 Korbinian Maria Riedhammer1,2
Korbinian Maria Riedhammer1,2 Corinna Siegel2Bader Alhaddad2Carmen Montoya3Reka Kovacs-Nagy2Matias Wagner2,4,5Thomas Meitinger2,4
Corinna Siegel2Bader Alhaddad2Carmen Montoya3Reka Kovacs-Nagy2Matias Wagner2,4,5Thomas Meitinger2,4 Julia Hoefele2*
Julia Hoefele2*
- 1Department of Nephrology, Klinikum rechts der Isar, Technical University of Munich, Munich, Germany
- 2Institute of Human Genetics, Klinikum rechts der Isar, Technical University of Munich, Munich, Germany
- 3KfH Center of Pediatric Nephrology, Children’s Hospital Munich Schwabing, Munich, Germany
- 4Institute of Human Genetics, Helmholtz Zentrum Munich, Neuherberg, Germany
- 5Institute of Neurogenomics, Helmholtz Zentrum Munich, Neuherberg, Germany
Introduction: Congenital anomalies of the kidney and urinary tract (CAKUT) represent the primary cause of chronic kidney disease in children. Many genes have been attributed to the genesis of this disorder. Recently, haploinsufficiency of PBX1 caused by microdeletions has been shown to result in bilateral renal hypoplasia and other organ malformations.
Materials and methods: Here, we report on a 14-year-old male patient with congenital bilateral dysplastic kidneys, cryptorchidism, hypoplastic clavicles, developmental delay, impaired intelligence, and minor dysmorphic features. Presuming a syndromic origin, we performed SNP array analysis to scan for large copy number variations (CNVs) followed by whole-exome sequencing (WES). Sanger sequencing was done to confirm the variant’s de novo status.
Results: SNP array analysis did not reveal any microdeletions or -duplications larger than 50 or 100 kb, respectively. WES identified a novel heterozygous 7-bp frameshift deletion in PBX1 (c.413_419del, p.Gly138Valfs*40) resulting in a loss-of-function. The de novo status could be confirmed by Sanger sequencing.
Discussion: By WES, we identified a novel heterozygous de novo 7-bp frameshift deletion in PBX1. Our findings expand the spectrum of causative variants in PBX1-related CAKUT. In this case, WES proved to be the apt technique to detect the variant responsible for the patient’s phenotype, as single gene testing is not feasible given the multitude of genes involved in CAKUT and SNP array analysis misses rare single-nucleotide variants and small Indels.
Introduction
Congenital anomalies of the kidney and urinary tract (CAKUT) represent the primary cause of chronic kidney disease in children. CAKUT is the collective term for many different renal and urinary tract malformations. In recent years, a multitude of monogenic disease-causing genes has been discovered (1). Disruption of the normal nephrogenesis by pathogenic variants in genes involved in kidney development is a basic principle of CAKUT (2).
When it comes to heterogeneous diseases like CAKUT, with many different genes involved, large-scale next-generation sequencing has become an extremely useful tool for the unbiased detection of pathogenic variants (3). In whole-exome sequencing (WES), the coding regions (the exome) of the human genome are enriched and sequenced. This has proven to be both an economic, as the exome only comprises 1% of the genome, and a pragmatic approach, as about 85% of disease-related variants can be found in the exome (4).
PBX1 encodes a transcription factor that has already been linked to nephrogenesis as shown by Pbx1-deficient mice (5). Additionally, earlier mouse models revealed its role in bone formation (6). In 2017, microdeletions comprising PBX1 as a minimal common region could be identified by microarray analysis in eight patients with syndromic CAKUT with predominantly renal hypoplasia. In the same publication, it was shown that PBX1 was strongly expressed in the fetal kidney and brain (7). Here, we report on a 14-year-old male patient presenting to the pediatric nephrology department with the predominant clinical features of bilateral dysplastic kidneys, hypoplastic clavicles, and developmental delay.
Materials and Methods
This study was approved by the local Ethics Committee of the Technical University of Munich and performed according to the standard of the Helsinki Declaration of 1975. Written informed consent was obtained from the parents of the participant for publication of this case report. Blood samples were collected after written informed consent.
