 Jahnavi Gollamudi1*
Jahnavi Gollamudi1* Kristine A. Karkoska1
Kristine A. Karkoska1 Oluwabukola T. Gbotosho2
Oluwabukola T. Gbotosho2 Wei Zou3
Wei Zou3 Hyacinth I. Hyacinth2
Hyacinth I. Hyacinth2 Steven L. Teitelbaum3
Steven L. Teitelbaum3
- 1Division of Hematology/Oncology, Department of Internal Medicine, University of Cincinnati College of Medicine, Cincinnati, OH, United States
- 2Department of Neurology and Rehabilitation Medicine, University of Cincinnati College of Medicine, Cincinnati, OH, United States
- 3Department of Medicine, Division of Bone and Mineral Diseases, and Department of Pathology and Immunology, Division of Anatomic and Molecular Pathology, Washington University School of Medicine, St. Louis, MO, United States
The bone is one of the most commonly affected organs in sickle cell disease (SCD). Repeated ischemia, oxidative stress and inflammation within the bone is largely responsible for promoting bone pain. As more individuals with SCD survive into adulthood, they are likely to experience a synergistic impact of both aging and SCD on their bone health. As bone health deteriorates, bone pain will likely exacerbate. Recent mechanistic and observational studies emphasize an intricate relationship between bone remodeling and the peripheral nervous system. Under pathological conditions, abnormal bone remodeling plays a key role in the propagation of bone pain. In this review, we first summarize mechanisms and burden of select bone complications in SCD. We then discuss processes that contribute to pathological bone pain that have been described in both SCD as well as non-sickle cell animal models. We emphasize the role of bone-nervous system interactions and pitfalls when designing new therapies especially for the sickle cell population. Lastly, we also discuss future basic and translational research in addressing questions about the complex role of stress erythropoiesis and inflammation in the development of SCD bone complications, which may lead to promising therapies and reduce morbidity in this vulnerable population.
Introduction
Sickle cell disease (SCD) is a monogenic red cell disorder that affects over 100,000 people in the United States and millions more worldwide (1, 2). A single base pair substitution at the sixth amino acid position of the beta globin gene, results in mutated hemoglobin (known as hemoglobin S, HbS) which polymerizes and changes the red cell shape (“sickle”) under hypoxic conditions. The hallmark complication of SCD is pain due to aggregation of sickled red cells within the microvasculature of the long bones, resulting in ischemia, and pain.
In addition to acute pain, repeated cycles of ischemia and infarction contribute to sickle cell bone disease (SBD). SBD refers to a combination of pathologies such as osteonecrosis, low bone mineral density, vertebral bone deformities, pathological fractures and osteomyelitis (3). SBD can contribute to chronic pain. For example, presence of osteonecrosis is associated with increased hospitalizations for vaso-occlusive episodes (VOE) requiring parenteral opiates (4).
As more individuals with SCD survive into adulthood, they are likely to experience a synergistic impact of both aging and sickle cell disease on their bone health. As bone complications accrue, the prevalence of chronic pain is also likely to increase (5). While opiates and non-steroidal anti-inflammatory drugs help with acute pain, these agents are less effective in treating chronic pain (6). Given the adverse effects of opiates including dependence (7), it is critical to understand the pathogenesis of SBD to catalyze the development of alternatives to opiates. Recently, several animal and clinical studies have elucidated contributors to the pathogenesis of chronic bone pain. While most of the studies are in non-SCD models, they may still be pertinent in SCD. In this review, we first provide a brief overview of select bone complications that result in chronic pain, and summarize mechanisms of bone pain, drawing particular attention to the role of bone remodeling, afferent nerve sensitization, and nerve sprouting. Finally, we briefly discuss tentative therapies that may have a role in bone pain in SCD.
Low bone mineral density
One of the most common bone complications of SCD is low bone mineral density (BMD) or low bone mass (3). Low bone mass is observed in more than 50% of children (8, 9) as well as over 70% of adults with SCD (10, 11). Furthermore, in children, low BMD associates with an increased risk of avascular necrosis (AVN) and chronic pain (9). In adults with SCD, low bone mass has been strongly linked to risk of fractures at an early age (12).
Mechanistic studies show low BMD in SCD is due to both accelerated bone loss and poor bone formation. Animal studies show increased marrow osteoclast precursors numbers and activity which is responsible for bone loss (13). In addition, defective terminal osteoblast differentiation results in poor bone formation (13–16). Markers of increased osteoclast activity such as tartrate-resistant acid phosphatase (TRAP) type 5b, have also been observed in sera of individuals with SCD (17–19). Preclinical, and clinical studies show osteoclast activation associates with both pathological bone loss and chronic bone pain (20–23). Interestingly, children with SCD and low BMD had improvement in chronic pain after receiving bisphosphates, an anti-resorptive medication, highlighting the role of OC activity in both low BMD and pain (24).
Fractures
Fractures are common n in children and young adults with SCD (11, 24). In one study, 46% of adults had evidence of vertebral fractures (11, 12). The mean age of adults with fractures is often less than 40 years (11, 12). Fractures can be seen in upper extremities, vertebral, pelvic, and femoral bone (11, 12, 25, 26). Risk factors for developing fractures, in addition to male sex and low vitamin D levels (27), include elevated lactate dehydrogenase (LDH) and elevated aspartate aminotransferase (AST) (12), suggesting a role for hemolysis. There was a lack of correlation with bone mineral density (11, 28). Furthermore, transfusions did not affect the risk of fractures (27). Presence of fractures and abnormal healing from fractures can worsen pre-existing pain and contribute to chronic pain (29). The current treatment for fractures is mostly conservative and surgeries such as laminectomy are reserved for when conservative management fails (30, 31).
Mechanistically, decreased bone strength results from loss of cortical, trabecular, and abnormal bone matrix composition known as bone quality (32). In two separate studies, the authors show femurs from sickle mice required less force to deform and had lower capacity to sustain high mechanical stress suggesting increased fragility and risk for fractures (14, 15). As in humans, there is a lack of correlation with BMD. This suggests that current imaging methods assessing BMD may be insufficient to inform fracture risk and capture complex changes in bone quality (33).
Avascular necrosis
Avascular necrosis or osteonecrosis is the second most common chronic complication of SCD (4). About 10%–22% of patients with SCD are affected; however, in reality, prevalence is likely to be much higher (17, 34).
