPGRIP1L as causative for Joubert syndrome in three families in 2007. This protein localizes to the basal body and interacts with NPHP4 (57).
NEK8
Otto et al. identified NEK8 as the causative gene for Joubert syndrome after observing that the jck mouse harbors a mutation in the highly conserved RCC1 domain of Nek8. They performed a mutational analysis of a worldwide cohort of patients and established the pathogenic role of NEK8 mutations in humans (13). More recently, NEK8 loss was implicated in increased DNA damage in the pathogenesis of NPHP (99). This established one of the first associations between the role DDR and cystic kidney disease. Grampa et al. have recently described the association of NEK8 with deregulation of the Hippo pathway and its role in severe syndromic renal cystic dysplasia (100). Al-Hamed et al. described a stillborn fetus with cystic kidneys, oligohydramnios, CVA and bilateral bowing of the femur secondary to NEK8 mutation (101).
SDCCAG8
SDCCAG8 was the first NPHP gene to be identified using next-generation sequencing approaches (58). Patients with mutations in this gene were diagnosed with Senior–Løken syndrome, but may also have features suggestive of BBS (102). The encoded protein SDCCAG8 localizes to centrioles and directly interacts with the ciliopathy-associated protein OFD1. A recently described murine model of SDCCAG8 has implicated elevated levels of DDR signaling as a potential mechanism of kidney disease (59).
TMEM67
Otto et al. screened a cohort of 62 patients with NPHP and liver fibrosis and found hypomorphic mutations in TMEM67 in 8% of the patients (61). TMEM67 has been implicated in the pathogenesis of Meckel–Gruber syndrome, Joubert syndrome, and COACH syndrome (cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, coloboma and congenital hepatic fibrosis) (33, 34). Liver fibrosis is a frequent feature of TMEM67 mutations, and any patient with NPHP along with liver involvement should have tests for mutations in TMEM67. In a cohort of 100 patients with Joubert syndrome, mutations in TMEM67 were most frequently associated with kidney disease (28).
TTC21B
Davis et al. reported the association of TTC21B mutations with both isolated NPHP and JS (37). TTC21B encodes the retrograde IFT protein IFT139, which has been shown to regulate Hedgehog signaling (103).
WDR19
WDR19 mutations have been reported in patients with ciliopathy syndromes including Sensenbrenner syndrome, Joubert syndrome, Senior–Løken syndrome, and isolated NPHP (38, 41, 104). WDR19 encodes for IFT144, a protein that participates in retrograde IFT and is important for ciliogenesis.
ZNF423
ZNF423 mutations have shown to cause Joubert syndrome with NPHP (63). The encoded protein ZNF423 is a nuclear protein which functions as a DNA-binding transcription factor and interacts with DDR protein PARP1 [poly (ADP-ribose) polymerase 1] and also CEP290 (63).
CEP164
Mutations in CEP164 may cause NPHP and related ciliopathy syndromes including Senior–Løken syndrome (63). The CEP164 protein is a regulator of ciliogenesis and is essential for the formation of the distal appendage of the centriole (105). Loss of CEP164 induces DNA damage (63).
ANKS6
ANKS6 mutations lead to NPHP. ANKS6 localizes to the inversin compartment and links the NPHP proteins NPHP2, NPHP3, and NPHP9 to NEK8. This functional role of ANKS6 in an NPHP module may explain the phenotypic overlap that includes abnormalities in heart and liver, seen in the patients carrying individual mutations in these genes (65, 66).
IFT172
Intraflagellar transport is vital in maintaining the cilium and in executing its functions. IFT-A module has six components, and mutations in genes encoding these proteins have all been associated with ciliopathy diseases. IFT-B has 14 components. Halbritter et al. established the first link between IFT-B component IFT172 and skeletal ciliopathies. Some patients in this cohort had NPHP (67). Mutations in IFT172 may also cause BBS syndrome (106).
CEP83
CEP83 mutations have recently been described to cause infantile NPHP (14). CEP83 encodes a centriolar distal appendage protein, CEP83. In the seven families so far described, the NPHP phenotype was early-onset (juvenile), and in some was also associated with hydrocephalus and learning difficulties (14).
DCDC2
Schueler et al. reported a novel mutation in the gene DCDC2, in patients presenting with an NPHP and hepatic fibrosis phenotype (69). DCDC2 localizes to the ciliary axoneme and the mitotic spindles. DCDC2 knockdown inhibits ciliogenesis. It interacts with DVL, and DCDC2 knockdown leads to defects in Wnt signaling and may contribute to the liver fibrosis (69).
MAPKBP1
Macia et al. have recently described a novel gene, MAPKBP1, in five families with eight individuals presenting with juvenile or late-onset NPHP with massive fibrosis. This gene encodes MAPKBP1, a scaffolding protein for JNK signaling. Interestingly, this protein does not localize to the primary cilium instead it localizes to the mitotic spindle pole. The authors also report increased DDR signaling in murine fibroblasts upon knockdown of Mapkbp1 (70).
AHI1
Mutations in AHI1 were initially described in patients with Joubert syndrome and no kidney involvement (71, 72). However, AHI1 mutations may cause NPHP phenotypes (73) and may cause multicystic dysplastic kidneys also (29). AHI1 is localized to the basal body and cell–cell junctions (74).
CC2D2A
Mutations in CC2D2A have been reported to cause Joubert syndrome with and without cystic kidney disease (92). CC2D2A is localized to the basal body and colocalizes with CEP290 (92). Mutations in CC2D2A may also cause antenatal cystic kidney disease phenotypes and severe brain phenotypes (typical of Meckel–Gruber syndrome) leading to fetal death (101).
Evidence of Oligogenicity and Triallelism in NPHP
Alongside the novel findings relating to gene discovery in NPHP has been the continued theme of wide phenotypic variability, especially in extrarenal manifestations. The type of mutation may influence the phenotype in certain circumstances. Examples include CC2D2A (107, 108) and TMEM67 (109) where two truncating mutations tend to lead to more severe phenotypes than missense mutations. With the now frequent sequencing of NPHP cohorts (110, 111) and the use of high-throughput genetic sequencing platforms (112), a few findings of oligogenicity and triallelism within NPHP have been reported; however, these are controversial as these findings are anecdotal. As an example, a heterozygous AHI1 mutation when inherited with biallelic NPHP1 mutations seems to lead to a more severe brain phenotype (111). Thus, a concept of mutation burden seems relevant to NPHP, and like BBS (113) it will be important that these variants are reported and that they are assessed in terms of their pathogenicity. Interestingly, NPHP1 mutations and copy number variants, as well as causing NPHP and Joubert syndrome, may also contribute to the mutational burden of BBS (114, 115). More recent next-generation sequencing data studying Joubert syndrome suggest that rare disease variants are frequently found in addition to the causal biallelic variants. Typically, over one-third of affected individuals carry rare disease variants in addition to the causal mutations but importantly they did not correlate with disease severity (116). This study also found no evidence or support for triallelism. Discordant phenotypes between affected siblings were observed in 60% of subjects who shared causal mutations, suggesting that modifier alleles are important but elusive (116). Therefore, using third alleles and other NPHP gene variants of uncertain significance to determine additional phenotypes and disease severity and inform genetic counseling is not presently advised, or should only be done with the utmost care.
