
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr. , 22 August 2017
Sec. Children and Health
Volume 5 - 2017 | https://doi.org/10.3389/fped.2017.00178
 Flavia Indrio1
Flavia Indrio1 Silvia Martini2*
Silvia Martini2* Ruggiero Francavilla1
Ruggiero Francavilla1 Luigi Corvaglia2Fernanda Cristofori1
Luigi Corvaglia2Fernanda Cristofori1 Salvatore Andrea Mastrolia3
Salvatore Andrea Mastrolia3 Josef Neu4Samuli Rautava5Giovanna Russo Spena6
Josef Neu4Samuli Rautava5Giovanna Russo Spena6 Francesco Raimondi6Giuseppe Loverro3
Francesco Raimondi6Giuseppe Loverro3
Epigenetic modifications are among the most important mechanisms by which environmental factors can influence early cellular differentiation and create new phenotypic traits during pregnancy and within the neonatal period without altering the deoxyribonucleic acid sequence. A number of antenatal and postnatal factors, such as maternal and neonatal nutrition, pollutant exposure, and the composition of microbiota, contribute to the establishment of epigenetic changes that can not only modulate the individual adaptation to the environment but also have an influence on lifelong health and disease by modifying inflammatory molecular pathways and the immune response. Postnatal intestinal colonization, in turn determined by maternal flora, mode of delivery, early skin-to-skin contact and neonatal diet, leads to specific epigenetic signatures that can affect the barrier properties of gut mucosa and their protective role against later insults, thus potentially predisposing to the development of late-onset inflammatory diseases. The aim of this review is to outline the epigenetic mechanisms of programming and development acting within early-life stages and to examine in detail the role of maternal and neonatal nutrition, microbiota composition, and other environmental factors in determining epigenetic changes and their short- and long-term effects.
Following the so-called “Developmental Origins” hypothesis, introduced by Barker and Osmond 30 years ago (1) and based on early developmental plasticity, the environmental influence on health and disease has been progressively explored over the last decades.
Research on this issue has eventually led to the discovery of a “second genome,” which comprises human microbiome and, via its metabolites, actively interacts with the genome derived by sperm and egg, resulting in far-reaching epigenetic modifications (2, 3).
The term “epigenetics,” which literally means “on top of genetics,” defines a variety of processes that cause mitotically and meiotically heritable changes in gene expression without modifying the deoxyribonucleic acid (DNA) sequence; particularly, DNA methylation, histone modification, and non-coding RNA are the main mechanisms underlying epigenetic modifications.
The period of life during which epigenetic DNA imprinting activity is the most active lasts from conception to the second anniversary, thus being referred to as “the 1,000 days period” (4). During this time interval, via epigenetic changes, early nutrition can play a key role in developmental programming, thereby possibly influencing the individual susceptibility to the later development of cardiovascular diseases, obesity, diabetes, and other non-communicable chronic conditions.
According to recent evidence, the microbial colonization begins far before birth; particularly, the microbial flora from amniotic fluid, placenta, and maternal gut can support the development of a prenatal microbiota (5–7) that is likely to have an influence on the developing embryo and fetus. Furthermore, early postnatal microbiota perturbations, resulting from skin contacts, mode of delivery and neonatal diet, have been proposed to play a role in the susceptibility to several late-onset diseases (i.e., obesity, diabetes, allergies, asthma, autoimmunity) by modulating the immune development through epigenetic modifications (8). Similar mechanisms might also underlie the increased risk of necrotizing enterocolitis associated with the use of antibiotics or histamine-2 receptor blockers in the neonatal population (9, 10).
Recent evidence has shown that some of the epigenetic changes ensuing from early nutrition and microbiome can be transgenerationally inherited, thus having a significant impact on evolution (11). Although preliminary data currently available on the role of epigenetic in the determination of long-term health and disease look promising, however, most of the underlying mechanisms still need to be clearly elucidated.
This review aims to provide a complete overview on the complex interactions between early nutrition, microbiome, and epigenome during the early phases of human development (summarized in Figure 1), examining current evidence in detail and shedding light on the complex epigenetic processes that have been identified so far.
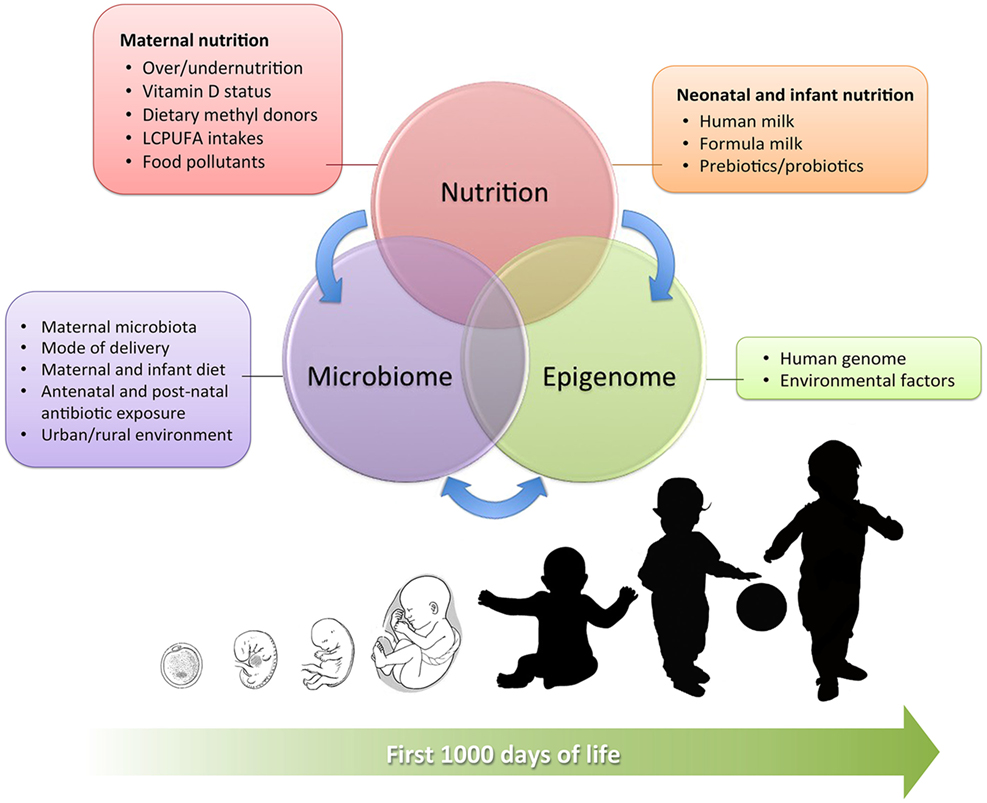
Figure 1. Interrelation between maternal and neonatal nutrition, gut microbiota, and epigenetics during the first 1,000 days of life. The main influencing factors are detailed in the boxes.
Throughout their lifespan, humans are exposed to several environmental hazards; nevertheless, the effects of these exposures may take decades for their phenotypic expression. It has been speculated that, in response to maternal homeostasis and intrauterine stimuli, the fetus undergoes predictive responses theoretically resulting in permanent adjustments of the homeostatic systems, aimed at improving adaptation to the postnatal environment. Nevertheless, a mismatch may occur, and these adjustments might ultimately become disadvantageous, resulting in heritable risk factors for future diseases (12).
In this section, we will review the effects of maternal nutrition, in its qualitative [i.e., micronutrients, long-chain polyunsaturated fatty acids (LCPUFAs), food pollutants] and quantitative (i.e., under- and overnutrition) aspects, on epigenetic fetal programming in both human and animal models. A detailed list of these effects on the offspring is provided in Table 1.
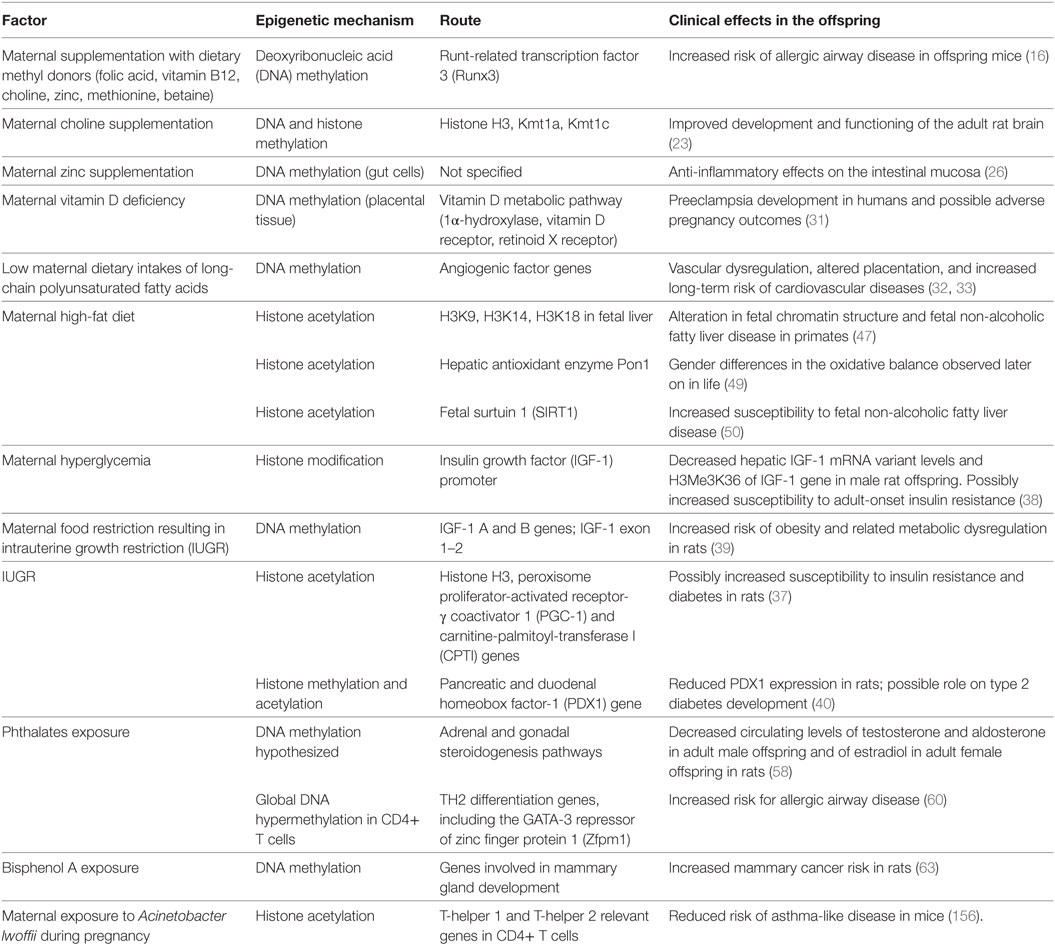
Table 1. Main antenatal factors associated with epigenetic modifications in the offspring.
