
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr. , 11 November 2014
Sec. Child and Adolescent Psychiatry
Volume 2 - 2014 | https://doi.org/10.3389/fped.2014.00121
This article is part of the Research Topic Experimental models of early exposure to alcohol: a way to unravel the neurobiology of mental retardation View all 11 articles
 Eric C. Olson1,2*
Eric C. Olson1,2*The development of the layered cerebral cortex starts with a process called preplate splitting. Preplate splitting involves the establishment of prospective cortical layer 6 (L6) neurons within a plexus of pioneer neurons called the preplate. The forming layer 6 splits the preplate into a superficial layer of pioneer neurons called the marginal zone and a deeper layer of pioneer neurons called the subplate. Disruptions of this early developmental event by toxin exposure or mutation are associated with neurological disease including severe intellectual disability. This review explores recent findings that reveal the dynamism of gene expression and morphological differentiation during this early developmental period. Over 1000 genes show expression increases of ≥2-fold during this period in differentiating mouse L6 neurons. Surprisingly, 88% of previously identified non-syndromic intellectual-disability (NS-ID) genes are expressed at this time and show an average expression increase of 1.6-fold in these differentiating L6 neurons. This changing genetic program must, in part, support the dramatic cellular reorganizations that occur during preplate splitting. While different models have been proposed for the formation of a layer of L6 cortical neurons within the preplate, original histological studies and more recent work exploiting transgenic mice suggest that the process is largely driven by the coordinated polarization and coalescence of L6 neurons rather than by cellular translocation or migration. The observation that genes associated with forms of NS-ID are expressed during very early cortical development raises the possibility of studying the relevant biological events at a time point when the cortex is small, contains relatively few cell types, and few functional circuits. This review then outlines how explant models may prove particularly useful in studying the consequence of toxin and mutation on the etiology of some forms of NS-ID.
Neocortical development in human is initiated in the seventh week of gestation by the appearance of a layer of pioneer neurons, called the preplate or primordial plexiform layer (1, 2). Preplate neurons lie underneath the meninges and ultimately these neurons cover both cerebral vesicles. At this time, the human cortical wall is only ~250 μm thick, the majority of which is ventricular zone (VZ) (neural precursor cells) and the remaining 20–30 μm contains these early differentiating neurons of the preplate (3, 4). For comparison, the mouse preplate stage corresponds to embryonic day 12.5 post conception (E12.5), a time when the cortical wall has a similar composition of cells and a similar thickness (~150 μm) to the human preplate stage cortex.
Starting at the seventh to eighth week of gestation in human or E13.5 in mouse, a process called preplate splitting initiates the formation of cortical layering. In the mouse, preplate splitting begins in the lateral neocortex and proceeds dorsally and caudally over the next embryonic day (5). Preplate splitting is an early event in cortical development and involves the establishment of an organized layer of cortical plate (CP) neurons within the preplate. The establishment of future layer 6 (L6) neurons splits the preplate into a superficial layer of pioneer neurons called the marginal zone (MZ) and a deeper layer of pioneer neurons called the subplate (SP) (6–10). Preplate splitting is the first step in the formation of the layered cortex and is followed by the successive migration and lamination of cortical layers 5–2 in an inside out fashion (11).
Both MZ neurons and SP neurons have essential roles in organizing the developing cortex (12, 13). MZ neurons, primarily Cajal–Retzius cells, secrete a critical chemotropic factor called Reelin (14) (discussed below) that is required for correct positioning of migrating CP neurons. SP neurons constitute a diverse group of cells (15, 16), which are essential for correct thalamocortical afferent targeting (17–19). Absent correct preplate splitting, the SP cells remain superficial in the cortex and both cortical layering (20) and thalamocortical targeting is disrupted (21, 22). Thus, preplate splitting is a fundamental event that enables the later assembly of the upper cortical layers and leads to a properly formed cerebral cortex (23–25).
The significance of preplate splitting is underscored by the large number of genes specifically upregulated during this period. A prior study used fluorescence activated cell sorting to purify genetically labeled, developing L6 neurons (26). Sorted cells from the transgenic Eomes:eGFP1 mouse embryos (27) were subjected to RNA extraction and Affymetrix gene chip analysis. In these embryos, enhanced green fluorescent protein (eGFP) expression is under the control of the Eomes (Tbr2) promoter. Eomes is a transcription factor that is selectively expressed by intermediate neural precursor cells of the glutamatergic cortical lineage (28, 29). In transgenic embryos, GFP expression is transient but persists for several days in immature post mitotic neurons of the excitatory cortical lineage. By comparing the GFP+ population, primarily immature neurons, to the GFP-population, primarily neural precursors, up and down regulated genes in the differentiating excitatory cortical lineage were identified. Approximately half the genome was expressed by these neurons and more than 1000 genes show expression increases ≥2-fold during the first ~24–36 h after cell cycle exit (26). Genes of interest could then be validated by comparison to the Genepaint in situ database2. This prior study validated, and grouped by spatial expression pattern, 317 genes that were upregulated ≥3-fold during early cortical neuron differentiation. Importantly, over half of these highly upregulated genes have been associated with neuronal disease (26).