Pathogenesis of NPHP
There are various theories behind the pathogenesis of the NPHP disease process. The very early hypotheses were based entirely on the histopathological description of the disease and led to the widespread belief that this disease was caused by some unknown nephrotoxic agent or an enzyme defect (8). The frequent finding of tubular basement membrane thickening led to a basement membrane hypothesis for the pathogenesis of NPHP. It was observed that nephrocystin-1, the protein product of NPHP1 had a high degree of sequence conservation with CRK (a focal adhesion protein) (117), contained an SH3 domain and interacted with other proteins including p130Cas and ACK1 (49, 118). Nephrocystin-1 was shown to localize to adherens junctions and focal adhesions. This supported a hypothesis that nephrocystin-1 has an important role in the maintenance of the tubular epithelium and that abnormal cell–cell and cell–matrix interactions were the underlying defect in NPHP. Many years later, the debate of the initial pathogenic defect in NPHP continues, with the focus on NPHP as a ciliopathy (119). This hypothesis is strongly supported by multiple gene discoveries in NPHP with nearly all the affected genes coding for the components of the cilia, basal body or centrosome. Defects in primary cilia associated with cystic kidney disease were initially noted in Ift88 mutant mice (120). This link between NPHP and cilia was confirmed in human disease established after the discovery that INVS mutations cause infantile NPHP and that the encoded protein inversin interacts with nephrocystin-1 and β-tubulin, colocalizing with them to the primary cilia of renal tubular cells (11). There is now almost universal agreement that the primary cilia are at the center of the disease process especially in terms of cystogenesis although it should not be forgotten that nephrocystins may have multiple subcellular localizations (54) and may play different roles in different tissues (1). There is an interesting overlap with the localization and function of NPHP genes and other “cystogenes” such as PKD1, PKD2, and the many other inherited causes of cystic kidney disease. The functional role of primary cilia in the human nephron is not fully understood. It was initially thought that the encoded proteins from PKD1 and PKD2, namely, polycystin-1 and polycystin-2, were able to sense luminal flow of urine, and ciliary deflection stimulated calcium entry into the cell via the polycystin proteins leading to downstream signaling cascades (121). However, more recent studies have challenged this hypothesis (122, 123).
A Comparison of NPHP and ADPKD Pathophysiology
A detailed discussion of the underlying pathophysiology of ADPKD has been recently published and is beyond the scope of this review (124). It is worth highlighting, however, that disease pathways in ADPKD involve cAMP, ciliary dysfunction, PCP and centrosome number as well as many others (124). Thus, these mechanisms of disease are shared with those of NPHP. Indeed, the development of tolvaptan, a vasopressin V2 receptor (V2R) antagonist for the use in patients with ADPKD was pioneered in murine models of NPHP (51). However, fluid secretion and proliferation seems less prominent in NPHP, while fibrosis and scarring are more prominent pathological features. The pairing of potential disease mechanisms in NPHP with targeted therapeutics will hopefully allow better treatments for NPHP in the near future (125).
Other Key Molecular Pathways Implicated in NPHP
Planar Cell Polarity
Planar cell polarity is an evolutionary conserved mechanism by which cells maintain their orientation in a plane perpendicular to the apical-basal polarity of a cell layer. This is achieved by the correct alignment and orientation of cell division, orchestrated by the mitotic spindle and centrosomes. The maintenance of tubular diameter is dependent upon PCP signaling, and when this is defective, tubular dilatation rather than elongation is thought to contribute to cystogenesis (126, 127). Non-canonical Wnt signaling is vital for these signaling events and mutations in INVS are thought to lead to defective regulation of this pathway (84, 85). Mutations in DCDC2, leading to NPHP type 19 have also been implicated in this pathway, lending weight to this mechanism of cytogenesis and NPHP. Loss of Dcdc2 in IMCD3 cells led to an activation of Wnt signaling, leading to a loss of cilia, which was amenable to treatment with Wnt inhibitor treatment (69). Other connections to the Wnt pathway in NPHP includes CEP164 which interacts with disheveled protein 3 (DVL3) (63). The disheveled protein is a key component of the Wnt pathway, and part of the switch between canonical and non-canonical Wnt signaling. Defective Wnt signaling has also been demonstrated in murine models of Joubert syndrome. Ahi1 mutant mice showed defect in cerebellar midline fusion in sites of reduced Wnt activity (128) while renal tissues from the same mice demonstrated abnormal Wnt signaling in late stages of NPHP (129). A murine gene trap model of Cep290 similarly showed Wnt pathway changes (reduced Tcf1 protein) only at later stages of the disease (murine kidney tissue aged 1 year) implicating this pathway in renal fibrosis (95). The relationship between Wnt signaling and cystic kidney disease has been recently reviewed (130).
cAMP Signaling
A huge amount of data have demonstrated the key role of elevated cAMP in mural epithelial cell proliferation and fluid secretion, which are the main drivers of cyst formation in polycystic kidney disease (131). However, some lines of evidence suggest that high cAMP levels are also implicated in junctional and polarity defects in NPHP. Levels of cAMP were found to be elevated in Nphp3-, Nphp6-, and Nphp8-stable knockdown mIMCD3 lines. When these cells were examined in a 3D spheroid culture system, they formed abnormal spheroids with no lumen and/or misaligned nuclei. Treatment with octreotide, an inhibitor of cAMP production, could rescue these structural abnormalities, linking high cAMP levels to cell polarity defects (132). Furthermore, it was shown that treatment with the cAMP analog 8-bromo-cAMP resulted in a dose-dependent loss of SDCCAG8 (NPHP10) protein in cell–cell junctions of the renal epithelial cell line MDCK-II, highlighting the potential role of high cAMP in perturbation of tissue architecture (58). Importantly, elevated cAMP levels and expression levels of the cAMP-dependent gene Aquaporin-2 were found in the renal tissue of Pcy mice that carry a missense mutation in Nphp3 (51).
mTOR Pathway
Increased mTOR (mechanistic target of rapamycin) activity was found in cystic kidney (133, 134) and in particular in cyst-lining epithelium (135) of several NPHP mouse models. mTOR is an atypical serine/threonine kinase that, by integrating a variety of signals from nutrients and growth factors, regulates cell growth and proliferation.
The detailed subcellular localization of mTOR pathway components is not clear but it has been showed that primary cilium is important for the regulation of mTOR pathway (136, 137). It has been proposed that the flow-dependent bending of primary cilium represents a mechanosensory signal that controls cell size through the regulation of mTOR activity (137). Consequently, cilia abnormalities may ultimately result in cell growth deregulation, which could be potentially critical for the tubular geometry of the kidney.
The Role of Cilia in Sonic Hedgehog Signaling and Cell Cycle
The Hedgehog (Hh) signaling pathway is a key developmental pathway and was first discovered in Drosophila (138). There are three mammalian Hh homologs, Desert, Indian, and Sonic. The sonic Hh (Shh) pathway is essential for development (139), patterning, organogenesis, and cell signaling (140). It acts as a morphogen and a mitogen and dysregulation of the pathway can lead to severe developmental defects and can give rise to various cancers (141). Shh signaling is intimately related to the primary cilium (141). The receptor Patched (Ptch1) is 12-pass transmembrane protein localized to the primary cilium. It has an inhibitory effect on the translocation of Smoothened (Smo, a G-protein-coupled-like receptor). The secreted ligand Shh binds to Ptch1 and triggers internalization of Ptch1 into endocytic vesicles. This allows the translocation of Smo into the primary cilium and its stepwise activation (142). Downstream effector Glioma proteins (Gli2,3) remain in a neutral state under the effect of suppressor of fused and by sequential phosphorylation by protein kinase A, glycogen synthase kinase 3β, and casein kinase 1, they undergo proteolytic conversion to their repressor form. Smo, when enriched in the primary cilium and activated, promote the conversion of Gli repressor (Gli3r) forms into full-length activator forms. The Gli activators (Gli3a) induce the expression of Hh target genes cyclin D1, Gli1, Gli2, N-myc, and Ptch1. An intact Hh pathway is important for ciliogenesis. The evidence implicating the defects of the Hh signaling in NPHP, renal development and cystogenesis is evolving (77, 103). Loss of the transcription factor Glis2 (Gli-similar zinc finger protein) causes NPHP type 7 (56), Shh knockout mouse embryos showed either renal agenesis or cystic dysplasia (143) and upregulated Indian Hh has been implicated in cystogenesis (144). A subset of BBS proteins has been shown to modulate Shh signaling and interact with IFT proteins (145). More recently, Hh signaling has been shown to be dysregulated in models of cystic kidney disease including Thm1, Pkd1, jck (103) and Cep290 (95).
