 Milos Jesenak
Milos Jesenak Peter Banovcin
Peter Banovcin Barbora Jesenakova
Barbora Jesenakova Eva Babusikova
Eva Babusikova- 1Center for Diagnosis and Treatment of Primary Immunodeficiencies, Department of Pediatrics, Jessenius Faculty of Medicine, Comenius University in Bratislava, Martin, Slovakia
- 2Department of Medical Biochemistry, Jessenius Faculty of Medicine, Comenius University in Bratislava, Martin, Slovakia
Primary immunodeficiencies (PIDs) are inherited disorders in which one or several components of immune system are decreased, missing, or of non-appropriate function. These diseases affect the development, function, or morphology of the immune system. The group of PID comprises more than 200 different disorders and syndromes and the number of newly recognized and revealed deficiencies is still increasing. Their clinical presentation and complications depend on the type of defects and there is a great variability in the relationship between genotypes and phenotypes. A variation of clinical presentation across various age categories is also presented and children could widely differ from adult patients with PID. Respiratory symptoms and complications present a significant cause of morbidity and also mortality among patients suffering from different forms of PIDs and they are observed both in children and adults. They can affect primarily either upper airways (e.g., sinusitis and otitis media) or lower respiratory tract [e.g., pneumonia, bronchitis, bronchiectasis, and interstitial lung diseases (ILDs)]. The complications from lower respiratory tract are usually considered to be more important and also more specific for PIDs and they determinate patients’ prognosis. The spectrum of the causal pathogens usually demonstrates typical pattern characteristic for each PID category. The respiratory signs of PIDs can be divided into infectious (upper and lower respiratory tract infections and complications) and non-infectious (ILDs, bronchial abnormalities – especially bronchiectasis, malignancies, and benign lymphoproliferation). Early diagnosis and appropriate therapy can prevent or at least slow down the development and course of respiratory complications of PIDs.
Introduction
Science of primary immunodeficiencies (PIDs) represents a fascinating and rapidly developing part of modern medicine and clinical immunology. PIDs are inherited disorders of immune system in which one or several immune components are decreased, missing, or of non-appropriate function. These diseases affect the development, function, or morphology of the immune system (1). Since the first official scientific publication of the PID case in 1952 (2), more than 200 other diseases were described and characterized. The heterogeneity of the PIDs, the variability of their clinical manifestations, inconsistence in the relationship between genotype and phenotype and involvement of whatever organ or tissue supports the interdisciplinary character of these diseases, which requires multidisciplinary approach in their management. Hand-in-hand, continual education of health professionals and laics as well is urgently needed. There are on-going efforts of the education of laic and medical community, which is aimed on the increase of public awareness of the PID. The early diagnosis is usually associated with better prognosis and sooner appropriate therapeutic strategies. From the symptomatic diagnosis that was based on the detection of various immune system defects after the onset of clinical symptoms and complications, we shifted toward complicated algorithms consisting from different immunological tests accompanied by molecular-genetic analysis in the selected cases (Figure 1). The biggest challenge of current immunology is to establish the diagnosis of PID even before the onset of the clinical symptoms, just to start the right therapy as soon as possible and to prevent the possible complications and consequences of non-treated or non-appropriately treated disease. Several countries have recently introduced into the praxis the pilot programs of neonatal screening for selected immunodeficiencies [especially for severe combined immunodeficiency (SCID) diseases, but also for ataxia telangiectasia and some severe humoral immunodeficiencies]. On the other hand, for the most severe diseases, the prenatal diagnosis in cases of positive family history for particular PID is fully recommended (3).
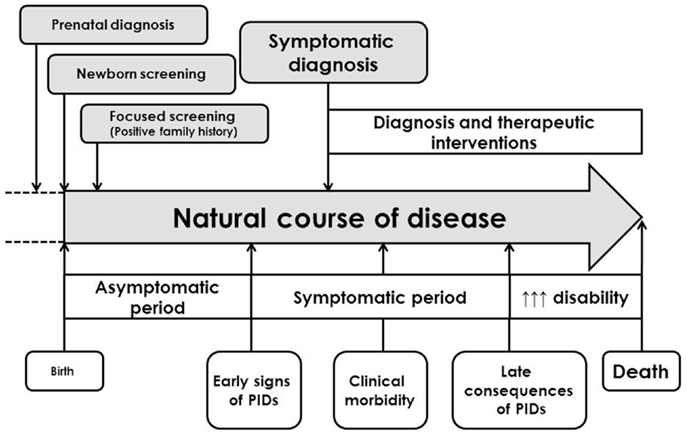
Figure 1. General clinical course of primary immunodeficiencies and different approaches in establishment of the diagnosis [adapted and modified from Ref. (4)].
The group of PIDs is getting more and more heterogeneous and the number of newly recognized and revealed deficiencies is increasing year by year. The majority of PIDs is monogenic, but in some cases, the polygenic basic is suspected. There are still many PIDs with unknown or not-fully described and characterized genetic background [e.g., common variable immunodeficiency (CVID) or selective deficiency of IgA]. PIDs are usually rare diseases with overall prevalence of 1:10,000 live births. However, some of the PIDs can be found quite frequently (e.g., selective deficiency of IgA, deficiencies of IgG subclasses, deficiency of mannose-binding lectin, etc.) (5). The most frequent immunodeficiencies yield usually mild course. Some of the frequent immunodeficiencies can be even asymptomatic during the lifetime and their clinical significant of questionable and discussed. In case of the combination of different “mild” defects in immunity, the clinical manifestation becomes more possible and evident.
The classification of PIDs underwent a long way and was elaborated regularly. The understanding of the particular diseases allowed the creation of the new categories and groups of related diseases of immune system. According to the current knowledge, the immunodeficiencies are classified into the eight major categories based on the primarily involved immune component and associated symptoms and signs (6):
1. predominantly humoral (antibody) deficiencies,
2. combined T-cell and B-cell immunodeficiencies,
3. other well defined immunodeficiency syndromes,
4. congenital defects of number and/or function of phagocytes,
5. complement deficiencies,
6. defects of immune dysregulation,
7. autoinflammatory disorders,
8. defects in innate immunity.
Among all the immunodeficiencies, antibody deficiencies are the most frequent and comprise approximately 70–75% of all PIDs. These patients are typically characterized by different respiratory symptoms and complications due to the inherited immune defect. In children, respiratory symptoms are typical initial presentation of various PIDs. However, also the other groups and classes of PIDs can be associated with significant respiratory morbidity and manifestations (Table 1). Through two simple widely available test – serum immunoglobulin concentration (IgG, IgA, IgM, and ±IgE) and differential leukocyte cell count – the majority of the PID can be detected and revealed. Therefore, these two simplex tests can be in general recommended as screening tools for PIDs in primary care.
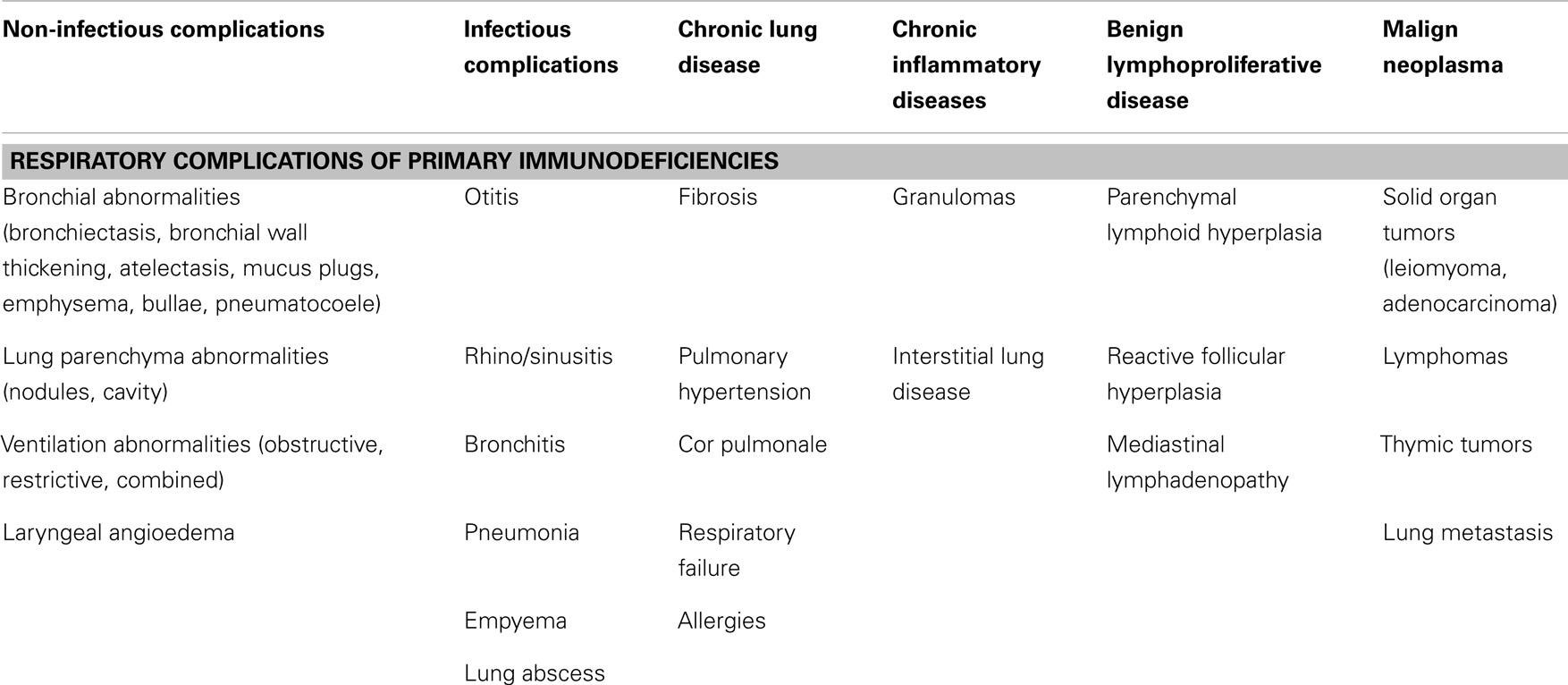
Table 1. Respiratory presentations and complications of primary immunodeficiencies [adapted according to Bierry et al. (7) and Touw et al. (8)].