Avascular necrosis results from a disruption of the blood supply to the ends of long bones, which results in death of bone tissue (35). In SCD, femoral or humeral heads are commonly affected. The femoral head is particularly susceptible to ischemia as it lacks a collateral blood supply. Risk factors include older age, male sex, high hemoglobin, body mass index, leukopenia, frequent VOEs, and a history of acute chest syndrome (36). Furthermore, genetic polymorphisms in genes with roles in vascular integrity, inflammation, and oxidant stress, such as Klotho, bone-morphogenic protein 6 (BMP-6), and annexin 2 (ANX2), confer an increased risk of AVN in SCD (37). While initially asymptomatic, AVN can rapidly progress to collapse and narrowing of the joint space (38), particularly in SCD. Treatment for AVN includes improving vitamin D status, physical therapy and/or surgical interventions, however the optimal treatment modality for early AVN is still debated (39).
Bone remodeling, innervation, and its functions
Bone is a dynamic tissue maintained in homeostasis by opposing actions of bone resorption and formation. The cells responsible for this homeostasis include osteoclasts, osteocytes, and osteoblasts (40). Osteoclasts arise from a myeloid lineage (32). Osteoblasts arise from mesenchymal stromal cells (MSC) and are primarily involved in bone formation at sites resorbed by osteoclasts (40). Osteocytes are terminally differentiated osteoblasts that form the bone matrix (40). The balance of bone resorption and formation is governed by cytokines, local bone environment and precise crosstalk between osteoclasts, osteoblasts, and osteocytes (41). For example, osteoblasts regulate formation, activation, and maturation of multinucleated osteoclasts from their precursors by secreting macrophage colony stimulating factor (M-CSF), receptor activator of nuclear factor-κB ligand (RANKL) and osteoprotegerin (OPG) (32). Osteocytes, which are the principal source of RANKL, send precise signals which allows for osteoclasts to resorb bone at designated locations (42). Osteoclasts can then resorb bone by secreting enzymes such as cathepsin K and hydrogen ions (41).
Bone is richly innervated (43). The skeletal nerves can be classified as sensory, sympathetic, or parasympathetic nerves (44). The density of the nerves is highest in the periosteum followed by bone marrow and lowest in mineralized bone (45). Sensory or afferent nerves are either thinly myelinated (A-delta) or unmyelinated (C-fibers) and transmit pain from bone and bone marrow to the dorsal root ganglion (DRG) (46–48). Together, A-delta and C-fibers account for most of the pain-transducing nerves in the bone (43). Specifically in SCD, skeletal pathologies such as fractures, marrow infarction, and inflammation may play a role in the initiation and propagation of bone pain.
Noxious stimuli activate pain receptors on sensory neurons such as acid-sensing ion channel 3 (ASIC3), transient receptor potential channel vanilloid subfamily member 1 (TRPV1), tetrodotoxin (TTX)-resistant sodium channels (Nav.1.8), purinergic receptor (P2X3), endothelin receptor (ETAR), prostaglandin (PG) among others (22, 49). Upon activation of these receptors, neurotransmitters such as calcitonin gene–related peptide (CGRP), substance P (SP), glutamate and pituitary adenylate cyclase-activating polypeptide (PACAP) are released from nerve terminals in the DRG (45, 50, 51). Skeletal afferent neurons also express tropomyosin receptor kinase A positive (TrkA+) (52). In fact, the majority of bone-innervating sensory fibers are CGRP and TrkA positive C-fiber neurons (53). CGRP and substance P signaling also affects bone mass (54, 55). For example, mice lacking CGRP have low bone mass, due to loss of CGRP mediated effects on osteoblasts (54, 56). Bone resorption and formation are also dependent on both sympathetic and parasympathetic signaling (44, 57). Osteoclasts and osteoblasts express α- and β-adrenergic receptors which are activated when bound to norepinephrine (NE) (58) which favors bone resorption in mice and humans (58, 59). As is the case with sensory nerves, neurotransmitters from sympathetic nerves can also modulate bone generation. Neuropeptide Y via its action on hypothalamic Y2 receptors has been shown to exert a negative effect on osteoblast activity and bone formation (60, 61). Of note, osteoblasts and to some extent osteoclasts, also secrete endocannabinoids such as 2-arachidonylglycerol (2-AG), which can reduce sympathetic signaling via CB1 receptors on skeletal sympathetic nerve endings (62). Lastly, neuropeptides from sensory nerves such as CGRP can also affect sympathetic signaling (63). Thus, there is an intricate and complex relationship between bone cells, the sensory and autonomic nervous systems. This has implications for therapies to treat bone pain, as complete inhibition of neuro-receptors may have unintended consequences on bone formation or resorption.
Emerging data suggests bone pain is due to pathologic remodeling, exuberant neurotransmitter release, and ectopic nerve growth. These abnormal changes occur with age, stress and/or systemic neuroinflammation, all of which are at play in SCD (22, 64, 65) (Figure 1). In the following sections, we provide evidence for these processes in bone pain described in SCD and non SCD models.
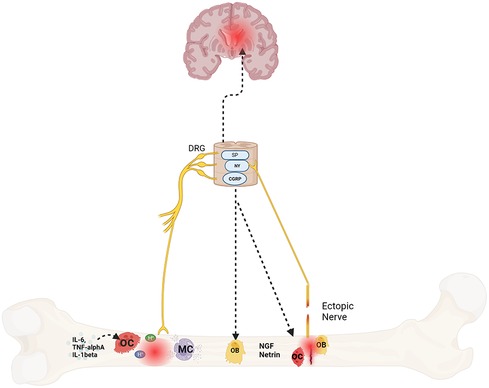
Figure 1. Mechanisms of bone pain in sickle cell disease. Protons secreted by osteoclasts in response to cytokines, fractures, or ischemia triggers afferent nerve signaling via nociceptors like acid-sensing ion channel 3 (ASIC3) and the transient receptor potential channel vanilloid subfamily member 1 (TRPV1) which communicate to the brain and spinal cord via neurotransmitters such as substance P (SP), neuropeptide Y (NY) and calcitonin gene–related peptide (CGRP). These neurotransmitters also sensitize the afferent nerves. In addition, they also play a role in bone formation and resorption as well. Lastly, mast cells (MC), osteoclasts and osteoblasts also release nerve growth factor (NGF) and netrin respectively that promotes ectopic nerve growth which further contributes to pain. Created with BioRender.com.
Bone remodeling and bone pain
In SCD, chronic and acute inflammation results in elevated levels of circulating interleukin 6 (IL-6), interleukin 1 beta (IL-1β), and tumor necrosis factor alpha (TNF-α) (66). These cytokines promote osteoclastogenesis (67–69). Tumor necrosis factor alpha, for example, can directly increase osteoclast activity with minimal RANKL secretion (67–69). The increased osteoclastic activity favors accelerated bone resorption. Osteoclasts resorb bone by acidifying their immediate bone environment (41). In addition to mobilizing minerals, low pH (acid) is a noxious stimulus, which is sensed by nociceptors such as ASICs and 1TRPV1 (51, 70). In preclinical models, a reduction in osteoclast activity was correlated with a decrease in ASICs and TRPV1 expression, activation of neurons in DRG and decreased pain behaviors (21). Furthermore, in this study and others, inhibition of ASIC3 and TRPV1 resulted in decreased pain and biomarkers for early osteoblast differentiation (71, 72). This is an important point as prolonged inhibition may potentially result in decreased bone formation and delayed fracture healing (72). Lastly, complete abrogation of these receptors, may have unintended side effects. For example, in an animal model of SCD, complete abrogation of TRPV1 resulted in worsening of VOEs by attenuating vasodilatory effects of CGRP (73).
