 Silvia Giovedí
Silvia Giovedí Anna Corradi
Anna Corradi Anna Fassio
Anna Fassio Fabio Benfenati
Fabio Benfenati- 1Department of Experimental Medicine, University of Genova, Genova, Italy
- 2Department of Neuroscience and Brain Technologies, Fondazione Istituto Italiano di Tecnologia, Genova, Italy
Autism spectrum disorders (ASDs) are heterogeneous neurodevelopmental disorders characterized by deficits in social interaction and social communication, restricted interests, and repetitive behaviors. Many synaptic protein genes are linked to the pathogenesis of ASDs, making them prototypical synaptopathies. An array of mutations in the synapsin (Syn) genes in humans has been recently associated with ASD and epilepsy, diseases that display a frequent comorbidity. Syns are pre-synaptic proteins regulating synaptic vesicle traffic, neurotransmitter release, and short-term synaptic plasticity. In doing so, Syn isoforms control the tone of activity of neural circuits and the balance between excitation and inhibition. As ASD pathogenesis is believed to result from dysfunctions in the balance between excitatory and inhibitory transmissions in neocortical areas, Syns are novel ASD candidate genes. Accordingly, deletion of single Syn genes in mice, in addition to epilepsy, causes core symptoms of ASD by affecting social behavior, social communication, and repetitive behaviors. Thus, Syn knockout mice represent a good experimental model to define synaptic alterations involved in the pathogenesis of ASD and epilepsy.
Autism spectrum disorders (ASDs) represent a wide array of neurodevelopmental disorders characterized by restricted interest, defective social interactions, repetitive behaviors, and deficit in language and verbal communication that manifest within the first 3 years of life (1, 2). The complexity of ASD is evident both at the levels of symptoms variability and of causative factors. An important genetic contribution has been observed for ASD, however, the mechanism of inheritance remains largely unknown (1). More than 500 genes have been associated with different forms of autism, but each of them account only for the minority of ASD cases and indeed environmental contributions and other modulating factors, as environment-genetic interplay and epigenetic modifications, are emerging as potential risk factors for ASDs (3).
Involvement of Synaptic Proteins in the Pathogenesis of ASDs
Autism spectrum disorders may result from mutations in a large array of genes having roles in various physiological processes such as chromatin remodeling, translation, metabolism, and synaptic functions. In experimental models of ASD, a common breakdown appears to occur at the level of synapse formation and stabilization, as well as of the ability of synapses to be modified by experience through plasticity mechanisms. Synapse dysfunctions are also at the convergence between ASD and other neuropsychiatric disorders with unknown etiology, such as schizophrenia and intellectual disabilities (4). In addition, ASDs frequently occur together with epilepsy and there may be common underlying mechanisms as well as common genetic and environmental risk factors. Synapse maturation and function rely on a vast array of compartmentalized protein–protein interactions that allow fidelity in neurotransmitter release and synaptic vesicle (SV) cycling at the pre-synaptic site and in neurotransmitter receptor localization and signaling at the post-synaptic site. Moreover, synaptic adhesion molecules link and stabilize pre- and post-synaptic sites and control synaptic modification induced by plasticity.
The synaptic theory for autism originally stems from the identification of ASD mutations in the Neuroligin genes [NLGN3 and NLGN4X; (5, 6)] coding for synaptic adhesion molecules expressed at the post-synaptic site. Since then, a growing repertoire of synaptic genes, coding for both pre- and post-synaptic proteins, have been implicated in non-syndromic ASDs: synaptic adhesion molecules [neuroligins, neurexins, cadherins, contactins and contactin-associated protein-like 2, or CNTNAP2; (7–9)], synaptic scaffold proteins [PROSAP/SHANK gene family; (10)], ion channels, and neurotransmitter receptors (11–13). Moreover, mutations in additional synaptic genes, such as the pre-synaptic RIMS3/NIM3 and the post-synaptic IL1RAPL1 and SynGAP1, involved in either SV organization or synapse formation, have been recently associated with ASD cases [(14–17); Figure 1 and Table 1]. The identification of synaptic genes implicated in ASDs is expanding together with the characterization of the respective animal models bearing mutations or deletions in these genes. These models allow a more systematic analysis to study the role of those genes in ASD etiology, to discover the biological mechanisms underlying autistic behaviors and evaluate the efficacy of new potential treatments (18–21).
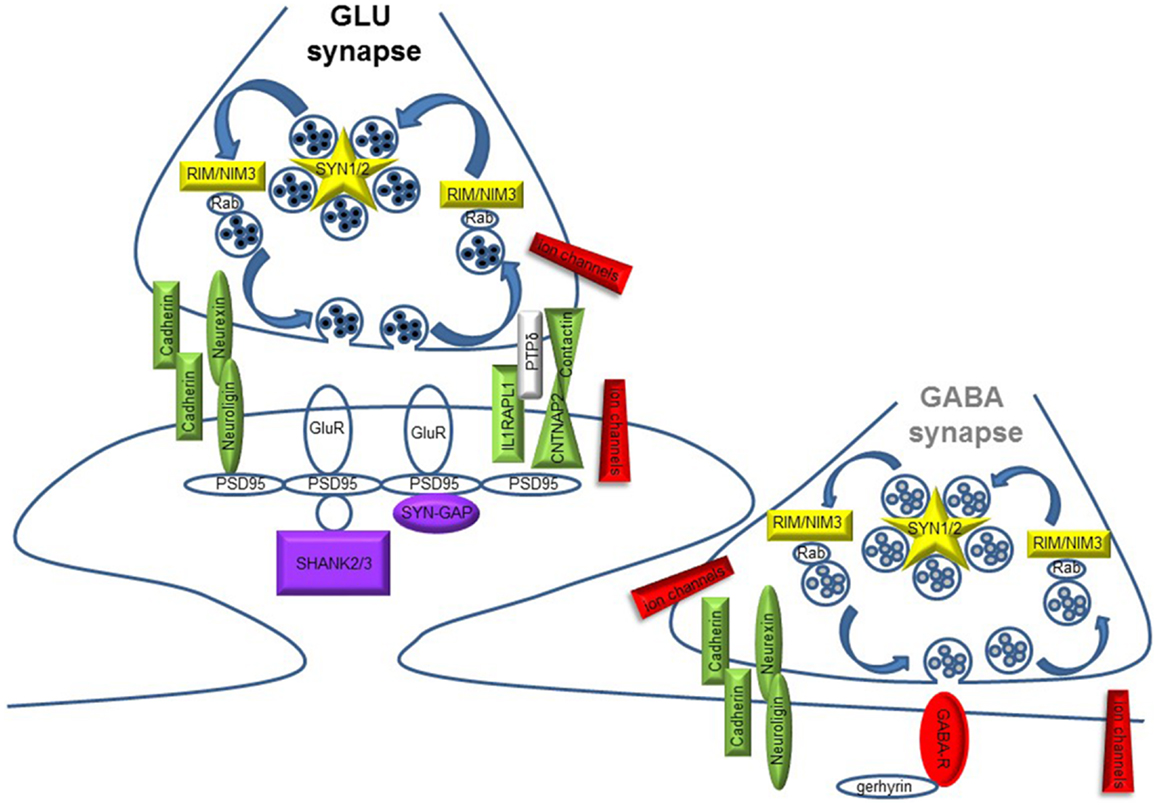
Figure 1. Schematic diagram illustrating pre- and post-synaptic gene products implicated in ASD. Glutamate (GLU) and GABA synapses are shown. Different colors code for synaptic function: yellow, synaptic vesicle cycling; green, synapse formation and maintenance; red, neuronal excitability and neurotransmission; violet, glutamate receptors (GluR) signaling/trafficking.