Respiratory Manifestations of Primary Immunodeficiencies
Respiratory symptoms and complications present a significant cause of morbidity and also mortality among patients suffering from different forms of PIDs (Table 2). Computed tomography (CT) in combination with other imaging techniques, clinical tests, and laboratory examinations play an important role in detecting, characterizing, and quantifying the extent and kind of lung damage (9). Another important role of the imaging techniques lies also in the evaluation of the lung pathology progression. The screening for lung complications should be performed regularly, especially in patients with antibody deficiencies. Based on the expected symptoms and complications, the so-called warning signs for primary immunodeficiencies were elaborated (Tables 3 and 4). Respiratory complications, especially infectious can be expressed very soon in the early life (Table 5). The non-infectious complications and manifestations usually appear during the course of PIDs in the adolescent or adult age. Among all age categories, respiratory symptoms present an important marker pointing the attention toward PIDs, although it should be assumed that the sensitivity of particular “warning” signs differs (10). The most relevant are these signs:
• positive family history for PIDs,
• more than 2-months antibiotic therapy for PIDs with the dysfunction of neutrophils,
• failure to thrive ± chronic diarrhea for T-cellular immunodeficiencies.
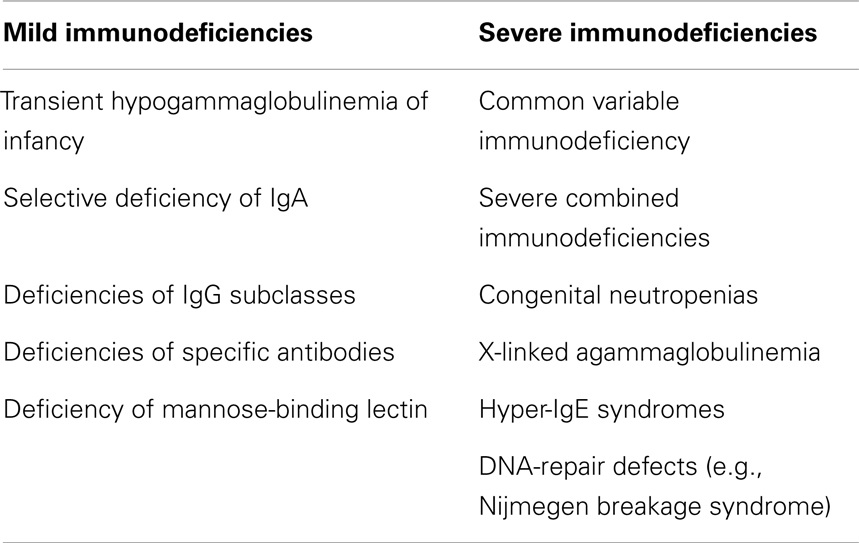
Table 2. The most important immunodeficiencies associated with respiratory complications in children.

Table 3. Warning signs for primary immunodeficiencies in children [adapted according to Arkwright and Gennery (11)].

Table 4. Warning signs for primary immunodeficiencies in adults.
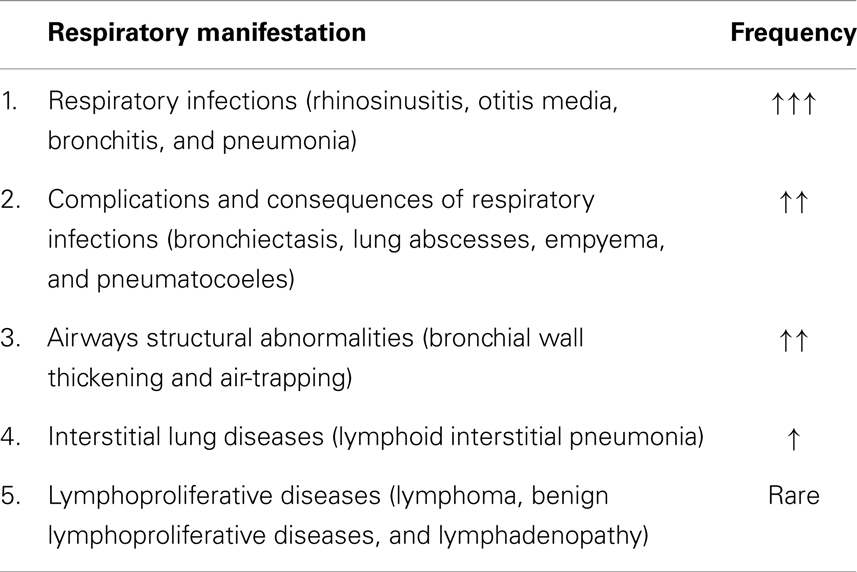
Table 5. Respiratory manifestations and complications of primary immunodeficiencies in childhood with estimated average frequency.
In the retrospective study in 64 children from Egypt, the most frequent symptoms were: need for intravenous antibiotic therapy, gastrointestinal symptoms, and recurrent pneumonias within 12 months (12). There are also some other potentially alarming signs and complications for PIDs:
• autoimmune disease of unknown etiology,
• opportunistic infections,
• complications after the vaccination with live attenuated vaccines (especially after BCG vaccination against tuberculosis),
• chronic graft-versus-host diseases (feto-maternal engraftment),
• systemic atypical mycobacteriosis,
• persistent and recalcitrant dermatitis in infants,
• delayed umbilical cord separation.
The respiratory symptoms and complications of PIDs can affect primarily either upper airways (e.g., sinusitis and otitis media) or lower respiratory tract [e.g., pneumonia, bronchiectasis, and interstitial lung diseases (ILDs)]. The complications from lower respiratory tract are usually considered to be more important and also more specific for PIDs and they determinate patients’ prognosis. The respiratory signs of PIDs can be divided into infectious and non-infectious (Figure 2). According to the other classification, they can be divided into several basic categories (9):
1. respiratory tract infections,
2. airways disease,
3. interstitial lung disease,
4. malignant diseases.
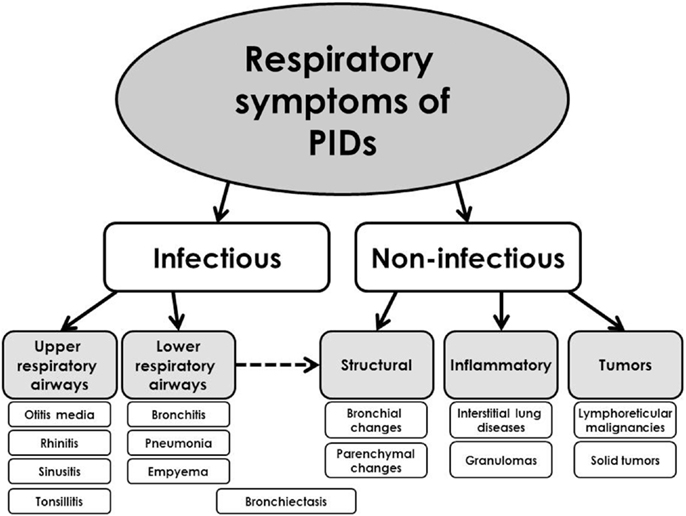
Figure 2. Simplified classification of the respiratory presentations of primary immunodeficiencies.
Infectious Respiratory Manifestations of Primary Immunodeficiencies
Respiratory infections are universal clinical problem and symptom across the whole childhood. One should discriminate among the child with normal susceptibility to infections, transient increased morbidity without any complications and consequences (so-called physiological respiratory morbidity) and the subjects with increased, severe, complicated respiratory morbidity, which evokes the possible immune defect (13, 14). The respiratory infectious complications show typical spectrum of etiological pathogens according to the immune defect, which can help in the diagnostic algorithm for particular type of PID (Table 6).