DNA was extracted from peripheral blood using the Gentra Puregene Blood Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The DNA sample of the patient was analyzed by using the SNP Array Affymetrix® CytoScanTM 750 K Array (Affymetrix® Inc., Santa Clara, CA, USA) with an average space between two oligonucleotides of 4 kb. Scanning was performed by the Affymetrix® GeneChip Scanner 3000 7G (resolution 0.51–2.5 µm). The data analysis was conducted using the Affymetrix® Chromosome Analysis Suite Software (ChAS), version 3.0, hg19.
Exome sequencing was performed using a Sure Select Human All Exon 60 Mb V6 Kit (Agilent) and a HiSeq4000 (Illumina) as previously described (8, 9). Reads were aligned to the UCSC human reference assembly (hg19) with BWA v.0.5.8. More than 98% of the exome was covered at least 20×. PBX1 was covered >20× to 100%. Single-nucleotide variants and small insertions and deletions were detected with SAMtools v.0.1.7. Variant prioritization was performed based on an autosomal recessive pattern of inheritance (homozygous or putative compound heterozygous variants with a minor allele frequency <0.1%) as well as an autosomal dominant pattern of inheritance (heterozygous variants with a minor allele frequency <0.001%).
Sanger sequencing was used to confirm the identified variant and to test the patient’s parents. Oligonucleotide primer sequences are available upon request.
Case Report and Results
The 14-year-old boy is the first child of healthy parents (Figure 1). He has one younger healthy brother and one younger healthy half-brother. The patient was born at a gestational age of 38 weeks [birth weight: 2,840 g (25th–50th percentile), birth length: 48 cm (10th–25th percentile)]. There were no obvious malformations noted at birth. During the neonatal period, a slender thorax and short clavicles were identified clinically. Body height was on the third percentile during infancy and early childhood (Figure 2) and a global developmental delay (including motor and speech delay) was diagnosed on regular pediatric screening examinations.
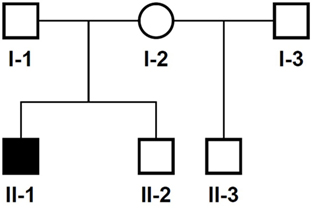
Figure 1. Pedigree of the family. Solid symbol, affected individual; circles, females; squares, males.
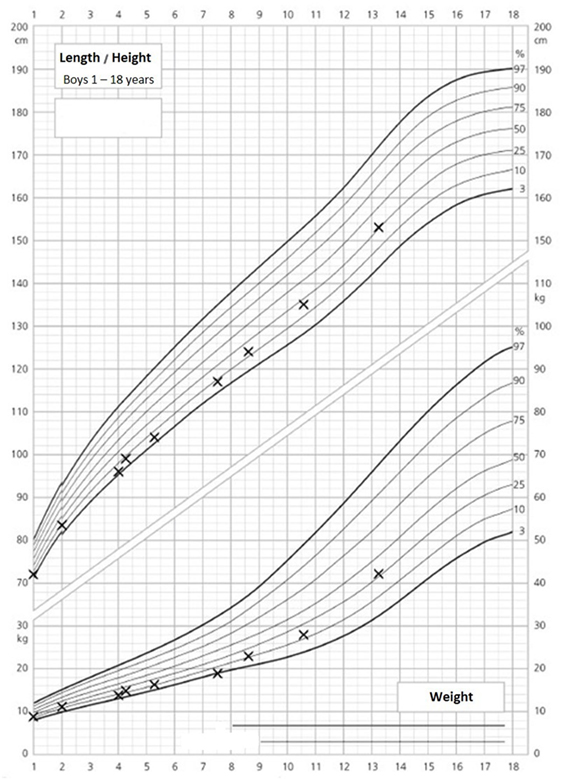
Figure 2. Growth and weight charts of the patient. These charts are gender specific and refer to boys. x-axis, age (years); y-axis, height (centimeter, upper part of the figure) and weight (kilograms, lower part of the figure).
An ultrasound of the kidneys at 3 months of age revealed small kidneys with hyperechogenic parenchyma. The boy was then regularly seen by a pediatric nephrologist. By the age of six, the right kidney had a length of 6.2 cm (<1st percentile, see Figure 3), the left kidney had a length of 5.8 cm (<1st percentile, see Figure 4). Kidney function ranged between eGFR 59 mL/min/1.73 m2 at 4 years of age, 90 mL/min/1.73 m2 at 8 years of age, and 69 mL/min/1.73 m2 at 13 years of age (Schwartz estimate, CKD II). Moreover, the patient had bilateral cryptorchidism for which he received hormonal therapy by the age of 2 years.
