
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Parasitol. , 27 January 2023
Sec. Immunity and Immune Evasion
Volume 2 - 2023 | https://doi.org/10.3389/fpara.2023.1113950
This article is part of the Research Topic Reviews in Immunology and Immune Evasion: 2022 View all 4 articles
 Jan R. Mead1,2*
Jan R. Mead1,2*Cryptosporidium spp. are opportunistic protozoan parasites that infect epithelial cells of the small intestine and cause diarrheal illness in both immunocompetent and immunodeficient individuals. These infections may be more severe in immunocompromised individuals and young children, especially in children under 2 in developing countries. The parasite has a global distribution and is an important cause of childhood diarrhea where it may result in cognitive impairment and growth deficits. Current therapies are limited with nitazoxanide being the only FDA-approved drug. However, it is not efficacious in immunocompromised patients. Additionally, there are no vaccines for cryptosporidiosis available. While acquired immunity is needed to clear Cryptosporidium parasites completely, innate immunity and early responses to infection are important in keeping the infection in check so that adaptive responses have time to develop. Infection is localized to the epithelial cells of the gut. Therefore, host cell defenses are important in the early response to infection and may be triggered through toll receptors or inflammasomes which induce a number of signal pathways, interferons, cytokines, and other immune mediators. Chemokines and chemokine receptors are upregulated which recruit immune cells such neutrophils, NK cells, and macrophages to the infection site to help in host cell defense as well as dendritic cells that are an important bridge between innate and adaptive responses. This review will focus on the host cell responses and the immune responses that are important in the early stages of infection.
Cryptosporidium spp. are opportunistic protozoan parasites that infect epithelial cells of the small intestine and cause diarrheal illness in both immunocompetent and immunodeficient individuals. Clinical signs vary considerably depending on the immunologic status of the host. In immunocompetent individuals, symptoms may range from asymptomatic to more severe signs of illness which may include a clinical presentation of watery diarrhea, abdominal pain (cramping), nausea, vomiting, anorexia, and mild fever. Typically, clinical signs abate in 1-2 weeks. In immunocompromised individuals such as AIDS patients, transplant recipients, and individuals undergoing chemotherapy, the disease can be severe and even fatal (Checkley et al., 2015). Infections may be protracted, and possibly involve extraintestinal sites of infection including the gall bladder, hepatobiliary ducts, pancreatic ducts, and respiratory tract (Shikani and Weiss, 2014).
The majority of human infections appear to be caused by two species: Cryptosporidium parvum and Cryptosporidium hominis. Most experimental studies relevant to humans have focused on the zoonotic species C. parvum which infects a number of animals as well as humans, including various immunocompromised and neonatal mice while adult wild-type mice are essentially refractory to infection. To a far lesser extent, studies have been performed with C. hominis, primarily due to limitations propagating the species (passage of C. hominis is accomplished in gnotobiotic pigs). Experimental work has also been performed with the murine species C. muris (McDonald et al., 1992) and C. tyzzeri (Ren et al., 2011; Sateriale et al., 2019). These species of Cryptosporidium are naturally found in mice, with a host range restricted mainly to rodents and are not considered human pathogens. The latter species has the advantage of primarily infecting the intestinal tract and for which a genetically modified traceable strain has been developed (Sateriale et al., 2019).
Two important immune mediators in response and recovery from cryptosporidiosis are IFN-γ production and CD4+ cells (Flanigan, 1994; Gomez Morales et al., 1996). In mice, experimental infections in nude (lacking T cells) and severe combined immunodeficiency (SCID) (lacking T and B cells) mice are chronic and lethal (Heine et al., 1984; Mead et al., 1991). In particular, CD4+ lymphocytes are crucial for the resolution of infection; mice depleted with anti-CD4+ antibody have markedly decreased immunity (Chen et al., 1993a). Adoptive transfer of CD4+ IELs (nonconventional lymphocytes located among the epithelial cells in the lumen) cells in SCID mice reduces parasite load and confers better protection against Cryptosporidium infections than CD8+ IELs (McDonald et al., 1996). CD8+ T-cells play an important though probably lesser role as these cells secrete IFN-γ early in infection (Leav et al., 2005) and may reduce the parasite load in the infected intestine via cytotoxicity by lysing infected intestinal epithelial cells (Pantenburg et al., 2010).
Antibodies also play a role in immune response as individuals with deficiencies such as X-linked hyper-IgM and IgA deficiencies are more susceptible to Cryptosporidium infection (Stiehm et al., 1986; Winkelstein et al., 2003; Wolska-Kusnierz et al., 2007). However, mice that lack B cells are able to clear infection (Chen et al., 2003). Following primary infections, antibody responses, specifically IgG and IgA, are mounted against the parasite (Ungar et al., 1986; Kassa et al., 1991). In the general population, the seropositivity rate in humans is high; reported to be anywhere from 25 to >60% depending on the location and population being surveyed (Priest et al., 2001; Frost et al., 2004). Antibodies provide some protection from reinfection (Fayer et al., 1990) and are directed against several immunodominant antigens that block parasite invasion in vitro or reduce parasite loads in mice (Arrowood et al., 1989; Riggs et al., 1989; Bjorneby et al., 1990; Petersen et al., 1992; Joe et al., 1998; Jenkins et al., 1999).
While exposure rates in North America are high, as indicated by high seropositivity rates (Ong et al., 2005), the parasite has a global distribution. It is second only to rotavirus as a cause of childhood diarrhea where it may result in cognitive impairment and growth deficits (Kotloff et al., 2013). The only FDA-approved anticryptosporidial drug is nitazoxanide, but it is not efficacious in immunocompromised patients (Abubakar et al., 2007). Additionally, there are no vaccines for cryptosporidiosis available. While adaptive immunity is critical for clearing the parasite completely, innate responses are important in reducing the number of parasites and in the initial stages. This review will highlight the host cell and early immune responses in the early stages that are important in controlling Cryptosporidium infections.
Cryptosporidial infections are usually initiated through ingestion of oocysts via contaminated food or water. Oocysts pass through the stomach to the small intestine where they excyst and release sporozoites. Sporozoites then invade the brush border of epithelial cells and subsequently differentiate into trophozoites (growing stage). Like other apicomplexan parasites, including Plasmodium and Toxoplasma, Cryptosporidium has a complex life cycle producing asexual and sexual stages, ultimately resulting in oocysts shed in the feces which are fully infectious upon leaving the host (Current and Reese, 1986).
The parasite occupies an unique niche in the epithelial cells in that the parasite is enclosed in a parasitophorous vacuole of both parasite and host cell origin. This location in the cell is described as “intracellular but extracytoplasmic” as the parasite resides inside the cell, i.e. beneath the host cell membrane. The base of the parasitophorous vacuole is adjacent to the host cell cytoplasm and a complex “feeder organelle” is located at the interface, presumably to facilitate transport into and out of the host cell cytoplasm. The parasitophorous vacuole has an apical surface facing the gastrointestinal (GI) lumen. Intestinal epithelial cells (IECs) and mucosa in the gut form a physical protective barrier against many pathogens and also serve as an interface between the lumen and underlying immune system. Injury to these barriers due to infection and inflammation may result in increases in the uptake of solutes and microbial antigens (Griffiths et al., 1994) or may alter tight junctions between the epithelial cells as levels of the tight junction proteins occludin and claudin are reportedly decreased following C. parvum infection of Caco-2 cells (Kumar et al., 2018). In addition to increased permeability of the epithelial barrier, induction of pro-inflammatory responses occurs. One main major path is through the inflammation transcription factor nuclear factor kappaB (NF-κB) (Chen et al., 2001).
Upon encountering microorganisms, intestinal epithelial cells are able to detect pathogens through innate receptors and respond by generating cytokines and upregulate chemokines which attract and activate other immune cells (Figure 1). C. parvum infection is reported to upregulate several chemokines including CXCL1 and CXCL8 (Laurent et al., 1997) that attracts neutrophils, CCL2 that attracts monocytes (Auray et al., 2007), CXCL10 that attracts dendritic cells (Lantier et al., 2013), CX3CL1 in biliary cells (Zhou et al., 2013) and CCL20 that have direct cytolytic activity against the free parasite (Guesdon et al., 2015). Additionally, adhesion molecules, such as beta-7 integrin (Mancassola et al., 2004) and ICAM-1 (Gong et al., 2011) that attract and activate other immune cells are up-regulated in C. parvum infection. Histocompatibility complex (MHC) class I and class II molecules are upregulated upon infection (Gomez Morales et al., 1995) and activation of Toll-like receptor (TLR) molecules have been reported in response to cryptosporidial infection (Chen et al., 2001; Chen et al., 2005). Nitric oxide produced through the induction of nitric oxide synthase (iNOS) of epithelial cells is significantly increased in C. parvum infection (Leitch and He, 1999; Gookin et al., 2004) and may be enhanced through dietary supplementation of L-arginine as demonstrated in several animals models (Leitch and He, 1994; Gookin et al., 2004; Castro et al., 2012). iNOS expression in C. parvum infection gives rise to increases in prostaglandin 2 (PGE2) synthesis which may maintain barrier function and promote clearance of infected epithelial cells (Gookin et al., 2004).