The most common epigenetic modification is DNA methylation, resulting from the addition of a methyl group to the cytosine of a cytosine–guanine pair. If this methylation is located in close proximity to a gene, it often results in lowered or abolished gene expression. As the one-carbon metabolism depends upon dietary methyl donors, DNA methylation can be influenced by nutrition during early life: this process involves a large number of enzymes with methyltransferase activity, cofactors including choline, methionine, vitamins B6 and B12, zinc, betaine and dietary micronutrients acting as methyl donors (13–15).
Folic acid is a well-known one-carbon donor for DNA methylation and synthesis; its role is crucial during early postnatal development, when rapid cell growth and proliferation take place. However, in animal models, in utero exposure to a maternal diet supplemented with methyl donors has also shown unexpected effects, such as an increased susceptibility to allergic airway disease. Particularly, mice born to mothers supplemented with folic acid, vitamin B12, methionine, zinc, betaine, and choline experienced significantly higher rates of allergic airway inflammation, ensuing from an excessive methylation of the runt-related transcription factor 3 (Runx3), a mediator of T-lymphocyte differentiation predisposing to asthma-like diseases (16). Hence, methylation can act as a double-edged sword, and this finding is consistent with human epidemiologic evidence of a significant association between perinatal folic acid supplementation and increased risk of wheezing at 18 months of age (17).
On the other hand, folic acid deficiency has been associated with an increased expression of inflammatory mediators, such as interleukin (IL)-β, IL-6, tumor necrosis factor-α (TNF-α), and monocyte chemoattractant protein-1 in the mouse monocyte cell line RAW264.7 (18); this is consistent with the beneficial role of folic acid in preventing inflammatory responses (19).
In addition to folic acid, imbalanced maternal concentrations of other micronutrients can affect DNA methylation patterns in the offspring. For example, increased maternal serum levels of vitamin B12 during pregnancy correlate with decreased global DNA methylation in newborns, while high vitamin B12 serum levels in newborns have been associated with reduced methylation of insulin-like growth factor-binding protein 3, a gene involved in intrauterine growth (20). Vitamin B12 deficiency, on the other hand, can result in global hypomethylation as, along with folic acid, this micronutrient is involved in the synthesis of methionine and S-adenosyl methionine, donors commonly required for the maintenance of DNA methylation patterns (21).
There is growing evidence that optimal dietary intakes of choline, which is involved in one-carbon transfer or methylation pathways as folic acid, support a successful completion of fetal development (22). In animal studies, maternal choline supplementation during pregnancy as been shown to modify histone and DNA methylation in fetal liver and brain, suggesting concerted epigenetic mechanisms that contribute to favorable long-term developmental effects (23).
Due to its role in DNA methylation, zinc status can exert a fundamental influence on the epigenome. Particularly, its deficiency during intrauterine life and childhood may contribute to alter promoter methylation, resulting in an immune dysregulation that could contribute to the development of chronic inflammatory diseases (24) and to increase cardiovascular risk (25). On the contrary, evidence from animal studies has shown that maternal zinc supplementation during pregnancy is associated with a lower degree of DNA methylation in gut cells, which, in turn, can have an anti-inflammatory effect on the intestinal mucosa (26).
In sum, these findings suggest that specific dietary interventions at key time points of fetal development can lead to different and unintended long-term consequences on health and disease.
Changes in vitamin D metabolism have been associated with altered methylation patterns in placental tissue, with a possible influence on pregnancy outcome and on the development of great obstetrical syndromes (27).
After oral ingestion, vitamin D3 undergoes hepatic and renal hydroxylation, being thus converted in its biologically active form (1,25-dihydroxyvitamin D3), which enters the cells and combines with the retinoid receptor, forming a heterodimer that binds to vitamin D-responsive genes and regulates their transcription and translation (28).
The placenta has a functional vitamin D endocrine system, expressing vitamin D receptors and allowing the local conversion of vitamin D in its active form (29). Of interest, a potential association between vitamin D insufficiency and increased risk of preeclampsia has been reported (27, 30). In order to better understand the possibly underlying epigenetic processes, Anderson et al. (31) analyzed DNA methylation patterns and protein expression of placental genes involved in vitamin D metabolism in relation to vitamin D intakes in women with preeclampsia, compared to those who remained normotensive throughout pregnancy. Though not significant, the incidence of vitamin D deficiency was higher in the preeclamptic (46%) than in the normotensive group (20%). Moreover, placental samples from pregnant women with preeclampsia showed increased DNA methylation of CYP27B1, vitamin D receptor and retinoid receptor genes, with lower protein expression levels of the latter. A possible interference between availability of vitamin D at the maternal–fetal interface, hypermethylation of key placental genes involved in vitamin D metabolism and placentation can be hypothesized; however, further studies are needed to clarify the exact mechanisms.
Long-chain polyunsaturated fatty acids are essential components of human diet known for their beneficial effects on health, growth and development. From an epigenetic point of view, LCPUFAs are rich in phospholipids, which are among the major methyl group acceptors in the one-carbon metabolic pathway, being thus involved in methylation reactions (32).
Recent studies investigating the role of LCPUFAs in determining gestational outcomes and influencing the offspring’s health have shown that inadequate intakes during pregnancy may result in aberrant DNA methylation patterns, affecting the expression of clinically relevant genes (e.g., angiogenic factor genes) (32). This can contribute not only to the vascular dysregulation associated with abnormal placentation but might also play detrimental effects on fetal programming, ensuing in an increased long-term risk of cardiovascular diseases (33).
High intakes of n-3 LCPUFAs, such as eicosapentaenoic acid, docosahexaenoic acid (DHA) and α-linolenic acid (18:3n23) are largely known to be associated with protective metabolic effects. In mice, high-dietary n-3 LCPUFAs have been shown to bring significant epigenetic changes in leptin promoter, resulting either from the inhibition of the enzymes catalyzing DNA methylation and histone modifications or from a reduced availability of the relative substrates (34). On the other hand, fish oil supplementation in animal models has been associated with decreased global DNA methylation in the liver persisting for two generations, lowered blood lipid concentrations, increased insulin-stimulated glucose uptake, and insulin sensitivity (35); however, the pathways involved still need to be completely elucidated.
Growth and development during fetal life and early childhood are greatly influenced by macronutrient intakes during pregnancy, suggesting the importance of an adequate maternal diet in this crucial phase. A poor maternal nutrition has been shown to modify epigenetic programming and have a negative impact on fetal gene expression, resulting in possible long-term consequences (36).
Maternal undernutrition (either global or protein-restricted) has been extensively used in rodent studies to induce intrauterine growth restriction (IUGR). In the setting of an altered early nutrition, organisms can achieve environmental adaptation by modulating their gene expression via epigenetic alterations of histone markers. In experimental models of IUGR, uteroplacental insufficiency has been shown to decrease postnatal insulin-like growth factor-1 (IGF1) mRNA variants and H3 acetylation of IGF1 gene (37). Similar patterns of histone modification on IGF-1 promoter have also been reported by Zinkhan et al. (38) following maternal gestational hyperglycemia. Moreover, different changes in hepatic IGF-1 mRNA expression and histone H3K4 methylation have also been reported in relation to early growth patterns following IUGR in the rat (39). One of the molecular phenotypes associated with IUGR rats is a decreased expression of pancreatic and duodenal homeobox factor-1 (PDX1), a key transcription factor regulating pancreatic development (40); similar findings have also been observed for the muscular glucose transporter GLUT4 (41).
Low-protein regimens at conception or during pregnancy bring additional evidence on the noxious epigenetics effects of maternal nutrition deficiency on the offspring health: in animal models, a protein-restricted maternal diet has been associated with impaired immune response (42), increased sensitivity to oxidative stress (43), metabolic abnormalities, and glucose dyshomeostasis (44).
The Dutch Winter Study provides a bright example on how famine during early and mid-gestation can lead to metabolic dysregulation later on in life, having been associated with hyperglycemia, higher incidence of coronary heart disease, a more atherogenic lipid profile, disturbed blood coagulation, increased stress responsiveness, and obesity (45). Remarkably, follow-up studies on the population exposed to the Dutch famine have shown increased rates of neonatal adiposity and poorer health among the offspring of women exposed to maternal undernutrition in utero, suggesting that the related detrimental effects are probably transmitted to subsequent generations through epigenetic modifications persisting during meiosis (46).
In addition to nutrient deficiency, overnutrition during pregnancy has also been demonstrated to cause detrimental effects on the offspring health either at early or late life stages.
Studies performed in primates have shown that a high-fat maternal diet can alter fetal chromatin structure via covalent histone modifications (47). Maternal high-fat dietary intakes have been associated with hormonal dysregulation and release of inflammatory cytokines that can predispose the offspring to various vascular diseases (48). Strakovsky et al. (49) showed that, by modifying histone acetylation, a maternal diet with high-fat contents contributes to alter the expression of neonatal hepatic antioxidant enzymes in a sex-specific manner, thus possibly contributing to the known gender differences in the oxidative balance observed later on in life. In primates, a maternal high-fat diet has been reported to modulate protein deacetylase activity and to modify fetal surtuin 1 (SIRT1) histone, which is a likely mediator of fetal epigenome and metabolome in the setting of maternal obesity (50).
As for maternal obesity, in further animal models it has been correlated with increased pancreatic beta cell mass and excessive insulin secretion (51) that, in the long-term, can predispose to the development of diabetes and non-alcoholic fatty pancreas disease in the offspring (52).
Eventually, of great interest is the effect of nutrition on microRNAs (miRNAs), which reciprocally interact with other epigenetic mechanisms, such as histone modification and DNA methylation, in order to modulate the expression of target genes (53). The expression of miRNAs can be controlled by DNA methylation and chromatin modifications; in turn, miRNAs affect the methylation machinery and the expression of proteins involved in histone modification. The combination of these mechanisms contributes to determine gene expression and the resultant phenotype.
Nutrition has been shown to modulate the expression of endogenous miRNAs, resulting in different serum miRNA profiles that may influence biological processes, including inflammation and metabolism. Overnutrition can actively regulate several miRNAs involved in immune function modulation, thus contributing to the development of chronic inflammation (54). Of interest, a maternal high-saturated-fat diet has been shown to induce inflammation pathways in the offspring in animal (55) and human studies (56).
Eventually, the intrauterine exposure to inflammatory mediators, whose levels are often increased in the context of obesity and metabolic dysregulation, has been shown to influence the adulthood risk of diseases with an inflammatory component (e.g., asthma, cardiovascular diseases, atherosclerosis) (48, 57). Although the causative mechanisms still need to be elucidated, a detrimental effect of maternal inflammation on the appropriate maintenance of fetal epigenetic profiles can be hypothesized.