The Ciliary Transition Zone and Links to NPHP
Between the basal body and the ciliary axoneme lies the transition zone, a physical barrier between the ciliary membrane and the apical plasma membrane of the cell. Several genes causing NPHP encode transition zone components, including NPHP1, RPGRIP1L, NPHP4, and CEP290. The transition zone controls the protein entry into and exit from the primary cilium and the composition of the ciliary membrane, which directly impacts ciliary signaling pathways such as Hedgehog signaling. The hedgehog signaling molecule Smoothened has been shown to accumulate in discrete clusters in the transition zone, and RPGRIP1L mutations disrupted this localization, leading to disrupted signaling (146). These data support the hypothesis of the transition zone as a gatekeeper of the cilium and that defects can account for phenotypes such as NPHP.
DDR Pathways and NPHP
The DDR signaling pathway allows the cell to detect DNA damage, apply an arrest in cell cycle and promote repair of the DNA. Repair of double stranded DNA breaks is particularly important for the maintenance of chromosome integrity. The DNA damage pathway ensures that damaged cells do not progress through S phase and into mitosis before repair is complete. Recently, several of the proteins implicated in NPHP including NEK8 (99), CEP164 (147), ZNF423 (63), SDCCAG8 (58, 59), and CEP290 (96) have been implicated in this pathway, suggesting a nuclear (non-ciliary) role. Following DNA damage, ZNF423, CEP164, and SDCCAG8 proteins have been shown to colocalize to nuclear foci positive for TIP60, a marker of sites of DNA damage and knockdown of CEP164 or ZNF423 causes increased sensitivity to DNA damaging agents (63). These observations provided a hypothesis that may explain why some NPHP genes with null mutations (such as NPHP3, CEP290, and RPGRIP1L) present as severe congenital-onset dysplasia and malformation in multiple organs including the kidney, brain and eye while hypomorphic mutations in the same genes produce milder phenotypes, which include late-onset degeneration and fibrosis leading to NPHP in the kidney and retinal degeneration in the eye. During periods of high proliferation and replication stress such as morphogenesis, DDR signaling is essential, and defects may lead to tissue dysplasia. By contrast, during maintenance of tissues in postnatal life, low replication stress would be expected, and defects would produce a degenerative phenotype. This hypothesis may go some way to explain the organ specific phenotypes seen in Joubert syndrome and other syndromes associated with NPHP (63). DDR defects and replication stress may also be an explanation for the fibrosis seen in association with NPHP and represents drugable target for the disease, which may be independent from and more reversible than cystogenesis (96, 148).
An Integration of Signaling Pathways in the Development of NPHP
It remains clear that given the genetic heterogeneity of NPHP and the numerous mechanistic pathways discussed that there is not one unifying pathology leading toward NPHP. The renal histology of NPHP points to a common endpoint of tubular damage and fibrosis, which may have multiple triggers. With each new gene discovery paper, there seems to be better clarity toward molecular diagnosis but more confusion regarding the signaling pathways underlying disease.
The clear themes concerning NPHP are that this disease is a manifestation of a renal ciliopathy with almost all NPHP-associated genes encoding gene products known to localize to primary cilia and regulate ciliary function and structure (149), with both the Hedgehog and Wnt signaling pathways implicated downstream from abnormal ciliary signaling. The function of the ciliary transition zone as a gatekeeper for ciliary protein entry and exit is clearly fundamental to ciliary signaling processes. Protein interaction studies of NPHP proteins now allow the proteins to be grouped into four distinct modules. These are the NPHP1–4–8 (NPHP1, NPHP4, and RPGRIP1L) module, the NPHP2–3–9-ANKS6 (INVS, NPHP3, NEK8, and ANKS6) module, the NPHP5–6 (IQCB1 and CEP290) module and the MKS module (MKS1, CC2D2A, and TCTN2). This points to the fact that each NPHP protein has a distinct localization and function within the centrosome/transition zone/cilium.
However, there is also growing evidence for a nuclear/DDR function of some NPHP-associated proteins, which may be important in disease initiation and progression. Whether this is independent of roles in the primary cilium is not known. It is possible that loss of ciliary function may be a downstream effect of nuclear events affecting cell cycle progression as a result of replication stress (148). The intimate relationship between ciliogenesis and DDR has recently been discussed (150). Centrosomal proteins including NEK8 and CEP290 that are mutated in ciliopathy disorders and are known to have functional roles in DDR are discussed in detail. Overall, it seems likely, given the body of evidence concerning cilia and cystic kidney disease that ciliary dysfunction is a relatively specific subcellular phenotype and final common pathway leading to NPHP, but other pathways may feed into this and be interrelated.
Treatment of NPHP
Nephronophthisis is incurable at present, but a range of potential therapeutic interventions has arisen from several lines of investigation into the pathogenesis of NPHP. Elevated renal cAMP levels were found associated with the cystic phenotype of NPHP and the modulation of cAMP production has been extensively explored as a potential strategy in the treatment of cystic kidney disease (51, 151–154). V2R antagonists are able to slow the rate of cAMP production by inhibiting V2R that, by coupling with G proteins, regulates the activity of adenylate cyclase and mediates urine concentration. Indeed, V2R antagonists OPC31260 and tolvaptan were shown to be effective in reducing renal accumulation of cAMP and rescuing the cystic kidney phenotype in Pcy mice (a model of NPHP3) (16, 51, 153). The use of tolvaptan has now moved successfully from preclinical models, through clinical trials (155) and into clinical practice (156). Its use in childhood ADPKD is currently being investigated in clinical trials.
As discussed, several lines of evidence support a direct link between DNA damage and the loss of NPHP proteins. In particular, both the NPHP type nine associated protein NEK8 and Cep290 were shown to be important regulators of DNA damage as their loss leads to increased sensitivity to replication stress and increased levels of CDKs (96, 99). Interestingly, CDK inhibition is able to suppress the DNA damage caused by loss of NEK8 or Cep290, therefore providing a rationale for CDK inhibition as a potential strategy in the treatment of NPHP (96, 99). Indeed, the CDK inhibitor roscovitine and its analog S-CR8 significantly halted the progression of cystic phenotype and attenuated loss of kidney function in jck mice (carrying a mutation in Nek8) (99, 157). Furthermore, roscovitine was able to ameliorate the ciliary phenotype of renal epithelial cells derived from a patient with NPHP secondary to a mutation in CEP290. Interestingly, it was shown that the treatment with purmorphamine, an agonist of the Shh pathway, is not only as effective as roscovitine in rescuing the ciliary defect, but is also able to decrease CDK5 protein levels in patient cells, suggesting a possible convergence of these signaling pathways (158).
Given the pivotal role played by the primary cilium in the context of Hh pathway, a manipulation of Hh signaling appears as an appealing strategy in the treatment of ciliopathies such as NPHP. It has been shown that genetic deletion of Gli2 can ameliorate the cystic kidney phenotype in an orthologous mouse model of TTC21B (56), while Hh agonism mediated by purmorphamine treatment is able to rescue the architectural defect displayed by 3D cultures of CEP290 renal epithelial cells (95).
Hyperactivation of mTOR (mechanistic target of rapamycin) pathway was found to be associated with cystic kidney disease and rapamycin has proven to be effective in several rodent [Han:SPRD rat (134, 159), LPK rat (135), Pcy mouse (133), and zebrafish models (invs, iqcb1, and cep290 morphant) of NPHP (160)].
The zebrafish models of NPHP are proving also to be extremely useful for high-throughput drug screens to determine their effect on kidney development (161).
There is hope therefore that these and other animal models of NPHP will provide valuable insights for future personalized medicine treatments of NPHP in affected patients (162). However, despite the great number of promising interventions that has arisen from preclinical studies, no clinical trials have yet been conducted to test their therapeutic potential in NPHP patients, most of whom eligible for treatment would be less than 18 years of age (125).
