 Dustin L. Higashi1
Dustin L. Higashi1 Jens Kreth
Jens Kreth Justin Merritt
Justin Merritt
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Oral. Health , 11 June 2024
Sec. Oral Infections and Microbes
Volume 5 - 2024 | https://doi.org/10.3389/froh.2024.1413842
This article is part of the Research Topic Proceedings of the 3rd International Conference on Oral Mucosal Immunity and Microbiome View all 8 articles
Inflammatory dysbiotic diseases present an intriguing biological paradox. Like most other infectious disease processes, the alarm bells of the host are potently activated by tissue-destructive pathobionts, triggering a cascade of physiological responses that ultimately mobilize immune cells like neutrophils to sites of active infection. Typically, these inflammatory host responses are critical to inhibit and/or eradicate infecting microbes. However, for many inflammatory dysbiotic diseases, inflammophilic pathobiont-enriched communities not only survive the inflammatory response, but they actually obtain a growth advantage when challenged with an inflammatory environment. This is especially true for those organisms that have evolved various strategies to resist and/or manipulate components of innate immunity. In contrast, members of the commensal microbiome typically experience a competitive growth disadvantage under inflammatory selective pressure, hindering their critical ability to restrict pathobiont proliferation. Here, we examine examples of bacteria-neutrophil interactions from both conventional pathogens and inflammophiles. We discuss some of the strategies utilized by them to illustrate how inflammophilic microbes can play a central role in the positive feedback cycle that exemplifies dysbiotic chronic inflammatory diseases.
Inflammation is a natural and necessary biological response to infection, injury, and/or antigen exposure. Clinically, the outward symptoms of inflammation include localized heat, redness, edema, pain, and the loss of proper function to the affected area. On a cellular level, the inflammatory process is highly complex and nuanced, but its overarching goal is to eliminate the insulting agent, remove damaged cells, and initiate wound healing.
Inflammation is a crucial protective response of the body when functioning properly. However, its dysregulation can result in an array of diseases, such as atherosclerosis, diabetes, irritable bowel syndrome, cancer, periodontal disease, and others (1, 2). Interestingly, in many of these cases, local inflammation is associated with a dysbiotic shift in the microbial community of the affected site (3). These population shifts not only serve to exacerbate pathogenesis, but they may also serve as an essential step in the sequence of events leading to the downward spiral of chronic inflammation (Figure 1).
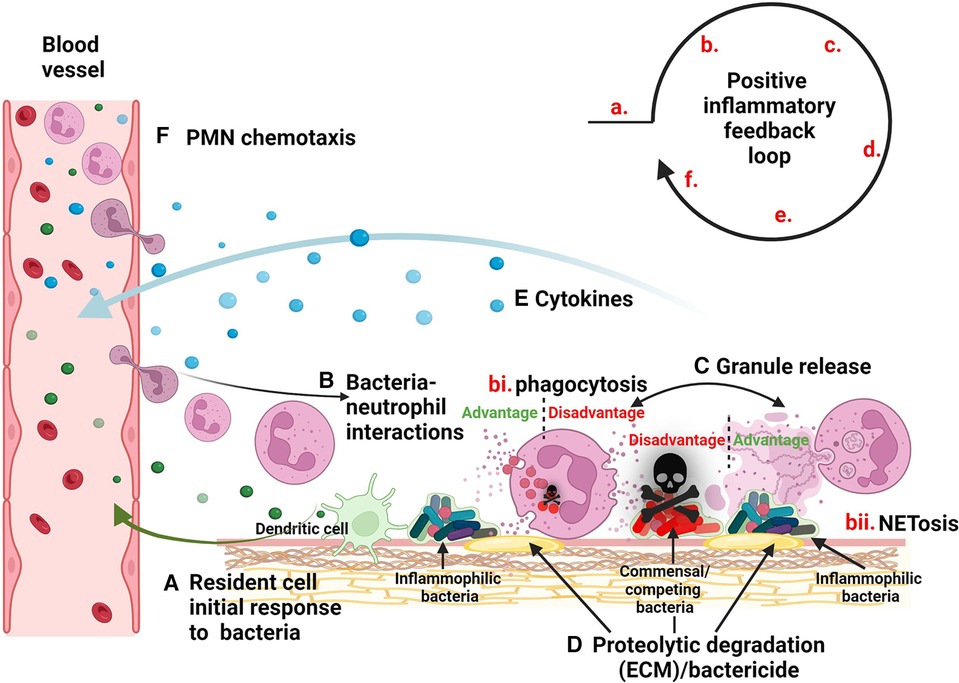
Figure 1. Model of inflammatory dysbiotic disease. (A) Inflammophilic bacteria evoke the migration of neutrophils to the site of infection via signaling through resident cells such as dendritic cells. (B) Neutrophils mobilize to sites of infection where they encounter bacteria leading to processes such as (bi) phagocytosis and degranulation or (bii) NETosis. (C) Granule release causes the (D) breakdown of the ECM as well as the elimination of competing micobes. (E) Bacteria stimulate neutrophils to release cytokines, (F) providing a positive feedback loop for mobilization of more neutrophils.