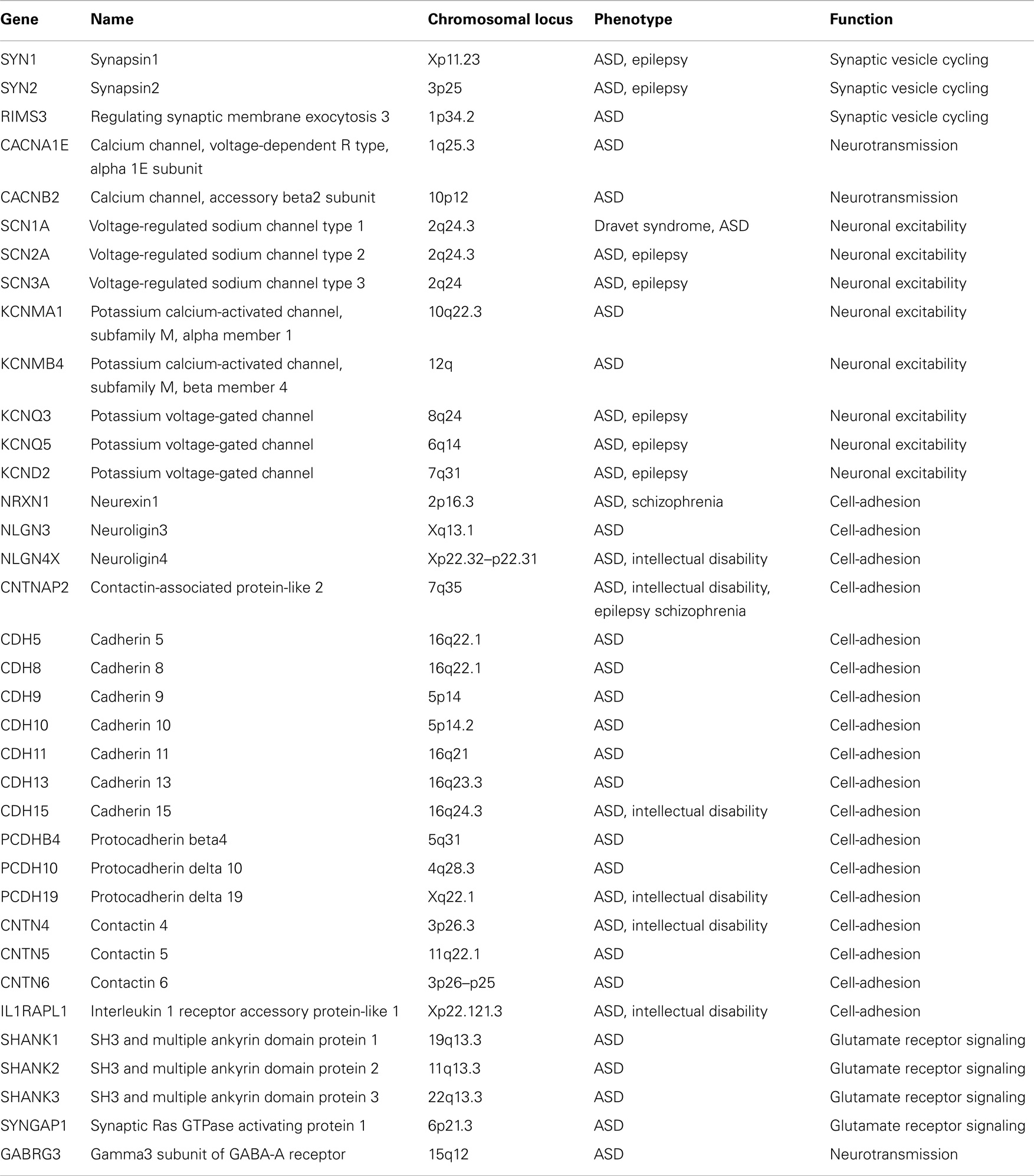
Table 1. Synaptic genes associated with ASD.
ASD is Associated with Dysfunctions in Cortical Circuits That also Predispose to Epilepsy
Autism spectrum disorder-like phenotypes are observed in a wide variety of neurological and neurodevelopmental disorders, including epilepsy, Rett syndrome, Fragile X Syndrome, Tuberous Sclerosis, or Fetal Anticonvulsant Syndrome, which are characterized by an imbalance of the excitatory/inhibitory tone. This aspect is consistent with the very high prevalence of epilepsy in autistic patients (about 25%) with respect to the average prevalence of 1% in the general population. Characteristic ASD phenotypes are associated with impairments or gains of GABAergic transmission (22–25).
