 Emily Stenke
Emily Stenke Billy Bourke1,2
Billy Bourke1,2 Ulla Knaus
Ulla Knaus
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pediatr. , 16 June 2017
Sec. Pediatric Gastroenterology, Hepatology and Nutrition
Volume 5 - 2017 | https://doi.org/10.3389/fped.2017.00141
This article is part of the Research Topic Pediatric Inflammatory Bowel Diseases: Looking to the Future View all 13 articles
Crohn’s disease (CD) is a lifelong inflammatory bowel disease with a rapidly rising incidence in the pediatric population. A common complication of CD is the development of fibrotic strictures, which may be present at initial diagnosis or develop many years later. Clinical presentation depends on stricture location and degree of obstruction, and strictures frequently contain a mixture of inflammatory and fibrotic tissue. Histological examination of Crohn’s strictures shows thickening of the muscular layers and the submucosa, where increased collagen deposition by activated myofibroblasts is concentrated around islands of smooth muscle cells and at the superficial margin of the muscularis propria. No antifibrotic therapies for Crohn’s strictures exist. Profibrotic transforming growth factor-β (TGFβ)/bone morphogenetic protein signaling stimulates myofibroblast differentiation and extracellular matrix deposition. Understanding and targeting TGFβ1 downstream signaling is the main focus of current research, raising the possibility of specific antifibrotic therapy in CD becoming available in the future.
The incidence of Crohn’s disease (CD) is rapidly rising in the pediatric population (1). Crohn’s inflammation is transmural and can occur throughout the gastrointestinal tract, unlike ulcerative colitis where mucosal inflammation is confined to the colon. CD is phenotyped according to disease behavior (inflammatory/penetrating/stricturing) and location using the Paris classification for pediatric patients (2), a modification of the Montreal classification (3). Fibrotic narrowing of the intestinal tract (stricturing disease) is present in approximately 10–17% of children at diagnosis (4), affecting up to 40% by 10 years after diagnosis (5–7). Strictures occur predominantly in the ileum, reflecting the common distribution of inflammation, but can arise throughout the digestive tract (8). Presenting features vary from overt intestinal obstruction to subacute and non-specific symptoms. The conventional conceptual framework of the pathobiology of fibrosis in CD is one of chronic inflammation leading to ongoing frustrated attempts at healing with formation of disorganized tissue. If this occurs circumferentially in a relatively narrow diameter organ such as the small intestine, the result is a fibrotic stricture. Here, we will describe current knowledge and recent advances regarding the diagnosis and pathogenesis of intestinal fibrosis and will review potential future therapies.
The clinical presentation of stricturing disease is highly variable, depending on the location and degree of obstruction. Presentations range from non-specific symptoms including abdominal discomfort, poor appetite, energy and/or growth, to overt obstruction with abdominal pain, vomiting, and reduced bowel movements. Strictures may be inflammatory, fibrotic, or more commonly a combination of both (8). Most strictures are in the small bowel, inaccessible to standard endoscopy, and even when accessible, endoscopy can only provide information (visual, histological) on the mucosa, while the fibrotic collagen deposition occurs submucosally. Capsule endoscopy can provide visual information but no tissue samples and is contraindicated in the presence of suspected strictures due to the risk of retention. Therefore, clinicians usually rely on radiological tools for the diagnosis of stenotic disease.
A fibrotic stricture is inferred by the presence of a narrowed lumen with proximal dilation. Computed tomography (CT) and magnetic resonance enterogram (MRE) are preferred to X-rays with enteral contrast [small-bowel follow through (SBFT)] as they allow assessment of extraintestinal as well as intestinal complications of CD and have similar sensitivity and accuracy. CT and MRE permit a transmural assessment of the bowel wall, enabling the classification of strictures as inflammatory, fibrotic, or mixed. Inflammation is suggested by avid enhancement and mesenteric inflammation (hypervascularity, fat stranding), whereas fibrosis is characterized by a thickened bowel wall with a featureless appearance, minimal or no enhancement and the absence of mesenteric inflammation. MRE has comparable (9–12) or superior (13) sensitivity and specificity to CT for the detection of fibrosis in the presence of inflammation in adult and pediatric patients, while MRE has greater sensitivity for the detection of fibrosis alone (10). A recent prospective pediatric study demonstrated 73% sensitivity and 81% specificity for the detection of strictures using MRE compared to 42% sensitivity and 68% specificity for CT enterograms (12). Due to its lack of ionizing radiation and its capacity to provide accurate diagnosis of both intra- and extraintestinal complications, MRE is the preferred modality in the pediatric population, although technical limitations (institutional access, need for general anesthesia) ensure a retained role for CT and SBFT. Recent advances in ultrasound elastography as a measurement of tissue stiffness have shown promising results in the differentiation between inflammatory and fibrotic strictures (14, 15), but have not yet reached routine clinical practice. As sensitivity does not reach 100% with any modality, clinical judgment is required in conjunction with imaging findings to decide about surgical exploration/intervention.
Fibrosis is defined as the permanent and abnormal deposition of extracellular matrix (ECM), primarily collagen, within tissues, resulting in a distortion of structure and impeding normal tissue and organ function. It is understood to be an aberrant response to ongoing inflammation, where tissue remodeling in response to injury, through ECM deposition and subsequent breakdown, becomes self-perpetuating. In the normal small bowel, the mucosa consists of a single layer of epithelium, lamina propria, and basement membrane. Deep to the intact mucosa is the muscularis mucosa, a thin layer of smooth muscle cells, and then the submucosa. The submucosa is a loose connective tissue layer with fibroblasts as main cell type within an ECM traversed by blood vessels and nerves (16). The ECM is a complex biochemical structure where collagen types I and III predominate (17). Deep to the submucosa is the muscularis propria with its inner circular and outer longitudinal layers of smooth muscle cells. The collagen strands of the submucosa tend to be concentrated at its border with the muscularis propria.