During health, the oral microbiome is typically enriched in commensal species, often organisms from genera such as Neisseria, Actinomyces, Rothia, Corynebacterium, and Streptococcus. These populations promote eubiosis with the host by limiting acidification of dental plaque, antagonizing the growth of pathobionts, and protecting the gingiva by dampening inflammation (4). For periodontitis, which is a chronic inflammatory disease that damages the supporting structures surrounding the tooth, disease is characterized by a dysbiotic shift in favor of inflammophilic microbes (i.e., those that thrive within an inflammatory environment). Genera such as Parvimonas, Porphyromonas, Fusobacterium, Prevotella, Tannerella, Treponema, and others predominate concurrently with chronic gingival inflammation, resorption of the alveolar bone, and the destruction of the connective tissues supporting the teeth (5–8). Presently, we only have a tenuous understanding of the polymicrobial aspects of dysbiotic immunopathology, but studies hint at highly coordinated and evolved processes.
For oral dysbiotic communities, members exhibit an exquisite reliance upon polymicrobial synergism to promote and maintain chronic inflammation. Through cooperation, microbes within these communities are able to enhance colonization, optimize nutrient acquisition, provide protection from environmental insults, and bolster pathogenicity (9). Accordingly, a number of studies have demonstrated how such synergism may occur among prominent inflammophilic oral pathobionts. For instance, Parvimonas micra has been shown to coaggregate with Treponema denticola, Porphyromonas gingivalis, and Fusobacterium nucleatum (10–12). Furthermore, P. micra can utilize soluble factors from these organisms to enhance its own growth, and can in turn, release factors that increase biofilm formation and the growth of P. gingivalis and F. nucleatum (11). Mixed biofilms of P. micra display increased resistance to sodium hypochlorite treatment as compared to single species biofilms (13). Coinfection with Prevotella intermedia was shown to increase the virulence of P. micra in a murine abscess model (14). P. micra may also influence the production of virulence factors from other members of the community. For example, P. micra is a potent stimulator of P. gingivalis gingipains, which are secreted proteases that serve as key virulence factors involved in host cell adhesion, nutrient acquisition, biofilm development, and immune evasion (15).
The strength of the symbiotic relationships between inflammophilic microbes is demonstrated by their frequent, if not typical, associations across numerous diseases. Indeed, many of the same species enriched in periodontitis are also highly prominent in acute infections such as odontogenic abscesses (16, 17). In malignant tumors, studies have demonstrated an enrichment of many of the same genera as those found in oral dysbiotic diseases, especially Parvimonas, Fusobacterium, and Prevotella (18–21). Co-infections at typically sterile sites throughout the body illustrate how members within these groups can survive extensive journeys together to establish new infections at distant extraoral sites (22, 23).
While it is now evident that inflammophilic bacterial communities are able to survive and thrive in inflammatory environments, the specific roles of each member remain largely unknown in the polymicrobial context. To understand the mechanisms of polymicrobial synergism in inflammatory dysbiotic disease, it is necessary to first reveal how individual species within these communities impact host immunity. Once this baseline understanding is established, the field can then compare how polymicrobial synergism modifies the expressed phenotypes of more complex assemblages. With this in mind, we recently presented new findings regarding neutrophil interactions with the inflammophilic pathobiont P. micra while at the 3rd International Conference on Oral Mucosal Immunity and Microbiome. However, these results represent only a small component of a much larger puzzle. Therefore, the following sections will describe some prominent examples of the interplay between neutrophils and inflammophilic microbes as well as conventional pathogens to demonstrate the broader implications for inflammatory dysbiotic disease, especially among complex polymicrobial communities. We discuss how aspects of inflammation provide a selective advantage, particularly for the inflammophilic members within these communities.
Polymorphonuclear leukocytes (neutrophils) are a central component of the innate immune system and make up the majority of white blood cells in humans. As one of the first defenders to sites of infection, neutrophils are key mediators that influence the host response and play a central role in pathogenesis and the resolution of disease (24–26). In recent years, it has become increasingly apparent that neutrophils engage in extensive crosstalk with both immune and non-immune cells (27). They have been shown to induce the production and secretion of cytokines from endothelial and epithelial cells (28). Neutrophils induce the activation and migration of macrophages and dendritic cells. They can also facilitate the activation, inhibition, and differentiation of T cells as well as promote B cell expansion (29). Amazingly, multiple studies have reported that T and B cell activation can be directly facilitated by a specialized subset of neutrophils capable of presenting antigens via both MHC class I and class II (30, 31).