Decreases in the GABA synthesizing enzyme GAD and a reduction in quantal size have been reported in the experimental model of Rett syndrome, the Mecp2 knockout (KO) mice (26). Various impairments in GABAergic function, including deficient GABAergic circuitry, decreased expression of GABAA receptor subunits (particularly α5 and γ subunits) and of the tonic GABA current, have been observed in mice lacking FMRP, a recognized experimental model of Fragile X Syndrome (27). The chromosomal region 15q11–q13, which is deleted or duplicated in 1–2% of idiopatic ASD patients, contains a cluster of genes encoding GABA receptor subunits [Gabra5, Gabrb3, and Gabrg3; (11)] and deletion of the Gabrb3 gene in mice, encoding for the GABAA β3 receptor subunit, leads to an ASD-like phenotype (28). Moreover, Reeler mice, lacking the protein reelin that is expressed in cortical interneurons, display an ASD phenotype that is associated with a decrease in GABA turnover (29, 30). Mice lacking synapsins (Syns) also display a primary impairment in GABA release dynamics that is associated with an ASD-like phenotype [see below; (31–35)]. Taken together, all these experimental data indicate that GABA systems are major actors in the development and functioning of cortical networks, and that their dysfunction can lead to altered development and/or function of cortical circuits resulting in epilepsy, ASD, or both. Moreover, the role of GABAergic dysfunctions in ASD pathogenesis is complex given the switch between excitatory and inhibitory GABA transmission that occurs during development and the multiple sites of action of GABA in mature neuronal networks, where it acts on predominantly post-synaptic GABAA receptors, extrasynaptic GABAA receptors regulating excitability and predominantly pre-synaptic GABAB receptors modulating glutamate release and short-term plasticity properties of excitatory synapses. Thus, disruption or dysfunction of GABAergic systems may delay critical periods in specific brain regions and perturb γ-oscillations implicated in high cognitive functions. Given the importance of the excitation/inhibition balance in the activity-dependent formation and plasticity of neocortical networks, we here review on the role of the Syns, a family of pre-synaptic proteins regulating release and plasticity in inhibitory and excitatory synapses, in the etiology of ASDs and the use of Syn KO animals as a model for these complex neuropsychiatric disorders.
Synapsins
The Syns are a family of abundant neuronal phosphoproteins that participate as regulators in synaptic transmission and plasticity, as well as in neuronal development [see Ref. (36, 37), for review]. The family is composed of 10 homologous proteins: Syn Ia–b, Syn IIa–b, and Syn IIIa–f (38–40), encoded in mammals by alternative splicing of three distinct genes (SYN1, SYN2, and SYN3) mapping on distinct chromosomes (chromosome X, 3, and 22, respectively) in human and mouse. Notably, Syn III is the most precociously expressed isoform that has a role in the early phases of neural development and is downregulated in mature neurons (40). On the other hand, Syn I and Syn II are expressed at low levels at birth and their expression progressively increases along synaptogenesis to reach a stable plateau at 1–2 months of life, approximately the time window epilepsy appears (41).
All Syn isoforms display a domain structure with the NH2-terminal region, highly conserved across isoforms and species, divided in domains A, B, and C, and the COOH-terminal portion, more divergent, composed of different spliced domains [D–I; (38)]. Many of these isoforms share consensus sequences for phosphorylation by several protein kinases, which all contribute to the modulation of Syn function (36). Domain A contains the phosphorylation site for PKA and CaMKI/IV that modulates the reversible association of Syn with SVs. Domain B, less conserved and considered as a link region, contains phosphorylation sites for MAPK/Erk, which also causes the redistribution of Syn from SVs to the cytosol. Domain C, a large central region of about 300 amino acids, mediates the interaction with actin filaments and SVs and promotes SV clustering by inducing Syn homo/hetero-dimerization (42, 43). This domain is phosphorylated by the tyrosine kinase Src (44) and contains residues mediating the binding to ATP (45). The sequences of Syn isoforms at the COOH-terminal region diverge (domain D in Syn Ia and Ib, domain G in Syn IIa and IIb, domain H in Syn IIa, and domain J in Syn IIIa), although they all bear proline-rich regions binding to several SH3-containing proteins (46, 47), and additional phosphorylation sites for CaMKII, MAPK/Erk, and cdk1/5, which affect the biochemical properties of Syn I resulting in a drastic reduction of its binding to both actin and SVs (48, 49). Finally domain E, highly conserved and common to all “a” isoforms, modulates Syn targeting to the pre-synaptic terminals and SV trafficking (50–53).
The best-characterized function of Syns is to control SV trafficking and modulate neurotransmitter release at the pre-synaptic terminal. The fine regulation of the balance between the reserve and the readily releasable pool of SVs is strictly controlled by Syn site-specific phosphorylation in response to stimulation, which modulates Syn association with SVs, actin cytoskeleton, and other synaptic proteins, and leaves SVs free to move close to the active zone and undergo fusion. Besides the function of Syns in these pre-docking stages of neurotransmission, recent data, supported by the fact that at least part of Syn do not dissociate from SVs upon fusion, strongly indicate that Syns play a role in the final post-docking stages of exocytosis, including SV priming, fusion, and recycling of the synaptic membrane in the area surrounding the active zone [see Ref. (36), for review].
Beyond the role in synaptic transmission, the various Syn isoforms play an important role in neuronal growth and synaptogenesis. Lack of Syn I or Syn II was shown to impair neurite outgrowth during the first days in vitro (54), while downregulation or ablation of Syn III caused an impairment in the development of axons at early stages in culture (55). Moreover, clear-cut structural and physiological defects were observed in the pre-synaptic terminals of Syn KO neurons (56–58), confirming the role of Syn isoforms in the modulation of synapse formation, maintenance, and rearrangement [see Ref. (37), for review].
Synapsin KO Mice are Epileptic
Knockout mice for either Syn gene are viable and fertile, have a normal life expectancy and brains of normal size and gross structure. As Syns are involved in the regulation of the excitability of neuronal networks, it is not surprising that the impairment of Syn function can result in epilepsy. Syn I KO, Syn II KO, Syn I/II double KO, and Syn I/II/III triple KO are all prone to epileptic seizures that appear approximately at 2–3 months of age, and progressively aggravate with aging and the number of Syn genes ablated [(59, 60); see Ref. (61) for review]. The fact that epilepsy does not appear at birth after ablation of Syn I or Syn II genes and that Syn III KO mice are not epileptic can be explained by the specific expression profile of the three Syn genes during development (55, 62). It is therefore likely that mature synapses require physiological levels of both Syn I and Syn II to achieve a stable excitation/inhibition balance during activity, while Syn III seems to be dispensable in this respect. In general, the loss of Syns disrupts the reserve pool of SV and alters release dynamics. However, the pre- and post-docking effects of the Syns differentially affect excitatory and inhibitory neurons. This, together with the selective distribution of the various Syn isoforms in distinct neuronal populations and the non-overlapping functions of Syn isoforms on neurotransmitter release, can result in an imbalance between excitatory and inhibitory synaptic transmission, both under conditions of basal activity and of high-frequency stimulation, potentially leading to epileptogenesis (31, 33, 34).