The epigenetic effects of prenatal exposure to chemical food contaminants, such as phthalates and bisphenol A (BPA), are in the early stages of elucidation.
Phthalates are ubiquitous plasticizers mainly used in the manufacture of polyvinyl chloride products; food packaging and contact materials are considered the major sources of food contamination. Phthalates have been shown to act as endocrine-disrupting compounds and are thought to interfere with epigenetic programming. In a rat model, fetal and postnatal contamination through maternal exposure and food sources, respectively, has been found to decrease circulating levels of testosterone and aldosterone in adult male offspring and of estradiol in females (58). Furthermore, recent murine evidence has contributed to shed light on the epigenetic mechanisms undergoing the increased risk for allergic airway disease observed after prenatal and early postnatal phthalate exposure (59), showing a significantly increased global DNA hypermethylation in CD4+ T cells of the offspring that resulted in a transcriptional downregulation of genes involved in T-helper 2 differentiation (60).
Contamination modalities for BPA are similar to those previously described for phthalates. Dietary exposure to BPA in the Agouti viable yellow (A(vy)) mouse model has been shown to hypomethylate metastable epialleles, an effect that can be counteracted with dietary supplementation of methyl donors or genistein (61). According to further evidence from animal models, antenatal and perinatal BPA exposure has been found to lead to specific epigenetic changes, resulting, among the others, in impaired stress response (62) and higher rates of mammary cancer (63); this contributes to highlight the clinical burden associated with chemical food contamination.
Among the postnatal factors that can contribute to determine lifelong health and disease via epigenetic mechanisms, infant feeding plays a key role. Evidence on the epigenetic effects of early nutrition on developmental programming, possibly resulting in later development of cardiovascular diseases, overweight, obesity, diabetes, and other chronic conditions (64), is constantly increasing.
Human breast milk (HBM) is universally considered the normative standard for infant feeding, as it confers unique nutritional and non-nutritional benefits that could be partly explained by epigenetics; although the majority of the underlying mechanisms are still unclear, a number of them, summarized in Table 2, have been progressively elucidated.
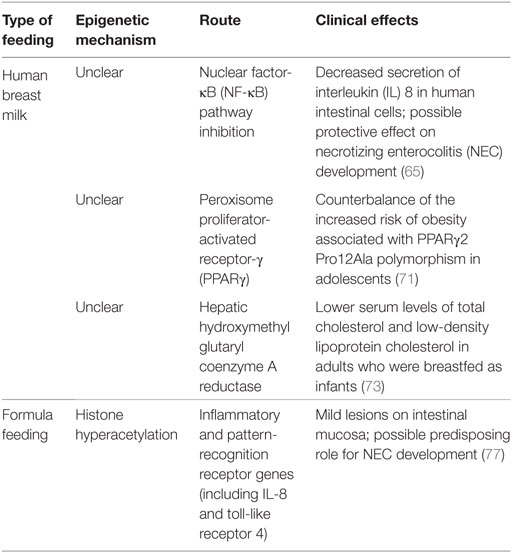
Table 2. Main epigenetic modifications associated with neonatal nutrition.
Lactoferrin is an abundant HBM protein that regulates gene expression by binding to pro-inflammatory bacterial DNA sequences in Peyer’s patches and intestinal mucosa. As a result, this binding inhibits the transcriptional activation of nuclear factor-κB (NF-κB) in human intestinal B-lymphocytes and downregulates IL-8 synthesis, which is involved in the pathogenesis of necrotizing enterocolitis (65).
Human milk oligosaccharides contained in HBM promote a healthier composition of gut microbiota, which plays a leading role in programming the infant’s immune phenotype and in preventing early and later diseases (66). Particularly, breastfeeding-induced microbiota has been proved to regulate the expression of genes involved in digestion, barrier function, and angiogenesis and enhance immunoglobulin-A secretion (67), thus possibly contributing to prevent necrotizing enterocolitis. Similar mechanisms involving gut microbiota have also been proposed to explain HBM beneficial effects in preventing infections and immune-mediated diseases, such as asthma and allergies (68).
The most striking evidence of nutritional programming, however, is observed for the protective effect of HBM on later obesity and metabolic diseases. The peroxisome proliferator-activated receptor-γ (PPARγ) transcription factor is highly expressed in adipocytes, where it regulates the maintenance of insulin sensitivity. In particular, PPARγ ligands contribute to restore normal levels of adipose-derived substances, such as leptin, adiponectin, and TNF-α, thus reversing insulin resistance syndrome, improving endothelial cell functions and reducing inflammation (69). Pro12Ala substitution at codon 12 is the most common variant identified in the PPARγ2 gene and has been associated with an increased risk of obesity in adulthood (70). Non-breastfed adolescents carrying Pro12Ala polymorphism showed higher body mass index, waist circumference, and higher skinfold thickness when compared to those who had been breastfed even for a short period. According to this finding, by inducing epigenetic modifications, breastfeeding could counterbalance the risk of obesity even in genetically predisposed adolescents (71). To this regard, the authors supposed a possible role of the natural PPARγ ligand contained in HBM in decreasing PPARγ2 transcriptional activity in Pro12ala carriers.
Human breast milk is particularly rich in n-3 LCPUFAs, such as DHA. In animal models, DHA has been shown to downregulate hepatic lipogenesis and cholesterol biosynthesis pathways (72), consistently with the lower levels of serum total cholesterol (TC) and of low-density lipoproteins (LDL) observed in adults who had been breastfed as infants (73). Paradoxically, however, increased TC and LDL levels have been observed in breastfed infants during their first year of life (73). This effect, which is rapidly reversible, is likely to be a direct consequence of the high cholesterol contents of HBM that, on the other hand, contribute to reduce TC and LDL levels at older ages by downregulating hepatic hydroxymethyl glutaryl coenzyme A reductase via epigenetic modifications.
The beneficial effects of HBM are not only limited to breastfed infants but are also relevant to breastfeeding mothers. An inverse correlation between breastfeeding duration and breast cancer risk has been previously established (74). This effect has also been observed in women carrying deleterious mutations in the BRCA1 gene who breastfed their children for more than 1 year (75). Although the epigenetic links occurring between HBM components and breast cancer prevention need to be further elucidated, a proposed underlying is the inhibiting effect that DHA exerts on breast cancer cell growth by modulating PPARβ mRNA expression (76).
While the beneficial epigenetic effects of HBM are well established, on the other hand, less is currently known about formula feeding.
Recent evidence from preterm piglets has shown that, compared to colostrum, formula feeding was associated with a significant upregulation of inflammatory and pattern-recognition receptor genes, including IL-8 and toll-like receptor (TLR)-4; this pro-inflammatory status corresponded to decompacted chromatin configurations and histone hyperacetylation in key inflammatory genes, thus suggesting underlying epigenetic modifications. These findings histologically correlated with mild mucosal lesions, and a possible predisposing role for NEC development has been hypothesized (77).
The gastrointestinal tract is the most important site of host–microbe interactions and the establishment of an indigenous intestinal microbiota during early life has been shown to have a major impact on human physiology.
Compelling evidence contradicts the dogma that the fetus resides in a sterile environment and that the newborn only attains its microbiota after extrauterine exposure (78). Although controversy still exists about the “sterile womb” versus the “in utero colonization” concept (79), it is difficult to dispute that the microbial environment of the fetus in utero has major implications for health and disease. For over 30 years, we have known that even without a ruptured amniotic sac, amniotic fluid frequently contains significant levels of bacteria as evaluated by quantitative culture techniques (80). Moreover, intrauterine “infection” and subsequent inflammatory responses link to prematurity, brain, lung, and eye disease (81, 82) and suggest that perturbations of critical maternal–fetal–microbial interactions lead to pathology.
The postnatal establishment of gut microbiota is influenced by several factors: mode of delivery, contact with the mother (such as with skin-to-skin care), composition of the diet, and administration of pharmacologic agents, especially antibiotics (83). Immediately after birth, the infant gut microbiome has relatively low-species diversity and high rates of bacterial flux. By 3 years of age, this flux begins to stabilize (84). Staphylococcus, Streptococcus, Escherichia coli, and Enterobacteria are thought to be the first colonizers of the gut. Facultative anaerobic bacteria subsequently replace these taxa and consist in large relative abundances of Actinobacteria and Firmicutes (85). This is influenced in large part by diet; for example, breastfeeding appears to stimulate the growth of Bifidobacteria species (86).
Hence, early environmental influences influencing gut microbiota during this crucial developmental period can modify its composition toward more pathogenic profiles that, in turn, can persist until adulthood and exert long-lasting effects on health and disease.
Among the possible mechanisms through which intestinal bacteria can influence human health, epigenetic modifications prevail. The direct potential of microbes to induce epigenetic changes in the host has been recently demonstrated by the evidence of microbe-specific patterns of epigenetic DNA modification after exposure to commensal or pathogenic organisms in immature human cells from intestinal epithelia (87). Interestingly, the same study also reported that prenatal glucocorticoid-induced epigenetic programming results in an altered gut microbiota composition in mice, suggesting the existence of complex interactions between microbiome and epigenome (87).
The role of microbiome as an epigenetic modulator is thus gaining increasing attention and, although the underlying mechanisms still need to be partially elucidated, current evidence supports a significant correlation between gut microbiome composition and epigenetic changes in genes relevant to immunological, metabolic, and neurological development and functions. The role of gut microbiota on each of these areas is addressed in the following paragraphs; moreover, a summary of the currently recognized epigenetic mechanisms associated with specific gut microbiota profiles is provided in Table 3.

Table 3. Epigenetic modifications associated with specific profiles of gut microbiota.
Gut microbiota plays a key role in the development of immune response since the early phases of life: by activating specific pathways of molecular signaling, it supports the maturation of gut-associated lymphoid tissue (88), promotes the conversion of CD4+ T-cells into T-regulatory cells (89), and influences the balance between T-helper 1 and 2, which is known to have significant effects on the development of allergic diseases (90). Furthermore, specific microbiota-induced patterns of TLR-2 and TLR-4 expression have been observed in gut cells; by modulating TLR expression, commensal intestinal bacteria may prime the host response to pathogenic threats and act as local immunomodulators, suppressing pro-inflammatory pathways and promoting the intestinal transcription of cytoprotective genes (91).
Intriguing data from experimental animal models have supported the existence of a causal relationship between early microbial contact and the development of immune system: particularly, mice reared in germ-free conditions failed to develop immune tolerance and were more prone to allergic-type immune responses (92).