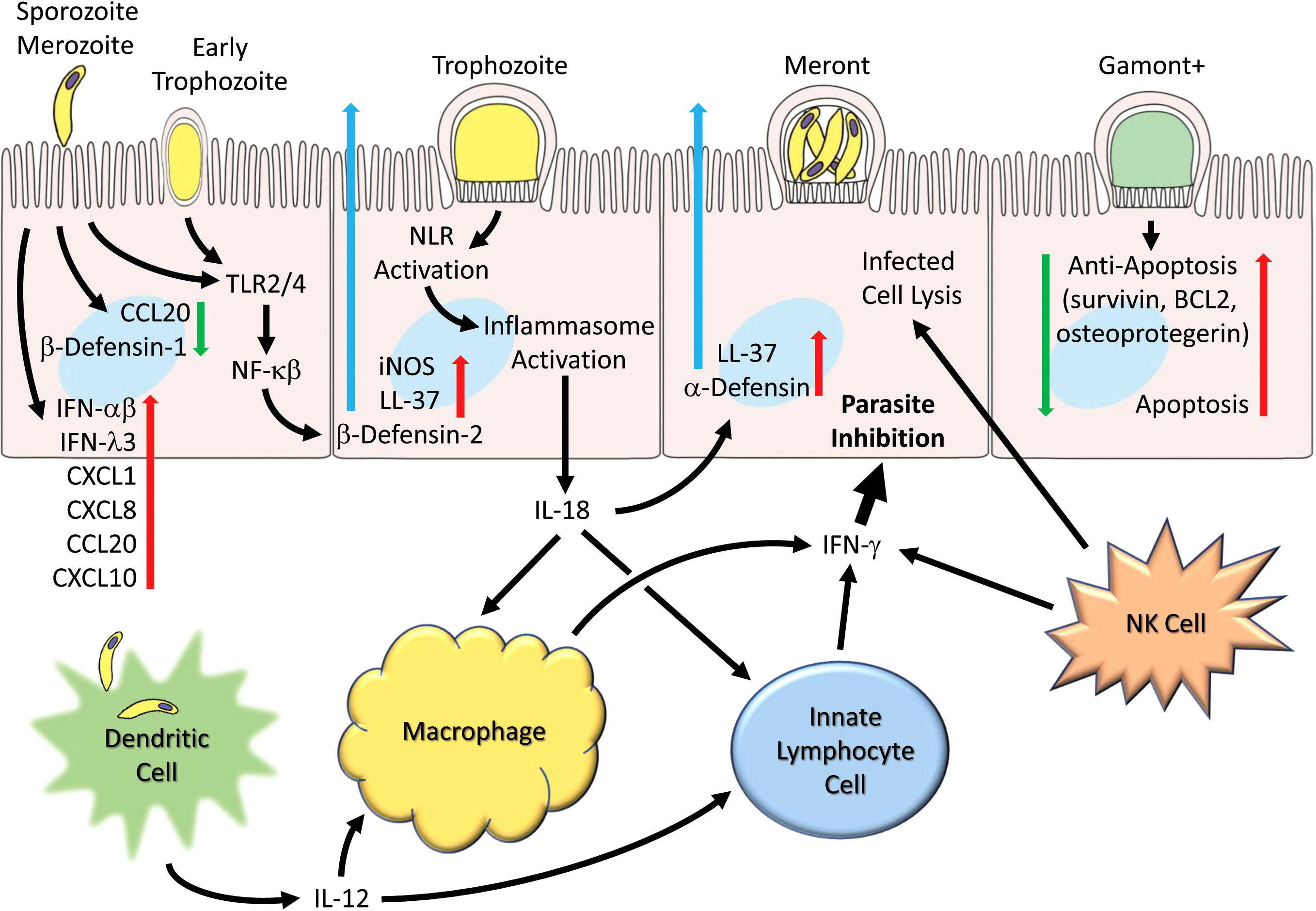
Figure 1 Generalized schematic illustrating early and host cell responses to Cryptosporidium infection. Parasite life cycle stage progression (left to right) following invasion by sporozoites (and merozoites) are shown as representative and the schematic pathways are not necessarily restricted to particular stages. Parasite infection leads to suppression of certain chemokines (e.g. CCL2) and β-defensin-1. Toll receptors (TLR2 and TLR4) are activated and lead to production and secretion of antimicrobial peptides (e.g. LL-37 and β-defensin-2). Inflammasome activation occurs and leads to IL-18 production. Numerous chemokines (e.g. CCL2, CXCL1, CXCL8, CXCL10), interferons (IFN-α, IFN-β and IFN-λ) and cytokines are released. Infection by C. parvum induces apoptosis but is countered by induction of anti-apoptotic genes (survivin, BCL2, and osteoprotegerin). Dendritic cells produce IL-12 in response to parasites or parasitic antigen and together with IL-18 released from epithelial cells helps to activate macrophages that generate IFN-γ. Innate lymphocyte cells (ILC1s) and NK cells also generate IFN-γ which can act on epithelial cells to inhibit the parasite.
Toll receptors (TLR), molecules on the surface of epithelial and other immune cells that recognize broad patterns of microbial molecules, are constitutively expressed on IECs and are important activators of innate immunity. Not surprisingly, they have been found to play a role in host responses to Cryptosporidium infection. Recognition of microbial proteins through toll receptors leads to the activation of the adaptor protein myeloid differentiation protein (MyD88) required for most TLR signaling. Mice lacking MyD88 are more susceptible to infection compared to wild-type mice and more so if the mice are made immunodeficient by IFN-γ neutralization (Rogers et al., 2006). Additionally, in human biliary epithelial cells, infections in vitro result in recruitment of TLR2 and TLR4 and are mediated by the activation of NF-κB activation, and result in the production of human β-defensin (Chen et al., 2005). When siRNA was used to knock down the expression of TLR2, TLR4, or MyD88, C. parvum-induced NF-κB activation was blocked, indicating the importance of Toll receptor pathways in defense (Chen et al., 2005). In a biliary model of cryptosporidiosis, infections in TLR4 deficient mice were more severe and unresolving compared to controls (O'Hara et al., 2011). Likewise, in a model of malnourished mice, increased susceptibility correlated to decreased expression of TLR2 and TLR4 in the intestinal tract (Costa et al., 2011). However, TLRs may be less prominent or less responsive in the intestinal environment or may be “model dependent” as deficiencies in TLR2 and TRL4 did not increase infection in a neonatal mouse model (Lantier et al., 2014).
TLR4 is up-regulated in response to C. parvum infection in infected cholangiocytes and is regulated by the microRNA, let7i. Suppression of the microRNA occurs soon after infection, resulting in the increased expression of TLR4 (Chen et al., 2007). Activation of TLR4 also promotes the release into the lumen of exosomes which contain antimicrobial peptides which can bind to the surface of sporozoites (Hu et al., 2013). The toll receptor TLR9 may also play some role as administration of the ligand CpG (which acts through TLR9) reduced infections in neonatal mice by 95% and increased IL-12 and IFN-γ compared to controls (Barrier et al., 2006). However, CpG was found to be less effective in an adult model of malnutrition (Costa et al., 2011). Toll receptors certainly play an important role in dendritic cell response to C. parvum infection, as dendritic cells from MyD88 knockout mice are unable to generate cytokines such as IL-12 in response to incubation with live sporozoites or antigen (Bedi and Mead, 2012).
Pathogens are also sensed intracellularly in the cytoplasm by NOD-like receptors (NLRs) which can activate immune defenses through inflammasome formation and may lead to the cleavage and activation of the proinflammatory cytokines IL-1β and IL-18. The latter cytokine, IL-18, is increased in infected mice (Ehigiator et al., 2005) and infected HCT-8 cells (McDonald et al., 2006). Additionally, IL-18 knockout mice are more susceptible to infection (Ehigiator et al., 2007).
The processing and secretion of mature IL-18 (and IL-1β) is mediated by caspase-1 which is activated within an inflammasome following the engagement of inflammasome-initiating sensors. In a report by McNair et al. (McNair et al., 2018), it was found that Casp-1-/-/Casp-11-/- knockout mice are over 35 times more susceptible to C. parvum infection than wild-type mice. Susceptibility correlated with a lack of IL-18 in caspase-1 and caspase1/11 knockout mice, as IL-18 is significantly elevated in wild-type mice. IL-1β was not generated in any significant amount following infection nor was any increased susceptibility observed in IL-1β nor IL-1βR knockout mice. These observations were corroborated in a recent study using a C. tyzzeri mouse model (Sateriale et al., 2021). This study further extended the previous work to show that mice lacking NLRP6, (an NLR known to induce IL-18 in the gut) were more susceptible to infection and had decreased levels of IL-18. Epithelial cells were shown to be the predominant cells generating IL-18. In contrast, mice lacking other NLRs such as NLRP3, NLRP1b, Aim2 and NLRC4 (which tend to induce an IL-1 response) were no more susceptible to infection than wild-type mice (McNair et al., 2018; Sateriale et al., 2021). It was also shown that the adapter protein ASC is important to susceptibility, and that the caspase-1 canonical inflammasome signaling pathway is the dominant pathway in C. parvum resistance (McNair et al., 2018) compared to the non-canonical pathway/caspase pathway activated in other pathogens like Leishmania (de Carvalho et al., 2019).
Type I interferons are mainly thought of as an innate defense mechanism to viral infections. However, many protozoan infections induce interferon α/β (IFN-α/β) in response to infection, including Cryptosporidium. In vitro, CMT-93 cells expressed IFN- α/β in response to C. parvum infection. Type I interferons were found to be protective as treatment with IFN-α or IFN-β before infection reduced parasite numbers significantly. In the same study, bone-marrow derived dendritic cells were also shown to generate IFN-α/β in response to live parasite (Barakat et al., 2009b). In vivo, Type I IFNs were expressed in the intestinal tissue of neonatal SCID mice 24 h after infection, while treatment with anti-IFN-α/β-neutralizing antibodies increased infection (Barakat et al., 2009b). The long non-coding RNA (NR_033736) is involved in the regulation of type I interferons and is increased in intestinal epithelial cells following C. parvum infection (Li et al., 2021).
Type III interferons, or IFN-λs, are important mediators of viral infections (Nice et al., 2018) and bacterial infections as well (Odendall et al., 2017). IFN-λs are particularly important in mucosal infections because of their preferential expression of the IFNLR1 receptor on epithelial cells. In a report by Ferguson et al., these genes were found to be upregulated in C. parvum-infected porcine epithelial cells (Ferguson et al., 2019). Monolayers pretreated with IFN-λ protected cells, resulting in significant reduction of infection by limiting invasion and decreasing maturation of parasites. Also, IFN-λ offset the loss of barrier function due to infection by decreasing paracellular permeability due to sodium. Administration of neutralizing antibodies against IFN-λ to neonatal mice increased C. parvum infection during peak infection. In another study, using a genetic screen (CRISPR-cas9 knockout) to identify human proteins involved in survival, it was found that infection of host cells resulted in an increase of predominately type III interferons compared to type I (Gibson et al., 2022). Additionally, production of type III interferons was shown to be dependent on TLR3 signaling as mice lacking TLR3 were more susceptible to infection and had significantly reduced levels of IFN-λ.
Other immune effectors generated in the epithelial cell in response to infection include antimicrobial peptides. In mammals, there are two large families of antimicrobial peptides: defensins and cathelicidins. The expression of β-defensins, which are widely expressed in epithelial cells, was studied using a Cryptosporidium-infected human colonic HT29 cell line (Zaalouk et al., 2004). Infection of these cells induced the expression of human β-defensin-1 (hBD-1), a partial reduction in the expression of human β-defensin-2 (hBD-2), but had no effect on human β-defensin-3 (hBD-3) gene expression. Antimicrobial peptides such as LL-37 (human) or murine cathelicidin related antimicrobial peptides (CRAMP) and β-defensins can result in the lysis of the parasitic cell membrane. Antimicrobial peptides have been found to lyse sporozoites in vitro (Arrowood et al., 1991; Carryn et al., 2012; McDonald et al., 2013). Exogenous administration of CRAMP decreased C. parvum infection in neonatal mice (Guesdon et al., 2020). Typically, CRAMP expression is higher in neonatal mice during the first 3 weeks of age to help protect neonatal mice from a number of intestinal infections. However, CRAMP expression was found to be reduced during C. parvum infection, while other antimicrobial peptides were upregulated (Guesdon et al., 2020), suggesting the possibility that the parasite may have a mechanism for downregulating this mediator. These antimicrobial peptides can be released through exosomes from epithelial cells (Hu et al., 2013). The activation of these exosomes is initiated through the TLR4 signaling pathway that downregulates microRNA Let-7, which in turn results in an increase of protein SNAP23 involved in vesicular exocytosis. Interestingly, other effector molecules (epithelial cell-derived proteins and cryptosporidial RNAs) have been shown to be released via exosomes and shuttle to the spleen, activating immune cells distal from the site of infection (Wang et al., 2019b).