Synapsin KO Mice Display an ASD-Like Phenotype
Although it is not an easy task to translate the complex symptoms of human ASD into mouse behaviors, the study of the phenotype of mice bearing deletions in the genes found to be mutated in ASD patients is fundamental for the understanding of how dysfunction of single components of the synaptic protein network may result in a general functional impairment that generates the disease. Such mouse models of ASDs should display decreased interest toward the environment, impaired sociability, and social interactions/communication, as well as repetitive behaviors.
Synapsin KO mice have generally preserved cognitive functions. A prospective study performed on Syn I and Syn II KO mice revealed that cognitive and emotional performances are not altered before the onset of epilepsy both in terms of spatial memory, object recognition, and emotional memory. Only later on, during aging and in the presence of an overt epileptic phenotype, behavioral deficits in emotional memory in both genotypes and spatial memory in Syn II KO mice emerged with respect to wild type controls, and were associated with neuronal loss and gliosis in the cortex and hippocampus (63). On the other hand, Syn III KO mice that are not epileptic, exhibit only minor alterations in spatial memory, object recognition, fear conditioning, and fear-potentiated startle (40, 64).
When an array of socially directed behaviors (social interaction and novelty, social recognition and social dominance, social transmission of food preference, and social memory) were investigated in Syn I, Syn II, and Syn III KO mice before (2-months old) and after (6-months old) the appearance of epilepsy (in Syn I and Syn II KO mice), it was immediately clear that mice presented various impairments in social behaviors and repetitive behaviors well before the appearance of the epileptic phenotype (65).
Synapsin III mice had the mildest phenotype and showed impairments only in social interactions with an intruder, a decreased social dominance and a decreased social transmission of food preference. Syn I KO mice had an intermediate phenotype and had deficits in social and environmental exploration, social transmission, and an increased social dominance. Finally, Syn II KO had the more severe behavioral phenotype and exhibited significant deficits in virtually all social behaviors tested (with the exception of the social transmission of food preference) together with an increased social dominance and repetitive self-grooming behaviors (Figure 2). These data indicate that Syn KO mice represent an interesting animal model for ASDs. The physio-pathological importance of this model for the understanding of ASD pathogenesis is underlined by the occurrence of ASD-linked loss-of-function mutations in human SYN genes, as described in the following section.
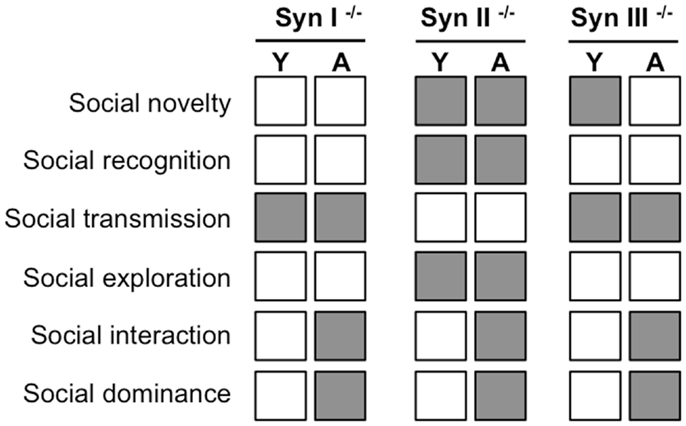
Figure 2. Summary of the abnormalities in social behavior (gray squares) observed in young (Y, 2-month old) and adult (A, 6-month old) Syn I, Syn II, and Syn III KO mice with respect to the behavior of age-matched controls sharing the same genetic background [data from Ref. (65)]. Adults, but not young, Syn I and Syn II KO mice are epileptic.
Mutations in the SYN Genes are Associated with ASD in Humans
Autism spectrum disorder-associated mutations were found in both human SYN1 and SYN2 genes. Mutations in SYN1 were mainly identified in patients affected by both epilepsy and autism, whereas, mutations in SYN2 were observed in cases of ASD without association with epilepsy. The identified mutations are schematized in Figure 3. Two SYN1 nonsense mutations, causing truncations at protein level (W356X and Q555X), were identified in two large families with epilepsy with recessive X-linked transmission. In addition to epilepsy, few males carrying the mutations in SYNI also presented learning difficulties, low average IQ and three of them meet criteria for ASD (66, 67). The SYN1 missense mutation A550T was isolated in four patients: two with epilepsy, one with autism, and one with both, whereas, the missense mutation T567A was isolated in two individuals with ASD only. A frameshift (A94fs199X) and two missense (Y236S and G464R) mutations were identified in the SYN2 gene in three males affected by ASD (68). The mutation was transmitted by the non-affected mother. Although this phenomenon was observed only in a limited number of individuals, it is consistent with a recent report on the autosomal SHANK1 gene deletions associated with ASD in males but not females (69). Autosomal sex-limited expression, in addition to the mutation in X-linked genes, may contribute to the increased prevalence of ASD in males with respect to females. The exact mechanism at the basis of the higher penetrance in males remains to be determined.
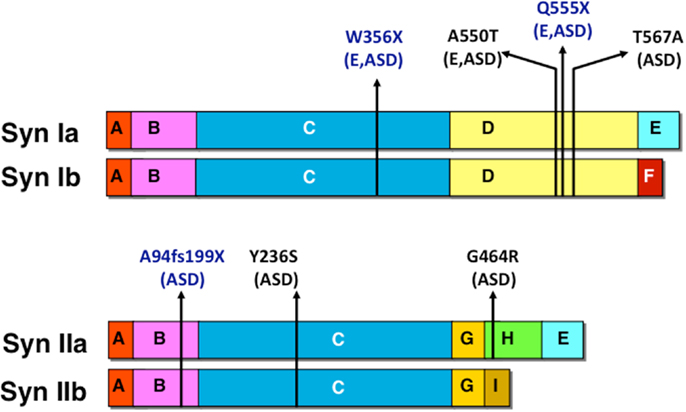
Figure 3. Human mutations in SYN1 and SYN2 associated with epilepsy and/or ASD. Nonsense and missense mutations are indicated in blue and black, respectively together with the associated pathology (E, epilepsy; ASD, autism spectrum disorder).
