 Alexandra E. Herzog
Alexandra E. Herzog Ritu Somayaji1
Ritu Somayaji1 Jacques E. Nör
Jacques E. Nör- 1Department of Cariology, Restorative Sciences, Endodontics, University of Michigan School of Dentistry, Ann Arbor, MI, United States
- 2Department of Otolaryngology – Head and Neck Surgery, University of Michigan Medical School; Ann Arbor, MI, United States
- 3Department of Biomedical Engineering, University of Michigan College of Engineering, Ann Arbor, MI, United States
- 4University of Michigan Rogel Cancer Center, Ann Arbor, MI, United States
Head and neck cancers are composed of a diverse group of malignancies, many of which exhibit an unacceptably low patient survival, high morbidity and poor treatment outcomes. The cancer stem cell (CSC) hypothesis provides an explanation for the substantial patient morbidity associated with treatment resistance and the high frequency of tumor recurrence/metastasis. Stem cells are a unique population of cells capable of recapitulating a heterogenous organ from a single cell, due to their capacity to self-renew and differentiate into progenitor cells. CSCs share these attributes, in addition to playing a pivotal role in cancer initiation and progression by means of their high tumorigenic potential. CSCs constitute only a small fraction of tumor cells but play a major role in tumor initiation and therapeutic evasion. The shift towards stem-like phenotype fuels many malignant features of a cancer cell and mediates resistance to conventional chemotherapy. Bmi-1 is a master regulator of stem cell self-renewal as part of the polycomb repressive complex 1 (PRC1) and has emerged as a prominent player in cancer stem cell biology. Bmi-1 expression is upregulated in CSCs, which is augmented by tumor-promoting factors and various conventional chemotherapies. Bmi-1+ CSCs mediate chemoresistance and metastasis. On the other hand, inhibiting Bmi-1 rescinds CSC function and re-sensitizes cancer cells to chemotherapy. Therefore, elucidating the functional role of Bmi-1 in CSC-mediated cancer progression may unveil an attractive target for mechanism-based, developmental therapeutics. In this review, we discuss the parallels in the role of Bmi-1 in stem cell biology of health and disease and explore how this can be leveraged to advance clinical treatment strategies for head and neck cancer.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common form of cancer worldwide with over 900,000 new cases and 400,000 deaths annually (1). HNSCC incidence is strongly correlated with alcohol and tobacco use, as well as human papillomavirus (HPV) infection. While there has been a marked decrease in HNSCC associated with tobacco use, the incidence and mortality rate of HPV-induced HNSCC has increased significantly (2). Current treatment modalities for HNSCC include surgery, radiation, chemotherapy, EGFR inhibitors (e.g. Cetuximab) and immunotherapy (e.g. Pembrolizumab) (3, 4). However, the modest improvement in overall survival rates achieved with current therapies emphasizes the significant need for further research in this area.
Salivary gland cancers account for approximately 6% of all head and neck cancers, and present significant treatment challenges due to their rarity and biological diversity (5). Mucoepidermoid carcinoma (MEC) is the most common subtype of salivary gland cancer, followed by adenoid cystic carcinoma (ACC) (6). Conventional chemotherapies are ineffective in salivary gland cancers, and currently no systemic or targeted therapy is approved (7, 8). Given the limited understanding of the underlying pathobiology of these diseases and lack of effective chemotherapeutic approaches, surgery remains the main treatment option for these patients. Considering that both HNSCC and malignant salivary gland cancers follow the cancer stem cell hypothesis, the understanding of the pathobiology of these cells may unveil new therapeutic targets for these tumors.
Cancer stem cells in head and neck cancer
The traditional, or stochastic, model of carcinogenesis postulates that tumor growth is initiated by a single cell harboring advantageous genetic mutations, which proliferates and dominates the tumor architecture (9). In this model, all subsequently formed tumor cells possess equal potential for tumorigenesis. Nowadays, it is widely accepted that a tumor is highly heterogenous, constituted by cells of varying biological characteristics. The hierarchical model of carcinogenesis suggests that only a unique subset of tumor cells is capable of tumorigenesis, namely cancer stem cells (CSCs) (10). The CSC hypothesis presents that these cells are endowed with the ability to self-renew and give rise to the various cell phenotypes of a heterogeneous tumor through asymmetric and symmetric cell division. CSCs and physiological stem cells share many attributes: the capacity for self-renewal and differentiation, the ability to survive for long periods of time, and strong resistance to harmful agents (11). Hence, the most-accepted hypothesis for the genesis of CSCs remains that they arise from physiological stem cells (12). Other hypotheses include that CSCs arise from physiological differentiated cells or progenitor cells.
Head and neck cancers have been shown to follow the CSC hypothesis and hierarchical model of carcinogenesis, as they are solid, heterogenous tumors consisting of both CSCs capable of tumorigenesis and bulk tumor cells. CSCs have been diligently studied and characterized in multiple types of head and neck cancers, including head and neck squamous cell carcinoma (HNSCC), mucoepidermoid carcinoma (MEC) and adenoid cystic carcinoma (ACC) (13–17). Head and neck CSCs are endowed with properties of invasiveness, quiescence, and epithelial-mesenchymal transition (EMT) – a crucial process in cancer metastasis (18). These cells have been found to reside in perivascular niches, with the majority residing within a 100 µm radius of blood vessels, from which endothelial cell-secreted factors enhance their self-renewal and promote their tumorigenicity (19, 20). This microenvironmental support, along with many other factors, contributes to the increased resistance to therapies observed in CSCs (21, 22).
The CSC hypothesis may explain the resistance to current cytotoxic treatments and propensity for recurrence and metastasis in head and neck squamous cell carcinoma, which are factors that have a negative impact on the long-term survival of these patients. CSCs are resistant to chemotherapies, because these agents generally target cells with high proliferation rates, whereas CSCs proliferate slowly and thus escape their cytotoxicity (23). Therefore, the modest progress of therapies against HNSCC can at least partially be attributed to CSCs, rendering them to be a therapeutic target of interest.
Biomarkers of cancer stem cells
In order to target CSCs, they must be identifiable by means of unique cellular markers and pathways, which is an area of active research in many cancers. Though differences in CSC biomarkers across cancer types exist, their identification relies heavily on intracellular enzymes, transcription factors, and cell surface molecules. Here, we briefly discuss some commonly used biomarkers of CSCs in head and neck cancer, which are illustrated in Figure 1.