Neutrophils are mini-armories, possessing a vast array of defensive and offensive weaponry to battle microbial infections. These include multiple types of granules—tiny membrane-bound organelles within neutrophils containing numerous components like complement receptor, lactoferrin, metalloproteases, lysozyme, elastase, collagenase, myeloperoxidase, defensins, and more (32). These cargos are intimately connected with almost every aspect of neutrophil biology and have a myriad of functions, including the digestion of microbes, nutrient sequestration, antimicrobial activity, and modulation of the adaptive immune response.
Granule contents are not only toxic to microbes, but they can also damage host tissues following extracellular degranulation (i.e., release of neutrophil granule contents to the extracellular milieu). Certain infecting microbes are particularly adept at provoking neutrophil degranulation. For example, a methicillin-resistant strain of Staphylococcus aureus was shown to utilize a phenol-soluble modulin to induce the phosphoinositide 3-kinase (PI3 K) dependent degranulation of neutrophils (33). Streptococcus pyogenes induces degranulation through an M protein-fibrinogen complex (34). Filifactor alocis engagement of TLR2 induces the release of secondary specific granules (35). In contrast, some microbes actively suppress neutrophil degranulation. Chlamydia trachomatis produces a protease-like activating factor (CPAF) that cleaves formyl peptide receptor 2 (FPR2) on neutrophils, preventing PI3K-induced degranulation. Importantly, a C. trachomatis CPAF mutant was demonstrated to be highly susceptible to neutrophil granules, unlike its parental wild-type (36).
How does extracellular degranulation influence disease? It is possible that certain microbes may actively induce granule release as an adjunctive strategy to impair susceptible competitors, usurping the antimicrobial arsenal of the host. For example, endodontic infections are seeded by the oral biofilm, yet persistent pressure from a neutrophil-rich immune response results in an abscess community composition that exhibits little resemblance to its original inoculum (16, 37). In particular, much of the commensal microbiome antagonists of oral pathobionts become depleted within the abscess environment, whereas these same species comprise a prominent fraction of normal dental plaque communities (38). Inducing degranulation could also provide nutrition for bacteria. Granules contain proteases that target not only bacteria but also extracellular matrix (ECM) proteins such as elastin and collagen. Release of these enzymes into the ECM facilitates the breakdown of host proteins into smaller peptides, making them available for bacterial consumption. It is worth noting that many of the commonly encountered inflammophilic pathobionts are also slow-growing fastidious organisms that prefer to metabolize amino acids, in contrast to the typical commensal species, which are primarily fast-growing saccharolytic organisms. Thus, inflammatory tissue destruction naturally provides a continual source of soluble free peptides, which are the preferred carbon sources for key inflammophilic organisms (39, 40). In the context of host signaling, degranulation is also pro-inflammatory, which serves to further promote an inflammatory growth environment favorable to inflammophilic microbes and unfavorable to many commensal species.
Neutrophils are professional phagocytes that possess an uncanny capacity to engulf and eliminate bacteria. After ingestion, neutrophil granules fuse with the bacterial phagosome resulting in the release of toxic bactericidal compounds. While an effective means to eliminate unwanted invaders, some pathogens have evolved mechanisms to circumvent these defenses.
One strategy involves targeting neutrophil phagocytosis at its initial stages. P. gingivalis signals neutrophils through TLR2 and C5aR leading to the proteasomal degradation of MyD88 and activating an alternate TLR2-Mal-PI3 K pathway that inhibits phagocytosis (41). Neisseria meningitidis avoids complement-mediated phagocytosis by binding host Factor H (42), whereas Yersinia pestis produces a F1 capsule which directly inhibits phagocytosis (43).
Following phagocytosis, the bacteria-containing phagosome matures through fusion with antimicrobial granules, resulting in the killing of the ingested microbe. A number of bacteria are able to prevent killing after ingestion by neutrophils. Neisseria gonorrhoeae can alter phase-specific production of the Opa protein to prevent the accumulation of primary granule proteins, thus enhancing their survival (42). The S. pyogenes M protein has been shown to selectively prevent the fusion of primary granules with phagosomes (44). Live F. alocis prevents primary granule recruitment to its phagosome compared to heat killed bacteria (45) and Mycobacterium smegmatis phagosomes fail to fuse with primary granules (46).
Microbes may benefit from the manipulation of phagocytosis and granule fusion in multiple ways. First, they avoid killing by preventing the engagement of neutrophil granules altogether. Extracellular bacteria remain unharmed while ingested bacteria are afforded protection from external threats. Furthermore, these intracellular bacteria could conceivably utilize the migrating neutrophil to disseminate to new sites of infection.
