 Zerina Hodzic
Zerina Hodzic Alexa M. Bolock
Alexa M. Bolock Misty Good
Misty Good
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 03 March 2017
Sec. Neonatology
Volume 5 - 2017 | https://doi.org/10.3389/fped.2017.00040
This article is part of the Research TopicThe Neonatal Immune System: A Unique Host-Microbial InterfaceView all 15 articles
Necrotizing enterocolitis (NEC) is the most devastating gastrointestinal disease of prematurity. Although the precise cause is not well understood, the main risk factors thought to contribute to NEC include prematurity, formula feeding, and bacterial colonization. Recent evidence suggests that NEC develops as a consequence of intestinal hyper-responsiveness to microbial ligands upon bacterial colonization in the preterm infant, initiating a cascade of aberrant signaling events, and a robust pro-inflammatory mucosal immune response. We now have a greater understanding of important mechanisms of disease pathogenesis, such as the role of cytokines, immunoglobulins, and immune cells in NEC. In this review, we will provide an overview of the mucosal immunity of the intestine and the relationship between components of the mucosal immune system involved in the pathogenesis of NEC, while highlighting recent advances in the field that have promise as potential therapeutic targets. First, we will describe the cellular components of the intestinal epithelium and mucosal immune system and their relationship to NEC. We will then discuss the relationship between the gut microbiota and cell signaling that underpins disease pathogenesis. We will conclude our discussion by highlighting notable therapeutic advancements in NEC that target the intestinal mucosal immunity.
Necrotizing enterocolitis (NEC) is notably the most lethal gastrointestinal disease of premature infants. The disease prevalence is approximately 7% of infants born between 500 and 1,500 g in the United States and Canada (1–3). Unfortunately, both the treatment approach and mortality have remained unchanged for decades with a mortality rate as high as 42% (4). At the same time, NEC represents a significant economic burden on the health system with an estimated annual cost upwards of one billion dollars in the United States (2, 5–7). These challenges fuel the need to understand disease pathogenesis so as to develop novel therapeutic and preventative strategies. Presently, it is believed that the immature intestine of premature infants exists in a hyper-active state due to abnormal bacterial colonization, which ultimately results in a robust inflammatory response and impairment of intestinal perfusion, thereby predisposing infants to NEC (2, 7, 8). Current research suggests the intestinal immune system is intricately involved in this process, which is comprised of the intestinal epithelium, immune cells, and commensal bacteria that maintain gastrointestinal homeostasis. Here, we will review the current knowledge of intestinal mucosal immunity in relation to NEC. First, we will discuss the cell types that comprise the intestinal immune system with attention to how these cells are involved in NEC. We will then describe the role of the innate immune system with specific attention to toll-like receptor 4 (TLR4) signaling in the pathogenesis of NEC. We will then review the role of gut microbiota in our current understanding of this disease. Finally, we will describe advancements in potential treatment strategies rooted in our current understanding of the relationship between mucosal immunity and the development of NEC.
In order to understand the pathogenesis of NEC, it is important to appreciate the role of the immune system in the maintenance of gastrointestinal homeostasis. We will first describe the role of epithelium and immune cells in mucosal immunity and then describe their role in the development of NEC with specific attention to the interplay of these cell types and the signaling pathways involved.
The epithelium represents the first layer of defense, comprised of at least seven differentiated cell types that together maintain barrier integrity and provide defense against pathogens with the presence of tight junctions (9). The epithelium has two distinct structures: the villus and the crypt. The villus contains enterocytes, goblet cells, enteroendocrine cells, and tuft cells, whereas the crypt houses transit amplifying cells, Paneth cells, and stem cells (Figure 1) (10). Stem cells expressing leucine-rich containing G protein-coupled receptor 5 (Lgr5) are capable of generating all cell types of the epithelium (11, 12). Together, these cells comprise the epithelium, which we will now discuss in further detail.
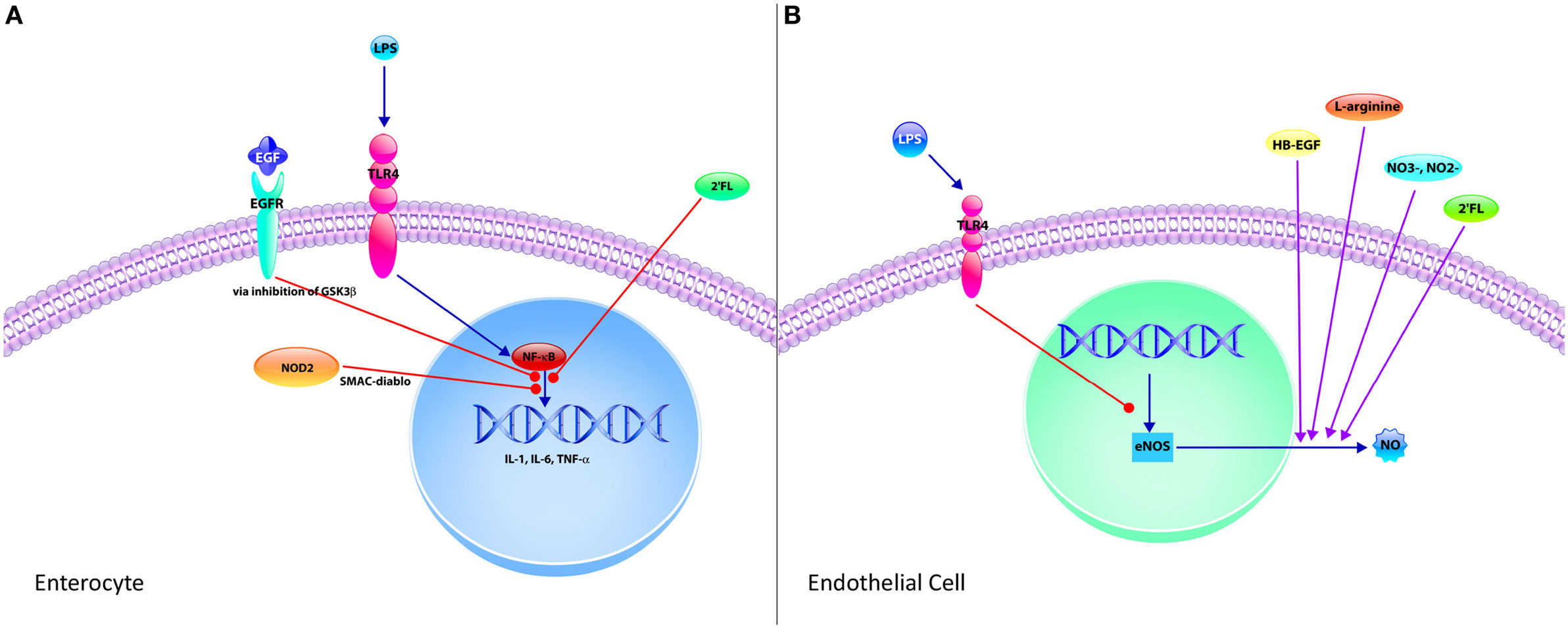
Figure 1. The neonatal intestinal immune system and its interaction in the pathogenesis of necrotizing enterocolitis (NEC). (A) The intestinal mucosal immune system is comprised of the cells of the epithelium, immune cells, and commensal bacteria. The epithelium consists of villi and crypts. Enterocytes, goblet cells, enteroendocrine cells, and tuft cells exist within the villi, whereas Paneth cells and stem cells occupy the base of the crypts. Immune cells consist of intraepithelial lymphocytes, T regulatory cells (Tregs), T cells, B cells, macrophages, and dendritic cells, which reside predominantly in the lamina propria underlying the epithelium. Commensal bacteria inhabit the lumen of the gut, which constantly interact with the epithelium and immune cells to help maintain protection against pathogenic bacteria. (B) In NEC, lipopolysaccharide from Gram-negative bacteria interact with toll-like receptor 4 (TLR4) expressed by predominantly by enterocytes, which results in the breakdown of the gut barrier, allowing for pathogenic bacterial translocation. A pro-inflammatory response follows resulting in increased production of pro-inflammatory cytokines (IL-6, IL-8, and TNF) as well as increased Th17 cells and decreased Tregs. The combination of these cellular responses with TLR4 signaling results in a profound inflammatory response and subsequently NEC.
Enterocytes (IECs) are the predominant absorptive cells of the epithelium defined by the presence of microvilli, but the role of enterocytes is not limited to nutrient absorption; rather, they are important cells in the preservation of intestinal integrity and mucosal immunity (13–16). IECs, as the most numerous cells of the epithelium, provide the physical barrier between the lumen of the gastrointestinal tract and the lamina propria via the maintenance of tight junctions (14). They originate from within the intestinal crypts and migrate along the villi, at which point they undergo apoptosis, renewing the epithelium every 3–5 days in a continuous cycle of IEC proliferation, migration, and apoptosis in mouse studies (17). This cycle is crucial for intestinal homeostasis; however, aberrancy in this process can lead to disastrous effects, such as bacterial translocation, which we will discuss in the context of NEC in a later section (18). IECs are notable for the presence of several pattern-recognition receptors (PRRs), such as toll-like receptors (TLRs) and nucleotide-binding oligomerization domains (NODs), thereby aiding in the clearance of pathogenic bacteria while maintaining a population of commensal bacteria (15, 16, 19, 20). We will describe TLRs and NODs more extensively in a later section with specific attention to the relationship between TLR4 expression by enterocytes and gut barrier integrity. Moreover, IECs also express major histocompatibility class (MHC) I and II molecules and non-classical MHC molecules, allowing IECs to process and present antigens to the immune cells of the intestine (21, 22). In doing so, there is a direct communication between the antigens within the lumen and the cells of the lamina propria (Figure 1). Accordingly, enterocytes are vital cells of the epithelium with the roles in the maintenance of the gut barrier and commensal bacteria, absorption of nutrients, and communication with the immune cells of the lamina propria.
Goblet cells are particularly important with their role in generating the mucus layer of the intestine, preventing the interaction between pathogenic bacteria and the epithelium, while providing support for commensal bacteria, antimicrobial peptides (AMPs), and secretory immunoglobulin A (IgA) (23). Moreover, goblet cells are also capable of delivering luminal antigens to a subset of underlying dendritic cells (DCs), CD103+ lamina propria DCs, which have tolerogenic properties, thereby assisting with the maintenance of commensal bacteria and intestinal homeostasis (24).
Goblet cell differentiation is determined by the activity of the Notch signaling pathway (25). Sodhi et al. (26) determined that the innate immune receptor TLR4 regulates Notch signaling and subsequent goblet cell development in the small intestine, such that TLR4 signaling prevented goblet cell differentiation independent from the influence of the microbiota. Furthermore, Notch signaling was found to be increased in mice as well as premature infants with NEC, whereas inhibition of the Notch pathway led to an increased number of goblet cells and attenuated experimental NEC in mice (26). This study highlights the regulation by TLR4 and Notch signaling in NEC pathogenesis.
Goblet cells secrete glycoproteins called mucins, of which, the Muc2 mucin is of critical importance in maintaining an inner mucus layer impervious to pathogenic bacteria, while simultaneously creating an outer mucus layer and providing an ideal habitat for commensal bacteria (27). Notably, ileal Muc2 is decreased in NEC and depletion of intestinal goblet cells increases susceptibility and severity of experimental NEC (28, 29). This subsequent decrease in mechanical defenses increases the vulnerability of the epithelium to pathogenic bacteria (26, 30), which can be further exacerbated by decreased intestinal motility in the setting of prematurity (31, 32). Abnormal goblet cell function is implicated in the development of NEC and mechanisms to enhance goblet cell production and/or function may provide a unique way to prevent the disease.
Paneth cells also provide a unique source of protection in the maintenance of the intestinal barrier. Paneth cells produce AMPs, lysozyme, secretory phospholipase A2, C-lectin RegIIIγ, α- and β-defensins, and angiogenin-4 to protect the host from pathogenic bacteria while shaping the composition of the microbiota (33, 34). Paneth cells and their AMPs, particularly α-defensins, have been implicated in diseases of the intestine through the use of several animal models (33). One noteworthy study utilized two genetic mouse models to study the role of α-defensins, including DEFA5-expressing transgenic mice and also mice that are deficient in matrix metalloproteinase 7 (MMP7), which is required to activate α-defensins (35). Using 16S rRNA sequencing, they found there was an α-defensin-dependent change in the composition of the microbiota, such that there was a correlation between α-defensin deficiency, decreased populations of bacteria from the phylum Bacteroidetes, and increased populations of bacteria from the phylum Firmicutes (35). Moreover, in DEFA5 transgenic mice, there was a loss of segmented filamentous bacteria and interleukin 17 (IL-17)-producing lamina propria T cells, substantiating the role of α-defensins and thus, Paneth cells in influencing the microbiota of the gut and modulating the intestinal immunologic response to pathogens (35).
The role of Paneth cells in disease has been well characterized in the studies on inflammatory bowel disease (IBD), specifically ileal Crohn’s disease (36). There are several susceptibility genes associated with Crohn’s disease, such as nucleotide-binding oligomerization domain-containing protein 2 (NOD2), which is expressed predominantly by Paneth cells in the small intestine (37–39). One such study evaluated known NOD2 loss-of-function gene mutations known to be risk factors for IBD in a group of very low birth weight infants (40). They found that the presence of two or more NOD2 genetic risk factors was an independently associated with the development of NEC and focal intestinal perforation (40). Moreover, NOD2-deficient mice develop ileal granulomatous inflammation in response to inoculation with Helicobacter hepaticus characterized by increased inflammatory cytokines and expression of Th1-related genes (41). This was restored with transgenic expression of α-defensin in Paneth cells (41). Moreover, in ileal Crohn’s disease, there is decreased expression of α-defensins, suggesting a relationship between the secretion of α-defensins by Paneth cells and the pathogenesis of Crohn’s disease. The involvement of Paneth cells in mediating protection against NEC is not well defined. However, animal models utilizing a Paneth-cell depletion method in the presence of Klebsiella pneumoniae have been shown to produce NEC-like intestinal injury, suggesting Paneth cells may have a role in NEC pathogenesis (42–44).