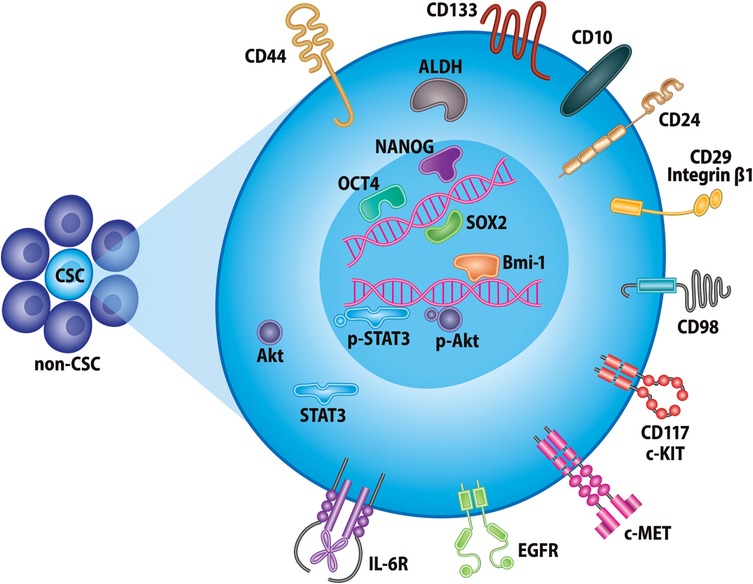
Figure 1. Markers of head and neck cancer stem cells. Selected proteins currently used for identification of CSCs in head and neck cancer. These include, but are not limited to, various cell-surface glycoproteins, receptor tyrosine kinases, intracellular enzymes, and transcription factors.
CSCs were first identified in HNSCC as expressing high levels of CD44, a cell-surface glycoprotein for hyaluronic acid that is involved in cell proliferation, survival, adhesion, migration, and intercellular interactions (13). CD44 is one of the most common CSC markers in several malignancies and has been shown to select for highly tumorigenic CSCs as compared to CD44− cells (24). However, since most cells in epithelia of the head and neck express CD44, other biomarkers have been established to refine identification of head and neck CSCs (25). Among these, aldehyde dehydrogenase (ALDH) activity has been accepted as a frequent marker of CSCs. In HNSCC patient-derived xenograft models and cell lines, ALDHhigh cells demonstrated increased tumorigenicity, therapy resistance, and EMT as compared to ALDHlow cells (26–28). As complementary markers, purified CD44+ ALDHhigh cells constitute an even more tumorigenic and invasive cancer cell population, as compared to the other combinations of both markers' expression status (15, 17, 19). Additionally, these cells positively correlate with decreased overall survival, disease grade, and treatment prognosis in patients with HNSCC (29, 30).
CD133, a cell surface glycoprotein, is another prominent yet more debated head and neck CSC marker (31). CD133+ cells exhibit increased invasiveness, tumorigenicity, and chemoresistance, but may present only a subpopulation of CSCs in oral squamous cell carcinoma cell lines and tissues (32). CD133 has also been shown to function as a regulatory switch of EMT and stemness properties (33). Multiple other cell surface proteins have been implicated as head and neck CSC markers: CD10 expression correlates with poorer overall survival, local recurrences, and therapeutic resistance (34, 35); CD24+ cells promote angiogenesis and tumor progression (36); CD29+ cells are highly invasive, migratory, and contribute to metastases (37); CD98+ cells are tumorigenic and demonstrate increased expression of DNA repair genes (38).
Various receptor tyrosine kinase (RTK) proteins have also been found to promote chemoresistance, metastasis, and CSC properties in head and neck cancer, with therapeutic targeting of these receptors providing clinical promise. CSCs responsible for Cisplatin-resistance and metastasis have been shown to express high levels of c-Met+, and a Phase 1 trial of selective c-MET inhibitor ARQ197 has shown early clinical success (16, 39). The epidermal-growth factor receptor (EGFR) is another example of RTK that is highly expressed in 90% of HNSCC patients and has been linked to treatment resistance, poor clinical outcomes, and higher fraction of CSCs, which, together with CD44, has been shown to promote tumor initiation and progression in vivo (40). The EGFR receptor is the target of Cetuximab, an FDA-approved antibody-based therapy currently accepted for treatment of HNSCC (41). The interleukin-6 receptor (IL-6R) is also strongly upregulated in head and neck CSCs and enhances tumorigenicity and self-renewal via STAT3 signaling (20). Lastly, CD117 is highly implicated in salivary gland CSCs, where it is also commonly used to isolate progenitor cells of the submandibular gland (42).
Other markers of CSCs are intracellular proteins vital to maintaining stemness such as Oct4, Sox2, Nanog, and Bmi-1. Oct 4, Sox2, and Nanog are markers of pluripotency in embryonic stem cells and crucial for these cells' property of self-renewal (43). CSCs from oral squamous cell carcinoma patient samples exhibited high expression of Oct4 and Nanog, along with CD133, which was correlated with greater tumor stage and worse overall survival prognosis (44). Sox2 expression in head and neck CSCs was responsible for their self-renewal, chemoresistance, invasion, and tumorigenicity in vitro and in vivo (45). A meta-analysis revealed that Sox2 could be utilized as an unfavorable prognostic factor for higher tumor grade, stage, and metastases (46). Bmi-1 is a polycomb group protein involved in the regulation of normal stem cells. Head and neck CSCs also exhibit high Bmi-1 expression, which has been shown to be required for sphere formation and self-renewal, indicating that Bmi-1 is an important cellular marker for CSC stemness (47). Interestingly, knockdown of Bmi-1 in ALDH+ CSCs was shown to also downregulate expression of Oct4, Nanog, Sox2, and c-Myc among other stemness markers in these cells (48). In MEC, intense CD44 and Bmi-1 expression was observed in the tumor invasive front, while Oct4 and Nanog was highly associated with perineural invasion in vivo (49).
Many of these cellular proteins have been investigated as potential therapeutic targets for HNSCC, converging in a common denominator of regulating cancer cell stemness (50). In this review, we specifically elaborate on the role and molecular regulatory network of Bmi-1 in mediating head and neck cancer stemness. Additionally, we discuss the current literature on Bmi-1 in promoting therapeutic evasion through chemo- and radioresistance, and the potential therapeutic implications of targeting this master regulator of stemness.
Bmi-1 and cancer stem cells
Physiological Bmi-1 function and regulation
Bmi-1 (B cell-specific Moloney murine leukemia virus Integration site 1) is a 37 kDA protein that consists of three domains: N-terminal RING domain, central domain, and C-terminal proline-serine domain. The RING domain at the N-terminal forms a complex with RING1B (51, 52). Bmi-1 is a member of the Polycomb repressive complex 1 (PRC-1) and is involved in H2A-K119 ubiquitination (53) facilitated in part through this interaction between Bmi-1 and RING1B. Polycomb group (PcG) proteins are a family of proteins involved in transcriptional regulation that form complexes such as PRC-1 to facilitate this regulation. The central domain of Bmi-1 contains a ubiquitin-like (UBL) fold that interacts with PHC2, a polyhomeotic protein that is a member of PRC-1; the UBL region also plays a role in the homo-oligomerization of Bmi-1 (54). Lastly, the C-terminal is a proline-serine rich domain that serves as a regulatory domain for Bmi-1 through negative regulation (55).