Neutrophils can also produce extracellular traps (NETs) in response to infection. Through a process called NETosis, neutrophils elaborate structures comprised of extracellular fibers of chromatin and granular proteins that are able to trap and kill pathogens (47). While an effective defense against most microbes, a number of bacteria have evolved strategies to subvert NET production or function (48).
S. pyogenes produces the protease SpyCEP which cleaves IL-8 to inhibit NET formation (49). S. aureus signaling through the phosphatase Wip1, inhibits calcium signaling to suppress NETosis (50). Pseudomonas aeruginosa adsorbs host sialoglycoproteins to engage siglec-9 and inhibit NET formation. S. pyogenes also engages siglec-9 for a similar outcome, but does so via its production of a high molecular weight hyaluronan capsule (51, 52). In group B Streptococcus, the cell wall β-protein impairs NET formation through engagement of siglec-5 (53).
In the presence of NETs, bacteria have evolved other means of survival. Prevotella intermedia can degrade NETs through the activity of NucA and NucD nucleases (54). Likewise, S. pyogenes produces Sda1 and SpnA nucleases to digest NETs and promote survival (55, 56). P. gingivalis produces the enzyme peptidylarginine deiminase (PPAD), which was shown to citrullinate histone H3 and the antimicrobial peptide LP9, facilitating bacterial escape and survival (57). Furthermore, P. gingivalis gingipain induces the activation of protease-activated receptor-2 (PAR-2) to induce NETs that lack bactericidal activity and also stimulate P. gingivalis growth (58).
The manipulation of NETosis and NETs by microbes demonstrates a highly evolved arms race which seems to involve most aspects of neutrophil biology. The ability of microbes to use compounds recognized as “self” molecules like hyaluronan and sialoglycoproteins supports signaling through host-specific pathways and demonstrates an ingenious form of molecular mimicry to undermine host defenses. Many microbes naturally promote NETosis as a consequence of their inherent pro-inflammatory nature (48). For those that have evolved ways to resist NET-dependent killing, the formation of NETs themselves may even be beneficial due to its aggravation of the inflammatory response (59). NETs contain many granular components including proteases like elastase. As posited above, the ability of these enzymes to digest the ECM may provide a peptide-rich food source for surrounding microbes, especially organisms that favor amino acid fermentation. Similarly, antimicrobial NETs may also target competing species like commensal organisms, further selecting for inflammophilic community development. NETs may also delay wound healing, which would encourage persistent infections (59).
Historically, neutrophils were not considered a major source of signaling molecules and were primarily viewed as strict responders to environment cues such as the chemokines produced by activated epithelial cells (60). However, it is now well established that neutrophils actively secrete numerous signal molecules, such as various cytokines (e.g., TNFα, IL-1β, IL-6, IL-8), colony-stimulating and angiogenic factors, as well as growth factors (61). Neutrophils treated with Mycobacterium tuberculosis secrete IL-8 and GRO-α (62). P. gingivalis, Peptoanaerobacter stomatis, and F. alocis induce neutrophil release of TNFα, IL-1β, and IL-1RA (63).
The abundance of neutrophils present in dysbiotic inflammatory infections suggests that bacteria having the ability to regulate anti- or pro-inflammatory signaling cascades have likely tapped into a potent feedback loop to actively modulate inflammation.
Inflammatory dysbiosis results from the disruption of normal microbiome ecology, resulting in the overgrowth of normally low abundance pathobionts. The species within these microbial communities synergize to support their survival and growth by actively promoting an ineffective inflammatory response. How do these bacteria thrive in such a hostile environment designed to inhibit bacterial growth? One explanation may lie in the fact that many inflammophilic pathobionts have naturally evolved to reside in such environments. For example, oral pathobionts like P. micra typically reside within the gingival sulcus, an environment continually bathed in gingival crevicular fluid (GCF), which is comprised of many innate host defenses like proteolytic enzymes, antibodies, complement, and neutrophils (64). It is conceivable that the same phenotypes allowing pathobionts to persist in the gingival sulcus could prove to be pathogenic to the host when expressed in a different environmental context, especially if such organisms were to achieve high numbers in the community. Studies on microbe-neutrophil interactions provide clues into the types of strategies employed by individual inflammophilic bacteria. It is clear that some microbes are able to commandeer and manipulate neutrophil functions for their benefit. The looming question is whether these same responses observed from individual species still occur similarly in a polymicrobial context. For example, some oral pathobionts provoke neutrophil NETosis, while others inhibit this process. What is the final outcome when these organisms coexist in polymicrobial communities? Is it simply a numbers game, with the greater abundance organism yielding the dominant effect upon surrounding neutrophils or is it an altogether unique neutrophil response that does not resemble the responses to the individual constituents of the community? One could ask a similar question regarding neutrophil production of signal molecules that direct downstream components of the immune response.