Over the last decade, increasing evidence has consistently shown that an abnormal composition of gut microbiota in the early phases of life is associated with the subsequent development of immune-mediated diseases (93, 94). The intrauterine period, as well as post-partum, is crucial for the establishment of immune response; therefore, different profiles of intestinal bacteria in this delicate phase can result in different long-term effects on immunological functions.
During vaginal delivery, infants receive a significant inoculum of colonizing microbes from maternal birth canal and intestine. Consequently, neonates born by C-section (CS) delivery exhibit aberrant gut colonization patterns (95), which may extend at least until the age of 7 years (96). According to an epidemiological study on 1.9 million subjects from Denmark (97), the incidence of immune-mediated disease including asthma, juvenile arthritis and inflammatory bowel disease (IBD) is significantly higher in CS-born children.
In a similar fashion, treatment with antibiotics is known to cause drastic changes in intestinal microecology and, although other potential confounding factors may be at play (98), early antibiotic exposure in human and animal studies has been associated with increased long-term risk of asthma (99), type 1 diabetes (T1D) (100), and IBD (101). However, the effects of microbiota composition in predisposing to non-communicable diseases are more likely to ensue from the concerted interactions occurring among intestinal bacterial profiles, their metabolites, and the host’s responsiveness rather than from the effects of single bacterial strains.
Recent data from pediatric patients with both ulcerative colitis and Crohn’s disease has shown a striking reduction in species richness and diversity in their microbiota, with particularly low abundance of Lactobacilli, Bifidobacteria, and other bacteria that are known to have a positive influence on gut homeostasis, such as Eubacterium rectale and Faecalibacterium prausnitzii (102). Given the substantial stability of intestinal microbiota after the first years of life (84), it is likely that these abnormal patterns may establish in earlier phases, contributing to predispose toward IBD development by inducing epigenetic changes. As an example, Lactobacilli and Bifidobacteria, whose concentration is lowered in children with IBD, exert well-known anti-inflammatory effects and support the integrity of intestinal barrier by producing butyrate through cross-feeding (103). Acting as a histone deacetylase (HDAC) inhibitor, butyrate can dampen gut inflammation by suppressing nuclear factor-B (NF-B) activation (88), upregulating the expression of PPARγ and decreasing interferon-γ production (104). In addition, Lactobacilli and Bifidobacteria can also affect DNA methylation by regulating methyl-donor availability through their production of folate (105). Hence, a decreased butyrate production and folate bioavailability ensuing from an altered microbiota may result in an increased expression of inflammatory pathways that, in turn, can predispose to intestinal and systemic inflammation (88).
Further evidence on how early modifications of gut microbiota can influence immune responses is provided by the non-obese diabetic mouse model of human T1D. Dysfunctional antigen-presenting cells (APCs), ensuing in aberrant immune tolerance, have been associated with T1D development (106). Recently, it has been shown that prenatal exposure to different antibiotics, altering gut bacterial composition at the earliest phases of life, is associated with significant differences in the autoantigen-presenting functions of APCs, resulting in either protective (107, 108) or more diabetogenic immune profiles (100). Of interest, the observed protective effects were heritable by the second-generation offspring and were also transmitted to other hosts via gut microbiota transfer, suggesting the presence of underlying epigenetic mechanisms (108).
The association between early antenatal and perinatal factors and metabolic profile later in life has been largely established. As an example, preterm birth has been extensively associated with the development of obesity, cardiovascular diseases, and type 2 diabetes (T2D) in adulthood (109, 110). A significant correlation among maternal and neonatal nutrition (73), antenatal (111) and postnatal antibiotic exposure (98), and later metabolic outcomes is also well-known, and a recent meta-analysis of epidemiological studies has shown that the risk of childhood obesity is significantly increased in infants born by CS, even after correcting for maternal weight (112).
Similar metabolic effects have been associated with early aberrant profiles of gut microbiota (113–115), thus suggesting that the impact of microbial contact on metabolic maturation is most profound during early life and healthy metabolic development depends on interactions with healthy microbiota. Of interest, the obese phenotype induced by early aberrant gut microbiota has been observed to persist even after gut microecology is restored (116), and it has also been shown that obesity may be transferred to germ-free mice by colonizing them with intestinal microbes from obese humans (117).
The role for microbiota-induced epigenetic modifications in mediating these effects is increasingly emerging; nevertheless, the mechanisms underlying the interactions between early-life microbiota and epigenetic programming of host metabolic physiology are only beginning to be unraveled.
According to Remely et al. (118), obese adults or subjects with T2D exhibited marked differences in gut microbiota composition compared to lean individuals. The metabolic state of the host correlated also with innate immune function, as significantly increased CpG methylation was discovered in the regulatory region of TLR4 gene in obese subjects compared to lean individuals; moreover, the promoter region of the TLR2 gene showed higher methylation levels in the diabetic group compared to healthy controls.
Deciphering the direction of causality between gut microbiota composition and the epigenetic modulation of microbial defense is difficult. Further insights into the sequence of events are provided in a report by Kumar et al. (119), according to which distinct DNA methylation profiles were detected in blood samples obtained from women 6 months after delivery, depending on the predominance of either Firmicutes or Bacteroidetes and Proteobacteria in the fecal microbiota during pregnancy. The potential clinical significance of this finding is highlighted by previous evidence indicating that the ratio of intestinal Bacteroidetes to Firmicutes is associated with metabolic disorders, and by the observation that the differences in methylation were located in genes whose function is linked to obesity, metabolism, and inflammation.
The first 1,000 days of life are crucial not only for the physiologic establishment of gut microbiota, but also for the development of central nervous system (CNS). While the reciprocal interaction between the brain and intestinal organs has been recognized long ago (120), evidence supporting a correlation between microbiota composition and altered neurocognitive and behavioral development has progressively emerged in the last decade, contributing to outlining the so-called “microbiota–gut–brain axis” (121).
Remarkable evidence on the effects of gut colonization on brain functions and development is provided by rodents raised in a sterile environment, which thus lack gut microbiota and are referred to as germ free (GF) (122). The most commonly reported phenotype in GF mice was an increased anxiety-related behavior (123). This altered behavioral response was accompanied by changes in the concentrations of neurotransmitters, their metabolites and neurotrophic factors involved in neural plasticity (124). Although the underlying molecular mechanisms are not well-understood, microbiota-related epigenetic regulation of gene expression and transcription in different brain regions has been hypothesized (125). Furthermore, increased basal levels of corticosterone and enhanced responses to stressors, which are known to negatively influence brain development (126), have also observed in GF animals, thus suggesting a possible role for gut microbiome in the regulation of hypothalamic–pituitary–adrenal axis (127).
Of interest, the reconstitution of normal microbiota patterns early in life in GF mice normalized both behavioral patterns and neurotransmission concentrations (125), whereas such an effect could not be established in adulthood (128, 129). Besides suggesting the role of microbial colonization in initiating signaling mechanisms that affect neuronal circuits involved in brain development and behavior (125), these results also suggest the existence of a critical window for intestinal microbes to influence developmental programming of long-lasting brain functions. In a similar fashion, GF mice have also shown deficits in social functioning, completely reversible after post-weaning microbial gut colonization (130).
Further supportive evidence on the role of gut microbiome in modulating brain development and behavior is provided by the evidence that specific microbial profiles resulting from infections, antibiotic treatment, or administration of probiotic bacteria have been associated with consistent behavioral changes in rodent studies. As for the bacterial strains possibly involved, preliminary evidence supports Bifidobacteria and Lactobacilli in exerting positive behavioral effects (131, 132).
Among the pathways through which gut microbiome may modulate CNS functions and development, which are just beginning to be unraveled, a possible role for immune signaling has been proposed (133). As previously discussed, the multiple antigenic stimuli provided by early gut colonization are fundamental for an appropriate immunological maturation and have been proved to modulate the expression of immune-related genes via epigenetic changes. In turn, cytokine receptors have been revealed on neurons and glial cells (134) and a significant contribution of immune signaling in normal brain function as well as during aging or in the context of neurodegenerative diseases (135–138) has been observed, thus contributing to support this hypothesis.
Recently, a possible correlation between autism-spectrum disorders (ASD) and abnormal gut microbiota composition and metabolism has been hypothesized, basing on observation that gastrointestinal symptoms are a common comorbidity in ASD children (139). However, current evidence from human (140–142) and rodent (130) studies is limited and controversial, and large prospective and randomized trials are needed to shed further light on this issue.
The most striking example supporting the epigenetics role in the context of microbiota–gut–brain axis is provided by irritable bowel syndrome (IBS). Increased visceral sensitivity, ensuing from abnormal brain responses to physiological visceral stimulation, has been widely proposed as one of the key mechanisms underlying IBS clinical manifestations (143). In order to investigate the possible influence of epigenetics in determining brain susceptibility to visceral stimuli, Tran et al. exposed mice from a rodent IBS-like model to cerebral injections of trichostatin A (TCA), a potent HDAC inhibitor (144). According with their results, TCA injections led to a significant improvement in visceral hypersensitivity, quantified by the number of gut contractions in response to graded colorectal distension, thus supporting a possible involvement of epigenetic mechanisms in modulating stress-induced visceral pain and hinting a potentially beneficial effect of HDAC inhibition in the treatment of IBS. In line with this, altered microbiota profiles, whose association with detrimental epigenetic changes has been previously discussed, are a common finding in both pediatric and adult IBS patients (145, 146) and therapeutic trials with specific probiotic strains, such as butyrate-producing Bifidobacteria, have been shown to ameliorate visceral hypersensitivity in animal (147) and human studies (148). However, further studies are needed to elucidate epigenetic modifications of genes relevant to visceral pain in relation to gut microbiome (123) and its influencers during the early phases of life.
In sum, according to the abovementioned evidence, a thorough understanding of how the microbiome–gut–brain axis operates during infancy may provide not only useful insights into early neurocognitive development with possible translational applications but also a greater awareness on the several modifiable factors influencing the establishment of infant microbiome during the first 1,000 days of life and their crucial long-term effects (149).
Evidence on epigenetic programming by gut microbiota may be interpreted to form a basis for a hypothesis according to which detrimental epigenetic modifications and, consequently, the development of disease, might be prevented by modulating microbial contact in early life. Modifying gut microbiota through prebiotic, probiotic, and symbiotic administration [the so-called “bacteriotherapy” (150)] might represent a promising approach to rebalance the homeostasis of systemic and mucosal immune systems.
So far, the most extensive evidence of early microbial interventions has been published in the case of reducing the risk of atopic disease using probiotics in high-risk populations. Maternal and infant probiotic supplementation from the last weeks of pregnancy until the offspring was 6 months of age has been reported to significantly reduce the incidence of atopic dermatitis (151). Interestingly, this protective effect was still detectable at the age of 7 years (152), thus suggesting the existence of immune programming mechanisms, even though still largely unknown. Another clinical trial demonstrated that a solely maternal probiotic intervention during pregnancy and breastfeeding can effectively reduce the incidence of atopic dermatitis in the child (153), and a recent meta-analysis of clinical trials (154) concluded that, in order to be effective in decreasing the risk of atopic dermatitis, the probiotic intervention should be commenced before birth.