Bmi-1 plays a direct role in cell cycle regulation and senescence as a negative regulator of the Ink4a locus that encodes p16Ink4a and p19Arf, tumor suppressor proteins. Downregulation of Bmi-1 resulted in an increase in p16Ink4a and p19Arf expression leading to senescence, while upregulation of Bmi-1 resulted in a decrease in p16Ink4a and p19Arf expression leading to tumor formation in vivo (56). p19Arf is an upstream regulator of p53, a key tumor suppressor protein that functions by blocking MDM2-induced p53 degradation (57). p16Ink4a is an upstream regulator of another tumor suppressor protein: the retinoblastoma (Rb) protein. The phosphorylation of Rb proteins by cyclin D and cyclin E-dependent kinases activates E2F transcription factors, which promotes cellular senescence through entry into the S phase of the cell cycle (58). Thus, Bmi-1 represses two tumor suppressor proteins, p16Ink4a and p19Arf, which function by activating senescence and apoptosis respectively.
Bmi-1 has also been implicated in several developmental signaling pathways including Hox, Hedgehog, and Sox2 pathways. The role of Bmi-1 in H2A-K119 ubiquitination has been linked to Hox gene silencing in mouse embryonic fibroblasts when bound to various Hox gene promoters, while Bmi-1 knockdown resulted in de-repression of these genes. This provided evidence that Hox genes are direct targets of Bmi-1-mediated transcriptional regulation and underlines the importance of Bmi-1 in regulation of key developmental processes in progenitor cells (53). Experiments conducted in mammary stem cells illustrated that activating Hedgehog (Hh) signaling upregulated self-renewal and Bmi-1 expression. In these cells within an in vivo mouse model, upregulation of Gli1 and Gli2, two downstream transcription factors of the Hh pathway, led to upregulation of Bmi-1 which suggests that Hedgehog signaling mediates stem cell self-renewal through Bmi-1 (59). Bmi-1 is also a downstream target of Sox2, a crucial transcription factor in maintaining stem cell pluripotency and stemness in concert with Wnt signaling. Sox2 inactivation leads to strong Bmi-1 downregulation in osteoblasts in vivo, whereas Sox2 overexpression causes Bmi-1 upregulation, and constitutive Bmi-1 expression rescues cell senescence promoted by Sox2 inactivation (60).
Bmi-1 is also regulated by the p38 mitogen-activated protein kinase (MAPK) and Akt pathways. Epidermal growth factor (EGF)-induced Akt activation directly phosphorylates and stabilizes Bmi-1, rendering it resistant to proteasomal degradation and allowing for its nuclear accumulation, whereas p38 inhibits Akt-induced phosphorylation, destabilizing Bmi-1 and promoting increased Bmi-1 degradation in neural stem cells in vivo (61). MAPKAP kinase 3 (3pk), a downstream convergence point of p38 and ERK signaling, also regulates Bmi-1 through phosphorylation. 3pK phosphorylation and activation of Bmi-1 resulted in chromatin dissociation and de-repression of Bmi-1 targets, one of which is p14ARF – a tumor suppressor by means of MDM2 inhibition and subsequent p53 stabilization, arresting cells in G1 cell cycle phase (62, 63). As Bmi-1 phosphorylation by 3pk illustrates, the phosphorylation status of Bmi-1 is inversely related to its chromatin association, allowing for fine-tuned regulation of Bmi-1 binding to chromatin throughout the cell cycle (64).
In addition to cell cycle regulation and the maintenance of the stem cell phenotype, Bmi-1 plays a role in reactive oxygen species (ROS) damage and DNA repair. The transcription factor FoxM1c is expressed highly in proliferating cells and was shown to protect them from oxidative stress-induced senescence by directly activating Bmi-1 expression via c-Myc in vitro and in vivo (65). Deletion of c-Myc lead to a decoupling in FoxM1c-induced Bmi-1 expression, emphasizing that c-Myc serves as a bona fide regulatory intermediate in Bmi-1 signaling. Similarly, Mel-18, another polycomb group ring finger protein, downregulates Bmi-1 expression through transcriptional repression of c-Myc in human fibroblasts (66). The absence of Bmi-1 in mice leads to an accumulation of ROS that subsequently triggers the DNA damage response (DDR) pathway (67). The p16Ink4a pathway, which is negatively regulated by Bmi-1, induces ROS accumulation to promote senescence (68). Bmi-1 is necessary for the DDR pathway and is recruited to DNA double-strand breaks, where it contributes to the repair of the DNA lesion with H2A ubiquitination (69). Thus, Bmi-1 contributes to DNA repair through the DDR pathway, but also by preventing elevated levels of ROS in the cell.
These reported findings suggest that Bmi-1-mediated repression is a finely regulated and dynamically controlled process, with all arrows pointing to Bmi-1 as a master regulator within the PcG-mediated transcriptional system (Figure 2).
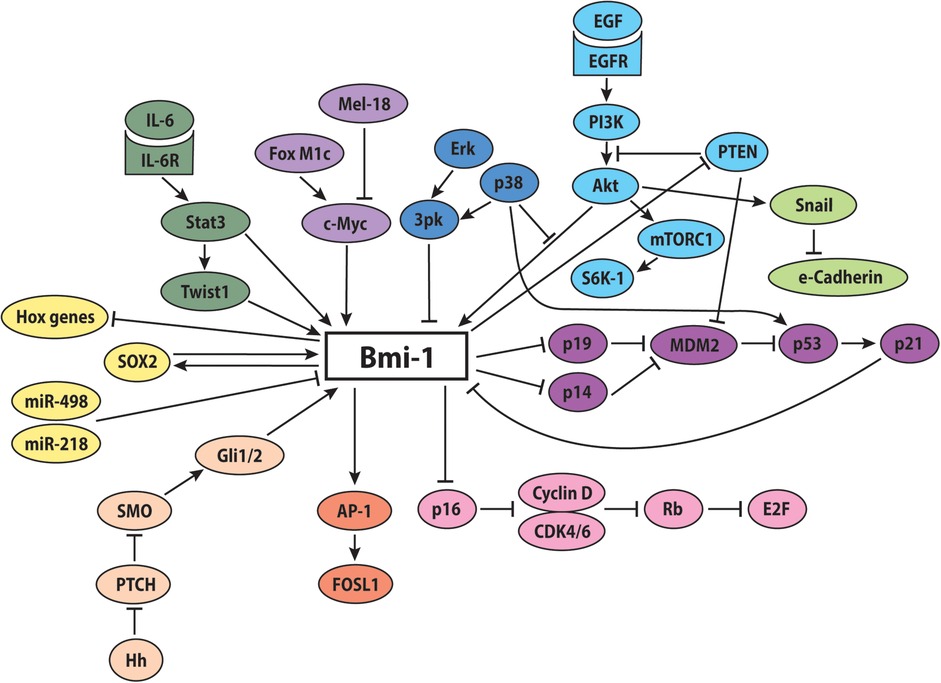
Figure 2. Potential involvement of Bmi-1 in key signaling pathways. The proposed molecular signaling network of Bmi-1 promotes increased stemness, self-renewal, and proliferation, while decreasing apoptosis, differentiation, and senescence.