Enteroendocrine cells encompass several cell types that are located throughout the gastrointestinal system and primarily act to secrete hormones in response to food stimuli (45). The role of enteroendocrine cells in the promotion of mucosal immunity is incompletely understood; however, there is interplay between the immune system and enteroendocrine cells (46). For example, enteroendocrine cells express TLRs, and accordingly, can release chemokines and defensins in response to bacterial antigens, suggesting these cells have a role in the maintenance of intestinal homeostasis with respect to bacterial colonization (47, 48). Interaction with the gut microbiota is not limited to the expression of TLRs by enteroendocrine cells. Rather, there are several mechanisms by which these cells cooperate with the microbiota (49). For example, L cells are a subset of enteroendocrine cells that secrete glucagon-like peptides 1 and 2 (GLP-1 and GLP-2) (45). Short-chain fatty acids (SCFAs) derived from gut microbiota can activate specific receptors of L cells, thereby influencing energy metabolism and gut barrier function via secretion of GLP-1 and GLP-2 (49). Accordingly, enteroendocrine cells have a unique interaction with the gut microbiota.
Furthermore, enteroendocrine cells have also been implicated in IBD. Friedrich et al. (50) found that patients have elevated serum and colonic pro-inflammatory IL-17c and discovered that enteroendocrine and to a lesser extent goblet cells were the main producers of IL-17c. This demonstrates a relationship between the neuroendocrine system of the gut and the Th17 pathway of the immune system (50). Nonetheless, more studies are necessary to understand the role of enteroendocrine cells during intestinal inflammation and more specifically NEC.
Finally, there is a unique cell type of the intestinal epithelium called tuft cells, which are thought to be taste-chemosensory cells found in both the respiratory tract and gastrointestinal tract (51, 52). Tuft cells were initially identified by electron microscopy due to their unique morphology with their tubulovesicular system and a tuft of long, blunt microvilli (51, 52). There is increasing evidence for tuft cell involvement in immunity (53). More specifically, there is a newly discovered role of tuft cells in parasitic infections, whereby, they generate type 2 helper T cell responses to promote immunity against such infections (54–56). This is accomplished by the ability to secrete IL-25 in a transient receptor potential cation channel subfamily M member 5 (Trpm5)-dependent mechanism (51, 54–56). IL-25 promotes the expansion of type 2 innate lymphoid cells, an important source of IL-13 (56). This is crucial because IL-13 acts to promote differentiation of both tuft and goblet cells contributing to the “weep and sweep” response of the intestine to parasitic helminth infections, which consists of goblet cell hyperplasia and increased smooth muscle contractility (56). Although there is currently no known role of tuft cells in the pathogenesis of NEC, these studies provide evidence that these cells are actively involved in the mucosal immunity of the intestine in addition to their chemosensory roles.
Immune cells that compose the innate and adaptive immunity of the intestine exist in the epithelium and lamina propria (57). The epithelium houses intraepithelial lymphocytes (IELs) and the cytoplasmic extensions of DCs that interdigitate between epithelial cells (23, 57). Several cell types inhabit the lamina propria, including but not limited to DCs, macrophages, neutrophils, immunocompetent T and B cells, and T regulatory cells (Tregs) in addition to mesenchymal cell types, such as endothelial cells (23, 57). We will now discuss the various immune cells implicated in NEC, including T cells, Tregs, Th17 cells, IELs, B cells, macrophages, and DCs.
There is increasing evidence that immune cells have a primary role in the development of NEC, including but not limited to T cells. In one study, NEC was induced in recombination activating gene 1-deficient (Rag1–/–) mice, which lack functional T and B cells (58, 59). Compared to wild-type counterparts, these animals exhibited significantly decreased intestinal injury and mucosal cytokine production, suggesting they are protected from NEC development (59). Demonstrating that these cells have a causative role in NEC pathogenesis, susceptibility to NEC was restored with the adoptive transfer of naïve CD4+ T cells (59). Furthermore, recruitment and differentiation of T cells were found to be mediated by activation of TLR4 via cognate chemokine ligand 25 (59). Thus, this study demonstrates the principal role of immune cells in NEC, specifically CD4+ T cells, and their relationship to TLR4 signaling.
The roles of immune cells in NEC are dynamic, and there is an imbalance of immune cells that favor a exaggerated pro-inflammatory state. This is remarkably demonstrated by the relationship between Tregs and Th17 cells. Forkhead box P3 (Foxp3)-expressing Treg cells are essential for the maintenance of a stable equilibrium in the gut (57, 60). Tregs are important in counterbalancing inflammation and promoting antigen-specific IgA responses so as to maintain commensalism with gut microbiota (57, 61). In humans, intestinal Tregs are present as early as 23 weeks of gestation, suggesting an early role in intestinal homeostasis (60). In the intestinal lamina propria of both mice and premature infants with NEC, there is decreased production of Tregs and increased production of CD4+ Th17 cells mediated by STAT3 (59). Additionally, expression of the IL-17 receptor, IL-17RA is significantly increased in a TLR4-mediated manner (59). Importantly, IL-17 release by CD4+ Th17 cells results in intestinal mucosal injury, as demonstrated by impaired enterocyte tight junctions, increased enterocyte apoptosis, and decreased enterocyte proliferation, all of which are hallmark features of NEC (59). Similarly, in a study analyzing the T cell populations in human NEC, there was decreased Foxp3+ Tregs relative to effector T cells compared to controls, which correlated with a mucosal cytokine expression profile indicative of inhibited Treg expression (62). With NEC resolution, Treg cell populations recovered, suggesting either the inflammatory response observed in NEC decreases Treg populations acutely or inflammation is attenuated as Treg populations increase with mucosal healing (62).
The premature human gut is notable for increased expression of pro-inflammatory cytokines, specifically IL-6, which is involved in T cells differentiating toward a Th17 phenotype during NEC development (59). Importantly, in a mouse model of NEC, oral administration of all-trans retinoic acid (ATRA), which binds to the nuclear retinoic acid receptor to stabilize transcription of Foxp3 and repress transcription of RAR-related orphan receptor γt, resulted in decreased NEC severity, increased Tregs, decreased CD4+ Th17 cells, and attenuation of IL-17 expression (59). This suggests that the Tregs serve to provide a protective role in intestinal homeostasis, which is disrupted in NEC.
Further studies demonstrated that Th17 recruitment and Treg depletion resulted in apoptosis of Lgr5+ intestinal stem cells within the crypts of Lieberkühn in an experimental model of NEC (63). After exposure to recombinant IL-17, enteroids derived from intestinal stem cells exhibited decreased proliferation, decreased differentiation, and increased apoptosis (63). In addition, depletion of Tregs led to increased intestinal crypt apoptosis, exacerbating the experimental murine NEC model (63). Importantly, these effects were reversed upon administration of ATRA with its ability to modulate lymphocyte populations toward a protective Treg phenotype (63). This highlights the potential use of retinoic acid as a therapeutic in the treatment and prevention of NEC. Taken together, NEC is an immune cell-mediated disease defined by an imbalance of lymphocytes shifted toward a Th17 cell phenotype with a decrease in Tregs, which is destructive in this specific setting.
Intraepithelial lymphocytes are a group of heterogeneous lymphocytes of the innate immune system in the distinctive position to interact dynamically with the local gut environment and the mucosal adaptive immune system to maintain intestinal homeostasis (64). This unique population of IELs bears the T cell receptor γδ (65). They differ from TCRαβ T-cells in that they do not require antigen processing for effector function (65). They are important in the discussion of the infant gut due to their early development in embryogenesis (66, 67). IELs are involved in a multifaceted approach in the maintenance and repair of the epithelium through tight junction preservation, recognition of stressors, and regulation of inflammation (65). They are also involved in a dynamic cross talk with commensal bacteria, as commensal bacteria direct expression of key immunomodulatory and antibacterial responses by influencing gene transcription of IELs, while IELs prevent opportunistic commensal bacteria overgrowth in the setting of mucosal injury (68). Weitkamp et al. (66) determined that γδ IELs are the predominant IEL subtype in the immature intestine of mice and premature infants. Furthermore, they showed that in the intestine of infants with NEC, there was a reduction in the γδ IEL subset as compared to non-NEC controls (66). This study highlights the important role of γδ IELs in intestinal barrier protection and suggests a potential cellular target for NEC prevention.
B cells are both important antigen-presenting cells (APCs) and integral members of adaptive immunity with their ability to secrete immunoglobulins as plasma cells, most notably IgA in the context of neonatal immunity. IgA can neutralize pathogenic bacteria to maintain intestinal homeostasis (9). The maternal IgA from colostrum and milk is an important source of immunity in the neonatal period. Secretory IgA (sIgA) is detected weeks after birth and increases progressively; however, preterm infants have decreased concentrations of sIgA compared to full-term infants (69, 70). One study found higher levels of sIgA from the colostrum and milk of mothers of preterm infants, suggesting immunological adaptation occurs in the setting of prematurity to enhance immunity with IgA and reinforces the benefits of breastfeeding in this context (71). There are both early and long-term benefits of early exposure to maternal sIgA (72). In one study, they found weanling mice exposed to sIgA-deficient breast milk displayed increased colonization of draining lymph nodes with bacteria, specifically aerobic bacteria and the opportunist pathogen Ochrobactrum anthropi (72). At both weaning and adulthood, there were significant differences in the gut microbiota and the pattern of epithelial gene expression of the intestine, including genes associated with IBD, between these mice compared to mice exposed to sIgA (72). Accordingly, exposure to sIgA in breast milk promotes intestinal homeostasis in the neonatal period with long-term implications (72). Taken together, IgA is important in intestinal immunity, and maternal IgA is crucial given the immaturity of the immune system during the neonatal period.
Macrophages are important phagocytic and bactericidal cells of the immune system. Macrophages are activated after exposure to lipopolysaccharide (LPS) and interferon-γ, which then release pro-inflammatory cytokines and nitric oxide (57). To counteract excessive inflammation, intestinal macrophages are inhibited by tumor growth factor-β (TGF-β) (57) and have been implicated in NEC pathogenesis (73). Intestinal macrophages from healthy term infants have increased phagocytic and bactericidal activity as well as minimal inflammatory cytokine production when exposed to bacterial products, which is attributed to the anti-inflammatory effects of transforming growth factor β2 (TGF-β2) (73). In contrast, the phenotype of macrophages during NEC is strongly inflammatory as demonstrated by increased expression of the gene, mothers against decapentaplegic homolog 7 (Smad7), an inhibitor of TGF-β2 (74, 75). Consequently, this interrupts TGF-β-mediated downregulation of the pro-inflammatory response by macrophages in NEC and thereby sensitizes macrophages to bacterial products, leading to a significant pro-inflammatory response (74). Taken together, the imbalance of macrophage effector responses toward an inflammatory state increases mucosal injury in NEC.
Dendritic cells are specialized APCs that have the delicate role of mediating protective adaptive immunity in response to pathogens while maintaining intestinal homeostasis and commensal bacteria (76). Although they are important in intestinal immunity, the role of DCs in NEC has not been fully elucidated. However, in one study using Cronobacter sakazakii to induce NEC in newborn mice, DCs were recruited from the lamina propria, resulting in intestinal barrier dysfunction via tight junction disruption and increased enterocyte apoptosis via TGF-β production (77). The study concluded that the presence C. sakazakii was able to modulate the activity of DCs to increase TGF-β production, which subsequently resulted in damage to the mucosal barrier integrity (77). This highlights the sensitive interaction between DCs and bacteria, which can be detrimental in the setting of NEC; however, more studies are necessary to further understand the role of DCs in NEC pathogenesis.
Neutrophils are important effector cells of the innate immune system distinguished by their ability to respond rapidly and robustly to tissue trauma, including the intestine. The role of neutrophils in NEC pathogenesis has not been fully elucidated, but there are several studies to note. Small for gestational age (SGA) neonates are more likely to have neutropenia during the first few days after birth, and these infants with neutropenia were at increased risk for developing NEC compared to SGA infants without neutropenia (78). Moreover, in a model of C. sakazakii-induced experimental NEC, depletion of neutrophils and macrophages in the lamina propria resulted in increased production of pro-inflammatory cytokines and increased enterocyte apoptosis, thereby exacerbating the disease (79). This suggests both macrophages and neutrophils are important in early infection and their absence intensifies the inflammatory response observed in NEC. One study utilizing TNBS-induced enterocolitis to model NEC found the leukocyte infiltrate relatively devoid of neutrophils in the intestinal tissue (80). Taken together, impairment of neutrophil function is one component of NEC; however, more studies are necessary to determine whether this is a primary observation in NEC or secondary to the disease.
Peyer’s patches are unique structures found primarily in the distal intestine that serve as secondary lymphoid tissue. Compared to other secondary lymphoid tissues, Peyer’s patches are distinguished by their significant exposure to a diverse group of antigens, from the gut microbiota to food antigens (81). Antigens are delivered by M cells of the epithelium, and upon activation, generate B cells and plasma cells to maintain mucosal immunity (81). Peyer’s patches appear in the intestine at 11 weeks, but continue to develop throughout gestation (82). Moreover, their numbers increase in proportion to gestational maturation, and thus, premature infants have less Peyer’s patch numbers and decreased maturation (82). It is unknown if this has clinical significance in the setting of NEC. Accordingly, more studies are necessary to determine if Peyer’s patches have a role in NEC as a consequence of prematurity.
The innate immune system of the intestine is intricately involved in the pathogenesis of NEC. An important aspect of the innate immune system is the expression of PRRs, including TLRs and NOD proteins, that can interact with the local gut environment and initiate several signaling pathways. We will review these essential components and their cellular responses with specific attention to aberrant TLR4 signaling, which is well studied in the pathogenesis of NEC.
Pattern-recognition receptors are expressed throughout the cells of the body as a means of detecting threats to local homeostasis. The cells of the intestinal epithelium and immune cells express TLRs and NOD proteins, which can detect pathogen-associated molecular patterns such as the LPS of Gram-negative bacteria and flagella and consequently initiate an appropriate response to these bacterial stressors (23).