Effects of Human SYN Mutants Expressed in SYN KO Neurons
To get insight into the molecular mechanisms of the pathogenesis of these diseases, the physiological effects of the SYN genetic variants associated with ASDs or epilepsy (or both) were analyzed in vitro by expressing the mutants in primary hippocampal neurons from Syn I KO or Syn II KO mice and their effects on neuronal development, nerve terminal targeting, dynamics of exo-endocytosis, and synaptic transmission were studied and compared with wild type Syn.
For the W356X mutation in Syn I, the presence of a premature stop codon in the human SYNI transcript leads to nonsense-mediated mRNA decay (NMD). The few transcripts escaping NMD process give rise to a mislocalized and non-functional protein (70). On the contrary, the second nonsense mutation (Q555X) in Syn I does not lead to NMD and a truncated form of Syn I is expressed that lacks about a half of the D domain and the COOH-terminal domains E/F. The lack of D domain impairs its binding to SVs, its phosphorylation by CaMKII and MAPK/Erk and its interactions with SH3 domain-containing proteins such as PI3K, Src, endophilin, and intersectin (67). Neurons expressing the Q555X-Syn I showed a transient impairment of axonal outgrowth, but normal dendritic arborization, nerve terminal targeting, and synaptic density (67). However, the exocytosis was impaired and the size of the readily releasable and recycling pools of SVs decreased. Electrophysiological recordings from neurons expressing Q555X-Syn I mutant in Syn I KO background showed that, while the basal excitatory and inhibitory transmissions were equally depressed by the mutant, a clear imbalance in short-term plasticity was present with excitatory synapses showing markedly increased paired-pulse facilitation and post-tetanic potentiation as well as faster recovery from depression, whereas, inhibitory synapses displaying an enhanced post-tetanic depression and synaptic depression during sustained high-frequency stimulation, as well as a marked slowed-down recovery from depression. This excitatory/inhibitory balance in the temporal domain of short-term synaptic plasticity produced a marked hyperexcitability and enhanced network bursting behavior at network level as demonstrated by multi-electrode array recordings, also underlining the key role of short-term plasticity at excitatory and inhibitory synapses in the regulation of network excitability (71). Two further missense mutations in Syn I (A550T and T567A) located in domain D did not significantly affected phosphorylation, molecular interactions of Syn I, or neuronal development. However, the mutants were not correctly targeted to nerve terminals and, in addition, the dynamics and sizes of the readily releasable and recycling pools of SVs at synaptic terminals were impaired (67).
The Syn II mutants had distinct physiological effects. The A94fs199X-Syn II mutant was not expressed in neurons, probably because fast degradation of the aberrant protein. Both missense mutants Y236S- and G464R-Syn II were correctly expressed in Syn II KO neurons and targeted to nerve terminals. However, both mutants impaired the size of the recycling pool of SVs, leaving the readily releasable pool unaffected. Moreover, the G464R-Syn II mutant also caused an impaired axonal growth and dendritic development of neurons (68). Similar defects in neuronal development and dendritic arborization were found in neurons silenced for the ASD-associated gene CNTNAP2, with a resulting impairment of neural circuit assembly and changes in network activity, possible causes of ASD pathogenesis (72).
Synapsin KO neurons expressing the genetic variants of Syns share common defects in SV pool dynamics. Syn I and Syn II are known to control the density of SVs at the nerve terminal and regulate their availability for release differentially, with Syn I affecting both the readily releasable and recycling pools of SVs, and Syn II only affecting the latter pool. The analysis of the dynamics of exo-endocytosis in neurons expressing genetic variants of Syns reflects these distinct effects. ASD manifestations begin in the second/third year of life, a period of intense refinement, remodeling, and experience-dependent plasticity of synapses. This periods overlaps with developmental expression pattern of Syns. Impairments in SV pool dynamics, associated with defects in short-term plasticity and/or neuronal development, may thereby destabilize the key processes of assembly of neuronal networks and the balance between excitation and inhibition.
Concluding Remarks
Synapsins are not essential for synaptic transmission, but play a key role in synaptic homeostasis and plasticity with direct consequences in network activity and excitatory/inhibitory balance. Based on the findings in human and mice, Syn genes may represent a common genetic basis for epilepsy and ASD and, accordingly, Syn KO mice can be considered a potentially interesting animal model for ASD. Similar to what occurs in children with ASDs and epilepsy, ASD-related behaviors in Syn I KO and Syn II KO mice precede the onset of seizures and epilepsy does not significantly affect the expression of the behavioral alterations in adult mice. Moreover, the non-epileptic Syn III KO mice also display some traits of social deficits. These observations lead to the idea that epilepsy and ASD follow distinct and independent pathogenic pathways, although the genetic basis appears to be largely shared by the two diseases. Although the mutations in the Syn genes found thus far account only for a limited number of ASD cases, they map into a “synaptic autism pathway” in which dysfunctions of any of the genes essential for the regulation of synapse formation, excitation/inhibition balance and activity-dependent plasticity can result in a similar ASD phenotype. Despite the inherent redundancy and robustness of mammalian biological systems, a focused dysfunction in one synaptic gene can induce secondary changes in the synaptic machinery impacting on synaptic plasticity, leading to complex dysfunctions at the circuit level associated with the appearance of the pathological phenotype. An example of this potential derangement of a complex machinery by genetic dysfunction of a single component is provided by the numerous and diverse genes implicated in phototransduction whose mutation converges toward the common clinical phenotype of Retinitis pigmentosa (73). In addition, it has been proposed that gene alterations and secondary dysfunctions may accumulate non-linearly in complex gene networks implicated in neural computation and higher brain functions, such as those constituting the synaptome (74, 75). In conclusion, although our map of ASD vulnerability genes is rapidly progressing, many challenges remain for the future, particularly concerning the interactions between genetic, epigenetic, and environmental factors to produce the complex ASD clinical manifestations.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Paul Greengard (The Rockefeller University, New York, NY, USA) for providing us with the Syn KO mice, for the fruitful discussions and the continuous support and to Dr. Patrick Cossette (Université de Montreal, Montreal, QC, Canada) for the fruitful collaboration on human SYN mutations. This study was supported by research grants from the Italian Ministry of University and Research (PRIN to Fabio Benfenati and Anna Corradi and FIRB 2010 “Futuro in Ricerca” to Silvia Giovedí), EU FP7 Integrating Project “Desire”, Jerome Lejeune Foundation (to Anna Fassio) and Cure Epilepsy “Advanced Innovator Award” (to Fabio Benfenati). The support of Telethon-Italy (Grants GGP09134 and GGP033 to Fabio Benfenati) is also acknowledged.