Bmi-1 regulation in cancer
In head and neck cancer, Bmi-1 is more highly expressed in tumorigenic cells as compared to normal cells. More specifically, elevated Bmi-1 expression is predominantly observed in ALDHhighCD44+ when compared to ALDHlowCD44− cells in vitro and in vivo (19, 70), and endothelial cell-secreted factors further induce Bmi-1 expression in the CSCs (19), revealing Bmi-1 as an important player in HNSCC CSC biology. Characterizing Bmi-1 as an oncogene is a novel, active area of research in head and neck cancer, with limited literature on the exact mechanism and signaling pathways in interplay with Bmi-1. Therefore, we will also review Bmi-1-associated signaling pathways in other types of cancer here.
As elaborated above, the Ink4a locus is a direct target of Bmi-1 in normal cells. In breast cancer and oral squamous cell carcinoma, changes in Bmi-1 expression did not affect p16Ink4a expression, suggesting that the oncogenic activity of Bmi-1 functions through a p16Ink4a-independent signaling pathway (71, 72). Conversely, in laryngeal cancer, colorectal cancer, gastrointestinal cancer, and non-small cell lung cancer a significant negative correlation between Bmi-1 and Ink4a locus gene expression was observed, suggesting that Bmi-1 promotes cellular renewal through the inhibition of senescence and apoptosis (73–76). In yet another example, Bmi-1-mediated tumorigenesis in liver cancer was not at all related to Ink4a/Arf expression but required for RasV12-driven tumor induction (77). These contrasting findings illuminate the complex dysregulation of Bmi-1 in cancer, highlighting the need for further research in this area.
Another paradoxical relationship has been observed between Bmi-1 and Hox signaling pathways in cancer. Typically, high expression of Bmi-1 results in lower expression of Hox signaling proteins. However, in both Ewing sarcoma and certain leukemias it was observed that despite the expected elevated levels of Bmi-1 expression, there were also elevated levels of Hox expression (78, 79). The underlying mechanism of these surprising findings is not clear, but one hypothesis suggests that this could be due to a mutation that unlinks Bmi-1 and Hox signaling (79) Unlike with Hox signaling, the direct relationship between Hh signaling and Bmi-1 observed in normal cells appears to be maintained in cancer cells. In breast cancer, Gli-1 and Gli-2 overexpression induced Bmi-1 expression, which was necessary to promote self-renewal of both normal and malignant mammary stem cells (59). In ovarian cancer, overexpression of various protein signaling effectors of the Hh pathway also induced Bmi-1 expression (80).
A direct relationship between the Akt pathway regulation of Bmi-1 is also observed in various cancer cells. In MEC, CSCs exhibit constitutive activation of mTOR, Akt, S6K1, and Bmi-1, and it was shown that phosphorylation of S6K1 presents a crucial step in regulation of Bmi-1 in vitro and in vivo (81). In pancreatic cancer, overexpression of Bmi-1 induced activation of the P13K/Akt pathway by negative regulation of PTEN in CSCs (82). In endometrial cancer, a direct correlation was found between Bmi-1 expression and Akt expression; interestingly, lower levels of both Bmi-1 and the Akt pathway were associated with more aggressive cancer phenotypes, which stands in contrast to most other cancers (83). In gastric cancer, the microRNAs miR-498 and miR-218 inhibited Bmi-1 function, as well as EMT and Akt signaling (84, 85). A similar finding in breast cancer showed that the PcG protein Mel-18 inhibited Bmi-1 and Akt expression, and that constitutively active Akt rescued the tumor-suppressive function of Mel-18 and Bmi-1 inhibition (86). As previously mentioned, Mel-18 suppresses Bmi-1 through inhibition of c-myc in normal cells. This relationship between c-myc and Bmi-1 was supported in a lymphoma mouse model: overexpression of both c-myc and Bmi-1 induced transformation primary embryo fibroblasts (87).
In salivary gland cancer, p53 has been shown to play a central role in regulating the CSC phenotype via Bmi-1 (70). Here, it was suggested that p53 reduces CSC stemness not by inducing apoptosis, but rather by regulating Bmi-1 expression via downstream p21 signaling and promoting CSC differentiation, and that this mechanism was independent of MDM2. In ACC, therapeutic inhibition of MDM2-p53 was shown to decrease the CSC fraction via apoptosis as well as an increase in cells within the G1 phase of the cell cycle in vitro and in vivo (88). Altogether, while Bmi-1 regulation in cancer isn't necessarily conserved as compared to its physiological regulation, these findings highlight Bmi-1 as an important player in tipping the scales between health and disease.
Bmi-1 in tumorigenesis & metastasis
As previously mentioned, Bmi-1 is highly expressed in head and neck CSCs, which drive tumorigenesis (13), and silencing Bmi-1 leads to a reduction in stemness and tumor formation in HNSCC (48, 47). Bmi-1+ CSCs were shown to mediate invasion and lymph node metastases in HNSCC, specifically through increased AP-1 activity and FOSL1 activation, as determined via lineage tracing and genetic ablation studies (47). Bmi-1+ tumor cells formed significantly more spheres and tumors than Bmi-1− cells, providing strong evidence for the direct role of Bmi-1 in tumorigenesis both in vitro and in vivo. In MEC, downregulation of p53 promoted tumor growth through expansion of the CSC population and upregulation of Bmi-1, providing evidence for not only the role of Bmi-1 in regulating cancer cell stemness, but also for p53 being a master regulator of cell fate within this context (70). Due to the high rate of metastasis and recurrence observed in head and neck cancer, these findings render the role of Bmi-1 particularly significant and may lead to new therapeutic strategies.