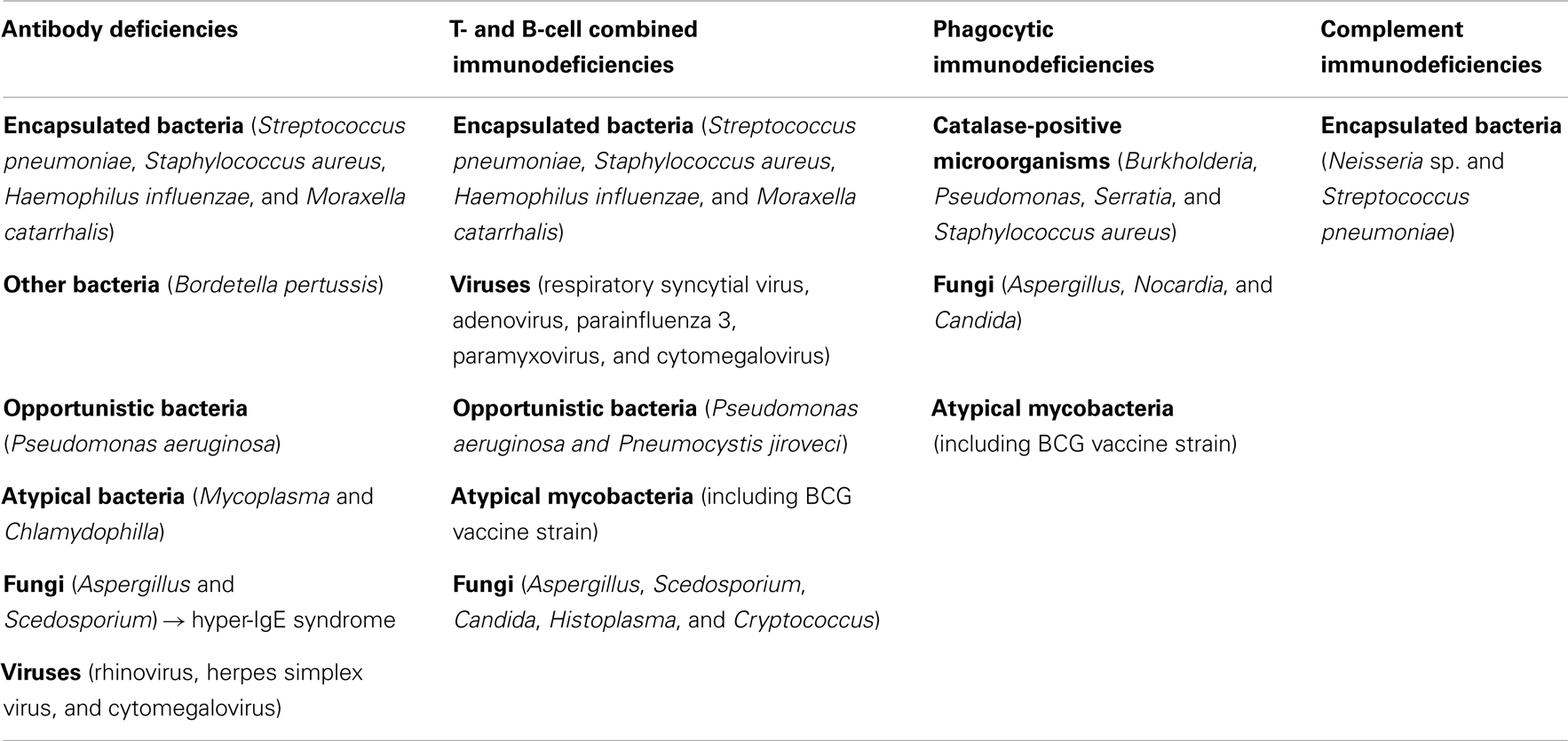
Table 6. Etiological agents of respiratory infections according to the PIDs category.
Predominantly humoral immunodeficiencies
Predominantly humoral immunodeficiencies represent clinically the most important and largest group of inherited immune defects. Their prevalence widely varied in different populations and geographical settings. The most frequent defects are: selective deficiency of IgA, deficiencies of IgG subclasses, and deficiency of specific antibodies. However, since the frequent diseases usually present clinically only with mild symptoms, the most clinically important disease from this PID category is CVID, which is typically presented by infectious symptoms, especially from respiratory or gastrointestinal tract, but is commonly associated with broad spectrum of different non-infectious complications. The first officially reported PID in the literature was X-linked (Bruton’s) agammaglobulinemia (XLA), which belongs also to this group and yields a broad spectrum of early respiratory complications. There are also some other defects associated with the antibody dysregulation and deficiency, but they were re-classified and now are involved in the other PIDs categories [e.g., Hyper-IgE syndrome (HIES), Hyper-IgM syndrome, etc.].
The most common clinical manifestation of predominant humoral (and combined immunodeficiencies with associated antibody defects) are recurrent and prolonged infections involving the respiratory tract, e.g., rhinosinusitis, otitis media, bronchitis, bronchiectasis, and pneumonias. Respiratory infections in PIDs patients are usually severe, persistent, caused by unusual, atypical, or opportunistic microorganisms and recurrent (acronym S.P.U.R.) in comparison with infections in non-PID patients. The clinical symptoms in humoral deficiencies, typically tend to occur after the first 6 months of life (after the disappearance of maternal IgG), however, recent studies indicate that sino-pulmonary infections may occur earlier (16). According to many studies, clinical history is the most important aspect of suspecting a diagnosis of primary humoral immunodeficiency. Therefore, patients at any age with recurrent upper or lower respiratory infections, where the frequency, severity, course of isolated pathogen is unusual or out of context (e.g., in non-smoking patients), should be investigated for possible humoral or other type immunodeficiency (17). The finding of bronchiectasis in young people should rise the suspicious for possible PIDs. During the examination for the possible primary immunodeficiency, the pathological susceptibility to infections can be demonstrated and identified. These patients have more than eight minor infections per year (until 3 years of age and beyond), severe major infections (pneumonia, sepsis, meningitis, encephalitis, osteomyelitis, septic arthritis, empyema, deep visceral, or skin abscesses) with chronic and relapsing course followed by residues. Typical are the relapses caused by the same pathogen and opportunistic infections are quite frequent (18). The frequency of chest infections including pneumonia varied between 37 and 90% across the studies with antibody deficiencies, and the frequency of recurrent sinus infections was estimated between 19 and 98% (17). In CVID, the recurrent pneumonia, sinusitis, and otitis media appear in the majority of the patients (19). In the recent big international study, which involved more than 2200 CVID patients, pneumonia was the most common clinical sign. Patients with pneumonias had lower trough levels of IgG compared to the patients without recurrent pneumonias (20). The un-controlled and recurrent lower respiratory tract infections in these patients usually lead to the chronic changes expressed by interstitial lung processes or the bronchiectasis development. The diagnosis of primary antibody deficiencies is often delayed, despite the presence of chronic respiratory symptoms (17). Chronic rhinosinusitis but also other infectious complications have a significant negative impact on the life quality of patients with humoral deficiencies (21).
Recurrent pneumonia is one of the most frequent, important, and characteristic sign of primary humoral deficiencies (Tables 2 and 3). The pneumonias in antibody deficiencies are typically caused by encapsulated bacteria: Streptococcus pneumonia, Haemophilus influenzae, and Staphylococcus sp. Since the primary antibody deficiencies are usually associated also with the impaired production of specific antibodies after the vaccinations, the etiological agents could be also the vaccine-preventable infections (e.g., Bordetella pertussis). Interestingly, in early disease, H. influenzae and S. pneumoniae typically cause the exacerbations of respiratory symptoms, while within the progression of lung damage, Pseudomonas and Staphylococcus aureus become more important and dominant (22). Mycoplasma can cause also chronic pneumonitis in X-linked agammaglobulinemia (23). Besides the bacteria, these patients have also the increased susceptibility to respiratory viral infections. There are also some differences in the natural course of particular infections, e.g., rhinoviral infections are frequent and prolonged. Severe infections caused by varicella-zoster virus, herpes simplex, and cytomegalovirus have been also reported (9). Some opportunistic pathogens (e.g., Pneumocystis jirovecii, Mycobacterium tuberculosis) can be also found in patients with antibody deficiencies. In patients with HIES, recurrent staphylococcal infections are very typical. The pneumonias in these patients lead to the bronchiectasis and pneumatocoele formation, which serve as locus minoris resistentiae for other infections, especially fungi. Other infectious pathologies, e.g., lung abscess and empyema have been also described (17, 24).
There are several differences between various forms of severe humoral PIDs regarding the frequency and type of respiratory symptoms and complications. As many as 75–84% of CVID patients have had at least one episode (often multiple episodes) of pneumonia before the diagnosis of PID (25). The risk of chronic lung disease is higher in patients with CVID than in those with XLA, probably due to the longer diagnostic delay in CVID (26, 27). In XLA, less than a third had a chronic lung disease. The IgG trough levels at around 500 mg/l are often inadequate to fully prevent chronic lung disease. The delayed diagnosis is associated with a higher risk of developing chronic lung disease (26, 28). The studies with pediatric patients with CVID have shown that CVID in children presents with the comparable symptoms and complications as in adults, but the later diagnosis significantly negatively influence growth and development. Compared to adults, higher frequency of otitis media can be observed. This is probably the consequence of the anatomic characteristics of upper airways in children (29).
The appropriate therapy using the immunoglobulin substitution and antibiotics usually leads to the significant decline of the frequency and severity of infections with the significant impact on the life quality and prognosis of these patients. However, the application of intravenous immunoglobulins can be on the other side associated rarely with pulmonary complications. Mild wheezing or dyspnea is not uncommon immediate reactions during the application and respiratory symptoms are constant part of systemic allergic reactions, which can also occur in association with this treatment. Serious but very rare pulmonary complications include pulmonary embolism, edema, pleural effusion, and transfusion-related lung injury with fever and hypotension. Despite these reactions, immunoglobulin therapy is highly effective and safe and in patients with reactions to intravenous application a shift to subcutaneous route with excellent safety profile is strongly recommended (30).
Combined and other well defined immunodeficiencies
Combined T- and B-cell immunodeficiencies represent usually very severe form of immune defects, which requires soon diagnosis and appropriate treatment. The most important disease from this PIDs category is SCID, which is caused by defects in nearly 20 genes. Combined immunodeficiency is typically associated with several complex syndromic immunodeficiencies, which are now involved in other PIDs categories, e.g., other well defined immunodeficiencies (e.g., immunodeficiencies with DNA-repair defects).
Patients with combined T- and B-cell immunodeficiencies have predisposition to infections caused by intracellular pathogens. SCID presents an immunological emergency with very soon presentation. A common feature of SCID infants is the lack of thymic shadow on chest X-ray. The respiratory tract is the most common site of infections in SCID and the most frequently involved microorganisms are P. jirovecii, cytomegalovirus, adenovirus, parainfluenza virus type 3, and respiratory syncytial virus (26). Another possible marker for SCID is chronic RSV or persistent bronchiolitis. The infections are severe, prolonged, and complicated (31). Pneumocystis pneumonia initially cause diffuse interstitial infiltrates, which progress to alveolar infiltrates, which can be focal and asymmetric (26). Although pneumonia caused by P. jirovecii is a common presentation feature of SCID, it is rarely recognized. The diagnosis should be estimated through bronchoalveolar lavage or lung biopsy. The diagnosis is suspected due to infiltrates on chest X-ray and presence of respiratory distress, especially in combination with other symptoms of PID such as diarrhea, failure to thrive, or thrush. Pneumocystis infection alone in child with SCID does not lessen the chances of successful hematopoietic stem cell transplantation (HSCT), which is the unique causal therapeutic option for these patients (32). Respiratory viruses, particularly paramyxoviruses and adenoviruses are common significant pathogens in these patients with significant worsening effect of BMT outcome. Aggressive treatment (virostatics, immunoglobulins – intravenous, subcutaneous, or nebulized, corticosteroids) may reduce viral replication, lung damage and may improve respiratory functions and outcome. No treatment can probably results in viral clearance without successful T-cell engraftment (33).
