 Agnieszka Swiatecka-Urban
Agnieszka Swiatecka-Urban- 1Department of Nephrology, Children’s Hospital of Pittsburgh, Pittsburgh, PA, USA
- 2Department of Cell Biology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
Endocytic trafficking couples cell signaling with the cytoskeletal dynamics by organizing a crosstalk between protein networks in different subcellular compartments. Proteins residing in the plasma membrane are internalized and transported as cargo in endocytic vesicles (i.e., endocytosis). Subsequently, cargo proteins can be delivered to lysosomes for degradation or recycled back to the plasma membrane. The slit diaphragm is a modified tight junction connecting foot processes of the glomerular epithelial cells, podocytes. Signaling at the slit diaphragm plays a critical role in the kidney while its dysfunction leads to glomerular protein loss (proteinuria), manifesting as nephrotic syndrome, a rare condition with an estimated incidence of 2–4 new cases per 100,000 each year. Relatively little is known about the role of endocytic trafficking in podocyte signaling and maintenance of the slit diaphragm integrity. This review will focus on the role of endocytic trafficking at the mature podocyte slit diaphragm.
Mechanisms of Endocytic Trafficking
All living cells process information by trafficking cargo from the plasma membrane in endocytic vesicles (i.e., endocytosis) and by returning much of the internalized membrane to the cell surface by a reciprocal process called recycling. The balance between endocytosis and recycling controls the plasma membrane composition and provides cells with information that has been resolved in time and space. Endocytic trafficking controls the supply of adaptor proteins and cargo, sorts the internalized cargo to specific intracellular compartments, and orchestrates the crosstalk in intracellular vesicles (1–3). As a result, cells turn over the equivalent of the entire plasma membrane one to five times per hour. Although endocytosis and recycling are ubiquitous, specific endocytic motifs and an assortment of protein adaptors guide cargo to diverse trafficking pathways. Defective endocytic trafficking has been associated with human disease, including congenital malformations, cancer, inflammation, and immunodeficiency (1). The mechanisms of endocytic entry of protein cargo, endocytic compartments, and mechanisms of cargo recycling were recently reviewed and will not be discussed here (4).
Endocytic Trafficking at the Mature Podocyte Slit Diaphragm
The kidney glomerulus is a filtering apparatus that allows passage of water and solute into the urinary space while retaining the vast majority of plasma proteins within the circulation. The functional unit of glomerular filtration is formed by the epithelial cells podocytes, glomerular basement membrane (GBM), and fenestrated capillary endothelial cells (5). The mature podocyte consists of the cell body, primary and secondary processes with microtubule and intermediate filament-based cytoskeleton, and the actin-based foot processes. Resting on the GBM, foot processes form interdigitating extensions linked by the slit diaphragm, which provides the only cell–cell contact between mature podocytes (6). Slit diaphragm is critical for (i) providing selective permeability between the blood and urinary space, (ii) separating the apical and basolateral plasma membrane, and (iii) serving as a signaling platform (4). Structurally, a network of proteins residing at the slit diaphragm membranes connects with the actin cytoskeleton through a juxtaposed cytoplasmic protein scaffold (7). Strong evidence demonstrates that the slit diaphragm is a dynamic unit (8, 9), suggesting that endocytic trafficking of membrane proteins plays an integral role in regulating the dynamics (Figure 1). Recent data provided genetic, functional, and high-resolution ultra-structural evidence highlighting a model of dynamic and multilayered architecture of the slit diaphragm (10). According to the model, the mammalian intercellular slit has a specific arrangement of the integral membrane proteins nephrin and Neph1 at a ratio of 1:2.5. The homophilic interactions between extracellular domains of nephrin are formed more apically, while the homophilic interactions of Neph1 localize closer to the basement membrane at the intercellular slit (10). Difference in the length of extracellular domain of nephrin and Neph1 results in a different length of the intercellular strands formed by the homophilic interactions that subsequently determine the width of the slit (10). Nephrin is an immunoglobulin-type cell adhesion molecule critical to the slit diaphragm function (11, 12). Mutations in the Nphs1 gene encoding nephrin protein lead to congenital nephrotic syndrome characterized by absence or profound impairment of the slit diaphragm and manifesting as severe proteinuria or nephrotic syndrome (13, 14). Although nephrin was shown to undergo endocytosis (15–19), relatively little is known how this process dynamically controls the slit diaphragm integrity. Data reviewed below demonstrate current understanding of the protein–protein interactions that regulate nephrin endocytic trafficking. The role of Neph1 is discussed in a separate section.