C. parvum can activate both the classical and lectin complement pathways, leading to the deposition of C3b on the parasite (Petry et al., 2008). However, increased susceptibility to the parasite could only be demonstrated in adult mice lacking mannose binding lectin (MBL) not in mice lacking C1q or in wild-type mice, indicating the importance of the lectin pathway and suggesting the alternative pathway does not play a major role in infection. MBL is a conserved protein that binds to microbial surfaces and promotes opsonophagocytosis. Studies have suggested the protective role of MBL may be most important in young children and individuals with immunodeficiencies (Kelly et al., 2000; Kirkpatrick et al., 2006). In 5% of the world’s population, polymorphisms in the MBL2 gene create low MBL-producing MBL2 genotypes, which may lead to an increased susceptibility to particular diseases, including cryptosporidiosis (Carmolli et al., 2009). In a recent study involving children in South India under the age of 3, polymorphism in the MBL2 gene was associated with susceptibility to cryptosporidial diarrhea (Liakath et al., 2021).
In general, intracellular pathogens induce a strong cell-mediated (Th1) response where IFN-γ plays a leading role in driving the cellular response and also in effector mechanisms inhibiting the parasite. IFN-γ is a key cytokine important in both human cryptosporidiosis and experimental animal studies (McDonald and Bancroft, 1994; Hayward et al., 2000). Depletion of the IFN-γ gene in mice or neutralization using anti-IFN-γ antibodies results in greater susceptibility to infection (Theodos et al., 1997; Griffiths et al., 1998; Mead and You, 1998). Sources of IFN-γ include NK cells, macrophages, and T cells. An early source of IFN-γ, produced soon after infection, are intestinal intraepithelial lymphocytes (iLEL), particularly CD8αTCRα cells which were found to generate IFN-γ as soon as 24 hours post infection (Leav et al., 2005). Most recently, innate lymphocyte cells (ILC1s) have been identified as a major source of IFN-γ (Gullicksrud et al., 2022).
While IFN-γ importance in infection is well established, the mechanism by which it aids resistance to infection is not fully understood. IFN-γ has a direct effect on infected epithelial cells resulting in decreased infection (Pollok et al., 2001). It has been suggested that this is due, in part, to depletion of available iron (Fe) in the cell as Fe+ supplementation in culture media can partially reverse the inhibition of parasite growth (Pollok et al., 2001). Transcriptional profiling of epithelial cells from infected mice has shown that an up-regulation of IFN-γ induced the effectors indoleamine 2,3-dioxygenase (IDO), guanylate binding protein (GBP) and immunity-related GTPases (IRG) (Gullicksrud et al., 2022). Additionally, while deletion in IDO and GBP did not increase susceptibility to C. parvum infection, mice lacking the GTPases, immunity-related GTPase family M (IRGM)1 and IRGM3 were more susceptible to infection compared to wild-type mice. In another study looking at changes in m6A mRNA methylation (which may regulate innate immune responses), expression levels of IRGM2 and IFN-γ induced GTPase IGTP or IRGM3 were found to be increased in infected cells (Xia et al., 2021).
Two key inducers of IFN-γ, IL-12 and IL-18, are important in primary immune responses to infection. Both cytokines work synergistically to produce IFN-γ (Choudhry et al., 2012). IL-12 was shown to be protective in mice (Urban et al., 1996) while lack of this cytokine leads to greater susceptibility and severity of infection (Ehigiator et al., 2003). Dendritic cells can generate IL-12 in response to C. parvum (Bedi and Mead, 2012). Specially, batf3-dependent CD103+ has been shown to be a source of IL-12 in neonatal mice (Potiron et al., 2019). IL-12 synergizes with IL-18 to stimulate effector cells (ILC1s) to produce IFN-γ (Gullicksrud et al., 2022). Additionally, IL-4 has also been shown to work synergistically with IFN-γ to enhance parasite killing (Lean et al., 2003).
TNF-α, a cytokine that is increased during infection, does reduce parasite numbers in infected epithelial cells in vitro through limiting invasion of host cells (Barakat et al., 2009b). However, the role for TNF-α in vivo may be less apparent as mice treated with anti-TNF-α or TNF-α deficient mice are no more susceptible than control mice (Chen et al., 1993b; Lean et al., 2006). However, in the absence of IFN-γ, protection was observed in IFN-γ knock out mice treated with TNF-α (Lacroix et al., 2001).
IL-10 and TGFβ are key regulators in the maintenance of immunological responses in the gastrointestinal tract. Both of these cytokines are increased during the resolution of cryptosporidial infections (Robinson et al., 2000; Lacroix et al., 2001). In particular, TGFβ has an inhibitory effect on IFN-γ (Lean et al., 2003).
The microbiome has become an important focus in the interactions of gut infection susceptibility and host response. Since Cryptosporidium infections are primarily localized to the epithelium of the intestine it is reasonable to assume that the local flora would impact cryptosporidial infection and the over-all disease state. In early studies, SCID mice with established conventional gut flora showed more resistance to infection by C. parvum than germ-free SCID mice in that oocyst production was delayed by several weeks (Harp et al., 1992). A TCR-α-deficient mouse model of cryptosporidiosis reportedly developed IBD-like lesions following infection (Sacco et al., 1998). Interestingly, TCR-α-deficient mice exhibited more severe mucosal lesions among infected germ-free versus mice bearing conventional gut flora (Sacco et al., 1998), suggesting microbiome influences on the pathology of disease. Additionally, gut flora may play a protective role through the activation of the TLR5-MyD88 pathway, stimulating dendritic cells and helping to generate cytokines, such as IL-12, and type I and type II IFNs (Lantier et al., 2014).
Differences in microbiome composition may correlate with differences in susceptibility and clinical symptoms. In one human study, stools from C. parvum infected volunteers showed changes in microbiota that correlated with differences in susceptibility to infection and also to shifts in metabolites (Chappell et al., 2016). In particular, fecal indole or indole producing bacteria were associated with protection. In another study, when comparing 72 infants with and without diarrheal symptoms, low Megasphaera abundance was associated with diarrheal symptoms prior to and at the time of Cryptosporidium detection (Carey et al., 2021). Interestingly, Megasphaera are known to synthesize short-chain fatty acids which help maintain the homeostasis of the intestinal tract.
Infections with C. parvum can also alter the microbiome (Ras et al., 2015). In animals, cryptosporidial infections in goat kids depleted butyrate-producing bacteria (Mammeri et al., 2020), changes in infected neonatal mice showed increased proportions of the bacterial phylum Bacteroidetes (Mammeri et al., 2020). Monkeys with active infection were found to have enriched taxa associated with dysbiosis, inflammation and rapid transit time thorough the gut (McKenney et al., 2017). Shifts in fungal microbiome in horses have also been observed after cryptosporidial infections (Wang et al., 2022).
Additionally, drugs and natural products may also alter the gut flora and influence infection (Gorla et al., 2014; Rahman et al., 2022). In a mouse model of cryptosporidiosis, increased severity of infection in antibiotic-treated mice depleted of primary Gram positive bacteria was associated with a decrease in short chain fatty acids (Charania et al., 2020). In animals, prebiotics (diet without fiber) altered the microbiome in C. parvum infected mice and enhanced infection, (Oliveira et al., 2019) whereas probiotics often used to bolster the microbiome were also found to increase infection (Oliveira and Widmer, 2018). Limited studies have been performed in humans. In one study, a probiotic was associated with enhanced resolution of disease (Pickerd and Tuthill, 2004) while another study showed improvement in barrier permeability among children infected with Cryptosporidium who were given Lactobacillus (Sindhu et al., 2014).
Bacteria produce many metabolites which can affect immune populations, such as T regulatory cells, or have a direct effect on epithelial cells themselves. In a study examining changes in flora and metabolites in neonatal mice, medium or long chain saturated fatty acids inhibited C. parvum growth while long-chain unsaturated fatty acids enhanced C. parvum invasion (VanDussen et al., 2020). Short chain fatty acids such as butyrate and propionate (produced mainly by Gram positive bacteria and depleted with certain antibiotics) were also found to have a direct inhibitory effect on C. parvum growth in vitro (Keelaghan et al., 2022). In another study looking at microbiome and metabolite changes during infection, bacteria such as Coriobacteriaceae and Lactobacillus were increased along with D-amino acids and short chain fatty acids while glycolysis/citrate cycle metabolites were depleted (Karpe et al., 2021).
Apoptosis is a form of programmed cell death that helps in the elimination of pathogens and abnormal cells. Apoptosis is initiated through either an intrinsic pathway (from within the cell) and or an extrinsic pathway (signals from other cells). Both of these pathways involve activating caspases which result in degrading proteins. There is an apparent interaction between the host and parasite genes with the host trying to eliminate the parasite through apoptosis and parasite induced suppression of some host apoptotic genes allowing for increased growth of the organism. Initially, apoptosis is up-regulated for the first few hours during invasion, perhaps as a way to dampen inflammatory responses (Mele et al., 2004). Anti-apoptotic genes are then up-regulated during development of the trophozoites at 24 hours (Mele et al., 2004). Inhibiting apoptosis occurs as a way to counter host cell response and to garner host metabolites in a dividing cell. These genes include BCL-2, osteoprotegerin and survivin (Mele et al., 2004; Castellanos-Gonzalez et al., 2008; Liu et al., 2008). When the latter was knocked down, an increase in apoptosis was observed which resulted in a decrease in parasite growth (Liu et al., 2008). At the same time (24 hours post infection), a moderate amount of apoptosis still occurs (McCole et al, 2000), reported mainly in non-infected adjacent cells (Chen et al., 2001; Mele et al., 2004) perhaps as a way to limit parasite expansion by the host. Induction of apoptosis occurs in the later stages of infection (48 hours post infection) perhaps as a way to facilitate escape by the parasite from the host (Chen et al., 1999; McCole et al., 2000; Buret et al., 2003). Alternatively, host-cell apoptosis may be advantageous for the host and a way to eliminate infected cells but in a controlled manner. In ex-vivo enterocytes from piglets, repression of apoptosis by the host cell has been reported to occur except at the villus tips allowing for elimination of infected cells in a more controlled manner while still maintaining integrity of the epithelial barrier (Foster et al., 2012). Studies with microRNA show that many different microRNAs are involved in apoptosis and anti-apoptosis pathways (Wang et al., 2019a). One microRNA, miR-942-5p, which is up-regulated in HCT-8 cells following activation of the TLR2/TLR4-NF-κB signaling pathway by C. parvum, has been implicated in regulation of host cell apoptosis (Zhang et al., 2020) through the TRAIL-dependent pathways (Xie et al., 2022).