Hyper-IgE syndrome (Job’s syndrome) is a complex combined immunodeficiency. Till today, at least three types of HIES were discriminated (autosomal dominant form – caused by mutation in signal transducer and activator of transcription, STAT3; and two autosomal recessive forms – caused by mutation in gene for tyrosine kinase 2, TYK2 or gene for dedicator of cytokinesis, DOCK8). In general, in HIES the lung symptoms are very common and early presentation of the disease. At the beginning, the recurrent sino-pulmonary infections are caused predominantly by S. aureus, and less frequently with S. pneumonia and H. influenzae. Recurrent pneumonias are typical clinical feature for all the three types of HIES. The healing after infections is usually aberrant and the result is the formation of bronchiectasis and pneumatocoeles, which are considered to be the pathogenic marker for autosomal dominant form of HIES (STAT3 mutation) whereas they were not reported in DOCK8 or TYK2 deficiencies. The pneumatocoeles can be occupied by Aspergillus or Scedosporium and are difficult to manage and treat. Pneumatocoeles are unusual in children infections and their appearance should alert to an unusual diagnosis are the very least (34, 35). Ones the parenchymal lung damage is present the spectrum of pulmonary pathogens shifts toward the spectrum seen in cystic fibrosis – Pseudomonas aeruginosa and non-tuberculous mycobacteria. The lung complication in HIES may be due to the impaired Th17 cell differentiation (36). Prior to pyogenic pneumonias, also HIES can be clinically presented by P. jirovecii pneumonia, however, in general this complication is rare. P. jirovecii can cause pneumonia in patients with HIES both with and without chronic lung diseases (37).
Patients with Hyper-IgM syndromes could have distinct clinical infectious complications based on the type of genetic background and exact type of the syndrome. While autosomal form of HIMS present as typical humoral PID, the X-linked form shows the spectrum of pathogens similar to the patients with combined immunodeficiencies. The underlying infectious cause of the pneumonias includes encapsulated bacteria, CMV, histoplasmosis, and P. jirovecii. Fungal pneumonias caused by Candida, Cryptococcus, and Histoplasma can be also found (38).
Pneumonia and chronic lung disease can be observed also in the patients with DNA-repair deficiencies (ataxia telangiectasia, Bloom syndrome, and Nijmegen breakage syndrome). The patients with ataxia telangiectasia are susceptible to recurrent viral and bacterial infections, but have also the increased risk of different malignancies, especially lymphoreticular. The increased sensitivity to ionizing radiation should be taken into account when indicating imaging in patients with DNA-repair defects (31).
Phagocytic immunodeficiencies
Phagocytic diseases are caused either by the decrease number and/or dysfunction of phagocytes. This group of PIDs is quite rare, however, in case of organ or deep skin abscesses, chronic dermatitis, persistent fungal infections, or delayed umbilical cord separation, one should think about them. The most important disease from this PIDs category is chronic granulomatous diseases (CGD), which is caused by the inability of the phagocytes to produce reactive oxygen species for intracellular killing of ingested microorganism. Another important group of phagocytic immunodeficiencies is presented by different form of congenital neutropenia (e.g., severe congenital neutropenia and cyclic neutropenia).
Patients with phagocytic diseases are at increased risk of recurrent respiratory infections. In CGD, the most important pathogens are S. aureus, Klebsiella, Aerobacter, Pseudomonas, Aspergillus, and Candida. The infections are difficult to treat, slow to resolve, and commonly recur (31). Also some rare and unusual pulmonary infectious complications have been reported in patients with CDG, e.g., pulmonary botryomycosis (39). In phagocytic diseases, the common pathogens are catalase-positive microorganisms (e.g., Pseudomonas, Burkholderia, and Serratia) or molds (especially Aspergillus) (15, 40). Invasive aspergillosis is the leading cause of mortality and morbidity in chronic granulomatous disease, which reflects the key role of the phagocytes NADPH oxidase in host defense against opportunistic fungi. Lung aspergillosis together with recurrent bacterial pneumonia was observed also in patients with severe congenital neutropenia. Their incidences have been significantly reduced as a result of the therapy with recombinant granulocyte colony-stimulating factor (41). In the early phase of disease, plain radiographs or CT scans may show non-specific, patchy, nodular opacities or segmental or lobar consolidation. Sometimes Aspergillus nodules may be very small and not visible on plain radiograph (42). They have a typical appearance on CT scans: halo of ground-glass attenuation representing pulmonary hemorrhage. Later on, the cavitation may develop within time with the “air crescent sign” in radiograph (26).
Complement immunodeficiencies
Complement immunodeficiencies are supposed to be the rarest type of PID; however, they are probably underreported and underdiagnosed. There is an increased risk for pyogenic infections with deficiencies of early components of classical pathway (C1–C4). Deficiencies of terminal complement components (C5–C9) are associated with increased susceptibility to Neisseria sp. Deficiency of C3 results in serious complications such as recurrent pneumonia (S. pneumoniae), meningitis, and peritonitis (43). Specific clinical presentation from the respiratory system is associated with hereditary angioedema, which is clinically manifested by recurrent angioedemas in different part of the body. Potentially life-threatening are the angioedemas located to larynx (15). The age of onset, frequency of attacks, and the factors triggering upper airway swelling are variable among different patients. To avoid a fatal outcome of laryngeal swelling, the therapy should be administered as soon as possible. The new available drugs for the treatment of acute attacks significantly improved the prognosis of these patients (44).
Non-Infectious Respiratory Complications of Primary Immunodeficiencies
Non-infections respiratory complications of PIDs can be the results of recurrent pulmonary infections or are the consequence of the PID itself. The recurrent pyogenic bacterial pulmonary infections lead to the air-trapping, bronchial wall thickening, atelectasis development, and bronchiectasis (9). Final development of chronic respiratory changes is the consequence of inter-play among different factors and mechanisms (Figure 3).

Figure 3. Pathophysiological substrates of the development of respiratory complications of primary immunodeficiencies.
Bronchiectasis
Bronchiectasis represents a common consequence of non-controlled chronic infections of lower airways. Among the most frequent causes of bronchiectasis in children, cystic fibrosis, primary ciliary dyskinesia, and immunodeficiencies should be listed (45). The commonest cause of bronchiectasis is cystic fibrosis, but neonatal screening reveals these children sooner before this clinical complication. Primary immunodeficiencies are among the most important causes for secondary bronchiectasis. Unfortunately, they are diagnosed late in the irreversible phase leading to the end-stage lung disease. On the other hand, their incidence in association with PID is much lower in children compared to adults. Bronchiectasis can be seen also in early-onset CVID in children as one of the main long-term complication in a comparable frequency than in adults (29). The regular screening for this important and mutilating complication is strongly recommended, especially in humoral and T- and B-cell combined immunodeficiencies. The bronchiectasis occurs earlier in X-linked agammaglobulinemia compared to CVID, probably due to the sooner onset of pulmonary infections in the first one. The bronchiectasis is usually in the lower or middle lobes. It seems that the majority of the hypogammaglobulinemic patients suffer from the mild type of bronchiectasis (46). Bronchiectasis and bronchial wall thickening occurs in 17–76% of antibody deficiency patients and are well recognized complication in these patients (17). Up to 73% of CVID patients develop chronic structural pulmonary complications, of which bronchiectasis and bronchial wall thickening are most frequently detected (8, 47). In milder form of humoral PIDs (e.g., IgA deficiency, IgG subclass deficiencies), the incidence of bronchiectasis is much lower than in XLA or CVID. Recent big study with CVID patients showed that bronchiectasis was not associated with any other complications (except for lobectomy). A possible explanation may be that bronchiectasis is the consequence of recurrent and un-controlled infections, whereas other non-infectious complications are the result of immune dysregulation (20).
Regarding the clinical characteristics of bronchiectasis, they are in general cylindrical, bilateral, and diffuse. It is most commonly found in the middle or lower lobes, and less frequently in the upper lobes (7). In the majority of the cases, tubular (cylindrical) bronchiectasis was observed, whereas varicose and cystic type is less common (48). Pathological bronchial findings are mostly observed in the proximal bronchi; meanwhile the involvement of distal bronchi is less common. Chest high-resolution computed tomography (HRCT) should be considered in all patients with chronic chest symptoms to monitor the disease progression, as the chest X-ray remains an insensitive tool for the diagnosis of early pathology (17). Characteristic findings of bronchiectasis on HRCT include bronchial dilatation, bronchial wall thickening, lack of tapering, and bronchi visible closer than 2 cm to the pleural surface (49). Today, regularly HRCT can be recommended as a screening tool for the lung complication of CVID and other antibody deficiencies.