To date, options for the treatment of NPHP remain supportive. Control of blood pressure is a priority in children and young adults affected. Management of complications arising from progressive renal failure such as anemia, symptoms of uremia and fluid overload are important alongside preparation for future renal replacement therapy. This disease does not recur in a transplant and renal transplantation remains the ideal mode of renal replacement therapy.
Conclusion
The clinical and pathological diagnosis of NPHP is important, given its progression to ESRD and its associated extrarenal manifestations. Molecular genetic investigations allows a diagnosis in around one-third of cases and can give insights into the associated disease features, the underlying mechanisms and hopefully pave the way for individualized treatments for the underlying kidney disease. As this review demonstrates, it is true that many genes cause NPHP and while most of the identified molecular causes implicate the primary cilium in the pathogenesis of NPHP, it has also become apparent that there are important differences in the underlying pathophysiology. The traditional descriptions of NPHP of infantile, juvenile, and adolescent may now seem dated; however, they highlight the fact that different genetic forms of the disease disrupt the kidney by different mechanisms, demanding a precision medicine approach to the diagnosis, understanding and treatment of NPHP and its associated syndromes.
Author Contributions
JS conceived, drafted, and wrote the manuscript. SS and EM drafted and revised the manuscript. SR drafted the manuscript and provided figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer KH and handling Editor declared their shared affiliation.
Acknowledgments
SS is a Kidney Research Clinical Training Fellow. EM is funded by Kids Kidney Research. JAS is funded by the MRC MR/M012212/1, the Newcastle upon Tyne Hospitals NHS Charity and Northern Counties Kidney Research Fund.
Funding
The authors gratefully acknowledge funding from Kidney Research UK, Kids Kidney Research, and Northern Counties Kidney Research Fund.
References
1. Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet (2007) 39(7):875–81. doi:10.1038/ng2039
2. Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol (2009) 20(1):23–35. doi:10.1681/ASN.2008050456
3. Simms RJ, Eley L, Sayer JA. Nephronophthisis. Eur J Hum Genet (2008) 17(4):406–16. doi:10.1038/ejhg.2008.238
4. Krishnan R, Eley L, Sayer JA. Urinary concentration defects and mechanisms underlying nephronophthisis. Kidney Blood Press Res (2008) 31(3):152–62. doi:10.1159/000129648
5. Hildebrandt F, Strahm B, Nothwang HG, Gretz N, Schnieders B, Singh-Sawhney I, et al. Molecular genetic identification of families with juvenile nephronophthisis type 1: rate of progression to renal failure. APN Study Group. Arbeitsgemeinschaft fur Padiatrische Nephrologie. Kidney Int (1997) 51(1):261–9. doi:10.1038/ki.1997.31
6. Zollinger HU, Mihatsch MJ, Edefonti A, Gaboardi F, Imbasciati E, Lennert T. Nephronophthisis (medullary cystic disease of the kidney). A study using electron microscopy, immunofluorescence, and a review of the morphological findings. Helv Paediatr Acta (1980) 35(6):509–30.
7. Gagnadoux MF, Bacri JL, Broyer M, Habib R. Infantile chronic tubulo-interstitial nephritis with cortical microcysts: variant of nephronophthisis or new disease entity? Pediatr Nephrol (1989) 3(1):50–5. doi:10.1007/BF00859626
8. Waldherr R, Lennert T, Weber HP, Fodisch HJ, Scharer K. The nephronophthisis complex. A clinicopathologic study in children. Virchows Arch A Pathol Anat Histol (1982) 394(3):235–54. doi:10.1007/BF00430668
9. Gibson AA, Arneil GC. Nephronophthisis-report of 8 cases from Great Britain. Arch Dis Child (1972) 47:84–9. doi:10.1136/adc.47.251.84
10. Hildebrandt F, Otto E. Molecular genetics of nephronophthisis and medullary cystic kidney disease. J Am Soc Nephrol (2000) 11(9):1753–61.
11. Otto EA, Schermer B, Obara T, O’Toole JF, Hiller KS, Mueller AM, et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet (2003) 34(4):413–20. doi:10.1038/ng1217
12. Tory K, Rousset-Rouviere C, Gubler MC, Moriniere V, Pawtowski A, Becker C, et al. Mutations of NPHP2 and NPHP3 in infantile nephronophthisis. Kidney Int (2009) 75(8):839–47. doi:10.1038/ki.2008.662
13. Otto EA, Trapp ML, Schultheiss UT, Helou J, Quarmby LM, Hildebrandt F. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J Am Soc Nephrol (2008) 19(3):587–92. doi:10.1681/ASN.2007040490
14. Failler M, Gee HY, Krug P, Joo K, Halbritter J, Belkacem L, et al. Mutations of CEP83 cause infantile nephronophthisis and intellectual disability. Am J Hum Genet (2014) 94(6):905–14. doi:10.1016/j.ajhg.2014.05.002
15. Wolf MT, Hildebrandt F. Nephronophthisis. Pediatr Nephrol (2011) 26(2):181–94. doi:10.1007/s00467-010-1585-z
16. Olbrich H, Fliegauf M, Hoefele J, Kispert A, Otto E, Volz A, et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet (2003) 34(4):455–9. doi:10.1038/ng1216
17. Georges B, Cosyns JP, Dahan K, Snyers B, Carlier B, Loute G, et al. Late-onset renal failure in Senior-Loken syndrome. Am J Kidney Dis (2000) 36(6):1271–5. doi:10.1053/ajkd.2000.19845
18. Hoefele J, Nayir A, Chaki M, Imm A, Allen SJ, Otto EA, et al. Pseudodominant inheritance of nephronophthisis caused by a homozygous NPHP1 deletion. Pediatr Nephrol (2011) 26(6):967–71. doi:10.1007/s00467-011-1761-9
19. Otto EA, Helou J, Allen SJ, O’Toole JF, Wise EL, Ashraf S, et al. Mutation analysis in nephronophthisis using a combined approach of homozygosity mapping, CEL I endonuclease cleavage, and direct sequencing. Hum Mutat (2008) 29(3):418–26. doi:10.1002/humu.20669
20. Halbritter J, Diaz K, Chaki M, Porath JD, Tarrier B, Fu C, et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J Med Genet (2012) 49(12):756–67. doi:10.1136/jmedgenet-2012-100973
21. Gee HY, Otto EA, Hurd TW, Ashraf S, Chaki M, Cluckey A, et al. Whole-exome resequencing distinguishes cystic kidney diseases from phenocopies in renal ciliopathies. Kidney Int (2014) 85(4):880–7. doi:10.1038/ki.2013.450
22. Salomon R, Saunier S, Niaudet P. Nephronophthisis. Pediatr Nephrol (2009) 24(12):2333–44. doi:10.1007/s00467-008-0840-z
23. Konig J, Kranz B, Konig S, Schlingmann KP, Titieni A, Tonshoff B, et al. Phenotypic spectrum of children with nephronophthisis and related ciliopathies. Clin J Am Soc Nephrol (2017) 12(12):1974–83. doi:10.2215/CJN.01280217
24. Senior B, Friedmann AI, Braudo JL. Juvenile familial nephropathy with tapetoretinal degeneration: a new oculorenal dystrophy. Am J Ophthalmol (1961) 52:625–33. doi:10.1016/0002-9394(61)90147-7
25. Løken AC, Hanssen O, Halvorsen S, Jølster NJ. Hereditary renal dysplasia and blindness. Acta Paediatr (1961) 50:177–84. doi:10.1111/j.1651-2227.1961.tb08037.x
26. Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genet (2007) 15(5):511–21. doi:10.1038/sj.ejhg.5201648