Specifically, in the case of TLR4 activation, signaling results in nuclear factor-κB (NF-κB) activation, subsequent cytokine production, resulting in an acute inflammatory response (23). TLRs are expressed by intestinal epithelial cells and immune cells of the lamina propria and are involved in epithelial cell proliferation, IgA production, maintenance of tight junctions, and AMP expression (23, 57, 83). TLR4 is of specific interest in the understanding of NEC pathogenesis, as its overexpression in the setting of prematurity yields a significant pro-inflammatory response and dysfunction of the epithelium (84). TLR4 is expressed by intestinal epithelial cells (85), which we will discuss in greater detail with respect to epithelial TLR4 expression and the development of NEC.
However, other TLRs have also been implicated in NEC pathogenesis. For example, TLR2 is expressed throughout the intestinal epithelium, including enterocytes, as well as various immune cells found in the lamina propria. Gram-positive bacteria in the intestine can engage TLR2, which can induce MyD88, which is a major adaptor for TLR2/TLR4, with subsequent NF-κB activation (86). TLR2 through the regulation of tight junctions is able to maintain gut barrier integrity, and thus, its deficiency can predispose the intestine to stress-induced injury (86). The role of TLR2 in NEC has not been fully elucidated thus far. However, in a rat model of NEC, investigators have found upregulation of ileal TLR4 and TLR2 in several studies, which precedes histological evidence of mucosal injury, suggesting a causative role in NEC pathogenesis (87, 88). Moreover, certain therapeutic interventions that reduced NEC severity in animal models downregulated the expression of TLR2 and TLR4, including glutamine (88) and probiotic Bifidobacterium (89). Thus, aberrancy in TLR4 signaling is not solely responsible for the pro-inflammatory response observed in NEC.
Moreover, NOD1 is also expressed by intestinal epithelial cells, which in response to the Gram-negative bacterial peptidoglycan, elicits an immune response to induce the formation of gut-associated lymphoid tissue, specifically Peyer’s patches (9, 57). Conversely, NOD2 is expressed highly by monocytes and Paneth cells at baseline, and by enterocytes when under stress (9, 57). NOD2 is important in the cross talk between T-cells and intestinal epithelium to downregulate inflammation by inhibiting TLR signaling (9, 57). One study demonstrated that NOD2 activation inhibited TLR4 in enterocytes, thereby decreasing enterocyte apoptosis and attenuating the severity of experimental NEC (90). In this study, NOD2 provided protection from TLR4-mediated enterocyte apoptosis via a novel SMAC-diablo pathway (90). This suggests that modulation of this cross talk may provide a potential means of decreasing NEC severity (83, 90). Furthermore, NOD2 is important in regulating commensal bacteria such that NOD2-deficient mice had decreased ability to prevent colonization by pathogenic bacteria in the intestine (91). Conversely, germ-free mice had significantly decreased NOD2 expression, but this expression was inducible with the introduction of commensal bacteria, demonstrating the feedback mechanism by which NOD2 regulates the gut microbiota (91). Upon ligand binding, both NOD1 and NOD2 activate NF-κB, and independently function by activating this signaling pathway, but can also modulate TLR4 signaling in this way (92). Thus, TLR and NOD signaling exemplify the delicate interaction between luminal bacteria and the innate immune system of the gut and how perturbations in these pathways may lead to NEC development.
One of the major cornerstones in understanding the development of NEC is the role that TLR4 signaling plays in the pathogenesis (59, 85, 93–100). Activation of TLR4 within the intestinal epithelium in the setting of prematurity results in decreased enterocyte proliferation, increased enterocyte apoptosis, disruption of intestinal barrier integrity, and bacterial translocation, resulting in a systemic inflammatory response (26, 84, 95, 97). As a result of bacterial translocation, TLR4 is activated on the endothelium of premature gut, leading to impaired blood flow and subsequent intestinal ischemia via reduction of endothelial nitric oxide synthase (eNOS) (100, 101). The differential response to stress between premature and full term neonates rests in the increased expression of TLR4 in prematurity (96). TLR4 is expressed at high levels in the developing intestine as it is involved in normal gut development in both mice and humans (8). In the setting of prematurity, TLR4 expression remains elevated, resulting in a hyper-active, exaggerated response to stressors upon colonization by bacteria (8).
Furthermore, TLR4 is necessary for the development of NEC, and its enhanced expression is not a consequence of the disease. In studies utilizing TLR4-mutant mice strains (C3H/HeJ mice), lack of functional TLR4 was protective against the development of NEC, such that wild-type mice had increased NEC severity, increased enterocyte apoptosis, reduced enterocyte proliferation, and impaired restitution compared to TLR4-mutant mice (97). These results were further supported utilizing mice with either global TLR4 deletion or intestinal-specific TLR4 deletion (26). Both groups of mice were protected from the development of experimental NEC with preservation of mucosal integrity and minimal elevation of pro-inflammatory cytokines (26). These studies highlight the causative role of aberrant TLR4 signaling expressed by IECs in NEC pathogenesis.
Aberrant TLR4 signaling also has a direct role in the breakdown of the gut barrier in NEC. In healthy mucosa, healing of the epithelium occurs in two phases, intestinal restitution followed by enterocyte proliferation, whereby healthy IECs migrate to injured mucosa followed by increased generation of IECs from stem cells of the intestinal crypts (18). TLR4 signaling impairs IEC migration, thus impairing restitution (102, 103). Moreover, enterocyte proliferation is significantly decreased in NEC, diminishing the ability to heal in the setting of mucosal injury (18, 95, 97–99). Importantly, autophagy is a response to cellular stress and has been found to be upregulated in NEC (104). Furthermore, TLR4 signaling induces autophagy of enterocytes in both mouse and human studies (105). An experiment utilizing intestinal epithelial-specific autophagy gene ATG7 conditional knockout mice demonstrated autophagy via ATG7 was required for NEC development as these mice were protected from NEC (105). TLR4-induced autophagy leads to impaired enterocyte migration, demonstrating the effects of TLR4 signaling on the epithelium are multifaceted and interconnected (105). Ultimately, the deficiency in mucosal repair via enterocyte restitution and proliferation in the setting of NEC weakens the integrity of gut, allowing for bacterial translocation and the downstream inflammatory response observed in this disease.
The study of genetic risk factors in NEC is an important component in understanding the signaling pathways that involve the innate immune system. There are several genetic association studies evaluating the relationship between specific genetic risk factors and the development of NEC (106). Genetic variation that affects TLR signaling increases the predisposition of the premature intestine to inflammatory aberrancy. The gene SIGIRR is important in the inhibition of LPS-induced inflammation (107). Loss-of-function mutations of SIGIRR result in unregulated TLR signaling, thereby predisposing infants to NEC (107). One study specifically evaluated the roles of single nucleotide polymorphisms of important genes in TLR signaling. They found in studying the blood samples of very low birth weight infants a relationship between NFKB1 and NFKBIA variants and the development of NEC, such that NFKB1 increased susceptibility to NEC, whereas NFKBIA decreased susceptibility to NEC (108). However, there was no association between TLR2, TLR4, TLR5, TLR9, IRAK1, and TIRAP genes and the development of NEC in this patient population (108). Genetic risk factors for NEC have not just been limited to TLR and NOD signaling pathways. For example, one study evaluating the autophagy gene ATG16L1 in premature infants found that hypomorphic variants conferred protection for the development of NEC (109). This study highlights the breadth of possible genetic risk factors in the NEC. Taken together, genetic predisposition to NEC is valid, and further large scale studies will likely reveal more genetic relationships and disease development.
The intricate relationship between gut microbiota and mucosal immunity is central to the discussion of NEC pathogenesis. The gut is exposed to a multitude of microbes, and it is the role of the intestinal immune system to distinguish commensal bacteria from pathogenic bacteria, particularly as the gut flora develops early in life. There are several variables that influence the microbial composition of the intestine; however, new evidence suggests that disruption of normal bacterial flora is involved in NEC pathogenesis. We will now explore the role of gut microbiota in the setting of prematurity.
There is a significant interest in understanding the composition of the gut microbiota and its relationship to the pathogenesis of NEC. Several factors influence the neonatal microbiota, including gestational age, mode of delivery (vaginal vs. cesarean section), antibiotic treatment, and diet (breast milk vs. formula feedings) (110). Colonization occurs in two waves, and the first wave is dependent on the mode of delivery (111). For example, one study found that compared to infants born vaginally, infants born via cesarean section had decreased populations of Bifidobacteria and Bacteroides, while there was an increased population of Clostridium difficile (112). The second wave is dependent on feeding method, which often differs between premature and term infants, as breast milk and formula feedings have different bacterial compositions and access to breast milk can be limited in prematurity with delayed initiation of enteral feeding in preterm newborns (113). More specifically, formula-fed infants have increased populations of Enterobacteriaceae, Bacteroides species, and C. difficile in the stool compared to infants fed breast milk (112, 114).
Moreover, duration of antibiotic exposure is associated with the development of NEC (115, 116). In one retrospective study investigating the association between antibiotic exposure and subsequent diagnosis of NEC in infants, they found that antibiotic exposure duration is associated with increased risk of developing NEC (115). When sepsis was eliminated as a potential confounder, the probability of developing NEC was increased 20% per day of antibiotic exposure (115). Strikingly, antibiotic exposure greater than 10 days in neonates resulted in an approximately threefold increased risk of developing NEC (115). Another retrospective study aiming to assess the association between initial antibiotic therapy in extremely low birth weight infants and NEC, found there was an increased risk of developing NEC after initial empiric antibiotic treatment in the first three postnatal days (116). Empiric antibiotic treatment for greater than 5 days, which the study defined as prolonged antibiotic treatment, was associated with the development of NEC and death in extremely low birth weight infants in the setting of sterile blood cultures (116). These studies highlight the need to be judicious in the implementation of empiric antibiotic treatment in preterm infants.
A recent study utilizing 16S rRNA gene pyrosequencing suggests the most important variable influencing the composition of premature gut microbiota is the degree of prematurity (117). Using 922 specimens from 58 subjects, they found an ordered, tightly controlled microbial progression of bacterial classes: Bacilli to Gammaproteobacteria to Clostridia (117). Other factors, including antibiotics, mode of delivery, and age influenced the pace of the progression of bacterial classes, but the particular sequence of development remained the same (117). This is important as more studies demonstrate the significance of dysbiosis and the development of NEC (113, 118–122).
The differences in microbial colonization between preterm infants and term infants suggests the possible role of dysbiosis on the pathogenesis of NEC. The onset of NEC occurs 2–6 weeks of life with the highest risk of NEC occurring at a corrected age of 29–33 weeks (123), only after microbial colonization of the gut (2, 124, 125), highlighting the role of gut microbiota in disease pathogenesis. To date, no specific microbial pathogen has been identified to be responsible for the development of NEC (119, 124, 125). However, resected intestinal tissue with active NEC demonstrated increased microbial burden and an abundance of strict anaerobes with a decrease in community diversity (126). Moreover, until recently, it was unclear if abnormal gut microbiota is a cause or consequence of NEC (127). A prospective case–control study by Warner et al. (127) helps to define the role that the microbiota plays in NEC development. In 166 very low birth weight infants, 3,586 stool samples were prospectively collected, and of these subjects, 46 developed NEC (127). Differences between the NEC cases and matched controls emerged after 1 month with a predominance of Gammaproteobacteria and decreased quantities of Negativicutes and Clostridia in infants that went on to develop NEC, with the strongest correlation between bacteria composition and development of NEC occurring prior to 27 weeks gestation (127). This study suggests an increased concentration of Gram-negative facultative bacteria is detrimental, possibly due to their ability to activate TLR4 via LPS (84, 122). Conversely, infants with NEC had significantly decreased obligate anaerobes (127), which interestingly produce anti-inflammatory SCFAs (128). These results suggest that interventions focused on modulation of the gut bacteria may play a role in preventing NEC.
With advancements in our knowledge of NEC, there are several potential preventative and therapeutic strategies that did not exist decades ago. We will review several of these strategies with respect to how they influence the intestinal immune system (Figure 2). These advancements target several aspects of the intestinal immune system, such as TLR4 signaling modulation, gut barrier integrity, immune cell composition, and the gut microbiota, all of which have been described with their individual roles in NEC. There have been several studies that demonstrate the importance of providing breast milk to premature infants instead of formula. Human breast milk contains several bioactive components beneficial to the neonate some of which include: epidermal growth factor (EGF), heparin-binding EGF-like growth factor, platelet-activating factor (PAF) acetylhydrolase, human milk oligosaccharides (HMOs), nitrates/nitrites, l-arginine, lactoferrin, and probiotics (8, 129, 130). We will review several breast milk components and the evidence supporting the mechanisms by which they impact the intestinal mucosal immunity during NEC. We will also discuss the use of antenatal corticosteroids and delayed umbilical cord clamping in the prevention of NEC.