In addition to inhibiting nociceptors, limiting osteoclast activity also reduces bone pain. Numerous studies in cancer and osteoporosis models show that bisphosphonates, which reduce osteoclast activity, improve bone pain (74–76). Pre-clinical studies in mouse models of SCD also show that bisphosphonates reduce markers of bone turnover and improve bone microarchitecture (13). A recent study in 46 children with SCD-related skeletal morbidity showed improvement in chronic pain after treatment with intravenous bisphosphates, highlighting the possible interaction between osteoclast activation, abnormal bone remodeling, and the development of bone pain (24). An ongoing prospective study is evaluating the effect of bisphosphonates on bone pain in adults with SCD (NCT05283148).
Afferent nerve sensitization and bone pain
A vast majority of the skeletal nerves express TrKA, whose ligand is nerve growth factor (NGF) (52). NGF promotes proliferation and survival of sympathetic and sensory afferent neurons during development (77). TrKA/NGF signaling also plays a critical role in bone fracture repair (78). However, abnormal TrkA/NGF signaling in the setting of exuberant inflammation results in peripheral nerve sensitization. Experimental evidence has shown that NGF is also released by mast cells (79, 80), lymphocytes (81), and monocytes/macrophages (82) in response to tissue inflammation. NGF propagates peripheral nerve sensitization through several mechanisms: (1) NGF binding to TrkA + neurons increases neuronal excitability by altering the activity of different ion channels in the nerve ending (83), which results in a lower threshold for neuronal depolarization. (2) NGF/TrKA complexes promote gene expression of nociceptors such as TRPV1, ASIC, Nav1.8 (sodium) channels, CaV (calcium channels), in addition to release of neuropeptides such as CGRP, and substance P, which can, in turn, increase the sensitivity of the afferent neuron to NGF (84). and (3) NGF induces inflammatory cells to release bradykinin, histamine, ATP, serotonin, and protons, which in turn activate receptors and ion channels thus propagating the pain feedback loop (85).
In rodent models of osteoarthritis or fracture repair, an increase in NGF levels associated with increased pain behaviors (86, 87). Furthermore, local, and systemic NGF administration into healthy humans induces deep pain and hypersensitivity that can last for several days (88, 89). Also, individuals with SCD and chronic pain also show elevated circulating levels of NGF (90). Thus, taken together, therapies that affect inflammatory cell release of NGF or inhibit NGF may be beneficial in decreasing bone pain.
Evidence from SCD and non-SCD models (91, 92) corroborate inhibition of NGF as a target to reduce pain. A murine model of SCD underscored the importance of mast cell activation, infiltration and NGF in pain hypersensitivity by showing that tryptase, secreted by mast cells, promotes release of substance P and CGRP in DRG (targets of NGF), and inhibition of mast cell activation by imatinib resulted in reduced musculoskeletal pain behaviors and reduced activation of neurons in the DRG (92). Animal models of osteoarthritis also show that increased mast cell activation and numbers resulted in worsening inflammation and pain which can be reversed with mast cell inhibition. Thus, mast cell inhibition may help with bone pain (93) and is a potential therapeutic target. Therapies such as monoclonal antibodies against NGF (94) showed improvement in OA induced pain in preclinical and clinical trials (95). However, clinical trials were halted early due to reports of rapidly progressive OA and of osteonecrosis among individuals receiving these agents. Preclinical studies suggest it could be due to autonomic dysfunction or decreased angiogenesis (52, 96) however, some studies speculate that it could be the natural progression of the disease itself (91).
Nerve growth sprouting and bone pain
Bone innervation and bone remodeling are closely linked (44). Physiologically, nerve growth is an important signal for ossification of newly formed bone (44). However, under pathological states, excessive nerve sprouting can result in pain hypersensitivity. Preclinical models of fractures or low back pain show that osteoblasts and osteoclasts secrete NGF and Netrin respectively, which are key proteins propagating nerve growth (78, 97, 98). In mammals, Netrins are either secreted (Netrin-1, -3, -4, and -5) or anchored to cell membrane (Netrin-G1 and -G2) (99–101). Netrin-1 is responsible for neuronal migration as well as axonal growth (102, 103). In addition to guiding neurons, Netrin-1 can also guide leukocytes to the site of injury and promote macrophage differentiation (103, 104). It is plausible netrin levels may be affected in SCD. To date, no study has surveyed the levels as well as impact of Netrin on chronic pain in preclinical models or patients with SCD.
In non-SCD model of low back pain, osteoclasts secrete Netrin-1 which supports the ingrowth of CGRP positive nerve fibers at the endplates of vertebral bodies. Elimination of osteoclasts attenuated pain behaviors (97). In an osteoarthritis model, osteoclasts were also shown to secrete Netrin-1 that induces growth of CGRP positive nerves and DRG activation (98). More importantly, both the genetic and pharmacological blockade of Netrin1 reduce pain behaviors (98) suggesting an important role for Netrin-1 inhibitors in bone pain. It is interesting to note that therapies affecting global netrin inhibition may affect other cell lines as netrin is important for hematopoietic stem cell (HSC) quiescence, and self-renewal (105). This is important as SCD is characterized by exuberant increase in HSC cells due to chronic stress erythropoiesis and inflammation resulting in premature aging of the HSC system (106).
Future directions
Bone is a dynamic organ. Bone formation and absorption are influenced by local as well as systemic factors including inflammation (107). In SCD, chronic hemolysis, inflammation, delayed puberty, and poor muscle mass may contribute to excessive bone remodeling and loss (3). Thus, an understanding of mechanisms leading to bone loss is important to understand bone pain.
Questions that still need answering include how stress erythropoiesis contributes to bone remodeling and pain. Individuals with thalassemia, a red cell disorder characterized by defective beta or alpha globin production and ineffective erythropoiesis, also exhibit low bone mass, increased risk of fractures, and bone pain (108). Erythropoiesis can also stimulate myelopoiesis, which in the right bone microenvironment, differentiate to osteoclasts (109). Marrow expansion can also activate mechanosensitive PIEZO receptors which may transduce pain (47). Other questions include if chronic hemolysis also contributes to osteoclastogenesis. In SCD, hemolysis results in release of free heme, which contributes to reactive oxidative stress (ROS) (110). In response to ROS, cells lining the blood vessels express heme oxygenase (HO-1) which attenuates inflammation. Non-SCD models show HO-1 regulates osteoclastogenesis by inhibiting RANKL- induced osteoclastic differentiation (111). Ferroptosis, iron induced cell death, could also play a role in osteoclastogenesis, however further research is needed (112, 113).