27. Bachmann-Gagescu R, Dempsey JC, Phelps IG, O’Roak BJ, Knutzen DM, Rue TC, et al. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet (2015) 52(8):514–22. doi:10.1136/jmedgenet-2015-103087
28. Vilboux T, Doherty DA, Glass IA, Parisi MA, Phelps IG, Cullinane AR, et al. Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center. Genet Med (2017) 19(8):875–82. doi:10.1038/gim.2016.204
29. Fleming LR, Doherty DA, Parisi MA, Glass IA, Bryant J, Fischer R, et al. Prospective evaluation of kidney disease in Joubert syndrome. Clin J Am Soc Nephrol (2017) 12(12):1962–73. doi:10.2215/CJN.05660517
30. Betz R, Rensing C, Otto E, Mincheva A, Zehnder D, Lichter P, et al. Children with ocular motor apraxia type Cogan carry deletions in the gene (NPHP1) for juvenile nephronophthisis. J Pediatr (2000) 136(6):828–31. doi:10.1067/mpd.2000.106225
31. Harris CM, Hodgkins PR, Kriss A, Chong WK, Thompson DA, Mezey LE, et al. Familial congenital saccade initiation failure and isolated cerebellar vermis hypoplasia. Dev Med Child Neurol (1998) 40(11):775–9. doi:10.1111/j.1469-8749.1998.tb12347.x
32. Bergmann C, Fliegauf M, Bruchle NO, Frank V, Olbrich H, Kirschner J, et al. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am J Hum Genet (2008) 82(4):959–70. doi:10.1016/j.ajhg.2008.02.017
33. Baala L, Audollent S, Martinovic J, Ozilou C, Babron MC, Sivanandamoorthy S, et al. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet (2007) 81(1):170–9. doi:10.1086/519494
34. Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S, et al. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet (2007) 80(1):186–94. doi:10.1086/510499
35. Frank V, den Hollander AI, Bruchle NO, Zonneveld MN, Nurnberg G, Becker C, et al. Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel-Gruber syndrome. Hum Mutat (2008) 29(1):45–52. doi:10.1002/humu.20614
36. Ellis DS, Heckenlively JR, Martin CL, Lachman RS, Sakati NA, Rimoin DL. Leber’s congenital amaurosis associated with familial juvenile nephronophthisis and cone-shaped epiphyses of the hands (the Saldino-Mainzer syndrome). Am J Ophthalmol (1984) 97(2):233–9. doi:10.1016/S0002-9394(14)76095-7
37. Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, Hartley J, et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet (2011) 43(3):189–96. doi:10.1038/ng.756
38. Coussa RG, Otto EA, Gee HY, Arthurs P, Ren H, Lopez I, et al. WDR19: an ancient, retrograde, intraflagellar ciliary protein is mutated in autosomal recessive retinitis pigmentosa and in Senior-Loken syndrome. Clin Genet (2013) 84(2):150–9. doi:10.1111/cge.12196
39. Fehrenbach H, Decker C, Eisenberger T, Frank V, Hampel T, Walden U, et al. Mutations in WDR19 encoding the intraflagellar transport component IFT144 cause a broad spectrum of ciliopathies. Pediatr Nephrol (2014) 29(8):1451–6. doi:10.1007/s00467-014-2762-2
40. Perrault I, Saunier S, Hanein S, Filhol E, Bizet AA, Collins F, et al. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am J Hum Genet (2012) 90(5):864–70. doi:10.1016/j.ajhg.2012.03.006
41. Bredrup C, Saunier S, Oud MM, Fiskerstrand T, Hoischen A, Brackman D, et al. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet (2011) 89(5):634–43. doi:10.1016/j.ajhg.2011.10.001
42. Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology (1969) 19(9):813–25. doi:10.1212/WNL.19.9.813
43. Pellegrino JE, Lensch MW, Muenke M, Chance PF. Clinical and molecular analysis in Joubert syndrome. Am J Med Genet (1997) 72(1):59–62. doi:10.1002/(SICI)1096-8628(19971003)72:1<59::AID-AJMG12>3.0.CO;2-T
44. Kulaga HM, Leitch CC, Eichers ER, Badano JL, Lesemann A, Hoskins BE, et al. Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat Genet (2004) 36(9):994–8. doi:10.1038/ng1418
45. Iannaccone A, Mykytyn K, Persico AM, Searby CC, Baldi A, Jablonski MM, et al. Clinical evidence of decreased olfaction in Bardet-Biedl syndrome caused by a deletion in the BBS4 gene. Am J Med Genet A (2005) 132A(4):343–6. doi:10.1002/ajmg.a.30512
46. McEwen DP, Koenekoop RK, Khanna H, Jenkins PM, Lopez I, Swaroop A, et al. Hypomorphic CEP290/NPHP6 mutations result in anosmia caused by the selective loss of G proteins in cilia of olfactory sensory neurons. Proc Natl Acad Sci U S A (2007) 104(40):15917–22. doi:10.1073/pnas.0704140104
47. Pluznick JL, Zou DJ, Zhang X, Yan Q, Rodriguez-Gil DJ, Eisner C, et al. Functional expression of the olfactory signaling system in the kidney. Proc Natl Acad Sci U S A (2009) 106(6):2059–64. doi:10.1073/pnas.0812859106
48. Ou Y, Ruan Y, Cheng M, Moser JJ, Rattner JB, van der Hoorn FA. Adenylate cyclase regulates elongation of mammalian primary cilia. Exp Cell Res (2009) 315(16):2802–17. doi:10.1016/j.yexcr.2009.06.028
49. Hildebrandt F, Otto E, Rensing C, Nothwang HG, Vollmer M, Adolphs J, et al. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet (1997) 17(2):149–53. doi:10.1038/ng1097-149
50. Fliegauf M, Horvath J, von Schnakenburg C, Olbrich H, Muller D, Thumfart J, et al. Nephrocystin specifically localizes to the transition zone of renal and respiratory cilia and photoreceptor connecting cilia. J Am Soc Nephrol (2006) 17(9):2424–33. doi:10.1681/ASN.2005121351
51. Gattone VH II, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med (2003) 9(10):1323–6. doi:10.1038/nm935
52. Otto E, Hoefele J, Ruf R, Mueller AM, Hiller KS, Wolf MT, et al. A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. Am J Hum Genet (2002) 71(5):1161–7. doi:10.1086/344395
53. Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan S, et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet (2005) 37(3):282–8. doi:10.1038/ng1520
54. Sayer JA, Otto EA, O’Toole JF, Nurnberg G, Kennedy MA, Becker C, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet (2006) 38(6):674–81. doi:10.1038/ng1786
55. Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet (2006) 38(6):623–5. doi:10.1038/ng1805
56. Attanasio M, Uhlenhaut NH, Sousa VH, O’Toole JF, Otto E, Anlag K, et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat Genet (2007) 39(8):1018–24. doi:10.1038/ng2072
57. Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, Gorden NT, et al. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet (2007) 39(7):882–8. doi:10.1038/ng2069
58. Otto EA, Hurd TW, Airik R, Chaki M, Zhou W, Stoetzel C, et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat Genet (2010) 42(10):840–50. doi:10.1038/ng.662
59. Airik R, Slaats GG, Guo Z, Weiss AC, Khan N, Ghosh A, et al. Renal-retinal ciliopathy gene Sdccag8 regulates DNA damage response signaling. J Am Soc Nephrol (2014) 25(11):2573–83. doi:10.1681/ASN.2013050565
60. Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S, et al. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Genet (2006) 38(2):191–6. doi:10.1038/ng1713
61. Otto EA, Tory K, Attanasio M, Zhou W, Chaki M, Paruchuri Y, et al. Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J Med Genet (2009) 46(10):663–70. doi:10.1136/jmg.2009.066613