This dataset is a valuable resource that can be queried for genes specifically linked to neurological disorders including non-syndromic intellectual disability (NS-ID) (30). Of 46 human NS-ID genes identified previously (30), 43 were represented within this dataset, i.e., represented on the mouse Affymetrix Gene 1.0 ST Array (Table 1). Of these 43, 38 are expressed above a stringent threshold of RMA = 7.0, and these genes display an average expression level of RMA = 9.5, placing them in the approximate top third of all expressed genes in these immature neurons. At the onset of preplate splitting (E13.5), the expressed genes display an average increased expression of 1.6-fold in GFP+ neurons versus GFP-precursors. Surprisingly, only 2 genes of the 38 (MAGT and ARX) were downregulated ≥1.5-fold in differentiating neurons (i.e., more highly expressed in neural precursors than in differentiating neurons). Thus, the majority of identified NS-ID genes are highly expressed and upregulated by differentiating CP neurons during this early differentiation period, well prior to synapse formation.
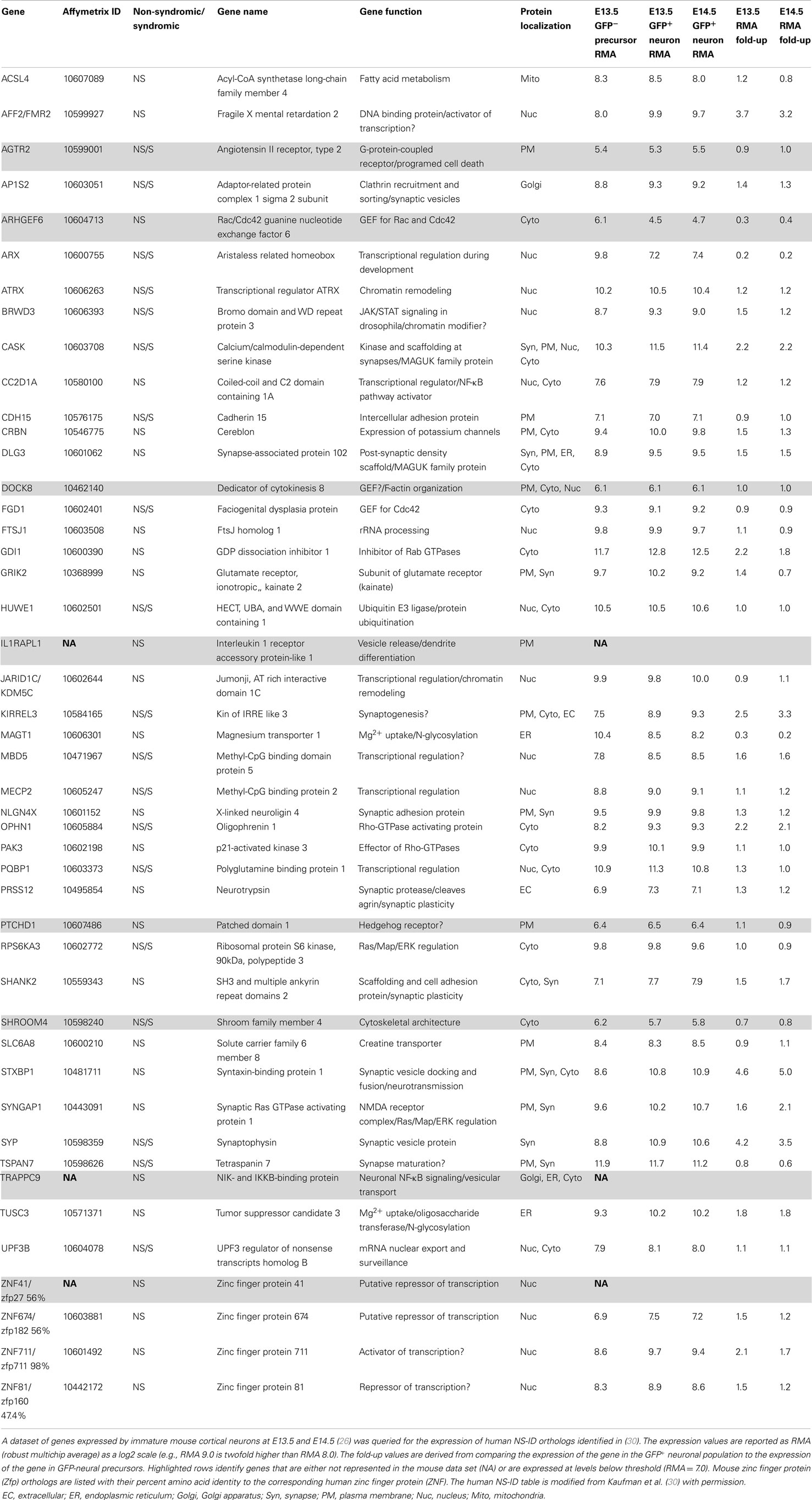
Table 1. List of non-syndromic intellectual disability (NS-ID) genes expressed in immature excitatory neurons.