References
1. Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet (2008) 9:341–55. doi: 10.1038/nrg2346
2. Levitt P, Campbell DB. The genetic and neurobiologic compass points toward common signaling dysfunctions in autism spectrum disorders. J Clin Invest (2009) 119:747–54. doi:10.1172/JCI37934
3. Persico AM, Napolioni V. Autism genetics. Behav Brain Res (2013) 251:95–112. doi:10.1016/j.bbr.2013.06.012
4. Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet (2014) 294:677–94. doi:10.1016/j.ajhg.2014.03.018
5. Jamain S, Quach H, Betancur C, Råstam M, Colineaux C, Gillberg IC, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet (2003) 34:27–9. doi:10.1038/ng1136
6. Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science (2003) 302:826–30. doi:10.1126/science.1089071
7. Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature (2008) 455:903–11. doi:10.1038/nature07456
8. Bentacur C, Sakurai T, Buxbaum JD. The emerging role of synaptic cell adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci (2009) 32:402–12. doi:10.1016/j.tins.2009.04.003
9. Redies C, Hertel N, Hübner CA. Cadherins and neuropsychiatric disorders. Brain Res (2012) 1470:130–44. doi:10.1016/j.brainres.2012.06.020
10. Jiang YH, Ehlers MD. Modeling autism by SHANK gene mutations in mice. Neuron (2013) 78:8–27. doi:10.1016/j.neuron.2013.03.016
11. Shao Y, Cuccaro ML, Hauser ER, Raiford KL, Menold MM, Wolpert CM, et al. Fine mapping of autistic disorder to chromosome 15q11-q13 by use of phenotypic subtypes. Am J Hum Genet (2003) 72:539–48. doi:10.1086/367846
12. Li X, Zou H, Brown WT. Genes associated with autism spectrum disorder. Brain Res Bull (2012) 88:543–52. doi:10.1016/j.brainresbull.2012.05.017
13. Schmunk G, Gargus JJ. Channelopathy pathogenesis in autism spectrum disorders. Front Genet (2013) 4:222. doi:10.3389/fgene.2013.00222
14. Piton A, Michaud JL, Peng H, Aradhya S, Gauthier J, Mottron L, et al. Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum Mol Genet (2008) 17:3965–74. doi:10.1093/hmg/ddn300
15. Kumar RA, Sudi J, Babatz TD, Brune CW, Oswald D, Yen M, et al. A de novo 1p34.2 microdeletion identifies the synaptic vesicle gene RIMS3 as a novel candidate for autism. J Med Genet (2010) 47:81–90. doi:10.1136/jmg.2008.065821
16. Hamdan FF, Daoud H, Piton A, Gauthier J, Dobrzeniecka S, Krebs MO, et al. De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol Psychiatry (2011) 69:898–901. doi:10.1016/j.biopsych.2010.11.015
17. Berryer MH, Hamdan FF, Klitten LL, Møller RS, Carmant L, Schwartzentruber J, et al. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum Mutat (2013) 34:385–94. doi:10.1002/humu.22248
18. Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol (2012) 4:a009886. doi:10.1101/cshperspect.a009886
19. Delorne R, Ely E, Toro R, Leboyer M, Gillberg C, Bourgeron T. Progress toward treatments for synaptic defects in autism. Nat Med (2013) 6:685–94. doi:10.1038/nm.3193
20. Banerjee S, Riordan M, Bhat MA. Genetic aspects of autism spectrum disorders: insight from animal models. Front Cell Neurosci (2014) 8:58. doi:10.3389/fncel.2014.00058
21. Kleijer KT, Schmeisser MJ, Krueger DD, Boeckers TM, Scheiffele P, Bourgeron T, et al. Neurobiology of autism gene products: towards pathogenesis and drug targets. Psychopharmacology (Berl) (2014) 231:1037–42. doi:10.1007/s00213-013-3403-3
22. Zoghbi HY. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature (2010) 468:263–9. doi:10.1038/nature09582
23. Ramamoorthi K, Lin Y. The contribution of GABAergic dysfunction to neurodevelopmental disorders. Trends Mol Med (2011) 17:452–62. doi:10.1016/j.molmed.2011.03.003
24. Pizzarelli R, Cherubini E. Alterations of GABAergic signaling in autism spectrum disorders. Neural Plast (2011) 2011:297153. doi:10.1155/2011/297153
25. Coghlan S, Horder J, Inkster B, Mendez MA, Murphy DG, Nutt DJ. GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci Biobehav Rev (2012) 36:2044–55. doi:10.1016/j.neubiorev.2012.07.005
26. Chao H-T, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature (2010) 468:263–9. doi:10.1038/nature09582
27. Selby L, Zhang C, Sun QQ. Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neurosci Lett (2007) 412:227–32. doi:10.1016/j.neulet.2006.11.062
28. DeLorey TM, Sahbaie P, Hashemi E, Homanics GE, Clark JD. Gabrb3 gene deficient mice exhibit impaired social and exploratory behaviors, deficits in non-selective attention and hypoplasia of cerebellar vermal lobules: a potential model of autism spectrum disorder. Behav Brain Res (2008) 187:207–20. doi:10.1016/j.bbr.2007.09.009
29. Bailey A, Luthert P, Dean A, Harding B, Janota I, Montgomery M, et al. A clinicopathological study of autism. Brain (1998) 121:889–905. doi:10.1093/brain/121.5.889
30. Carboni G, Tueting P, Tremolizzo L, Sugaya I, Davis J, Costa E, et al. Enhanced dizocilpine efficacy in heterozygous reeler mice relates to GABA turnover downregulation. Neuropharmacology (2004) 46:1070–81. doi:10.1016/j.neuropharm.2004.02.001
31. Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, et al. Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci (2004) 24:11368–80. doi:10.1523/JNEUROSCI.3795-04.2004