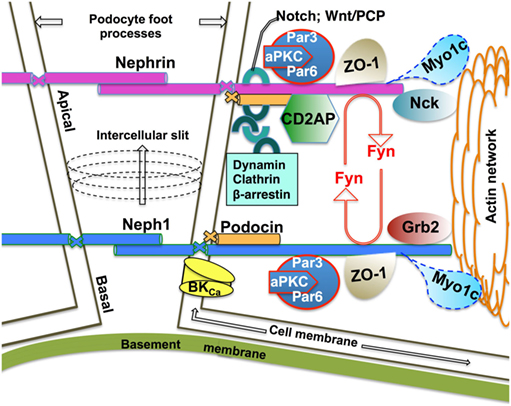
Figure 1. Schematic of the functional Neph1–nephrin protein complex at the mature mammalian slit diaphragm. Homophilic interactions of the integral membrane proteins nephrin and Neph1 form the mature mammalian slit diaphragm with a ratio of 1:2.5, respectively (10). Nephrin homophilic interactions are located more apically while the Neph1 strands localize more basally at the intercellular slit (10). The number of Ig repeats in the extracellular domain (9 and 5 for nephrin and Neph1, respectively) determines, at least in part, the width of the intercellular slit (10). The PHB domain-containing protein podocin anchors nephrin and Neph1 at the cell membrane. The intracellular domain of nephrin and Neph1 interacts with the cortical actin cytoskeleton via the juxtaposed cytoplasmic protein networks of junctional scaffolding proteins, channels, adaptor proteins, including endocytic adaptors, protein kinases, and motor proteins. Fyn-mediated phosphorylation of nephrin and Neph1 is essential for the podocyte architecture and signaling. Growing evidence supports a model where endocytic trafficking of the integral membrane proteins dynamically controls podocyte architecture and signaling. While the endocytic itineraries of nephrin are better understood, very little is known about the Neph1 endocytic mechanisms. Published data demonstrate that the motor protein Myo1c recruits Neph1 to the podocyte membrane and controls the cell membrane turnover of Neph1 in an actin-dependent manner (20, 21).
Nephrin localization at the slit diaphragm is determined by podocin, a lipid raft-resident protein encoded by the Nphs2 gene (22, 23). The prohibitin homology (PHB) domain—a lipid recognition motif present in podocin may target the podocin–nephrin complex to the lipid raft domains of slit diaphragm membranes (24, 25). Certainly, the Nphs2 gene mutations that mislocalize podocin also mislocalize nephrin and lead to steroid resistant, hereditary, and sporadic nephrotic syndrome (22, 26). The podocin C-terminal T339VV motif regulates the cell surface localization as well as the lipid raft-independent podocin stability (27). While tagged podocin resides specifically in the late endosomal/lysosomal compartment (27), mislocalized endogenous podocin co-localizes with an early endosomal marker Rab5 in the puromycin nephrosis rat model (28). Despite the above data, a large gap in understanding of the podocin endocytic itineraries still exists. Flotillins are members of the PHB domain-containing protein family. PHB domains contain hairpin-forming, hydrophobic regions that help to insert a protein into the inner leaflet of plasma membrane. The PHB domain and a region upstream of the N-terminus, modified posttranslationally by palmitoylation and myristoylation, mediates binding of flotillins to the inner leaflet of plasma membranes (24). The predicted topology of mouse podocin has the intracellular N- and C-terminal regions, a transmembrane domain, and the PHB domain, which unlike the PHB of flotillins, is not involved in hairpin formation (22). Flotillins form homo- and heterophillic interactions mediated via the C-terminal region conserved only within the flotillin family while in podocin both the N- and C-terminal domains are predicted to mediate homooligomerization (22, 24). As of now, nothing is known about the molecular organization of the podocin hairpin domain or whether podocin undergoes palmitoylation and/or myristoylation. The PHB domain of flotillins plays an essential role in endocytic trafficking of several receptors, including those activated by the Src family kinase Fyn (29). Nephrin is phosphorylated by Fyn, and the phosphorylation augments nephrin interaction with podocin and facilitates nephrin endocytosis via a clathrin-independent pathway, activating nephrin signaling (15, 30). By contrast, dephosphorylation of the nephrin-Y1193 increases nephrin endocytosis mediated by β-arrestin, decreases nephrin signaling, and impairs the slit diaphragm integrity (31). These data suggest that different endocytic pathways may play opposite roles in modulating nephrin function and that the endocytic itineraries of nephrin are regulated by the phosphorylation/dephosporylation cycle (see below).
PKC-α, stimulated by the high glucose concentration, induces nephrin endocytosis and leads to proteinuria while depletion of PKC-α stabilizes nephrin in diabetic nephropathy (32, 33). PKC-α phosphorylates two residues in the intracellular domain of nephrin, T1120 and T1125, and facilitates nephrin interaction with β-arrestin in murine podocytes (16). These data suggest that PKC-α promotes β-arrestin-mediated nephrin endocytosis, most likely via the clathrin-dependent pathway. Following clathrin-dependent endocytosis, receptors are transported either to the fast or slow recycling route, depending on the stability of interaction between the receptor and β-arrestin. In addition to regulating endocytosis, β-arrestin controls post-endocytic itineraries of several receptors. Nothing is currently known about post-endocytic sorting of nephrin. Understanding the structural basis and kinetics of binding between nephrin and β-arrestin may help to elucidate the fate of internalized nephrin, whether it undergoes lysosomal degradation or recycling to the plasma membrane or both.