Another mechanism through which host cells help eliminate intracellular pathogens is through the process of autophagy. In order to maintain homeostasis, the host cell recycles degraded macromolecules and organelles. Alternatively, autophagy processes may be used to target intracellular pathogens by autophagosome formation around the free pathogen or pathogen within a vacuole, fusing with lysosomes and ultimately the degradation of the pathogen. There is also some evidence to suggest that immunity-related GTPases are linked to the autophagosome and may aid in disrupting pathogen vacuoles (Evans et al., 2018). In a recent study, autophagy was induced in C. parvum-infected CaCo2 cells. This was demonstrated through increased LC3II/I ratio (increase in this ratio suggests autophagy induction) and enhanced autophagosome formation (Priyamvada et al., 2021). While some intracellular protozoa such as T. gondii and T. cruzi have mechanisms to interfere with the autophagy process of the host cell (Muniz-Feliciano et al., 2013; Portillo et al., 2017; Onizuka et al., 2017), this has not yet been shown for C. parvum. However, silencing of the autophagy ATG7 gene results in decreased expressing of two barrier proteins, occludin and claudin, suggested that there may be a link between C. parvum induced autophagy and barrier integrity (Priyamvada et al., 2021). Interestingly, in humans, single nucleotide polymorphisms (SNP) in the gene ATG16L1 impairs autophagy. SNPs in the ATG16L1 cause greater survival of intracellular pathogens (Evans et al., 2018). EL-Refai et al. were able to show that patients that had this SNP had greater severity of disease and increased risk of infection to C. parvum (El-Refai et al., 2021).
Neutrophils are important first responders to active infection sites, especially for bacteria but also for other pathogens as well. They provide important defense mechanisms such as phagocytosis and the production of reactive oxygen reactive species. Their role, however, in cryptosporidiosis may be less direct and more in augmentation of other cell populations. In cryptosporidiosis, neutrophils are recruited in the lamina propria soon after infection (Genta et al., 1993; Zadrozny et al., 2006). In piglets, depletion of neutrophils using an anti-CD18 antibody did not alter lipid peroxidation or peroxynitrite formation, suggesting these cells were not a significant source of free radicals nor did they have an influence on the severity or pathology of infection (Zadrozny et al., 2006). However, they were found to enhance barrier function (Zadrozny et al., 2006). SCID-beige mice lacking either macrophages or neutrophils have more severe infections when infected with C. parvum (Takeuchi et al., 2008). Adoptive transfer of either macrophages or neutrophils alone into these mice could not rescue the mice from succumbing to the infection but rather required both cell types. It was suggested that there may be a cooperative interaction been the two cell types (Takeuchi et al., 2008). In the same study, polymorphonuclear leukocyte cells (PMN)s isolated from C. parvum-infected mice were shown to play a role in converting resident macrophages to M1 macrophages. Another possible mechanism is through NETosis, an effector mechanism by which extracellular fibers (primarily DNA) from neutrophils bind pathogens (Hasheminasab et al., 2022). In one study, neutrophil extracellular traps (NETs) were found to form when bovine neutrophils were incubated with either sporozoites or oocysts. This led to the entrapment of 15% of the sporozoites (Munoz-Caro et al., 2015). PMNs exposed to sporozoites had increased levels of the chemokine CXCL8, TNF-α, IL-6, and the growth factor GM-CSF.
Macrophages are a source of IFN-γ and contribute to clearance of cryptosporidiosis through phagocytosis of the parasite. Intact and partially digested parasites have been found in macrophages associated with M cells that overlay Peyer’s patches in guinea pigs (Marcial and Madara, 1986). Mice depleted of macrophages using clodronate-containing liposomes showed higher levels of infection than controls and were unable to mount a significant IFN-γ response suggesting macrophages are an important source of this cytokine (Choudhry et al., 2012). In neonatal mice, inflammatory Ly6C+ monocytes recruited into the subepithelial intestine and macrophages are shifted to a proinflammatory phenotype during infection, produce TNF-α and IL-1β. These cytokines, while enhancing other immune functions, also contribute to decreasing barrier function and increasing gut permeability (de Sablet et al., 2016). While macrophages generate IFN-γ and other immune factors they also require appropriate signals to be stimulated. Macrophages can be activated in vitro in the presence of C. parvum but require a combination of IL-18 and IL12 to generate significant amounts of IFN-γ as neither of these cytokines alone result in activation (Choudhry et al., 2012).
Dendritic cells are important in the early stages of Cryptosporidium infection, as mice depleted of these cells are more susceptible to infection (Lantier et al., 2013; Bedi et al., 2014). They are recruited in the ileum of C. parvum-infected mice (Auray et al., 2007) and express important mediators such as IFN-α and IFN-β (Barakat et al., 2009b) and IL-12, IL-1β and IL-6 (Bedi and Mead, 2012) in response to live sporozoites or soluble sporozoite antigen. The former (live sporozoites) generated a better response as parasite burden was reduced with longer protection observed in mice adoptively transferred with dendritic cells stimulated with live sporozoites (Bedi et al., 2014). DCs capture parasites or phagocytized infected epithelial cells and transport them to the mesenteric lymph nodes (Ponnuraj and Hayward, 2001; Auray et al., 2007; Perez-Cordon et al., 2014). Gut flora may help in synergizing responses of DCs (through TLR5 signaling) as expression of IFN-α and IFN-β, as induced by poly(I:C), increased in the presence of commensal bacteria (Lantier et al., 2014). Additionally, they aid in the initiation of adaptive responses and activation of T cells, resulting in IFN-γ production (Bedi and Mead, 2012). In a study that examined different subsets of dendritic cells, CD103+ were found to control infections better than CD103- DCs (Potiron et al., 2019). Since neonatal mice have fewer CD103+ cells, it has been suggested that this is one reason neonates are more susceptible to C. parvum infection (Potiron et al., 2019). Additionally, neonatal mice that lacked transcription factor Batf3 (Batf3 drives the development of CD103+ DCs) were more susceptible to infection and could not clear infection. These mice had a lower ability to generate IL-12 and were presumably not able to contribute to IFN-γ production. Lastly, mice deficient in cDC1 dendritic cells were found to be more susceptible to C. Tyzzeri infections. Infections in these mice generated skewed T cell responses that were predominately Th17 and Treg responses compared to IFN-γ -producing Th1 response generated by wild-type mice (Russler-Germain et al., 2021) demonstrating the importance of these cells in initiating the appropriate Th1 response.
NK cells contribute to innate immunity to cryptosporidiosis through lysis of infected epithelial cells or through production of IFN-γ. In neonatal lambs, NK cells (CR1+/CD16+ and NCR1+/CD16- populations) increase in response to infection (Olsen et al., 2015). They are activated by the cytokine IL-15 and have cytolytic activity against C. parvum-infected epithelial cells in vitro (Dann et al., 2005). Susceptibility to disease and severity of disease does increase in mouse models deficient in NK cells. Infections in beige mice (lacking NK cells) were slightly more susceptible (detectable by microscopy) than other strains of wild-type mice (Enriquez and Sterling, 1991). Infections in NIH III mice (SCID mice deficient in NK cells) were found to be more disseminated than SCID mice with normal NK cell function (Mead et al., 1991) and infections were more severe in Rag2-/- gammaC-/- mice (T and B cells, lack NK cells) than Rag2-/- mice (Barakat et al., 2009a). However, in experimental mouse models, depletion of NK cells using α-asialo-GM1 antibody against NK cells did not alter susceptibility or severity of infection (Rohlman et al., 1993; McDonald and Bancroft, 1994; McDonald et al., 2013), while depletion using anti-NK1.1 antibody did increase severity (Korbel et al., 2011), perhaps because the α-asialo-GM1 antibody does not completely deplete all target cells, including tissue NK cells (Victorino et al., 2015). In a recent study, Rag2-/- mice were treated with α-asialo-GM1 which, once again, was found to deplete NK spleen cells but did not deplete innate lymphoid cells (ILCs), a type of NK cell that resides in the gut mucosal (Gullicksrud et al., 2022). Additionally, mice that lack T, B and splenic NK cells remained more resistant, with less severe infections, compared to Rag2−/−/Il2rg−/− that lacked all 4 cell populations (T, B, NK and ILC cells). Innate lymphoid cell type 1s (ILC1s) were found to be protective in the early stages of cryptosporidiosis and an important source of IFN-γ (Gullicksrud et al., 2022).
Cryptosporidium parvum has developed strategies to evade or alter the host cell response. The parasite’s unique location (intracellular but surrounded by host cell membrane) may play a role in evasion. While the intracellular location may help in avoiding exposure to immune effector mechanisms such as antibodies and phagocytosis, the extracytoplasmic location avoids host cellular defense mechanisms such as Guanylate Binding Proteins (GBP)s that interact with vacuoles that are completely intracellular. This unique location may also limit exposure of parasite antigens to immune cells in the lamina propria.
Another potential mechanism of immune evasion involves inhibiting signal transduction pathways in the host cell. STAT-1α is a major component of IFN-γ signaling and important in the immune response as mice with STAT-1α knocked out are more susceptible to infection (Ehigiator et al., 2007). C. parvum infection in CMT-93 cells was shown to decrease several IFN-γ-dependent genes, including indoleamine 2,3-dioxygenase (IDO) which catabolizes tryptophan to kynurenine (Choudhry et al., 2009). Investigating possible mechanisms, it was found that C. parvum infection caused depletion of the STAT-1α protein in infected host cells, leading to the down-regulation of IFN-γ (Choudhry et al., 2009). Likewise, p38 MAPK signaling is downregulated (in particular Mk2 and Mk3) in infected cells (He et al., 2021). MAP kinase-activated protein kinases are important for a number of immune responses.
As described above, C. parvum inhibits apoptosis early in infection allowing it to establish itself but pro-apoptotic gene expression appeared to be favored at late stages (Liu et al., 2009). The chemokine CCL20, a chemokine involved in dendritic cell recruitment, was found to be down-regulated after C. parvum infection, independently of an IFN-γ response or the presence of microbiota (Guesdon et al., 2015). Additionally, C. parvum has been shown to deliver transcripts into the nucleus of host cells, resulting in the modulation of a number of host genes (Wang et al., 2017). In particular, delivery of the parasite RNA transcript, cdg7_FLc_1000 to the host cell down regulates the β-defensin-1 gene, contributing to evasion early in infection (Ming et al., 2018).
