 Gina M. Gehling
Gina M. Gehling Keesha Powell-Roach
Keesha Powell-Roach Diana J. Wilkie
Diana J. Wilkie Jennifer R. Dungan
Jennifer R. Dungan- 1College of Nursing, University of Florida, Gainesville, FL, United States
- 2College of Nursing, Department of Community and Population Health, University of Tennessee Health Science Center, Memphis, TN, United States
Background: Scientists have speculated genetic variants may contribute to an individual's unique pain experience. Although research exists regarding the relationship between single nucleotide polymorphisms and sickle cell disease-related pain, this literature has not been synthesized to help inform future precision health research for sickle cell disease-related pain. Our primary aim of this systematic review was to synthesize the current state of scientific literature regarding single nucleotide polymorphisms and their association with sickle cell disease-related pain.
Methods: Using the Prisma guidelines, we conducted our search between December 2021–April 2022. We searched PubMed, Web of Science, CINAHL, and Embase databases (1998–2022) and selected all peer-reviewed articles that included reports of associations between single nucleotide polymorphisms and sickle cell disease-related pain outcomes.
Results: Our search yielded 215 articles, 80 of which were duplicates, and after two reviewers (GG, JD) independently screened the 135 non-duplicate articles, we retained 22 articles that met the study criteria. The synthesis of internationally generated evidence revealed that this scientific area remains predominantly exploratory in nature, with only three studies reporting sufficient power for genetic association. Sampling varied across studies with a range of children to older adults with SCD. All of the included articles (n = 22) examined acute pain, while only nine of those studies also examined chronic pain.
Conclusion: Currently, the evidence implicating genetic variation contributing to acute and chronic sickle cell disease-related pain is characterized by modestly powered candidate-gene studies using rigorous SCD-pain outcomes. Effect sizes and directions vary across studies and are valuable for informing the design of future studies. Further research is needed to replicate these associations and extend findings with hypothesis-driven research to inform precision health research.
1. Introduction
Sickle Cell Disease (SCD) is the most common single-gene hemoglobinopathy affecting around 6 million people globally, with an estimated 100,000 living in the United States (U.S.) (1). Pain is a hallmark of SCD pathophysiology, with recent studies demonstrating that adults with SCD suffer from multidimensional pain related to recurrent episodes of acute pain, commonly referred to as vaso-occlusive crisis (VOC), as well as ongoing chronic pain (2, 3). Pain as a result of VOC is the leading complication associated with SCD. VOCs have a sudden onset and occur as a result of polymerization of abnormal sickle hemoglobin, inflammation, and adhesion (4). VOC events are the most frequent cause of hospitalization and one of the most common predictors of death among people with SCD (5). Other sources of pain in SCD result from secondary disease-related pathophysiology such as splenic sequestration, gall bladder issues, necrosis of femur, ulcers, and more. There is evidence to support that mismanaged pain leads to inadequate pain control (6), an increase in mortality (7), and the need for health care utilization resulting in billions of dollars spent annually for people with SCD (6, 8).
Precision biomarker solutions for the management of SCD pain may reduce complications that lead to the excess morbidity, hospitalization, and mortality among this vulnerable population. Yet the underlying genetic contributions to SCD-related pain remain largely unknown. Evidence exists supporting a genetic link between an individual's unique experience with pain and the presence of SNPs (9). Moreover, scientists have discovered that a person's genetic makeup can play a critical role in pain medication metabolism and effectiveness (10). Yet, no current systematic review exists that characterizes the candidate genes associated with SCD-related pain pheno- and sub-phenotypes. Heritability estimates for chronic pain phenotypes indicate that genetic variation contributes to the presence and experience of pain (any chronic pain, = .16 ± .07, p = 0.02, severe chronic pain = .30 ± .13, p = 0.007) (11).
While heritability estimates are lacking specifically for SCD pain phenotypes, significant candidate gene associations for pain phenotypes in SCD patients have been reported. Given the large variability in pain among people with SCD, others have hypothesized genetic contribution to pain perception in SCD, as tested by genetic association studies in the literature (12, 13). Although research exists regarding the relationship between single nucleotide polymorphisms (SNPs) and SCD-related pain, the quality and strength of this literature has not been synthesized to describe key scientific gaps and to help inform future precision health research for SCD-related pain. Since pain is a highly complex and variable subjective phenotype, understanding the state-of-the-science regarding the SNPs and specific phenotypes and sub-phenotypes that have been studied will be useful to guide future precision biomarker research and clinical applications.
The purpose of this article is to report a systematic review of the candidate gene SNP association studies among patients with SCD-related pain phenotypes. Specifically, across the series of studies identified, we sought to characterize the state of the science in this field in terms of study designs, population characteristics, measures of the SCD-pain phenotypes, candidate genes and specific SNPs studied, associations between SNPs and the SCD-pain phenotypes, and an evaluation of strength of evidence. Synthesis of this body of literature will be useful to guide future research and for implementation of the knowledge in clinical settings to control the pain of SCD.