CD2AP may control nephrin endocytic trafficking by regulating actin assembly and vesicle sorting. The importance of CD2AP in podocytes is demonstrated by glomerulosclerosis and foot process effacement, leading to renal failure in mice lacking CD2AP (34). CD2AP localizes in the slit diaphragm cytoplasmic region juxtaposed to the lipid raft membranes and co-localizes with the actin-related protein 2 (ARP2) and ARP3 (Arp2/3) and cortactin. CD2AP regulates endosomal sorting and vesicle trafficking by regulating actin assembly (35). Moreover, CD2AP associates with the dynamic actin pool and co-localizes with the endocytic adaptors Rab5, Rab4, the clathrin-dependent endocytic adaptor assembly polypeptide-2 complex (AP-2), and participates in formation of multivesicular bodies (35, 36). CD2AP interacts with podocin and nephrin, anchoring the proteins to the actin cytoskeleton (37, 38). CD2AP and nephrin interact with the p85 regulatory subunit of the phosphoinositide 3-OH kinase (PI3K), leading to its recruitment to the plasma membrane (39). PI3K plays an essential role in endocytic trafficking, including regulation of membrane lipid composition, release of clathrin-coated vesicles, and the Rab5 recruitment (40). The interactions of PI3K with nephrin, CD2AP, and other slit diaphragm proteins suggest that PI3K plays an important role in endocytic trafficking at the slit diaphragm (41).
CIN85/RukL is a closely related homolog of CD2AP (42). In podocytes, CD2AP blocks CIN85/RukL expression by sumoylation while CD2AP depletion increases CIN85/RukL abundance and leads to CIN85/RukL-mediated nephrin ubiquitination and endocytosis (43, 44). CIN85/RukL depletion has the opposite effect and preserves nephrin expression at the slit diaphragm membranes and reduces proteinuria in diabetic mice (45). Studies showed that unlike CD2AP, CIN85 does not contain an actin-binding domain (43, 46). Absence of CD2AP may facilitate nephrin interaction with CIN85, impair nephrin partitioning to lipid rafts, and induce nephrin endocytosis. However, another study has shown that CIN85 interacts directly with actin and together with CD2AP bundles actin filaments and modulates podocyte migration (47). Moreover, CIN85 clusters Src family kinases, providing a scaffold for Arp2/3-mediated actin assembly, and regulates cell polarization and motility. Expression of multiple CIN85 splice variants in different cell types may explain the reported difference of CIN85 function.
Notch activation alters foot process architecture in mature podocytes and induces proteinuria by stimulating the dynamin-dependent and lipid raft-independent nephrin endocytosis (17). The Wnt/planar cell polarity (PCP) pathway increases clathrin/β-arrestin-dependent nephrin endocytosis, and depletion of the PCP protein Vngl2 increases nephrin abundance at the cell surface and disturbs glomerular maturation (19).
The following protein networks have been shown to play a role in endocytic trafficking in podocytes, either directly or by regulating actin dynamics but their role in nephrin trafficking has not been demonstrated. α-Actinin-4 provides structural stability at the slit diaphragm by cross-linking and connecting actin filaments with nephrin and membrane associated guanylate kinase inverted (MAGI-1) (7, 48). The α-actinin-4 gene mutations are associated with nephrotic syndrome called focal segmental glomerulosclerosis (49). Direct interactions between members of the multi-protein scaffold organized by α-actinin-4, MAGI-1, and the endocytic adaptor megalin indicate a role of the scaffold in endocytic trafficking at the slit diaphragm (50, 51). As a member of the cytoskeleton-associated recycling or transport (CART): an Hrs/actinin-4/BERP/myosin V, α-actinin-4 mediates constitutive recycling of transmembrane receptors via the rapid recycling route on actin filaments (52). The role of α-actinin-4 or other members of the CART complex in cargo recycling at the slit diaphragm remains unknown. Endocytic adaptors, synaptojamin 1, and endophilin 1–3 are critical for the foot process formation, while dynamin I and II play a role in the foot process maintenance (18).
Structural Basis for Nephrin Endocytic Trafficking
A protein frequently utilizes different endocytic itineraries to diversify its function (1). Clathrin-dependent endocytosis is one of the most important internalization routes in eukaryotic cells (1). Endocytic adaptors recognize linear internalization signals located in the intracellular C-terminal tail domains of transmemembrane proteins and recruit these proteins as cargo to clathrin-coated pits, which are plasma membrane deformities coated with clathrin and clathrin-dependent endocytic adaptors. Subsequently, additional adaptors cleave off clathrin-coated pits and release them into the cell interior as clathrin-coated vesicles [reviewed in Ref (4)]. Clathrin-independent endocytosis includes a diverse group of internalization mechanisms sharing a requirement for free cholesterol, proteins, and lipids that reside in sphingolipid-rich lipid raft membranes (53).