The presence of bronchiectasis at diagnosis predicts poor prognosis, while early diagnosis and aggressive management predict good outcome (22). The diagnostic delay in patients with bronchiectasis is significantly higher than those without the developed bronchiectasis. Some authors suggested that the bronchiectasis secondary to PID in childhood is not always a progressive condition and there is a potential to slow or prevent disease progression with appropriate treatment (49). A significant correlation was found between severe pneumonia/sepsis and the development of bronchiectasis (50). The severity of developed bronchiectasis significantly correlates with clinical symptoms and impaired life quality (51). The majority of CVID patients with bronchiectasis have mild to severe vitamin D deficiency, which could be therefore recommended to be assessed in these patients with subsequent substitution. Vitamin D deficiency could aggravate the risk and length of respiratory infections in patients with CVID (52). The aggressive antibiotic treatment, physiotherapy, and substitution therapy with immunoglobulins in appropriate dose are the most important preventive strategies. The appropriate treatment (immunoglobulins and antibiotics) may delay the development and also alter the natural course of bronchiectasis. The earlier the immunoglobulin substitution is started, the lower probability of bronchiectasis development and need for invasive surgical interventions can be seen at least in some patients (29, 53). Recent study has showed that bronchiectasis secondary to PID in childhood is not always a progressive condition and appropriate treatment can slow or even prevent disease progression (49). However, several authors reported development of bronchiectasis despite IVIG therapy, probably due to persistent local inflammation and mucous obstruction (54, 55). Anyway, efficient immunoglobulin substitution supported by antibiotics when required seems to promote normal growth and to inhibit the development of disabling lung disease in PID patients (56). In general, there are several options and approaches how to treat both infectious and non-infectious lung complications of PIDs (Table 7).

Table 7. Therapeutic possibilities for the respiratory complications of primary immunodeficiencies.
Interstitial lung diseases and PIDs
Interstitial lung diseases represent one of the most important complications of PIDs and belong to their late-onset symptoms. Their occurrence in childhood is quite rare. The chronic non-infectious complications are the best characterized in patients with CVID, where the lungs are affected with the specific CVID-associated entity called granulomatous-lymphocytic interstitial lung disease (GLILD), which currently constitutes an important cause of morbidity and mortality. The lung pathology in sense of ILD includes lymphocytic interstitial pneumonia, follicular bronchiolitis, granulomatous lung disease, and organizing pneumonia. Follicular bronchiolitis, nodular lymphoid hyperplasia, reactive lymphoid infiltrates, and lymphocytic interstitial pneumonia are all forms of pulmonary lymphoid hyperplasia, which is included within the umbrella term GLILD. GLILD presents the pathologic combination of granulomas and lymphoid hyperplasia (57). GLILD appears to be distinct from the bronchiectasis secondary to recurrent infections and possesses some similarities (but also striking differences) with sarcoidosis (58).
The structural airway disease and ILD in CVID display dissimilar clinical and immunological characteristics, which may influence the diagnosis and follow-up of lung pathology in these patients in the future. The pathogenesis of these two entities differs. While airway structural disease is mainly the cumulative effect of recurrent infections followed by subsequent cicatrization of lung tissue, ILD usually results from immune dysregulation (59). However, some forms of “post-infectious ILD” have been also described (60). Some patients with CVID may display the coincidence of both entities and the prevalence of combined lung disease is higher in adults (59). In a subgroup of CVID patients, the development of granulomatous disease can be observed that may cause ILD in approximately 10% of the cases (so-called sarcoid-like disease). The incidence of pulmonary nodules in a population of CVID patients is high (approximately 20–40%), correlates with splenomegaly and autoimmune phenomena (61, 62). The presence of GLILD is associated with a worse prognosis and increased prevalence of lymphoproliferative disorders (63, 64). Lymphoid interstitial pneumonitis may also be isolated (26). Although the lungs are the most common organ system affected by granulomatous disease in CVID, granulomas can be found also in other organs including skin, liver, spleen, and gastrointestinal tract. The type and severity of lung lesions do not correlate with the type of immunodeficiency or with the severity of the sinusal involvement (65). Whereas the recurrent bacterial pneumonia and bronchial suppuration are the most frequent complications of CVID, reactive interstitial pneumonitis and pulmonary lymphoma are less frequent among these patients (19). ILD appears to be asymptomatic at the initial stage, and therefore, the screening of all (including asymptomatic) CVID patients for lung pathologies should be strongly recommended to facilitate early detection and prevent progression of this disease (59). The etiology of granulomatous diseases is still unknown (66). Many different possible factors inducing the development of GLILD have been evaluated, but one of the most relevant is probably the infection with human herpes virus type 8 (64). Other viruses such as EBV or CMV could be also involved (67). The silent radiological features of GLILD include diffuse interstitial infiltrates on plain radiograph, consolidation, ground-glass opacities, and reticular abnormalities on HRCT (17, 68). GLILD presents with CT findings distinct from the usual airway abnormalities most commonly associated with CVID – pulmonary micronodules, thoracic lymphadenopathy, interlobular septal thickening, and multifocal pulmonary consolidation (69). Common physical, radiographic, and laboratory abnormalities in patients with CVID and granulomatous disease include splenomegaly, hilar, and mediastinal lymphadenopathy with ground glass or nodular opacities in lung parenchyma and reduced number and functions of T-cells.
Patients with other antibody deficiencies may also have radiologic and functional evidence of ILD, but their frequency and type are incompletely appreciated. Several histological patterns have been reported including lymphocytic interstitial pneumonitis (LIP), granulomatous interstitial pneumonitis, bronchiolitis obliterans organizing pneumonia (BOOP), and usual interstitial pneumonia (UIP) (60). In patients with antibody deficiencies, especially with repeated respiratory infections ILD is frequent and much more prevalent than expected from general population. The most common immunological abnormality associated with ILD is the deficiency of IgG subclasses. There is no particular histologic or radiologic feature consistently related to a particular immunodeficiency (60). As most of patients with IgA deficiency can produce IgG antibodies, they are less prone to bacterial infections so bronchiectasis is not as common as in XLA or CVID (43). Anyway, there are several reports of chronic lung disease also in selective IgA deficiency, especially when associated with IgG2 deficiency (70).
Pneumatocoeles are the typical clinical radiological finding in the patients with HIES. They are typical warning characteristic sign for autosomal dominant HIES and they can present the locus for other possible infections, e.g., fungal.
Tumors of respiratory tract in primary immunodeficiencies
Malignancy is after infections the second leading cause of death in PIDs. The majority of the tumors is associated with the Epstein–Barr virus (30–60% of cases). In general, the risk of developing malignancies varies from 1 to 25%, but the children with CVID and Wiskott–Aldrich syndrome are at greater risk (31). We can discriminate between benign lymphoproliferative diseases (parenchymal lymphoid hyperplasia, reactive follicular hyperplasia) and malignancies. The enlargement of mediastinal lymph node can occasionally lead to superior vena caval syndrome and CVID was recommended to be included in differential diagnosis of ILDs and hilar lymphadenopathy (71).
Regarding the solid lung tumors, there are only few case reports in the literature (leiomyoma, pulmonary adenocarcinoma) and this respiratory complication is considered to be in general rare in PID patients (72, 73). Pulmonary comprise due to metastasis (origin from, e.g., gastric carcinoma) is more often seen than primary pulmonary malignancy (7). Thymoma may occur in combined immunodeficiency and this rare entity is called Good syndrome. Good syndrome occurs in 1–6% of patients with primary humoral immunodeficiencies (74). However, pulmonary lymphoma seems to be a serious complication associated with CVID or other PIDs (19, 75). Patients with CVID frequently develop lymphoproliferative disease and the risk for malignant lymphoma is increased by more than 300-fold. Non-Hodgkin’s lymphoma (especially high-grade B-cell lymphoma) is more common than Hodgkin’s lymphoma. Approximately 8% patients with CVID develop non-Hodgkin’s lymphoma and in general, <1% of patients with humoral PID develop Hodgkin’s lymphoma (76). There is also a possibility of benign lymphoproliferative disease in PID, e.g., parenchymal lymphoid hyperplasia of the lungs (77). Some benign conditions (e.g., lymphocytic interstitial pneumonia) can transform into malignant lymphoma (78).
Other respiratory complications of PIDs
In patients with humoral immunodeficiencies, the respiratory allergic or allergy-like symptoms such as dyspnea, rhinitis, or asthma can be frequently seen (29). Many patients despite the appropriate treatment present with chronic productive cough, which is usually the hallmark of chronic sinusitis or bronchitis (79). Despite some similarities in the clinical picture of primary ciliary dyskinesia and CVID, these two entities require different therapeutic approach. There is a case report in the literature with both diseases in one patient. Therefore, in a patient with already established diagnosis of chronic lung disease with the deterioration of clinical course, the search for secondary diagnosis should be recommended (80, 81).
Several cases of primary pulmonary hypertension were also described in association with primary immunodeficiency (82).
Primary pulmonary dysgenesis or even agenesis was discussed to be considered as a part of the spectrum anomalies associated with velocardiofacial syndrome (83).
Recently, a new immunodeficiency syndrome called pulmonary alveolar proteinosis has been described. It is characterized by accumulation of pulmonary surfactant, respiratory insufficiency and increased infections due to associated immunodeficiency. This syndrome belongs to the category of phagocytic diseases and is a result of the disturbed surfactant catabolism in alveolar macrophages. It can be either acquired (due to autoantibodies against GM-CSF) or congenital caused by CSF2RA mutations. CSF2RA encodes the GM-CSF receptor α protein. These patients have increased susceptibility to opportunistic microbial pathogens and increased mortality from un-controlled infections (84). Another new syndrome in two 46XX sisters with fatal lung fibrosis, profound combined immunodeficiency and gonadal dysgenesis was also recently described. Comparative genome hybridization and analysis of genes known to be associated with severe immune defects in infancy or gonadal dysgenesis showed no abnormality (85).
The delay in the diagnosis of PID results in the poorer prognosis and considerable morbidity, particularly recurrent pneumonias, resulting in structural lung damage such as bronchiectasis, pulmonary hypertension, and ultimately cor pulmonale (17). The end-stage lung disease with the development of cor pulmonale and respiratory insufficiency has been documented as the most common cause of morbidity in primary humoral immunodeficiencies (25). However, non-infectious lung diseases may occur despite optimal immunoglobulin therapy (28). Therefore, patients developing chronic respiratory symptoms should be managed in a multidisciplinary team, including chest physician, as progression of lung disease may occur despite apparently optimal immunoglobulin therapy (17).