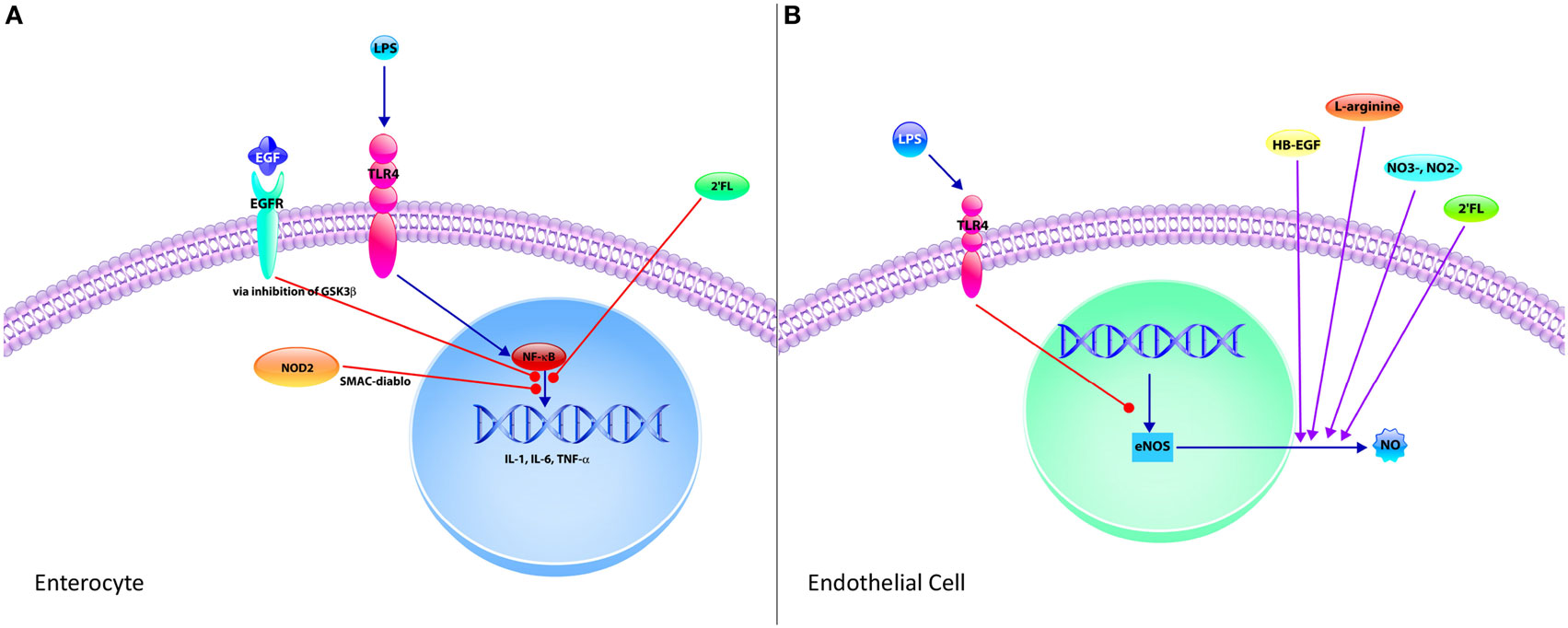
Figure 2. Mechanisms of protective strategies in necrotizing enterocolitis (NEC). (A) The enterocyte is the predominant cell type of the epithelium and significantly contributes to the exaggerated pro-inflammatory response observed in NEC upon activation of toll-like receptor 4 (TLR4) by Gram-negative bacteria lipopolysaccharide (LPS). TLR4-mediated activation of nuclear factor-κB (NF-κB) results in increased expression of pro-inflammatory cytokines, such as IL-1, IL-6, and TNF-α. However, this signaling pathway can be attenuated through various mechanisms. Epidermal growth factor (EGF), which is found in breast milk and amniotic fluid, inhibits this pathway via inhibition of glycogen synthase kinase-3 beta (GSK3β). The human milk oligosaccharide 2′-fucosyllactose (2′FL) also attenuates the TLR4 pro-inflammatory signaling pathway. Finally, intracellular pattern recognition receptor nucleotide-binding oligomerization domain-containing protein 2 upon stimulation by pathogen associated molecular patterns via second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI (SMAC-diablo). (B) Endothelial cells have an integral role in the pathogenesis of NEC. Endothelial cells express TLR4, and upon stimulation by LPS following gut barrier dysfunction, decrease the expression of endothelial nitric oxide synthase (eNOS), thereby reducing the formation of nitric oxide (NO), an important vasodilator. This results in intestinal ischemia and subsequent NEC. However, several protective strategies increase the production of NO, including heparin-binding EGF-like growth factor (HB-EGF) and 2′FL by increasing expression of eNOS. Nitrates and nitrites can be found in breast milk and are precursors to NO, thus increasing NO production. Finally, l-arginine is required for NOS-dependent formation of NO. Supplementation with HB-EGF, nitrates, nitrites, and l-arginine are protective in NEC with their ability to improve intestinal blood supply via NO-mediated vasodilation.
Breast milk has a well-established role in the prevention of NEC and clinically represents one of the most effective strategies in decreasing the incidence and progression of NEC (130). New pre-clinical evidence has demonstrated that breast milk is capable of attenuating the TLR4-mediated pro-inflammatory response integral to NEC pathogenesis by activating the receptor for epidermal growth factor (EGFR), revealing the interplay between the EGF pathway and the hallmark TLR4 signaling of NEC (95). These inhibitory effects on TLR4 signaling were mediated by breast milk preventing the activity of the downstream target glycogen synthase kinase 3β, resulting in enhanced mucosal healing, intestinal stem cell proliferation and decreased enterocyte apoptosis (95). In seeking to determine the particular component in breast milk that mediated the protective effects, when EGF was either removed or its receptor, EGFR was inhibited, the protection on experimental NEC and TLR4 signaling was abolished (95). Taken together, these mechanistic studies describe one of the ways breast milk protects the intestinal epithelium against NEC.
Several other studies have looked at the protective role of EGF itself against NEC. In a neonatal rat model of NEC, administration of EGF alone reduced both the incidence and severity of the disease (131). EGF administration resulted in significantly decreased epithelial permeability, normalized expression of tight junction proteins, increased goblet cells (28), and inhibited enterocyte apoptosis (132), all of which are protective hallmarks in NEC (26, 59, 95). Moreover, another mechanism by which EGF provides protection against NEC is inhibition of autophagy (133), a lysosomal pathway of self-digestion that has been shown to be activated in mice and infants with NEC (105). Additionally, a recent study demonstrates that a hypomorphic variant in an autophagy-related gene, ATG16L1 is associated with NEC in premature infants (108). This further underlies the importance of determining which infants are the most susceptible to NEC, so that protective strategies including EGF administration may be tailored to those individuals.
As discussed above, EGF administration mediates several protective effects on the intestine and is found in breast milk as well as amniotic fluid. We demonstrated that amniotic fluid inhibited TLR4-mediated inflammatory signaling in the fetal and neonatal intestinal epithelium in a manner dependent on EGFR (94). Intestinal EGFR expression was low in premature infants with NEC compared to that of a fetus or at the time of reanastamosis after NEC had resolved (94). The protective effects of amniotic fluid have been described in experimental NEC models in several species including neonatal mice (94), rats (134), and piglets (135). Other investigators have examined amniotic fluid outside the setting of EGF in the study of NEC. In a neonatal rat model of NEC, administration of amniotic fluid stem cells attenuated NEC by increasing enterocyte proliferation and decreasing apoptosis in a cyclooxygenase 2 dependent mechanism (136). In other studies, the administration of amniotic fluid resulted in differences in gut microbiota and reduced intestinal permeability compared to controls (137). Taken together, the ingredients in both breast milk and amniotic fluid provide important protective mechanisms to counter-regulate the detrimental pro-inflammatory effects in the intestine afflicted by NEC.
Heparin-binding EGF-like growth factor is found in breast milk and amniotic fluid and provide numerous protective effects in models of intestinal injury and NEC (138). In mice, overexpression of the HB-EGF gene is protective against the development of NEC (139), whereas deletion of HB-EGF increases susceptibility to experimental NEC (140). As described previously, an important component in the pathogenesis of NEC is the disruption of gut perfusion and decreased expression of the vasodilatory molecule eNOS (100). In neonatal mice subjected to experimental NEC, impairment of intestinal microvascular blood flow was improved, and there was decreased epithelial injury with administration of HB-EGF (141). Moreover, HB-EGF promotes angiogenesis with upregulation of eNOS and subsequent production of nitric oxide via the PI3K pathway (142, 143).
Heparin-binding EGF-like growth factor is also unique in its ability to modulate the intestinal immune system. For example, there are subtypes of macrophages, which can either be pro-inflammatory (M1) or anti-inflammatory (M2) (144). Macrophage infiltration in NEC was marked by a predominance of M1 pro-inflammatory macrophages and treatment with HB-EGF resulted in increased M2 anti-inflammatory macrophages, thereby protecting against experimental NEC. Moreover, HB-EGF also helps in maintaining gut barrier integrity with increased enterocyte proliferation (145) and migration (146) as well as decreased apoptosis of enterocytes (147), which is a common theme among the potential protective strategies in NEC.
Platelet-activating factor is a potent phospholipid inflammatory mediator involved in the pathogenesis of NEC in human and animal studies (148, 149). Activation results in epithelial cell damage, increased apoptosis, increased mucosal permeability, disruption of tight junctions, leukocyte and platelet aggregation, and vasoconstriction, disrupting mucosal integrity (148, 149). Importantly, PAF-AH is capable of catabolizing PAF and decreases the destructive properties of PAF in the intestine (150). Additionally, breast milk contains PAF-AH, which is one component in breast milk thought to provide protection against the development of NEC (150, 151). The clinical relevance of PAF is demonstrated by the findings that compared to controls, infants with NEC display increased plasma and stool PAF concentrations and decreased plasma PAF-AH, which is responsible for PAF breakdown (152–154). In neonatal mouse and rat models, NEC susceptibility decreased with inhibition of PAF and increased with PAF-AH depletion (155–157). Of note, PAF was found to induce TLR expression in the intestine, providing a connection between two pathways involved in NEC pathogenesis, thereby suggesting luminal PAF in the preterm intestine may upregulate TLR expression and subsequently promote a pro-inflammatory state (158). Taken together, PAF-AH may serve as an important target by inhibiting intestinal inflammation and epithelial disruption.
Human milk oligosaccharides are carbohydrates recently heralded as protective components within breast milk. There are several means by which HMOs are protective in the setting of NEC. We recently demonstrated that 2′-fucosyllactose (2′FL) is an abundant HMO that is protective against NEC in mice via modulation of the vasodilatory molecule, eNOS expression and subsequently enhancing intestinal perfusion (101), which has been previously shown to be impaired in NEC (100). Moreover, 2′FL is capable of attenuating inflammation, and we demonstrated that 2′FL decreased the expression of several pro-inflammatory markers including IL-6, IL-1β, inducible nitric oxide synthase, and TLR4 (101). Importantly, HMOs have been shown to influence bacterial colonization in the intestine during the critical neonatal period in several studies (159–165). In another study, the HMO 2′FL inhibited the release of IL-8 by IECs in the setting of bacterial infection by attenuating the expression of CD14 (166). However, this effect was not observed in IECs that were not exposed to the bacterial pathogen, showing that HMOs are capable of modulating specific inflammatory pathways during infection (166). Another HMO, disialyllacto-N-tetraose, was capable of reducing the NEC severity in neonatal rats (167). Taken together, these studies advance our understanding of the effects of HMOs in preventing against NEC and may provide a nutritional preventative strategy worth pursuing.
The breast milk composition of HMOs varies among mothers, which in turn influences the microbiota of their offspring. Specifically, mothers with inactive alleles of the gene fucosyltransferase 2 (FUT2) are referred to as non-secretor mothers (168), and this mutation leads to changes of the microbiota of their infants. Specifically, infants in this setting had delayed establishment of the genus Bifidobacterium, which are early colonizers in breastfed infants (168). This suggests HMOs can enrich certain beneficial populations of bacteria. Moreover, since the individual composition of breast milk varies widely, attention to these details may provide a novel strategy of personalized breast milk fortification to prevent NEC in preterm infants.
One of the hallmarks of NEC pathogenesis is the disruption of the microcirculatory perfusion to the gut, which is regulated by endothelial expression of TLR4 (100). Specifically, endothelial TLR4 signaling impairs intestinal perfusion and decreases eNOS expression and accordingly nitric oxide (NO), such that mice with selective endothelial TLR4 deletion exhibited preserved mucosal integrity and decreased intestinal ischemia (100). In this context, TLR4 signaling promotes bacterial translocation with the disruption of the epithelial barrier, intensified by impaired intestinal perfusion by decreased expression of eNOS with the activation of endothelial TLR4.
Human breast milk contains sodium nitrate, which is a precursor to nitrite and NO production, a key vasodilatory molecule involved in maintaining intestinal perfusion (100). Interestingly, the concentration of sodium nitrate is higher in breast milk as compared to formula (100), and when compared to adults, infants in the NICU receive significantly less dietary nitrites and nitrates (169). Bacterial conversion of nitrates to nitrites first occurs in saliva, which can then be converted to nitric oxide via both NOS-independent mechanisms in the stomach and NOS-dependent mechanisms in the intestine from the amino acid l-arginine (170). This illustrates the interaction of bacteria and the gastrointestinal system and also highlights the importance of having dietary nitrates and nitrites in the form of breast milk during the critical neonatal period. This further suggests there is the opportunity to improve the deficits in dietary nitrites and nitrates inherent to neonates to prevent NEC. Moreover, studies have shown that enteral supplementation of l-arginine was not only safe but was able to reduce incidence and severity of NEC in very low birth weight preterm infants (171, 172). Accordingly, l-arginine, nitrates, and nitrites in the neonatal period are not only important but given their ability to modulate the microcirculation of the preterm intestine, they have utility as a preventative strategy for NEC.
Lactoferrin is found in secretory fluids including breast milk and colostrum, where it acts an important member of mucosal immunity with its antimicrobial properties (173), ability to modulate the gut microbiota (174), and maturation of the intestine by inducing enterocyte growth and proliferation (175). Accordingly, the use of lactoferrin supplementation in preterm infants to prevent complications has been studied. The most recent Cochrane Review suggests that there is utility in the administration of oral lactoferrin prophylaxis due to the decrease in the development of NEC and late-onset sepsis without adverse effects (176–180). Another study evaluated the use of recombinant human lactoferrin (talactoferrin or TLf) in infants with a birth weight of 750–1,500 mg as means of reducing infection (181). There was no associated toxicity in the administration of TLf, and there was a trend toward less infectious morbidity in infants treated with TLf (181). Thus, lactoferrin administration in low birth weight infants is a promising preventative strategy against NEC with no adverse effects reported thus far.