Little is known about linear endocytic motifs in nephrin or other integral membrane proteins at the slit diaphragm. For example, the nephrin-Y1193 and subsequent amino acid residues D–E–V conform to a canonical, tyrosine-based endocytic signal of the YxxØ type, which is essential for clathrin-mediated endocytosis. Phosphorylation of the tyrosine residue inhibits the interaction of the YxxØ motif with the μ2 subunit of AP-2 and prevents endocytosis. By contrast, dephosphorylation of the tyrosine residue allows the YxxØ interaction with AP-2 and facilitates endocytosis. Thus, the phosphorylation state of the tyrosine residue serves as a regulatory switch controlling protein retention at the plasma membrane or its endocytosis via the clathrin-dependent pathway (54). Consistent with this model, Quack et al. demonstrated that β-arrestin mediates clathrin-dependent endocytosis of nephrin dephosphorylated at the conserved Y1193 residue (31). By contrast, phosphorylation of nephrin-Y1193 by Fyn augments nephrin interaction with podocin, prevents nephrin interaction with β-arrestin, attenuates β-arrestin-mediated nephrin endocytosis, and augments nephrin signaling (31). Moreover, the Fyn-mediated phosphorylation facilitates nephrin endocytosis via the clathrin-independent pathway (15, 30). These data demonstrate that the phosphorylation state of nephrin-Y1193 regulates nephrin signaling by directing nephrin to different endocytic pathways, either the clathrin-independent endocytosis to augment signaling or the clathrin-dependent endocytic to attenuate nephrin signaling. Although β-arrestin is known to interact predominantly with the clathrin-dependent adaptors, it was also found to modulate the clathrin-independent pathway by interacting with Arf6, and it may also utilize ubiquitination to inactivate receptor signaling (55, 56). At this time, the role of β-arrestin in nephrin ubuquitination or clathrin-independent internalization is unknown. Presence of several sequences conforming to the tyrosine-based endocytic motifs in nephrin cytoplasmic C-terminal domain suggests that regulation of nephrin endocytic itineraries is even more complex. The complexity is further increased by additional protein–protein interactions between nephrin, Fyn, and the actin cytoskeleton. Nephrin tyrosine phosphorylation is critical for recruitment of actin adaptors, such as p85/PI3K, Cas/Crk, and Nck, facilitating cytoskeletal dynamics in the podocyte foot processes (39, 41, 57–60). Nck facilitates Fyn-mediated nephrin phosphorylation, while Nck depletion leads to decreased nephrin tyrosine phosphorylation and foot process effacement (61, 62). Three conserved, Nck-binding Y–D–x–V motifs in the C-terminal nephrin tail, Y1176DEV, Y1193DEV, and Y1217DQV mediate these effects (63). Replacing the Y1176, Y1193, and Y1217 residues with the non-phosphorylated tyrosine mimic phenylalanine leads to proteinuria associated with foot process effacement, irregular thickening of the GBM, and dilated capillary loops in a mouse model (63).
The Essential Role of Neph1 at the Mammalian Slit Diaphragm
Neph1, a transmembrane protein partially homologous to nephrin is another key component of the mammalian slit diaphragm (10, 64). Nephrotic syndrome resulting from mutations in the neph1 gene in humans as well as severe proteinuria, foot process effacement, and early postnatal death in neph1 KO-mice demonstrate its importance for the glomerular function (64). Although the extracellular domain of Neph1 can interact with nephrin, recent data show that such heterophilic interactions are rare while the hemophilic Neph1 and nephrin interactions are critical for the mammalian slit diaphragm architecture (10, 65). Similar to nephrin, the Neph1 cytoplasmic domain engages directly with podocin, anchoring the integral membrane proteins at the cell membrane and the scaffolding protein ZO-1 that connects with the cortical actin cytoskeleton (66, 67) (Figure 1). Upon signaling engagement, Neph1 and nephrin form a phosphorylation-dependent unit transmitting outside-in signaling through a multi-protein scaffold of junctional proteins to the podocyte actin network (68, 69). While the Fyn-mediated tyrosine phosphorylation of the Neph1 intracellular domain leads to recruitment of protein adaptor Grb2 necessary for the Neph1-mediated actin polymerization, tyrosine phosphorylation of nephrin intracellular domain leads to recruitment of Nck, necessary for nephrin-mediated actin polymerization (58, 69–71). Activation of the Neph1–nephrin functional complex occurs during junction formation and during podocyte injury, both requiring intense signaling and actin cytoskeleton reorganization (69). One study demonstrated that inhibiting Neph1 signaling in podocyte culture preserves podocyte architecture and function in the puromycin aminoglycoside (PAN) injury model, while maintaining high Neph1 levels protects podocytes from injury in in vivo zebrafish PAN and adriamycin injury models (72). Another group demonstrated that renal ischemia induces a rapid loss of Neph1 interaction with ZO-1 in an in vivo rat ischemic model (73). Induction of injury by ATP depletion in podocyte culture resulted in rapid loss of Neph1 and ZO-1 interaction and redistribution of both proteins from the cell membrane to the cytoplasm while recovery from the injury resulted in increased Neph1 tyrosine phosphorylation, restoring Neph1 and ZO-1 membrane localization and interaction (73). The structural basis for the Fyn-mediated Neph1–ZO-1 interaction essential for the podocyte function has been recently elucidated. The ZO-1 PDZ1 domain interacts directly with the PDZ-binding region in the C-terminus of Neph1 where the T787HV and L761T residues are critical for protein–protein binding and its stability (67, 74). The functional Neph1–nephrin unit also engages with the Par3–Par6–atypical protein kinase C (aPKC) complex to maintain a three-dimensional foot process architecture and integrity of the slit diaphragm (75). Neph1 may influence localization of other proteins in the foot process. For example, Neph1 interaction with the large-conductance Ca2+-activated (BKCa) channel affects membrane localization of the channel (76). Data reviewed above attest to a dynamic role that Neph1 plays in the slit diaphragm remodeling. Yet, little is known about Neph1 trafficking itineraries. Emerging data demonstrate that Neph1 is recruited to the podocyte membrane by a motor protein Myo1c that interacts with Neph1 intracellular region directly via cargo-binding, C-terminal domain in an actin-dependent manner (20, 21). Moreover, Myo1c may control the cell membrane turnover of Neph1 (21). Myo1c also binds nephrin directly. However, by contrast to the effect on Neph1, Myo1c reduces nephrin localization at the podocyte membrane (20). It remain unknown how Myo1c exerts opposite effects on the membrane localization of Neph1 and nephrin.