In the recent years, much research has focused on host cell responses and innate immunity to cryptosporidiosis. Advances have been made in understanding how the control of those responses occurs which is often through nucleic acid molecules such as miRNA (Ming et al., 2017) and long non-coding (linc) RNA (Mathy et al., 2022) which help in the regulation of various immune mediators. Additionally, studies on less studied mucosal cell types such as innate lymphocytes will remain an area of interest. The microbiome and metabolites will continue to be a focus as it is discovered how these bacteria and other microbes affect host cell and parasite interactions. New in vitro models including 2D and 3D culture systems, organoid/enteroid (Zhang et al., 2016; DeCicco RePass et al., 2017; Heo et al., 2018) and stem cell cultures (Wilke et al., 2019) will facilitate extended or continuous culture of the parasite and support the study of hard to propagate strains such as C. hominis. Cell culture models of greater complexity and control may make it possible to study interactions of individual cell types of the immune systems with epithelial cells and other components (e.g. microbiome, metabolites of the intestinal environment) in an in vitro setting.
The author confirms being the sole contributor of this work and has approved it for publication.
This work was supported by grants from the National Institutes of Health, R21AI157730 and the Foundation for Atlanta Veterans Education and Research (FAVER).
The author would like to thank Michael Arrowood for his help on the illustration.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abubakar I., Aliyu S. H., Arumugam C., Usman N. K., Hunter P. R. (2007). Treatment of cryptosporidiosis in immunocompromised individuals: Systematic review and meta-analysis. Br. J. Clin. Pharmacol. 63, 387–393.
Arrowood M. J., Jaynes J. M., Healey M. C. (1991). In vitro activities of lytic peptides against the sporozoites of Cryptosporidium parvum. Antimicrob. Agents Chemother. 35, 224–227.
Arrowood M. J., Mead J. R., Mahrt J. L., Sterling C. R. (1989). Effects of immune colostrum and orally administered antisporozoite monoclonal antibodies on the outcome of Cryptosporidium parvum infections in neonatal mice. Infect. Immun. 57, 2283–2288.
Auray G., Lacroix-Lamande S., Mancassola R., Dimier-Poisson I., Laurent F. (2007). Involvement of intestinal epithelial cells in dendritic cell recruitment during C. parvum infection. Microbes Infect. 9, 574–582.
Barakat F. M., Mcdonald V., Di Santo J. P., Korbel D. S. (2009a). Roles for NK cells and an NK cell-independent source of intestinal gamma interferon for innate immunity to Cryptosporidium parvum infection. Infect. Immun. 77, 5044–5049.
Barakat F. M., Mcdonald V., Foster G. R., Tovey M. G., Korbel D. S. (2009b). Cryptosporidium parvum infection rapidly induces a protective innate immune response involving type I interferon. J. Infect. Dis. 200, 1548–1555.
Barrier M., Lacroix-Lamande S., Mancassola R., Auray G., Bernardet N., Chausse A. M., et al. (2006). Oral and intraperitoneal administration of phosphorothioate oligodeoxynucleotides leads to control of Cryptosporidium parvum infection in neonatal mice. J. Infect. Dis. 193, 1400–1407.
Bedi B., Mcnair N. N., Mead J. R. (2014). Dendritic cells play a role in host susceptibility to Cryptosporidium parvum infection. Immunol. Lett. 158, 42–51.
Bedi B., Mead J. R. (2012). Cryptosporidium parvum antigens induce mouse and human dendritic cells to generate Th1-enhancing cytokines. Parasite Immunol. 34, 473–485.
Bjorneby J. M., Riggs M. W., Perryman L. E. (1990). Cryptosporidium parvum merozoites share neutralization-sensitive epitopes with sporozoites. J. Immunol. 145, 298–304.
Buret A. G., Chin A. C., Scott K. G. (2003). Infection of human and bovine epithelial cells with Cryptosporidium andersoni induces apoptosis and disrupts tight junctional ZO-1: effects of epidermal growth factor. Int. J. Parasitol. 33, 1363–1371.
Carey M. A., Medlock G. L., Alam M., Kabir M., Uddin M. J., Nayak U., et al. (2021). Megasphaera in the stool microbiota is negatively associated with diarrheal cryptosporidiosis. Clin. Infect. Dis. 73, e1242–e1251.
Carmolli M., Duggal P., Haque R., Lindow J., Mondal D., Petri W. A. Jr., et al. (2009). Deficient serum mannose-binding lectin levels and MBL2 polymorphisms increase the risk of single and recurrent Cryptosporidium infections in young children. J. Infect. Dis. 200, 1540–1547.
Carryn S., Schaefer D. A., Imboden M., Homan E. J., Bremel R. D., Riggs M. W. (2012). Phospholipases and cationic peptides inhibit Cryptosporidium parvum sporozoite infectivity by parasiticidal and non-parasiticidal mechanisms. J. Parasitol. 98, 199–204.
Castellanos-Gonzalez A., Yancey L. S., Wang H. C., Pantenburg B., Liscum K. R., Lewis D. E., et al. (2008). Cryptosporidium infection of human intestinal epithelial cells increases expression of osteoprotegerin: A novel mechanism for evasion of host defenses. J. Infect. Dis. 197, 916–923.
Castro I. C., Oliveira B. B., Slowikowski J. J., Coutinho B. P., Siqueira F. J., Costa L. B., et al. (2012). Arginine decreases Cryptosporidium parvum infection in undernourished suckling mice involving nitric oxide synthase and arginase. Nutrition 28, 678–685.
Chappell C. L., Darkoh C., Shimmin L., Farhana N., Kim D. K., Okhuysen P. C., et al. (2016). Fecal indole as a biomarker of susceptibility to Cryptosporidium infection. Infect. Immun. 84, 2299–2306.
Charania R., Wade B. E., Mcnair N. N., Mead J. R. (2020). Changes in the microbiome of Cryptosporidium-infected mice correlate to differences in susceptibility and infection levels. Microorganisms 8, 879.
Checkley W., White A. C. Jr., Jaganath D., Arrowood M. J., Chalmers R. M., Chen X. M., et al. (2015). A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for Cryptosporidium. Lancet Infect. Dis. 15, 85–94.
Chen X. M., Gores G. J., Paya C. V., Larusso N. F. (1999). Cryptosporidium parvum induces apoptosis in biliary epithelia by a Fas/Fas ligand-dependent mechanism. Am. J. Physiol. 277, G599–G608.
Chen W. X., Harp J. A., Harmsen A. G. (1993a). Requirements for CD4+ cells and gamma interferon in resolution of established Cryptosporidium parvum infection in mice. Infect. Immun. 61, 3928–3932.
Chen W., Harp J. A., Harmsen A. G. (2003). Cryptosporidium parvum infection in gene-targeted b cell-deficient mice. J. Parasitol. 89, 391–393.
Chen W. X., Harp J. A., Harmsen A. G., Havell E. A. (1993b). Gamma-interferon functions in resistance to Cryptosporidium parvum infection in severe combined immunodeficient mice. Infect. Immun. 61, 3548–3551.
Chen X. M., Levine S. A., Splinter P. L., Tietz P. S., Ganong A. L., Jobin C., et al. (2001). Cryptosporidium parvum activates nuclear factor kappaB in biliary epithelia preventing epithelial cell apoptosis. Gastroenterology 120, 1774–1783.
Chen X. M., O'hara S. P., Nelson J. B., Splinter P. L., Small A. J., Tietz P. S., et al. (2005). Multiple TLRs are expressed in human cholangiocytes and mediate host epithelial defense responses to Cryptosporidium parvum via activation of NF-kappaB. J. Immunol. 175, 7447–7456.
Chen X. M., Splinter P. L., O'hara S. P., Larusso N. F. (2007). A cellular micro-RNA, let-7i, regulates toll-like receptor 4 expression and contributes to cholangiocyte immune responses against Cryptosporidium parvum infection. J. Biol. Chem. 282, 28929–28938.
Choudhry N., Korbel D. S., Edwards L. A., Bajaj-Elliott M., Mcdonald V. (2009). Dysregulation of interferon-gamma-mediated signalling pathway in intestinal epithelial cells by Cryptosporidium parvum infection. Cell. Microbiol. 11, 1354–1364.
Choudhry N., Petry F., Van Rooijen N., Mcdonald V. (2012). A protective role for interleukin 18 in interferon gamma-mediated innate immunity to Cryptosporidium parvum that is independent of natural killer cells. J. Infect. Dis. 206, 117–124.
Costa L. B., Johnbull E. A., Reeves J. T., Sevilleja J. E., Freire R. S., Hoffman P. S., et al. (2011). Cryptosporidium-malnutrition interactions: mucosal disruption, cytokines, and TLR signaling in a weaned murine model. J. Parasitol. 97, 1113–1120.
Current W. L., Reese N. C. (1986). A comparison of endogenous development of three isolates of Cryptosporidium in suckling mice. J. Protozool. 33, 98–108.
Dann S. M., Wang H. C., Gambarin K. J., Actor J. K., Robinson P., Lewis D. E., et al. (2005). Interleukin-15 activates human natural killer cells to clear the intestinal protozoan Cryptosporidium. J. Infect. Dis. 192, 1294–1302.
de Carvalho R. V. H., Andrade W. A., Lima-Junior D. S., Dilucca M., De Oliveira C. V., Wang K., et al. (2019). Leishmania lipophosphoglycan triggers caspase-11 and the non-canonical activation of the NLRP3 inflammasome. Cell Rep. 26, 429–437.e5.
DeCicco RePass M. A., Chen Y., Lin Y., Zhou W., Kaplan D. L., Ward H. D. (2017). Novel bioengineered three-dimensional human intestinal model for long-term infection of Cryptosporidium parvum. Infect. Immun. 85, e00731–16.
de Sablet T., Potiron L., Marquis M., Bussiere F. I., Lacroix-Lamande S., Laurent F. (2016). Cryptosporidium parvum increases intestinal permeability through interaction with epithelial cells and IL-1β and TNFα released by inflammatory monocytes. Cell. Microbiol. 18, 1871–1880.
Ehigiator H. N., Mcnair N., Mead J. R. (2003). IL-12 knockout C57BL/6 mice are protected from re-infection with Cryptosporidium parvum after challenge. J. Eukaryot. Microbiol. 50 Suppl, 539–541.
Ehigiator H. N., Mcnair N., Mead J. R. (2007). Cryptosporidium parvum: the contribution of Th1-inducing pathways to the resolution of infection in mice. Exp. Parasitol. 115, 107–113.