These questions portend necessary future investigations of inflammophilic synergism as a crucial next frontier in dysbiotic pathogenesis research. A better understanding of the mechanisms used by inflammophilic microbes to manipulate neutrophils during infection may reveal new therapeutic strategies for the treatment of dysbiotic inflammatory diseases. Indeed, therapeutic approaches targeting neutrophil functions have already been proposed for the treatment of ailments such as cancer, pulmonary disease, and sepsis, with active clinical trials investigating a number of medical indications (65). For inflammatory oral diseases like periodontitis, therapeutic approaches targeting pathobiont-induced neutrophil immunopathologies may ultimately restrict pathobiont growth, thereby reducing inflammation, tissue damage, and bone loss.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
DH: Writing – original draft, Writing – review & editing. HQ: Writing – review & editing. CB: Writing – review & editing. JK: Writing – review & editing. JM: Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
JK acknowledges the support of NIH-NIDCR grants DE029612 and DE029492 and JM of grant DE028252.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bennett JM, Reeves G, Billman GE, Sturmberg JP. Inflammation-nature’s way to efficiently respond to all types of challenges: implications for understanding and managing “the epidemic” of chronic diseases. Front Med (Lausanne). (2018) 5:316. doi: 10.3389/fmed.2018.00316
2. Higashi DL, Krieger MC, Qin H, Zou Z, Palmer EA, Kreth J, et al. Who is in the driver’s seat? Parvimonas micra: an understudied pathobiont at the crossroads of dysbiotic disease and cancer. Environ Microbiol Rep. (2023) 4:254–64. doi: 10.1111/1758-2229.13153
3. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. (2019) 25(12):1822–32. doi: 10.1038/s41591-019-0675-0
4. Sedghi L, DiMassa V, Harrington A, Lynch SV, Kapila YL. The oral microbiome: role of key organisms and complex networks in oral health and disease. Periodontol 2000. (2021) 87(1):107–31. doi: 10.1111/prd.12393
5. Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, et al. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. (2013) 7(5):1016–25. doi: 10.1038/ismej.2012.174
6. Boutin S, Hagenfeld D, Zimmermann H, El Sayed N, Hopker T, Greiser HK, et al. Clustering of subgingival microbiota reveals microbial disease ecotypes associated with clinical stages of periodontitis in a cross-sectional study. Front Microbiol. (2017) 8:340. doi: 10.3389/fmicb.2017.00340
7. Kirst ME, Li EC, Alfant B, Chi YY, Walker C, Magnusson I, et al. Dysbiosis and alterations in predicted functions of the subgingival microbiome in chronic periodontitis. Appl Environ Microbiol. (2015) 81(2):783–93. doi: 10.1128/AEM.02712-14
8. Lourenco TG, Heller D, Silva-Boghossian CM, Cotton SL, Paster BJ, Colombo AP. Microbial signature profiles of periodontally healthy and diseased patients. J Clin Periodontol. (2014) 41(11):1027–36. doi: 10.1111/jcpe.12302
9. Lamont RJ, Koo H, Hajishengallis G. The oral Microbiota: dynamic communities and host interactions. Nat Rev Microbiol. (2018) 16(12):745–59. doi: 10.1038/s41579-018-0089-x
10. Cogoni V, Morgan-Smith A, Fenno JC, Jenkinson HF, Dymock D. Treponema denticola chymotrypsin-like proteinase (ctlp) integrates Spirochaetes within oral microbial communities. Microbiology (Reading). (2012) 158(Pt 3):759–70. doi: 10.1099/mic.0.055939-0
11. Horiuchi A, Kokubu E, Warita T, Ishihara K. Synergistic biofilm formation by Parvimonas micra and Fusobacterium nucleatum. Anaerobe. (2020) 62:102100. doi: 10.1016/j.anaerobe.2019.102100
12. Kremer BH, van Steenbergen TJ. Peptostreptococcus micros coaggregates with Fusobacterium nucleatum and non-encapsulated Porphyromonas gingivalis. FEMS Microbiol Lett. (2000) 182(1):57–62. doi: 10.1111/j.1574-6968.2000.tb08873.x