Effective therapies against bone pain also have to balance their targets precisely, such that bone health is not impaired. For example, CGRP inhibitors, used effectively against migraines, may help with bone pain, however it could mediate increased vaso-occlusive crisis seen in animal models of SCD (73, 114). Thus, there is an unmet need for therapies that treat bone pain that are safe and do not further impact bone health in SCD.
Conclusion
Bone complications and bone pain often starts early in childhood (4, 9) and results in much disability, missed school or work days and poor quality of life due to lack of effective treatments. The etiology is poorly understood and is likely multifactorial with contributions from hypoxia, inflammation, nutritional status, and growth delays. Interestingly, disease modifying therapies such as chronic red cell exchanges and even hematopoietic stem-cell transplant does not seem to reverse chronic pain suggesting a role for peripheral and/or central nerve sensitization (64, 115). While opiates are often used for this indication, their adverse effects limit their use.
To identify novel therapeutics, a deeper understanding of pathogenesis of bone pain is needed. Specifically, the role of systemic inflammation, central sensitization, growth delays, nutritional interventions and physical activity are lacking. Such research, for example, could catalyze the use of existing anti-inflammatory treatments often used to treat bone pain in inflammatory arthritis (116). While newer studies are investigating the role of bisphosphonates on bone pain, given complex pathogenesis of bone pain, a monotherapy may not be sufficient. Instead, an integrated and interdisciplinary approach that involves patient education, hematologists, pain specialists, nutritionists, and psychologists may be needed to help with bone pain.
Author contributions
JG: Conceptualization, Data curation, Writing – original draft, Writing – review & editing. KK: Writing – review & editing. OG: Writing – review & editing. WZ: Writing – review & editing. HH: Writing – review & editing. ST: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This manuscript was supported by grant R01HL156024 to HIH and R01DK111389 to SLT.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. (2010) 38(4 Suppl):S512–521. doi: 10.1016/j.amepre.2009.12.022
2. Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010–2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. (2013) 10(7):e1001484. doi: 10.1371/journal.pmed.1001484
3. Almeida A, Roberts I. Bone involvement in sickle cell disease. Br J Haematol. (2005) 129(4):482–90. doi: 10.1111/j.1365-2141.2005.05476.x
4. Yu T, Campbell T, Ciuffetelli I, Haywood C Jr., Carroll CP, Resar L, et al. Symptomatic avascular necrosis: an understudied risk factor for acute care utilization by patients with SCD. South Med J. (2016) 109(9):519–24. doi: 10.14423/SMJ.0000000000000512
5. Smith WR, Penberthy LT, Bovbjerg VE, McClish DK, Roberts JD, Dahman B, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med. (2008) 148(2):94–101. doi: 10.7326/0003-4819-148-2-200801150-00004
6. Tran H, Gupta M, Gupta K. Targeting novel mechanisms of pain in sickle cell disease. Hematol Am Soc Hematol Educ Program. (2017) 2017(1):546–55. doi: 10.1182/asheducation-2017.1.546
7. Ruta NS, Ballas SK. The opioid drug epidemic and sickle cell disease: guilt by association. Pain Med. (2016) 17(10):1793–8. doi: 10.1093/pm/pnw074
8. Buison AM, Kawchak DA, Schall JI, Ohene-Frempong K, Stallings VA, Leonard MB, et al. Bone area and bone mineral content deficits in children with sickle cell disease. Pediatrics. (2005) 116(4):943–9. doi: 10.1542/peds.2004-2582
9. Adesina OO, Gurney JG, Kang G, Villavicencio M, Hodges JR, Chemaitilly W, et al. Height-corrected low bone density associates with severe outcomes in sickle cell disease: SCCRIP cohort study results. Blood Adv. (2019) 3(9):1476–88. doi: 10.1182/bloodadvances.2018026047
10. Miller RG, Segal JB, Ashar BH, Leung S, Ahmed S, Siddique S, et al. High prevalence and correlates of low bone mineral density in young adults with sickle cell disease. Am J Hematol. (2006) 81(4):236–41. doi: 10.1002/ajh.20541
11. de Alcantara Pedro PP, Castro CHM, Pinheiro MM, Goncalves LM, Figueiredo MS, Szejnfeld VL. Vertebral fractures and low lean mass in young men with sickle cell disease: lack of association with bone mineral density and clinical characteristics. Br J Haematol. (2023) 202(5):e46–9. doi: 10.1111/bjh.18954
12. De Franceschi L, Gabbiani D, Giusti A, Forni G, Stefanoni F, Pinto VM, et al. Development of algorithm for clinical management of sickle cell bone disease: evidence for a role of vertebral fractures in patient follow-up. J Clin Med. (2020) 9(5):1601. doi: 10.3390/jcm9051601
13. Dalle Carbonare L, Matte A, Valenti MT, Siciliano A, Mori A, Schweiger V, et al. Hypoxia-reperfusion affects osteogenic lineage and promotes sickle cell bone disease. Blood. (2015) 126(20):2320–8. doi: 10.1182/blood-2015-04-641969
14. Selma J, Song H, Rivera C, Douglas S, Akella A, Bollavaram K, et al. Sickle cell disease promotes sex-dependent pathological bone loss through enhanced cathepsin proteolytic activity in mice. Blood Adv. (2022) 6(5):1381–93. doi: 10.1182/bloodadvances.2021004615
15. Green M, Akinsami I, Lin A, Banton S, Ghosh S, Chen B, et al. Microarchitectural and mechanical characterization of the sickle bone. J Mech Behav Biomed Mater. (2015) 48:220–8. doi: 10.1016/j.jmbbm.2015.04.019