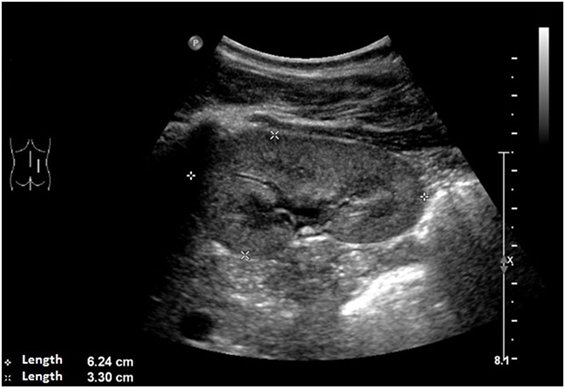
Figure 3. Ultrasound of the right kidney with a length of 6.2 cm (<1st percentile).
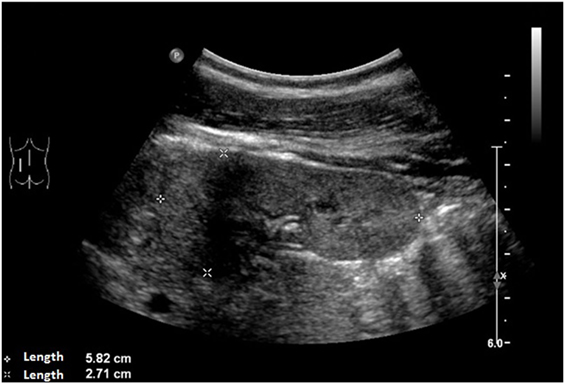
Figure 4. Ultrasound of the left kidney with a length of 5.8 cm (<1st percentile).
Psychological intelligence testing by the age of 13 revealed a below average speech comprehension (IQ 81), a reduced information processing speed (IQ 74) and an impaired auditory working memory (IQ 77).
On orthopedic examination the patient had a slim shoulder profile, impaired abduction of the arms, and a hunched back. Thoracic X-ray revealed hypoplastic clavicles (Figure 5, only left clavicle visible). He also had some dysmorphic features (wide nasal bridge, short neck, bilateral overfolding of the helix, and bilateral clinodactyly).
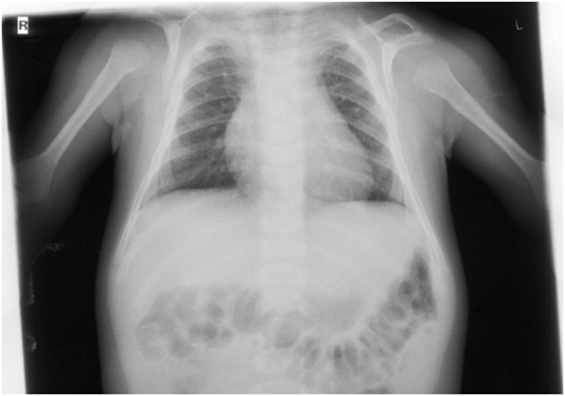
Figure 5. X-ray (detail of a babygram) of the patient at the age of 7 months showing a hook-shaped hypoplastic left clavicle.
In this child, we presumed a syndromic origin and initially performed SNP array analysis. However, no microdeletions or duplications (copy number variations, CNVs) larger than 50 or 100 kb, respectively, could be detected. As CAKUT have been associated with a large number of genes, we then performed WES. By WES, we could identify a novel heterozygous 7-bp deletion in PBX1 leading to a frameshift (c.413_419delGGGCAGG, p.Gly138Valfs*40), resulting in either nonsense-mediated decay of the mRNA or a truncated protein lacking the DNA-binding domain. The variant is not listed in 60,000 control individuals of the Exome Aggregation Consortium (ExAC) browser. The ExAC browser does not list any high-confidence loss-of-function variants in PBX1 indicating that PBX1 is intolerant to loss-of-function variants. To verify the de novo status of the variant, we performed Sanger sequencing in the patient and his parents. The variant could not be detected in the blood DNA of both parents (Figure 6).