The tumor microenvironment plays a crucial role in supporting cancer cell growth, with microenvironment-associated cytokines and growth factors defining tumorigenic potential of CSCs. In HNSCC, endothelial cell-secreted IL-6 has been shown to promote tumorigenicity of CSCs through STAT3 signaling activation and Bmi-1 expression (89, 20). In fact, endothelial cells were shown to produce a chemotactic gradient through secreted IL-6, which enhances survival and motility of tumorigenic head and neck CSCs (20).
The epithelial-mesenchymal transition (EMT), a process through which a cell shifts from an epithelial phenotype to a more migratory mesenchymal phenotype, is a key feature of invasive cancers and metastases. In nasopharyngeal carcinoma cells, silencing Bmi-1 resulted in a reversal of EMT, exhibited by a shift in epithelial and mesenchymal markers and a reduction in metastases, indicating that Bmi-1 induces EMT resulting in a more migratory and aggressive phenotype in vitro (90). This occurs via the underlying mechanism of Bmi-1 repressing PTEN, a negative regulator of the PI3K/Akt pathway, which activates this pathway and down-regulates E-cadherin in a Snail-dependent manner. Likewise, upregulating Bmi-1 in ALDH− head and neck CSCs promotes stemness properties, tumorigenicity, and migration by upregulating Snail, an EMT regulatory protein (91). A direct regulatory link has also been established between Bmi-1 and another EMT regulatory protein, Twist1. Twist1 directly binds to the regulatory region of Bmi-1, and both interdependently promote EMT especially under hypoxic conditions (92). Endothelial cell-secreted EGF and IL-6 were also found to induce EMT to enhance the invasive capacity of head and neck CSCs (20, 93). The human telomerase reverse transcriptase catalytic subunit (hTERT) is involved in maintaining the telomeres of cells, thereby prolonging cell life, and also contributes to EMT. Bmi-1 expression mirrored that of hTERT and was required for hTERT-induced EMT marker expression of oral epithelial cells via suppression of p16INK4a (94). Altogether, these findings demonstrate the direct role Bmi-1 plays in EMT and therefore, to the more aggressive stem-like and migratory phenotype of tumor cells.
Bmi-1 in cancer therapeutics
Bmi-1 in chemoresistance
The lack of progress in HNSCC, MEC, and ACC treatment can largely be attributed to therapeutic resistance of each of these cancer types, which can lead to both metastasis and recurrence; notably, CSCs are particularly resistant to therapies compared to bulk tumor cells. In the context of head and neck CSCs, Bmi-1 has been strongly implicated in therapy resistance (Figure 3).
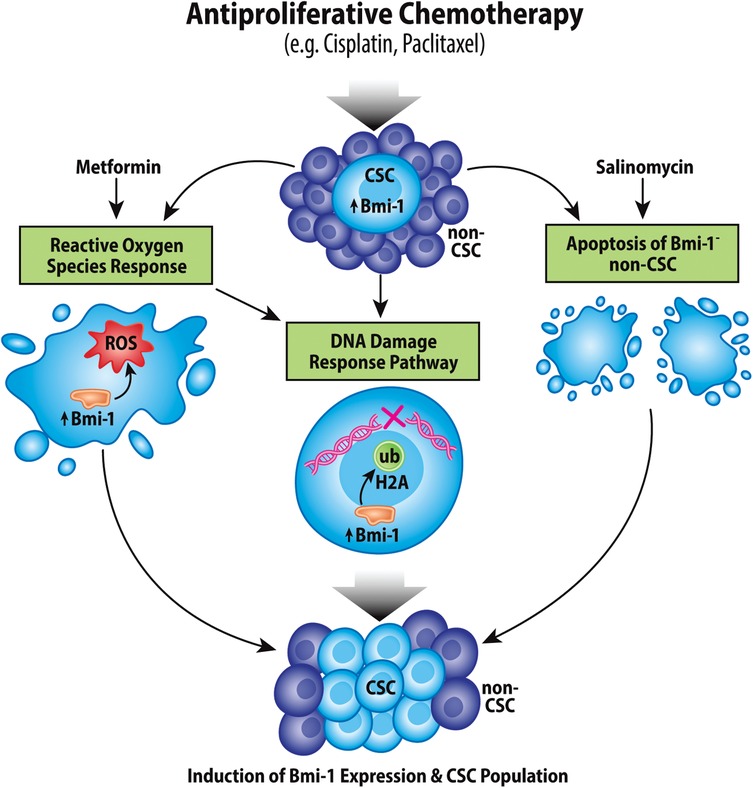
Figure 3. Chemotherapeutic induction of Bmi-1 increases the cancer stem cell population. Antiproliferative chemotherapeutic agents currently used for treatment of head and neck cancers have been shown to increase the CSC fraction in tumors via multiple mechanisms, which include inducing the reactive oxygen species response, the DNA damage response pathway, and apoptosis of Bmi-1− CSCs.
Cisplatin is still the most common chemotherapy agent used in treatment of HNSCC, as well as many other cancers. However, it has been shown that treatment with Cisplatin actually increases the CSC fraction in HNSCC tumors, and Cisplatin-resistant cells intrinsically express elevated levels of Bmi-1; there is also a direct association between Cisplatin dosage or resistance and Bmi-1 expression in vitro and in vivo (22, 95). Interestingly, it has been demonstrated that Cisplatin-induced apoptosis mainly occurred in Bmi-1− tumor cells, and that in HNSCC recurrence specifically Bmi-1+ CSC lineages were maintained in these tumors (47). As has been previously mentioned, the IL-6/STAT3 pathway is highly upregulated in head and neck CSCs, and IL-6 augments the Cisplatin-induction of Bmi-1 expression and CSC fraction (22, 89). Inhibition of IL-6 signaling decreased the CSC fraction in vitro and in vivo, as well as suppressed Cisplatin-induction of Bmi-1 expression and the CSC fraction (20, 22, 95). Similarly, a combination treatment of an AP-1 inhibitor and Cisplatin resulted in inhibition of Bmi-1+ CSCs and a reduction in metastases, suggesting a possible mechanistic pathway for chemoresistance via Bmi-1 (47). Altogether, these results suggest a clear link between Cisplatin resistance and Bmi-1 expression and support the therapeutic strategy to include the use of either a direct Bmi-1 inhibitor or an inhibitor of a Bmi-1-associated pathway, such as those mentioned above.