2. Methods
2.1. Search strategy
We conducted our search for articles published between December 2021–April 2022. We searched PubMed, Web of Science, CINAHL, and Embase databases (1998–2022) and selected all peer-reviewed articles that reported on associations between SNPs and SCD-related pain. Our search strategy included the following search terms: “sickle cell disease” OR “SCD” OR “beta-plus thalassemia” OR “beta-zero thalassemia” OR “hemoglobin-c” OR “sickle cell anemia” OR “sickle cell anaemia” OR “hemoglobinopathy” AND “pain character*” OR “pain phenotype” OR “pain*” OR “pain expression” OR “pain experience” OR “pain cris*” (iterations related to crisis) AND “genetic varia*” (iterations related to variation) OR “genetic mutation” OR “single nucleotide*” OR “SNP.” The complete search strategy appears in Supplementary Data S1. Because there is limited research on this topic, and because we wanted to understand the full scope of this research area, we decided not to limit the number of years included in the study. However, we expected that a majority of articles containing SNP associations would be published after 2003, when the Human Genome Project was completed, and the nature of this research expanded rapidly.
All articles from the search phase were then exported into a Prisma-guided literature review management software that facilitates selection and extraction among multiple team members (Covidence systematic review software, Veritas Health Innovation, Melbourne, Australia, 2022).
2.2. Selection criteria
We selected articles based on criteria established by the team before initiating the screening. Inclusion criteria were as follows: (1) must include human participants with a diagnosis of SCD, which is inclusive of sickle cell anemia, (2) full article available in English, (3) focused on the association of SNPs or genetic variants and SCD-related pain phenotypes, and (4) published in a peer-reviewed journal. We included sickle cell crisis or vaso-occlusive crisis-focused articles if they specifically acknowledged a pain-related component (e.g., pain or painful crisis, pain event). Articles were excluded if they were: (1) literature reviews, (2) SNPs or genetic variants focused on SCD-related complications not specific to pain phenotype (e.g., acute chest syndrome, stroke), (3) full article unavailable, (4) animal study or model, (5) conference or meeting abstracts, or (6) there was a triangulation focus to a pain phenotype (e.g., SNPs with evidence supporting an association with fetal hemoglobin in heterogeneity of pain severity). Two authors (GG and JD) independently screened all articles and met to resolve any conflicts. After the title and abstract screening process, the two reviewers’ decisions to include or exclude an article had an inter-rater reliability proportionate agreement of 90.2% (Cohen's kappa of 0.78). During the full-text screening, the two investigators achieved a proportionate agreement of 84.4% (Cohen's kappa of 0.63). Figure 1 shows the PRISMA diagram indicating the flow of the article selection process. From the 215 identified articles, 22 articles met the inclusion criteria for further analysis.
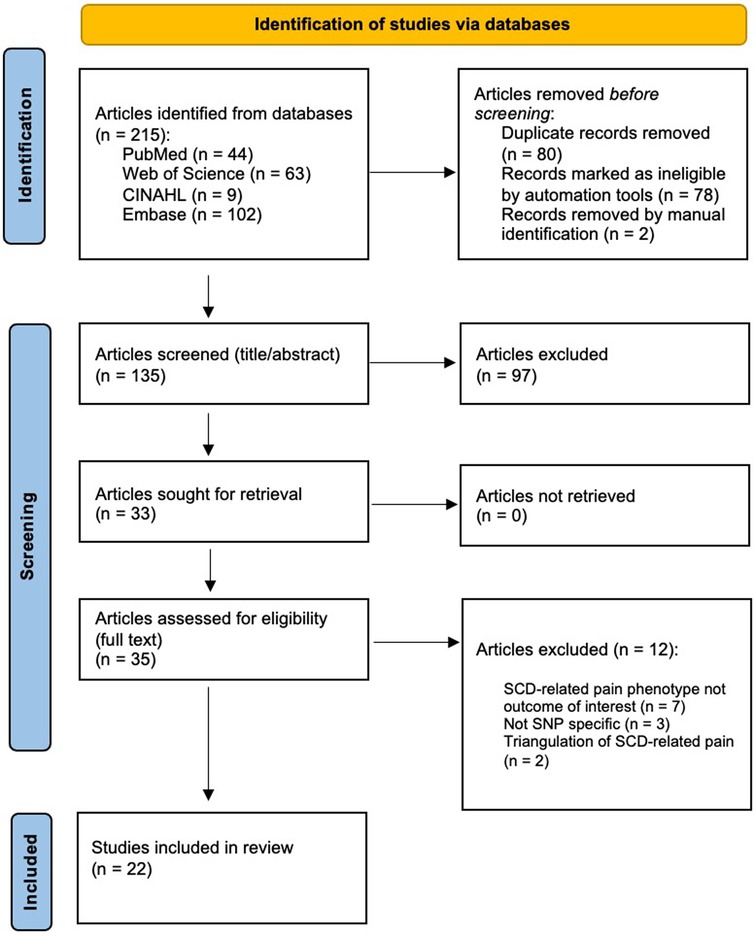
Figure 1. PRISMA diagram.