62. Adams M, Simms RJ, Abdelhamed Z, Dawe HR, Szymanska K, Logan CV, et al. A meckelin–filamin A interaction mediates ciliogenesis. Hum Mol Genet (2012) 21(6):1272–86. doi:10.1093/hmg/ddr557
63. Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell (2012) 150(3):533–48. doi:10.1016/j.cell.2012.06.028
64. Cheng LE, Zhang J, Reed RR. The transcription factor Zfp423/OAZ is required for cerebellar development and CNS midline patterning. Dev Biol (2007) 307(1):43–52. doi:10.1016/j.ydbio.2007.04.005
65. Hoff S, Halbritter J, Epting D, Frank V, Nguyen TM, van Reeuwijk J, et al. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat Genet (2013) 45(8):951–6. doi:10.1038/ng.2681
66. Taskiran EZ, Korkmaz E, Gucer S, Kosukcu C, Kaymaz F, Koyunlar C, et al. Mutations in ANKS6 cause a nephronophthisis-like phenotype with ESRD. J Am Soc Nephrol (2014) 25(8):1653–61. doi:10.1681/ASN.2013060646
67. Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, Gee HY, et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet (2013) 93(5):915–25. doi:10.1016/j.ajhg.2013.09.012
68. Stinchcombe JC, Randzavola LO, Angus KL, Mantell JM, Verkade P, Griffiths GM. Mother centriole distal appendages mediate centrosome docking at the immunological synapse and reveal mechanistic parallels with ciliogenesis. Curr Biol (2015) 25(24):3239–44. doi:10.1016/j.cub.2015.10.028
69. Schueler M, Braun DA, Chandrasekar G, Gee HY, Klasson TD, Halbritter J, et al. DCDC2 mutations cause a renal-hepatic ciliopathy by disrupting Wnt signaling. Am J Hum Genet (2015) 96(1):81–92. doi:10.1016/j.ajhg.2014.12.002
70. Macia MS, Halbritter J, Delous M, Bredrup C, Gutter A, Filhol E, et al. Mutations in MAPKBP1 cause juvenile or late-onset cilia-independent nephronophthisis. Am J Hum Genet (2017) 100(2):372. doi:10.1016/j.ajhg.2017.01.025
71. Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet (2004) 36(9):1008–13. doi:10.1038/ng1419
72. Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet (2004) 75(6):979–87. doi:10.1086/425985
73. Utsch B, Sayer JA, Attanasio M, Pereira RR, Eccles M, Hennies HC, et al. Identification of the first AHI1 gene mutations in nephronophthisis-associated Joubert syndrome. Pediatr Nephrol (2006) 21(1):32–5. doi:10.1007/s00467-005-2054-y
74. Tuz K, Hsiao YC, Juarez O, Shi B, Harmon EY, Phelps IG, et al. The Joubert syndrome-associated missense mutation (V443D) in the Abelson-helper integration site 1 (AHI1) protein alters its localization and protein-protein interactions. J Biol Chem (2013) 288(19):13676–94. doi:10.1074/jbc.M112.420786
75. Bachmann-Gagescu R, Dona M, Hetterschijt L, Tonnaer E, Peters T, de Vrieze E, et al. The ciliopathy protein CC2D2A associates with NINL and functions in RAB8-MICAL3-regulated vesicle trafficking. PLoS Genet (2015) 11(10):e1005575. doi:10.1371/journal.pgen.1005575
76. O’Toole JF, Liu Y, Davis EE, Westlake CJ, Attanasio M, Otto EA, et al. Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronophthisis-like nephropathy. J Clin Invest (2010) 120(3):791–802. doi:10.1172/JCI40076
77. Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell (2011) 145(4):513–28. doi:10.1016/j.cell.2011.04.019
78. Hurd TW, Otto EA, Mishima E, Gee HY, Inoue H, Inazu M, et al. Mutation of the Mg2+ transporter SLC41A1 results in a nephronophthisis-like phenotype. J Am Soc Nephrol (2013) 24(6):967–77. doi:10.1681/ASN.2012101034
79. Saunier S, Calado J, Benessy F, Silbermann F, Heilig R, Weissenbach J, et al. Characterization of the NPHP1 locus: mutational mechanism involved in deletions in familial juvenile nephronophthisis. Am J Hum Genet (2000) 66(3):778–89. doi:10.1086/302819
80. Sayer JA, Simms RJ. The challenges and surprises of a definitive molecular genetic diagnosis. Kidney Int (2014) 85(4):748–9. doi:10.1038/ki.2013.432
81. Donaldson JC, Dise RS, Ritchie MD, Hanks SK. Nephrocystin-conserved domains involved in targeting to epithelial cell-cell junctions, interaction with filamins, and establishing cell polarity. J Biol Chem (2002) 277(32):29028–35. doi:10.1074/jbc.M111697200
82. Donaldson JC, Dempsey PJ, Reddy S, Bouton AH, Coffey RJ, Hanks SK. Crk-associated substrate p130(Cas) interacts with nephrocystin and both proteins localize to cell-cell contacts of polarized epithelial cells. Exp Cell Res (2000) 256(1):168–78. doi:10.1006/excr.2000.4822
83. Benzing T, Gerke P, Hopker K, Hildebrandt F, Kim E, Walz G. Nephrocystin interacts with Pyk2, p130(Cas), and tensin and triggers phosphorylation of Pyk2. Proc Natl Acad Sci U S A (2001) 98(17):9784–9. doi:10.1073/pnas.171269898
84. Lienkamp S, Ganner A, Walz G. Inversin, Wnt signaling and primary cilia. Differentiation (2012) 83(2):S49–55. doi:10.1016/j.diff.2011.11.012
85. Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet (2005) 37(5):537–43. doi:10.1038/ng1552
86. Werner ME, Ward HH, Phillips CL, Miller C, Gattone VH, Bacallao RL. Inversin modulates the cortical actin network during mitosis. Am J Physiol Cell Physiol (2013) 305(1):C36–47. doi:10.1152/ajpcell.00279.2012
87. Omran H, Fernandez C, Jung M, Haffner K, Fargier B, Villaquiran A, et al. Identification of a new gene locus for adolescent nephronophthisis, on chromosome 3q22 in a large Venezuelan pedigree. Am J Hum Genet (2000) 66(1):118–27. doi:10.1086/302705
88. Mollet G, Silbermann F, Delous M, Salomon R, Antignac C, Saunier S. Characterization of the nephrocystin/nephrocystin-4 complex and subcellular localization of nephrocystin-4 to primary cilia and centrosomes. Hum Mol Genet (2005) 14(5):645–56. doi:10.1093/hmg/ddi061
89. Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz-Font A, Rix S, et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet (2008) 40(4):443–8. doi:10.1038/ng.97
90. Brancati F, Iannicelli M, Travaglini L, Mazzotta A, Bertini E, Boltshauser E, et al. MKS3/TMEM67 mutations are a major cause of COACH syndrome, a Joubert syndrome related disorder with liver involvement. Hum Mutat (2009) 30(2):E432–42. doi:10.1002/humu.20924
91. Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M, Bates D, et al. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J Med Genet (2010) 47(1):8–21. doi:10.1136/jmg.2009.067249
92. Gorden NT, Arts HH, Parisi MA, Coene KL, Letteboer SJ, van Beersum SE, et al. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am J Hum Genet (2008) 83(5):559–71. doi:10.1016/j.ajhg.2008.10.002
93. Tsang WY, Bossard C, Khanna H, Peranen J, Swaroop A, Malhotra V, et al. CP110 suppresses primary cilia formation through its interaction with CEP290, a protein deficient in human ciliary disease. Dev Cell (2008) 15(2):187–97. doi:10.1016/j.devcel.2008.07.004
94. Singla V, Reiter JF. The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science (2006) 313(5787):629–33. doi:10.1126/science.1124534