What functions might these NS-ID genes be performing during this early period? Expression analysis identified 15 out of 38 (40%) of these early expressed NS-ID gene products as being localized to the nucleus with most of these genes having functions in transcription, chromosomal remodeling, or RNA transport (Table 1) (30). An additional 10 of 38 (26%) of the predicted gene products localize to the plasma membrane, where they perform diverse functions as synaptic proteins, adhesion proteins, transporters, and receptors. This group includes Cadherin15 (CDH15) and a subunit of an ionotropic glutamate receptor (GRIK2). Four out of five NS-ID gene products that localize to the cytoplasm are involved in the regulation of small GTPases, namely, Rho, Cdc42, and Rab1, which coordinate cytoskeletal remodeling and vesicular transport, respectively. The five most upregulated NS-ID genes (STXBP1, SYP, FMR2, KIRREL3, and CASK) encode proteins with likely synaptic function. This was a surprise, since there are no morphologically identified synapses on L6 neurons at this time in development. While some of these early expressed mRNAs may not encode functional proteins, it is likely that many NS-ID genes have important roles during this very early period of cortical development.
This dynamic transcriptional profile may underlie the coincident processes of cortical neuron migration and molecular differentiation. Before achieving their mature form, cortical neurons are known to transition through multiple morphological states: from multipolar neuron to radial glial-associated migrating neuron to post migratory differentiating neuron (31–34). Immediately after cell cycle exit, the immature neuron adopts a multipolar morphology and migrates slowly through intermediate zone (IZ) (33, 34) while simultaneously initiating an axon (35, 36). The multipolar neuron, trailing an axon, continues migration until it reaches the SP, the layer of pioneer neurons that underlie the forming CP. At the SP, migrating neurons change from the multipolar shape to a bipolar shape coincident with their attachment to a radial glial fiber (37). The neuron, now apposed to the radial glial fiber, migrates through the developing CP in a saltatory (stepwise) fashion (38). As the neuron approaches the top of the CP, the neuron detaches from the radial glial fiber and translocates into position underneath the MZ (future layer 1) (38) where it elaborates an apical dendrite and becomes excitable.
The sequence of morphological changes is less understood during the earlier period of preplate splitting. Two models have been proposed to account for the appearance of L6 neurons within the preplate. The first model posits direct somal translocation of the immature neuron from the VZ into the preplate (38, 39). This translocation involves the rapid movement of the nucleus into the leading process of the neuron and is thought to occur independent of radial glial guidance or attachment (38). Thus, as more L6 neurons translocate into the preplate, the preplate is split into the MZ and SP. This model, however, appears inconsistent with prior histological observations using electron microscopy (7, 40) and the Golgi stain method (7, 40), or with more recent observations made from the Eomes:eGFP embryonic cortex (10). In these animals, GFP expressing neurons of the excitatory lineage were found intermixed with Calretinin expressing preplate neurons, prior to preplate splitting (Figures 1A–C). Furthermore, below this mixture of preplate and L6 neurons lies a thick IZ composed of multipolar neurons that do not show translocating morphology (i.e., highly elongated in the radial direction). Thus, the GFP+ cells that are poised to enter the developing CP are not translocating from the VZ. Instead, preplate splitting appears to be initiated by polarized dendritic growth of L6 neurons and the concurrent coalescing of these L6 neurons into an organized and recognizable CP. The calcium-binding protein Calretinin is a marker for subsets of both MZ and SP neurons during early rodent cortical development (41, 42) and therefore the separation of preplate Calretinin+ cells into the MZ and SP groups is a hallmark of preplate splitting. In this model, Calretinin+ MZ neurons stay in place and Calretinin+ SP neurons either actively migrate away (43) from or are passively displaced by the coalescing L6 neurons. Thus, the initial phase of preplate splitting is driven by active reorganization of these L6 neurons, rather than their translocation. Future imaging studies should help resolve these two models. In both models, however, the period of preplate splitting represents a period of dynamic cellular transformations.