2.3. Data extraction
GG was responsible for data extraction from the 22 articles that met study criteria into a literature matrix that KPR, DW, and JD independently reviewed for accuracy. Data extracted from each study appears in our literature matrix which can be found in Supplementary Data S2. Within our matrix are select sample characteristics (i.e., geographic location, age, gender, sickle cell genotype), SNPs and pain phenotype of interest, measures used to extract and sequence DNA, select significant statistical results, and a brief summary of significant study findings.
3. Results
3.1. Study characteristics
Of the 22 studies, 13 studies were conducted exclusively in the U.S., and one exclusively in the United Kingdom. Three multi-center international studies were conducted between the U.S. and another foreign country (n = 2 in the UK, n = 1 in Brazil), two in Africa (n = 1 Cameroon, n = 1 Tunisia), two in the Middle East (Bahrain), and one in India (Jabalpur). Very few investigators (n = 4) reported performing a power analysis, revealing majority of these studies were exploratory in nature (n = 18). Of those, three of the four studies reported sufficient power (79%–84%) to detect associations between SNPs of interest and an SCD-related pain phenotype (13–16).
3.2. Population characteristics
Sample sizes ranged from 107 to 1,275. Five studies sampled only children (14–18) under the age of 18 with a median sample size of n = 771. Four studies sampled only adults (19, 20–22) aged 18–70 years with a median sample size of n = 200. Thirteen studies sampled both children and adults (23–35) with a median sample size of n = 134. Of the 21 articles providing sufficient age data, we calculated a mean age of 24.8 ± 11.1 years (range 5–70) for most of the participants included in our review. There was a higher proportion of female participants than male participants (female 55.88% vs. male 44.12%).
3.3. Measures of SCD-related pain phenotypes
Pain phenotype characterization was most notably addressed within the pain measures. Since pain is subjective, self-report is considered the Gold Standard and most accurate measure of pain. Pain proxies are considered measures of pain which do not involve the patient's self-report, such as medical record review.
3.3.1. Acute SCD pain phenotype
We found consistency among articles in phenotyping SCD-related pain through the use of proxies. All 22 studies used some form of health care utilization as a proxy for acute pain in SCD. Ten studies used acute health care utilization as a result of pain crisis obtained through medical record review and biweekly telephone calls (19, 24–29, 31–33). Other researchers used acute care utilization as a surrogate for acute pain defined by any visits to an acute care center, emergency center, or hospitalization. For example, five studies used vaso-occlusive crisis or pain crisis as a surrogate marker for acute pain (14, 20–23). Six studies used characteristics of hospitalization or provider intervention (i.e., utilization events) and pain management (e.g., required or did not require pharmaceutical intervention) as part of their proposed definition for pain (15, 18, 22, 23, 30, 35).
3.3.2. Chronic SCD pain phenotype
Nine studies used self-reported pain at a routine clinic visit as a measure for chronic pain (24–29, 31–33). In these studies, investigators used the Composite Pain Index (CPI) score obtained from PAINReportIt as a self-report measure of the multidimensional pain experience (location, intensity, quality, pattern). Notably, CPI has been previously shown to be both a valid and reliable measure of chronic pain in SCD (36). In another study, investigators used the average pain intensity score (API) from PAINReportIt to identify an association between chronic pain heterogeneity and COMT SNPs (19).
3.4. Genetic associations with SCD-related pain
Synthesis of the 22 articles revealed evidence of statistically suggestive associations between SCD-related acute pain heterogeneity in 20 gene regions with 48 respective SNPs (see Table 1) (14, 16, 20, 22, 23, 25, 26, 28, 29, 31–35). Additionally, there is evidence suggestive of a significant association between SCD-related chronic pain heterogeneity in 4 gene regions with 13 respective SNPs (see Table 2) (24, 27, 28, 33). Two genes, ADRB2 (28) and GCH1 (33) have preliminary evidence indicating they may play a role in variation among both acute and chronic pain in SCD. Figure 2 displays all genes reported to have significant associations with SCD pain phenotypes.

Table 1. Reported significant gene associations with SCD-related acute pain.
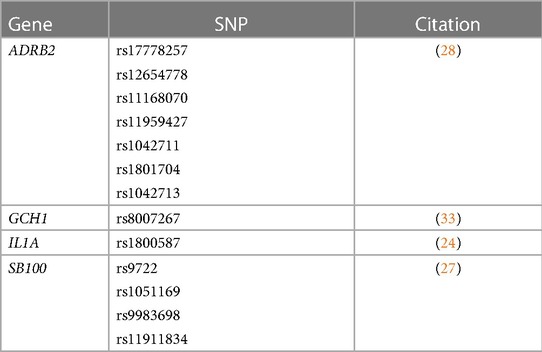
Table 2. Reported significant gene associations with SCD-related chronic pain.