Histological examination of CD strictures reveals abnormalities in both the submucosal space and muscular layers (Figure 1). The submucosa is increased in volume and density, with increased collagen deposition being concentrated around islands of smooth muscle cells and at the superficial margin of the muscularis propria. Studies have shown an increase in total protein and collagen content, especially collagen subtypes I, III, and V in these strictures. Although increased in absolute terms, the relative proportions of types I and III collagen appear to be comparable between strictures and healthy intestine, and although the proportion of type V collagen is relatively amplified, type I collagen remains dominant (16, 17). Myofibroblasts, differentiated from fibroblasts, are the primary source of ECM production and secretion. Increased numbers of local fibroblasts and myofibroblasts in fibrosis have been attributed to a variety of mechanisms including the proliferation of existing fibroblasts in the local area (18), the induction of epithelial-to-mesenchymal transition (19), the recruitment and differentiation of bone marrow-derived fibrocytes (20), as well as endothelial-to-mesenchymal transition (21), but the relative contribution of each process is unknown. Thickening of the muscularis mucosa and muscularis propria as well as smooth muscle cell proliferation within the submucosa itself have been observed. In some instances, the proliferation of smooth muscle cells within the submucosa can be so pronounced that it results in the obliteration of the submucosa (22). The combined overall effect of increased ECM deposition and muscular hypertrophy is transmural thickening and stiffening, which when occurring circumferentially causes narrowing and obstruction of the intestinal lumen.
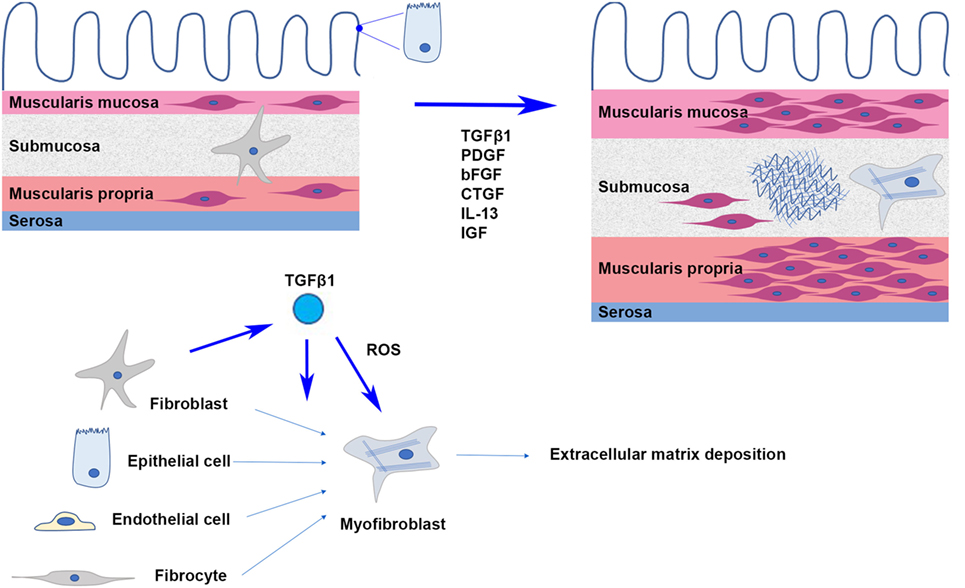
Figure 1. In intestinal fibrosis, hypertrophy and hyperplasia of smooth muscle cells cause thickening of the muscularis mucosa and muscularis propria; smooth muscle cells proliferate in the thickened submucosa, where activated myofibroblasts secrete extracellular matrix proteins.
Various growth factors and cytokines have been implicated in the development of fibrosis including IL-13, platelet-derived growth factor (PDGF), connective tissue growth factor, basic fibroblast growth factor, insulin-like growth factor, bone morphogenetic proteins (BMPs), and transforming growth factor-β (TGFβ) (23). The TGFβ family is secreted by a wide variety of cells throughout the body and its effects are highly varied and complex. As well as being responsible for cell proliferation and differentiation, anti-inflammatory control, wound healing, tumor suppression in healthy tissues and cancer progression in neoplastic tissues, TGFβ is the primary cytokine driving the development of fibrosis in tissues throughout the body. Differences in TGFβ subtype, receptors, cofactors, and signaling pathways allow this group of cytokines to display such versatility (24). TGFβ1 is the most common subtype and its role in the development of tissue fibrosis through the recruitment of fibroblasts, transdifferentiation to myofibroblasts, and stimulation of ECM secretion has been convincingly demonstrated in many organs including in intestinal fibrosis (25–28).
TGFβ1 is a homodimeric signaling molecule produced by myofibroblasts and inflammatory cells (e.g., macrophages), platelets, and parenchymal cells during the hemostasis and inflammation phases of tissue injury and healing. This para- and autocrine signaling is anti-inflammatory and supports wound healing but drives fibrosis. The TGFβ receptor is a transmembrane complex of two dimers: two type I receptors (ALK5, also called TGFBR1 or TβTI) and two type II receptors (TGFBR2); both are serine/threonine kinases and binding of TGFβ results in phosphorylation of ALK5 by TGFBR2 with subsequent binding and phosphorylation of downstream signaling proteins SMAD2 and SMAD3 by ALK5 (Figure 2). Once phosphorylated, SMAD2 and SMAD3 form a trimer with SMAD4, which translocates to the nucleus and binds to target DNA. The transcriptional targeting, nuclear translocation, and longevity of these SMAD transcription factors is modulated by the binding of a wide variety of effector molecules. SMAD2/3 binding to ALK5 is antagonized by SMAD7. SMAD7 antisense oligonucleotides (mongersen) showed promise in the treatment of inflammatory CD by restoring the TGFβ1–SMAD2/3 pathway (29); no associated increase in fibrosis has been observed but longer term follow-up is required. Several non-canonical TGFβ signaling pathways have been described (30). Phosphorylated TGFBR1 and TGFBR2 lead to activation of mitogen-activated protein kinases (MAPKs), which regulate multiple pathways including those leading to transcription. MAPKs also regulate the canonical TGFβ pathway by phosphorylating active SMAD to promote its proteasomal degradation. TGFβ activates phosphoinositide 3-kinase, leading to downstream activation of Akt, mTOR, and upregulation of protein translation. Akt interacts directly with SMAD3 to prevent its activation and indirectly inhibits SMAD-mediated transcription through phosphorylation of FoxO transcription factors, thus blocking their translocation to the nucleus. A more detailed review of the complexity of TGFβ signaling can be found in the study by Massague (24).