32. Baldelli P, Fassio A, Valtorta F, Benfenati F. Lack of synapsin I reduces the readily releasable pool of synaptic vesicles at central inhibitory synapses. J Neurosci (2007) 27:13520–31. doi:10.1523/JNEUROSCI.3151-07.2007
33. Chiappalone M, Casagrande S, Tedesco M, Valtorta F, Baldelli P, Martinoia S, et al. Opposite changes in glutamatergic and GABAergic transmission underlie the diffuse hyperexcitability of synapsin I-deficient cortical networks. Cereb Cortex (2008) 19:1422–39. doi:10.1093/cercor/bhn182
34. Farisello P, Boido D, Nieus T, Medrihan L, Cesca F, Valtorta F, et al. Synaptic and extrasynaptic origin of the excitation/inhibition imbalance in the hippocampus of synapsin I/II/III knockout mice. Cereb Cortex (2013) 23:581–93. doi:10.1093/cercor/bhs041
35. Medrihan L, Cesca F, Raimondi A, Lignani G, Baldelli P, Benfenati F. Synapsin II desynchronizes neurotransmitter release at inhibitory synapses by interacting with presynaptic calcium channels. Nat Commun (2013) 4:1512. doi:10.1038/ncomms2515
36. Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol (2010) 91:313–48. doi:10.1016/j.pneurobio.2010.04.006
37. Fornasiero EF, Bonanomi D, Benfenati F, Valtorta F. The role of synapsins in neuronal development. Cell Mol Life Sci (2010) 67:1383–96. doi:10.1007/s00018-009-0227-8
38. Südhof TC, Czernik AJ, Kao H-T, Takei K, Johnston PA, Horiuchi A, et al. Synapsins: mosaics of shared and individual domains in a family of synaptic vesicle phosphoproteins. Science (1989) 245:1474–80. doi:10.1126/science.2506642
39. Porton B, Kao H-T, Greengard P. Characterization of transcripts from the synapsin III gene locus. J Neurochem (1999) 73:2266–71. doi:10.1046/j.1471-4159.1999.0732266.x
40. Porton B, Wetsel WC, Kao H-T. Synapsin III: role in neuronal plasticity and disease. Semin Cell Dev Biol (2011) 22:416–24. doi:10.1016/j.semcdb.2011.07.007
41. Bogen IL, Jensen V, Hvalby O, Walaas SI. Synapsin-dependent development of glutamatergic synaptic vesicles and presynaptic plasticity in postnatal mouse brain. Neuroscience (2009) 158:231–41. doi:10.1016/j.neuroscience.2008.05.055
42. Benfenati F, Valtorta F, Rossi MC, Onofri F, Sihra T, Greengard P. Interactions of synapsin I with phospholipids: possible role in synaptic vesicle clustering and in the maintenance of bilayer structures. J Cell Biol (1993) 123:1845–55. doi:10.1083/jcb.123.6.1845
43. Hosaka M, Südhof TC. Homo- and heterodimerization of synapsins. J Biol Chem (1999) 274:16747–53. doi:10.1074/jbc.274.24.16747
44. Onofri F, Messa M, Matafora V, Bonanno G, Corradi A, Bachi A, et al. Synapsin phosphorylation by SRC tyrosine kinase enhances SRC activity in synaptic vesicles. J Biol Chem (2007) 282:15754–67. doi:10.1074/jbc.M701051200
45. Esser L, Wang CR, Hosaka M, Smagula CS, Südhof TC, Deisenhofer J. Synapsin I is structurally similar to ATP-utilizing enzymes. EMBO J (1998) 17:977–84. doi:10.1093/emboj/17.4.977
46. Onofri F, Giovedí S, Kao H-T, Valtorta F, Bongiorno Borbone L, De Camilli P, et al. Specificity of the binding of synapsin I to SRC homology 3 domains. J Biol Chem (2000) 275:29857–67. doi:10.1074/jbc.M006018200
47. Giovedí S, Darchen F, Valtorta F, Greengard P, Benfenati F. Synapsin is a novel Rab3 effector protein on small synaptic vesicles. II. Functional effects of the Rab3A-synapsin I interaction. J Biol Chem (2004) 279:43769–79. doi:10.1074/jbc.M404168200
48. Benfenati F, Valtorta F, Rubenstein JL, Gorelick FS, Greengard P, Czernik AJ. Synaptic vesicle-associated Ca2+/calmodulin-dependent protein kinase II is a binding protein for synapsin I. Nature (1992) 359:417–20. doi:10.1038/359417a0
49. Verstegen AM, Tagliatti E, Lignani G, Marte A, Stolero T, Atias M, et al. Phosphorylation of synapsin I by cyclin-dependent kinase-5 sets the ratio between the resting and recycling pools of synaptic vesicles at hippocampal synapses. J Neurosci (2014) 34:7266–80. doi:10.1523/JNEUROSCI.3973-13.2014
50. Hilfiker S, Schweizer FE, Kao H-T, Czernik AJ, Greengard P, Augustine GJ. Two sites of action for synapsin domain E in regulating neurotransmitter release. Nat Neurosci (1998) 1:29–35. doi:10.1038/1147
51. Gitler D, Xu Y, Kao H-T, Lin D, Lim S, Feng J, et al. Molecular determinants of synapsin targeting to presynaptic terminals. J Neurosci (2004) 224:3711–20. doi:10.1523/JNEUROSCI.5225-03.2004
52. Fassio A, Merlo D, Mapelli J, Menegon A, Corradi A, Mete M, et al. The synapsin domain E accelerates the exoendocytotic cycle of synaptic vesicles in cerebellar Purkinje cells. J Cell Sci (2006) 119:4257–68. doi:10.1242/jcs.03194
53. Monaldi I, Vassalli M, Bachi A, Giovedí S, Millo E, Valtorta F, et al. The highly conserved synapsin domain E mediates synapsin dimerization and phospholipid vesicle clustering. Biochem J (2010) 426:55–64. doi:10.1042/BJ20090762
54. Chin LS, Li L, Ferreira A, Kosik KS, Greengard P. Impairment of axonal development and of synaptogenesis in hippocampal neurons of synapsin I-deficient mice. Proc Natl Acad Sci U S A (1995) 92:9230–4. doi:10.1073/pnas.92.20.9230
55. Ferreira A, Kao H-T, Feng J, Rapoport M, Greengard P. Synapsin III: developmental expression, subcellular localization, and role in axon formation. J Neurosci (2000) 20:3736–44.