16. Xiao L, Andemariam B, Taxel P, Adams DJ, Zempsky WT, Dorcelus V, et al. Loss of bone in sickle cell trait and sickle cell disease female mice is associated with reduced IGF-1 in bone and serum. Endocrinology. (2016) 157(8):3036–46. doi: 10.1210/en.2015-2001
17. Adesina OO, Jenkins IC, Wu QV, Fung EB, Narla RR, Lipkin EW, et al. Urinary cross-linked carboxyterminal telopeptide, a bone resorption marker, decreases after vaso-occlusive crises in adults with sickle cell disease. Blood Cells Mol Dis. (2020) 80:102369. doi: 10.1016/j.bcmd.2019.102369
18. Schundeln MM, Goretzki SC, Hauffa PK, Wieland R, Bauer J, Baeder L, et al. Impairment of bone health in pediatric patients with hemolytic anemia. PLoS One. (2014) 9(10):e108400. doi: 10.1371/journal.pone.0108400
19. Nouraie M, Cheng K, Niu X, Moore-King E, Fadojutimi-Akinsi MF, Minniti CP, et al. Predictors of osteoclast activity in patients with sickle cell disease. Haematologica. (2011) 96(8):1092–8. doi: 10.3324/haematol.2011.042499
20. Schett G, Teitelbaum SL. Osteoclasts and arthritis. J Bone Miner Res. (2009) 24(7):1142–6. doi: 10.1359/jbmr.090533
21. Nagae M, Hiraga T, Wakabayashi H, Wang L, Iwata K, Yoneda T. Osteoclasts play a part in pain due to the inflammation adjacent to bone. Bone. (2006) 39(5):1107–15. doi: 10.1016/j.bone.2006.04.033
22. Zhen G, Fu Y, Zhang C, Ford NC, Wu X, Wu Q, et al. Mechanisms of bone pain: progress in research from bench to bedside. Bone Res. (2022) 10(1):44. doi: 10.1038/s41413-022-00217-w
23. Kanaya K, Iba K, Abe Y, Dohke T, Okazaki S, Matsumura T, et al. Acid-sensing ion channel 3 or P2X2/3 is involved in the pain-like behavior under a high bone turnover state in ovariectomized mice. J Orthop Res. (2016) 34(4):566–73. doi: 10.1002/jor.23047
24. Grimbly C, Escagedo PD, Jaremko JL, Bruce A, Alos N, Robinson ME, et al. Sickle cell bone disease and response to intravenous bisphosphonates in children. Osteoporos Int. (2022) 33(11):2397–408. doi: 10.1007/s00198-022-06455-2
25. Bahebeck J, Ngowe Ngowe M, Monny Lobe M, Sosso M, Hoffmeyer P. Stress fracture of the femur: a rare complication of sickle cell disease. Rev Chir Orthop Reparatrice Appar Mot. (2002) 88(8):816–8.12503024
26. Fung EB, Harmatz PR, Milet M, Coates TD, Thompson AA, Ranalli M, et al. Fracture prevalence and relationship to endocrinopathy in iron overloaded patients with sickle cell disease and thalassemia. Bone. (2008) 43(1):162–8. doi: 10.1016/j.bone.2008.03.003
27. Arlet JB, Courbebaisse M, Chatellier G, Eladari D, Souberbielle JC, Friedlander G, et al. Relationship between vitamin D deficiency and bone fragility in sickle cell disease: a cohort study of 56 adults. Bone. (2013) 52(1):206–11. doi: 10.1016/j.bone.2012.10.005
28. Kutlar A, Embury SH. Cellular adhesion and the endothelium: P-selectin. Hematol Oncol Clin North Am. (2014) 28(2):323–39. doi: 10.1016/j.hoc.2013.11.007
29. Mitchell SAT, Majuta LA, Mantyh PW. New insights in understanding and treating bone fracture pain. Curr Osteoporos Rep. (2018) 16(4):325–32. doi: 10.1007/s11914-018-0446-8
30. Terzi S, Griffoni C, Babbi L, Barbanti Brodano G. Surgical treatment of thoraco-lumbar fractures in sickle cell disease: a case report. Eur Rev Med Pharmacol Sci. (2014) 18(1 Suppl):84–8.24825049
31. Rudy HL, Yang D, Nam AD, Cho W. Review of sickle cell disease and spinal pathology. Global Spine J. (2019) 9(7):761–6. doi: 10.1177/2192568218799074
32. Teitelbaum SL. Bone resorption by osteoclasts. Science. (2000) 289(5484):1504–8. doi: 10.1126/science.289.5484.1504
33. Li N, Li XM, Xu L, Sun WJ, Cheng XG, Tian W. Comparison of QCT and DXA: osteoporosis detection rates in postmenopausal women. Int J Endocrinol. (2013) 2013:895474. doi: 10.1155/2013/895474
34. Milner PF, Kraus AP, Sebes JI, Sleeper LA, Dukes KA, Embury SH, et al. Sickle cell disease as a cause of osteonecrosis of the femoral head. N Engl J Med. (1991) 325(21):1476–81. doi: 10.1056/NEJM199111213252104
35. Adesina O, Brunson A, Keegan THM, Wun T. Osteonecrosis of the femoral head in sickle cell disease: prevalence, comorbidities, and surgical outcomes in California. Blood Adv. (2017) 1(16):1287–95. doi: 10.1182/bloodadvances.2017005256
36. Aguilar C, Vichinsky E, Neumayr L. Bone and joint disease in sickle cell disease. Hematol Oncol Clin North Am. (2005) 19(5):929–941, viii. doi: 10.1016/j.hoc.2005.07.001
37. Baldwin C, Nolan VG, Wyszynski DF, Ma QL, Sebastiani P, Embury SH, et al. Association of klotho, bone morphogenic protein 6, and annexin A2 polymorphisms with sickle cell osteonecrosis. Blood. (2005) 106(1):372–5. doi: 10.1182/blood-2005-02-0548
38. Mont MA, Zywiel MG, Marker DR, McGrath MS, Delanois RE. The natural history of untreated asymptomatic osteonecrosis of the femoral head: a systematic literature review. J Bone Joint Surg Am. (2010) 92(12):2165–70. doi: 10.2106/JBJS.I.00575
39. Neumayr LD, Aguilar C, Earles AN, Jergesen HE, Haberkern CM, Kammen BF, et al. Physical therapy alone compared with core decompression and physical therapy for femoral head osteonecrosis in sickle cell disease. Results of a multicenter study at a mean of three years after treatment. J Bone Joint Surg Am. (2006) 88(12):2573–82. doi: 10.2106/JBJS.E.01454
40. Bianco P, Robey PG. Skeletal stem cells. Development. (2015) 142(6):1023–7. doi: 10.1242/dev.102210
41. Veis DJ, O'Brien CA. Osteoclasts, master sculptors of bone. Annu Rev Pathol. (2023) 18:257–81. doi: 10.1146/annurev-pathmechdis-031521-040919