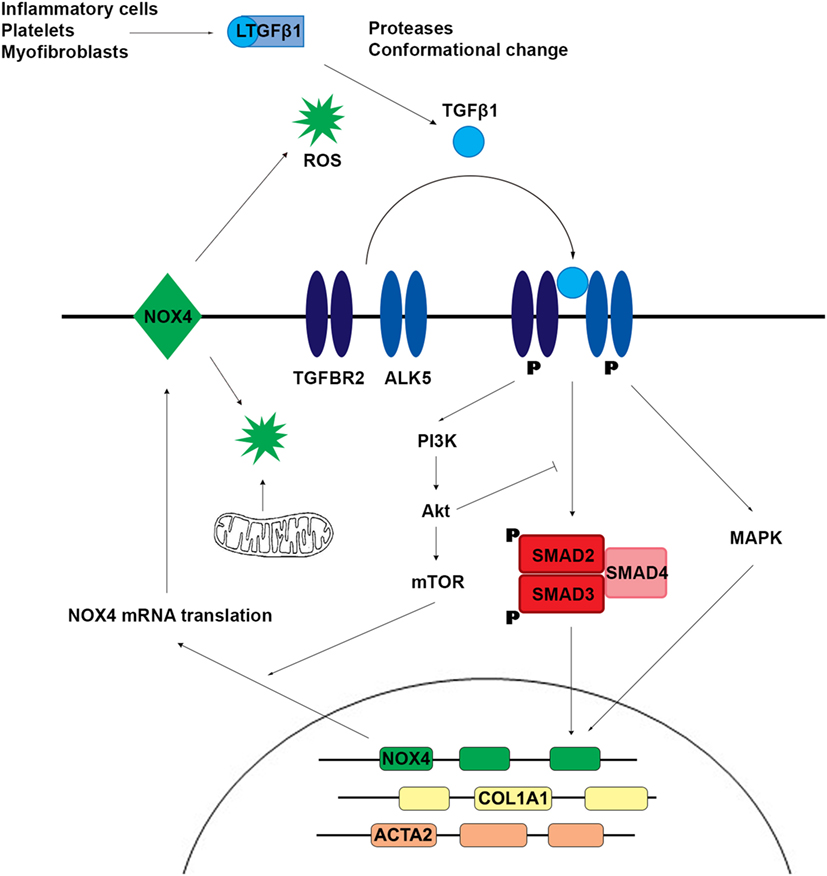
Figure 2. TGFβ1 signaling induces SMAD2 and SMAD3 phosphorylation and activation of non-canonical mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) pathways, which upregulate the expression of profibrotic genes including NADPH oxidase 4 (NOX4). Reactive oxygen species (ROS) generated by NOX4 and/or mitochondria promote further release of active TGFβ1.
The pleiotropic and multifunctional effects of TGFβ1 signaling make it an unattractive therapeutic target in fibrosis. Substantial effort has been directed at characterizing downstream signaling pathways of TGFβ where fibrosis-specific pharmacologic intervention might be achievable. Reactive oxygen species (ROS) have been implicated as mediators of the profibrotic effect of TGFβ1. ROS can be produced non-enzymatically (e.g., by radiation, toxic chemicals) or enzymatically. Most cellular ROS is produced in the mitochondria during cellular respiration. Other ROS sources include the cytochrome P450 family, cyclooxygenases, peroxisomal oxidases, xanthine oxidoreductase, and the family of NADPH oxidases (NOX). The NOX family is unique as its only known biological role is ROS production, and NOX enzymes appear to be intimately involved in fibrogenesis. NADPH oxidase 4 (NOX4) is a constitutively active, transcriptionally regulated producer of H2O2. An accumulating body of evidence suggests that TGFβ1 upregulates NOX4 expression and that NOX4, through the production of H2O2, promotes myofibroblast differentiation and secretion of ECM proteins (31–33). In a rat model of renal fibrosis and in cell-based assay systems, TGFβ1 stimulation was associated with increased NOX4 expression and H2O2 production, while reduced NOX4 expression by siRNA-mediated knockdown decreased ROS production and expression of profibrotic proteins including collagen, α-SMA, and fibronectin (34). These findings have been replicated using cell-based assays, mouse models, and patient samples in the lung (35–38), skin (39), and liver (40–42). A study by Jung et al. using fibroblasts and a mouse model of renal fibrosis suggested that NOX4 upregulation by TGFβ1 depends on activation of SMAD2 and Akt (43). Latella and coworkers reported that SMAD3 was necessary for the development of intestinal fibrosis in a chronic TNBS model (44), suggesting that NOX4 mediates the effect of TGFβ1 through canonical and non-canonical pathways. Somewhat contradictorily, NOX4 was protective in a renal fibrosis model (45), and there is evidence from mice and patient data that NOX4 may be protective against atherosclerosis (46–49). Another mouse model showed preserved myofibroblast differentiation but impaired wound healing in NOX4−/− mice (50), suggesting that the requirement of NOX4 for myofibroblast differentiation and collagen secretion may be tissue and/or context dependent (acute versus chronic insult). Despite the growing evidence for a fundamental role of NOX4 in fibrosis, only limited studies exist for the intestine. Hotta and coworkers demonstrated a reduction in TGFβ1-dependent collagen I production by murine intestinal myofibroblasts treated with a pan-NOX inhibitor or NOX4 siRNA (51). Data from RNA-Seq analysis of intestinal fibroblasts showed variable upregulation of NOX4 transcription in three patients with CD compared to three healthy controls (52). BMPs belong to the TGFβ superfamily and signal via phosphorylation and complex formation of SMAD 1, 5, and 8 (53). BMP7 may protect against colitis and prevent fibrosis by antagonizing TGFβ signaling (54–56). Angiopoietin-like protein 2 (ANGPTL2) modulates BMP signaling and initial studies suggest that organ damage in ANGPTL2 knockdown mice is linked to NOX4 (57, 58).