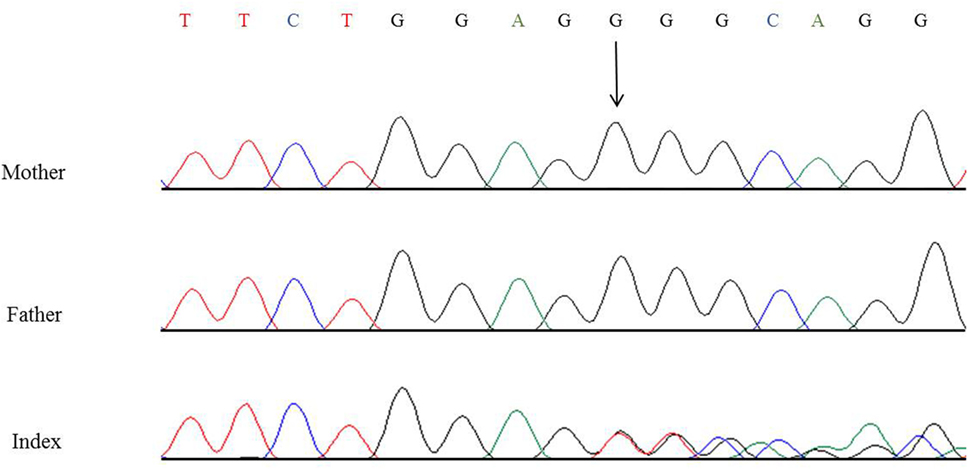
Figure 6. Partial nucleotide sequence of exon 3 of PBX1 of the patient and his parents showing the heterozygous de novo variant c.413_419delGGGCAGG, p.Gly138Valfs*40. Shown reference sequence: TTCTGGAGGGGCAGG. Genomic position of the variant: chr1:164761876–164761882 (hg19, transcript NM_002585.3).
Discussion
CAKUT represent the primary cause of chronic kidney disease in children and many genes have been attributed to the genesis of this disorder with both dominant and recessive modes of inheritance. CAKUT mainly occur as part of (multi-organ) syndromes but there are also isolated cases described in the literature (1, 2, 10). Just recently, haploinsufficiency of PBX1 caused by microdeletions was shown to result in bilateral renal hypoplasia and other organ malformations (7). Furthermore, a targeted exome sequencing of 330 genes in 204 unrelated CAKUT patients could identify five novel de novo heterozygous loss-of-function variants/deletions in PBX1 (11).
PBX1 encodes a transcription factor which promotes protein–protein interaction and is important for organogenesis (12). Pbx1−/− mice die at an embryonic age and show extensive organ malformations including hypoplastic kidneys with unilateral agenesis (5). In a further publication, Pbx1-deficient mice exhibited—besides multiple organ malformations—a pronounced skeletal phenotype with a slender thorax, hunched posture, and axial malformations. PBX1 is highly expressed in proliferating chondrocytes (6). Additionally, there is strong PBX1 expression in the fetal brain (7), and it regulates patterning of the cerebral cortex in progenitor neurons in mice (13). To date, two publications reported CAKUT phenotypes related to pathogenic PBX1 variants/microdeletions; however, only two of the eight patients published by Le Tanno et al. had heterozygous microdeletions only encompassing PBX1 (7). The phenotype of these patients involved, among other things, bilateral renal hypoplasia with hyperechogenic parenchyma, cryptorchidism, skeletal malformations, and developmental delay. The other patients in this publication had larger deletions involving a more extensive set of genes contributing to the phenotype. The five patients with novel loss-of-function variants/deletions in PBX1 identified in a targeted exome sequencing study (11) lacked a detailed genotype–phenotype correlation, as only limited information on the extrarenal phenotype was provided. See Table 1 for a detailed summary of the genetic changes in PBX1 and clinical features described so far.