42. Xiong J, O'Brien CA. Osteocyte RANKL: new insights into the control of bone remodeling. J Bone Miner Res. (2012) 27(3):499–505. doi: 10.1002/jbmr.1547
43. Jimenez-Andrade JM, Mantyh WG, Bloom AP, Xu H, Ferng AS, Dussor G, et al. A phenotypically restricted set of primary afferent nerve fibers innervate the bone versus skin: therapeutic opportunity for treating skeletal pain. Bone. (2010) 46(2):306–13. doi: 10.1016/j.bone.2009.09.013
44. Tao R, Mi B, Hu Y, Lin S, Xiong Y, Lu X, et al. Hallmarks of peripheral nerve function in bone regeneration. Bone Res. (2023) 11(1):6. doi: 10.1038/s41413-022-00240-x
45. Mantyh P. Bone cancer pain: causes, consequences, and therapeutic opportunities. Pain. (2013) 154(Suppl 1):S54–62. doi: 10.1016/j.pain.2013.07.044
46. Nencini S, Ringuet M, Kim DH, Chen YJ, Greenhill C, Ivanusic JJ. Mechanisms of nerve growth factor signaling in bone nociceptors and in an animal model of inflammatory bone pain. Mol Pain. (2017) 13:1744806917697011. doi: 10.1177/1744806917697011
47. Nencini S, Ivanusic J. Mechanically sensitive Adelta nociceptors that innervate bone marrow respond to changes in intra-osseous pressure. J Physiol. (2017) 595(13):4399–415. doi: 10.1113/JP273877
48. Castaneda-Corral G, Jimenez-Andrade JM, Bloom AP, Taylor RN, Mantyh WG, Kaczmarska MJ, et al. The majority of myelinated and unmyelinated sensory nerve fibers that innervate bone express the tropomyosin receptor kinase A. Neuroscience. (2011) 178:196–207. doi: 10.1016/j.neuroscience.2011.01.039
49. Oostinga D, Steverink JG, van Wijck AJM, Verlaan JJ. An understanding of bone pain: a narrative review. Bone. (2020) 134:115272. doi: 10.1016/j.bone.2020.115272
50. Elefteriou F. Impact of the autonomic nervous system on the Skeleton. Physiol Rev. (2018) 98(3):1083–112. doi: 10.1152/physrev.00014.2017
51. Gupta K, Jahagirdar O, Gupta K. Targeting pain at its source in sickle cell disease. Am J Physiol Regul Integr Comp Physiol. (2018) 315(1):R104–12. doi: 10.1152/ajpregu.00021.2018
52. Tomlinson RE, Li Z, Zhang Q, Goh BC, Li Z, Thorek DLJ, et al. NGF-TrkA signaling by sensory nerves coordinates the vascularization and ossification of developing endochondral bone. Cell Rep. (2016) 16(10):2723–35. doi: 10.1016/j.celrep.2016.08.002
53. Brain SD, Williams TJ. Substance P regulates the vasodilator activity of calcitonin gene-related peptide. Nature. (1988) 335(6185):73–5. doi: 10.1038/335073a0
54. Schinke T, Liese S, Priemel M, Haberland M, Schilling AF, Catala-Lehnen P, et al. Decreased bone formation and osteopenia in mice lacking alpha-calcitonin gene-related peptide. J Bone Miner Res. (2004) 19(12):2049–56. doi: 10.1359/jbmr.040915
55. Niedermair T, Schirner S, Seebroker R, Straub RH, Grassel S. Substance P modulates bone remodeling properties of murine osteoblasts and osteoclasts. Sci Rep. (2018) 8(1):9199. doi: 10.1038/s41598-018-27432-y
56. Mrak E, Guidobono F, Moro G, Fraschini G, Rubinacci A, Villa I. Calcitonin gene-related peptide (CGRP) inhibits apoptosis in human osteoblasts by beta-catenin stabilization. J Cell Physiol. (2010) 225(3):701–8. doi: 10.1002/jcp.22266
57. Tomlinson RE, Christiansen BA, Giannone AA, Genetos DC. The role of nerves in skeletal development, adaptation, and aging. Front Endocrinol (Lausanne). (2020) 11:646. doi: 10.3389/fendo.2020.00646
58. Kajimura D, Hinoi E, Ferron M, Kode A, Riley KJ, Zhou B, et al. Genetic determination of the cellular basis of the sympathetic regulation of bone mass accrual. J Exp Med. (2011) 208(4):841–51. doi: 10.1084/jem.20102608
59. Khosla S, Drake MT, Volkman TL, Thicke BS, Achenbach SJ, Atkinson EJ, et al. Sympathetic beta1-adrenergic signaling contributes to regulation of human bone metabolism. J Clin Invest. (2018) 128(11):4832–42. doi: 10.1172/JCI122151
60. Baldock PA, Sainsbury A, Couzens M, Enriquez RF, Thomas GP, Gardiner EM, et al. Hypothalamic Y2 receptors regulate bone formation. J Clin Invest. (2002) 109(7):915–21. doi: 10.1172/JCI0214588
61. Allison SJ, Baldock P, Sainsbury A, Enriquez R, Lee NJ, Lin E-J, et al. Conditional deletion of hypothalamic Y2 receptors reverts gonadectomy-induced bone loss in adult mice. J Biol Chem. (2006) 281(33):23436–44. doi: 10.1074/jbc.M604839200
62. Tam J, Ofek O, Fride E, Ledent C, Gabet Y, Muller R, et al. Involvement of neuronal cannabinoid receptor CB1 in regulation of bone mass and bone remodeling. Mol Pharmacol. (2006) 70(3):786–92. doi: 10.1124/mol.106.026435
63. Oh-hashi Y, Shindo T, Kurihara Y, Imai T, Wang Y, Morita H, et al. Elevated sympathetic nervous activity in mice deficient in alphaCGRP. Circ Res. (2001) 89(11):983–90. doi: 10.1161/hh2301.100812
64. Aich A, Jones MK, Gupta K. Pain and sickle cell disease. Curr Opin Hematol. (2019) 26(3):131–8. doi: 10.1097/MOH.0000000000000491
65. Sharma D, Brandow AM. Neuropathic pain in individuals with sickle cell disease. Neurosci Lett. (2020) 714:134445. doi: 10.1016/j.neulet.2019.134445
66. Conran N, Belcher JD. Inflammation in sickle cell disease. Clin Hemorheol Microcirc. (2018) 68(2-3):263–99. doi: 10.3233/CH-189012
67. Nguyen L, Dewhirst FE, Hauschka PV, Stashenko P. Interleukin-1 beta stimulates bone resorption and inhibits bone formation in vivo. Lymphokine Cytokine Res. (1991) 10(1-2):15–21.1873357
68. Kitaura H, Sands MS, Aya K, Zhou P, Hirayama T, Uthgenannt B, et al. Marrow stromal cells and osteoclast precursors differentially contribute to TNF-alpha-induced osteoclastogenesis in vivo. J Immunol. (2004) 173(8):4838–46. doi: 10.4049/jimmunol.173.8.4838