TGFβ1 signaling stimulated the production of mitochondrial ROS (mtROS), possibly by inhibition of complex III and IV and via the mTOR signaling pathway, with subsequent increase in profibrotic gene expression. Reduction of mtROS by the antioxidant MitoQ reduced TGFβ1 expression, SMAD2 and SMAD3 activation, and collagen deposition in a liver fibrosis model (59). mtROS and NOX4 may interact through a positive feedback loop to promote TGFβ1-driven fibrosis. NOX4-derived ROS caused mitochondrial dysfunction and increased mtROS, while mtROS amplified the TGFβ1-mediated increase in NOX4 expression (60). Additionally, increased TGFβ1 inhibited the antioxidant response, thereby exacerbating the prooxidant shift and further driving fibrosis (61).
Key enzymes in cross-linking and stabilizing the network of collagen fibrils are H2O2-generating lysyl oxidase (LOX) and the lysyl oxidase-like proteins (LOXL1–4), which are copper-dependent amine oxidases that oxidatively modify the ε-amino group of lysine side chains in collagen and elastin for formation of inter- and intrachain cross-links. Clinical and animal-based studies in the liver and myocardium demonstrate that LOXs promote tissue stiffening through cross-linking of existing collagen and elastin fibrils and that inhibition of LOXL2 may inhibit and even reverse fibrosis (62–65). Studies in rat and human lung fibroblasts, human trabecular cells, and human osteoblasts suggest that TGFβ1 upregulates LOXs, which in turn modify the actions of TGFβ1 (66–69). It is important to note that TGFβ1 is secreted and stored extracellularly bound to a latent TGF-β-binding protein and a latency-associated peptide. This inactive complex is bound to the ECM via integrins, and active TGFβ is released by protease cleavage or conformational changes caused by increased stiffness of the ECM (61, 70). This provides a possible explanation for TGFβ1’s effects in the absence of active inflammation (71, 72) as well as the finding that a stiff matrix is required for myofibroblast differentiation (62, 73).
Although improved management of CD inflammation by anti-TNFα therapy (infliximab, adalimumab) appears to reduce the rate of stricture development (74), there is currently no medical therapy directly targeting fibrosis in CD. Patients whose strictures fail to respond to anti-inflammatory therapies (aimed at any inflammation and edema coexisting with fibrosis) require surgical intervention. Endoscopic balloon dilatation (EBD) is an option for single, short, and uncomplicated strictures accessible by endoscopy, for instance, stricture recurrence at ileocecal anastomoses. Although technical success rates are high, with a low rate of complications, retrospective data from adult patients demonstrate that 42–70% of patients will require repeat EBD or surgical intervention by 5 years (75–77). Pediatric data are limited, but support the feasibility and safety of EBD (78, 79). Given the predominance of mixed fibrotic/inflammatory strictures over purely fibrotic strictures, intrastricture injection of corticosteroids has been proposed as an adjunct to balloon dilation. A prospective randomized control trial including nine adult patients showed no difference in stricture recurrence rates at 1 year (80), while a prospective RCT including 29 pediatric patients showed earlier stricture recurrence in patients treated with placebo (79). Due to the small trials and number of patients studied, the benefit of intralesional steroid injection has not been confirmed (81). Surgery is the mainstay of treatment for fixed Crohn’s strictures. Simple, short strictures can be treated with bowel preserving strictureplasty; longer, multiple, or complicated strictures (e.g., significant inflammation, penetrating disease, and suspected cancer) are treated with resection and primary anastomosis. Resection carries its own risks, including anastomotic dehiscence and disease recurrence at the site of the anastomosis, malabsorptive issues following terminal ileal resection including vitamin B12 deficiency and bile salt malabsorption, and, in cases requiring repeated resections for recurrent strictures, short bowel syndrome leading to a dependence on parenteral nutrition.
Medical therapies to prevent and reverse fibrosis are eagerly sought and much of the focus for new therapies has been for pulmonary and hepatic disease. As our understanding of the pathophysiology of fibrosis improves, we are discovering more potential drug targets. Pirfenidone, a growth factor inhibitor, has been licensed for use in idiopathic pulmonary fibrosis (IPF), based on positive results from Phase 2 and 3 trials (82, 83). Nintedanib, a kinase inhibitor that acts on vascular endothelial growth factor receptors, PDGF receptors, and fibroblast growth factor receptors, showed efficacy and safety in Phase 2 and 3 trials and is licensed for use in IPF (83). Specific NOX4 inhibitors are still not available, albeit a NOX inhibitor (GKT137831) performed well in preclinical models (35, 42) and a Phase 1 clinical trial (84). A Phase 2 trial confirmed safety but not efficacy in the treatment of diabetic kidney disease; recruitment for a Phase 2 trial in patients with primary biliary cirrhosis is ongoing. Integrin αvβ6 is another target of interest; it mediates the conformational release of active TGFβ from its latent complex (72) and Phase 2 trials of a monoclonal antibody in the treatment of IPF are ongoing (85). Given the complex interaction between TGFβ secretion and tissue stiffness mediated by myofibroblast contraction, ECM deposition, and cross-linking with further TGFβ release, it is possible that the effective treatment of fibrosis will require combination therapy. For example, both Simtuzumab, a monoclonal antibody to LOXL2, and Relaxin (an inhibitor of myofibroblast contraction) have individually lacked efficacy for fibrotic disease in Phase 2/3 trials (86). However, a recent preclinical trial demonstrated a reduction in airway fibrosis using Relaxin and anti-LOXL2 antibody together (87).
Clinical trials assessing antifibrotic efficacy of currently available drugs in CD have not yet been initiated. Three times daily oral doses of pirfenidone commenced at time of transplantation and continued for 7 days reduced TGFβ expression and intestinal fibrosis in an intestinal transplant mouse model (88), suggesting that it may prevent fibrosis in CD. Animal models of intestinal fibrosis will provide an opportunity for preclinical testing of future drugs that target signaling pathways (23, 89), and therapies to reverse as well as prevent fibrosis will be required (35). A separate but related challenge still to be addressed will be the development of biomarkers for the accurate categorization of patients at risk of, or in the early stages of fibrosis, in order to intervene in CD at an early stage with existing or future antifibrotic drugs.
ES: conception and drafting of article. ES, BB, and UK: revision and final approval of article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was funded by the Health Research Board, Ireland (HPF 2016-1677) (ES) and the Science Foundation Ireland (UK).