Summary
Recent data confirm that endocytic trafficking allows rapid regulation of the signaling strength and duration at the slit diaphragm. Identifying endocytic motifs of the integral membrane proteins and examining the structural basis of the protein–protein interactions that control endocytic trafficking may unravel novel mechanisms and enrich our understanding of the role of this essential biological function at the podocyte slit diaphragm, as well as teach about the mechanisms of nephrotic syndrome and measures to correct it.
Author Contributions
AS-U reviewed the literature and wrote the manuscript.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by the American Heart Association grant 15GRNT25700019 and the National Institutes of Health grant R56HL127202 (to AS-U).
References
1. Sigismund S, Confalonieri S, Ciliberto A, Polo S, Scita G, Di Fiore PP. Endocytosis and signaling: cell logistics shape the eukaryotic cell plan. Physiol Rev (2012) 92:273–366. doi:10.1152/physrev.00005.2011
2. Canton I, Battaglia G. Endocytosis at the nanoscale. Chem Soc Rev (2012) 41:2718–39. doi:10.1039/c2cs15309b
3. Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol (2009) 10:597–608. doi:10.1038/nrm2755
4. Swiatecka-Urban A. Membrane trafficking in podocyte health and disease. Pediatr Nephrol (2013) 28:1723–37. doi:10.1007/s00467-012-2281-y
5. Haraldsson B, Jeansson M. Glomerular filtration barrier. Curr Opin Nephrol Hypertens (2009) 18:331–5. doi:10.1097/MNH.0b013e32832c9dba
6. Asanuma K, Mundel P. The role of podocytes in glomerular pathobiology. Clin Exp Nephrol (2003) 7:255–9. doi:10.1007/s10157-003-0259-6
7. Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol (2007) 17:428–37. doi:10.1016/j.tcb.2007.06.006
8. Steed E, Balda MS, Matter K. Dynamics and functions of tight junctions. Trends Cell Biol (2010) 20:142–9. doi:10.1016/j.tcb.2009.12.002
9. Tang VW, Brieher WM. alpha-Actinin-4/FSGS1 is required for Arp2/3-dependent actin assembly at the adherens junction. J Cell Biol (2012) 196:115–30. doi:10.1083/jcb.201103116
10. Grahammer F, Wigge C, Schell C, Kretz O, Patrakka J, Schneider S, et al. A flexible, multilayered protein scaffold maintains the slit in between glomerular podocytes. JCI Insight (2016) 1:e86177. doi:10.1172/jci.insight.86177
11. Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, et al. Positionally cloned gene for a novel glomerular protein – nephrin – is mutated in congenital nephrotic syndrome. Mol Cell (1998) 1:575–82.
12. Wartiovaara J, Ofverstedt LG, Khoshnoodi J, Zhang J, Makela E, Sandin S, et al. Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. J Clin Invest (2004) 114:1475–83. doi:10.1172/JCI22562
13. Lenkkeri U, Mannikko M, McCready P, Lamerdin J, Gribouval O, Niaudet PM, et al. Structure of the gene for congenital nephrotic syndrome of the Finnish type (NPHS1) and characterization of mutations. Am J Hum Genet (1999) 64:51–61.
14. Patrakka J, Kestila M, Wartiovaara J, Ruotsalainen V, Tissari P, Lenkkeri U, et al. Congenital nephrotic syndrome (NPHS1): features resulting from different mutations in Finnish patients. Kidney Int (2000) 58:972–80. doi:10.1046/j.1523-1755.2000.00254.x
15. Qin XS, Tsukaguchi H, Shono A, Yamamoto A, Kurihara H, Doi T. Phosphorylation of nephrin triggers its internalization by raft-mediated endocytosis. J Am Soc Nephrol (2009) 20:2534–45. doi:10.1681/ASN.2009010011
16. Quack I, Woznowski M, Potthoff SA, Palmer R, Konigshausen E, Sivritas S, et al. PKC alpha mediates beta-arrestin2-dependent nephrin endocytosis in hyperglycemia. J Biol Chem (2011) 286:12959–70. doi:10.1074/jbc.M110.204024
17. Waters AM, Wu MY, Huang YW, Liu GY, Holmyard D, Onay T, et al. Notch promotes dynamin-dependent endocytosis of nephrin. J Am Soc Nephrol (2012) 23:27–35. doi:10.1681/ASN.2011010027