Probiotics have generated significant interest in the prevention of NEC given the increasing data regarding the relationship between the gut microbiota and disease development. Prophylactic enteral probiotics show significant promise in the prevention of NEC. A Cochrane Review concluded probiotics in the setting of prematurity prevent severe NEC and decrease all-cause mortality (182). A more recent systematic review and meta-analysis utilizing additional studies supported these findings (183). As probiotics continue to be investigated, it is important to recognize there is significant heterogeneity with organisms chosen and dosing regimens across the studies. Bifidobacterium lactis has been studied in isolation in clinical studies. In a prospective randomized case–control study, administration of B. lactis-supplemented formula resulted in decreased intestinal permeability as measured by a sugar absorption test, suggesting B. lactis is important in maintaining epithelial integrity (184). In another clinical study, B. lactis Bb12 supplementation influenced the gut microbiota such that supplementation increased the number of Bifidobacterium spp. and reduced the number of Enterobacteriaceae and Clostridium spp., which may be implicated in NEC pathogenesis, in stool samples of preterm infants (185). It is important to note that the largest clinical trial for a specific probiotic, Bifidobacterium breve BBG-001, found no benefit in the administration of this probiotic in preterm infants to prevent NEC (186). More clinical studies evaluating the utility of specific organisms within probiotic regimens are necessary.
Moreover, there are several mechanisms by which probiotics can prevent the development of NEC (187). Two studies highlight our present knowledge of NEC pathogenesis as related to mucosal immunology. Bifidobacterium adolescentis is able to alter TLR4, TOLLIP, and SIGIRR expression in preterm neonatal rats, suggesting it is able to downregulate the TLR4-mediated pro-inflammatory response observed in NEC (188). Oral administration of Lactobacillus rhamnosus HN001 attenuated NEC severity in both premature piglets and newborn mice and the mechanism mediating this effect was via TLR9 activation (189). Lactobacillus reuteri in neonatal mice was able to increase enterocyte migration, enterocyte proliferation, and crypt height of the epithelium. This counters the impairment in enterocyte migration and proliferation that serve as hallmarks of NEC-mediated gut barrier dysfunction (190). These studies suggest probiotics are able to interact with the mucosal immunity of the gastrointestinal tract, thereby offering a viable preventative strategy in clinical NEC management.
Corticosteroids are often administered in the antenatal period to reduce mortality and morbidities associated with prematurity. Based on a Cochrane Review, the use of a single course of antenatal corticosteroids is recommended for all women at risk for preterm birth (191). In addition to accelerating fetal lung maturation, antenatal corticosteroids are associated with decreased likelihood for the development of necrotizing enterocolitis analyzing 8 studies with 1,675 infants total (191). A more recent clinical study supports these findings, in which there were significant differences between infants that received full course antenatal steroids and infants that did not, such that prompt administration of a full course of antenatal corticosteroids decreased the incidence of NEC and mortality (192). Presently, it is unknown the mechanism by which corticosteroids are protective in NEC. However, with our knowledge of the primary role of immune cells in the pro-inflammatory response in NEC, corticosteroids serve as an interest of study with their ability to modulate the immune system.
There is debate on the optimal interval of time between infant delivery and umbilical cord clamping in the setting of prematurity. Delayed cord clamping provides more placental transfusion between the placenta and the newborn, whereas immediate cord clamping allows for immediate resuscitation of the premature newborn by a neonatologist (193). A Cochrane Review found that delayed cord clamping decreased the incidence of NEC (193); however, larger studies are necessary to evaluate this finding as well as the long-term effects of immediate vs. delayed cord clamping. In a more recent study, umbilical cord milking, which utilizes the same principle behind delayed umbilical cord milking also conferred protection against the development of NEC in preterm infants (194). The mechanisms by which umbilical cord clamping and umbilical cord milking provide protection against NEC are incompletely understood and warrant further investigation.
The roles of B cells and immunoglobulins in the setting of NEC are not fully understood in the context of disease development and preventative strategies. A trial of oral immunoglobulins administration has been tried for their presumed immunoprotective effects as prophylaxis in preterm infants (195). However, based on a recent Cochrane Review of three randomized trials, there is no protective role against NEC with the oral administration of immunoglobulins in the neonatal period, specifically with IgG or a combination of IgG/IgA, as these studies did not demonstrate a significant reduction in NEC incidence (195–198). A trial of oral IgA alone powered for the prevention of NEC in low birth weight neonates would be beneficial given the known beneficial properties of sIgA in breast milk (195). Nonetheless, more studies are necessary to appreciate the role of IgA in NEC pathogenesis and as a viable prophylactic or treatment strategy.
Necrotizing enterocolitis is a devastating disease of prematurity and recent evidence demonstrates incredible promise for prevention with continuous advancements in the field. These advancements are not limited to mucosal immunology as recent attention to the interplay of intestinal immune system, which includes epithelial integrity, immune cell composition, TLR4 signaling, intestinal microbiota, has yielded expansion of our understanding of the disease and how to best prevent its development. One of the most important themes of this review is the dynamic interaction of the components that comprise the mucosal immunity of the intestine. Several aspects are integral to maintain intestinal homeostasis, and there is no one component singularly responsible for the development of NEC. Rather, it is an intricate balance of many variables, which yield numerous therapeutic potentials in treating and preventing this disease. The goal of this review was to highlight these recent innovations and mechanistic insights into how administration of various treatments impacts the intestinal immune system and may alleviate NEC in premature infants.
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
MG is supported by K08DK101608 from the National Institutes of Health, the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital, and the Department of Pediatrics at Washington University School of Medicine, St. Louis.
1. Guillet R, Stoll BJ, Cotten CM, Gantz M, McDonald S, Poole WK, et al. Association of H2-blocker therapy and higher incidence of necrotizing enterocolitis in very low birth weight infants. Pediatrics (2006) 117(2):e137–42. doi:10.1542/peds.2005-1543
2. Lau K, Benitez P, Ardissone A, Wilson TD, Collins EL, Lorca G, et al. Inhibition of type 1 diabetes correlated to a Lactobacillus johnsonii N6.2-mediated Th17 bias. J Immunol (2011) 186(6):3538–46. doi:10.4049/jimmunol.1001864
3. Holman RC, Stoll BJ, Curns AT, Yorita KL, Steiner CA, Schonberger LB. Necrotising enterocolitis hospitalisations among neonates in the United States. Paediatr Perinat Epidemiol (2006) 20(6):498–506. doi:10.1111/j.1365-3016.2006.00756.x
4. Fitzgibbons SC, Ching Y, Yu D, Carpenter J, Kenny M, Weldon C, et al. Mortality of necrotizing enterocolitis expressed by birth weight categories. J Pediatr Surg (2009) 44(6):1072–5; discussion 1075–6. doi:10.1016/j.jpedsurg.2009.02.013
5. Bisquera JA, Cooper TR, Berseth CL. Impact of necrotizing enterocolitis on length of stay and hospital charges in very low birth weight infants. Pediatrics (2002) 109(3):423–8. doi:10.1542/peds.109.3.423
6. Stey A, Barnert ES, Tseng CH, Keeler E, Needleman J, Leng M, et al. Outcomes and costs of surgical treatments of necrotizing enterocolitis. Pediatrics (2015) 135(5):e1190–7. doi:10.1542/peds.2014-1058
7. Neu J, Walker WA. Necrotizing enterocolitis. N Engl J Med (2011) 364(3):255–64. doi:10.1056/NEJMra1005408
8. Nino DF, Sodhi CP, Hackam DJ. Necrotizing enterocolitis: new insights into pathogenesis and mechanisms. Nat Rev Gastroenterol Hepatol (2016) 13(10):590–600. doi:10.1038/nrgastro.2016.119
9. Santaolalla R, Fukata M, Abreu MT. Innate immunity in the small intestine. Curr Opin Gastroenterol (2011) 27(2):125–31. doi:10.1097/MOG.0b013e3283438dea
10. van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol (2009) 71:241–60. doi:10.1146/annurev.physiol.010908.163145
11. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature (2007) 449(7165):1003–7. doi:10.1038/nature06196
12. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature (2009) 459(7244):262–5. doi:10.1038/nature07935
13. Snoeck V, Goddeeris B, Cox E. The role of enterocytes in the intestinal barrier function and antigen uptake. Microbes Infect (2005) 7(7–8):997–1004. doi:10.1016/j.micinf.2005.04.003
14. Vereecke L, Beyaert R, van Loo G. Enterocyte death and intestinal barrier maintenance in homeostasis and disease. Trends Mol Med (2011) 17(10):584–93. doi:10.1016/j.molmed.2011.05.011
15. Vitale S, Picascia S, Gianfrani C. The cross-talk between enterocytes and intraepithelial lymphocytes. Mol Cell Pediatr (2016) 3(1):20. doi:10.1186/s40348-016-0048-4
16. Miron N, Cristea V. Enterocytes: active cells in tolerance to food and microbial antigens in the gut. Clin Exp Immunol (2012) 167(3):405–12. doi:10.1111/j.1365-2249.2011.04523.x
17. Podolsky DK. Healing the epithelium: solving the problem from two sides. J Gastroenterol (1997) 32(1):122–6. doi:10.1007/BF01213309
18. Hackam DJ, Good M, Sodhi CP. Mechanisms of gut barrier failure in the pathogenesis of necrotizing enterocolitis: toll-like receptors throw the switch. Semin Pediatr Surg (2013) 22(2):76–82. doi:10.1053/j.sempedsurg.2013.01.003
19. Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol (2010) 10(2):131–44. doi:10.1038/nri2707
20. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi:10.1016/j.cell.2010.01.022
21. Hershberg RM, Mayer LF. Antigen processing and presentation by intestinal epithelial cells – polarity and complexity. Immunol Today (2000) 21(3):123–8. doi:10.1016/s0167-5699(99)01575-3
22. Hershberg RM, Framson PE, Cho DH, Lee LY, Kovats S, Beitz J, et al. Intestinal epithelial cells use two distinct pathways for HLA class II antigen processing. J Clin Invest (1997) 100(1):204–15. doi:10.1172/JCI119514
23. Tanner SM, Berryhill TF, Ellenburg JL, Jilling T, Cleveland DS, Lorenz RG, et al. Pathogenesis of necrotizing enterocolitis: modeling the innate immune response. Am J Pathol (2015) 185(1):4–16. doi:10.1016/j.ajpath.2014.08.028
24. McDole JR, Wheeler LW, McDonald KG, Wang B, Konjufca V, Knoop KA, et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature (2012) 483(7389):345–9. doi:10.1038/nature10863
25. Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis-Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature (2005) 435(7044):964–8. doi:10.1038/nature03589
26. Sodhi CP, Neal MD, Siggers R, Sho S, Ma C, Branca MF, et al. Intestinal epithelial toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology (2012) 143(3):e1–5. doi:10.1053/j.gastro.2012.05.053
27. Johansson ME, Larsson JM, Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci U S A (2011) 108(Suppl 1):4659–65. doi:10.1073/pnas.1006451107
28. Clark JA, Doelle SM, Halpern MD, Saunders TA, Holubec H, Dvorak K, et al. Intestinal barrier failure during experimental necrotizing enterocolitis: protective effect of EGF treatment. Am J Physiol Gastrointest Liver Physiol (2006) 291(5):G938–49. doi:10.1152/ajpgi.00090.2006
29. Martin NA, Mount Patrick SK, Estrada TE, Frisk HA, Rogan DT, Dvorak B, et al. Active transport of bile acids decreases mucin 2 in neonatal ileum: implications for development of necrotizing enterocolitis. PLoS One (2011) 6(12):e27191. doi:10.1371/journal.pone.0027191
30. Deplancke B, Gaskins HR. Microbial modulation of innate defense: goblet cells and the intestinal mucus layer. Am J Clin Nutr (2001) 73(6):1131S–41S.