1. Hope B, Shahdadpuri R, Dunne C, Broderick AM, Grant T, Hamzawi M, et al. Rapid rise in incidence of Irish paediatric inflammatory bowel disease. Arch Dis Child (2012) 97(7):590–4. doi: 10.1136/archdischild-2011-300651
2. Levine A, Griffiths A, Markowitz J, Wilson DC, Turner D, Russell RK, et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: the Paris classification. Inflamm Bowel Dis (2011) 17(6):1314–21. doi:10.1002/ibd.21493
3. Silverberg MS, Satsangi J, Ahmad T, Arnott ID, Bernstein CN, Brant SR, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a working party of the 2005 Montreal World Congress of gastroenterology. Can J Gastroenterol (2005) 19(Suppl A):5a–36a. doi:10.1155/2005/269076
4. Shaoul R, Karban A, Reif S, Weiss B, Shamir R, Tamir A, et al. Disease behavior in children with Crohn’s disease: the effect of disease duration, ethnicity, genotype, and phenotype. Dig Dis Sci (2009) 54(1):142–50. doi:10.1007/s10620-008-0326-7
5. Louis E, Collard A, Oger AF, Degroote E, Aboul Nasr El Yafi FA, Belaiche J. Behaviour of Crohn’s disease according to the Vienna classification: changing pattern over the course of the disease. Gut (2001) 49(6):777–82. doi:10.1136/gut.49.6.777
6. Duricova D, Fumery M, Annese V, Lakatos PL, Peyrin-Biroulet L, Gower-Rousseau C. The natural history of Crohn’s disease in children: a review of population-based studies. Eur J Gastroenterol Hepatol (2017) 29(2):125–34. doi:10.1097/meg.0000000000000761
7. Cosnes J, Cattan S, Blain A, Beaugerie L, Carbonnel F, Parc R, et al. Long-term evolution of disease behavior of Crohn’s disease. Inflamm Bowel Dis (2002) 8(4):244–50. doi:10.1097/00054725-200207000-00002
8. Rieder F, Latella G, Magro F, Yuksel ES, Higgins PD, Di Sabatino A, et al. European Crohn’s and colitis organisation topical review on prediction, diagnosis and management of fibrostenosing Crohn’s disease. J Crohns Colitis (2016) 10(8):873–85. doi:10.1093/ecco-jcc/jjw055
9. Seung SL, Ah YK, Yang S-K, Chung J-W, So YK, Seong HP, et al. Crohn disease of the small bowel: comparison of CT enterography, MR enterography, and small-bowel follow-through as diagnostic techniques. Radiology (2009) 251(3):751–61. doi:10.1148/radiol.2513081184
10. Gee MS, Nimkin K, Hsu M, Israel EJ, Biller JA, Katz AJ, et al. Prospective evaluation of MR enterography as the primary imaging modality for pediatric Crohn disease assessment. AJR Am J Roentgenol (2011) 197(1):224–31. doi:10.2214/AJR.10.5970
11. Jensen MD, Kjeldsen J, Rafaelsen SR, Nathan T. Diagnostic accuracies of MR enterography and CT enterography in symptomatic Crohn’s disease. Scand J Gastroenterol (2011) 46(12):1449–57. doi:10.3109/00365521.2011.613947
12. Quencer KB, Nimkin K, Mino-Kenudson M, Gee MS. Detecting active inflammation and fibrosis in pediatric Crohn’s disease: prospective evaluation of MR-E and CT-E. Abdom Imaging (2013) 38(4):705–13. doi:10.1007/s00261-013-9981-z
13. Fiorino G, Bonifacio C, Peyrin-Biroulet L, Minuti F, Repici A, Spinelli A, et al. Prospective comparison of computed tomography enterography and magnetic resonance enterography for assessment of disease activity and complications in ileocolonic Crohn’s disease. Inflamm Bowel Dis (2011) 17(5):1073–80. doi:10.1002/ibd.21533
14. Fraquelli M, Branchi F, Cribiu FM, Orlando S, Casazza G, Magarotto A, et al. The role of ultrasound elasticity imaging in predicting ileal fibrosis in Crohn’s disease patients. Inflamm Bowel Dis (2015) 21(11):2605–12. doi:10.1097/mib.0000000000000536
15. Sconfienza LM, Cavallaro F, Colombi V, Pastorelli L, Tontini G, Pescatori L, et al. In-vivo axial-strain sonoelastography helps distinguish acutely-inflamed from fibrotic terminal ileum strictures in patients with Crohn’s disease: preliminary results. Ultrasound Med Biol (2016) 42(4):855–63. doi:10.1016/j.ultrasmedbio.2015.11.023
16. Shelley-Fraser G, Borley NR, Warren BF, Shepherd NA. The connective tissue changes of Crohn’s disease. Histopathology (2012) 60(7):1034–44. doi:10.1111/j.1365-2559.2011.03911.x
17. Graham MF, Diegelmann RF, Elson CO, Lindblad WJ, Gotschalk N, Gay S, et al. Collagen content and types in the intestinal strictures of Crohn’s disease. Gastroenterology (1988) 94(2):257–65. doi:10.1016/0016-5085(88)90411-8
18. Lawrance IC, Maxwell L, Doe W. Altered response of intestinal mucosal fibroblasts to profibrogenic cytokines in inflammatory bowel disease. Inflamm Bowel Dis (2001) 7(3):226–36. doi:10.1097/00054725-200108000-00008
19. Flier SN, Tanjore H, Kokkotou EG, Sugimoto H, Zeisberg M, Kalluri R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J Biol Chem (2010) 285(26):20202–12. doi:10.1074/jbc.M110.102012
20. Uehara H, Nakagawa T, Katsuno T, Sato T, Isono A, Noguchi Y, et al. Emergence of fibrocytes showing morphological changes in the inflamed colonic mucosa. Dig Dis Sci (2010) 55(2):253–60. doi:10.1007/s10620-009-0730-7
21. Rieder F, Kessler SP, West GA, Bhilocha S, de la Motte C, Sadler TM, et al. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol (2011) 179(5):2660–73. doi:10.1016/j.ajpath.2011.07.042