Interestingly, Salinomycin, a commonly used antibiotic, successfully targeted CSCs in HNSCC, resulting in reduced Bmi-1 expression and invasive phenotypes of CSCs. When used in combination with Cisplatin or Paclitaxel, Salinomycin greatly increased overall cell death. However, it was found to increase EMT markers, Akt, and mTor signaling, which may correlate with cancer cell stemness (96). As mentioned previously, the PI3K/Akt pathway is highly upregulated in head and neck CSCs and therapeutic inhibition of the mechanistic target of rapamycin (mTOR) has shown clinical success in head and neck cancer (97). mTOR inhibition ablates Cisplatin-induced stemness and blocks Bmi-1 expression in MEC (98). As opposed to in HNSCC, p53 is not oftentimes mutated in salivary gland cancers, and a major therapeutic strategy that has since been translated into clinical trials involves targeting the MDM2-p53 interaction. A small-molecule inhibitor of MDM2-p53 complex (MI-773) has been shown to potently decrease the CSC fraction, Bmi-1 expression, and tumor recurrence in both MEC and ACC (70, 88, 99, 100). Inhibition of MDM2-p53 triggered G1 cell cycle arrest, and sensitized tumors to Cisplatin chemotherapy (99, 100). Altogether, these findings illuminate a variety of strategies to overcoming CSC-mediated chemotherapeutic resistance to Cisplatin.
Comparable to the effect of Cisplatin, treatment with Metformin (diabetes drug that has exhibited anticancer properties in various other cancers) lead to a reduction in bulk tumor cells but an increase in CSCs and Bmi-1 expression in HNSCC. Metformin increased expression of Bmi-1, Oct4, Nanog, and CD44, and was revealed to bind to mitochondrial complex III, suggesting a possible role of Metformin in mediating ROS (101). Interestingly, in prostate cancer, Cisplatin functions by elevating intracellular ROS through NADPH oxidase activation (102). Indeed, mechanistic studies revealed that Cisplatin induces a mitochondria-dependent ROS response in conjunction with, but independent from, its well-known cytotoxic effect through inducing DNA damage (103). Therefore, it is hypothesized that many cytotoxic effects of chemotherapeutic agents function through ROS, eliciting protective effects on CSCs and Bmi-1 function. This relates to results described above regarding the role Bmi-1 plays in maintenance of low ROS levels and in the DDR pathway. By elevating ROS and DNA damage levels in the cell, Cisplatin is likely activating Bmi-1 for response to these stimuli, inadvertently activating cancer cell stemness properties.
Thus, further investigation into targeted therapies is necessary, but the treatment for head and neck cancers will likely entail a combination of systemic cytotoxic therapies and CSC-targeted therapies such as small molecule inhibitors of Bmi-1.
Bmi-1 in radioresistance
In parallel to chemoresistance, radioresistance poses another obstacle in successful treatment of head and neck cancers. In HNSCC, silencing Bmi-1 activity in ALDH+ CSCs led to increased apoptotic activity as detected via Annexin V staining, resulting in decreased radioresistance and an overall higher survival rate in a mouse model (48). In nasopharyngeal cancer, silencing Bmi-1 resulted in re-sensitization to radiation therapy through increased apoptotic activity of p53 and increased production of ROS (104). These findings implicated Bmi-1 in contributing to HNSCC CSC radioresistance.
Further literature on the role of Bmi-1 in head and neck cancer radioresistance is limited. However, notable findings have been reported in other cancers. Elevated levels of Bmi-1 were also observed in adaptively radioresistant esophageal squamous cell carcinoma (ESCC) cells, where Bmi-1 silencing led to re-sensitization to radiation therapy (105). Similar to the observations made in nasopharyngeal cancer, Bmi-1 conferred adaptive radioresistance to ESCC cells, and Bmi-1-depleted cells treated with radiotherapy expressed elevated levels of ROS and impaired DNA repair capacities, further supporting a common mechanism by which Bmi-1 mediates therapeutic resistance (105). In breast cancer cells, Bmi-1 expression was also indicative of radioresistance. Upon Bmi-1 knockdown, increased DNA double strand breaks, reduced DNA repair, and increased apoptosis through elevated p53, p21 and Bax protein expression was observed (106). In glioblastomas, radiation therapy primarily functions by halting senescence and it was shown that Bmi-1 confers radioresistance by inhibiting cell senescence through the p16 signaling pathway (107). Reduction of Bmi-1 expression by overexpression of microRNA-128 in glioma cells also promoted radiosensitivity and prevalence of senescent cells (108).
Collectively, these studies suggest that Bmi-1 promotes radioresistance through decreased levels of ROS, increased DNA repair, and suppression of senescence through mechanisms resembling those that arbitrate chemoresistance. This also strongly implicates Bmi-1 as a powerful point of convergence in multiple therapeutic resistance mechanisms of different treatment modalities across many cancers.
Bmi-1 as a prognostic factor
We have reviewed the prominent impact Bmi-1 has on tumorigenesis, metastasis, and therapy resistance of head and neck cancers. Yet, there are many unanswered questions: How does this translate to overall patient survival? How can we leverage this knowledge to better make predictions about patient prognoses and treatment outcomes?
Few research studies to date have robustly investigated Bmi-1 expression patterns in head and neck cancer. Bmi-1 expression in tumor tissue of oropharyngeal squamous cell carcinoma was significantly higher as compared to normal mucosa, but no difference in expression was observed between the primary tumor and lymph node metastases (109). This expression pattern was observed in both HPV-negative and HPV-positive samples (n = 12). In another study, HPV-positive human oropharyngeal squamous cell carcinoma specimens showed lower Bmi-1 expression than HPV-negative tumors (n = 202) (110). In human tissue specimens, Bmi-1 expression was significantly higher in oral squamous cell carcinoma but showed no difference between normal mucosa and oral dysplasia (n = 129) (111). Thus, more elaborate studies are needed to determine the relevance of Bmi-1 expression in head and neck cancer, especially in relation to HPV status and disease progression.
In one study analyzing CSC markers as prognostic factors for HNSCC, it was observed that both Bmi-1 and CD44 are indicators for poorer prognosis of overall and disease-free survival in patients receiving primary radio-chemotherapy irrespective of HPV status (n = 85) (112). Conversely, in SCC of the tongue, a strong correlation was observed between low Bmi-1 expression and poor patient prognosis (n = 73) (113), and in a separate meta-analysis (n = 2143), Bmi-1 did not impact overall HNSCC survival significantly (114). In another study, Bmi-1 expression in patient samples (n = 216) was correlated with poor prognosis of recurrence-free survival, but not overall survival (95). These observations may indeed be explained by the CSC hypothesis, since the small CSC fraction may not significantly contribute to overall tumor growth and survival, but to tumor recurrence or metastases. This ambiguity illustrates a definitive need for further research on Bmi-1 as a prognostic factor in HNSCC.