Ehigiator H. N., Romagnoli P., Borgelt K., Fernandez M., Mcnair N., Secor W. E., et al. (2005). Mucosal cytokine and antigen-specific responses to Cryptosporidium parvum in IL-12p40 KO mice. Parasite Immunol. 27, 17–28.
El-Refai S. A., Helwa M. A., Rakha E. B., Atia A. F. (2021). Autophagy - related 16 - like 1 single nucleotide gene polymorphism increases the risk and severity of Cryptosporidium parvum infection. Mol. Biochem. Parasitol. 245, 111407.
Enriquez F. J., Sterling C. R. (1991). Cryptosporidium infections in inbred strains of mice. J. Protozool. 38, S100–S102.
Evans R. J., Sundaramurthy V., Frickel E. M. (2018). The interplay of host autophagy and eukaryotic pathogens. Front. Cell Dev. Biol. 6, 118.
Fayer R., Guidry A., Blagburn B. L. (1990). Immunotherapeutic efficacy of bovine colostral immunoglobulins from a hyperimmunized cow against cryptosporidiosis in neonatal mice. Infect. Immun. 58, 1962–2965.
Ferguson S. H., Foster D. M., Sherry B., Magness S. T., Nielsen D. M., Gookin J. L. (2019). Interferon-lambda3 promotes epithelial defense and barrier function against Cryptosporidium parvum infection. Cell. Mol. Gastroenterol. Hepatol. 8, 1–20.
Flanigan T. P. (1994). Human immunodeficiency virus infection and cryptosporidiosis: protective immune responses. Am. J. Trop. Med. Hyg. 50, 29–35.
Foster D. M., Stauffer S. H., Stone M. R., Gookin J. L. (2012). Proteasome inhibition of pathologic shedding of enterocytes to defend barrier function requires X-linked inhibitor of apoptosis protein and nuclear factor kappaB. Gastroenterology 143, 133–44.e4.
Frost F. J., Muller T. B., Calderon R. L., Craun G. F. (2004). Analysis of serological responses to Cryptosporidium antigen among NHANES III participants. Ann. Epidemiol. 14, 473–478.
Genta R. M., Chappell C. L., White A. C. Jr., Kimball K. T., Goodgame R. W. (1993). Duodenal morphology and intensity of infection in AIDS-related intestinal cryptosporidiosis. Gastroenterology 105, 1769–1775.
Gibson A. R., Sateriale A., Dumaine J. E., Engiles J. B., Pardy R. D., Gullicksrud J. A., et al. (2022). A genetic screen identifies a protective type III interferon response to Cryptosporidium that requires TLR3 dependent recognition. PloS Pathog. 18, e1010003.
Gomez Morales M. A., Ausiello C. M., Guarino A., Urbani F., Spagnuolo M. I., Pignata C., et al. (1996). Severe, protracted intestinal cryptosporidiosis associated with interferon gamma deficiency: pediatric case report. Clin. Infect. Dis. 22, 848–850.
Gomez Morales M. A., Ausiello C. M., Urbani F., Pozio E. (1995). Crude extract and recombinant protein of Cryptosporidium parvum oocysts induce proliferation of human peripheral blood mononuclear cells in vitro. J. Infect. Dis. 172, 211–216.
Gong A. Y., Hu G., Zhou R., Liu J., Feng Y., Soukup G. A., et al. (2011). MicroRNA-221 controls expression of intercellular adhesion molecule-1 in epithelial cells in response to Cryptosporidium parvum infection. Int. J. Parasitol. 41, 397–403.
Gookin J. L., Duckett L. L., Armstrong M. U., Stauffer S. H., Finnegan C. P., Murtaugh M. P., et al. (2004). Nitric oxide synthase stimulates prostaglandin synthesis and barrier function in C. parvum-infected porcine ileum. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G571–G581.
Gorla S. K., Mcnair N. N., Yang G., Gao S., Hu M., Jala V. R., et al. (2014). Validation of IMP dehydrogenase inhibitors in a mouse model of cryptosporidiosis. Antimicrob. Agents Chemother. 58, 1603–1614.
Griffiths J. K., Moore R., Dooley S., Keusch G. T., Tzipori S. (1994). Cryptosporidium parvum infection of caco-2 cell monolayers induces an apical monolayer defect, selectively increases transmonolayer permeability, and causes epithelial cell death. Infect. Immun. 62, 4506–4514.
Griffiths J. K., Theodos C., Paris M., Tzipori S. (1998). The gamma interferon gene knockout mouse: a highly sensitive model for evaluation of therapeutic agents against Cryptosporidium parvum. J. Clin. Microbiol. 36, 2503–2508.
Guesdon W., Auray G., Pezier T., Bussiere F. I., Drouet F., Le Vern Y., et al. (2015). CCL20 displays antimicrobial activity against Cryptosporidium parvum, but its expression is reduced during infection in the intestine of neonatal mice. J. Infect. Dis. 212, 1332–1340.
Guesdon W., Pezier T., Menard S., Nicolosi A., Le Vern Y., Silvestre A., et al. (2020). Cryptosporidium parvum subverts antimicrobial activity of CRAMP by reducing its expression in neonatal mice. Microorganisms (8):1635.
Gullicksrud J. A., Sateriale A., Engiles J. B., Gibson A. R., Shaw S., Hutchins Z. A., et al. (2022). Enterocyte-innate lymphoid cell crosstalk drives early IFN-gamma-mediated control of Cryptosporidium. Mucosal Immunol. 15, 362–372.
Harp J. A., Chen W., Harmsen A. G. (1992). Resistance of severe combined immunodeficient mice to infection with Cryptosporidium parvum: The importance of intestinal microflora. Infect. Immun. 60, 3509–3512.
Hasheminasab S. S., Conejeros I., Velásquez Z. D., Borggrefe T., Gartner U., Kamena F., et al. (2022). ATP purinergic receptor P2X1-dependent suicidal NETosis induced by Cryptosporidium parvum under physioxia conditions. Biol. (Basel) 11, 442.
Hayward A. R., Chmura K., Cosyns M. (2000). Interferon-gamma is required for innate immunity to Cryptosporidium parvum in mice. J. Infect. Dis. 182, 1001–1004.
Heine J., Moon H. W., Woodmansee D. B. (1984). Persistent Cryptosporidium infection in congenitally athymic (nude) mice. Infect. Immun. 43, 856–859.
He W., Li J., Gong A. Y., Deng S., Li M., Wang Y., et al. (2021). Cryptosporidial infection suppresses intestinal epithelial cell MAPK signaling impairing host anti-parasitic defense. Microorganisms 9, 151.
Heo I., Dutta D., Schaefer D. A., Iakobachvili N., Artegiani B., Sachs N., et al. (2018). Modelling Cryptosporidium infection in human small intestinal and lung organoids. Nat. Microbiol. 3, 814–823.
Hu G., Gong A. Y., Roth A. L., Huang B. Q., Ward H. D., Zhu G., et al. (2013). Release of luminal exosomes contributes to TLR4-mediated epithelial antimicrobial defense. PloS Pathog. 9, e1003261.
Jenkins M. C., O'brien C., Trout J., Guidry A., Fayer R. (1999). Hyperimmune bovine colostrum specific for recombinant Cryptosporidium parvum antigen confers partial protection against cryptosporidiosis in immunosuppressed adult mice. Vaccine 17, 2453–2460.
Joe A., Verdon R., Tzipori S., Keusch G. T., Ward H. D. (1998). Attachment of Cryptosporidium parvum sporozoites to human intestinal epithelial cells. Infect. Immun. 66, 3429–3432.
Karpe A. V., Hutton M. L., Mileto S. J., James M. L., Evans C., Shah R. M., et al. (2021). Cryptosporidiosis modulates the gut microbiome and metabolism in a murine infection model. Metabolites 11, 380.
Kassa M., Comby E., Lemeteil D., Brasseur P., Ballet J. J. (1991). Characterization of anti-Cryptosporidium IgA antibodies in sera from immunocompetent individuals and HIV-infected patients. J. Protozool. 38, S179–S180.
Keelaghan A. P., Charania R., Mead J. R. (2022). The effect of short-chain fatty acids on growth of Cryptosporidium parvum in vitro. Microorganisms 10, 1822.
Kelly P., Jack D. L., Naeem A., Mandanda B., Pollok R. C., Klein N. J., et al. (2000). Mannose-binding lectin is a component of innate mucosal defense against Cryptosporidium parvum in AIDS. Gastroenterology 119, 1236–1242.
Kirkpatrick B. D., Huston C. D., Wagner D., Noel F., Rouzier P., Pape J. W., et al. (2006). Serum mannose-binding lectin deficiency is associated with cryptosporidiosis in young Haitian children. Clin. Infect. Dis. 43, 289–294.
Korbel D. S., Barakat F. M., Di Santo J. P., Mcdonald V. (2011). CD4+ T cells are not essential for control of early acute Cryptosporidium parvum infection in neonatal mice. Infect. Immun. 79, 1647–1653.
Kotloff K. L., Nataro J. P., Blackwelder W. C., Nasrin D., Farag T. H., Panchalingam S., et al. (2013). Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the global enteric multicenter study, GEMS): A prospective, case-control study. Lancet 382, 209–222.
Kumar A., Chatterjee I., Anbazhagan A. N., Jayawardena D., Priyamvada S., Alrefai W. A., et al. (2018). Cryptosporidium parvum disrupts intestinal epithelial barrier function via altering expression of key tight junction and adherens junction proteins. Cell. Microbiol. 20, e12830.
Lacroix S., Mancassola R., Naciri M., Laurent F. (2001). Cryptosporidium parvum-specific mucosal immune response in C57BL/6 neonatal and gamma interferon-deficient mice: role of tumor necrosis factor alpha in protection. Infect. Immun. 69, 1635–1642.
Lantier L., Drouet F., Guesdon W., Mancassola R., Metton C., Lo-Man R., et al. (2014). Poly(I:C)-induced protection of neonatal mice against intestinal Cryptosporidium parvum infection requires an additional TLR5 signal provided by the gut flora. J. Infect. Dis. 209, 457–467.
Lantier L., Lacroix-Lamande S., Potiron L., Metton C., Drouet F., Guesdon W., et al. (2013). Intestinal CD103+ dendritic cells are key players in the innate immune control of Cryptosporidium parvum infection in neonatal mice. PloS Pathog. 9, e1003801.
Laurent F., Eckmann L., Savidge T. C., Morgan G., Theodos C., Naciri M., et al. (1997). Cryptosporidium parvum infection of human intestinal epithelial cells induces the polarized secretion of c-X-C chemokines. Infect. Immun. 65, 5067–5073.