22. Koukoulis G, Ke Y, Henley JD, Cummings OW. Obliterative muscularization of the small bowel submucosa in Crohn disease: a possible mechanism of small bowel obstruction. Arch Pathol Lab Med (2001) 125(10):1331–4. doi:10.1043/0003-9985(2001)125<1331:omotsb>2.0.co;2
23. Bettenworth D, Rieder F. Medical therapy of stricturing Crohn’s disease: what the gut can learn from other organs – a systematic review. Fibrogenesis Tissue Repair (2014) 7(1):5. doi:10.1186/1755-1536-7-5
24. Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol (2012) 13(10):616–30. doi:10.1038/nrm3434
25. Medina C, Santos-Martinez MJ, Santana A, Paz-Cabrera MC, Johnston MJ, Mourelle M, et al. Transforming growth factor-beta type 1 receptor (ALK5) and Smad proteins mediate TIMP-1 and collagen synthesis in experimental intestinal fibrosis. J Pathol (2011) 224(4):461–72. doi:10.1002/path.2870
26. Beddy D, Mulsow J, Watson RW, Fitzpatrick JM, O’Connell PR. Expression and regulation of connective tissue growth factor by transforming growth factor beta and tumour necrosis factor alpha in fibroblasts isolated from strictures in patients with Crohn’s disease. Br J Surg (2006) 93(10):1290–6. doi:10.1002/bjs.5431
27. Di Sabatino A, Jackson CL, Pickard KM, Buckley M, Rovedatti L, Leakey NA, et al. Transforming growth factor beta signalling and matrix metalloproteinases in the mucosa overlying Crohn’s disease strictures. Gut (2009) 58(6):777–89. doi:10.1136/gut.2008.149096
28. Vallance BA, Gunawan MI, Hewlett B, Bercik P, Van Kampen C, Galeazzi F, et al. TGF-beta1 gene transfer to the mouse colon leads to intestinal fibrosis. Am J Physiol Gastrointest Liver Physiol (2005) 289(1):G116–28. doi:10.1152/ajpgi.00051.2005
29. Ardizzone S, Bevivino G, Monteleone G. Mongersen, an oral Smad7 antisense oligonucleotide, in patients with active Crohn’s disease. Therap Adv Gastroenterol (2016) 9(4):527–32. doi:10.1177/1756283x16636781
30. Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res (2009) 19(1):128–39. doi:10.1038/cr.2008.328
31. Jiang F, Liu GS, Dusting GJ, Chan EC. NADPH oxidase-dependent redox signaling in TGF-beta-mediated fibrotic responses. Redox Biol (2014) 2:267–72. doi:10.1016/j.redox.2014.01.012
32. Crestani B, Besnard V, Boczkowski J. Signalling pathways from NADPH oxidase-4 to idiopathic pulmonary fibrosis. Int J Biochem Cell Biol (2011) 43(8):1086–9. doi:10.1016/j.biocel.2011.04.003
33. Sampson N, Berger P, Zenzmaier C. Redox signaling as a therapeutic target to inhibit myofibroblast activation in degenerative fibrotic disease. Biomed Res Int (2014) 2014:131737. doi:10.1155/2014/131737
34. Zhou B, Mu J, Gong Y, Lu C, Zhao Y, He T, et al. Brd4 inhibition attenuates unilateral ureteral obstruction-induced fibrosis by blocking TGF-beta-mediated Nox4 expression. Redox Biol (2016) 11:390–402. doi:10.1016/j.redox.2016.12.031
35. Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med (2014) 6(231):231ra47. doi:10.1126/scitranslmed.3008182
36. Thannickal VJ. Mechanisms of pulmonary fibrosis: role of activated myofibroblasts and NADPH oxidase. Fibrogenesis Tissue Repair (2012) 5(Suppl 1):S23. doi:10.1186/1755-1536-5-s1-s23
37. Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med (2009) 15(9):1077–81. doi:10.1038/nm.2005
38. Sato N, Takasaka N, Yoshida M, Tsubouchi K, Minagawa S, Araya J, et al. Metformin attenuates lung fibrosis development via NOX4 suppression. Respir Res (2016) 17(1):107. doi:10.1186/s12931-016-0420-x
39. Dosoki H, Stegemann A, Taha M, Schnittler H, Luger TA, Schroder K, et al. Targeting of NADPH oxidase in vitro and in vivo suppresses fibroblast activation and experimental skin fibrosis. Exp Dermatol (2017) 26(1):73–81. doi:10.1111/exd.13180
40. Sancho P, Mainez J, Crosas-Molist E, Roncero C, Fernandez-Rodriguez CM, Pinedo F, et al. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS One (2012) 7(9):e45285. doi:10.1371/journal.pone.0045285
41. Liang S, Kisseleva T, Brenner DA. The role of NADPH oxidases (NOXs) in liver fibrosis and the activation of myofibroblasts. Front Physiol (2016) 7:17. doi:10.3389/fphys.2016.00017
42. Jiang JX, Chen X, Serizawa N, Szyndralewiez C, Page P, Schroder K, et al. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic Biol Med (2012) 53(2):289–96. doi:10.1016/j.freeradbiomed.2012.05.007
43. Jung KJ, Min KJ, Park JW, Park KM, Kwon TK. Carnosic acid attenuates unilateral ureteral obstruction-induced kidney fibrosis via inhibition of Akt-mediated Nox4 expression. Free Radic Biol Med (2016) 97:50–7. doi:10.1016/j.freeradbiomed.2016.05.020
44. Latella G, Vetuschi A, Sferra R, Zanninelli G, D’Angelo A, Catitti V, et al. Smad3 loss confers resistance to the development of trinitrobenzene sulfonic acid-induced colorectal fibrosis. Eur J Clin Invest (2009) 39(2):145–56. doi:10.1111/j.1365-2362.2008.02076.x
45. Nlandu Khodo S, Dizin E, Sossauer G, Szanto I, Martin PY, Feraille E, et al. NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J Am Soc Nephrol (2012) 23(12):1967–76. doi:10.1681/asn.2012040373
46. Craige SM, Kant S, Reif M, Chen K, Pei Y, Angoff R, et al. Endothelial NADPH oxidase 4 protects ApoE−/− mice from atherosclerotic lesions. Free Radic Biol Med (2015) 89:1–7. doi:10.1016/j.freeradbiomed.2015.07.004