95. Hynes AM, Giles RH, Srivastava S, Eley L, Whitehead J, Danilenko M, et al. Murine Joubert syndrome reveals Hedgehog signaling defects as a potential therapeutic target for nephronophthisis. Proc Natl Acad Sci U S A (2014) 111(27):9893–8. doi:10.1073/pnas.1322373111
96. Slaats GG, Saldivar JC, Bacal J, Zeman MK, Kile AC, Hynes AM, et al. DNA replication stress underlies renal phenotypes in CEP290-associated Joubert syndrome. J Clin Invest (2015) 125(9):3657–66. doi:10.1172/JCI80657
97. Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol (2011) 26(7):1039–56. doi:10.1007/s00467-010-1731-7
98. Lu D, Rauhauser A, Li B, Ren C, McEnery K, Zhu J, et al. Loss of Glis2/NPHP7 causes kidney epithelial cell senescence and suppresses cyst growth in the Kif3a mouse model of cystic kidney disease. Kidney Int (2016) 89(6):1307–23. doi:10.1016/j.kint.2016.03.006
99. Choi HJ, Lin JR, Vannier JB, Slaats GG, Kile AC, Paulsen RD, et al. NEK8 links the ATR-regulated replication stress response and S phase CDK activity to renal ciliopathies. Mol Cell (2013) 51(4):423–39. doi:10.1016/j.molcel.2013.08.006
100. Grampa V, Delous M, Zaidan M, Odye G, Thomas S, Elkhartoufi N, et al. Novel NEK8 mutations cause severe syndromic renal cystic dysplasia through YAP dysregulation. PLoS Genet (2016) 12(3):e1005894. doi:10.1371/journal.pgen.1005894
101. Al-Hamed MH, Kurdi W, Alsahan N, Alabdullah Z, Abudraz R, Tulbah M, et al. Genetic spectrum of Saudi Arabian patients with antenatal cystic kidney disease and ciliopathy phenotypes using a targeted renal gene panel. J Med Genet (2016) 53(5):338–47. doi:10.1136/jmedgenet-2015-103469
102. Schaefer E, Zaloszyc A, Lauer J, Durand M, Stutzmann F, Perdomo-Trujillo Y, et al. Mutations in SDCCAG8/NPHP10 cause Bardet-Biedl syndrome and are associated with penetrant renal disease and absent polydactyly. Mol Syndromol (2011) 1(6):273–81. doi:10.1159/000331268
103. Tran PV, Talbott GC, Turbe-Doan A, Jacobs DT, Schonfeld MP, Silva LM, et al. Downregulating Hedgehog signaling reduces renal cystogenic potential of mouse models. J Am Soc Nephrol (2014) 25(10):2201–12. doi:10.1681/ASN.2013070735
104. Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, et al. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum Genet (2013) 132(8):865–84. doi:10.1007/s00439-013-1297-0
105. Graser S, Stierhof YD, Lavoie SB, Gassner OS, Lamla S, Le Clech M, et al. Cep164, a novel centriole appendage protein required for primary cilium formation. J Cell Biol (2007) 179(2):321–30. doi:10.1083/jcb.200707181
106. Schaefer E, Stoetzel C, Scheidecker S, Geoffroy V, Prasad MK, Redin C, et al. Identification of a novel mutation confirms the implication of IFT172 (BBS20) in Bardet-Biedl syndrome. J Hum Genet (2016) 61(5):447–50. doi:10.1038/jhg.2015.162
107. Mougou-Zerelli S, Thomas S, Szenker E, Audollent S, Elkhartoufi N, Babarit C, et al. CC2D2A mutations in Meckel and Joubert syndromes indicate a genotype-phenotype correlation. Hum Mutat (2009) 30(11):1574–82. doi:10.1002/humu.21116
108. Bachmann-Gagescu R, Ishak GE, Dempsey JC, Adkins J, O’Day D, Phelps IG, et al. Genotype-phenotype correlation in CC2D2A-related Joubert syndrome reveals an association with ventriculomegaly and seizures. J Med Genet (2012) 49(2):126–37. doi:10.1136/jmedgenet-2011-100552
109. Iannicelli M, Brancati F, Mougou-Zerelli S, Mazzotta A, Thomas S, Elkhartoufi N, et al. Novel TMEM67 mutations and genotype-phenotype correlates in meckelin-related ciliopathies. Hum Mutat (2010) 31(5):E1319–31. doi:10.1002/humu.21239
110. Hoefele J, Wolf MT, O’Toole JF, Otto EA, Schultheiss U, Deschenes G, et al. Evidence of oligogenic inheritance in nephronophthisis. J Am Soc Nephrol (2007) 18(10):2789–95. doi:10.1681/ASN.2007020243
111. Tory K, Lacoste T, Burglen L, Moriniere V, Boddaert N, Macher MA, et al. High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J Am Soc Nephrol (2007) 18(5):1566–75. doi:10.1681/ASN.2006101164
112. Hopp K, Heyer CM, Hommerding CJ, Henke SA, Sundsbak JL, Patel S, et al. B9D1 is revealed as a novel Meckel syndrome (MKS) gene by targeted exon-enriched next-generation sequencing and deletion analysis. Hum Mol Genet (2011) 20(13):2524–34. doi:10.1093/hmg/ddr151
113. Muller J, Stoetzel C, Vincent MC, Leitch CC, Laurier V, Danse JM, et al. Identification of 28 novel mutations in the Bardet-Biedl syndrome genes: the burden of private mutations in an extensively heterogeneous disease. Hum Genet (2010) 127(5):583–93. doi:10.1007/s00439-010-0804-9
114. Lindstrand A, Davis EE, Carvalho CM, Pehlivan D, Willer JR, Tsai IC, et al. Recurrent CNVs and SNVs at the NPHP1 locus contribute pathogenic alleles to Bardet-Biedl syndrome. Am J Hum Genet (2014) 94(5):745–54. doi:10.1016/j.ajhg.2014.03.017
115. Katsanis N. The continuum of causality in human genetic disorders. Genome Biol (2016) 17(1):233. doi:10.1186/s13059-016-1107-9
116. Phelps IG, Dempsey JC, Grout ME, Isabella CR, Tully HM, Doherty D, et al. Interpreting the clinical significance of combined variants in multiple recessive disease genes: systematic investigation of Joubert syndrome yields little support for oligogenicity. Genet Med (2017). doi:10.1038/gim.2017.94
117. Hildebrandt F. Identification of a gene for nephronophthisis. Nephrol Dial Transplant (1998) 13(6):1334–6. doi:10.1093/ndt/13.6.1334
118. Eley L, Moochhala SH, Simms R, Hildebrandt F, Sayer JA. Nephrocystin-1 interacts directly with Ack1 and is expressed in human collecting duct. Biochem Biophys Res Commun (2008) 371(4):877–82. doi:10.1016/j.bbrc.2008.05.016
120. Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol (2000) 151(3):709–18. doi:10.1083/jcb.151.3.709
121. Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet (2003) 33(2):129–37. doi:10.1038/ng1076
122. Norris DP, Jackson PK. Cell biology: calcium contradictions in cilia. Nature (2016) 531(7596):582–3. doi:10.1038/nature17313
123. Delling M, Indzhykulian AA, Liu X, Li Y, Xie T, Corey DP, et al. Primary cilia are not calcium-responsive mechanosensors. Nature (2016) 531(7596):656–60. doi:10.1038/nature17426
124. Chang MY, Ong ACM. Targeting new cellular disease pathways in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant (2017) 32(12):2144. doi:10.1093/ndt/gfx289
125. Molinari E, Sayer JA. Emerging treatments and personalised medicine for ciliopathies associated with cystic kidney disease. Expert Opin Orphan Drugs (2017) 5(10):785–98. doi:10.1080/21678707.2017.1372282
126. Fischer E, Legue E, Doyen A, Nato F, Nicolas JF, Torres V, et al. Defective planar cell polarity in polycystic kidney disease. Nat Genet (2006) 38(1):21–3. doi:10.1038/ng1701
128. Lancaster MA, Gopal DJ, Kim J, Saleem SN, Silhavy JL, Louie CM, et al. Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat Med (2011) 17(6):726–31. doi:10.1038/nm.2380
129. Lancaster MA, Louie CM, Silhavy JL, Sintasath L, Decambre M, Nigam SK, et al. Impaired Wnt-beta-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nat Med (2009) 15(9):1046–54. doi:10.1038/nm.2010