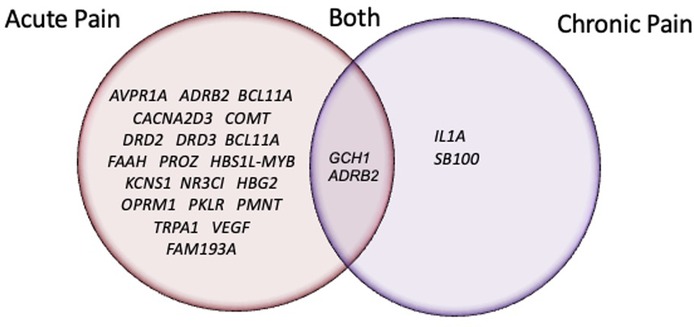
Figure 2. Venn-Diagram of genes with SNPs reported to have a significant association with an SCD pain-related phenotype.
Moreover, we compiled a summary of magnitude and direction of effect for candidate genes identified as having a possible association with SCD-related pain during our review (see Table 3). As an additional point of reference, we have provided a summary gene annotation table which includes information on gene function for candidate genes identified during our review (Supplementary Data S3).
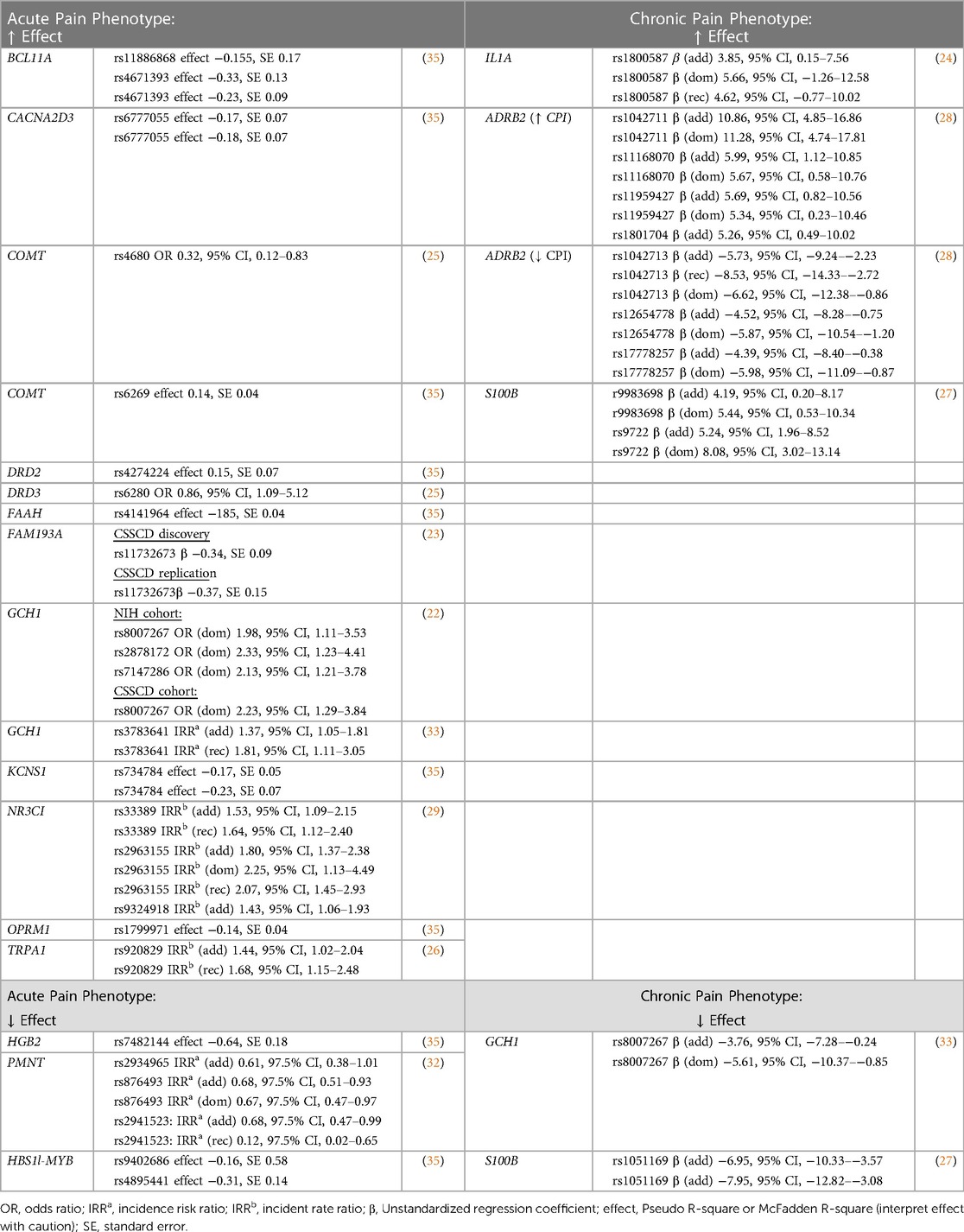
Table 3. Summary table reporting magnitude of effect for single SNPs with reported evidence suggestive of association with SCD-related pain phenotypes.