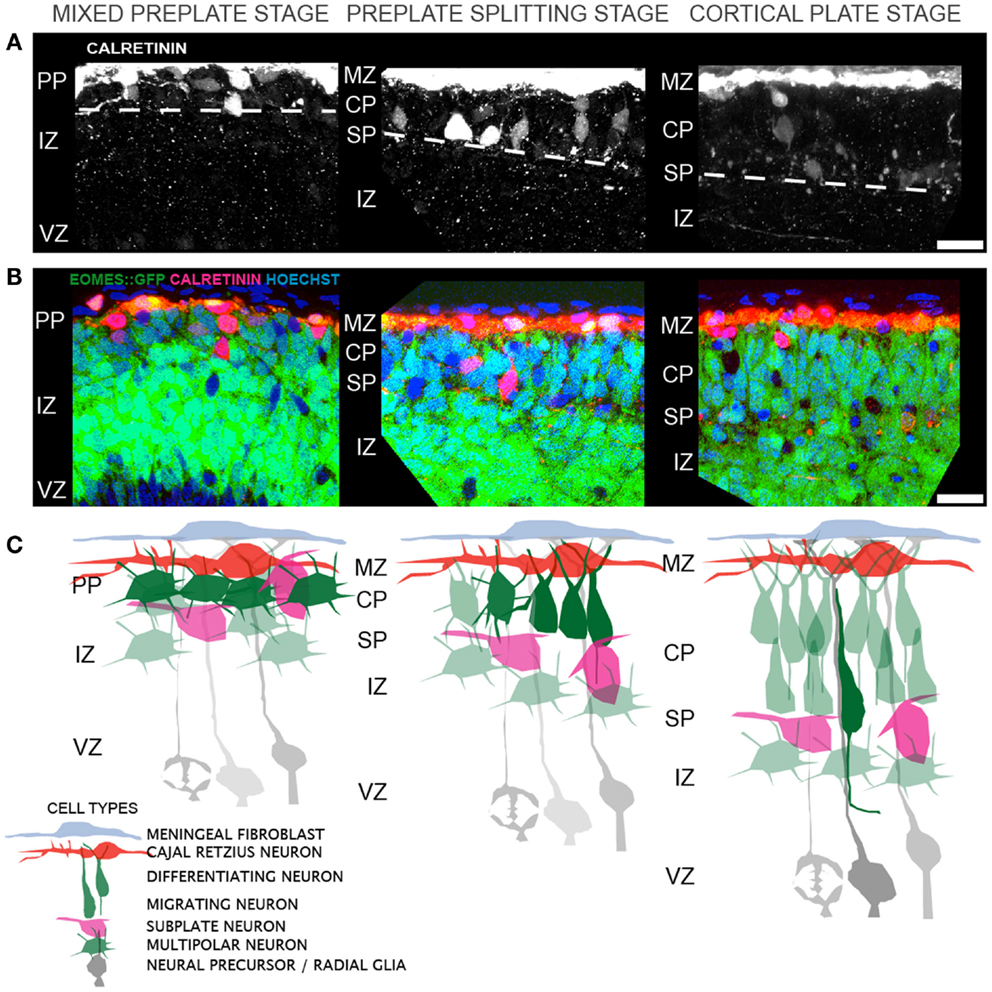
Figure 1. Cellular events that contribute to cortical layer 6 formation and preplate splitting. (A) Distribution of Calretinin immunopositive preplate neurons prior to (left), during (middle), and after (right) preplate splitting. (B) A single confocal slice through the Eomes:eGFP cortex at the same regions as in (A), showing the distribution of GFP+ excitatory cortical neurons (green) and Calretinin+ preplate neurons (red) and Hoechst+ nuclei (blue). (C) Model of the corresponding cellular rearrangements during the mixed preplate stage, where L6 neurons are intermingled with preplate neurons, the preplate splitting stage, where L6 neurons become radially oriented and coalesce into a recognizable cellular layer and the cortical plate stage, where migrating neurons enter the CP on radial glial fibers. Abbreviations: marginal zone (MZ), preplate (PP), cortical plate (CP), subplate (SP), intermediate zone (IZ), ventricular zone (VZ). Scale bar is 25 μm in (A,B).
Disruptions of preplate splitting either by toxin or mutation (44) are associated with serious neurological disability including mental retardation, epilepsy (45), and possibly autism (46). Prenatal exposure to alcohol is a leading cause of mental retardation and intellectual disability (47, 48). The CDC estimates that 0.2–1.5 per 1000 live births are children with fetal alcohol syndrome (FAS), a syndrome defined by mental dysfunction (49). The cognitive deficits caused by prenatal exposure to EtOH are likely reflected in the specific functional and structural abnormalities found in brains of alcohol-exposed children (50, 51).
EtOH exposure is known to impact neuronal plasticity and these disruptions range from the short term (e.g., memory deficits caused by binge drinking) (52–54) to long term [e.g., disruption in memory and cognition associated with alcoholism (55)] to permanent [e.g., structural changes and intellectual disability associated with FASD (56, 57)] or chronic alcoholism (58). The disruptions caused by ethanol exposure vary with time period of exposure (59, 60). This differential sensitivity to ethanol may reflect the major underlying cellular processes occurring at the time of exposure (61).
Although EtOH exposure strongly promotes apoptosis during the synaptic formation period (62), EtOH can also target multiple events prior to synapse formation including neurogenesis, neuronal migration (63), axonal outgrowth (64, 65), and dendritic development (66–68). These biologically important processes can be assayed using early embryonic cortical explants. At this time, the cortex is small, composed of relatively few cell types and have few synapses. Nevertheless, these explants captures critical organotypic interactions including signals derived from other neurons as well as non-neuronal elements including radial glia (69, 70), blood vessels (71), meninges (72), and associated extracellular matrix (14). This organotypic environment provides the multiple substrates and signals that allow cortical neurons to mature through intermediate stages and to finally adopt appropriate form and function. Understanding how EtOH disrupts these signaling systems may be required for a fuller picture of the etiology FASD and the development of NS-ID.
A whole hemisphere explant procedure that permits 2 days of organotypic growth and encompasses the period of preplate splitting has been valuable in understanding the cellular transformations of preplate splitting (10, 73). In this procedure, entire embryonic cortices are isolated with the meninges intact and are then cultured on collagen filters as is done with slice explants (74, 75). Keeping the meninges intact helps preserve the organization of the basal lamina, the radial glial endfeet as well as the pioneer neurons that are found in the MZ. Disruptions of the meninges during development, through mutation (76, 77) or injury (78) can cause focal heterotopia and disrupt underlying cortical layering. Therefore, keeping the meninges as intact as possible is desirable and allows for continuous growth and lamination of the CP during the in vitro period. The CP is organized and shows appropriate expression of the transcription factors Tbr1 and Ctip2. Similarly, the radial glial network is intact evidenced by appropriate expression of the intermediate filament protein Nestin (73).