69. Devlin RD, Reddy SV, Savino R, Ciliberto G, Roodman GD. IL-6 mediates the effects of IL-1 or TNF, but not PTHrP or 1,25(OH)2D3, on osteoclast-like cell formation in normal human bone marrow cultures. J Bone Miner Res. (1998) 13(3):393–9. doi: 10.1359/jbmr.1998.13.3.393
70. Hillery CA, Kerstein PC, Vilceanu D, Barabas ME, Retherford D, Brandow AM, et al. Transient receptor potential vanilloid 1 mediates pain in mice with severe sickle cell disease. Blood. (2011) 118(12):3376–83. doi: 10.1182/blood-2010-12-327429
71. Hanaka M, Iba K, Dohke T, Kanaya K, Okazaki S, Yamashita T. Antagonists to TRPV1, ASICs and P2X have a potential role to prevent the triggering of regional bone metabolic disorder and pain-like behavior in tail-suspended mice. Bone. (2018) 110:284–94. doi: 10.1016/j.bone.2018.02.006
72. He LH, Liu M, He Y, Xiao E, Zhao L, Zhang T, et al. TRPV1 Deletion impaired fracture healing and inhibited osteoclast and osteoblast differentiation. Sci Rep. (2017) 7:42385. doi: 10.1038/srep42385
73. Xu C, Gulinello M, Frenette PS. Nociceptors protect sickle cell disease mice from vaso-occlusive episodes and chronic organ damage. J Exp Med. (2021) 218(1):e20200065. doi: 10.1084/jem.20200065
74. Honore P, Luger NM, Sabino MA, Schwei MJ, Rogers SD, Mach DB, et al. Osteoprotegerin blocks bone cancer-induced skeletal destruction, skeletal pain and pain-related neurochemical reorganization of the spinal cord. Nat Med. (2000) 6(5):521–8. doi: 10.1038/74999
75. Abe Y, Iba K, Sasaki K, Chiba H, Kanaya K, Kawamata T, et al. Inhibitory effect of bisphosphonate on osteoclast function contributes to improved skeletal pain in ovariectomized mice. J Bone Miner Metab. (2015) 33(2):125–34. doi: 10.1007/s00774-014-0574-x
76. Ringe JD, Body JJ. A review of bone pain relief with ibandronate and other bisphosphonates in disorders of increased bone turnover. Clin Exp Rheumatol. (2007) 25(5):766–74.18078631
77. Barker PA, Mantyh P, Arendt-Nielsen L, Viktrup L, Tive L. Nerve growth factor signaling and its contribution to pain. J Pain Res. (2020) 13:1223–41. doi: 10.2147/JPR.S247472
78. Li Z, Meyers CA, Chang L, Lee S, Li Z, Tomlinson R, et al. Fracture repair requires TrkA signaling by skeletal sensory nerves. J Clin Invest. (2019) 129(12):5137–50. doi: 10.1172/JCI128428
79. Bienenstock J, Tomioka M, Matsuda H, Stead RH, Quinonez G, Simon GT, et al. The role of mast cells in inflammatory processes: evidence for nerve/mast cell interactions. Int Arch Allergy Appl Immunol. (1987) 82(3-4):238–43. doi: 10.1159/000234197
80. Skaper SD. Nerve growth factor: a neuroimmune crosstalk mediator for all seasons. Immunology. (2017) 151(1):1–15. doi: 10.1111/imm.12717
81. Torcia M, Bracci-Laudiero L, Lucibello M, Nencioni L, Labardi D, Rubartelli A, et al. Nerve growth factor is an autocrine survival factor for memory B lymphocytes. Cell. (1996) 85(3):345–56. doi: 10.1016/S0092-8674(00)81113-7
82. Bracci-Laudiero L, Aloe L, Caroleo MC, Buanne P, Costa N, Starace G, et al. Endogenous NGF regulates CGRP expression in human monocytes, and affects HLA-DR and CD86 expression and IL-10 production. Blood. (2005) 106(10):3507–14. doi: 10.1182/blood-2004-10-4055
83. Nicol GD, Vasko MR. Unraveling the story of NGF-mediated sensitization of nociceptive sensory neurons: oN or OFF the trks? Mol Interv. (2007) 7(1):26–41. doi: 10.1124/mi.7.1.6
84. Donnerer J, Schuligoi R, Stein C. Increased content and transport of substance P and calcitonin gene-related peptide in sensory nerves innervating inflamed tissue: evidence for a regulatory function of nerve growth factor in vivo. Neuroscience. (1992) 49(3):693–8. doi: 10.1016/0306-4522(92)90237-V
85. Jiang H, Takeda K, Lazarovici P, Katagiri Y, Yu ZX, Dickens G, et al. Nerve growth factor (NGF)-induced calcium influx and intracellular calcium mobilization in 3T3 cells expressing NGF receptors. J Biol Chem. (1999) 274(37):26209–16. doi: 10.1074/jbc.274.37.26209
86. Brodie C. Platelet activating factor induces nerve growth factor production by rat astrocytes. Neurosci Lett. (1995) 186(1):5–8. doi: 10.1016/0304-3940(95)11267-Z
87. Cahill CM, Dray A, Coderre TJ. Intrathecal nerve growth factor restores opioid effectiveness in an animal model of neuropathic pain. Neuropharmacology. (2003) 45(4):543–52. doi: 10.1016/S0028-3908(03)00192-8
88. Dyck PJ, Peroutka S, Rask C, Burton E, Baker MK, Lehman KA, et al. Intradermal recombinant human nerve growth factor induces pressure allodynia and lowered heat-pain threshold in humans. Neurology. (1997) 48(2):501–5. doi: 10.1212/WNL.48.2.501
89. Svensson P, Cairns BE, Wang K, Arendt-Nielsen L. Injection of nerve growth factor into human masseter muscle evokes long-lasting mechanical allodynia and hyperalgesia. Pain. (2003) 104(1-2):241–7. doi: 10.1016/S0304-3959(03)00012-5
90. Albo C, Kumar S, Pope M, Kidwell KM, Xu H, Bowman L, et al. Characteristics and potential biomarkers of adult sickle cell patients with chronic pain. Eur J Haematol. (2020) 105(4):419–25. doi: 10.1111/ejh.13461
91. Norman BH, McDermott JS. Targeting the nerve growth factor (NGF) pathway in drug discovery. Potential applications to new therapies for chronic pain. J Med Chem. (2017) 60(1):66–88. doi: 10.1021/acs.jmedchem.6b00964
92. Vincent L, Vang D, Nguyen J, Gupta M, Luk K, Ericson ME, et al. Mast cell activation contributes to sickle cell pathobiology and pain in mice. Blood. (2013) 122(11):1853–62. doi: 10.1182/blood-2013-04-498105