13. Ozok AR, Wu MK, Luppens SB, Wesselink PR. Comparison of growth and susceptibility to sodium hypochlorite of mono- and dual-species biofilms of Fusobacterium nucleatum and Peptostreptococcus (micromonas) micros. J Endod. (2007) 33(7):819–22. doi: 10.1016/j.joen.2007.03.008
14. Araki H, Kuriyama T, Nakagawa K, Karasawa T. The microbial synergy of Peptostreptococcus micros and Prevotella intermedia in a murine abscess model. Oral Microbiol Immunol. (2004) 19(3):177–81. doi: 10.1111/j.0902-0055.2004.00138.x
15. Neilands J, Davies JR, Bikker FJ, Svensater G. Parvimonas micra stimulates expression of gingipains from Porphyromonas gingivalis in multi-Species communities. Anaerobe. (2019) 55:54–60. doi: 10.1016/j.anaerobe.2018.10.007
16. Colombo AP, Boches SK, Cotton SL, Goodson JM, Kent R, Haffajee AD, et al. Comparisons of subgingival microbial profiles of refractory periodontitis, severe periodontitis, and periodontal health using the human oral microbe identification microarray. J Periodontol. (2009) 80(9):1421–32. doi: 10.1902/jop.2009.090185
17. Gomes BP, Lilley JD, Drucker DB. Associations of endodontic symptoms and signs with particular combinations of specific bacteria. Int Endod J. (1996) 29(2):69–75. doi: 10.1111/j.1365-2591.1996.tb01164.x
18. Dai Z, Coker OO, Nakatsu G, Wu WKK, Zhao L, Chen Z, et al. Multi-cohort analysis of colorectal cancer metagenome identified altered bacteria across populations and universal bacterial markers. Microbiome. (2018) 6(1):70. doi: 10.1186/s40168-018-0451-2
19. Yang CY, Yeh YM, Yu HY, Chin CY, Hsu CW, Liu H, et al. Oral Microbiota community dynamics associated with oral squamous cell carcinoma staging. Front Microbiol. (2018) 9:862. doi: 10.3389/fmicb.2018.00862
20. Yu J, Feng Q, Wong SH, Zhang D, Liang QY, Qin Y, et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut. (2017) 66(1):70–8. doi: 10.1136/gutjnl-2015-309800
21. Zhao H, Chu M, Huang Z, Yang X, Ran S, Hu B, et al. Variations in oral microbiota associated with oral cancer. Sci Rep. (2017) 7(1):11773. doi: 10.1038/s41598-017-11779-9
22. Durovic A, Eberhard N, Scharen S, Widmer AF. Parvimonas micra as a rare cause of spondylodiscitis - case series from a single centre. Swiss Med Wkly. (2020) 150:w20272. doi: 10.4414/smw.2020.20272
23. Murdoch DA, Mitchelmore IJ, Tabaqchali S. Peptostreptococcus micros in polymicrobial abscesses. Lancet. (1988) 1(8585):594. doi: 10.1016/s0140-6736(88)91393-1
24. Burn GL, Foti A, Marsman G, Patel DF, Zychlinsky A. The neutrophil. Immunity. (2021) 54(7):1377–91. doi: 10.1016/j.immuni.2021.06.006
25. Hajishengallis G, Chavakis T, Hajishengallis E, Lambris JD. Neutrophil homeostasis and inflammation: novel paradigms from studying periodontitis. J Leukoc Biol. (2015) 98(4):539–48. doi: 10.1189/jlb.3VMR1014-468R
26. Uriarte SM, Hajishengallis G. Neutrophils in the periodontium: interactions with pathogens and roles in tissue homeostasis and inflammation. Immunol Rev. (2023) 314(1):93–110. doi: 10.1111/imr.13152
27. Selders GS, Fetz AE, Radic MZ, Bowlin GL. An overview of the role of neutrophils in innate immunity, inflammation and host-biomaterial integration. Regen Biomater. (2017) 4(1):55–68. doi: 10.1093/rb/rbw041
28. Owen CA, Campbell EJ. The cell biology of leukocyte-mediated proteolysis. J Leukoc Biol. (1999) 65(2):137–50. doi: 10.1002/jlb.65.2.137
29. Shrestha S, Hong CW. Extracellular mechanisms of neutrophils in immune cell crosstalk. Immune Netw. (2023) 23(5):e38. doi: 10.4110/in.2023.23.e38
30. Mysore V, Cullere X, Mears J, Rosetti F, Okubo K, Liew PX, et al. Fcgammar engagement reprograms neutrophils into antigen cross-presenting cells that elicit acquired anti-tumor immunity. Nat Commun. (2021) 12(1):4791. doi: 10.1038/s41467-021-24591-x
31. Rosales C. Neutrophils at the crossroads of innate and adaptive immunity. J Leukoc Biol. (2020) 108(1):377–96. doi: 10.1002/JLB.4MIR0220-574RR