Using the whole hemisphere explant model, it was found that cellular orientation and apical dendritic growth was disrupted by single dose ethanol exposure, with an increase in primary dendrite number detected within 4 h of exposure (67). This dendritic alteration was accompanied by a morphological compaction of the Golgi apparatus, a key support organelle for the growing dendrite (79), as well as a slower reduction in cytoskeletal F-actin and the microtubule associated protein MAP2 content (67). These disruptions are remarkably similar to, but less severe than, disruptions caused by disruption of the Reelin-signaling pathway (80). Reelin is a large glycoprotein that is secreted by Cajal–Retzius cells in the MZ, during the period of preplate splitting (14, 81). Without Reelin, the preplate fails to split (9, 74, 75) and the subsequently generated cortical layers pile up underneath L6 leading to an inversion of cortical layering (20). In human beings, Reelin deficiency leads to mild epilepsy and severe mental retardation (45). At the cellular level in Reelin-deficient (reeler) cortical explants, neurons were tangentially oriented rather than radially oriented. The dendritic arbor was simplified and these neurons displayed more primary processes and a compact Golgi apparatus (10, 80). In addition dendritic expression of F-actin and MAP2 was reduced in reeler mutants compared to wild-type controls (10). Thus, studies using early cortical explants are showing a potential convergence of cellular phenotypes underlying two etiologically distinct forms of intellectual disability.
Early cortical development is a period of remarkable dynamism with large scale changes in the pattern of gene expression, significant tissue growth and a surprising amount of neuronal differentiation. Disruption of early cortical development by exposure to toxin (e.g., EtOH) or mutation (e.g., Reln) can lead to intellectual disability. Explant models of early cortical development provide a bridge between dissociated culture studies and in vivo studies. The relatively small size and simple cellular composition allow for the study of disease relevant biology in the absence of synapses and functional circuits.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
I thank members of the Developmental Exposure Alcohol Research Center (DEARC) and my colleagues in my laboratory as well as Drs. Rick Matthews, Brian Howell, and David Cameron for valuable discussions. I thank Dr. John Vincent (Centre for Addiction and Mental Health, Toronto) for permission to use the Table of NS-ID genes from reference (30). The work was supported from NIAAA P50AA015850 (DEARC) and NINDS R01NS066071 grants.
1. Marin-Padilla M. Dual origin of the mammalian neocortex and evolution of the cortical plate. Anat Embryol (Berl) (1978) 152:109–26. doi:10.1007/BF00315920
2. Marin-Padilla M. Ontogenesis of the pyramidal cell of the mammalian neocortex and developmental cytoarchitectonics: a unifying theory. J Comp Neurol (1992) 321:223–40. doi:10.1002/cne.903210205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Meyer G, Schaaps JP, Moreau L, Goffinet AM. Embryonic and early fetal development of the human neocortex. J Neurosci (2000) 20:1858–68.
4. Marin-Padilla M. Structural organization of the human cerebral cortex prior to the appearance of the cortical plate. Anat Embryol (Berl) (1983) 168:21–40. doi:10.1007/BF00305396
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Takahashi T, Goto T, Miyama S, Nowakowski RS, Caviness VS Jr. Sequence of neuron origin and neocortical laminar fate: relation to cell cycle of origin in the developing murine cerebral wall. J Neurosci (1999) 19:10357–71.
6. Goffinet AM, Lyon G. Early histogenesis in the mouse cerebral cortex: a Golgi study. Neurosci Lett (1979) 14:61–6. doi:10.1016/0304-3940(79)95344-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Goffinet AM. An early development defect in the cerebral cortex of the reeler mouse. A morphological study leading to a hypothesis concerning the action of the mutant gene. Anat Embryol (Berl) (1979) 157:205–16. doi:10.1007/BF00305160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Derer P, Nakanishi S. Extracellular matrix distribution during neocortical wall ontogenesis in “normal” and “reeler” mice. J Hirnforsch (1983) 24:209–24.
9. Sheppard AM, Pearlman AL. Abnormal reorganization of preplate neurons and their associated extracellular matrix: an early manifestation of altered neocortical development in the reeler mutant mouse. J Comp Neurol (1997) 378:173–9. doi:10.1002/(SICI)1096-9861(19970210)378:2<173::AID-CNE2>3.0.CO;2-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Nichols AJ, Olson EC. Reelin promotes neuronal orientation and dendritogenesis during preplate splitting. Cereb Cortex (2010) 20:2213–23. doi:10.1093/cercor/bhp303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Angevine JB, Sidman RL. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature (1961) 192:766–8. doi:10.1038/192766b0
12. Martinez-Cerdeno V, Noctor SC. Cajal, Retzius, and Cajal-Retzius cells. Front Neuroanat (2014) 8:48. doi:10.3389/fnana.2014.00048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Ayoub AE, Kostovic I. New horizons for the subplate zone and its pioneering neurons. Cereb Cortex (2009) 19:1705–7. doi:10.1093/cercor/bhp025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature (1995) 374:719–23. doi:10.1038/374719a0
15. Osheroff H, Hatten ME. Gene expression profiling of preplate neurons destined for the subplate: genes involved in transcription, axon extension, neurotransmitter regulation, steroid hormone signaling, and neuronal survival. Cereb Cortex (2009) 19(Suppl 1):i126–34. doi:10.1093/cercor/bhp034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Hoerder-Suabedissen A, Molnar Z. Molecular diversity of early-born subplate neurons. Cereb Cortex (2013) 23:1473–83. doi:10.1093/cercor/bhs137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Shatz CJ, Luskin MB. The relationship between the geniculocortical afferents and their cortical target cells during development of the cat’s primary visual cortex. J Neurosci (1986) 6:3655–68.