130. Goggolidou P. Wnt and planar cell polarity signaling in cystic renal disease. Organogenesis (2014) 10(1):86–95. doi:10.4161/org.26766
131. Calvet JP. (2015). “The role of calcium and cyclic AMP in PKD,” in Polycystic Kidney Disease [Internet], ed. X. Li (Brisbane, AU: Codon Publications). doi:10.15586/codon.pkd.2015.ch8
132. Ghosh AK, Hurd T, Hildebrandt F. 3D spheroid defects in NPHP knockdown cells are rescued by the somatostatin receptor agonist octreotide. Am J Physiol Renal Physiol (2012) 303(8):F1225–9. doi:10.1152/ajprenal.00135.2012
133. Gattone VH II, Sinders RM, Hornberger TA, Robling AG. Late progression of renal pathology and cyst enlargement is reduced by rapamycin in a mouse model of nephronophthisis. Kidney Int (2009) 76(2):178–82. doi:10.1038/ki.2009.147
134. Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant (2006) 21(3):598–604. doi:10.1093/ndt/gfi181
135. Ta MH, Schwensen KG, Foster S, Korgaonkar M, Ozimek-Kulik JE, Phillips JK, et al. Effects of TORC1 inhibition during the early and established phases of polycystic kidney disease. PLoS One (2016) 11(10):e0164193. doi:10.1371/journal.pone.0164193
136. Bell PD, Fitzgibbon W, Sas K, Stenbit AE, Amria M, Houston A, et al. Loss of primary cilia upregulates renal hypertrophic signaling and promotes cystogenesis. J Am Soc Nephrol (2011) 22(5):839–48. doi:10.1681/ASN.2010050526
137. Boehlke C, Kotsis F, Patel V, Braeg S, Voelker H, Bredt S, et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol (2010) 12(11):1115–22. doi:10.1038/ncb2117
138. Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature (1980) 287(5785):795–801. doi:10.1038/287795a0
139. McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of Hedgehog signaling. Curr Top Dev Biol (2003) 53:1–114. doi:10.1016/S0070-2153(03)53002-2
140. Wong SY, Reiter JF. The primary cilium at the crossroads of mammalian Hedgehog signaling. Curr Top Dev Biol (2008) 85:225–60. doi:10.1016/S0070-2153(08)00809-0
141. Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet (2010) 11(5):331–44. doi:10.1038/nrg2774
142. Milenkovic L, Scott MP, Rohatgi R. Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. J Cell Biol (2009) 187(3):365–74. doi:10.1083/jcb.200907126
143. Hu MC, Mo R, Bhella S, Wilson CW, Chuang PT, Hui CC, et al. GLI3-dependent transcriptional repression of Gli1, Gli2 and kidney patterning genes disrupts renal morphogenesis. Development (2006) 133(3):569–78. doi:10.1242/dev.02220
144. Chan SK, Riley PR, Price KL, McElduff F, Winyard PJ, Welham SJ, et al. Corticosteroid-induced kidney dysmorphogenesis is associated with deregulated expression of known cystogenic molecules, as well as Indian Hedgehog. Am J Physiol Renal Physiol (2010) 298(2):F346–56. doi:10.1152/ajprenal.00574.2009
145. Zhang Q, Seo S, Bugge K, Stone EM, Sheffield VC. BBS proteins interact genetically with the IFT pathway to influence SHH-related phenotypes. Hum Mol Genet (2012) 21(9):1945–53. doi:10.1093/hmg/dds004
146. Shi X, Garcia G III, Van De Weghe JC, McGorty R, Pazour GJ, Doherty D, et al. Super-resolution microscopy reveals that disruption of ciliary transition-zone architecture causes Joubert syndrome. Nat Cell Biol (2017) 19(10):1178–88. doi:10.1038/ncb3622
147. Slaats GG, Ghosh AK, Falke LL, Le Corre S, Shaltiel IA, van de Hoek G, et al. Nephronophthisis-associated CEP164 regulates cell cycle progression, apoptosis and epithelial-to-mesenchymal transition. PLoS Genet (2014) 10(10):e1004594. doi:10.1371/journal.pgen.1004594
148. Slaats GG, Giles RH. Are renal ciliopathies (replication) stressed out? Trends Cell Biol (2015) 25(6):317–9. doi:10.1016/j.tcb.2015.03.005
149. Braun DA, Hildebrandt F. Ciliopathies. Cold Spring Harb Perspect Biol (2017) 9(3):a028191. doi:10.1101/cshperspect.a028191
150. Johnson CA, Collis SJ. Ciliogenesis and the DNA damage response: a stressful relationship. Cilia (2016) 5:19. doi:10.1186/s13630-016-0040-6
151. Torres VE, Wang X, Qian Q, Somlo S, Harris PC, Gattone VH II. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med (2004) 10(4):363–4. doi:10.1038/nm1004
152. Wang X, Gattone V II, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol (2005) 16(4):846–51. doi:10.1681/ASN.2004121090
153. Aihara M, Fujiki H, Mizuguchi H, Hattori K, Ohmoto K, Ishikawa M, et al. Tolvaptan delays the onset of end-stage renal disease in a polycystic kidney disease model by suppressing increases in kidney volume and renal injury. J Pharmacol Exp Ther (2014) 349(2):258–67. doi:10.1124/jpet.114.213256
154. Friedlander G, Amiel C. Somatostatin and alpha 2-adrenergic agonists selectively inhibit vasopressin-induced cyclic AMP accumulation in MDCK cells. FEBS Lett (1986) 198(1):38–42. doi:10.1016/0014-5793(86)81180-2
155. Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med (2012) 367(25):2407–18. doi:10.1056/NEJMoa1205511
156. Müller R-U, Haas CS, Sayer JA. Practical approaches to the management of autosomal dominant polycystic kidney disease patients in the era of tolvaptan. Clin Kidney J (2017):sfx071. doi:10.1093/ckj/sfx071
157. Husson H, Moreno S, Smith LA, Smith MM, Russo RJ, Pitstick R, et al. Reduction of ciliary length through pharmacologic or genetic inhibition of CDK5 attenuates polycystic kidney disease in a model of nephronophthisis. Hum Mol Genet (2016) 25(11):2245–55. doi:10.1093/hmg/ddw093
158. Srivastava S, Ramsbottom SA, Molinari E, Alkanderi S, Filby A, White K, et al. A human patient-derived cellular model of Joubert syndrome reveals ciliary defects which can be rescued with targeted therapies. Hum Mol Genet (2017) 26(23):4657–67. doi:10.1093/hmg/ddx347
159. Tao Y, Kim J, Schrier RW, Edelstein CL. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J Am Soc Nephrol (2005) 16(1):46–51. doi:10.1681/ASN.2004080660
160. Tobin JL, Beales PL. Restoration of renal function in zebrafish models of ciliopathies. Pediatr Nephrol (2008) 23(11):2095–9. doi:10.1007/s00467-008-0898-7
161. Westhoff JH, Giselbrecht S, Schmidts M, Schindler S, Beales PL, Tonshoff B, et al. Development of an automated imaging pipeline for the analysis of the zebrafish larval kidney. PLoS One (2013) 8(12):e82137. doi:10.1371/journal.pone.0082137
Keywords: ciliopathy, molecular genetics, nephronophthisis, cilia, centrosome, DNA damage, cyclic adenosine monophosphate, Joubert syndrome
Citation: Srivastava S, Molinari E, Raman S and Sayer JA (2018) Many Genes—One Disease? Genetics of Nephronophthisis (NPHP) and NPHP-Associated Disorders. Front. Pediatr. 5:287. doi: 10.3389/fped.2017.00287
Received: 16 October 2017; Accepted: 14 December 2017;
Published: 05 January 2018
Edited by:
Max Christoph Liebau, Universitätsklinikum Köln, GermanyReviewed by:
Ruxandra Bachmann-Gagescu, University of Zurich, SwitzerlandKatja Höpker, Universitätsklinikum Köln, Germany
Copyright: © 2018 Srivastava, Molinari, Raman and Sayer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John A. Sayer, am9obi5zYXllckBuY2wuYWMudWs=