Inflammatory bowel disease and T1D are other examples of immune-mediated diseases that would possibly benefit from early probiotic interventions. As different bacteria can induce different immune responses, gut microbiota would represent an optimal target for preventive and therapeutic strategies aimed, for example, at generating self-tolerogenic APCs with protective effects toward T1D development (107), or at hindering the establishment of bacterial profiles that are known to upregulate pro-inflammatory pathways and predispose to IBD development (88).
Epigenetic programming is known to take place during fetal life, and microbial contact might have an impact on disease risk even before birth. This notion is consistent with observations according to which children whose mothers have lived in a farming environment during pregnancy, being thus more exposed to microbial antigens as compared to urban mothers, display a lower risk for asthma (155). This protective effect may be mediated by epigenetic mechanisms, since gestational exposure to Acinetobacter lwoffii F78, a microbe isolated from cowsheds, resulted in modulation of histone acetylation of key immune mediators and protected the offspring from the development of asthma-like disease in an experimental mouse model (156).
The novel evidence suggesting that microbial gut colonization may begin in utero (80) open new areas of research aiming both at understanding microbial epigenetic programming during fetal life and at devising maternal interventions to modify disease risk in the offspring, with potentially useful clinical implications.
Evidence on the role of maternal diet, early nutrition, and gut microbiota in the establishment of lifelong health and disease by determining epigenetic modifications that can be transgenerationally inherited has progressively spread over the last decades.
In addition to a qualitative assessment, aimed at identifying the molecular pathways and the bacterial patterns of gut colonization involved, a quantitative evaluation would help to establish the threshold levels of exposure to nutrients deficiencies or to other noxious environmental factors that can lead to clinically relevant epigenetic changes. Moreover, a better understanding of the underlying mechanisms may be fundamental to improve the approach to disease prevention. Nevertheless, further experimental research linking together technology advances and bioinformatics analyses is needed to identify new key markers for translational studies of disease prediction and treatment in the human population.
FI, SM, RF, LC, FC, GL, SAM, JN, SR, GRS, and FR contributed to draft the paper, critically reviewed its content, and approved the final version submitted for publication.
LC, FC, GL, SM, SAM, SR, GS, and FR have no conflict of interest to declare; FI has participated as a clinical investigator and/or consultant and/or speaker for Arla Food, Biogaia, Noos, Nestlè, and Nestlè Nutrition Institute, Wyeth, Danone. RF serves as a speaker for Biogaia and Plasmon Italia; JN is a Consultant for Infant Microbial Therapeutics and is in Scientific Advisory Board of Medela.
The authors would like to thank Tiziana Tursi for the help in editing and revising the manuscript.
The authors declare that no funding was received in support of the present manuscript and no sponsor was involved.
APC, antigen-presenting cell; ASD, autism-spectrum disorders; BPA, bisphenol A; CNS, central nervous system; CS, C-section; DHA: docosahexaenoic acid; DNA, deoxyribonucleic acid; GF, germ free; HBM, human breast milk; HDAC, histone deacetylase; IBD, intestinal bowel diseases; IBS, irritable bowel syndrome; IL, interleukin; IUGR, intrauterine growth restriction; LCPUFAs, long-chain polyunsaturated fatty acids; LDL, low-density lipoproteins; miRNAs, microRNAs; TC, total cholesterol; TCA, trichostatin A; TLR, toll-like receptor.
1. Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet (1986) 1:1077–81. doi:10.1016/S0140-6736(86)91340-1
2. Grice EA, Segre JA. The human microbiome: our second genome. Annu Rev Genomics Hum Genet (2012) 13:151–70. doi:10.1146/annurev-genom-090711-163814
3. Celluzzi A, Masotti A. How our other genome controls our epi-genome. Trends Microbiol (2016) 24:777–87. doi:10.1016/j.tim.2016.05.005
4. Bhutta ZA, Das JK, Rizvi A, Gaffey MF, Walker N, Horton S, et al. Evidence-based interventions for improvement of maternal and child nutrition: what can be done and at what cost? Lancet (2013) 382:452–77. doi:10.1016/S0140-6736(13)60996-4
5. Collado MC, Rautava S, Aakko J, Isolauri E, Salminen S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci Rep (2016) 6:23129. doi:10.1038/srep23129
6. Neu J. The microbiome during pregnancy and early postnatal life. Semin Fetal Neonatal Med (2016) 21:373–9. doi:10.1016/j.siny.2016.05.001
7. Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Bäckhed HK, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell (2012) 150:470–80. doi:10.1016/j.cell.2012.07.008
8. Mueller NT, Mao G, Bennet WL, Hourigan SK, Dominguez-Bello MG, Appel LJ, et al. Does vaginal delivery mitigate or strengthen the intergenerational association of overweight and obesity? Findings from the Boston Birth Cohort. Int J Obes (Lond) (2017) 41:497–501. doi:10.1038/ijo.2016.219
9. Torrazza RM, Ukhanova M, Wang X, Sharma R, Hudak ML, Neu J, et al. Intestinal microbial ecology and environmental factors affecting necrotizing enterocolitis. PLoS One (2013) 8:e83304. doi:10.1371/journal.pone.0083304
10. Greenwood C, Morrow AL, Lagomarcino AJ, Altaye M, Taft DH, Yu Z, et al. Early empiric antibiotic use in preterm infants is associated with lower bacterial diversity and higher relative abundance of Enterobacter. J Pediatr (2014) 165:23–9. doi:10.1016/j.jpeds.2014.01.010
11. Miller WB. The eukaryotic microbiome: origins and implications for fetal and neonatal life. Front Pediatr (2016) 4:96. doi:10.3389/fped.2016.00096
12. Gluckman PD, Hanson MA, Beedle AS, Spencer HG. Predictive adaptive responses in perspective. Trends Endocrinol Metab (2008) 19:109–10; author reply 112. doi:10.1016/j.tem.2008.02.002
13. MacLennan NK, James SJ, Melnyk S, Piroozi A, Jernigan S, Hsu JL, et al. Uteroplacental insufficiency alters DNA methylation, one-carbon metabolism, and histone acetylation in IUGR rats. Physiol Genomics (2004) 18:43–50. doi:10.1152/physiolgenomics.00042.2004
14. Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature (2010) 466:253–7. doi:10.1038/nature09165
15. Vanhees K, Vonhögen IGC, van Schooten FJ, Godschalk RWL. You are what you eat, and so are your children: the impact of micronutrients on the epigenetic programming of offspring. Cell Mol Life Sci (2014) 71:271–85. doi:10.1007/s00018-013-1427-9
16. Hollingsworth JW, Maruoka S, Boon K, Garantziotis S, Li Z, Tomfohr J, et al. In utero supplementation with methyl donors enhances allergic airway disease in mice. J Clin Invest (2008) 118:3462–9. doi:10.1172/JCI34378
17. Håberg SE, London SJ, Stigum H, Nafstad P, Nystad W. Folic acid supplements in pregnancy and early childhood respiratory health. Arch Dis Child (2009) 94:180–4. doi:10.1136/adc.2008.142448
18. Kolb AF, Petrie L. Folate deficiency enhances the inflammatory response of macrophages. Mol Immunol (2013) 54:164–72. doi:10.1016/j.molimm.2012.11.012
19. McCarty MF, Barroso-Aranda J, Contreras F. High-dose folate and dietary purines promote scavenging of peroxynitrite-derived radicals – clinical potential in inflammatory disorders. Med Hypotheses (2009) 73:824–34. doi:10.1016/j.mehy.2008.09.058
20. McKay JA, Groom A, Potter C, Coneyworth LJ, Ford D, Mathers JC, et al. Genetic and non-genetic influences during pregnancy on infant global and site specific DNA methylation: role for folate gene variants and vitamin B12. PLoS One (2012) 7:e33290. doi:10.1371/journal.pone.0033290
21. Fenech M. The role of folic acid and vitamin B12 in genomic stability of human cells. Mutat Res (2001) 475:57–67. doi:10.1016/S0027-5107(01)00069-0
22. Zeisel SH. Importance of methyl donors during reproduction. Am J Clin Nutr (2009) 89:673S–7S. doi:10.3945/ajcn.2008.26811D
23. Davison JM, Mellott TJ, Kovacheva VP, Blusztajn JK. Gestational choline supply regulates methylation of histone H3, expression of histone methyltransferases G9a (Kmt1c) and Suv39h1 (Kmt1a), and DNA methylation of their genes in rat fetal liver and brain. J Biol Chem (2009) 284:1982–9. doi:10.1074/jbc.M807651200
24. Wong CP, Rinaldi NA, Ho E. Zinc deficiency enhanced inflammatory response by increasing immune cell activation and inducing IL6 promoter demethylation. Mol Nutr Food Res (2015) 59:991–9. doi:10.1002/mnfr.201400761
25. Tomat AL, de los Ángeles Costa M, Arranz CT. Zinc restriction during different periods of life: influence in renal and cardiovascular diseases. Nutrition (2011) 27:392–8. doi:10.1016/j.nut.2010.09.010
26. Li C, Guo S, Gao J, Guo Y, Du E, Lv Z, et al. Maternal high-zinc diet attenuates intestinal inflammation by reducing DNA methylation and elevating H3K9 acetylation in the A20 promoter of offspring chicks. J Nutr Biochem (2015) 26:173–83. doi:10.1016/j.jnutbio.2014.10.005
27. Burris HH, Rifas-Shiman SL, Huh SY, Kleinman K, Litonjua AA, Oken E, et al. Vitamin D status and hypertensive disorders in pregnancy. Ann Epidemiol (2014) 24:399–403.e1. doi:10.1016/j.annepidem.2014.02.001
28. Orlov I, Rochel N, Moras D, Klaholz BP. Structure of the full human RXR/VDR nuclear receptor heterodimer complex with its DR3 target DNA. EMBO J (2012) 31:291–300. doi:10.1038/emboj.2011.445
29. Avila E, Díaz L, Barrera D, Halhali A, Méndez I, González L, et al. Regulation of vitamin D hydroxylases gene expression by 1,25-dihydroxyvitamin D3 and cyclic AMP in cultured human syncytiotrophoblasts. J Steroid Biochem Mol Biol (2007) 103:90–6. doi:10.1016/j.jsbmb.2006.07.010
30. Robinson CJ, Wagner CL, Hollis BW, Baatz JE, Johnson DD. Association of maternal vitamin D and placenta growth factor with the diagnosis of early onset severe preeclampsia. Am J Perinatol (2013) 30:167–72. doi:10.1055/s-0032-1322514
31. Anderson CM, Ralph JL, Johnson L, Scheett A, Wright ML, Taylor JY, et al. First trimester vitamin D status and placental epigenomics in preeclampsia among Northern Plains primiparas. Life Sci (2015) 129:10–5. doi:10.1016/j.lfs.2014.07.012
32. Khot V, Chavan-Gautam P, Joshi S. Proposing interactions between maternal phospholipids and the one carbon cycle: a novel mechanism influencing the risk for cardiovascular diseases in the offspring in later life. Life Sci (2015) 129:16–21. doi:10.1016/j.lfs.2014.09.026