18. Kostovic I, Rakic P. Developmental history of the transient subplate zone in the visual and somatosensory cortex of the macaque monkey and human brain. J Comp Neurol (1990) 297:441–70. doi:10.1002/cne.902970309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Ghosh A, Antonini A, McConnell SK, Shatz CJ. Requirement for subplate neurons in the formation of thalamocortical connections. Nature (1990) 347:179–81. doi:10.1038/347179a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Caviness VS Jr, Sidman RL. Time of origin or corresponding cell classes in the cerebral cortex of normal and reeler mutant mice: an autoradiographic analysis. J Comp Neurol (1973) 148:141–51. doi:10.1002/cne.901480202
21. Caviness VS Jr, Korde MG. Monoaminergic afferents to the neocortex: a developmental histofluorescence study in normal and reeler mouse embryos. Brain Res (1981) 209:1–9. doi:10.1016/0006-8993(81)91167-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Molnar Z, Adams R, Goffinet AM, Blakemore C. The role of the first postmitotic cortical cells in the development of thalamocortical innervation in the reeler mouse. J Neurosci (1998) 18:5746–65.
23. Aboitiz F, Montiel J. Origin and evolution of the vertebrate telencephalon, with special reference to the mammalian neocortex. Adv Anat Embryol Cell Biol (2007) 193:1–112.
24. Aboitiz F, Montiel J, Garcia RR. Ancestry of the mammalian preplate and its derivatives: evolutionary relicts or embryonic adaptations? Rev Neurosci (2005) 16:359–76. doi:10.1515/REVNEURO.2005.16.4.359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Aboitiz F. The origin of isocortical development. Trends Neurosci (2001) 24:202–3. doi:10.1016/S0166-2236(00)01771-9
26. Cameron DA, Middleton FA, Chenn A, Olson EC. Hierarchical clustering of gene expression patterns in the Eomes + lineage of excitatory neurons during early neocortical development. BMC Neurosci (2012) 13:90. doi:10.1186/1471-2202-13-90
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Arnold SJ, Sugnaseelan J, Groszer M, Srinivas S, Robertson EJ. Generation and analysis of a mouse line harboring GFP in the Eomes/Tbr2 locus. Genesis (2009) 47:775–81. doi:10.1002/dvg.20562
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, et al. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci (2005) 25:247–51. doi:10.1523/JNEUROSCI.2899-04.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Kowalczyk T, Pontious A, Englund C, Daza RA, Bedogni F, Hodge R, et al. Intermediate neuronal progenitors (basal progenitors) produce pyramidal-projection neurons for all layers of cerebral cortex. Cereb Cortex (2009) 19(10):2439–50. doi:10.1093/cercor/bhn260
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Kaufman L, Ayub M, Vincent JB. The genetic basis of non-syndromic intellectual disability: a review. J Neurodev Disord (2010) 2:182–209. doi:10.1007/s11689-010-9055-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. O’Rourke NA, Dailey ME, Smith SJ, McConnell SK. Diverse migratory pathways in the developing cerebral cortex. Science (1992) 258:299–302. doi:10.1126/science.1411527
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Nadarajah B, Alifragis P, Wong RO, Parnavelas JG. Neuronal migration in the developing cerebral cortex: observations based on real-time imaging. Cereb Cortex (2003) 13:607–11. doi:10.1093/cercor/13.6.607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Tabata H, Nakajima K. Multipolar migration: the third mode of radial neuronal migration in the developing cerebral cortex. J Neurosci (2003) 23:9996–10001.
34. Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci (2004) 7(2):136–44. doi:10.1038/nn1172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. de Anda FC, Meletis K, Ge X, Rei D, Tsai LH. Centrosome motility is essential for initial axon formation in the neocortex. J Neurosci (2010) 30:10391–406. doi:10.1523/JNEUROSCI.0381-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Hatanaka Y, Yamauchi K. Excitatory cortical neurons with multipolar shape establish neuronal polarity by forming a tangentially oriented axon in the intermediate zone. Cereb Cortex (2013) 23:105–13. doi:10.1093/cercor/bhr383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Jossin Y, Cooper JA. Reelin, Rap1 and N-cadherin orient the migration of multipolar neurons in the developing neocortex. Nat Neurosci (2011) 14:697–703. doi:10.1038/nn.2816
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci (2001) 4:143–50. doi:10.1038/83967
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Cooper JA. A mechanism for inside-out lamination in the neocortex. Trends Neurosci (2008) 31:113–9. doi:10.1016/j.tins.2007.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Goffinet AM. The cerebral cortex of the reeler mouse embryo. An electron microscopic analysis. Anat Embryol (Berl) (1980) 159:199–210. doi:10.1007/BF00304978
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Fonseca M, del Rio JA, Martinez A, Gomez S, Soriano E. Development of calretinin immunoreactivity in the neocortex of the rat. J Comp Neurol (1995) 361:177–92. doi:10.1002/cne.903610114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. del Rio JA, Martinez A, Fonseca M, Auladell C, Soriano E. Glutamate-like immunoreactivity and fate of Cajal-Retzius cells in the murine cortex as identified with calretinin antibody. Cereb Cortex (1995) 5:13–21. doi:10.1093/cercor/5.1.13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Schneider S, Gulacsi A, Hatten ME. Lrp12/Mig13a reveals changing patterns of preplate neuronal polarity during corticogenesis that are absent in reeler mutant mice. Cereb Cortex (2011) 21:134–44. doi:10.1093/cercor/bhq070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Rakic S, Davis C, Molnar Z, Nikolic M, Parnavelas JG. Role of p35/Cdk5 in preplate splitting in the developing cerebral cortex. Cereb Cortex (2006) 16(Suppl 1):i35–45. doi:10.1093/cercor/bhj172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet (2000) 26:93–6. doi:10.1038/79246
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell (2013) 155:997–1007. doi:10.1016/j.cell.2013.10.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Jones KL, Smith DW. Recognition of the fetal alcohol syndrome in early infancy. Lancet (1973) 302:999–1001. doi:10.1016/S0140-6736(73)91092-1
48. Jones KL, Smith DW. The fetal alcohol syndrome. Teratology (1975) 12:1–10. doi:10.1002/tera.1420120102
49. Stratton S, Howe C, Battaglia F. Fetal Alcohol Syndrome: Diagnosis, Epidemiology, Prevention and Treatment. Washington, DC: National Academy Press (1996).
50. Yang Y, Roussotte F, Kan E, Sulik KK, Mattson SN, Riley EP, et al. Abnormal cortical thickness alterations in fetal alcohol spectrum disorders and their relationships with facial dysmorphology. Cereb Cortex (2012) 22:1170–9. doi:10.1093/cercor/bhr193
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Roussotte FF, Sulik KK, Mattson SN, Riley EP, Jones KL, Adnams CM, et al. Regional brain volume reductions relate to facial dysmorphology and neurocognitive function in fetal alcohol spectrum disorders. Hum Brain Mapp (2012) 33:920–37. doi:10.1002/hbm.21260
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Goodwin DW, Crane JB, Guze SB. Phenomenological aspects of the alcoholic “blackout”. Br J Psychiatry (1969) 115:1033–8. doi:10.1192/bjp.115.526.1033
53. Goodlett CR, Johnson TB. Neonatal binge ethanol exposure using intubation: timing and dose effects on place learning. Neurotoxicol Teratol (1997) 19:435–46. doi:10.1016/S0892-0362(97)00062-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Heffernan T, O’Neill T. Time based prospective memory deficits associated with binge drinking: evidence from the cambridge prospective memory test (CAMPROMPT). Drug Alcohol Depend (2012) 123:207–12. doi:10.1016/j.drugalcdep.2011.11.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Dingwall KM, Maruff P, Cairney S. Similar profile of cognitive impairment and recovery for aboriginal Australians in treatment for episodic or chronic alcohol use. Addiction (2011) 106:1419–26. doi:10.1111/j.1360-0443.2011.03434.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Steinhausen HC, Willms J, Spohr HL. Long-term psychopathological and cognitive outcome of children with fetal alcohol syndrome. J Am Acad Child Adolesc Psychiatry (1993) 32:990–4. doi:10.1097/00004583-199309000-00016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Kerns KA, Don A, Mateer CA, Streissguth AP. Cognitive deficits in nonretarded adults with fetal alcohol syndrome. J Learn Disabil (1997) 30:685–93. doi:10.1177/002221949703000612
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Kril JJ, Halliday GM, Svoboda MD, Cartwright H. The cerebral cortex is damaged in chronic alcoholics. Neuroscience (1997) 79:983–98. doi:10.1016/S0306-4522(97)00083-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Maier SE, Chen WJ, Miller JA, West JR. Fetal alcohol exposure and temporal vulnerability regional differences in alcohol-induced microencephaly as a function of the timing of binge-like alcohol exposure during rat brain development. Alcohol Clin Exp Res (1997) 21:1418–28. doi:10.1097/00000374-199711000-00011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Goodlett CR, Horn KH, Zhou FC. Alcohol teratogenesis: mechanisms of damage and strategies for intervention. Exp Biol Med (2005) 230:394–406.