Lean I. S., Lacroix-Lamande S., Laurent F., Mcdonald V. (2006). Role of tumor necrosis factor alpha in development of immunity against Cryptosporidium parvum infection. Infect. Immun. 74, 4379–4382.
Lean I. S., Mcdonald S. A., Bajaj-Elliott M., Pollok R. C., Farthing M. J., Mcdonald V. (2003). Interleukin-4 and transforming growth factor beta have opposing regulatory effects on gamma interferon-mediated inhibition of Cryptosporidium parvum reproduction. Infect. Immun. 71, 4580–4585.
Leav B. A., Yoshida M., Rogers K., Cohen S., Godiwala N., Blumberg R. S., et al. (2005). An early intestinal mucosal source of gamma interferon is associated with resistance to and control of Cryptosporidium parvum infection in mice. Infect. Immun. 73, 8425–8428.
Leitch G. J., He Q. (1994). Arginine-derived nitric oxide reduces fecal oocyst shedding in nude mice infected with Cryptosporidium parvum. Infect. Immun. 62, 5173–5176.
Leitch G. J., He Q. (1999). Reactive nitrogen and oxygen species ameliorate experimental cryptosporidiosis in the neonatal BALB/c mouse model. Infect. Immun. 67, 5885–5891.
Liakath F. B., Varatharajan S., Premkumar P. S., Syed C., Ward H., Kang G., et al. (2021). Toll-like receptors and mannose binding lectin gene polymorphisms associated with cryptosporidial diarrhea in children in southern India. Am. J. Trop. Med. Hyg. 105, 1706–1711.
Li J., Jin K., Li M., Mathy N. W., Gong A. Y., Deng S., et al. (2021). A host cell long noncoding RNA NR_033736 regulates type I interferon-mediated gene transcription and modulates intestinal epithelial anti-Cryptosporidium defense. PloS Pathog. 17, e1009241.
Liu J., Deng M., Lancto C. A., Abrahamsen M. S., Rutherford M. S., Enomoto S. (2009). Biphasic modulation of apoptotic pathways in Cryptosporidium parvum-infected human intestinal epithelial cells. Infect. Immun. 77, 837–849.
Liu J., Enomoto S., Lancto C. A., Abrahamsen M. S., Rutherford M. S. (2008). Inhibition of apoptosis in Cryptosporidium parvum-infected intestinal epithelial cells is dependent on survivin. Infect. Immun. 76, 3784–3792.
Mammeri M., Obregon D. A., Chevillot A., Polack B., Julien C., Pollet T., et al. (2020). Cryptosporidium parvum infection depletes butyrate producer bacteria in goat kid microbiome. Front. Microbiol. 11, 548737.
Mancassola R., Lacroix-Lamande S., Barrier M., Naciri M., Salmon H., Laurent F. (2004). Increased susceptibility of beta7-integrin-deficient neonatal mice in the early stage of Cryptosporidium parvum infection. Infect. Immun. 72, 3634–3637.
Marcial M. A., Madara J. L. (1986). Cryptosporidium: cellular localization, structural analysis of absorptive cell-parasite membrane-membrane interactions in guinea pigs, and suggestion of protozoan transport by m cells. Gastroenterology 90, 583–594.
Mathy N. W., Deng S., Gong A. Y., Li M., Wang Y., Burleigh O., et al. (2022). The long non-coding RNA nostrill regulates transcription of Irf7 through interaction with NF-kappaB p65 to enhance intestinal epithelial defense against Cryptosporidium parvum. Front. Immunol. 13, 863957.
McCole D. F., Eckmann L., Laurent F., Kagnoff M. F. (2000). Intestinal epithelial cell apoptosis following Cryptosporidium parvum infection. Infect. Immun. 68, 1710–1713.
McDonald V., Bancroft G. J. (1994). Mechanisms of innate and acquired resistance to Cryptosporidium parvum infection in SCID mice. Parasite Immunol. 16, 315–320.
McDonald V., Deer R., Uni S., Iseki M., Bancroft G. J. (1992). Immune responses to Cryptosporidium muris and Cryptosporidium parvum in adult immunocompetent or immunocompromised (nude and SCID) mice. Infect. Immun. 60, 3325–3331.
McDonald V., Korbel D. S., Barakat F. M., Choudhry N., Petry F. (2013). Innate immune responses against Cryptosporidium parvum infection. Parasite Immunol. 35, 55–64.
McDonald V., Pollok R. C., Dhaliwal W., Naik S., Farthing M. J., Bajaj-Elliott M. (2006). A potential role for interleukin-18 in inhibition of the development of Cryptosporidium parvum. Clin. Exp. Immunol. 145, 555–562.
McDonald V., Robinson H. A., Kelly J. P., Bancroft G. J. (1996). Immunity to Cryptosporidium muris infection in mice is expressed through gut CD4+ intraepithelial lymphocytes. Infect. Immun. 64, 2556–2562.
McKenney E. A., Greene L. K., Drea C. M., Yoder A. D. (2017). Down for the count: Cryptosporidium infection depletes the gut microbiome in coquerel's sifakas. Microb. Ecol. Health Dis. 28, 1335165.
McNair N. N., Bedi C., Shayakhmetov D. M., Arrowood M. J., Mead J. R. (2018). Inflammasome components caspase-1 and adaptor protein apoptosis-associated speck-like proteins are important in resistance to Cryptosporidium parvum. Microbes Infect. 20, 369–375.
Mead J. R., Arrowood M. J., Sidwell R. W., Healey M. C. (1991). Chronic Cryptosporidium parvum infections in congenitally immunodeficient SCID and nude mice. J. Infect. Dis. 163, 1297–1304.
Mead J. R., You X. (1998). Susceptibility differences to Cryptosporidium parvum infection in two strains of gamma interferon knockout mice. J. Parasitol. 84, 1045–1048.
Mele R., Gomez Morales M. A., Tosini F., Pozio E. (2004). Cryptosporidium parvum at different developmental stages modulates host cell apoptosis in vitro. Infect. Immun. 72, 6061–6067.
Ming Z., Gong A. Y., Wang Y., Zhang X. T., Li M., Dolata C. E., et al. (2018). Trans-suppression of defense DEFB1 gene in intestinal epithelial cells following Cryptosporidium parvum infection is associated with host delivery of parasite Cdg7_FLc_1000 RNA. Parasitol. Res. 117, 831–840.
Ming Z., Zhou R., Chen X. M. (2017). Regulation of host epithelial responses to Cryptosporidium infection by microRNAs. Parasite Immunol. 39.
Muniz-Feliciano L., Van Grol J., Portillo J. A., Liew L., Liu B., Carlin C. R., et al. (2013). Toxoplasma gondii-induced activation of EGFR prevents autophagy protein-mediated killing of the parasite. PloS Pathog. 9, e1003809.
Munoz-Caro T., Lendner M., Daugschies A., Hermosilla C., Taubert A. (2015). NADPH oxidase, MPO, NE, ERK1/2, p38 MAPK and Ca2+ influx are essential for Cryptosporidium parvum-induced NET formation. Dev. Comp. Immunol. 52, 245–254.
Nice T. J., Robinson B. A., Van Winkle J. A. (2018). The role of interferon in persistent viral infection: Insights from murine norovirus. Trends Microbiol. 26, 510–524.
O'Hara S. P., Bogert P. S., Trussoni C. E., Chen X., Larusso N. F. (2011). TLR4 promotes Cryptosporidium parvum clearance in a mouse model of biliary cryptosporidiosis. J. Parasitol. 97, 813–821.
Odendall C., Voak A. A., Kagan J. C. (2017). Type III IFNs are commonly induced by bacteria-sensing TLRs and reinforce epithelial barriers during infection. J. Immunol. 199, 3270–3279.
Oliveira B. C. M., Bresciani K. D. S., Widmer G. (2019). Deprivation of dietary fiber enhances susceptibility of mice to cryptosporidiosis. PloS Negl. Trop. Dis. 13, e0007411.
Oliveira B. C. M., Widmer G. (2018). Probiotic product enhances susceptibility of mice to cryptosporidiosis. Appl. Environ. Microbiol. 84.
Olsen L., Akesson C. P., Storset A. K., Lacroix-Lamande S., Boysen P., Metton C., et al. (2015). The early intestinal immune response in experimental neonatal ovine cryptosporidiosis is characterized by an increased frequency of perforin expressing NCR1(+) NK cells and by NCR1(-) CD8(+) cell recruitment. Veterinary Res. 46, 28.
Ong C. S., Li A. S., Priest J. W., Copes R., Khan M., Fyfe M. W., et al. (2005). Enzyme immunoassay of Cryptosporidium-specific immunoglobulin G antibodies to assess longitudinal infection trends in six communities in British Columbia, Canada. Am. J. Trop. Med. Hyg. 73, 288–295.
Onizuka Y., Takahashi C., Uematsu A., Shinjo S., Seto E., Nakajima-Shimada J. (2017). Inhibition of autolysosome formation in host autophagy by Trypanosoma cruzi infection. Acta Trop. 170, 57–62.
Pantenburg B., Castellanos-Gonzalez A., Dann S. M., Connelly R. L., Lewis D. E., Ward H. D., et al. (2010). Human CD8(+) T cells clear Cryptosporidium parvum from infected intestinal epithelial cells. Am. J. Trop. Med. Hyg. 82, 600–607.
Perez-Cordon G., Yang G., Zhou B., Nie W., Li S., Shi L., et al. (2014). Interaction of Cryptosporidium parvum with mouse dendritic cells leads to their activation and parasite transportation to mesenteric lymph nodes. Pathog. Dis. 70, 17–27.
Petersen C., Gut J., Doyle P. S., Crabb J. H., Nelson R. G., Leech J. H. (1992). Characterization of a > 900,000-m(r) Cryptosporidium parvum sporozoite glycoprotein recognized by protective hyperimmune bovine colostral immunoglobulin. Infect. Immun. 60, 5132–5138.
Petry F., Jakobi V., Wagner S., Tessema T. S., Thiel S., Loos M. (2008). Binding and activation of human and mouse complement by Cryptosporidium parvum (Apicomplexa) and susceptibility of C1q- and MBL-deficient mice to infection. Mol. Immunol. 45, 3392–3400.
Pickerd N., Tuthill D. (2004). Resolution of cryptosporidiosis with probiotic treatment. Postgrad. Med. J., 80, 112–113.
Pollok R. C., Farthing M. J., Bajaj-Elliott M., Sanderson I. R., Mcdonald V. (2001). Interferon gamma induces enterocyte resistance against infection by the intracellular pathogen Cryptosporidium parvum. Gastroenterology 120, 99–107.