33. Khot VV, Chavan-Gautam P, Mehendale S, Joshi SR. Variable methylation potential in preterm placenta: implication for epigenetic programming of the offspring. Reprod Sci (2017) 24:891–901. doi:10.1177/1933719116671001
34. Shen W, Wang C, Xia L, Fan C, Dong H, Deckelbaum RJ, et al. Epigenetic modification of the leptin promoter in diet-induced obese mice and the effects of N-3 polyunsaturated fatty acids. Sci Rep (2014) 4:5282. doi:10.1038/srep05282
35. Hirabara SM, Folador A, Fiamoncini J, Lambertucci RH, Rodrigues CF, Rocha MS, et al. Fish oil supplementation for two generations increases insulin sensitivity in rats. J Nutr Biochem (2013) 24:1136–45. doi:10.1016/j.jnutbio.2012.08.014
36. Li CCY, Maloney CA, Cropley JE, Suter CM. Epigenetic programming by maternal nutrition: shaping future generations. Epigenomics (2010) 2:539–49. doi:10.2217/epi.10.33
37. Fu Q, McKnight RA, Yu X, Wang L, Callaway CW, Lane RH. Uteroplacental insufficiency induces site-specific changes in histone H3 covalent modifications and affects DNA-histone H3 positioning in day 0 IUGR rat liver. Physiol Genomics (2004) 20:108–16. doi:10.1152/physiolgenomics.00175.2004
38. Zinkhan EK, Fu Q, Wang Y, Yu X, Callaway CW, Segar JL, et al. Maternal hyperglycemia disrupts histone 3 lysine 36 trimethylation of the IGF-1 gene. J Nutr Metab (2012) 2012:930364. doi:10.1155/2012/930364
39. Tosh DN, Fu Q, Callaway CW, McKnight RA, McMillen IC, Ross MG, et al. Epigenetics of programmed obesity: alteration in IUGR rat hepatic IGF1 mRNA expression and histone structure in rapid vs. delayed postnatal catch-up growth. Am J Physiol Gastrointest Liver Physiol (2010) 299:G1023–9. doi:10.1152/ajpgi.00052.2010
40. Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest (2008) 118:2316–24. doi:10.1172/JCI33655
41. Raychaudhuri N, Raychaudhuri S, Thamotharan M, Devaskar SU. Histone code modifications repress glucose transporter 4 expression in the intrauterine growth-restricted offspring. J Biol Chem (2008) 283:13611–26. doi:10.1074/jbc.M800128200
42. He ZX, Sun ZH, Yang WZ, Beauchemin KA, Tang SX, Zhou CS, et al. Effects of maternal protein or energy restriction during late gestation on immune status and responses to lipopolysaccharide challenge in postnatal young goats. J Anim Sci (2014) 92:4856–64. doi:10.2527/jas.2014-7904
43. de Sousa SM, Braz GRF, Freitas de Moura C, de Santana DF, Sellitti DF, Fernandes MP, et al. Oxidative injuries induced by maternal low-protein diet in female brainstem. Nutr Neurosci (2017) 1–9. doi:10.1080/1028415X.2017.1325974
44. Zheng J, Xiao X, Zhang Q, Wang T, Yu M, Xu J. Maternal low-protein diet modulates glucose metabolism and hepatic microRNAs expression in the early life of offspring †. Nutrients (2017) 9:205. doi:10.3390/nu9030205
45. Roseboom T, de Rooij S, Painter R. The Dutch famine and its long-term consequences for adult health. Early Hum Dev (2006) 82:485–91. doi:10.1016/j.earlhumdev.2006.07.001
46. Painter R, Osmond C, Gluckman P, Hanson M, Phillips D, Roseboom T. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG (2008) 115:1243–9. doi:10.1111/j.1471-0528.2008.01822.x
47. Aagaard-Tillery KM, Grove K, Bishop J, Ke X, Fu Q, McKnight R, et al. Developmental origins of disease and determinants of chromatin structure: maternal diet modifies the primate fetal epigenome. J Mol Endocrinol (2008) 41:91–102. doi:10.1677/JME-08-0025
48. Stupin J, Arabin B. Overweight and obesity before, during and after pregnancy. Geburtshilfe Frauenheilkd (2014) 74:639–45. doi:10.1055/s-0034-1368486
49. Strakovsky RS, Zhang X, Zhou D, Pan Y-X. The regulation of hepatic Pon1 by a maternal high-fat diet is gender specific and may occur through promoter histone modifications in neonatal rats. J Nutr Biochem (2014) 25:170–6. doi:10.1016/j.jnutbio.2013.09.016
50. Suter MA, Chen A, Burdine MS, Choudhury M, Harris RA, Lane RH, et al. A maternal high-fat diet modulates fetal SIRT1 histone and protein deacetylase activity in nonhuman primates. FASEB J (2012) 26:5106–14. doi:10.1096/fj.12-212878
51. Ford SP, Zhang L, Zhu M, Miller MM, Smith DT, Hess BW, et al. Maternal obesity accelerates fetal pancreatic beta-cell but not alpha-cell development in sheep: prenatal consequences. Am J Physiol Regul Integr Comp Physiol (2009) 297:R835–43. doi:10.1152/ajpregu.00072.2009
52. Oben JA, Patel T, Mouralidarane A, Samuelsson AM, Matthews P, Pombo J, et al. Maternal obesity programmes offspring development of non-alcoholic fatty pancreas disease. Biochem Biophys Res Commun (2010) 394:24–8. doi:10.1016/j.bbrc.2010.02.057
53. Hawkins PG, Morris KV. RNA and transcriptional modulation of gene expression. Cell Cycle (2008) 7:602–7. doi:10.4161/cc.7.5.5522
54. Cui J, Zhou B, Ross SA, Zempleni J. Nutrition, microRNAs, and human health. Adv Nutr (2017) 8:105–12. doi:10.3945/an.116.013839
55. Yan X, Huang Y, Wang H, Du M, Hess BW, Ford SP, et al. Maternal obesity induces sustained inflammation in both fetal and offspring large intestine of sheep. Inflamm Bowel Dis (2011) 17:1513–22. doi:10.1002/ibd.21539
56. Sasaki A, de Vega W, Sivanathan S, St-Cyr S, McGowan PO. Maternal high-fat diet alters anxiety behavior and glucocorticoid signaling in adolescent offspring. Neuroscience (2014) 272:92–101. doi:10.1016/j.neuroscience.2014.04.012
57. Leibowitz KL, Moore RH, Ahima RS, Stunkard AJ, Stallings VA, Berkowitz RI, et al. Maternal obesity associated with inflammation in their children. World J Pediatr (2012) 8:76–9. doi:10.1007/s12519-011-0292-6
58. Martinez-Arguelles DB, Papadopoulos V. Prenatal phthalate exposure: epigenetic changes leading to lifelong impact on steroid formation. Andrology (2016) 4:573–84. doi:10.1111/andr.12175
59. Ku HY, Su PH, Wen HJ, Sun HL, Wang CJ, Chen HY, et al. Prenatal and postnatal exposure to phthalate esters and asthma: a 9-year follow-up study of a Taiwanese birth cohort. PLoS One (2015) 10:e0123309. doi:10.1371/journal.pone.0123309
60. Jahreis S, Trump S, Bauer M, Bauer T, Thürmann L, Feltens R, et al. Maternal phthalate exposure promotes allergic airway inflammation over 2 generations through epigenetic modifications. J Allergy Clin Immunol (2017). doi:10.1016/j.jaci.2017.03.017
61. Bernal AJ, Jirtle RL. Epigenomic disruption: the effects of early developmental exposures. Birth Defects Res A Clin Mol Teratol (2010) 88:938–44. doi:10.1002/bdra.20685
62. Kitraki E, Nalvarte I, Alavian-Ghavanini A, Rüegg J. Developmental exposure to bisphenol A alters expression and DNA methylation of Fkbp5, an important regulator of the stress response. Mol Cell Endocrinol (2015) 417:191–9. doi:10.1016/j.mce.2015.09.028
63. Leung Y-K, Govindarajah V, Cheong A, Veevers J, Song D, Gear R, et al. Gestational high-fat diet and bisphenol A exposure heightens mammary cancer risk. Endocr Relat Cancer (2017) 24(7):365–78. doi:10.1530/ERC-17-0006
64. Koletzko B, Brands B, Poston L, Godfrey K, Demmelmair H; Early Nutrition Project. Early nutrition programming of long-term health. Proc Nutr Soc (2012) 71:371–8. doi:10.1017/S0029665112000596
65. Minekawa R, Takeda T, Sakata M, Hayashi M, Isobe A, Yamamoto T, et al. Human breast milk suppresses the transcriptional regulation of IL-1beta-induced NF-kappaB signaling in human intestinal cells. Am J Physiol Cell Physiol (2004) 287:C1404–11. doi:10.1152/ajpcell.00471.2003
66. Weng M, Walker WA. The role of gut microbiota in programming the immune phenotype. J Dev Orig Health Dis (2013) 4:203–14. doi:10.1017/S2040174412000712
67. Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science (2001) 291:881–4. doi:10.1126/science.291.5505.881
68. Sjögren YM, Tomicic S, Lundberg A, Böttcher MF, Björkstén B, Sverremark-Ekström E, et al. Influence of early gut microbiota on the maturation of childhood mucosal and systemic immune responses. Clin Exp Allergy (2009) 39:1842–51. doi:10.1111/j.1365-2222.2009.03326.x
69. Sharma AM, Staels B. Review: peroxisome proliferator-activated receptor gamma and adipose tissue – understanding obesity-related changes in regulation of lipid and glucose metabolism. J Clin Endocrinol Metab (2007) 92:386–95. doi:10.1210/jc.2006-1268
70. Mansoori A, Amini M, Kolahdooz F, Seyedrezazadeh E. Obesity and Pro12Ala polymorphism of peroxisome proliferator-activated receptor-gamma gene in healthy adults: a systematic review and meta-analysis. Ann Nutr Metab (2015) 67:104–18. doi:10.1159/000439285
71. Verier C, Meirhaeghe A, Bokor S, Breidenassel C, Manios Y, Molnár D, et al. Breast-feeding modulates the influence of the peroxisome proliferator-activated receptor-gamma (PPARG2) Pro12Ala polymorphism on adiposity in adolescents: the Healthy Lifestyle in Europe by Nutrition in Adolescence (HELENA) cross-sectional study. Diabetes Care (2010) 33:190–6. doi:10.2337/dc09-1459
72. Rossmeisl M, Medrikova D, van Schothorst EM, Pavlisova J, Kuda O, Hensler M, et al. Omega-3 phospholipids from fish suppress hepatic steatosis by integrated inhibition of biosynthetic pathways in dietary obese mice. Biochim Biophys Acta (2014) 1841:267–78. doi:10.1016/j.bbalip.2013.11.010
73. Owen CG, Whincup PH, Odoki K, Gilg JA, Cook DG. Infant feeding and blood cholesterol: a study in adolescents and a systematic review. Pediatrics (2002) 110:597–608. doi:10.1542/peds.110.3.597
74. Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and breastfeeding: collaborative reanalysis of individual data from 47 epidemiological studies in 30 countries, including 50302 women with breast cancer and 96973 women without the disease. Lancet (2002) 360:187–95. doi:10.1016/S0140-6736(02)09454-0
75. Jernström H, Lubinski J, Lynch HT, Ghadirian P, Neuhausen S, Isaacs C, et al. Breast-feeding and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst (2004) 96:1094–8. doi:10.1093/jnci/djh211
76. Wannous R, Bon E, Mahéo K, Goupille C, Chamouton J, Bougnoux P, et al. PPARβ mRNA expression, reduced by n-3 PUFA diet in mammary tumor, controls breast cancer cell growth. Biochim Biophys Acta (2013) 1831:1618–25. doi:10.1016/j.bbalip.2013.07.010
77. Willems R, Krych L, Rybicki V, Jiang P, Sangild PT, Shen RL, et al. Introducing enteral feeding induces intestinal subclinical inflammation and respective chromatin changes in preterm pigs. Epigenomics (2015) 7:553–65. doi:10.2217/epi.15.13
78. Funkhouser LJ, Bordenstein SR. Mom knows best: the universality of maternal microbial transmission. PLoS Biol (2013) 11:e1001631. doi:10.1371/journal.pbio.1001631
79. Perez-Muñoz ME, Arrieta M-C, Ramer-Tait AE, Walter J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: implications for research on the pioneer infant microbiome. Microbiome (2017) 5:48. doi:10.1186/s40168-017-0268-4