4. Discussion
Prevalence of pain-related genes and associated variants found in individuals with SCD was previously reported by Jhun et al. (14). Yet, to our knowledge, no study has reported on the state of the science regarding the levels of evidence and study characteristics of associations between SNPs and pain phenotypes in SCD. We identified 22 articles across 2008–2022 reporting SNP associations with SCD-related pain. Statistically suggestive evidence associates SCD-related acute pain heterogeneity with 21 gene regions and 42 respective SNPs. Evidence for chronic pain gene associations is limited to reports of 2 gene regions with 5 respective SNPs. Moreover, 20 SNPs within 2 candidate genes had suggestive associations with both acute and chronic pain outcomes.
Acute health care utilization for pain management related to vaso-occlusive episodes or events were the most commonly tested acute pain phenotypes. Investigators obtained this information through patient or caretaker self-report and medical record review of acute care utilization for pain control or hospitalization rates. Since the high prevalence of SCD pain caused by sickling and VOC often leads to ineffective self-pain management and results in healthcare seeking behaviors, this method of measuring acute pain in SCD is widely accepted by SCD research experts. According to Ballas & Dampier, roughly 60% of individuals with SCD visit the hospital for VOC treatment at least once a year and close to 25% require hospital-based VOC treatment five or more times annually (6). Of note, Prestia and colleagues reported healthcare visits for active vaso-occlusive crisis account for 95% of hospitalizations with an average length of stay of 4.5 days (37).
An estimated 30%–40% of adolescents and adults with SCD live with chronic pain (38). Our synthesis on the state of the science revealed the chronic pain phenotype within this population varies widely. Some researchers have investigated the relationship between SNPs and chronic SCD pain using a composite pain index (CPI) from a well-validated tablet-based pain measurement tool, PAINReportIt (24–29, 31–33). Other investigators developed a chronic pain phenotype using average pain intensity scores (API) from PAINReportIt and a standard quantitative sensory testing protocol for heat, cold, and pressure thresholds, shown to be safe in those with SCD (39, 40). SCD pain is multidimensional in nature, with inconsistent frequency and severity, even in individuals with the same genotype. As such, the reviewed studies assumed the common disease-common variant hypothesis and studied a-priori candidate genes with biologic plausibility. For example, epinephrine, a neurotransmitter in pain signaling, has been shown to target the beta-2 adrenergic receptor (ADRB2) and polymorphisms in the ADRB2 gene have been linked to chronic and acute pain in SCD, along with other conditions such as functional gastrointestinal and temporomandibular disorders (28, 35, 41, 42). Polymorphisms of the GCH1 (GTP Cyclohydrolase 1) gene (22, 33), which encodes GTP cyclohydrolase enzyme and has been associated with pain sensitivity, has been shown to contribute to both chronic and acute pain by increasing or decreasing tetrahydrobiopterin (BH4) production, an essential enzyme cofactor responsible for the biosynthesis of neurotransmitters such as serotonin, dopamine, epinephrine, and norepinephrine.
Key gaps were identified in this synthesis. At this time, we could not find any studies exploring the genetic relationship between SNPs and neuropathic pain in SCD. In a quantitative sensory testing study on people with SCD who had previously diagnosed chronic pain, 32% of participants were identified as having allodynia, hyperalgesia, or both, all of which are widely accepted indicators of neuropathic pain among scientists in the pain research community (2). While there is preliminary evidence to support some individuals with SCD may experience neuropathic pain (NP), there is no objective measure for NP in this population. Providers primarily rely on the visual analog scale, pain descriptors and intensity (43–45). It is unclear if the NP observed in other conditions is equivalent to NP in SCD.
This study is not without its own limitations. A majority of the gene association studies were performed within the U.S., despite the predominance of SCD internationally. A lack of geographical representation hinders our ability to generalize our findings on a global scale. Smaller sample sizes and the potential for population-specific genetic effects are limitations of the reviewed work. Additionally, since much of the research conducted was exploratory in nature, definitive conclusions cannot be made at this time about the relationship between these candidate genes and SCD-related pain phenotypes. Even so, it is clear there is a need for future replication studies and hypothesis-driven work among individuals with SCD if we are to understand the genetic implications of SCD-related pain. Of note, we chose to focus specifically on human-derived candidate genes and acknowledge the value of experimental animal models and their relevance to future study in this area.
5. Conclusion
Without further investigation into SNPs and their association with SCD-related pain, morbidity and health care utilization for these individuals may remain high. Gene association research in SCD-related pain has been primarily focused on increasing our understanding of the role of genetic variants on acute pain in SCD. There is a clear need for replication and hypothesis-driven gene association studies. Moreover, we believe there is an essential need to investigate genetic associations within all subtypes of pain in the sickle cell population. Further research has the potential to inform precision health practices on how best to manage all pain phenotypes in SCD.