Diagnosis of Respiratory Complications of PIDs
Due to the chronic involvement of the respiratory tract (bacterial pneumonia, abscesses, fungal lung disease, and interstitial pneumonia) in PIDs, the changes of lung functions could be logically expected. Although different patterns are typically associated with distinct type of immune defects, there is a substantial overlap in imaging findings (26). Screening examinations, such as lung function testing (LFT) and HRCT of the chest, should be used to evaluate pulmonary status in PID patients (86).
Lung function testing in PIDs
Several studies evaluated the changes in pulmonary functions. Normal lung functions can be seen in 45–74% patients. In general, the ventilatory disturbances evaluated by lung function tests can be observed in the majority of the patients with CVID or XLA (87). There are some inter-disease differences, e.g., patients with CVID have more commonly abnormal lung tests compared to the patients with X-linked agammaglobulinemia (88). An obstructive pattern is reported most frequently, although restrictive pulmonary disease is well recognized (17). Small airways involvement leads to ventilation abnormalities and chronic obstructive disease, which are usually irreversible (7). A reduced rate of carbon monoxide uptake and restrictive ventilatory pattern is typically found in CVID patients with ILD (63). A significant correlation between the HRCT score and the predicted values in pulmonary function test has been confirmed in several studies (46), while the others did not find such associations (49, 87). There is a negative correlation between a number of episodes of pneumonia and lung function tests. Recent study has shown that the longitudinal decline of lung functions can be observed in patients with PID. Chronic persistent cough can be used as a marker of the reduction of lung functions (87). One study reported an improvement of lung function tests despite bronchiectasis progression on HRCT in patients after the initiation of immunoglobulin treatment and authors concluded that LFT may serve as a toll for evaluation of the clinical response to immunoglobulin therapy (89). The changes in lung function could be used as an indicator of immunoglobulin treatment efficacy. Based on the significant association between the dose of IVIG and spirometric indices, the use of higher IVIG doses as a protection of lung function decline was recommended (90). Lung functional tests can be used to monitor patients at a more regular basis, although featured by decreased sensitivity for complications (8).
Imaging techniques in PIDs
There is an essential role of different imaging techniques, especially CT in the identification and monitoring of pulmonary changes and complications in CVID or some other specific immunodeficiencies. One of the most important diagnostic tools for the detection, quantifying, and characterizing the extent and kind of lung damage is CT. HRCT is the reference standard for the detection of bronchiectasis. The early sign of the possible development of bronchiectasis is the bronchial wall thickening. HRCT appears to be more sensitive for the detection of pneumopathies (with special emphasis on bronchial changes and interstitial lesions) in PID patients than chest X-ray (55, 91). Both HRCT and helical CT proved to be useful tools for monitoring of lung changes associated with PID, especially in symptomatic patients with negative radiographic findings (92).
The patients with severe PID should be regularly examined for the possible respiratory symptoms and complications. In general, LFT can be recommended every 6–12 months with repeated chest X-ray. CT examination should be repeated within 5–10 years after initial presentation of PID unless there is a specific indication or changes in clinical status. If significant abnormalities are found, a repeated CT should be performed every 3–5 years depending on the type of abnormality or clinical stability. If patients are prescribed potentially toxic therapies, e.g., high-dose steroids for GLILD, CT examination should be performed approximately 6 months after the initiation of therapy. Subsequent CT examinations depend on clinical, X-ray, and lung function test changes, but in general should be repeated annually for 1–2 years followed by a decreased frequency is stability is achieved (9, 79).
A special approach should be given to the patients with increased radiation sensitivity (DNA-repair defects, also the part of CVID patients). In these cases, an alternative radiation-free technique alternative to CT scan or chest X-ray could be MRI evaluation of the pulmonary changes and alterations. In recent study, CT and MRI findings were comparable for moderate to severe degrees of bronchial and parenchymal alterations, but weaker concordance between CT and MRI scan was found for lower scores of bronchial abnormalities. Therefore, it should be admitted that CT allows better identification of peripheral airways changes. However, further studies are needed to image quality and involvement of this method into the diagnostic algorithm in PID (93). Actually, low-dose HRCT is still considered to be the standard and the most sensitive method for identification of structural abnormalities and pulmonary complications in CVID at the time of diagnosis and at regular time-points during follow-up, however, with the proper follow-up interval yet to be determined. Annual testing (both spirometry and transfer factor) is useful in the assessment of PID patients, and should not be confined to those with radiological evidence of lung disease (27, 94).
In a study with 58 pediatric patients with PID or cancer (12 with PID) suffering from different respiratory symptoms, the bronchoscopy and bronchoalveolar lavage was shown to be clinically useful and established an overall diagnostic rate 94% of patients. Infection rate was 74.2% and infectious agent was isolated in 53% of the cases. In PID patients, different agents were isolated (Pseudomonas, Enterobacter, E. coli, Acinetobacter, Proteus, and Aspergillus). P. jirovecii was identified only in patients without prophylactic therapy with trimethoprim–sulfamethoxazole. Authors stated that these two methods should be considered as an initial diagnostic tool in pediatric PID patients (95).
Conclusion
Respiratory system is the most common site of the clinical manifestations of different PID both in adults and in children. Infectious and non-infectious respiratory complications determinate the patients’ prognosis. To reduce the morbidity associated with PID the greater awareness of respiratory complications for such patients should be raised. Regular examinations by the appropriate tests should reveal the respiratory pathologies in early stages and should be used also for the monitoring of already existing abnormalities. In general, due to the raising awareness of PIDs the prognosis of these patients gradually improves.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported by the projects: Measurement of kinetics of cilia in respiratory tract (ITMS 262202200019) and VEGA 1/0252/14.
References
1. Madkaikar M, Mishra A, Ghosh K. Diagnostic approach to primary immunodeficiency disorders. Indian Pediatr (2013) 50:579–86. doi: 10.1007/s13312-013-0171-4
3. Puck JM. The case for newborn screening for severe combined immunodeficiency and related disorders. Ann N Y Acad Sci (2011) 1246:108–17. doi:10.1111/j.1749-6632.2011.06346.x
4. Lindegren ML, Kobrynski L, Rasmussen SA, Moore CA, Grosse SD, Vanderford ML, et al. Applying public health strategies to primary immunodeficiency disorders. MMWR Recomm Rep (2004) 53(RR01):1–28.
5. Stiehm RE. The four most common immunodeficiencies. Adv Exp Med Biol (2007) 601:15–26. doi:10.1007/978-0-387-72005-0_2
6. Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol (2014) 5:162. doi:10.3389/fimmu.2014.00162
7. Bierry G, Boileau J, Barnig C, Gasser B, Korganow AS, Buy X, et al. Thoracis manifestations of primary humoral immunodeficiency: a comprehensive review. Radiographics (2009) 29:1909–20. doi:10.1148/rg.297095717
8. Touw CM, van de Ven AA, de Jong PA, Terheggen-Lagro S, Beek E, Sanders EA, et al. Detection of pulmonary complications in common variable immunodeficiency. Pediatr Allergy Immunol (2010) 21:793–805. doi:10.1111/j.1399-3038.2009.00963.x
9. Hampson FA, Chandra A, Screaton NJ, Condliffe A, Kumararatne DS, Exley AR, et al. Respiratory disease in common variable immunodeficiency and other primary immunodeficiency disorders. Clin Radiol (2012) 67:587–95. doi:10.1016/j.crad.2011.10.028
10. O’Sullivan MD, Cant AJ. The 10 warning signs: a time for a change? Curr Opin Allergy Clin Immunol (2012) 12:588–94. doi:10.1097/ACI.0b013e3283591534
11. Arkwright PD, Gennery AR. Ten warning signs of primary immunodeficiency: a new paradigm is needed for the 21st century. Ann N Y Acad Sci (2011) 1238:7–14. doi:10.1111/j.1749-6632.2011.06206.x
12. Reda SM, Afifi HM, Amine MM. Primary immunodeficiency diseases in Egyptian children: a single-center study. J Clin Immunol (2009) 29:343–51. doi:10.1007/s10875-008-9260-x
13. Jesenak M, Ciljakova M, Rennerova Z, Babusikova E, Banovcin P. Recurrent respiratory infections in children – definition, diagnostic approach, treatment and prevention. In: Martin-Loeches I, editor. Bronchitis. Rijeka: InTech (2011). p. 119–48.
14. Jesenak M, Majtan J, Rennerova Z, Kyselovic J, Banovcin P, Hrubisko M. Immunomodulatory effect of pleuran (β-glucan from Pleurotus ostreatus) in children with recurrent respiratory tract infections. Int Immunopharmacol (2013) 15:395–9. doi:10.1016/j.intimp.2012.11.020
15. Jesenak M, Banovcin P, Freiberger T. Hereditary angioedema in paediatric and adolescent age – in centre experience. Lek Obz (2013) 62:245–9.
16. Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (2006) 85:193–202. doi:10.1097/01.md.0000229482.27398.ad
17. Wood P, Stanworth S, Burton J, Jones A, Peckham DG, Green T, et al. Recognition, clinical diagnosis and management of patients with primary antibody deficiencies: a systemic review. Clin Exp Immunol (2007) 149:410–23. doi:10.1111/j.1365-2249.2007.03432.x
18. Wahn V. When is susceptibility to infections abnormal? Pediatr Allergy Immunol (2011) 22:650–1. doi:10.1111/j.1399-3038.2011.01217.x
19. Cadranel J, Bouvry D, Wislez M. Manifestations respiratoires au cours du deficit immunitaire commun variable de l’adulte. Rev Mal Respir (2003) 20: 126–33.
20. Gathmann B, Mahlaoui N, Gerard L, Oksenhendler E, Warnatz K, Schulze I, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol (2014) 134:116–26. doi:10.1016/j.jaci.2013.12.1077
21. Mendieta-Flores E, Del-Rivero-Hernandez LG, Zavala-Perez M, Segura-Mendez NH. Rhinosinusitis and its impact on the quality of life in patients with common variable immunodeficiency. Rev Alerg Mex (2012) 59:60–4.