80. Bobitt JR, Ledger WJ. Unrecognized amnionitis and prematurity: a preliminary report. J Reprod Med (1977) 19:8–12.
81. Dammann O, Leviton A. Inflammation, brain damage and visual dysfunction in preterm infants. Semin Fetal Neonatal Med (2006) 11:363–8. doi:10.1016/j.siny.2006.02.003
82. Dammann O, Kuban KCK, Leviton A. Perinatal infection, fetal inflammatory response, white matter damage, and cognitive limitations in children born preterm. Ment Retard Dev Disabil Res Rev (2002) 8:46–50. doi:10.1002/mrdd.10005
83. Neu J. Developmental aspects of maternal-fetal, and infant gut microbiota and implications for long-term health. Matern Health Neonatol Perinatol (2015) 1:6. doi:10.1186/s40748-015-0007-4
84. Bergstrom A, Skov TH, Bahl MI, Roager HM, Christensen LB, Ejlerskov KT, et al. Establishment of intestinal microbiota during early life: a longitudinal, explorative study of a large cohort of Danish infants. Appl Environ Microbiol (2014) 80:2889–900. doi:10.1128/AEM.00342-14
85. Turroni F, Peano C, Pass DA, Foroni E, Severgnini M, Claesson MJ, et al. Diversity of Bifidobacteria within the infant gut microbiota. PLoS One (2012) 7:e36957. doi:10.1371/journal.pone.0036957
86. Jost T, Lacroix C, Braegger CP, Chassard C. New insights in gut microbiota establishment in healthy breast fed neonates. PLoS One (2012) 7:e44595. doi:10.1371/journal.pone.0044595
87. Cortese R, Lu L, Yu Y, Ruden D, Claud EC. Epigenome-microbiome crosstalk: a potential new paradigm influencing neonatal susceptibility to disease. Epigenetics (2016) 11:205–15. doi:10.1080/15592294.2016.1155011
88. Fofanova TY, Petrosino JF, Kellermayer R. Microbiome-epigenome interactions and the environmental origins of inflammatory bowel diseases. J Pediatr Gastroenterol Nutr (2016) 62:208–19. doi:10.1097/MPG.0000000000000950
89. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A (2010) 107:12204–9. doi:10.1073/pnas.0909122107
90. McLoughlin RM, Mills KHG. Influence of gastrointestinal commensal bacteria on the immune responses that mediate allergy and asthma. J Allergy Clin Immunol (2011) 127:1097–107. doi:10.1016/j.jaci.2011.02.012
91. Patel RM, Lin PW. Developmental biology of gut-probiotic interaction. Gut Microbes (2010) 1:186–95. doi:10.4161/gmic.1.3.12484
92. Sudo N, Sawamura S, Tanaka K, Aiba Y, Kubo C, Koga Y. The requirement of intestinal bacterial flora for the development of an IgE production system fully susceptible to oral tolerance induction. J Immunol (1997) 159:1739–45.
93. Kalliomäki M, Kirjavainen P, Eerola E, Kero P, Salminen S, Isolauri E. Distinct patterns of neonatal gut microflora in infants in whom atopy was and was not developing. J Allergy Clin Immunol (2001) 107:129–34. doi:10.1067/mai.2001.111237
94. Kalliomäki M, Collado MC, Salminen S, Isolauri E. Early differences in fecal microbiota composition in children may predict overweight. Am J Clin Nutr (2008) 87:534–8.
95. Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A (2010) 107:11971–5. doi:10.1073/pnas.1002601107
96. Salminen S, Gibson GR, McCartney AL, Isolauri E. Influence of mode of delivery on gut microbiota composition in seven year old children. Gut (2004) 53:1388–9. doi:10.1136/gut.2004.041640
97. Sevelsted A, Stokholm J, Bønnelykke K, Bisgaard H. Cesarean section and chronic immune disorders. Pediatrics (2015) 135:e92–8. doi:10.1542/peds.2014-0596
98. Turta O, Rautava S. Antibiotics, obesity and the link to microbes – what are we doing to our children? BMC Med (2016) 14:57. doi:10.1186/s12916-016-0605-7
99. Semic-Jusufagic A, Belgrave D, Pickles A, Telcian AG, Bakhsoliani E, Sykes A, et al. Assessing the association of early life antibiotic prescription with asthma exacerbations, impaired antiviral immunity, and genetic variants in 17q21: a population-based birth cohort study. Lancet Respir Med (2014) 2:621–30. doi:10.1016/S2213-2600(14)70096-7
100. Candon S, Perez-Arroyo A, Marquet C, Valette F, Foray A-P, Pelletier B, et al. Antibiotics in early life alter the gut microbiome and increase disease incidence in a spontaneous mouse model of autoimmune insulin-dependent diabetes. PLoS One (2015) 10:e0125448. doi:10.1371/journal.pone.0125448
101. Hviid A, Svanström H, Frisch M. Antibiotic use and inflammatory bowel diseases in childhood. Gut (2011) 60:49–54. doi:10.1136/gut.2010.219683
102. Knoll RL, Forslund K, Kultima JR, Meyer CU, Kullmer U, Sunagawa S, et al. Gut microbiota differs between children with inflammatory bowel disease and healthy siblings in taxonomic and functional composition: a metagenomic analysis. Am J Physiol Gastrointest Liver Physiol (2017) 312:G327–39. doi:10.1152/ajpgi.00293.2016
103. Falony G, Vlachou A, Verbrugghe K, De Vuyst L. Cross-feeding between Bifidobacterium longum BB536 and acetate-converting, butyrate-producing colon bacteria during growth on oligofructose. Appl Environ Microbiol (2006) 72:7835–41. doi:10.1128/AEM.01296-06
104. Berni Canani R, Di Costanzo M, Leone L. The epigenetic effects of butyrate: potential therapeutic implications for clinical practice. Clin Epigenetics (2012) 4:4. doi:10.1186/1868-7083-4-4
105. Rossi M, Amaretti A, Raimondi S. Folate production by probiotic bacteria. Nutrients (2011) 3:118–34. doi:10.3390/nu3010118
106. Jin Y, Chen X, Podolsky R, Hopkins D, Makala LHC, Muir A, et al. APC dysfunction is correlated with defective suppression of T cell proliferation in human type 1 diabetes. Clin Immunol (2009) 130:272–9. doi:10.1016/j.clim.2008.10.005
107. Hu Y, Jin P, Peng J, Zhang X, Wong FS, Wen L. Different immunological responses to early-life antibiotic exposure affecting autoimmune diabetes development in NOD mice. J Autoimmun (2016) 72:47–56. doi:10.1016/j.jaut.2016.05.001
108. Hu Y, Peng J, Tai N, Hu C, Zhang X, Wong FS, et al. Maternal antibiotic treatment protects offspring from diabetes development in nonobese diabetic mice by generation of tolerogenic APCs. J Immunol (2015) 195:4176–84. doi:10.4049/jimmunol.1500884
109. Evensen KAI, Steinshamn S, Tjønna AE, Stølen T, Høydal MA, Wisløff U, et al. Effects of preterm birth and fetal growth retardation on cardiovascular risk factors in young adulthood. Early Hum Dev (2009) 85:239–45. doi:10.1016/j.earlhumdev.2008.10.008
110. Singhal A, Fewtrell M, Cole TJ, Lucas A. Low nutrient intake and early growth for later insulin resistance in adolescents born preterm. Lancet (2003) 361:1089–97. doi:10.1016/S0140-6736(03)12895-4
111. Mueller NT, Whyatt R, Hoepner L, Oberfield S, Dominguez-Bello MG, Widen EM, et al. Prenatal exposure to antibiotics, cesarean section and risk of childhood obesity. Int J Obes (2015) 39:665–70. doi:10.1038/ijo.2014.180
112. Kuhle S, Tong OS, Woolcott CG. Association between caesarean section and childhood obesity: a systematic review and meta-analysis. Obes Rev (2015) 16:295–303. doi:10.1111/obr.12267
113. Korpela K, Zijlmans MAC, Kuitunen M, Kukkonen K, Savilahti E, Salonen A, et al. Childhood BMI in relation to microbiota in infancy and lifetime antibiotic use. Microbiome (2017) 5:26. doi:10.1186/s40168-017-0245-y
114. Lemas DJ, Yee S, Cacho N, Miller D, Cardel M, Gurka M, et al. Exploring the contribution of maternal antibiotics and breastfeeding to development of the infant microbiome and pediatric obesity. Semin Fetal Neonatal Med (2016) 21:406–9. doi:10.1016/j.siny.2016.04.013
115. Paun A, Danska JS. Modulation of type 1 and type 2 diabetes risk by the intestinal microbiome. Pediatr Diabetes (2016) 17:469–77. doi:10.1111/pedi.12424
116. Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell (2014) 158:705–21. doi:10.1016/j.cell.2014.05.052
117. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science (2013) 341:1241214. doi:10.1126/science.1241214
118. Remely M, Aumueller E, Jahn D, Hippe B, Brath H, Haslberger AG. Microbiota and epigenetic regulation of inflammatory mediators in type 2 diabetes and obesity. Benef Microbes (2014) 5:33–43. doi:10.3920/BM2013.006
119. Kumar H, Lund R, Laiho A, Lundelin K, Ley RE, Isolauri E, et al. Gut microbiota as an epigenetic regulator: pilot study based on whole-genome methylation analysis. MBio (2014) 5:e2113–4. doi:10.1128/mBio.02113-14