Author contributions
GG and DW developed the search strategy and GG compiled content for review by the study team. GG and JD screened all articles for inclusion. GG wrote the manuscript under the advisement of KP, DW, and JD. All authors approved the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpain.2023.1223309/full#supplementary-material
References
1. Merchant E. Life Expectancy in the U.S. for the Population with Sickle Cell Disease. Population Association of America. (2021). Available at: https://www.populationassociation.org/blogs/emily-merchant1/2021/01/31/life-expectancy-sickle-cell (Accessed November 21, 2021).
2. Molokie RE, Wang ZJ, Yao Y, Powell-Roach KL, Schlaeger JM, Suarez ML, et al. Sensitivities to thermal and mechanical stimuli: adults with sickle cell disease compared to healthy, pain-free African American controls. J Pain. (2020) 21:957–67. doi: 10.1016/j.jpain.2019.11.002
3. Dyal BW, Ezenwa MO, Yoon SL, Fillingim RB, Yao Y, Schlaeger JM, et al. A QST-based pain phenotype in adults with sickle cell disease: sensitivity and specificity of quality descriptors. Pain Pract. (2020) 20:168–78. doi: 10.1111/papr.12841
4. Darbari DS, Sheehan VA, Ballas SK. The vaso-occlusive pain crisis in sickle cell disease: definition, pathophysiology, and management. Eur J Haematol. (2020) 105:237–46. doi: 10.1111/ejh.13430
5. Ballas SK, Gupta K, Adams-Graves P. Sickle cell pain: a critical reappraisal. Blood. (2012) 120:3647–56. doi: 10.1182/blood-2012-04-383430
6. Ballas SK, Dampier C. Risk factors associated with increased emergency department utilization in patients with sickle cell disease: a systematic literature review. Ann Hematol. (2020) 99:2483–95. doi: 10.1007/s00277-020-04205-0
7. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. (1994) 330:1639–44. doi: 10.1056/NEJM199406093302303
8. Lee S, Vania DK, Bhor M, Revicki D, Abogunrin S, Sarri G. Patient-reported outcomes and economic burden of adults with sickle cell disease in the United States: a systematic review. Int J Gen Med. (2020) 13:361–77. doi: 10.2147/IJGM.S257340
9. Naureen Z, Lorusso L, Manganotti P, Caruso P, Mazzon G, Cecchin S, et al. Genetics of pain: from rare Mendelian disorders to genetic predisposition to pain. Acta Biomed. (2020) 91:e2020010. doi: 10.23750/abm.v91i13-S.10682
10. Chenoweth MJ, Giacomini KM, Pirmohamed M, Hill SL, Van Schaik RHN, Schwab M, et al. Global pharmacogenomics within precision medicine: challenges and opportunities. Clin Pharmacol Ther. (2020) 107:57–61. doi: 10.1002/cpt.1664
11. Hocking LJ, Morris AD, Dominiczak AF, Porteous DJ, Smith BH. Heritability of chronic pain in 2195 extended families. Eur J Pain. (2012) 16:1053–63. doi: 10.1002/j.1532-2149.2011.00095.x
12. Gupta K, Jahagirdar O, Gupta K. Targeting pain at its source in sickle cell disease. Am J Physiol Regul Integr Comp Physiol. (2018) 315:R104–12. doi: 10.1152/ajpregu.00021.2018
13. Jhun EH, Yao Y, He Y, Kyle MA, Wilkie DJ, Molokie RE, et al. Prevalence of pain-related single nucleotide polymorphisms in patients of African origin with sickle cell disease. Pharmacogenomics. (2015) 16:1795–806. doi: 10.2217/pgs.15.126
14. Al-Habboubi HH, Mahdi N, Abu-Hijleh TM, Abu-Hijleh FM, Sater MS, Almawi WY. The relation of vascular endothelial growth factor (VEGF) gene polymorphisms on VEGF levels and the risk of vasoocclusive crisis in sickle cell disease. Eur J Haematol. (2012) 89:403–9. doi: 10.1111/ejh.12003
15. Rampersaud E, Kang G, Palmer LE, Rashkin SR, Wang S, Bi W, et al. A polygenic score for acute vaso-occlusive pain in pediatric sickle cell disease. Blood Adv. (2021) 5:2839–51. doi: 10.1182/bloodadvances.2021004634
16. Mahdi N, Abu-Hijleh TM, Abu-Hijleh FM, Sater MS, Al-Ola K, Almawi WY. Protein Z polymorphisms associated with vaso-occlusive crisis in young sickle cell disease patients. Ann Hematol. (2012) 91:1215–20. doi: 10.1007/s00277-012-1474-6