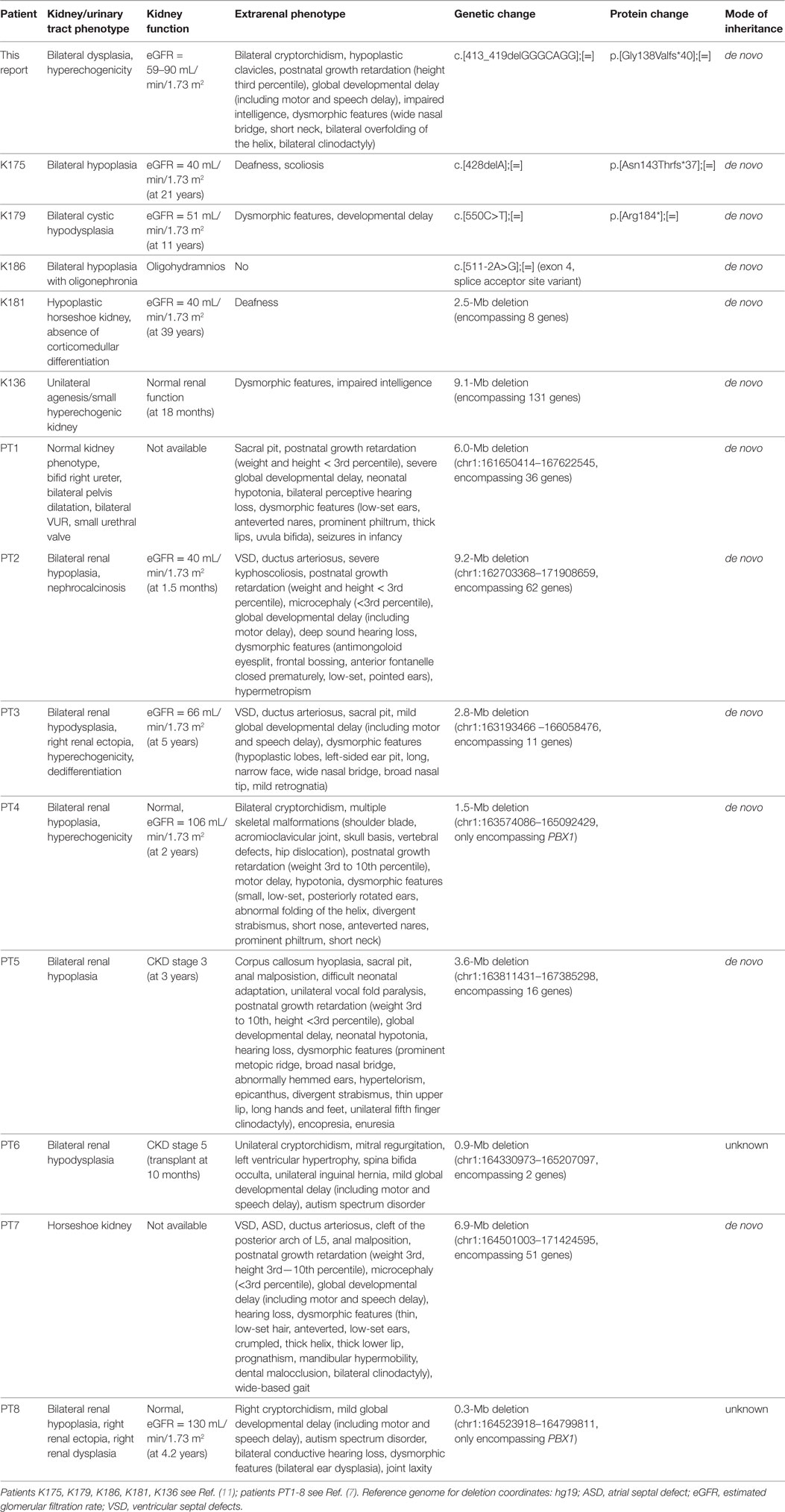
Table 1. Summary of genetic changes in PBX1 and clinical features reported so far.
The patient in our report displayed a complex clinical picture with kidney and skeletal malformations and a neuronal phenotype with developmental delay and impairment of intelligence. In addition to the previously published data from PBX1 mouse models, microdeletions and loss-of-function variants/deletions mentioned above, we provide a detailed description of the phenotype and make the case for an improved diagnostic approach in CAKUT: in this patient our diagnostic algorithm involved SNP array analysis which did not yield a positive result. We then performed WES and identified a novel heterozygous de novo 7-bp frameshift deletion in PBX1 (c.413_419del, p.Gly138Valfs*40). For future CAKUT cases, we recommend directly employing whole-exome or whole-genome sequencing, as these are the apt techniques to identify new pathogenic variants/CNVs in this genetically heterogeneous syndrome. This is especially true for syndromal and familial CAKUT. In patients with isolated CAKUT, however, diagnostic yield is probably rather low, as less than 10% carry variants in 20 known CAKUT genes (2).
Ethics Statement
This study was carried out in accordance with the recommendations of the Ethics Committee of the Technical University of Munich with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Ethics Committee of the Technical University of Munich.