18. Soda K, Balkin DM, Ferguson SM, Paradise S, Milosevic I, Giovedi S, et al. Role of dynamin, synaptojanin, and endophilin in podocyte foot processes. J Clin Invest (2012) 122:4401–11. doi:10.1172/JCI65289
19. Babayeva S, Rocque B, Aoudjit L, Zilber Y, Li J, Baldwin C, et al. Planar cell polarity pathway regulates nephrin endocytosis in developing podocytes. J Biol Chem (2013) 288(33):24035–48. doi:10.1074/jbc.M113.452904
20. Arif E, Wagner MC, Johnstone DB, Wong HN, George B, Pruthi PA, et al. Motor protein Myo1c is a podocyte protein that facilitates the transport of slit diaphragm protein Neph1 to the podocyte membrane. Mol Cell Biol (2011) 31:2134–50. doi:10.1128/MCB.05051-11
21. Arif E, Sharma P, Solanki A, Mallik L, Rathore YS, Twal WO, et al. Structural analysis of the Myo1c and Neph1 complex provides insight into the intracellular movement of Neph1. Mol Cell Biol (2016) 36:1639–54. doi:10.1128/MCB.00020-16
22. Huber TB, Simons M, Hartleben B, Sernetz L, Schmidts M, Gundlach E, et al. Molecular basis of the functional podocin-nephrin complex: mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum Mol Genet (2003) 12:3397–405. doi:10.1093/hmg/ddg360
23. Roselli S, Gribouval O, Boute N, Sich M, Benessy F, Attie T, et al. Podocin localizes in the kidney to the slit diaphragm area. Am J Pathol (2002) 160:131–9. doi:10.1016/S0002-9440(10)64357-X
24. Otto GP, Nichols BJ. The roles of flotillin microdomains – endocytosis and beyond. J Cell Sci (2011) 124:3933–40. doi:10.1242/jcs.092015
25. Morrow IC, Parton RG. Flotillins and the PHB domain protein family: rafts, worms and anaesthetics. Traffic (2005) 6:725–40. doi:10.1111/j.1600-0854.2005.00318.x
26. Roselli S, Moutkine I, Gribouval O, Benmerah A, Antignac C. Plasma membrane targeting of podocin through the classical exocytic pathway: effect of NPHS2 mutations. Traffic (2004) 5:37–44. doi:10.1046/j.1600-0854.2003.00148.x
27. Godel M, Ostendorf BN, Baumer J, Weber K, Huber TB. A novel domain regulating degradation of the glomerular slit diaphragm protein podocin in cell culture systems. PLoS One (2013) 8:e57078. doi:10.1371/journal.pone.0057078
28. Fukuda H, Hidaka T, Takagi-Akiba M, Ichimura K, Oliva Trejo JA, Sasaki Y, et al. Podocin is translocated to cytoplasm in puromycin aminonucleoside nephrosis rats and in poor-prognosis patients with IgA nephropathy. Cell Tissue Res (2015) 360:391–400. doi:10.1007/s00441-014-2100-9
29. Zhao F, Zhang J, Liu YS, Li L, He YL. Research advances on flotillins. Virol J (2011) 8:479. doi:10.1186/1743-422X-8-479
30. Li H, Lemay S, Aoudjit L, Kawachi H, Takano T. SRC-family kinase Fyn phosphorylates the cytoplasmic domain of nephrin and modulates its interaction with podocin. J Am Soc Nephrol (2004) 15:3006–15. doi:10.1097/01.ASN.0000146689.88078.80
31. Quack I, Rump LC, Gerke P, Walther I, Vinke T, Vonend O, et al. beta-Arrestin2 mediates nephrin endocytosis and impairs slit diaphragm integrity. Proc Natl Acad Sci U S A (2006) 103:14110–5. doi:10.1073/pnas.0602587103
32. Tossidou I, Teng B, Menne J, Shushakova N, Park JK, Becker JU, et al. Podocytic PKC-alpha is regulated in murine and human diabetes and mediates nephrin endocytosis. PLoS One (2010) 5:e10185. doi:10.1371/journal.pone.0010185
33. Menne J, Meier M, Park JK, Boehne M, Kirsch T, Lindschau C, et al. Nephrin loss in experimental diabetic nephropathy is prevented by deletion of protein kinase C alpha signaling in-vivo. Kidney Int (2006) 70:1456–62. doi:10.1038/sj.ki.5001830
34. Shih NY, Li J, Karpitskii V, Nguyen A, Dustin ML, Kanagawa O, et al. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science (1999) 286:312–5.