32. Eichelberger KR, Goldman WE. Manipulating neutrophil degranulation as a bacterial virulence strategy. PLoS Pathog. (2020) 16(12):e1009054. doi: 10.1371/journal.ppat.1009054
33. Li L, Pian Y, Chen S, Hao H, Zheng Y, Zhu L, et al. Phenol-soluble modulin alpha4 mediates Staphylococcus aureus-associated vascular leakage by stimulating heparin-binding protein release from neutrophils. Sci Rep. (2016) 6:29373. doi: 10.1038/srep29373
34. Herwald H, Cramer H, Morgelin M, Russell W, Sollenberg U, Norrby-Teglund A, et al. M protein, a classical bacterial virulence determinant, forms complexes with fibrinogen that induce vascular leakage. Cell. (2004) 116(3):367–79. doi: 10.1016/s0092-8674(04)00057-1
35. Armstrong CL, Miralda I, Neff AC, Tian S, Vashishta A, Perez L, et al. Filifactor alocis promotes neutrophil degranulation and chemotactic activity. Infect Immun. (2016) 84(12):3423–33. doi: 10.1128/IAI.00496-16
36. Rajeeve K, Das S, Prusty BK, Rudel T. Chlamydia trachomatis paralyses neutrophils to evade the host innate immune response. Nat Microbiol. (2018) 3(7):824–35. doi: 10.1038/s41564-018-0182-y
37. Chen J, Wu X, Zhu D, Xu M, Yu Y, Yu L, et al. Microbiota in human periodontal abscess revealed by 16s rdna sequencing. Front Microbiol. (2019) 10:1723. doi: 10.3389/fmicb.2019.01723
38. Bottger S, Zechel-Gran S, Schmermund D, Streckbein P, Wilbrand JF, Knitschke M, et al. Microbiome of odontogenic abscesses. Microorganisms. (2021) 9(6):1307. doi: 10.3390/microorganisms9061307
39. Murdoch DA, Mitchelmore IJ. The laboratory identification of gram-positive anaerobic cocci. J Med Microbiol. (1991) 34(5):295–308. doi: 10.1099/00222615-34-5-295
40. Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol. (2010) 10(10):712–23. doi: 10.1038/nri2852
41. Maekawa T, Krauss JL, Abe T, Jotwani R, Triantafilou M, Triantafilou K, et al. Porphyromonas gingivalis manipulates complement and tlr signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe. (2014) 15(6):768–78. doi: 10.1016/j.chom.2014.05.012
42. Allen LH, Criss AK. Cell intrinsic functions of neutrophils and their manipulation by pathogens. Curr Opin Immunol. (2019) 60:124–9. doi: 10.1016/j.coi.2019.05.004
43. Dudte SC, Hinnebusch BJ, Shannon JG. Characterization of Yersinia pestis interactions with human neutrophils in vitro. Front Cell Infect Microbiol. (2017) 7:358. doi: 10.3389/fcimb.2017.00358
44. Staali L, Bauer S, Morgelin M, Bjorck L, Tapper H. Streptococcus pyogenes bacteria modulate membrane traffic in human neutrophils and selectively inhibit azurophilic granule fusion with phagosomes. Cell Microbiol. (2006) 8(4):690–703. doi: 10.1111/j.1462-5822.2005.00662.x
45. Edmisson JS, Tian S, Armstrong CL, Vashishta A, Klaes CK, Miralda I, et al. Filifactor alocis modulates human neutrophil antimicrobial functional responses. Cell Microbiol. (2018) 20(6):e12829. doi: 10.1111/cmi.12829
46. Cougoule C, Constant P, Etienne G, Daffe M, Maridonneau-Parini I. Lack of fusion of azurophil granules with phagosomes during phagocytosis of Mycobacterium smegmatis by human neutrophils is not actively controlled by the bacterium. Infect Immun. (2002) 70(3):1591–8. doi: 10.1128/IAI.70.3.1591-1598.2002
47. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
48. Baz AA, Hao H, Lan S, Li Z, Liu S, Chen S, et al. Neutrophil extracellular traps in bacterial infections and evasion strategies. Front Immunol. (2024) 15:1357967. doi: 10.3389/fimmu.2024.1357967
49. Zinkernagel AS, Timmer AM, Pence MA, Locke JB, Buchanan JT, Turner CE, et al. The il-8 protease spycep/scpc of group a Streptococcus promotes resistance to neutrophil killing. Cell Host Microbe. (2008) 4(2):170–8. doi: 10.1016/j.chom.2008.07.002
50. Chen Y, Zhao C, Guo H, Zou W, Zhang Z, Wei D, et al. Wip1 inhibits neutrophil extracellular traps to promote abscess formation in mice by directly dephosphorylating coronin-1a. Cell Mol Immunol. (2023) 20(8):941–54. doi: 10.1038/s41423-023-01057-2