31. Berseth CL. Gut motility and the pathogenesis of necrotizing enterocolitis. Clin Perinatol (1994) 21(2):263–70.
32. Hackam DJ, Upperman JS, Grishin A, Ford HR. Disordered enterocyte signaling and intestinal barrier dysfunction in the pathogenesis of necrotizing enterocolitis. Semin Pediatr Surg (2005) 14(1):49–57. doi:10.1053/j.sempedsurg.2004.10.025
33. Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol (2011) 9(5):356–68. doi:10.1038/nrmicro2546
34. Clevers HC, Bevins CL. Paneth cells: maestros of the small intestinal crypts. Annu Rev Physiol (2013) 75:289–311. doi:10.1146/annurev-physiol-030212-183744
35. Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol (2010) 11(1):76–83. doi:10.1038/ni.1825
36. Wehkamp J, Stange EF. Paneth’s disease. J Crohns Colitis (2010) 4(5):523–31. doi:10.1016/j.crohns.2010.05.010
37. Ogura Y. Expression of NOD2 in Paneth cells: a possible link to Crohn’s ileitis. Gut (2003) 52(11):1591–7. doi:10.1136/gut.52.11.1591
38. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature (2001) 411(6837):603–6. doi:10.1038/35079114
39. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature (2001) 411(6837):599–603. doi:10.1038/35079107
40. Hartel C, Hartz A, Pagel J, Rupp J, Stein A, Kribs A, et al. NOD2 loss-of-function mutations and risks of necrotizing enterocolitis or focal intestinal perforation in very low-birth-weight infants. Inflamm Bowel Dis (2016) 22(2):249–56. doi:10.1097/MIB.0000000000000658
41. Biswas A, Liu YJ, Hao L, Mizoguchi A, Salzman NH, Bevins CL, et al. Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proc Natl Acad Sci U S A (2010) 107(33):14739–44. doi:10.1073/pnas.1003363107
42. McElroy SJ, Underwood MA, Sherman MP. Paneth cells and necrotizing enterocolitis: a novel hypothesis for disease pathogenesis. Neonatology (2013) 103(1):10–20. doi:10.1159/000342340
43. Zhang C, Sherman MP, Prince LS, Bader D, Weitkamp JH, Slaughter JC, et al. Paneth cell ablation in the presence of Klebsiella pneumoniae induces necrotizing enterocolitis (NEC)-like injury in the small intestine of immature mice. Dis Model Mech (2012) 5(4):522–32. doi:10.1242/dmm.009001
44. Underwood MA. Paneth cells and necrotizing enterocolitis. Gut Microbes (2012) 3(6):562–5. doi:10.4161/gmic.21738
45. Gribble FM, Reimann F. Enteroendocrine cells: chemosensors in the intestinal epithelium. Annu Rev Physiol (2016) 78:277–99. doi:10.1146/annurev-physiol-021115-105439
46. Worthington JJ. The intestinal immunoendocrine axis: novel cross-talk between enteroendocrine cells and the immune system during infection and inflammatory disease. Biochem Soc Trans (2015) 43(4):727–33. doi:10.1042/BST20150090
47. Palazzo M, Balsari A, Rossini A, Selleri S, Calcaterra C, Gariboldi S, et al. Activation of enteroendocrine cells via TLRs induces hormone, chemokine, and defensin secretion. J Immunol (2007) 178(7):4296–303. doi:10.4049/jimmunol.178.7.4296
48. Bogunovic M, Dave SH, Tilstra JS, Chang DT, Harpaz N, Xiong H, et al. Enteroendocrine cells express functional toll-like receptors. Am J Physiol Gastrointest Liver Physiol (2007) 292(6):G1770–83. doi:10.1152/ajpgi.00249.2006
49. Cani PD, Everard A, Duparc T. Gut microbiota, enteroendocrine functions and metabolism. Curr Opin Pharmacol (2013) 13(6):935–40. doi:10.1016/j.coph.2013.09.008
50. Friedrich M, Diegelmann J, Schauber J, Auernhammer CJ, Brand S. Intestinal neuroendocrine cells and goblet cells are mediators of IL-17A-amplified epithelial IL-17C production in human inflammatory bowel disease. Mucosal Immunol (2015) 8(4):943–58. doi:10.1038/mi.2014.124
51. Gerbe F, Jay P. Intestinal tuft cells: epithelial sentinels linking luminal cues to the immune system. Mucosal Immunol (2016) 9(6):1353–9. doi:10.1038/mi.2016.68
53. Harris N. IMMUNOLOGY. The enigmatic tuft cell in immunity. Science (2016) 351(6279):1264–5. doi:10.1126/science.aaf5215
54. Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science (2016) 351(6279):1329–33. doi:10.1126/science.aaf1648
55. Gerbe F, Sidot E, Smyth DJ, Ohmoto M, Matsumoto I, Dardalhon V, et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature (2016) 529(7585):226–30. doi:10.1038/nature16527
56. von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature (2016) 529(7585):221–5. doi:10.1038/nature16161
57. McElroy SJ, Weitkamp JH. Innate immunity in the small intestine of the preterm infant. Neoreviews (2011) 12(9):e517–26. doi:10.1542/neo.12-9-e517
58. Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell (1992) 68(5):869–77. doi:10.1016/0092-8674(92)90030-g
59. Egan CE, Sodhi CP, Good M, Lin J, Jia H, Yamaguchi Y, et al. Toll-like receptor 4-mediated lymphocyte influx induces neonatal necrotizing enterocolitis. J Clin Invest (2015) 126(2):495–508. doi:10.1172/JCI83356
60. Weitkamp JH, Rudzinski E, Koyama T, Correa H, Matta P, Alberty B, et al. Ontogeny of FOXP3(+) regulatory T cells in the postnatal human small intestinal and large intestinal lamina propria. Pediatr Dev Pathol (2009) 12(6):443–9. doi:10.2350/08-09-0533.1
61. Cong Y, Feng T, Fujihashi K, Schoeb TR, Elson CO. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc Natl Acad Sci U S A (2009) 106(46):19256–61. doi:10.1073/pnas.0812681106
62. Weitkamp JH, Koyama T, Rock MT, Correa H, Goettel JA, Matta P, et al. Necrotising enterocolitis is characterised by disrupted immune regulation and diminished mucosal regulatory (FOXP3)/effector (CD4, CD8) T cell ratios. Gut (2013) 62(1):73–82. doi:10.1136/gutjnl-2011-301551
63. Nino DF, Sodhi CP, Egan CE, Zhou Q, Lin J, Lu P, et al. Retinoic acid improves incidence and severity of necrotizing enterocolitis by lymphocyte balance restitution and repopulation of LGR5+ intestinal stem cells. Shock (2016) 47(1):22–32. doi:10.1097/SHK.0000000000000713
64. Cheroutre H, Lambolez F, Mucida D. The light and dark sides of intestinal intraepithelial lymphocytes. Nat Rev Immunol (2011) 11(7):445–56. doi:10.1038/nri3007
65. Sheridan BS, Lefrancois L. Intraepithelial lymphocytes: to serve and protect. Curr Gastroenterol Rep (2010) 12(6):513–21. doi:10.1007/s11894-010-0148-6
66. Weitkamp JH, Rosen MJ, Zhao Z, Koyama T, Geem D, Denning TL, et al. Small intestinal intraepithelial TCRgammadelta+ T lymphocytes are present in the premature intestine but selectively reduced in surgical necrotizing enterocolitis. PLoS One (2014) 9(6):e99042. doi:10.1371/journal.pone.0099042
67. Gibbons DL, Haque SF, Silberzahn T, Hamilton K, Langford C, Ellis P, et al. Neonates harbour highly active gammadelta T cells with selective impairments in preterm infants. Eur J Immunol (2009) 39(7):1794–806. doi:10.1002/eji.200939222
68. Ismail AS, Behrendt CL, Hooper LV. Reciprocal interactions between commensal bacteria and gamma delta intraepithelial lymphocytes during mucosal injury. J Immunol (2009) 182(5):3047–54. doi:10.4049/jimmunol.0802705
69. Borges MC, Sesso ML, Roberti LR, de Menezes Oliveira MA, Nogueira RD, Geraldo-Martins VR, et al. Salivary antibody response to streptococci in preterm and fullterm children: a prospective study. Arch Oral Biol (2015) 60(1):116–25. doi:10.1016/j.archoralbio.2014.08.003
70. Nogueira RD, Sesso ML, Borges MC, Mattos-Graner RO, Smith DJ, Ferriani VP. Salivary IgA antibody responses to Streptococcus mitis and Streptococcus mutans in preterm and fullterm newborn children. Arch Oral Biol (2012) 57(6):647–53. doi:10.1016/j.archoralbio.2011.11.011
71. Araújo ED, Gonçalves AK, da Conceição Cornetta M, Cunha H, Cardoso ML, Morais SS, et al. Evaluation of the secretory immunoglobulin A levels in the colostrum and milk of mothers of term and pre-term newborns. Braz J Infect Dis (2005) 9(5):357–62. doi:10.1590/S1413-86702005000500002
72. Rogier EW, Frantz AL, Bruno ME, Wedlund L, Cohen DA, Stromberg AJ, et al. Secretory antibodies in breast milk promote long-term intestinal homeostasis by regulating the gut microbiota and host gene expression. Proc Natl Acad Sci U S A (2014) 111(8):3074–9. doi:10.1073/pnas.1315792111
73. Maheshwari A, Kelly DR, Nicola T, Ambalavanan N, Jain SK, Murphy-Ullrich J, et al. TGF-beta2 suppresses macrophage cytokine production and mucosal inflammatory responses in the developing intestine. Gastroenterology (2011) 140(1):242–53. doi:10.1053/j.gastro.2010.09.043
74. MohanKumar K, Namachivayam K, Chapalamadugu KC, Garzon SA, Premkumar MH, Tipparaju SM, et al. Smad7 interrupts TGF-beta signaling in intestinal macrophages and promotes inflammatory activation of these cells during necrotizing enterocolitis. Pediatr Res (2016) 79(6):951–61. doi:10.1038/pr.2016.18
75. Namachivayam K, Blanco CL, MohanKumar K, Jagadeeswaran R, Vasquez M, McGill-Vargas L, et al. Smad7 inhibits autocrine expression of TGF-beta2 in intestinal epithelial cells in baboon necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol (2013) 304(2):G167–80. doi:10.1152/ajpgi.00141.2012
76. Steimle A, Frick JS. Molecular mechanisms of induction of tolerant and tolerogenic intestinal dendritic cells in mice. J Immunol Res (2016) 2016:1958650. doi:10.1155/2016/1958650
77. Emami CN, Mittal R, Wang L, Ford HR, Prasadarao NV. Recruitment of dendritic cells is responsible for intestinal epithelial damage in the pathogenesis of necrotizing enterocolitis by Cronobacter sakazakii. J Immunol (2011) 186(12):7067–79. doi:10.4049/jimmunol.1100108
78. Christensen RD, Yoder BA, Baer VL, Snow GL, Butler A. Early-onset neutropenia in small-for-gestational-age infants. Pediatrics (2015) 136(5):e1259–67. doi:10.1542/peds.2015-1638
79. Emami CN, Mittal R, Wang L, Ford HR, Prasadarao NV. Role of neutrophils and macrophages in the pathogenesis of necrotizing enterocolitis caused by Cronobacter sakazakii. J Surg Res (2012) 172(1):18–28. doi:10.1016/j.jss.2011.04.019
80. MohanKumar K, Kaza N, Jagadeeswaran R, Garzon SA, Bansal A, Kurundkar AR, et al. Gut mucosal injury in neonates is marked by macrophage infiltration in contrast to pleomorphic infiltrates in adult: evidence from an animal model. Am J Physiol Gastrointest Liver Physiol (2012) 303(1):G93–102. doi:10.1152/ajpgi.00016.2012
81. Reboldi A, Cyster JG. Peyer’s patches: organizing B-cell responses at the intestinal frontier. Immunol Rev (2016) 271(1):230–45. doi:10.1111/imr.12400
82. Maheshwari A. Immunologic and hematological abnormalities in necrotizing enterocolitis. Clin Perinatol (2015) 42(3):567–85. doi:10.1016/j.clp.2015.04.014
83. Lu P, Sodhi CP, Hackam DJ. Toll-like receptor regulation of intestinal development and inflammation in the pathogenesis of necrotizing enterocolitis. Pathophysiology (2014) 21(1):81–93. doi:10.1016/j.pathophys.2013.11.007
84. Hackam DJ, Afrazi A, Good M, Sodhi CP. Innate immune signaling in the pathogenesis of necrotizing enterocolitis. Clin Dev Immunol (2013) 2013:475415. doi:10.1155/2013/475415
85. Neal MD, Leaphart C, Levy R, Prince J, Billiar TR, Watkins S, et al. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J Immunol (2006) 176(5):3070–9. doi:10.4049/jimmunol.176.5.3070
86. Cario E. Barrier-protective function of intestinal epithelial toll-like receptor 2. Mucosal Immunol (2008) 1(Suppl 1):S62–6. doi:10.1038/mi.2008.47
87. Liu Y, Zhu L, Fatheree NY, Liu X, Pacheco SE, Tatevian N, et al. Changes in intestinal toll-like receptors and cytokines precede histological injury in a rat model of necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol (2009) 297(3):G442–50. doi:10.1152/ajpgi.00182.2009
88. Zhou W, Li W, Zheng XH, Rong X, Huang LG. Glutamine downregulates TLR-2 and TLR-4 expression and protects intestinal tract in preterm neonatal rats with necrotizing enterocolitis. J Pediatr Surg (2014) 49(7):1057–63. doi:10.1016/j.jpedsurg.2014.02.078
89. Zhou W, Lv H, Li MX, Su H, Huang LG, Li J, et al. Protective effects of Bifidobacteria on intestines in newborn rats with necrotizing enterocolitis and its regulation on TLR2 and TLR4. Genet Mol Res (2015) 14(3):11505–14. doi:10.4238/2015.September.28.2
90. Richardson WM, Sodhi CP, Russo A, Siggers RH, Afrazi A, Gribar SC, et al. Nucleotide-binding oligomerization domain-2 inhibits toll-like receptor-4 signaling in the intestinal epithelium. Gastroenterology (2010) 139(3):e1–6. doi:10.1053/j.gastro.2010.05.038
91. Petnicki-Ocwieja T, Hrncir T, Liu YJ, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A (2009) 106(37):15813–8. doi:10.1073/pnas.0907722106
92. Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol (2006) 6(1):9–20. doi:10.1038/nri1747
93. Afrazi A, Branca MF, Sodhi CP, Good M, Yamaguchi Y, Egan CE, et al. Toll like receptor 4-mediated endoplasmic reticulum stress in intestinal crypts induces necrotizing enterocolitis. J Biol Chem (2014) 289(14):9584–99. doi:10.1074/jbc.M113.526517
94. Good M, Siggers RH, Sodhi CP, Afrazi A, Alkhudari F, Egan CE, et al. Amniotic fluid inhibits toll-like receptor 4 signaling in the fetal and neonatal intestinal epithelium. Proc Natl Acad Sci U S A (2012) 109(28):11330–5. doi:10.1073/pnas.1200856109