47. Langbein H, Brunssen C, Hofmann A, Cimalla P, Brux M, Bornstein SR, et al. NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. Eur Heart J (2016) 37(22):1753–61. doi:10.1093/eurheartj/ehv564
48. Gray SP, Di Marco E, Kennedy K, Chew P, Okabe J, El-Osta A, et al. Reactive oxygen species can provide atheroprotection via NOX4-dependent inhibition of inflammation and vascular remodeling. Arterioscler Thromb Vasc Biol (2016) 36(2):295–307. doi:10.1161/atvbaha.115.307012
49. Di Marco E, Gray SP, Kennedy K, Szyndralewiez C, Lyle AN, Lassegue B, et al. NOX4-derived reactive oxygen species limit fibrosis and inhibit proliferation of vascular smooth muscle cells in diabetic atherosclerosis. Free Radic Biol Med (2016) 97:556–67. doi:10.1016/j.freeradbiomed.2016.07.013
50. Levigne D, Modarressi A, Krause KH, Pittet-Cuenod B. NADPH oxidase 4 deficiency leads to impaired wound repair and reduced dityrosine-crosslinking, but does not affect myofibroblast formation. Free Radic Biol Med (2016) 96:374–84. doi:10.1016/j.freeradbiomed.2016.04.194
51. Hotta Y, Takagi T, Tanaka M, Higashimura Y, Mizushima K, Okayama T, et al. Mo1765 the role of NADPH oxidase (NOX) 4 expressed in pericryptal myofibroblasts for intestinal fibrosis. Gastroenterology (2015) 148(4):S–706. doi:10.1016/S0016-5085(15)32395-7
52. Sadler T, Bhasin JM, Xu Y, Barnholz-Sloan J, Chen Y, Ting AH, et al. Genome-wide analysis of DNA methylation and gene expression defines molecular characteristics of Crohn’s disease-associated fibrosis. Clin Epigenetics (2016) 8:30. doi:10.1186/s13148-016-0193-6
53. Munoz-Felix JM, Gonzalez-Nunez M, Martinez-Salgado C, Lopez-Novoa JM. TGF-beta/BMP proteins as therapeutic targets in renal fibrosis. Where have we arrived after 25 years of trials and tribulations? Pharmacol Ther (2015) 156:44–58. doi:10.1016/j.pharmthera.2015.10.003
54. Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med (2003) 9(7):964–8. doi:10.1038/nm888
55. Maric I, Kucic N, Turk Wensveen T, Smoljan I, Grahovac B, Zoricic Cvek S, et al. BMP signaling in rats with TNBS-induced colitis following BMP7 therapy. Am J Physiol Gastrointest Liver Physiol (2012) 302(10):G1151–62. doi:10.1152/ajpgi.00244.2011
56. Maric I, Poljak L, Zoricic S, Bobinac D, Bosukonda D, Sampath KT, et al. Bone morphogenetic protein-7 reduces the severity of colon tissue damage and accelerates the healing of inflammatory bowel disease in rats. J Cell Physiol (2003) 196(2):258–64. doi:10.1002/jcp.10275
57. Horiguchi H, Endo M, Kawane K, Kadomatsu T, Terada K, Morinaga J, et al. ANGPTL2 expression in the intestinal stem cell niche controls epithelial regeneration and homeostasis. EMBO J (2017) 36(4):409–24. doi:10.15252/embj.201695690
58. Martel C, Raignault A, Yu C, Gillis MA, Mamarbachi M, Bousette N, et al. Abstract 16267. Circulation (2015) 132(Suppl_3):A16267.
59. Rehman H, Liu Q, Krishnasamy Y, Shi Z, Ramshesh VK, Haque K, et al. The mitochondria-targeted antioxidant MitoQ attenuates liver fibrosis in mice. Int J Physiol Pathophysiol Pharmacol (2016) 8(1):14–27.
60. Liu R-M, Desai LP. Reciprocal regulation of TGF-β and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol (2015) 6:565–77. doi:10.1016/j.redox.2015.09.009
61. Richter K, Kietzmann T. Reactive oxygen species and fibrosis: further evidence of a significant liaison. Cell Tissue Res (2016) 365(3):591–605. doi:10.1007/s00441-016-2445-3
62. Perepelyuk M, Terajima M, Wang AY, Georges PC, Janmey PA, Yamauchi M, et al. Hepatic stellate cells and portal fibroblasts are the major cellular sources of collagens and lysyl oxidases in normal liver and early after injury. Am J Physiol Gastrointest Liver Physiol (2013) 304(6):G605–14. doi:10.1152/ajpgi.00222.2012
63. Yang J, Savvatis K, Kang JS, Fan P, Zhong H, Schwartz K, et al. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat Commun (2016) 7:13710. doi:10.1038/ncomms13710
64. Ikenaga N, Peng ZW, Vaid KA, Liu SB, Yoshida S, Sverdlov DY, et al. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut (2017). doi:10.1136/gutjnl-2016-312473
65. Lopez B, Gonzalez A, Hermida N, Valencia F, de Teresa E, Diez J. Role of lysyl oxidase in myocardial fibrosis: from basic science to clinical aspects. Am J Physiol Heart Circ Physiol (2010) 299(1):H1–9. doi:10.1152/ajpheart.00335.2010
66. Atsawasuwan P, Mochida Y, Katafuchi M, Kaku M, Fong KS, Csiszar K, et al. Lysyl oxidase binds transforming growth factor-beta and regulates its signaling via amine oxidase activity. J Biol Chem (2008) 283(49):34229–40. doi:10.1074/jbc.M803142200
67. Roy R, Polgar P, Wang Y, Goldstein RH, Taylor L, Kagan HM. Regulation of lysyl oxidase and cyclooxygenase expression in human lung fibroblasts: interactions among TGF-beta, IL-1 beta, and prostaglandin E. J Cell Biochem (1996) 62(3):411–7. doi:10.1002/(SICI)1097-4644(199609)62:3<411::AID-JCB11>3.3.CO;2-I
68. Sethi A, Mao W, Wordinger RJ, Clark AF. Transforming growth factor-beta induces extracellular matrix protein cross-linking lysyl oxidase (LOX) genes in human trabecular meshwork cells. Invest Ophthalmol Vis Sci (2011) 52(8):5240–50. doi:10.1167/iovs.11-7287