93. Wang Q, Lepus CM, Raghu H, Reber LL, Tsai MM, Wong HH, et al. IgE-mediated mast cell activation promotes inflammation and cartilage destruction in osteoarthritis. Elife. (2019) 9(1):2449. doi: 10.7554/eLife.39905
94. Lindsay RM, Harmar AJ. Nerve growth factor regulates expression of neuropeptide genes in adult sensory neurons. Nature. (1989) 337(6205):362–4. doi: 10.1038/337362a0
95. Scarpi D, Cirelli D, Matrone C, Castronovo G, Rosini P, Occhiato EG, et al. Low molecular weight, non-peptidic agonists of TrkA receptor with NGF-mimetic activity. Cell Death Dis. (2012) 3(7):e339. doi: 10.1038/cddis.2012.80
96. Zanone MM, Banga JP, Peakman M, Edmonds M, Watkins PJ. An investigation of antibodies to nerve growth factor in diabetic autonomic neuropathy. Diabet Med. (1994) 11(4):378–83. doi: 10.1111/j.1464-5491.1994.tb00289.x
97. Ni S, Ling Z, Wang X, Cao Y, Wu T, Deng R, et al. Sensory innervation in porous endplates by netrin-1 from osteoclasts mediates PGE2-induced spinal hypersensitivity in mice. Nat Commun. (2019) 10(1):5643. doi: 10.1038/s41467-019-13476-9
98. Zhu S, Zhu J, Zhen G, Hu Y, An S, Li Y, et al. Subchondral bone osteoclasts induce sensory innervation and osteoarthritis pain. J Clin Invest. (2019) 129(3):1076–93. doi: 10.1172/JCI121561
99. Koch M, Murrell JR, Hunter DD, Olson PF, Jin W, Keene DR, et al. A novel member of the netrin family, beta-netrin, shares homology with the beta chain of laminin: identification, expression, and functional characterization. J Cell Biol. (2000) 151(2):221–34. doi: 10.1083/jcb.151.2.221
100. Nakashiba T, Ikeda T, Nishimura S, Tashiro K, Honjo T, Culotti JG, et al. Netrin-G1: a novel glycosyl phosphatidylinositol-linked mammalian netrin that is functionally divergent from classical netrins. J Neurosci. (2000) 20(17):6540–50. doi: 10.1523/JNEUROSCI.20-17-06540.2000
101. Cirulli V, Yebra M. Netrins: beyond the brain. Nat Rev Mol Cell Biol. (2007) 8(4):296–306. doi: 10.1038/nrm2142
102. Miloudi K, Binet F, Wilson A, Cerani A, Oubaha M, Menard C, et al. Truncated netrin-1 contributes to pathological vascular permeability in diabetic retinopathy. J Clin Invest. (2016) 126(8):3006–22. doi: 10.1172/JCI84767
103. Xu K, Wu Z, Renier N, Antipenko A, Tzvetkova-Robev D, Xu Y, et al. Neural migration. Structures of netrin-1 bound to two receptors provide insight into its axon guidance mechanism. Science. (2014) 344(6189):1275–9. doi: 10.1126/science.1255149
104. Claro V, Ferro A. Netrin-1: focus on its role in cardiovascular physiology and atherosclerosis. JRSM Cardiovasc Dis. (2020) 9:2048004020959574. doi: 10.1177/2048004020959574
105. Renders S, Svendsen AF, Panten J, Rama N, Maryanovich M, Sommerkamp P, et al. Niche derived netrin-1 regulates hematopoietic stem cell dormancy via its receptor neogenin-1. Nat Commun. (2021) 12(1):608. doi: 10.1038/s41467-020-20801-0
106. Tolu SS, Wang K, Yan Z, Zhang S, Roberts K, Crouch AS, et al. Characterization of hematopoiesis in sickle cell disease by prospective isolation of stem and progenitor cells. Cells. (2020) 9(10):2159. doi: 10.3390/cells9102159
107. Feng X, Teitelbaum SL. Osteoclasts: new insights. Bone Res. (2013) 1(1):11–26. doi: 10.4248/BR201301003
108. Piga A. Impact of bone disease and pain in thalassemia. Hematol Am Soc Hematol Educ Program. (2017) 2017(1):272–7. doi: 10.1182/asheducation-2017.1.272
109. Gbotosho OT, Kapetanaki MG, Ghosh S, Villanueva FS, Ofori-Acquah SF, Kato GJ. Heme induces IL-6 and cardiac hypertrophy genes transcripts in sickle cell mice. Front Immunol. (2020) 11:1910. doi: 10.3389/fimmu.2020.01910
110. Belcher JD, Mahaseth H, Welch TE, Otterbein LE, Hebbel RP, Vercellotti GM. Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J Clin Invest. (2006) 116(3):808–16. doi: 10.1172/JCI26857
111. Zwerina J, Tzima S, Hayer S, Redlich K, Hoffmann O, Hanslik-Schnabel B, et al. Heme oxygenase 1 (HO-1) regulates osteoclastogenesis and bone resorption. FASEB J. (2005) 19(14):2011–3. doi: 10.1096/fj.05-4278fje
112. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. (2020) 11(2):88. doi: 10.1038/s41419-020-2298-2
113. Liu P, Wang W, Li Z, Li Y, Yu X, Tu J, et al. Ferroptosis: a new regulatory mechanism in osteoporosis. Oxid Med Cell Longev. (2022) 2022:2634431. doi: 10.1155/2022/2634431
114. Wattiez AS, Sowers LP, Russo AF. Calcitonin gene-related peptide (CGRP): role in migraine pathophysiology and therapeutic targeting. Expert Opin Ther Targets. (2020) 24(2):91–100. doi: 10.1080/14728222.2020.1724285
115. Curtis SA, Raisa BM, Roberts JD, Hendrickson JE, Starrels J, Lesley D, et al. Non-crisis related pain occurs in adult patients with sickle cell disease despite chronic red blood cell exchange transfusion therapy. Transfus Apher Sci. (2022) 61(2):103304. doi: 10.1016/j.transci.2021.103304
Keywords: sickle cell disease, pain, nervous system, inflammation, bone remodeling, hemolytic anemia
Citation: Gollamudi J, Karkoska KA, Gbotosho OT, Zou W, Hyacinth HI and Teitelbaum SL (2024) A bone to pick-cellular and molecular mechanisms of bone pain in sickle cell disease. Front. Pain Res. 4:1302014. doi: 10.3389/fpain.2023.1302014
Received: 25 September 2023; Accepted: 4 December 2023;
Published: 4 January 2024.
Edited by:
Keesha Roach, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Jacqueline Harris, University of Tennessee Health Science Center (UTHSC), United States© 2024 Gollamudi, Karkoska, Gbotosho, Zou, Hyacinth and Teitelbaum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jahnavi Gollamudi Z29sbGFtamlAdWNtYWlsLnVjLmVkdQ==