17. Lettre G, Sankaran VG, Bezerra MA, Araujo AS, Uda M, Sanna S, et al. DNA Polymorphisms at the BCL11A, HBS1l-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci U S A. (2008) 105:11869–74. doi: 10.1073/pnas.0804799105
18. Bean CJ, Boulet SL, Yang G, Payne AB, Ghaji N, Pyle ME, et al. Acute chest syndrome is associated with single nucleotide polymorphism-defined beta globin cluster haplotype in children with sickle cell anaemia. Br J Haematol. (2013) 163:268–76. doi: 10.1111/bjh.12507
19. Powell-Roach KL, Yao Y, Wallace MR, Chamala S, Cruz-Almeida Y, Jhun E, et al. HUMAN STUDY COMT and DRD3 haplotype-associated pain intensity and acute care utilization in adult sickle cell disease. Exp Biol Med (Maywood, N.J.). (2022) 247(17):1601–8. doi: 10.1177/15353702221080716
20. Zhang Y, Belfer I, Nouraie M, Zeng Q, Goel R, Chu Y, et al. Association of genetic variation in COMT gene with pain related to sickle cell disease in patients from the walk-PHaSST study. J Pain Res. (2018) 11:537–43. doi: 10.2147/JPR.S149958
21. Kalai M, Chaouch L, Mansour IB, Hafsia R, Ghanem A, Abbes S. Frequency of three polymorphisms of the CCL5 gene (RS2107538, Rs2280788 and Rs2280789) and their implications for the phenotypic expression of sickle cell anemia in Tunisia. Pol J Pathol. (2013a) 64:84–9. doi: 10.5114/pjp.2013.36012
22. Belfer I, Youngblood V, Darbari DS, Wang Z, Diaw L, Freeman L, et al. A GCH1 haplotype confers sex-specific susceptibility to pain crises and altered endothelial function in adults with sickle cell anemia. Am J Hematol. (2014) 89:187–93. doi: 10.1002/ajh.23613
23. Galarneau G, Coady S, Garrett ME, Jeffries N, Puggal M, Paltoo D, et al. Gene-centric association study of acute chest syndrome and painful crisis in sickle cell disease patients. Blood. (2013) 122:434–42. doi: 10.1182/blood-2013-01-478776
24. Hu X, Jhun E, Yao Y, He Y, Molokie R, Wilkie D, et al. Interleukin 1alpha RS1800587 associates with chronic non-crisis pain in sickle cell disease. J Pain. (2016) 17:S46. doi: 10.1016/j.jpain.2016.01.186
25. Jhun E, He Y, Yao Y, Molokie RE, Wilkie DJ, Jim Wang Z. Dopamine D3 receptor Ser9Gly and catechol-O-methyltransferase Val158Met polymorphisms and acute pain in sickle cell disease. Anesth Analg. (2014) 119:1201–7. doi: 10.1213/ANE.0000000000000382
26. Jhun EH, X HU, Sadhu N, Yao Y, He Y, Wilkie DJ, et al. Transient receptor potential polymorphism and haplotype associate with crisis pain in sickle cell disease. Pharmacogenomics. (2018a) 19:401–11. doi: 10.2217/pgs-2017-0198
27. Jhun EH, Sadhu N, He Y, Yao Y, Wilkie DJ, Molokie RE, et al. S100b single nucleotide polymorphisms exhibit sex-specific associations with chronic pain in sickle cell disease in a largely African-American cohort. PLoS One. (2020) 15:e0232721. doi: 10.1371/journal.pone.0232721
28. Jhun EH, Sadhu N, Hu X, Yao Y, He Y, Wilkie DJ, et al. Beta2-adrenergic receptor polymorphisms and haplotypes associate with chronic pain in sickle cell disease. Front Pharmacol. (2019) 10:84. doi: 10.3389/fphar.2019.00084
29. Jhun EH, Sadhu N, Yao Y, He Y, Molokie RE, Wilkie DJ, et al. Glucocorticoid receptor single nucleotide polymorphisms are associated with acute crisis pain in sickle cell disease. Pharmacogenomics. (2018b) 19:1003–11. doi: 10.2217/pgs-2018-0064
30. Kumar R, Yadav R, Mishra S, Singh M, Gwal A, Bharti PK, et al. Kruppel-like factor 1 (KLF1) gene single nucleotide polymorphisms in sickle cell disease and its association with disease-related morbidities. Ann Hematol. (2021) 100:365–73. doi: 10.1007/s00277-020-04381-z
31. Powell-Roach KL, Yao Y, Jhun EH, He Y, Suarez ML, Ezenwa MO, et al. Vasopressin SNP pain factors and stress in sickle cell disease. PLoS One. (2019) 14:e0224886. doi: 10.1371/journal.pone.0224886
32. Sadhu N, Jhun EH, Posen A, Yao Y, He Y, Molokie RE, et al. Phenylethanolamine N-methyltransferase gene polymorphisms associate with crisis pain in sickle cell disease patients. Pharmacogenomics. (2020) 21:269–78. doi: 10.2217/pgs-2019-0096