61. Lindsley TA, Comstock LL, Rising LJ. Morphologic and neurotoxic effects of ethanol vary with timing of exposure in vitro. Alcohol (2002) 28:197–203. doi:10.1016/S0741-8329(02)00279-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Olney JW. Fetal alcohol syndrome at the cellular level. Addict Biol (2004) 9:137–49; discussion 151. doi:10.1080/13556210410001717006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Miller MW. Effects of alcohol on the generation and migration of cerebral cortical neurons. Science (1986) 233:1308–11. doi:10.1126/science.3749878
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Lindsley TA, Kerlin AM, Rising LJ. Time-lapse analysis of ethanol’s effects on axon growth in vitro. Brain Res Dev Brain Res (2003) 147:191–9. doi:10.1016/j.devbrainres.2003.10.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Chen S, Charness ME. Ethanol disrupts axon outgrowth stimulated by netrin-1, GDNF, and L1 by blocking their convergent activation of Src family kinase signaling. J Neurochem (2012) 123:602–12. doi:10.1111/j.1471-4159.2012.07954.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Yanni PA, Rising LJ, Ingraham CA, Lindsley TA. Astrocyte-derived factors modulate the inhibitory effect of ethanol on dendritic development. Glia (2002) 38:292–302. doi:10.1002/glia.10071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Powrozek TA, Olson EC. Ethanol-induced disruption of Golgi apparatus morphology, primary neurite number and cellular orientation in developing cortical neurons. Alcohol (2012) 46:619–27. doi:10.1016/j.alcohol.2012.07.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Granato A, Palmer LM, De Giorgio A, Tavian D, Larkum ME. Early exposure to alcohol leads to permanent impairment of dendritic excitability in neocortical pyramidal neurons. J Neurosci (2012) 32:1377–82. doi:10.1523/JNEUROSCI.5520-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Anton ES, Marchionni MA, Lee KF, Rakic P. Role of GGF/neuregulin signaling in interactions between migrating neurons and radial glia in the developing cerebral cortex. Development (1997) 124:3501–10.
70. Rakic P. Neuronal migration and contact guidance in the primate telencephalon. Postgrad Med J (1978) 54(Suppl 1):25–40.
71. Stubbs D, DeProto J, Nie K, Englund C, Mahmud I, Hevner R, et al. Neurovascular congruence during cerebral cortical development. Cerebral cortex (2009) 19(Suppl 1):i32–41. doi:10.1093/cercor/bhp040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Siegenthaler JA, Pleasure SJ. We have got you “covered”: how the meninges control brain development. Curr Opin Genet Dev (2011) 21:249–55. doi:10.1016/j.gde.2010.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Nichols AJ, O’Dell RS, Powrozek TA, Olson EC. Ex utero electroporation and whole hemisphere explants: a simple experimental method for studies of early cortical development. J Vis Exp (2013) 74. doi:10.3791/50271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Jossin Y, Ogawa M, Metin C, Tissir F, Goffinet AM. Inhibition of SRC family kinases and non-classical protein kinases C induce a reeler-like malformation of cortical plate development. J Neurosci (2003) 23:9953–9.
75. Jossin Y, Ignatova N, Hiesberger T, Herz J, Lambert de Rouvroit C, Goffinet AM. The central fragment of reelin, generated by proteolytic processing in vivo, is critical to its function during cortical plate development. J Neurosci (2004) 24:514–21. doi:10.1523/JNEUROSCI.3408-03.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Yang Y, Zhang P, Xiong Y, Li X, Qi Y, Hu H. Ectopia of meningeal fibroblasts and reactive gliosis in the cerebral cortex of the mouse model of muscle-eye-brain disease. J Comp Neurol (2007) 505:459–77. doi:10.1002/cne.21474
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Hu H, Yang Y, Eade A, Xiong Y, Qi Y. Breaches of the pial basement membrane and disappearance of the glia limitans during development underlie the cortical lamination defect in the mouse model of muscle-eye-brain disease. J Comp Neurol (2007) 501:168–83. doi:10.1002/cne.21238
78. Peiffer AM, Fitch RH, Thomas JJ, Yurkovic AN, Rosen GD. Brain weight differences associated with induced focal microgyria. BMC Neurosci (2003) 4:12. doi:10.1186/1471-2202-4-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Horton AC, Racz B, Monson EE, Lin AL, Weinberg RJ, Ehlers MD. Polarized secretory trafficking directs cargo for asymmetric dendrite growth and morphogenesis. Neuron (2005) 48:757–71. doi:10.1016/j.neuron.2005.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. O’Dell RS, Ustine CJ, Cameron DA, Lawless SM, Williams RR, Zipfel WR, et al. Layer 6 cortical neurons require reelin-Dab1 signaling for cellular orientation, Golgi deployment, and directed neurite growth into the marginal zone. Neural Dev (2012) 7:25. doi:10.1186/1749-8104-7-25
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: preplate, reelin, fetal alcohol spectrum disorders, dendritogenesis, Golgi apparatus
Citation: Olson EC (2014) Analysis of preplate splitting and early cortical development illuminates the biology of neurological disease. Front. Pediatr. 2:121. doi: 10.3389/fped.2014.00121
Received: 03 September 2014; Accepted: 23 October 2014;
Published online: 11 November 2014.
Edited by:
Alberto Granato, Catholic University, ItalyReviewed by:
Alessandro Vercelli, Università degli Studi di Torino, ItalyCopyright: © 2014 Olson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric C. Olson, Department of Neuroscience and Physiology, State University of New York Upstate Medical University, 4703 NRB, 505 Irving Avenue, Syracuse, NY 13210, USA e-mail:b2xzb25lQHVwc3RhdGUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.