69. Boak AM, Roy R, Berk J, Taylor L, Polgar P, Goldstein RH, et al. Regulation of lysyl oxidase expression in lung fibroblasts by transforming growth factor-beta 1 and prostaglandin E2. Am J Respir Cell Mol Biol (1994) 11(6):751–5. doi:10.1165/ajrcmb.11.6.7946403
70. Jobling MF, Mott JD, Finnegan MT, Jurukovski V, Erickson AC, Walian PJ, et al. Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat Res (2006) 166(6):839–48. doi:10.1667/rr0695.1
71. Hinz B. Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Curr Rheumatol Rep (2009) 11(2):120. doi:10.1007/s11926-009-0017-1
72. Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol (2007) 179(6):1311–23. doi:10.1083/jcb.200704042
73. Li Z, Dranoff JA, Chan EP, Uemura M, Sevigny J, Wells RG. Transforming growth factor-beta and substrate stiffness regulate portal fibroblast activation in culture. Hepatology (2007) 46(4):1246–56. doi:10.1002/hep.21792
74. Cosnes J, Nion-Larmurier I, Beaugerie L, Afchain P, Tiret E, Gendre JP. Impact of the increasing use of immunosuppressants in Crohn’s disease on the need for intestinal surgery. Gut (2005) 54(2):237–41. doi:10.1136/gut.2004.045294
75. Gustavsson A, Magnuson A, Blomberg B, Andersson M, Halfvarson J, Tysk C. Endoscopic dilation is an efficacious and safe treatment of intestinal strictures in Crohn’s disease. Aliment Pharmacol Ther (2012) 36(2):151–8. doi:10.1111/j.1365-2036.2012.05146.x
76. Hassan C, Zullo A, De Francesco V, Ierardi E, Giustini M, Pitidis A, et al. Systematic review: endoscopic dilatation in Crohn’s disease. Aliment Pharmacol Ther (2007) 26(11–12):1457–64. doi:10.1111/j.1365-2036.2007.03532.x
77. Thienpont C, D’Hoore A, Vermeire S, Demedts I, Bisschops R, Coremans G, et al. Long-term outcome of endoscopic dilatation in patients with Crohn’s disease is not affected by disease activity or medical therapy. Gut (2010) 59(3):320–4. doi:10.1136/gut.2009.180182
78. Di Nardo G, Oliva S, Aloi M, Rossi P, Casciani E, Masselli G, et al. Usefulness of single-balloon enteroscopy in pediatric Crohn’s disease. Gastrointest Endosc (2012) 75(1):80–6. doi:10.1016/j.gie.2011.06.021
79. Di Nardo G, Oliva S, Passariello M, Pallotta N, Civitelli F, Frediani S, et al. Intralesional steroid injection after endoscopic balloon dilation in pediatric Crohn’s disease with stricture: a prospective, randomized, double-blind, controlled trial. Gastrointest Endosc (2010) 72(6):1201–8. doi:10.1016/j.gie.2010.08.003
80. van der Have M, Noomen C, Oldenburg B, Walter D, Houben MH, Wasser MN, et al. Balloon dilatation with or without intralesional and oral corticosteroids for anastomotic Crohn’s disease strictures. J Gastrointestin Liver Dis (2015) 24(4):537–9. doi:10.15403/jgld.2014.1121.244.hav
81. East JE, Brooker JC, Rutter MD, Saunders BP. A pilot study of intrastricture steroid versus placebo injection after balloon dilatation of Crohn’s strictures. Clin Gastroenterol Hepatol (2007) 5(9):1065–9. doi:10.1016/j.cgh.2007.04.013
82. Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet (2011) 377(9779):1760–9. doi:10.1016/s0140-6736(11)60405-4
83. Hughes G, Toellner H, Morris H, Leonard C, Chaudhuri N. Real world experiences: pirfenidone and nintedanib are effective and well tolerated treatments for idiopathic pulmonary fibrosis. J Clin Med (2016) 5(9):E78. doi:10.3390/jcm5090078
84. Wiesel P, Hovsepian L, Mutch PJ, Herve J, Heitz F, Page P. Safety and pharmacokinetics of single and multiple doses of a first in class dual NADPH oxidase 1 and 4 inhibitor administered orally in healthy subjects [Abstract]. J Am Soc Nephrol (2012) 23:559A.
85. Liu Y-M, Nepali K, Liou J-P. Idiopathic pulmonary fibrosis: current status, recent progress, and emerging targets. J Med Chem (2017) 60(2):527–53. doi:10.1021/acs.jmedchem.6b00935
86. Raghu G, Brown KK, Collard HR, Cottin V, Gibson KF, Kaner RJ, et al. Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: a randomised, double-blind, controlled, phase 2 trial. Lancet Respir Med (2017) 5(1):22–32. doi:10.1016/s2213-2600(16)30421-0
87. Lin YC, Sung YK, Jiang X, Peters-Golden M, Nicolls MR. Simultaneously targeting myofibroblast contractility and extracellular matrix cross-linking as a therapeutic concept in airway fibrosis. Am J Transplant (2017) 17(5):1229–41. doi:10.1111/ajt.14103
88. Meier R, Lutz C, Cosin-Roger J, Fagagnini S, Bollmann G, Hunerwadel A, et al. Decreased fibrogenesis after treatment with pirfenidone in a newly developed mouse model of intestinal fibrosis. Inflamm Bowel Dis (2016) 22(3):569–82. doi:10.1097/mib.0000000000000716
Keywords: Crohn’s disease, inflammatory bowel disease, fibrosis, intestine, reactive oxygen species, NADPH oxidase
Citation: Stenke E, Bourke B and Knaus U (2017) Crohn’s Strictures—Moving Away from the Knife. Front. Pediatr. 5:141. doi: 10.3389/fped.2017.00141
Received: 16 March 2017; Accepted: 02 June 2017;
Published: 16 June 2017
Edited by:
Jeff Critch, Memorial University of Newfoundland, CanadaReviewed by:
Andrew S. Day, University of Otago, New ZealandCopyright: © 2017 Stenke, Bourke and Knaus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emily Stenke, ZW1pbHkuc3RlbmtlQHVjZGNvbm5lY3QuaWU=;
Ulla Knaus, dWxsYS5rbmF1c0B1Y2QuaWU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.