95. Good M, Sodhi CP, Egan CE, Afrazi A, Jia H, Yamaguchi Y, et al. Breast milk protects against the development of necrotizing enterocolitis through inhibition of toll-like receptor 4 in the intestinal epithelium via activation of the epidermal growth factor receptor. Mucosal Immunol (2015) 8(5):1166–79. doi:10.1038/mi.2015.30
96. Gribar SC, Sodhi CP, Richardson WM, Anand RJ, Gittes GK, Branca MF, et al. Reciprocal expression and signaling of TLR4 and TLR9 in the pathogenesis and treatment of necrotizing enterocolitis. J Immunol (2009) 182(1):636–46. doi:10.4049/jimmunol.182.1.636
97. Leaphart CL, Cavallo J, Gribar SC, Cetin S, Li J, Branca MF, et al. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J Immunol (2007) 179(7):4808–20. doi:10.4049/jimmunol.179.7.4808
98. Neal MD, Sodhi CP, Jia H, Dyer M, Egan CE, Yazji I, et al. Toll-like receptor 4 is expressed on intestinal stem cells and regulates their proliferation and apoptosis via the p53 up-regulated modulator of apoptosis. J Biol Chem (2012) 287(44):37296–308. doi:10.1074/jbc.M112.375881
99. Sodhi CP, Shi XH, Richardson WM, Grant ZS, Shapiro RA, Prindle T Jr, et al. Toll-like receptor-4 inhibits enterocyte proliferation via impaired beta-catenin signaling in necrotizing enterocolitis. Gastroenterology (2010) 138(1):185–96. doi:10.1053/j.gastro.2009.09.045
100. Yazji I, Sodhi CP, Lee EK, Good M, Egan CE, Afrazi A, et al. Endothelial TLR4 activation impairs intestinal microcirculatory perfusion in necrotizing enterocolitis via eNOS-NO-nitrite signaling. Proc Natl Acad Sci U S A (2013) 110(23):9451–6. doi:10.1073/pnas.1219997110
101. Good M, Sodhi CP, Yamaguchi Y, Jia H, Lu P, Fulton WB, et al. The human milk oligosaccharide 2’-fucosyllactose attenuates the severity of experimental necrotising enterocolitis by enhancing mesenteric perfusion in the neonatal intestine. Br J Nutr (2016) 116(7):1175–87. doi:10.1017/S0007114516002944
102. Cetin S, Ford HR, Sysko LR, Agarwal C, Wang J, Neal MD, et al. Endotoxin inhibits intestinal epithelial restitution through activation of Rho-GTPase and increased focal adhesions. J Biol Chem (2004) 279(23):24592–600. doi:10.1074/jbc.M313620200
103. Qureshi FG, Leaphart C, Cetin S, Li J, Grishin A, Watkins S, et al. Increased expression and function of integrins in enterocytes by endotoxin impairs epithelial restitution. Gastroenterology (2005) 128(4):1012–22. doi:10.1053/j.gastro.2005.01.052
104. Yu Y, Shiou SR, Guo Y, Lu L, Westerhoff M, Sun J, et al. Erythropoietin protects epithelial cells from excessive autophagy and apoptosis in experimental neonatal necrotizing enterocolitis. PLoS One (2013) 8(7):e69620. doi:10.1371/journal.pone.0069620
105. Neal MD, Sodhi CP, Dyer M, Craig BT, Good M, Jia H, et al. A critical role for TLR4 induction of autophagy in the regulation of enterocyte migration and the pathogenesis of necrotizing enterocolitis. J Immunol (2013) 190(7):3541–51. doi:10.4049/jimmunol.1202264
106. Cuna A, Sampath V. Genetic alterations in necrotizing enterocolitis. Semin Perinatol (2016). doi:10.1053/j.semperi.2016.09.019
107. Sampath V, Menden H, Helbling D, Li K, Gastonguay A, Ramchandran R, et al. SIGIRR genetic variants in premature infants with necrotizing enterocolitis. Pediatrics (2015) 135(6):e1530–4. doi:10.1542/peds.2014-3386
108. Sampath V, Le M, Lane L, Patel AL, Cohen JD, Simpson PM, et al. The NFKB1 (g.-24519delATTG) variant is associated with necrotizing enterocolitis (NEC) in premature infants. J Surg Res (2011) 169(1):e51–7. doi:10.1016/j.jss.2011.03.017
109. Sampath V, Bhandari V, Berger J, Merchant D, Zhang L, Ladd M, et al. A functional ATG16L1 (T300A) variant is associated with necrotizing enterocolitis in premature infants. Pediatr Res (2017). doi:10.1038/pr.2016.260
110. Collado MC, Cernada M, Neu J, Perez-Martinez G, Gormaz M, Vento M. Factors influencing gastrointestinal tract and microbiota immune interaction in preterm infants. Pediatr Res (2015) 77(6):726–31. doi:10.1038/pr.2015.54
111. Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A (2010) 107(26):11971–5. doi:10.1073/pnas.1002601107
112. Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, et al. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics (2006) 118(2):511–21. doi:10.1542/peds.2005-2824
113. Elgin TG, Kern SL, McElroy SJ. Development of the neonatal intestinal microbiome and its association with necrotizing enterocolitis. Clin Ther (2016) 38(4):706–15. doi:10.1016/j.clinthera.2016.01.005
114. Penders J, Vink C, Driessen C, London N, Thijs C, Stobberingh EE. Quantification of Bifidobacterium spp., Escherichia coli and Clostridium difficile in faecal samples of breast-fed and formula-fed infants by real-time PCR. FEMS Microbiol Lett (2005) 243(1):141–7. doi:10.1016/j.femsle.2004.11.052
115. Alexander VN, Northrup V, Bizzarro MJ. Antibiotic exposure in the newborn intensive care unit and the risk of necrotizing enterocolitis. J Pediatr (2011) 159(3):392–7. doi:10.1016/j.jpeds.2011.02.035
116. Cotten CM, Taylor S, Stoll B, Goldberg RN, Hansen NI, Sanchez PJ, et al. Prolonged duration of initial empirical antibiotic treatment is associated with increased rates of necrotizing enterocolitis and death for extremely low birth weight infants. Pediatrics (2009) 123(1):58–66. doi:10.1542/peds.2007-3423
117. La Rosa PS, Warner BB, Zhou Y, Weinstock GM, Sodergren E, Hall-Moore CM, et al. Patterned progression of bacterial populations in the premature infant gut. Proc Natl Acad Sci U S A (2014) 111(34):12522–7. doi:10.1073/pnas.1409497111
118. Arboleya S, Ang L, Margolles A, Yiyuan L, Dongya Z, Liang X, et al. Deep 16S rRNA metagenomics and quantitative PCR analyses of the premature infant fecal microbiota. Anaerobe (2012) 18(3):378–80. doi:10.1016/j.anaerobe.2012.04.013
119. Carlisle EM, Morowitz MJ. The intestinal microbiome and necrotizing enterocolitis. Curr Opin Pediatr (2013) 25(3):382–7. doi:10.1097/MOP.0b013e3283600e91
120. Torrazza RM, Neu J. The altered gut microbiome and necrotizing enterocolitis. Clin Perinatol (2013) 40(1):93–108. doi:10.1016/j.clp.2012.12.009
121. Vongbhavit K, Underwood MA. Prevention of necrotizing enterocolitis through manipulation of the intestinal microbiota of the premature infant. Clin Ther (2016) 38(4):716–32. doi:10.1016/j.clinthera.2016.01.006
122. Patel RM, Denning PW. Intestinal microbiota and its relationship with necrotizing enterocolitis. Pediatr Res (2015) 78(3):232–8. doi:10.1038/pr.2015.97
123. Yee WH, Soraisham AS, Shah VS, Aziz K, Yoon W, Lee SK, et al. Incidence and timing of presentation of necrotizing enterocolitis in preterm infants. Pediatrics (2012) 129(2):e298–304. doi:10.1542/peds.2011-2022
124. Neu J. Preterm infant nutrition, gut bacteria, and necrotizing enterocolitis. Curr Opin Clin Nutr Metab Care (2015) 18(3):285–8. doi:10.1097/MCO.0000000000000169
125. Coggins SA, Wynn JL, Weitkamp JH. Infectious causes of necrotizing enterocolitis. Clin Perinatol (2015) 42(1):133–54,ix. doi:10.1016/j.clp.2014.10.012
126. Brower-Sinning R, Zhong D, Good M, Firek B, Baker R, Sodhi CP, et al. Mucosa-associated bacterial diversity in necrotizing enterocolitis. PLoS One (2014) 9(9):e105046. doi:10.1371/journal.pone.0105046
127. Warner BB, Deych E, Zhou Y, Hall-Moore C, Weinstock GM, Sodergren E, et al. Gut bacteria dysbiosis and necrotising enterocolitis in very low birthweight infants: a prospective case-control study. Lancet (2016) 387(10031):1928–36. doi:10.1016/s0140-6736(16)00081-7
128. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341(6145):569–73. doi:10.1126/science.1241165
129. Lonnerdal B. Bioactive proteins in human milk: mechanisms of action. J Pediatr (2010) 156(2 Suppl):S26–30. doi:10.1016/j.jpeds.2009.11.017
130. Good M, Sodhi CP, Hackam DJ. Evidence-based feeding strategies before and after the development of necrotizing enterocolitis. Expert Rev Clin Immunol (2014) 10(7):875–84. doi:10.1586/1744666X.2014.913481
131. Dvorak B, Halpern MD, Holubec H, Williams CS, McWilliam DL, Dominguez JA, et al. Epidermal growth factor reduces the development of necrotizing enterocolitis in a neonatal rat model. Am J Physiol Gastrointest Liver Physiol (2002) 282(1):G156–64. doi:10.1152/ajpgi.00196.2001
132. Clark JA, Lane RH, Maclennan NK, Holubec H, Dvorakova K, Halpern MD, et al. Epidermal growth factor reduces intestinal apoptosis in an experimental model of necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol (2005) 288(4):G755–62. doi:10.1152/ajpgi.00172.2004
133. Maynard AA, Dvorak K, Khailova L, Dobrenen H, Arganbright KM, Halpern MD, et al. Epidermal growth factor reduces autophagy in intestinal epithelium and in the rat model of necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol (2010) 299(3):G614–22. doi:10.1152/ajpgi.00076.2010
134. Jain SK, Baggerman EW, Mohankumar K, Namachivayam K, Jagadeeswaran R, Reyes VE, et al. Amniotic fluid-borne hepatocyte growth factor protects rat pups against experimental necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol (2014) 306(5):G361–9. doi:10.1152/ajpgi.00272.2013
135. Siggers J, Ostergaard MV, Siggers RH, Skovgaard K, Molbak L, Thymann T, et al. Postnatal amniotic fluid intake reduces gut inflammatory responses and necrotizing enterocolitis in preterm neonates. Am J Physiol Gastrointest Liver Physiol (2013) 304(10):G864–75. doi:10.1152/ajpgi.00278.2012
136. Zani A, Cananzi M, Fascetti-Leon F, Lauriti G, Smith VV, Bollini S, et al. Amniotic fluid stem cells improve survival and enhance repair of damaged intestine in necrotising enterocolitis via a COX-2 dependent mechanism. Gut (2014) 63(2):300–9. doi:10.1136/gutjnl-2012-303735
137. Ostergaard MV, Shen RL, Stoy AC, Skovgaard K, Krych L, Leth SS, et al. Provision of amniotic fluid during parenteral nutrition increases weight gain with limited effects on gut structure, function, immunity, and microbiology in newborn preterm pigs. JPEN J Parenter Enteral Nutr (2016) 40(4):552–66. doi:10.1177/0148607114566463
138. Yang J, Su Y, Zhou Y, Besner GE. Heparin-binding EGF-like growth factor (HB-EGF) therapy for intestinal injury: application and future prospects. Pathophysiology (2014) 21(1):95–104. doi:10.1016/j.pathophys.2013.11.008
139. Radulescu A, Zhang HY, Yu X, Olson JK, Darbyshire AK, Chen Y, et al. Heparin-binding epidermal growth factor-like growth factor overexpression in transgenic mice increases resistance to necrotizing enterocolitis. J Pediatr Surg (2010) 45(10):1933–9. doi:10.1016/j.jpedsurg.2010.05.002
140. Radulescu A, Yu X, Orvets ND, Chen Y, Zhang HY, Besner GE. Deletion of the heparin-binding epidermal growth factor-like growth factor gene increases susceptibility to necrotizing enterocolitis. J Pediatr Surg (2010) 45(4):729–34. doi:10.1016/j.jpedsurg.2009.06.035
141. Yu X, Radulescu A, Zorko N, Besner GE. Heparin-binding EGF-like growth factor increases intestinal microvascular blood flow in necrotizing enterocolitis. Gastroenterology (2009) 137(1):221–30. doi:10.1053/j.gastro.2009.03.060
142. Mehta VB, Besner GE. HB-EGF promotes angiogenesis in endothelial cells via PI3-kinase and MAPK signaling pathways. Growth Factors (2007) 25(4):253–63. doi:10.1080/08977190701773070
143. Mehta VB, Zhou Y, Radulescu A, Besner GE. HB-EGF stimulates eNOS expression and nitric oxide production and promotes eNOS dependent angiogenesis. Growth Factors (2008) 26(6):301–15. doi:10.1080/08977190802393596
144. Wei J, Besner GE. M1 to M2 macrophage polarization in heparin-binding epidermal growth factor-like growth factor therapy for necrotizing enterocolitis. J Surg Res (2015) 197(1):126–38. doi:10.1016/j.jss.2015.03.023
145. Feng J, Besner GE. Heparin-binding epidermal growth factor-like growth factor promotes enterocyte migration and proliferation in neonatal rats with necrotizing enterocolitis. J Pediatr Surg (2007) 42(1):214–20. doi:10.1016/j.jpedsurg.2006.09.055
146. Su Y, Besner GE. Heparin-binding EGF-like growth factor (HB-EGF) promotes cell migration and adhesion via focal adhesion kinase. J Surg Res (2014) 189(2):222–31. doi:10.1016/j.jss.2014.02.055
147. Feng J, El-Assal ON, Besner GE. Heparin-binding epidermal growth factor-like growth factor reduces intestinal apoptosis in neonatal rats with necrotizing enterocolitis. J Pediatr Surg (2006) 41(4):742–7; discussion 742–7. doi:10.1016/j.jpedsurg.2005.12.020
148. Frost BL, Caplan MS. Necrotizing enterocolitis: pathophysiology, platelet-activating factor, and probiotics. Semin Pediatr Surg (2013) 22(2):88–93. doi:10.1053/j.sempedsurg.2013.01.005
149. Caplan MS, Simon D, Jilling T. The role of PAF, TLR, and the inflammatory response in neonatal necrotizing enterocolitis. Semin Pediatr Surg (2005) 14(3):145–51. doi:10.1053/j.sempedsurg.2005.05.002
150. Furukawa M, Narahara H, Yasuda K, Johnston JM. Presence of platelet-activating factor-acetylhydrolase in milk. J Lipid Res (1993) 34(9):1603–9.