In other cancers, Bmi-1 has been a more promising negative prognosticator. Bmi-1 overexpression has been reported in a plethora of cancers, including gastric, ovarian, breast, head and neck, pancreatic, lung, hepatocellular, and endometrial carcinoma and correlated with a variety of indicators of poor prognoses as described elsewhere (82). A meta-analysis of non-small cell lung cancer revealed a correlation between elevated Bmi-1 expression and increased tumor size, metastasis, and lower overall survival rates (115). Bmi-1 was also found to be a negative prognostic factor in gastric cancer and endometrial adenocarcinoma, each demonstrating heightened Bmi-1 expression to be indicative of worse clinical stage, lymph node metastases, and overall survival (116, 117). Based on these varied and contradictory findings, the utility of Bmi-1 as a prognostic factor remains unclear. One plausible explanation for this may be the lack of expression analyses specific to a tumor subsite or clonal cell population. As mentioned previously, tumor cells as well as a plethora of stromal cells from the tumor microenvironment express Bmi-1, which highly clouds the prognostic value of this marker. While these studies do not yet provide convincing evidence for Bmi-1 serving as a possible way to prognosticate treatment response, Bmi-1 is undoubtedly an important master regulator of cancer cell stemness and therapeutic resistance, rendering it a putative therapeutic target, nonetheless.
Therapeutic targeting of Bmi-1
Bmi-1 has emerged as an attractive therapeutic target in CSC-focused, mechanism-based cancer treatments. Therapeutic inhibition of Bmi-1 was first described in a primary colorectal cancer xenograft model, where it inhibited CSC self-renewal and thus abrogated their tumorigenic potential (118). The anti-PD-1 immunotherapies Nivolumab and Pembrolizumab have been approved as first-line therapies for HNSCC and are commonly combined with Cisplatin treatment. In an in vivo mouse model of HNSCC, Bmi-1+ CSCs were enriched in tumors following treatment with Cisplatin and anti-PD-1 therapy, but treatment with the Bmi-1 inhibitor PTC209 prevented induction of these cells and tumor progression (119). In this study, Bmi-1 inhibition was also shown to promote CD8+ T-cell infiltration by removing repressive H2A ubiquitination to induce transcription of chemokines necessary for their recruitment. Interestingly, Bmi-1 may play a significant role as immune modifier in several in vivo studies: Bmi-1 inhibition restored B-cell-mediated humoral immune responses via increased antibody function (120). Immune escape of pancreatic cancer cells from NK cell-mediated elimination in a hyperglycemic tumor microenvironment was also shown to be mediated by upregulation of Bmi-1 and subsequent MICA/B inhibition and GATA2 promotion (121). In a murine myeloma model, Bmi-1 inhibition eliminated tumor-associated macrophages and mediated chemoresistance (122).
The therapeutic potential of Bmi-1 inhibitors has also been investigated in early human clinical trials. In a Phase 1 multi-center, open-label study in patients with advanced solid tumors, the second-generation Bmi-1 inhibitor PTC596 was determined to be tolerable with manageable side effects (123). Notably, PTC596 was shown to be successful in the treatment of acute leukemia in an in vitro study irrespective of p53 mutational status, which could provide highly beneficial in the treatment of salivary gland cancers which typically demonstrate high mutational burden of p53 (124). Another Bmi-1 inhibitor, PRT4165, prevents accumulation of all detectable H2A ubiquitination sites around DNA double-stranded breaks in an osteosarcoma model, which could be highly relevant in combination with antiproliferative chemotherapies that propagate the DNA damage response as mentioned in Figure 3 (125). In an ovarian cancer model, the Bmi-1 inhibitor PTC028 was shown to selectively inhibit cancer cell growth while leaving normal cells unaffected, which could present a unique benefit as compared to conventional chemotherapeutic agents that do not selectively eliminate tumor cells (126). While there are limited published studies on the efficacy of Bmi-1 inhibitors in head and neck cancers, the success of these small molecule drugs in treatment of other cancers in preclinical and clinical models strongly supports their therapeutic potential.
Conclusion
There has been modest progress in the outcome of head and neck cancer patients, in part due to therapy resistance which can be attributed to the function of CSCs in tumorigenesis and tumor dissemination. This emphasizes the need for further research into the underlying mechanisms regulating the CSC phenotype. Bmi-1, a polycomb complex protein, has been established as a pivotal player in controlling cancer cell stemness. It has been implicated in many major signaling pathways such as the p16Ink4a/p19Arf, PI3K/Akt, MAPK, STAT3, and Hedgehog pathways. It also plays vital roles in cellular processes responding to ROS and DNA damage. In cancer, it has been widely linked to increased stemness, tumor formation and metastasis, as well as therapeutic resistance. This review attempted to synthesize the current evidence on Bmi-1 within the context of head and neck cancer stem cells, and to provide support for future research aimed at targeting this master regulator of cancer cell stemness using novel therapeutic approaches.