35. Welsch T, Endlich N, Gokce G, Doroshenko E, Simpson JC, Kriz W, et al. Association of CD2AP with dynamic actin on vesicles in podocytes. Am J Physiol Renal Physiol (2005) 289:F1134–43. doi:10.1152/ajprenal.00178.2005
36. Kim JM, Wu H, Green G, Winkler CA, Kopp JB, Miner JH, et al. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science (2003) 300:1298–300. doi:10.1126/science.1081068
37. Shih NY, Li J, Cotran R, Mundel P, Miner JH, Shaw AS. CD2AP localizes to the slit diaphragm and binds to nephrin via a novel C-terminal domain. Am J Pathol (2001) 159:2303–8. doi:10.1016/S0002-9440(10)63080-5
38. Kawachi H, Miyauchi N, Suzuki K, Han GD, Orikasa M, Shimizu F. Role of podocyte slit diaphragm as a filtration barrier. Nephrology (Carlton) (2006) 11:274–81. doi:10.1111/j.1440-1797.2006.00583.x
39. Huber TB, Hartleben B, Kim J, Schmidts M, Schermer B, Keil A, et al. Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol Cell Biol (2003) 23:4917–28. doi:10.1128/MCB.23.14.4917-4928.2003
40. Mellor P, Furber LA, Nyarko JN, Anderson DH. Multiple roles for the p85alpha isoform in the regulation and function of PI3K signalling and receptor trafficking. Biochem J (2012) 441:23–37. doi:10.1042/BJ20111164
41. Zhu J, Sun N, Aoudjit L, Li H, Kawachi H, Lemay S, et al. Nephrin mediates actin reorganization via phosphoinositide 3-kinase in podocytes. Kidney Int (2008) 73:556–66. doi:10.1038/sj.ki.5002691
42. Watanabe S, Take H, Takeda K, Yu ZX, Iwata N, Kajigaya S. Characterization of the CIN85 adaptor protein and identification of components involved in CIN85 complexes. Biochem Biophys Res Commun (2000) 278:167–74. doi:10.1006/bbrc.2000.3760
43. Tossidou I, Teng B, Drobot L, Meyer-Schwesinger C, Worthmann K, Haller H, et al. CIN85/RukL is a novel binding partner of nephrin and podocin and mediates slit diaphragm turnover in podocytes. J Biol Chem (2010) 285:25285–95. doi:10.1074/jbc.M109.087239
44. Tossidou I, Niedenthal R, Klaus M, Teng B, Worthmann K, King BL, et al. CD2AP regulates SUMOylation of CIN85 in podocytes. Mol Cell Biol (2012) 32:1068–79. doi:10.1128/MCB.06106-11
45. Teng B, Schroder P, Muller-Deile J, Schenk H, Staggs L, Tossidou I, et al. CIN85 deficiency prevents nephrin endocytosis and proteinuria in diabetes. Diabetes (2016) 65(12):3667–79. doi:10.2337/db16-0081
46. Lehtonen S, Zhao F, Lehtonen E. CD2-associated protein directly interacts with the actin cytoskeleton. Am J Physiol Renal Physiol (2002) 283:F734–43. doi:10.1152/ajprenal.00312.2001
47. Gaidos G, Soni S, Oswald DJ, Toselli PA, Kirsch KH. Structure and function analysis of the CMS/CIN85 protein family identifies actin-bundling properties and heterotypic-complex formation. J Cell Sci (2007) 120:2366–77. doi:10.1242/jcs.004333
48. Patrie KM, Drescher AJ, Welihinda A, Mundel P, Margolis B. Interaction of two actin-binding proteins, synaptopodin and alpha-actinin-4, with the tight junction protein MAGI-1. J Biol Chem (2002) 277:30183–90. doi:10.1074/jbc.M203072200
49. Khurana S, Chakraborty S, Lam M, Liu Y, Su YT, Zhao X, et al. Familial focal segmental glomerulosclerosis (FSGS)-linked alpha-Actinin 4 (ACTN4) protein mutants lose ability to activate transcription by nuclear hormone receptors. J Biol Chem (2012) 287:12027–35. doi:10.1074/jbc.M112.345421
50. Prabakaran T, Nielsen R, Larsen JV, Sorensen SS, Feldt-Rasmussen U, Saleem MA, et al. Receptor-mediated endocytosis of alpha-galactosidase A in human podocytes in Fabry disease. PLoS One (2011) 6:e25065. doi:10.1371/journal.pone.0025065
51. Ni J, Bao S, Johnson RI, Zhu B, Li J, Vadaparampil J, et al. MAGI-1 interacts with nephrin to maintain slit diaphragm structure through enhanced Rap1 activation in podocytes. J Biol Chem (2016) 291:24406–17. doi:10.1074/jbc.M116.745026
52. Yan Q, Sun W, Kujala P, Lotfi Y, Vida TA, Bean AJ. CART: an Hrs/actinin-4/BERP/myosin V protein complex required for efficient receptor recycling. Mol Biol Cell (2005) 16:2470–82. doi:10.1091/mbc.E04-11-1014
53. Gong Q, Huntsman C, Ma D. Clathrin-independent internalization and recycling. J Cell Mol Med (2008) 12:126–44. doi:10.1111/j.1582-4934.2007.00148.x
54. Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem (2003) 72:395–447. doi:10.1146/annurev.biochem.72.121801.161800
55. Shenoy SK, Lefkowitz RJ. Trafficking patterns of beta-arrestin and G protein-coupled receptors determined by the kinetics of beta-arrestin deubiquitination. J Biol Chem (2003) 278:14498–506. doi:10.1074/jbc.M209626200
56. Shukla AK, Xiao K, Lefkowitz RJ. Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci (2011) 36:457–69. doi:10.1016/j.tibs.2011.06.003
57. George B, Verma R, Soofi AA, Garg P, Zhang J, Park TJ, et al. Crk1/2-dependent signaling is necessary for podocyte foot process spreading in mouse models of glomerular disease. J Clin Invest (2012) 122:674–92. doi:10.1172/JCI60070