120. Mayer EA. Gut feelings: the emerging biology of gut-brain communication. Nat Rev Neurosci (2011) 12:453–66. doi:10.1038/nrn3071
121. O’ Mahony SM, Stilling RM, Dinan TG, Cryan JF. The microbiome and childhood diseases: focus on brain-gut axis. Birth Defects Res C Embryo Today (2015) 105:296–313. doi:10.1002/bdrc.21118
122. Yi P, Li L. The germfree murine animal: an important animal model for research on the relationship between gut microbiota and the host. Vet Microbiol (2012) 157:1–7. doi:10.1016/j.vetmic.2011.10.024
123. Stilling RM, Dinan TG, Cryan JF. Microbial genes, brain & behaviour – epigenetic regulation of the gut-brain axis. Genes Brain Behav (2014) 13:69–86. doi:10.1111/gbb.12109
124. Maqsood R, Stone TW. The gut-brain axis, BDNF, NMDA and CNS disorders. Neurochem Res (2016) 41:2819–35. doi:10.1007/s11064-016-2039-1
125. Heijtz RD, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A, et al. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci U S A (2011) 108:3047–52. doi:10.1073/pnas.1010529108
126. Carrion VG, Wong SS. Can traumatic stress alter the brain? Understanding the implications of early trauma on brain development and learning. J Adolesc Health (2012) 51:S23–8. doi:10.1016/j.jadohealth.2012.04.010
127. Dinan TG, Cryan JF. Regulation of the stress response by the gut microbiota: implications for psychoneuroendocrinology. Psychoneuroendocrinology (2012) 37:1369–78. doi:10.1016/j.psyneuen.2012.03.007
128. Clarke G, Grenham S, Scully P, Fitzgerald P, Moloney RD, Shanahan F, et al. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol Psychiatry (2013) 18:666–73. doi:10.1038/mp.2012.77
129. Foster JA, McVey Neufeld K-A. Gut-brain axis: how the microbiome influences anxiety and depression. Trends Neurosci (2013) 36:305–12. doi:10.1016/j.tins.2013.01.005
130. Desbonnet L, Clarke G, Shanahan F, Dinan TG, Cryan JF. Microbiota is essential for social development in the mouse. Mol Psychiatry (2014) 19:146–8. doi:10.1038/mp.2013.65
131. Collins S, Reid G. Distant site effects of ingested prebiotics. Nutrients (2016) 8:523. doi:10.3390/nu8090523
132. Savignac HM, Kiely B, Dinan TG, Cryan JF. Bifidobacteria exert strain-specific effects on stress-related behavior and physiology in BALB/c mice. Neurogastroenterol Motil (2014) 26:1615–27. doi:10.1111/nmo.12427
133. Keunen K, van Elburg RM, van Bel F, Benders MJNL. Impact of nutrition on brain development and its neuroprotective implications following preterm birth. Pediatr Res (2015) 77:148–55. doi:10.1038/pr.2014.171
134. Deverman BE, Patterson PH. Cytokines and CNS development. Neuron (2009) 64:61–78. doi:10.1016/j.neuron.2009.09.002
135. Collins LM, Toulouse A, Connor TJ, Nolan YM. Contributions of central and systemic inflammation to the pathophysiology of Parkinson’s disease. Neuropharmacology (2012) 62:2154–68. doi:10.1016/j.neuropharm.2012.01.028
136. Lampron A, Elali A, Rivest S. Innate immunity in the CNS: redefining the relationship between the CNS and its environment. Neuron (2013) 78:214–32. doi:10.1016/j.neuron.2013.04.005
137. Soliman ML, Combs CK, Rosenberger TA. Modulation of inflammatory cytokines and mitogen-activated protein kinases by acetate in primary astrocytes. J Neuroimmune Pharmacol (2013) 8:287–300. doi:10.1007/s11481-012-9426-4
138. Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature (2011) 477:90–4. doi:10.1038/nature10357
139. Li Q, Han Y, Dy ABC, Hagerman RJ. The gut microbiota and autism spectrum disorders. Front Cell Neurosci (2017) 11:120. doi:10.3389/fncel.2017.00120
140. Inoue R, Sakaue Y, Sawai C, Sawai T, Ozeki M, Romero-Pérez GA, et al. A preliminary investigation on the relationship between gut microbiota and gene expressions in peripheral mononuclear cells of infants with autism spectrum disorders. Biosci Biotechnol Biochem (2016) 80:2450–8. doi:10.1080/09168451.2016.1222267
141. Son JS, Zheng LJ, Rowehl LM, Tian X, Zhang Y, Zhu W, et al. Comparison of fecal microbiota in children with autism spectrum disorders and neurotypical siblings in the Simons simplex collection. PLoS One (2015) 10:e0137725. doi:10.1371/journal.pone.0137725
142. Gondalia SV, Palombo EA, Knowles SR, Cox SB, Meyer D, Austin DW. Molecular characterisation of gastrointestinal microbiota of children with autism (with and without gastrointestinal dysfunction) and their neurotypical siblings. Autism Res (2012) 5:419–27. doi:10.1002/aur.1253
143. Keszthelyi D, Troost FJ, Masclee AA. Irritable bowel syndrome: methods, mechanisms, and pathophysiology. Methods to assess visceral hypersensitivity in irritable bowel syndrome. Am J Physiol Gastrointest Liver Physiol (2012) 303:G141–54. doi:10.1152/ajpgi.00060.2012
144. Tran L, Chaloner A, Sawalha AH, Greenwood Van-Meerveld B. Importance of epigenetic mechanisms in visceral pain induced by chronic water avoidance stress. Psychoneuroendocrinology (2013) 38:898–906. doi:10.1016/j.psyneuen.2012.09.016
145. Jeffery IB, O’Toole PW, Öhman L, Claesson MJ, Deane J, Quigley EMM, et al. An irritable bowel syndrome subtype defined by species-specific alterations in faecal microbiota. Gut (2012) 61:997–1006. doi:10.1136/gutjnl-2011-301501
146. Shankar V, Agans R, Holmes B, Raymer M, Paliy O. Do gut microbial communities differ in pediatric IBS and health? Gut Microbes (2013) 4:347–52. doi:10.4161/gmic.24827
147. Mckernan DP, Fitzgerald P, Dinan TG, Cryan JF. The probiotic Bifidobacterium infantis 35624 displays visceral antinociceptive effects in the rat. Neurogastroenterol Motil (2010) 22:1029–35, e268. doi:10.1111/j.1365-2982.2010.01520.x
148. Clarke G, Cryan JF, Dinan TG, Quigley EM. Review article: probiotics for the treatment of irritable bowel syndrome – focus on lactic acid bacteria. Aliment Pharmacol Ther (2012) 35:403–13. doi:10.1111/j.1365-2036.2011.04965.x
149. Yang I, Corwin EJ, Brennan PA, Jordan S, Murphy JR, Dunlop A. The infant microbiome. Nurs Res (2016) 65:76–88. doi:10.1097/NNR.0000000000000133
150. Patel R, DuPont HL. New approaches for bacteriotherapy: prebiotics, new-generation probiotics, and synbiotics. Clin Infect Dis (2015) 60:S108–21. doi:10.1093/cid/civ177
151. Kalliomäki M, Salminen S, Arvilommi H, Kero P, Koskinen P, Isolauri E. Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet (2001) 357:1076–9. doi:10.1016/S0140-6736(00)04259-8
152. Kalliomäki M, Salminen S, Poussa T, Isolauri E. Probiotics during the first 7 years of life: a cumulative risk reduction of eczema in a randomized, placebo-controlled trial. J Allergy Clin Immunol (2007) 119:1019–21. doi:10.1016/j.jaci.2006.12.608
153. Rautava S, Kainonen E, Salminen S, Isolauri E. Maternal probiotic supplementation during pregnancy and breast-feeding reduces the risk of eczema in the infant. J Allergy Clin Immunol (2012) 130:1355–60. doi:10.1016/j.jaci.2012.09.003
154. Panduru M, Panduru NM, Sălăvăstru CM, Tiplica G-S. Probiotics and primary prevention of atopic dermatitis: a meta-analysis of randomized controlled studies. J Eur Acad Dermatol Venereol (2015) 29:232–42. doi:10.1111/jdv.12496
155. Douwes J, Cheng S, Travier N, Cohet C, Niesink A, McKenzie J, et al. Farm exposure in utero may protect against asthma, hay fever and eczema. Eur Respir J (2008) 32:603–11. doi:10.1183/09031936.00033707
Keywords: epigenetic regulation, fetal programming, overnutrition, undernutrition, micronutrients, human milk, microbiome, disease origin
Citation: Indrio F, Martini S, Francavilla R, Corvaglia L, Cristofori F, Mastrolia SA, Neu J, Rautava S, Russo Spena G, Raimondi F and Loverro G (2017) Epigenetic Matters: The Link between Early Nutrition, Microbiome, and Long-term Health Development. Front. Pediatr. 5:178. doi: 10.3389/fped.2017.00178
Received: 08 June 2017; Accepted: 07 August 2017;
Published: 22 August 2017
Edited by:
Richard Eugene Frye, Arkansas Children’s Hospital Research Institute, United StatesReviewed by:
Fernando Lizcano, Universidad de La Sabana, ColombiaCopyright: © 2017 Indrio, Martini, Francavilla, Corvaglia, Cristofori, Mastrolia, Neu, Rautava, Russo Spena, Raimondi and Loverro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Martini, c2lsdmlhLm1hcnRpbmk0QGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.