56. Li L, Chin LS, Shupliakov O, Brodin L, Sihra TS, Hvalby O, et al. Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc Natl Acad Sci U S A (1995) 92:9235–9. doi:10.1073/pnas.92.20.9235
57. Takei Y, Harada A, Takeda S, Kobayashi K, Terada S, Noda T, et al. Synapsin I deficiency results in the structural change in the presynaptic terminals in the murine nervous system. J Cell Biol (1995) 131:1789–800. doi:10.1083/jcb.131.6.1789
58. Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, et al. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature (1995) 375:488–93. doi:10.1038/375488a0
59. Cambiaghi M, Cursi M, Monzani E, Benfenati F, Comi G, Minicucci F, et al. Temporal evolution of neurophysiological and behavioral features of synapsin I/II/III triple knock-out mice. Epilepsy Res (2013) 103:153–60. doi:10.1016/j.eplepsyres.2012.07.012
60. Etholm L, Bahonjic E, Heggelund P. Sensitive and critical periods in the development of handling induced seizures in mice lacking synapsins: differences between synapsin I and synapsin II knockouts. Exp Neurol (2013) 247:59–65. doi:10.1016/j.expneurol.2013.03.025
61. Fassio A, Raimondi A, Lignani G, Benfenati F, Baldelli P. Synapsins: from synapse to network hyperexcitability and epilepsy. Semin Cell Dev Biol (2011) 22:408–15. doi:10.1016/j.semcdb.2011.07.005
62. Feng J, Chi P, Blanpied TA, Xu Y, Magarinos AM, Ferreira A, et al. Regulation of neurotransmitter release by synapsin III. J Neurosci (2002) 22:4372–80.
63. Corradi A, Zanardi A, Giacomini C, Onofri F, Valtorta F, Zoli M, et al. Synapsin-I- and synapsin-II-null mice display an increased age-dependent cognitive impairment. J Cell Sci (2008) 121:3042–51. doi:10.1242/jcs.035063
64. Porton B, Rodriguiz RM, Phillips LE, Gilbert JW IV, Feng J, Greengard P, et al. Mice lacking synapsin III show abnormalities in explicit memory and conditioned fear. Genes Brain Behav (2010) 9:257–68. doi:10.1111/j.1601-183X.2009.00555.x
65. Greco B, Managò F, Tucci V, Kao H-T, Valtorta F, Benfenati F. Autism-related behavioral abnormalities in synapsin knockout mice. Behav Brain Res (2013) 251:65–74. doi:10.1016/j.bbr.2012.12.015
66. Garcia CC, Blair HJ, Seager M, Coulthard A, Tennant S, Buddles M, et al. Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J Med Genet (2004) 41:183–6. doi:10.1136/jmg.2003.013680
67. Fassio A, Patry L, Congia S, Onofri F, Piton A, Gauthier J, et al. SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum Mol Genet (2011) 20:2297–307. doi:10.1093/hmg/ddr122
68. Corradi A, Fadda M, Piton A, Patry L, Marte A, Rossi P, et al. SYN2 is an autism predisposing gene: loss-of-function mutations alter synaptic vesicle cycling and axon outgrowth. Hum Mol Genet (2014) 23:90–103. doi:10.1093/hmg/ddt401
69. Sato D, Lionel AC, Leblond CS, Prasad A, Pinto D, Walker S, et al. Shank1 deletions in males with autism spectrum disorder. Am J Hum Genet (2012) 90:879–87. doi:10.1016/j.ajhg.2012.03.017
70. Giannandrea M, Guarnieri FC, Gehring NH, Monzani E, Benfenati F, Kulozik AE, et al. Nonsense-mediated mRNA decay and loss-of-function of the protein underlie the X-linked epilepsy associated with the W356X mutation in synapsin I. PLoS One (2013) 8:e67724. doi:10.1371/journal.pone.0067724
71. Lignani G, Raimondi A, Ferrea E, Rocchi A, Paonessa F, Cesca F, et al. Epileptogenic Q555X SYN1 mutant triggers imbalances in release dynamics and short-term plasticity. Hum Mol Genet (2013) 22:2186–99. doi:10.1093/hmg/ddt071
72. Anderson GR, Galfin T, Xu W, Aoto J, Malenka RC, Sudhof TC. Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc Natl Acad Sci U S A (2012) 109:18120–5. doi:10.1073/pnas.1216398109
73. Ferrari S, Di Iorio E, Barbaro V, Ponzin D, Sorrentini FS, Parmeggiani F. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics (2011) 12:238–49. doi:10.2174/138920211795860107
74. Lips ES, Cornelisse LN, Toonen RF, Min JL, Hultman CM, International Schizophrenia Consortium X, et al. Functional gene group analysis identifies synaptic gene groups as risk factor for schizophrenia. Mol Psychiatry (2012) 17:996–1006. doi:10.1038/mp.2011.117
Keywords: autism, synaptopathies, synaptic vesicles, synaptic transmission, social behavior, human mutations, knockout mice
Citation: Giovedí S, Corradi A, Fassio A and Benfenati F (2014) Involvement of synaptic genes in the pathogenesis of autism spectrum disorders: the case of synapsins. Front. Pediatr. 2:94. doi: 10.3389/fped.2014.00094
Received: 11 June 2014; Accepted: 21 August 2014;
Published online: 04 September 2014.
Edited by:
Yuri Bozzi, University of Trento, ItalyReviewed by:
A. Kimberley McAllister, University of California, USALaurie Doering, McMaster University, Canada
Copyright: © 2014 Giovedí, Corradi, Fassio and Benfenati. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabio Benfenati, Department of Neuroscience and Brain Technologies, Fondazione Istituto Italiano di Tecnologia, Via Morego 30, 16163 Genoa, Italy e-mail:ZmFiaW8uYmVuZmVuYXRpQGlpdC5pdA==
†Silvia Giovedí and Anna Corradi have contributed equally to this work.