Ponnuraj E. M., Hayward A. R. (2001). Intact intestinal mRNAs and intestinal epithelial cell esterase, but not Cryptosporidium parvum, reach mesenteric lymph nodes of infected mice. J. Immunol. 167, 5321–5328.
Portillo J. C., Muniz-Feliciano L., Lopez Corcino Y., Lee S. J., Van Grol J., Parsons S. J., et al. (2017). Toxoplasma gondii induces FAK-Src-STAT3 signaling during infection of host cells that prevents parasite targeting by autophagy. PloS Pathog. 13, e1006671.
Potiron L., Lacroix-Lamande S., Marquis M., Levern Y., Fort G., Franceschini I., et al. (2019). Batf3-dependent intestinal dendritic cells play a critical role in the control of Cryptosporidium parvum infection. J. Infect. Dis. 219, 925–935.
Priest J. W., Li A., Khan M., Arrowood M. J., Lammie P. J., Ong C. S., et al. (2001). Enzyme immunoassay detection of antigen-specific immunoglobulin g antibodies in longitudinal serum samples from patients with cryptosporidiosis. Clin. Diagn. Lab. Immunol. 8, 415–423.
Priyamvada S., Jayawardena D., Bhalala J., Kumar A., Anbazhagan A. N., Alrefai W. A., et al. (2021). Cryptosporidium parvum infection induces autophagy in intestinal epithelial cells. Cell. Microbiol. 23, e13298.
Rahman S. U., Zhou K., Zhou S., Sun T., Mi R., Huang Y., et al. (2022). Curcumin mitigates Cryptosporidium parvum infection through modulation of gut microbiota and innate immune-related genes in immunosuppressed neonatal mice. Microb. Pathog. 164, 105424.
Ras R., Huynh K., Desoky E., Badawy A., Widmer G. (2015). Perturbation of the intestinal microbiota of mice infected with Cryptosporidium parvum. Int. J. Parasitol. 45, 567–573.
Ren X., Zhao J., Zhang L., Ning C., Jian F., Wang R., et al. (2011). Cryptosporidium tyzzeri n. sp. (Apicomplexa: Cryptosporidiidae) in domestic mice (Mus musculus). Exp. Parasitol. 130, 274–281.
Riggs M. W., Mcguire T. C., Mason P. H., Perryman L. E. (1989). Neutralization-sensitive epitopes are exposed on the surface of infectious Cryptosporidium parvum sporozoites. J. Immunol. 143, 1340–1345.
Robinson P., Okhuysen P. C., Chappell C. L., Lewis D. E., Shahab I., Lahoti S., et al. (2000). Transforming growth factor beta1 is expressed in the jejunum after experimental Cryptosporidium parvum infection in humans. Infect. Immun. 68, 5405–5407.
Rogers K. A., Rogers A. B., Leav B. A., Sanchez A., Vannier E., Uematsu S., et al. (2006). MyD88-dependent pathways mediate resistance to Cryptosporidium parvum infection in mice. Infect. Immun. 74, 549–556.
Rohlman V. C., Kuhls T. L., Mosier D. A., Crawford D. L., Greenfield R. A. (1993). Cryptosporidium parvum infection after abrogation of natural killer cell activity in normal and severe combined immunodeficiency mice. J. Parasitol. 79, 295–297.
Russler-Germain E. V., Jung J., Miller A. T., Young S., Yi J., Wehmeier A., et al. (2021). Commensal Cryptosporidium colonization elicits a cDC1-dependent Th1 response that promotes intestinal homeostasis and limits other infections. Immunity 54, 2547–2564.e7.
Sacco R. E., Haynes J. S., Harp J. A., Waters W. R., Wannemuehler M. J. (1998). Cryptosporidium parvum initiates inflammatory bowel disease in germfree T cell receptor-alpha-deficient mice. Am. J. Pathol. 153, 1717–1722.
Sateriale A., Gullicksrud J. A., Engiles J. B., Mcleod B. I., Kugler E. M., Henao-Mejia J., et al. (2021). The intestinal parasite Cryptosporidium is controlled by an enterocyte intrinsic inflammasome that depends on NLRP6. Proc. Natlonal Acad. Sci. U.S.A. 118, e2007807118.
Sateriale A., Slapeta J., Baptista R., Engiles J. B., Gullicksrud J. A., Herbert G. T., et al. (2019). A genetically tractable, natural mouse model of cryptosporidiosis offers insights into host protective immunity. Cell Host Microbe 26, 135–146.e5.
Shikani H., Weiss L. M. (2014). “Human cryptosporidiosis: A clinical perspective,” in Cryptosporidium: parasite and disease. Eds. Cacciò S. M., Widmer G. (Vienna: Springer Vienna).
Sindhu K. N., Sowmyanarayanan T. V., Paul A., Babji S., Ajjampur S. S., Priyadarshini S., et al. (2014). Immune response and intestinal permeability in children with acute gastroenteritis treated with Lactobacillus rhamnosus GG: a randomized, double-blind, placebo-controlled trial. Clin. Infect. Dis. 58, 1107–1115.
Stiehm E. R., Chin T. W., Haas A., Peerless A. G. (1986). Infectious complications of the primary immunodeficiencies. Clin. Immunol. Immunopathol. 40, 69–86.
Takeuchi D., Jones V. C., Kobayashi M., Suzuki F. (2008). Cooperative role of macrophages and neutrophils in host antiprotozoan resistance in mice acutely infected with Cryptosporidium parvum. Infect. Immun. 76, 3657–3663.
Theodos C. M., Sullivan K. L., Griffiths J. K., Tzipori S. (1997). Profiles of healing and nonhealing Cryptosporidium parvum infection in C57BL/6 mice with functional b and T lymphocytes: the extent of gamma interferon modulation determines the outcome of infection. Infect. Immun. 65, 4761–4769.
Ungar B. L. P., Soave R., Fayer R., Nash T. E. (1986). Enzyme immunoassay detection of immunoglobulin m and G antibodies to Cryptosporidium in immunocompetent and immunocompromised persons. J. Infect. Dis. 153, 570–578.
Urban J. F. Jr., Fayer R., Chen S. J., Gause W. C., Gately M. K., Finkelman F. D. (1996). IL-12 protects immunocompetent and immunodeficient neonatal mice against infection with Cryptosporidium parvum. J. Immunol. 156, 263–268.
VanDussen K. L., Funkhouser-Jones L. J., Akey M. E., Schaefer D. A., Ackman K., Riggs M. W., et al. (2020). Neonatal mouse gut metabolites influence Cryptosporidium parvum infection in intestinal epithelial cells. mBio 11, e02582-20.
Victorino F., Sojka D. K., Brodsky K. S., Mcnamee E. N., Masterson J. C., Homann D., et al. (2015). Tissue-resident NK cells mediate ischemic kidney injury and are not depleted by anti-asialo-GM1 antibody. J. Immunol. 195, 4973–4985.
Wang Y., Gong A. Y., Ma S., Chen X., Li Y., Su C. J., et al. (2017). Delivery of parasite RNA transcripts into infected epithelial cells during Cryptosporidium infection and its potential impact on host gene transcription. J. Infect. Dis. 215, 636–643.
Wang Y., Li X., Chen X., Kulyar M. F., Duan K., Li H., et al. (2022). Gut fungal microbiome responses to natural Cryptosporidium infection in horses. Front. Microbiol. 13, 877280.
Wang C., Liu L., Zhu H., Zhang L., Wang R., Zhang Z., et al. (2019a). MicroRNA expression profile of HCT-8 cells in the early phase of Cryptosporidium parvum infection. BMC Genom. 20, 37.
Wang Y., Shen Y., Liu H., Yin J., Zhang X. T., Gong A. Y., et al. (2019b). Induction of inflammatory responses in splenocytes by exosomes released from intestinal epithelial cells following Cryptosporidium parvum infection. Infect. Immun. 87, e00705-18.
Wilke G., Funkhouser-Jones L. J., Wang Y., Ravindran S., Wang Q., Beatty W. L., et al. (2019). A stem-cell-derived platform enables complete Cryptosporidium development in vitro and genetic tractability. Cell Host Microbe 26, 123–134.e8.
Winkelstein J. A., Marino M. C., Ochs H., Fuleihan R., Scholl P. R., Geha R., et al. (2003). The X-linked hyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine 82, 373–384.
Wolska-Kusnierz B., Bajer A., Caccio S., Heropolitanska-Pliszka E., Bernatowska E., Socha P., et al. (2007). Cryptosporidium infection in patients with primary immunodeficiencies. J. Pediatr. Gastroenterol. Nutr. 45, 458–464.
Xia Z., Xu J., Lu E., He W., Deng S., Gong A. Y., et al. (2021). m6A mRNA methylation regulates epithelial innate antimicrobial defense against cryptosporidial infection. Front. Immunol. 12, 705232.
Xie F., Zhang Y., Li J., Sun L., Zhang L., Qi M., et al. (2022). MiR-942-5p targeting the IFI27 gene regulates HCT-8 cell apoptosis via a TRAIL-dependent pathway during the early phase of Cryptosporidium parvum infection. Parasitol. Vectors 15, 291.
Zaalouk T. K., Bajaj-Elliott M., George J. T., Mcdonald V. (2004). Differential regulation of beta-defensin gene expression during Cryptosporidium parvum infection. Infect. Immun. 72, 2772–2779.
Zadrozny L. M., Stauffer S. H., Armstrong M. U., Jones S. L., Gookin J. L. (2006). Neutrophils do not mediate the pathophysiological sequelae of Cryptosporidium parvum infection in neonatal piglets. Infect. Immun. 74, 5497–5505.
Zhang X. T., Gong A. Y., Wang Y., Chen X., Lim S. S., Dolata C. E., et al. (2016). Cryptosporidium parvum infection attenuates the ex vivo propagation of murine intestinal enteroids. Physiol. Rep. 4, e13060.
Zhang G., Zhang Y., Niu Z., Wang C., Xie F., Li J., et al. (2020). Cryptosporidium parvum upregulates miR-942-5p expression in HCT-8 cells via TLR2/TLR4-NF-kappaB signaling. Parasitol. Vectors 13, 435.
Keywords: Cryptosporidium, innate immunity, dendritic cells, interferon, epithelial cells, TLRs, inflammasomes, cytokines
Citation: Mead JR (2023) Early immune and host cell responses to Cryptosporidium infection. Front. Parasitol. 2:1113950. doi: 10.3389/fpara.2023.1113950
Received: 01 December 2022; Accepted: 13 January 2023;
Published: 27 January 2023.
Edited by:
Danielle Stanisic, Griffith University, AustraliaReviewed by:
Attinder Chadha, University of California, San Francisco, United StatesCopyright © 2023 Mead. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan R. Mead, am1lYWRAZW1vcnkuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.