33. Sadhu N, Jhun EH, Yao Y, He Y, Molokie RE, Wilkie DJ, et al. Genetic variants of GCH1 associate with chronic and acute crisis pain in African Americans with sickle cell disease. Exp Hematol. (2018) 66:42–9. doi: 10.1016/j.exphem.2018.07.004
34. Wang X, Gardner K, Tegegn MB, Dalgard CL, Alba C, Menzel S, et al. Genetic variants of PKLR are associated with acute pain in sickle cell disease. Blood Adv. (2022) 6(11):3535–40. doi: 10.1182/bloodadvances.2021006668
35. Wonkam A, Mnika K, Ngo Bitoungui VJ, Chetcha Chemegni B, Chimusa ER, Dandara C, et al. Clinical and genetic factors are associated with pain and hospitalisation rates in sickle cell anaemia in Cameroon. Br J Haematol. (2018) 180:134–46. doi: 10.1111/bjh.15011
36. Wilkie DJ, Molokie RE, Suarez ML, Ezenwa MO, Wang ZJ. Composite pain index: reliability, validity, and sensitivity of a patient-reported outcome for research. Pain Med. (2015) 16:1341–8. doi: 10.1111/pme.12703
37. Prestia B, Ramzan T, Waldron C, Malik A, Pallay R, Murbach C, et al. Utilization of patient-controlled analgesia reduces length of stay of sickle cell crisis hospitalizations. HCA Healthc J Med. (2021) 2(4):12. doi: 10.36518/2689-0216.1275
38. Brandow AM, Carroll CP, Creary S, Edwards-Elliott R, Glassberg J, Hurley RW, et al. American society of hematology 2020 guidelines for sickle cell disease: management of acute and chronic pain. Blood Adv. (2020) 4:2656–701. doi: 10.1182/bloodadvances.2020001851
39. Powell-Roach KL, Yao Y, Rutherford JN, Schlaeger JM, Patil CL, Suarez ML, et al. Thermal and mechanical quantitative sensory testing values among healthy African American adults. J Pain Res. (2019) 12:2511–27. doi: 10.2147/JPR.S211855
40. Ezenwa MO, Molokie RE, Wang ZJ, Yao Y, Suarez ML, Pullum C, et al. Safety and utility of quantitative sensory testing among adults with sickle cell disease: indicators of neuropathic pain? Pain Pract. (2016) 16:282–93. doi: 10.1111/papr.12279
41. Kushnir VM, Cassell B, Gyawali CP, Newberry RD, Kibe P, Nix BD, et al. Genetic variation in the beta-2 adrenergic receptor (ADRB2) predicts functional gastrointestinal diagnoses and poorer health-related quality of life. Aliment Pharmacol Ther. (2013) 38:313–23. doi: 10.1111/apt.12378
42. Bonato LL, Quinelato V, De Felipe Cordeiro PC, Vieira AR, Granjeiro JM, Tesch R, et al. Polymorphisms in COMT, ADRB2 and HTR1A genes are associated with temporomandibular disorders in individuals with other arthralgias. CRANIO®. (2021) 39:351–61. doi: 10.1080/08869634.2019.1632406
43. Molokie RE, Wang ZJ, Wilkie DJ. Presence of neuropathic pain as an underlying mechanism for pain associated with cold weather in patients with sickle cell disease. Med Hypotheses. (2011) 77:491–3. doi: 10.1016/j.mehy.2011.06.018
44. Wang ZJ, Wilkie DJ, Molokie R. Neurobiological mechanisms of pain in sickle cell disease. Hematology Am Soc Hematol Educ Program. (2010) 2010:403–8. doi: 10.1182/asheducation-2010.1.403
45. Wilkie DJ, Molokie R, Boyd-Seal D, Suarez ML, Kim YO, Zong S, et al. Patient-reported outcomes: descriptors of nociceptive and neuropathic pain and barriers to effective pain management in adult outpatients with sickle cell disease. J Natl Med Assoc. (2010) 102:18–27. doi: 10.1016/s0027-9684(15)30471-5
Keywords: genetic variant, single nucleotide polymorphism, sickle cell disease, acute pain, chronic pain
Citation: Gehling GM, Powell-Roach K, Wilkie DJ and Dungan JR (2023) Single nucleotide polymorphisms and sickle cell disease-related pain: a systematic review. Front. Pain Res. 4:1223309. doi: 10.3389/fpain.2023.1223309
Received: 16 May 2023; Accepted: 21 August 2023;
Published: 14 September 2023.
Edited by:
Bradley Kerr, University of Alberta, CanadaReviewed by:
Kalpna Gupta, University of California, Irvine, United StatesErin E. Young, University of Kansas School of Medicine, KU Medical Center, United States
© 2023 Gehling, Powell-Roach, Wilkie and Dungan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer R. Dungan anJkdW5nYW5AdWZsLmVkdQ==