58. Verma R, Kovari I, Soofi A, Nihalani D, Patrie K, Holzman LB. Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J Clin Invest (2006) 116:1346–59. doi:10.1172/JCI27414
59. Jones N, Blasutig IM, Eremina V, Ruston JM, Bladt F, Li H, et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature (2006) 440:818–23. doi:10.1038/nature04662
60. Li H, Zhu J, Aoudjit L, Latreille M, Kawachi H, Larose L, et al. Rat nephrin modulates cell morphology via the adaptor protein Nck. Biochem Biophys Res Commun (2006) 349:310–6. doi:10.1016/j.bbrc.2006.08.053
61. Jones N, New LA, Fortino MA, Eremina V, Ruston J, Blasutig IM, et al. Nck proteins maintain the adult glomerular filtration barrier. J Am Soc Nephrol (2009) 20:1533–43. doi:10.1681/ASN.2009010056
62. New LA, Keyvani Chahi A, Jones N. Direct regulation of nephrin tyrosine phosphorylation by Nck adaptor proteins. J Biol Chem (2013) 288:1500–10. doi:10.1074/jbc.M112.439463
63. New LA, Martin CE, Scott RP, Platt MJ, Keyvani Chahi A, Stringer CD, et al. Nephrin tyrosine phosphorylation is required to stabilize and restore podocyte foot process architecture. J Am Soc Nephrol (2016) 27:2422–35. doi:10.1681/ASN.2015091048
64. Donoviel DB, Freed DD, Vogel H, Potter DG, Hawkins E, Barrish JP, et al. Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol Cell Biol (2001) 21:4829–36. doi:10.1128/MCB.21.14.4829-4836.2001
65. Gerke P, Huber TB, Sellin L, Benzing T, Walz G. Homodimerization and heterodimerization of the glomerular podocyte proteins nephrin and NEPH1. J Am Soc Nephrol (2003) 14:918–26. doi:10.1097/01.ASN.0000057853.05686.89
66. Sellin L, Huber TB, Gerke P, Quack I, Pavenstadt H, Walz G. NEPH1 defines a novel family of podocin interacting proteins. FASEB J (2003) 17:115–7. doi:10.1096/fj.02-0242fje
67. Huber TB, Schmidts M, Gerke P, Schermer B, Zahn A, Hartleben B, et al. The carboxyl terminus of Neph family members binds to the PDZ domain protein zonula occludens-1. J Biol Chem (2003) 278:13417–21. doi:10.1074/jbc.C200678200
68. Liu G, Kaw B, Kurfis J, Rahmanuddin S, Kanwar YS, Chugh SS. Neph1 and nephrin interaction in the slit diaphragm is an important determinant of glomerular permeability. J Clin Invest (2003) 112:209–21. doi:10.1172/JCI18242
69. Garg P, Verma R, Nihalani D, Johnstone DB, Holzman LB. Neph1 cooperates with nephrin to transduce a signal that induces actin polymerization. Mol Cell Biol (2007) 27:8698–712. doi:10.1128/MCB.00948-07
70. Harita Y, Kurihara H, Kosako H, Tezuka T, Sekine T, Igarashi T, et al. Neph1, a component of the kidney slit diaphragm, is tyrosine-phosphorylated by the Src family tyrosine kinase and modulates intracellular signaling by binding to Grb2. J Biol Chem (2008) 283:9177–86. doi:10.1074/jbc.M707247200
71. Verma R, Wharram B, Kovari I, Kunkel R, Nihalani D, Wary KK, et al. Fyn binds to and phosphorylates the kidney slit diaphragm component Nephrin. J Biol Chem (2003) 278:20716–23. doi:10.1074/jbc.M301689200
72. Arif E, Rathore YS, Kumari B, Ashish F, Wong HN, Holzman LB, et al. Slit diaphragm protein Neph1 and its signaling: a novel therapeutic target for protection of podocytes against glomerular injury. J Biol Chem (2014) 289:9502–18. doi:10.1074/jbc.M113.505743
73. Wagner MC, Rhodes G, Wang E, Pruthi V, Arif E, Saleem MA, et al. Ischemic injury to kidney induces glomerular podocyte effacement and dissociation of slit diaphragm proteins Neph1 and ZO-1. J Biol Chem (2008) 283:35579–89. doi:10.1074/jbc.M805507200
74. Mallik L, Arif E, Sharma P, Rathore YS, Wong HN, Holzman LB, et al. Solution structure analysis of cytoplasmic domain of podocyte protein Neph1 using small/wide angle x-ray scattering (SWAXS). J Biol Chem (2012) 287:9441–53. doi:10.1074/jbc.M111.284927
75. Hartleben B, Schweizer H, Lubben P, Bartram MP, Moller CC, Herr R, et al. Neph-Nephrin proteins bind the Par3-Par6-atypical protein kinase C (aPKC) complex to regulate podocyte cell polarity. J Biol Chem (2008) 283:23033–8. doi:10.1074/jbc.M803143200
Keywords: nephrotic syndrome, proteinuria, endocytic trafficking, endocytosis, podocytes, nephrin, podocin, glomerular slit diaphragm
Citation: Swiatecka-Urban A (2017) Endocytic Trafficking at the Mature Podocyte Slit Diaphragm. Front. Pediatr. 5:32. doi: 10.3389/fped.2017.00032
Received: 03 October 2016; Accepted: 03 February 2017;
Published: 24 February 2017
Edited by:
Miriam Schmidts, Radboud University Nijmegen, NetherlandsReviewed by:
Michal Malina, NH Hospital, CzechiaMax Christoph Liebau, University Hospital of Cologne, Germany
Copyright: © 2017 Swiatecka-Urban. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agnieszka Swiatecka-Urban, YXN1cmJhbkBwaXR0LmVkdQ==