22. Tarzi MD, Grigoriadou S, Carr SB, Kuitert LM, Longhurst HJ. Clinical immunology review series: an approach to the management of pulmonary disease in primary antibody deficiency. Clin Exp Immunol (2008) 155:147–55. doi:10.1111/j.1365-2249.2008.03851.x
23. Furr PM, Taylor-Robinson D, Webster AD. Mycoplasmas and ureaplasmas in patients with hypogammaglobulinaemia and their role in arthritis: microbiological observations over twenty years. Ann Rheum Dis (1994) 53:183–7. doi:10.1136/ard.53.3.183
24. Havlicekova Z, Jesenak M, Freiberger T, Banovcin P. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub (2013). doi:10.5507/bp.2013.011
25. Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol (1999) 92:34–48. doi:10.1006/clim.1999.4725
26. Notarangelo LD, Plebani A, Mazzolari E, Soresina A, Bondioni MP. Genetic causes of bronchiectasis: primary immune deficiencies and the lung. Respiration (2007) 74:264–75. doi:10.1159/000101784
27. Aghamohammadi A, Allahverdi A, Abolhassani H, Moazzami K, Alizadeh H, Gharagozlou M, et al. Comparison of pulmonary diseases in common variable immunodeficiency and X-linked agammaglobulinaemia. Respirology (2010) 15:289–95. doi:10.1111/j.1440-1843.2009.01679.x
28. Quartier P, Debre M, De Blic J, De Sauverzac R, Sayegh N, Jabado N, et al. Early and prolonged intravenous immunoglobulin replacement therapy in childhood agammaglobulinemia: a retrospective survey of 31 patients. J Pediatr (1999) 134:589–96. doi:10.1016/S0022-3476(99)70246-5
29. Urschel S, Kayikci L, Wintergerst U, Notheis G, Jansson A, Belohradsky BH. Common variable immunodeficiency in children: delayed diagnosis despite typical clinical presentation. J Pediatr (2009) 154:888–94. doi:10.1016/j.jpeds.2008.12.020
30. Stiehm RE. Adverse effects of human immunoglobulin therapy. Transfus Med Rev (2013) 27:171–8. doi:10.1016/j.tmrv.2013.05.004
31. Jeanes AC, Owens CM. Chest imaging in the immunocompromised child. Paediatr Respir Rev (2002) 3:59–69. doi:10.1053/prrv.2002.0186
32. Berrington JE, Flood TJ, Abinun M, Galloway A, Cant AJ. Unsuspected Pneumocystis carinii pneumonia at presentation of severe primary immunodeficiency. Arch Dis Child (2000) 82:144–7. doi:10.1136/adc.82.2.144
33. Crooks BN, Taylor CE, Turner AJ, Osman HK, Abinun M, Flood TJ, et al. Respiratory viral infections in primary immune deficiencies: significance and relevance to clinical outcome in a single BMT unit. Bone Marrow Transplant (2000) 26:1097–102. doi:10.1038/sj.bmt.1702656
34. Cant A, Battersby A. When to think of immunodeficiency? In: Curtis N, editor. Hot Topics in Infection and Immunity in Children. New York, NY: Springer Science+Business Media (2013). p. 167–77.
35. Almyroudis NG, Holland SM, Segal BH. Invasive aspergillosis in primary immunodeficiencies. Med Mycol (2005) 43(Suppl 1):S247–59. doi:10.1080/13693780400025203
36. Sowerwine KJ, Holland SM, Freeman AF. Hyper-IgE syndrome update. Ann N Y Acad Sci (2012) 1250:25–32. doi:10.1111/j.1749-6632.2011.06387.x
37. Freeman AF, Davis J, Anderson VJ, Barson W, Darnell DN, Puck JM, et al. Pneumocystis jiroveci infection in patients with hyper-immunoglobulin E syndrome. Pediatrics (2006) 118:e1271–5. doi:10.1542/peds.2006-0311
38. Dosanjh A. Chronic pediatric pulmonary disease and primary humoral antibody based immune disease. Respir Med (2011) 105:511–4. doi:10.1016/j.rmed.2010.11.013
39. Paz HL, Little BJ, Ball WC, Winkelstein JA. Primary pulmonary botryomycosis – a manifestation of chronic granulomatous disease. Chest (1992) 101:1160–2. doi:10.1378/chest.101.4.1160
40. Jesenak M, Havlicekova Z, Banovcin P, Stasia MJ. Chronic granulomatous disease caused by a novel mutation in a 2-month-old boy with multifocal splenic abscesses. J Investig Allergol Clin Immunol (2013) 23:137–8.
41. Dale DC, Cottle TE, Fier CJ, Bolyard AA, Bonilla MA, Boxer LA, et al. Severe chronic neutropenia: treatment and follow-up of patients in the severe chronic neutropenia international registry. Am J Hematol (2003) 72:82–93. doi:10.1002/ajh.10255
42. Copley SJ. Application of computed tomography in childhood respiratory infections. Br Med Bull (2002) 61:263–79. doi:10.1093/bmb/61.1.263
43. Buckley RH. Pulmonary complications of primary immunodeficiencies. Paediatr Respir Rev (2004) 5(Suppl A):225–33. doi:10.1016/S1526-0542(04)90043-7
44. Papadopoulou-Alataki E. Upper airway considerations in hereditary angioedema. Curr Opin Allergy Clin Immunol (2010) 10:20–5. doi:10.1097/ACI.0b013e328334f629
45. Bouyahia O, Essadem L, Matoussi N, Gharsallah L, Fitouri Z, Mrad Mazigh S, et al. Etiology and outcome of bronchiectasis in children: a study of 41 patients. Tunis Med (2008) 86:996–9.
46. Gharagozlou M, Ebrahimi FA, Farhoudi A, Aghamohammadi A, Bemanian MH, Chavoshzadeh Z, et al. Pulmonary complications in primary hypogammaglobulinemia: a survey by high resolution CT scan. Monaldi Arch Chest Dis (2006) 65:69–74.
47. Thickett MK, Kumararatne DS, Banerjee AK, Dudley R, Stableforth DE. Common variable immune deficiency: respiratory manifestations, pulmonary function and high-resolution CT scan findings. Q J Med (2002) 95:655–62. doi:10.1093/qjmed/95.10.655
48. Obregon RG, Lynch DA, Kaske T, Newell JD, Kirkpatrick CH. Radiologic findings of adult primary immunodeficiency disorders. Contribution of CT. Chest (1994) 106:490–5. doi:10.1378/chest.106.2.490
49. Haidopoulou K, Calder A, Jones A, Jaffe A, Sonnappa S. Bronchiectasis secondary to primary immunodeficiency in children: longitudinal changes in structure and function. Pediatr Pulmonol (2009) 44:669–75. doi:10.1002/ppul.21036
50. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood (2008) 112:277–86. doi:10.1182/blood-2007-11-124545
51. Galindo-Pacheco LV, Amaya-Mejia AS, O’Farrill-Romanillos PM, Del Rivero-Hernandez LG, Segura-Mendez NH. Quality of life in adults with variable common immunodeficiency and bronchiectasis. Rev Alerg Mex (2013) 60: 123–8.
52. Amaya-Mejia AS, O’Farrill-Romanillos PM, Galindo-Pacheco LV, Vargas-Ortega G, Mendoza-Zubieta V, Del Rivero-Hernandez LG, et al. Vitamin D deficiency in patients with common variable immunodeficiency, with autoimmune diseases and bronchiectasis. Rev Alerg Mex (2013) 60:110–6.
53. Cantani A, Ferrara M. The lung and primary immunodeficiency. Allergol Immunopathol (Madr) (1988) 16:429–37.
54. Quinti I, Soresina A, Spadaro G, Martino S, Donnano S, Agostani C, et al. Long-term follow-up and outcome of a large cohort of patient with common variable immunodeficiency. J Clin Immunol (2007) 27:308–16. doi:10.1007/s10875-007-9075-1
55. Mogica Martinez MD, Garcia Lara S, Silva Very R, Montano Velazquez B, Cruz Merida A, Santibanez Bustamante J, et al. Neumopathies in patients with primary immunodeficiencies in treatment with intravenous gammaglobulin. Rev Alerg Mex (2007) 54:14–9.
56. Bjorkander J, Bake B, Hanson LA. Primary hypogammaglobulinaemia: impaired lung function and body growth with delayed diagnosis and inadequate treatment. Eur J Respir Dis (1984) 65:529–36.
57. Maglione PJ, Ko HM, Beasley MB, Strauchen JA, Cunningham-Rundles C. Tertiary lymphoid neogenesis is a component of pulmonary hyperplasia in patients with common variable immunodeficiency. J Allergy Clin Immunol (2014) 133:535–42. doi:10.1016/j.jaci.2013.08.022
58. Bianchi MP, Letovanec I, Spertini F, Nicod LP, Lazor R. Granulomatous lymphocytic interstitial lung disease in common variable immunodeficiency. Rev Med Suisse (2013) 9:2175–80.