151. Moya FR, Eguchi H, Zhao B, Furukawa M, Sfeir J, Osorio M, et al. Platelet-activating factor acetylhydrolase in term and preterm human milk: a preliminary report. J Pediatr Gastroenterol Nutr (1994) 19(2):236–9. doi:10.1097/00005176-199408000-00015
152. Caplan MS, Sun XM, Hseuh W, Hageman JR. Role of platelet activating factor and tumor necrosis factor-alpha in neonatal necrotizing enterocolitis. J Pediatr (1990) 116(6):960–4. doi:10.1016/S0022-3476(05)80661-4
153. Amer MD, Hedlund E, Rochester J, Caplan MS. Platelet-activating factor concentration in the stool of human newborns: effects of enteral feeding and neonatal necrotizing enterocolitis. Biol Neonate (2004) 85(3):159–66. doi:10.1159/000075306
154. Rabinowitz SS, Dzakpasu P, Piecuch S, Leblanc P, Valencia G, Kornecki E. Platelet-activating factor in infants at risk for necrotizing enterocolitis. J Pediatr (2001) 138(1):81–6. doi:10.1067/mpd.2001.110132
155. Caplan MS, Hedlund E, Adler L, Lickerman M, Hsueh W. The platelet-activating factor receptor antagonist WEB 2170 prevents neonatal necrotizing enterocolitis in rats. J Pediatr Gastroenterol Nutr (1997) 24(3):296–301. doi:10.1097/00005176-199703000-00012
156. Caplan MS, Lickerman M, Adler L, Dietsch GN, Yu A. The role of recombinant platelet-activating factor acetylhydrolase in a neonatal rat model of necrotizing enterocolitis. Pediatr Res (1997) 42(6):779–83. doi:10.1203/00006450-199712000-00010
157. Lu J, Pierce M, Franklin A, Jilling T, Stafforini DM, Caplan M. Dual roles of endogenous platelet-activating factor acetylhydrolase in a murine model of necrotizing enterocolitis. Pediatr Res (2010) 68(3):225–30. doi:10.1203/00006450-201011001-00439
158. Soliman A, Michelsen KS, Karahashi H, Lu J, Meng FJ, Qu X, et al. Platelet-activating factor induces TLR4 expression in intestinal epithelial cells: implication for the pathogenesis of necrotizing enterocolitis. PLoS One (2010) 5(10):e15044. doi:10.1371/journal.pone.0015044
159. Yu ZT, Chen C, Kling DE, Liu B, McCoy JM, Merighi M, et al. The principal fucosylated oligosaccharides of human milk exhibit prebiotic properties on cultured infant microbiota. Glycobiology (2013) 23(2):169–77. doi:10.1093/glycob/cws138
160. Yu ZT, Chen C, Newburg DS. Utilization of major fucosylated and sialylated human milk oligosaccharides by isolated human gut microbes. Glycobiology (2013) 23(11):1281–92. doi:10.1093/glycob/cwt065
161. Weichert S, Jennewein S, Hufner E, Weiss C, Borkowski J, Putze J, et al. Bioengineered 2’-fucosyllactose and 3-fucosyllactose inhibit the adhesion of Pseudomonas aeruginosa and enteric pathogens to human intestinal and respiratory cell lines. Nutr Res (2013) 33(10):831–8. doi:10.1016/j.nutres.2013.07.009
162. Lane JA, Mehra RK, Carrington SD, Hickey RM. The food glycome: a source of protection against pathogen colonization in the gastrointestinal tract. Int J Food Microbiol (2010) 142(1–2):1–13. doi:10.1016/j.ijfoodmicro.2010.05.027
163. Ruiz-Palacios GM, Cervantes LE, Ramos P, Chavez-Munguia B, Newburg DS. Campylobacter jejuni binds intestinal H(O) antigen (Fuc alpha 1, 2Gal beta 1, 4GlcNAc), and fucosyloligosaccharides of human milk inhibit its binding and infection. J Biol Chem (2003) 278(16):14112–20. doi:10.1074/jbc.M207744200
164. Newburg DS. Neonatal protection by an innate immune system of human milk consisting of oligosaccharides and glycans. J Anim Sci (2009) 87(13 Suppl):26–34. doi:10.2527/jas.2008-1347
165. Pacheco AR, Barile D, Underwood MA, Mills DA. The impact of the milk glycobiome on the neonate gut microbiota. Annu Rev Anim Biosci (2015) 3:419–45. doi:10.1146/annurev-animal-022114-111112
166. He Y, Liu S, Kling DE, Leone S, Lawlor NT, Huang Y, et al. The human milk oligosaccharide 2’-fucosyllactose modulates CD14 expression in human enterocytes, thereby attenuating LPS-induced inflammation. Gut (2016) 65(1):33–46. doi:10.1136/gutjnl-2014-307544
167. Jantscher-Krenn E, Zherebtsov M, Nissan C, Goth K, Guner YS, Naidu N, et al. The human milk oligosaccharide disialyllacto-N-tetraose prevents necrotising enterocolitis in neonatal rats. Gut (2012) 61(10):1417–25. doi:10.1136/gutjnl-2011-301404
168. Lewis ZT, Totten SM, Smilowitz JT, Popovic M, Parker E, Lemay DG, et al. Maternal fucosyltransferase 2 status affects the gut bifidobacterial communities of breastfed infants. Microbiome (2015) 3:13. doi:10.1186/s40168-015-0071-z
169. Jones JA, Ninnis JR, Hopper AO, Ibrahim Y, Merritt TA, Wan KW, et al. Nitrite and nitrate concentrations and metabolism in breast milk, infant formula, and parenteral nutrition. JPEN J Parenter Enteral Nutr (2013) 38(7):856–66. doi:10.1177/0148607113496118
170. Jones JA, Hopper AO, Power GG, Blood AB. Dietary intake and bio-activation of nitrite and nitrate in newborn infants. Pediatr Res (2015) 77(1–2):173–81. doi:10.1038/pr.2014.168
171. Amin HJ, Zamora SA, McMillan DD, Fick GH, Butzner JD, Parsons HG, et al. Arginine supplementation prevents necrotizing enterocolitis in the premature infant. J Pediatr (2002) 140(4):425–31. doi:10.1067/mpd.2002.123289
172. Polycarpou E, Zachaki S, Tsolia M, Papaevangelou V, Polycarpou N, Briana DD, et al. Enteral l-arginine supplementation for prevention of necrotizing enterocolitis in very low birth weight neonates: a double-blind randomized pilot study of efficacy and safety. JPEN J Parenter Enteral Nutr (2013) 37(5):617–22. doi:10.1177/0148607112471561
173. Sherman MP. Lactoferrin and necrotizing enterocolitis. Clin Perinatol (2013) 40(1):79–91. doi:10.1016/j.clp.2012.12.006
174. Hu W, Zhao J, Wang J, Yu T, Wang J, Li N. Transgenic milk containing recombinant human lactoferrin modulates the intestinal flora in piglets. Biochem Cell Biol (2012) 90(3):485–96. doi:10.1139/o2012-003
175. Buccigrossi V, de Marco G, Bruzzese E, Ombrato L, Bracale I, Polito G, et al. Lactoferrin induces concentration-dependent functional modulation of intestinal proliferation and differentiation. Pediatr Res (2007) 61(4):410–4. doi:10.1203/pdr.0b013e3180332c8d
176. Pammi M, Abrams SA. Oral lactoferrin for the prevention of sepsis and necrotizing enterocolitis in preterm infants. Cochrane Database Syst Rev (2015) (2):CD007137. doi:10.1002/14651858.CD007137.pub4
177. Manzoni P, Rinaldi M, Cattani S, Pugni L, Romeo MG, Messner H, et al. Bovine lactoferrin supplementation for prevention of late-onset sepsis in very low-birth-weight neonates: a randomized trial. JAMA (2009) 302(13):1421–8. doi:10.1001/jama.2009.1403
178. Akin IM, Atasay B, Dogu F, Okulu E, Arsan S, Karatas HD, et al. Oral lactoferrin to prevent nosocomial sepsis and necrotizing enterocolitis of premature neonates and effect on T-regulatory cells. Am J Perinatol (2014) 31(12):1111–20. doi:10.1055/s-0034-1371704
179. Manzoni P, Meyer M, Stolfi I, Rinaldi M, Cattani S, Pugni L, et al. Bovine lactoferrin supplementation for prevention of necrotizing enterocolitis in very-low-birth-weight neonates: a randomized clinical trial. Early Hum Dev (2014) 90(Suppl 1):S60–5. doi:10.1016/S0378-3782(14)70020-9
180. Manzoni P, Stolfi I, Messner H, Cattani S, Laforgia N, Romeo MG, et al. Bovine lactoferrin prevents invasive fungal infections in very low birth weight infants: a randomized controlled trial. Pediatrics (2012) 129(1):116–23. doi:10.1542/peds.2011-0279
181. Sherman MP, Adamkin DH, Niklas V, Radmacher P, Sherman J, Wertheimer F, et al. Randomized controlled trial of talactoferrin oral solution in preterm infants. J Pediatr (2016) 175:68–73e3. doi:10.1016/j.jpeds.2016.04.084
182. AlFaleh K, Anabrees J. Probiotics for prevention of necrotizing enterocolitis in preterm infants. Cochrane Database Syst Rev (2014) (4):CD005496. doi:10.1002/14651858.CD005496.pub4
183. Sawh SC, Deshpande S, Jansen S, Reynaert CJ, Jones PM. Prevention of necrotizing enterocolitis with probiotics: a systematic review and meta-analysis. Peer J (2016) 4:e2429. doi:10.7717/peerj.2429
184. Stratiki Z, Costalos C, Sevastiadou S, Kastanidou O, Skouroliakou M, Giakoumatou A, et al. The effect of a bifidobacter supplemented bovine milk on intestinal permeability of preterm infants. Early Hum Dev (2007) 83(9):575–9. doi:10.1016/j.earlhumdev.2006.12.002
185. Mohan R, Koebnick C, Schildt J, Schmidt S, Mueller M, Possner M, et al. Effects of Bifidobacterium lactis Bb12 supplementation on intestinal microbiota of preterm infants: a double-blind, placebo-controlled, randomized study. J Clin Microbiol (2006) 44(11):4025–31. doi:10.1128/JCM.00767-06
186. Costeloe K, Bowler U, Brocklehurst P, Hardy P, Heal P, Juszczak E, et al. A randomised controlled trial of the probiotic Bifidobacterium breve BBG-001 in preterm babies to prevent sepsis, necrotising enterocolitis and death: the Probiotics in Preterm infantS (PiPS) trial. Health Technol Assess (2016) 20(66):1–194. doi:10.3310/hta20660
187. Bron PA, Kleerebezem M, Brummer RJ, Cani PD, Mercenier A, MacDonald TT, et al. Can probiotics modulate human disease by impacting intestinal barrier function? Br J Nutr (2017) 117(1):93–107. doi:10.1017/S0007114516004037
188. Wu W, Wang Y, Zou J, Long F, Yan H, Zeng L, et al. Bifidobacterium adolescentis protects against necrotizing enterocolitis and upregulates TOLLIP and SIGIRR in premature neonatal rats. BMC Pediatr (2017) 17(1):1. doi:10.1186/s12887-016-0759-7
189. Good M, Sodhi CP, Ozolek JA, Buck RH, Goehring KC, Thomas DL, et al. Lactobacillus rhamnosus HN001 decreases the severity of necrotizing enterocolitis in neonatal mice and preterm piglets: evidence in mice for a role of TLR9. Am J Physiol Gastrointest Liver Physiol (2014) 306(11):G1021–32. doi:10.1152/ajpgi.00452.2013
190. Preidis GA, Saulnier DM, Blutt SE, Mistretta TA, Riehle KP, Major AM, et al. Probiotics stimulate enterocyte migration and microbial diversity in the neonatal mouse intestine. FASEB J (2012) 26(5):1960–9. doi:10.1096/fj.10-177980
191. Roberts D, Dalziel S. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev (2006) (3):CD004454. doi:10.1002/14651858.CD004454.pub2
192. Chawla S, Natarajan G, Shankaran S, Pappas A, Stoll BJ, Carlo WA, et al. Association of neurodevelopmental outcomes and neonatal morbidities of extremely premature infants with differential exposure to antenatal steroids. JAMA Pediatr (2016) 170(12):1164–72. doi:10.1001/jamapediatrics.2016.1936
193. Rabe H, Diaz-Rossello JL, Duley L, Dowswell T. Effect of timing of umbilical cord clamping and other strategies to influence placental transfusion at preterm birth on maternal and infant outcomes. Cochrane Database Syst Rev (2012) (8):CD003248. doi:10.1002/14651858.CD003248.pub3
194. Dang D, Zhang C, Shi S, Mu X, Lv X, Wu H. Umbilical cord milking reduces need for red cell transfusions and improves neonatal adaptation in preterm infants: meta-analysis. J Obstet Gynaecol Res (2015) 41(6):890–5. doi:10.1111/jog.12657
195. Foster JP, Seth R, Cole MJ. Oral immunoglobulin for preventing necrotizing enterocolitis in preterm and low birth weight neonates. Cochrane Database Syst Rev (2016) 4:CD001816. doi:10.1002/14651858.CD001816.pub3
196. Eibl MM, Wolf HM, Furnkranz H, Rosenkranz A. Prevention of necrotizing enterocolitis in low-birth-weight infants by IgA-IgG feeding. N Engl J Med (1988) 319(1):1–7. doi:10.1056/NEJM198807073190101
197. Rubaltelli FF, Benini F, Sala M. Prevention of necrotizing enterocolitis in neonates at risk by oral administration of monomeric IgG. Dev Pharmacol Ther (1991) 17(3–4):138–43.
Keywords: necrotizing enterocolitis, innate immunity, mucosa, intestine, toll-like receptor 4, human milk oligosaccharide, microbiota, prematurity
Citation: Hodzic Z, Bolock AM and Good M (2017) The Role of Mucosal Immunity in the Pathogenesis of Necrotizing Enterocolitis. Front. Pediatr. 5:40. doi: 10.3389/fped.2017.00040
Received: 21 December 2016; Accepted: 15 February 2017;
Published: 03 March 2017
Edited by:
James Lawrence Wynn, University of Florida, USAReviewed by:
Eric Giannoni, Centre hospitalier universitaire vaudois, SwitzerlandCopyright: © 2017 Hodzic, Bolock and Good. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Misty Good, bWdvb2RAa2lkcy53dXN0bC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.