51. Khatua B, Bhattacharya K, Mandal C. Sialoglycoproteins adsorbed by Pseudomonas aeruginosa facilitate their survival by impeding neutrophil extracellular trap through siglec-9. J Leukoc Biol. (2012) 91(4):641–55. doi: 10.1189/jlb.0511260
52. Secundino I, Lizcano A, Roupe KM, Wang X, Cole JN, Olson J, et al. Host and pathogen hyaluronan signal through human siglec-9 to suppress neutrophil activation. J Mol Med (Berl). (2016) 94(2):219–33. doi: 10.1007/s00109-015-1341-8
53. Carlin AF, Chang YC, Areschoug T, Lindahl G, Hurtado-Ziola N, King CC, et al. Group B Streptococcus suppression of phagocyte functions by protein-mediated engagement of human siglec-5. J Exp Med. (2009) 206(8):1691–9. doi: 10.1084/jem.20090691
54. Doke M, Fukamachi H, Morisaki H, Arimoto T, Kataoka H, Kuwata H. Nucleases from Prevotella intermedia can degrade neutrophil extracellular traps. Mol Oral Microbiol. (2017) 32(4):288–300. doi: 10.1111/omi.12171
55. Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, et al. Dnase expression allows the pathogen group a Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol. (2006) 16(4):396–400. doi: 10.1016/j.cub.2005.12.039
56. Chang A, Khemlani A, Kang H, Proft T. Functional analysis of Streptococcus pyogenes nuclease a (spna), a novel group a streptococcal virulence factor. Mol Microbiol. (2011) 79(6):1629–42. doi: 10.1111/j.1365-2958.2011.07550.x
57. Stobernack T, du Teil Espina M, Mulder LM, Palma Medina LM, Piebenga DR, Gabarrini G, et al. A secreted bacterial peptidylarginine deiminase can neutralize human innate immune defenses. mBio. (2018) 9(5):1704–18. doi: 10.1128/mBio.01704-18
58. Bryzek D, Ciaston I, Dobosz E, Gasiorek A, Makarska A, Sarna M, et al. Triggering netosis via protease-activated receptor (par)-2 signaling as a mechanism of hijacking neutrophils function for pathogen benefits. PLoS Pathog. (2019) 15(5):e1007773. doi: 10.1371/journal.ppat.1007773
59. Castanheira FVS, Kubes P. Neutrophils and nets in modulating acute and chronic inflammation. Blood. (2019) 133(20):2178–85. doi: 10.1182/blood-2018-11-844530
60. Groeger S, Meyle J. Oral mucosal epithelial cells. Front Immunol. (2019) 10:208. doi: 10.3389/fimmu.2019.00208
61. Tamassia N, Bianchetto-Aguilera F, Arruda-Silva F, Gardiman E, Gasperini S, Calzetti F, et al. Cytokine production by human neutrophils: revisiting the “dark Side of the moon”. Eur J Clin Invest. (2018) 48(Suppl 2):e12952. doi: 10.1111/eci.12952
62. Riedel DD, Kaufmann SH. Chemokine secretion by human polymorphonuclear granulocytes after stimulation with Mycobacterium tuberculosis and lipoarabinomannan. Infect Immun. (1997) 65(11):4620–3. doi: 10.1128/iai.65.11.4620-4623.1997
63. Vashishta A, Jimenez-Flores E, Klaes CK, Tian S, Miralda I, Lamont RJ, et al. Putative periodontal pathogens, Filifactor alocis and peptoanaerobacter stomatis, induce differential cytokine and chemokine production by human neutrophils. Pathogens. (2019) 8(2):59. doi: 10.3390/pathogens8020059
64. Subbarao KC, Nattuthurai GS, Sundararajan SK, Sujith I, Joseph J, Syedshah YP. Gingival crevicular fluid: an overview. J Pharm Bioallied Sci. (2019) 11(Suppl 2):S135–S9. doi: 10.4103/JPBS.JPBS_56_19
Keywords: inflammation, pathobionts, neutrophils (PMNs), pathogenesis, microbiome & dysbiosis, innate immunity
Citation: Higashi DL, Qin H, Borland C, Kreth J and Merritt J (2024) An inflammatory paradox: strategies inflammophilic oral pathobionts employ to exploit innate immunity via neutrophil manipulation. Front. Oral. Health 5:1413842. doi: 10.3389/froh.2024.1413842
Received: 8 April 2024; Accepted: 28 May 2024;
Published: 11 June 2024.
Edited by:
Ramiro Mendonça Murata, East Carolina University, United StatesReviewed by:
Marcelo Henrique Napimoga, São Leopoldo Mandic School, Brazil© 2024 Higashi, Qin, Borland, Kreth and Merritt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Justin Merritt, bWVycml0dGpAb2hzdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.