59. van de Ven AA, de Jong PA, Hoytema van Konijnenburg DP, Kessels OA, Boes M, Sanders EA, et al. Airway and interstitial lung disease are distinct entities in paediatric common variable immunodeficiency. Clin Exp Immunol (2011) 165:235–42. doi:10.1111/j.1365-2249.2011.04425.x
60. Popa V, Colby TV, Reich SB. Pulmonary interstitial disease in Ig deficiency. Chest (2002) 122:1594–603. doi:10.1378/chest.122.5.1594
61. Bondioni MP, Soresina A, Lougaris V, Gatta D, Plebani A, Maroldi R. Common variable immunodeficiency: computed tomography evaluation of bronchopulmonary changes including nodular lesions in 40 patients. Correlation with clinical and immunological data. J Comput Assist Tomogr (2010) 34:395–401. doi:10.1097/RCT.0b013e3181cad9da
62. Park JE, Beal I, Dilworth JP, Tormey V, Haddock J. The HRCT appearances of granulomatous pulmonary disease in common variable immune deficiency. Eur J Radiol (2005) 54:359–64. doi:10.1016/j.ejrad.2004.09.005
63. Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol (2004) 114:415–21. doi:10.1016/j.jaci.2004.05.057
64. Wheat WH, Cool CD, Morimoto Y, Rai PR, Kirkpatrick CH, Lindenbaum BA, et al. Possible role of human herpesvirus 8 in the lymphoproliferative disorders in common variable immunodeficiency. J Exp Med (2005) 202:479–84. doi:10.1084/jem.20050381
65. Bondioni MP, Duse M, Plebani A, Soresina A, Notarangelo LD, Berlucchi M, et al. Pulmonary and sinusal changes in 45 patients with primary immunodeficiencies: computed tomography evaluation. J Comput Assist Tomogr (2007) 31:620–8. doi:10.1097/RCT.0b013e31802e3c11
66. Morimoto Y, Routes JM. Granulomatous disease in common variable immunodeficiency. Curr Allergy Asthma Rep (2005) 5:370–5. doi:10.1007/s11882-005-0008-x
67. Stewart JP, Egan JJ, Ross AJ, Kelly BG, Lok SS, Hasleton PS, et al. The detection of Epstein-Barr virus DNA in lung tissue from patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (1999) 159:1336–41. doi:10.1164/ajrccm.159.4.9807077
68. Park JH, Levinson AI. Granulomatous-lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Immunol (2010) 134:97–103. doi:10.1016/j.clim.2009.10.002
69. Torigian DA, LaRosa DF, Levinson AI, Litzky LA, Miller WT Jr. Granulomatous-lymphocytic interstitial lung disease associated with common variable immunodeficiency: CT findings. J Thorac Imaging (2008) 23:162–9. doi:10.1097/RTI.0b013e318166d32f
70. Bjorkander J, Bake B, Oxelius VA, Hanson LA. Impaired lung function in patients with IgA deficiency and low levels of IgG2 or IgG3. N Engl J Med (1985) 313:720–4. doi:10.1056/NEJM198509193131203
71. Sacco O, Fragonese B, Picco P, Faraci M, Facchetti P, Pistoia V, et al. Common variable immunodeficiency presenting in a girl as lung infiltrates and mediastinal adenopathies leading to severe “superior vena caval” syndrome. Eur Respir J (1996) 9:1958–61. doi:10.1183/09031936.96.09091958
72. User IR, Dogru D, Talim B, Orhan D, Karnak I. Endobronchial, pulmonary and liver leiomyomata in a child with primary immune deficiency. Eur J Pediatr Surg (2010) 20:423–5. doi:10.1055/s-0030-1254144
73. Oztop I, Demirkan B, Tarhan O, Kayahan H, Yilmaz U, Kargi A, et al. The development of pulmonary adenocarcinoma in a patient with Job’s syndrome, a rare immunodeficiency condition. Tumori (2004) 90:132–5.
74. Agarwal S, Cunningham-Rundles C. Thymoma and immunodeficiency (Good syndrome): a report of 2 unusual cases and review of the literature. Ann Allergy Asthma Immunol (2007) 98:185–90. doi:10.1016/S1081-1206(10)60695-3
75. Bal A, Gupta A, Sodhi KS, Das A, Singh S. Multifocal extranodal non-Hodgkin lymphoma involving both the lungs and brain in a child with primary immunodeficiency. J Pediatr Hematol Oncol (2008) 30(2008):317–9. doi:10.1097/MPH.0b013e318161aa3b
76. Cunningham-Rundles C, Lieberman P, Hellman G, Chaganti RS. Non-Hodgkin lymphoma in common variable immunodeficiency. Am J Hematol (1991) 37:69–74. doi:10.1002/ajh.2830370202
77. Kajiwara S, Sakai S, Soeda H, Takahashi N, Okafuji T, Yoshimitsu K, et al. Multifocal nodular lymphoid hyperplasia of the lung. J Thorac Imaging (2005) 20:239–41. doi:10.1097/01.rti.0000158404.40711.26
78. Fishback N, Koss M. Update on lymphoid interstitial pneumonitis. Curr Opin Pulm Med (1996) 2:429–33. doi:10.1097/00063198-199609000-00014
79. Rusconi F, Panisi C, Dellepiane RM, Cardinale F, Chini L, Martire B, et al. Pulmonary and sinus diseases in primary humoral immunodeficiencies with chronic productive cough. Arch Dis Child (2003) 88:1101–5. doi:10.1136/adc.88.12.1101
80. Skorpinski EW, Kung SJ, Yousef E, McGeady SJ. Diagnosis of common variable immunodeficiency in a patients with primary ciliary dyskinesia. Pediatrics (2007) 119:e1203–5. doi:10.1542/peds.2006-2396
81. Modrzewska K, Wiatr E, Langfort R, Oniszh K, Roszkowski-Sliz K. Common variable immunodeficiency in a patient with suspected sarcoidosis. Pneumonol Alergol Pol (2009) 77:91–6.
82. Bolkova NV. Primary immunodeficiency state in a child with the pulmonary hypertension syndrome. Arkh Patol (1985) 47:63–6.
83. Cunningham ML, Perry RJ, Eby PR, Gibson RL, Opheim KE, Manning SC. Primary pulmonary dysgenesis in velocardiofacial syndrome: a second patient. Am J Med Genet A (2003) 121A:177–9. doi:10.1002/ajmg.a.20142
84. Trapnell BC, Carey BC, Uchida K, Suzuki T. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr Opin Immunol (2009) 21:514–21. doi:10.1016/j.coi.2009.09.004
85. Somech R, Somers GR, Chitayat D, Grunebaum E, Atkinson A, Kolomietz E, et al. Fatal lung fibrosis associated with immunodeficiency and gonadal dysgenesis in 46XX sisters – a new syndrome. Am J Med Genet A (2008) 146A:8–14. doi:10.1002/ajmg.a.32014
86. Busse PJ, Farzan S, Cunningham-Rundles C. Pulmonary complications of common variable immunodeficiency. Ann Allergy Asthma Immunol (2007) 98:1–8. doi:10.1016/S1081-1206(10)60853-8
87. Costa-Carvalho BR, Wandalsen GF, Pulici G, Aranda CS, Sole D. Pulmonary complications in patients with antibody deficiency. Allergol Immunopathol (Madr) (2011) 39:128–32. doi:10.1016/j.aller.2010.12.003
88. Sweinberg SK, Wodell RA, Grodofsky MP, Greene JM, Conley ME. Retrospective analysis of the incidence of pulmonary disease in hypogammaglobulinemia. J Allergy Clin Immunol (1991) 88:96–104. doi:10.1016/0091-6749(91)90306-9
89. de Gracia J, Vendrell M, Alvarez A, Pallisa E, Rodrigo MJ, de la Rosa D, et al. Immunoglobulin therapy to control lung damage in patients with common variable immunodeficiency. Int Immunopharmacol (2004) 4:745–53. doi:10.1016/j.intimp.2004.02.011
90. Chen Y, Stirling RG, Paul E, Hore-Lacy F, Thompson BR, Douglass JA. Longitudinal decline in lung function in patients with primary immunoglobulin deficiencies. J Allergy Clin Immunol (2011) 127:1414–7. doi:10.1016/j.jaci.2011.03.041
91. Feydy A, Sibilia J, De Kerviler E, Zagdanski AM, Chevret S, Fermand JP, et al. Chest high resolution CT in adults with primary humoral immunodeficiency. Br J Radiol (1996) 69:1108–16. doi:10.1259/0007-1285-69-828-1108
92. Gattoni F, Tagliaferri B, Boioli F, Mazzoleni C, Uslenghi CM. Computerized tomography of the lungs in patients with congenital immunodeficiency. Comparison with clinicoradiologic assessment. Radiol Med (1999) 98: 26–35.
93. Serra G, Milito C, Mitrevski M, Granata G, Martini H, Pesce AM, et al. Lung MRI as a possible alternative to CT scan for patients with primary immune deficiencies and increased radiosensitivity. Chest (2011) 140:1581–9. doi:10.1378/chest.10-3147
94. Rich AL, Le Jeune IR, McDermott L, Kinnear WJ. Serial lung function tests in primary immune deficiency. Clin Exp Immunol (2007) 151:110–3. doi:10.1111/j.1365-2249.2007.03550.x
Keywords: infectious complications, inheritance, immune system dysregulation, interstitial lung diseases, respiratory tract, non-infectious complications, primary immunodeficiencies
Citation: Jesenak M, Banovcin P, Jesenakova B and Babusikova E (2014) Pulmonary manifestations of primary immunodeficiency disorders in children. Front. Pediatr. 2:77. doi: 10.3389/fped.2014.00077
Received: 24 February 2014; Accepted: 07 July 2014;
Published online: 25 July 2014.
Edited by:
Luis Garcia-Marcos, University of Murcia, SpainReviewed by:
Michael B. Anthracopoulos, University Hospital of Patras, GreeceKelvin D. MacDonald, Oregon Health & Science University, USA
Copyright: © 2014 Jesenak, Banovcin, Jesenakova and Babusikova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Milos Jesenak and Eva Babusikova, Center for Diagnosis and Treatment of Primary Immunodeficiencies, Departments of Pediatrics and Medical Biochemistry, Jessenius Faculty of Medicine, Comenius University in Bratislava, Kollarova 2, Martin 036 59, Slovakia e-mail:amVzZW5ha0BnbWFpbC5jb20=;ZXZhLmJhYnVzaWtvdmFAamZtZWQudW5pYmEuc2s=