Author Contributions
KR, MW, TM, and JH were responsible for writing and revision of the manuscript. CS and CM cared for the patient and provided the clinical data. KR, BA, RK-N, and MW did the exome analysis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the family for participation and Dr. M. Steinborn from the Department of Diagnostic and Pediatric Radiology, Schwabing Hospital, Munich, Germany, for permission to use the X-ray. This work was supported by the German Research Foundation (DFG) and the Technical University of Munich (TUM) in the framework of the Open Access Publishing Program.
References
1. Vivante A, Kohl S, Hwang DY, Dworschak GC, Hildebrandt F. Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans. Pediatr Nephrol (2014) 29:695–704. doi:10.1007/s00467-013-2684-4
2. Capone VP, Morello W, Taroni F, Montini G. Genetics of congenital anomalies of the kidney and urinary tract: the current state of play. Int J Mol Sci (2017) 18. doi:10.3390/ijms18040796
3. Petersen BS, Fredrich B, Hoeppner MP, Ellinghaus D, Franke A. Opportunities and challenges of whole-genome and -exome sequencing. BMC Genet (2017) 18:14. doi:10.1186/s12863-017-0479-5
4. Majewski J, Schwartzentruber J, Lalonde E, Montpetit A, Jabado N. What can exome sequencing do for you? J Med Genet (2011) 48:580–9. doi:10.1136/jmedgenet-2011-100223
5. Schnabel CA, Godin RE, Cleary ML. Pbx1 regulates nephrogenesis and ureteric branching in the developing kidney. Dev Biol (2003) 254:262–76. doi:10.1016/S0012-1606(02)00038-6
6. Selleri L, Depew MJ, Jacobs Y, Chanda SK, Tsang KY, Cheah KS, et al. Requirement for Pbx1 in skeletal patterning and programming chondrocyte proliferation and differentiation. Development (2001) 128:3543–57.
7. Le Tanno P, Breton J, Bidart M, Satre V, Harbuz R, Ray PF, et al. PBX1 haploinsufficiency leads to syndromic congenital anomalies of the kidney and urinary tract (CAKUT) in humans. J Med Genet (2017) 54:502–10. doi:10.1136/jmedgenet-2016-104435
8. Haack TB, Gorza M, Danhauser K, Mayr JA, Haberberger B, Wieland T, et al. Phenotypic spectrum of eleven patients and five novel MTFMT mutations identified by exome sequencing and candidate gene screening. Mol Genet Metab (2014) 111:342–52. doi:10.1016/j.ymgme.2013.12.010
9. Wortmann SB, Zietkiewicz S, Kousi M, Szklarczyk R, Haack TB, Gersting SW, et al. CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am J Hum Genet (2015) 96:245–57. doi:10.1016/j.ajhg.2014.12.013
10. Weber S. Novel genetic aspects of congenital anomalies of kidney and urinary tract. Curr Opin Pediatr (2012) 24:212–8. doi:10.1097/MOP.0b013e32834fdbd4
11. Heidet L, Moriniere V, Henry C, De Tomasi L, Reilly ML, Humbert C, et al. Targeted exome sequencing identifies PBX1 as involved in monogenic congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol (2017) 28:2901–14. doi:10.1681/ASN.2017010043
12. Yu J, Mcmahon AP, Valerius MT. Recent genetic studies of mouse kidney development. Curr Opin Genet Dev (2004) 14:550–7. doi:10.1016/j.gde.2004.07.009
Keywords: CAKUT, PBX1, dysplastic kidneys, hypoplastic clavicles, developmental delay
Citation: Riedhammer KM, Siegel C, Alhaddad B, Montoya C, Kovacs-Nagy R, Wagner M, Meitinger T and Hoefele J (2017) Identification of a Novel Heterozygous De Novo 7-bp Frameshift Deletion in PBX1 by Whole-Exome Sequencing Causing a Multi-Organ Syndrome Including Bilateral Dysplastic Kidneys and Hypoplastic Clavicles. Front. Pediatr. 5:251. doi: 10.3389/fped.2017.00251
Received: 18 August 2017; Accepted: 07 November 2017;
Published: 24 November 2017
Edited by:
Miriam Schmidts, Radboud University Nijmegen, NetherlandsReviewed by:
Kirsten Renkema, University Medical Center Utrecht, NetherlandsJan Halbritter, Leipzig University, Germany
Copyright: © 2017 Riedhammer, Siegel, Alhaddad, Montoya, Kovacs-Nagy, Wagner, Meitinger and Hoefele. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julia Hoefele, julia.hoefele@tum.de