Author contributions
AEH, RS, and JEN were jointly responsible for the preparation, writing, and editing of the manuscript and figures. All authors contributed to the article and approved the submitted version. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by NIH/NIDCR F30-DE029097 (AEH), R01-DE21139 (JEN), and R01-DE23220 (JEN).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. Ca Cancer J Clin. (2021) 71(1):7–33. doi: 10.3322/caac.21654
2. Zaravinos A. An updated overview of HPV-associated head and neck carcinomas. Oncotarget. (2014) 5(12):3956. doi: 10.18632/oncotarget.1934
3. Moskovitz J, Moy J, Ferris RL. Immunotherapy for head and neck squamous cell carcinoma. Curr Oncol Rep. (2018) 20(2):1–7. doi: 10.1007/s11912-018-0654-5
4. Cramer JD, Burtness B, Le QT, Ferris RL. The changing therapeutic landscape of head and neck cancer. Nat Rev Clin Oncol. (2019) 16(11):669–83. doi: 10.1038/s41571-019-0227-z
5. Seethala RR, Stenman G. Update from the 4th edition of the world health organization classification of head and neck tumours: tumors of the salivary gland. Head Neck Pathol. (2017) 11(1):55–67. doi: 10.1007/s12105-017-0795-0
6. Speight PM, Barrett AW. Salivary gland tumours. Oral Dis. (2002) 8(5):229–40. doi: 10.1034/j.1601-0825.2002.02870.x
7. Chintakuntlawar AV, Okuno SH, Price KA. Systemic therapy for recurrent or metastatic salivary gland malignancies. Cancers Head Neck. (2016) 1(1):1–9. doi: 10.1186/s41199-016-0011-z
8. Sahara S, Herzog AE, Nör JE. Systemic therapies for salivary gland adenoid cystic carcinoma. Am J Cancer Res. (2021) 11(9):4092.34659878
9. Nowell PC. The clonal evolution of tumor cell populations: acquired genetic lability permits stepwise selection of variant sublines and underlies tumor progression. Science. (1976) 194(4260):23–8. doi: 10.1126/science.959840
10. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. (2001) 414(6859):105–11. doi: 10.1038/35102167
11. Sell S. Stem cell origin of cancer and differentiation therapy. Crit Rev Oncol Hematol. (2004) 51(1):1–28. doi: 10.1016/j.critrevonc.2004.04.007
12. Costea DE, Tsinkalovsky O, Vintermyr OK, Johannessen AC, Mackenzie IC. Cancer stem cells–new and potentially important targets for the therapy of oral squamous cell carcinoma. Oral Dis. (2006) 12(5):443–54. doi: 10.1111/j.1601-0825.2006.01264.x
13. Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA. (2007) 104(3):973–8. doi: 10.1073/pnas.0610117104
14. Krishnamurthy S, Nör JE. Head and neck cancer stem cells. J Dent Res. (2012) 91(4):334–40. doi: 10.1177/0022034511423393
15. Adams A, Warner K, Pearson AT, Zhang Z, Kim HS, Mochizuki D, et al. ALDH/CD44 identifies uniquely tumorigenic cancer stem cells in salivary gland mucoepidermoid carcinomas. Oncotarget. (2015) 6(29):26633. doi: 10.18632/oncotarget.5782
16. Sun S, Wang Z. Head neck squamous cell carcinoma c-Met+ cells display cancer stem cell properties and are responsible for cisplatin-resistance and metastasis. Int J Cancer. (2011) 129(10):2337–48. doi: 10.1002/ijc.25927
17. Keysar SB, Eagles JR, Miller B, Jackson BC, Chowdhury FN, Reisinger J, et al. Salivary gland cancer patient-derived xenografts enable characterization of cancer stem cells and new gene events associated with tumor progression salivary cancer stem cells increase with disease progression. Clin Cancer Res. (2018) 24(12):2935–43. doi: 10.1158/1078-0432.CCR-17-3871
18. Chen C, Wei Y, Hummel M, Hoffmann TK, Gross M, Kaufmann AM, et al. Evidence for epithelial-mesenchymal transition in cancer stem cells of head and neck squamous cell carcinoma. PloS One. (2011) 6(1):e16466. doi: 10.1371/journal.pone.0016466
19. Krishnamurthy S, Dong Z, Vodopyanov D, Imai A, Helman JI, Prince ME, et al. Endothelial cell-initiated signaling promotes the survival and self-renewal of cancer stem cells head and neck cancer stem cell niche. Cancer Res. (2010) 70(23):9969–78. doi: 10.1158/0008-5472.CAN-10-1712
20. Kim HS, Chen YC, Nör F, Warner KA, Andrews A, Wagner VP, et al. Endothelial-derived interleukin-6 induces cancer stem cell motility by generating a chemotactic gradient towards blood vessels. Oncotarget. (2017) 8(59):100339. doi: 10.18632/oncotarget.22225
21. Reid PA, Wilson P, Li Y, Marcu LG, Bezak E. Current understanding of cancer stem cells: review of their radiobiology and role in head and neck cancers. Head Neck. (2017) 39(9):1920–32. doi: 10.1002/hed.24848
22. Nör C, Zhang Z, Warner KA, Bernardi L, Visioli F, Helman JI, et al. Cisplatin induces Bmi-1 and enhances the stem cell fraction in head and neck cancer. Neoplasia. (2014) 16(2):137–W8. doi: 10.1593/neo.131744
23. Sayed SI, Dwivedi RC, Katna R, Garg A, Pathak KA, Nutting CM, et al. Implications of understanding cancer stem cell (CSC) biology in head and neck squamous cell cancer. Oral Oncol. (2011) 47(4):237–43. doi: 10.1016/j.oraloncology.2011.02.009
24. Facompre N, Nakagawa H, Herlyn M, Basu D. Stem-like cells and therapy resistance in squamous cell carcinomas. Adv Pharmacol. (2012) 65:235–65. doi: 10.1016/B978-0-12-397927-8.00008-7
25. Mack B, Gires O. CD44s And CD44v6 expression in head and neck epithelia. PloS One. (2008) 3(10):e3360. doi: 10.1371/journal.pone.0003360
26. Clay MR, Tabor M, Owen JH, Carey TE, Bradford CR, Wolf GT, et al. Single-marker identification of head and neck squamous cell carcinoma cancer stem cells with aldehyde dehydrogenase. Head Neck. (2010) 32(9):1195–201. doi: 10.1002/hed.21315
27. Bertrand G, Maalouf M, Boivin A, Battiston-Montagne P, Beuve M, Levy A, et al. Targeting head and neck cancer stem cells to overcome resistance to photon and carbon ion radiation. Stem Cell Rev Rep. (2014) 10(1):114–26. doi: 10.1007/s12015-013-9467-y
28. Chen YC, Chen YW, Hsu HS, Tseng LM, Huang PI, Lu KH, et al. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem Biophys Res Commun. (2009) 385(3):307–13. doi: 10.1016/j.bbrc.2009.05.048
29. Kokko LL, Hurme S, Maula SM, Alanen K, Grénman R, Kinnunen I, et al. Significance of site-specific prognosis of cancer stem cell marker CD44 in head and neck squamous-cell carcinoma. Oral Oncol. (2011) 47(6):510–6. doi: 10.1016/j.oraloncology.2011.03.026
30. Dong Y, Ochsenreither S, Cai C, Kaufmann AM, Albers AE, Qian X. Aldehyde dehydrogenase 1 isoenzyme expression as a marker of cancer stem cells correlates to histopathological features in head and neck cancer: a meta-analysis. PloS One. (2017) 12(11):e0187615. doi: 10.1371/journal.pone.0187615
31. Chen D, Wang CY. Targeting cancer stem cells in squamous cell carcinoma. Precision Clin Med. (2019) 2(3):152–65. doi: 10.1093/pcmedi/pbz016
32. Zhang Q, Shi S, Yen Y, Brown J, Ta JQ, Le AD. A subpopulation of CD133 + cancer stem-like cells characterized in human oral squamous cell carcinoma confer resistance to chemotherapy. Cancer Lett. (2010) 289(2):151–60. doi: 10.1016/j.canlet.2009.08.010
33. Chen YS, Wu MJ, Huang CY, Lin SC, Chuang TH, Yu CC, et al. CD133/Src Axis mediates tumor initiating property and epithelial-mesenchymal transition of head and neck cancer. PloS One. (2011) 6(11):e28053. doi: 10.1371/journal.pone.0028053
34. Fukusumi T, Ishii H, Konno M, Yasui T, Nakahara S, Takenaka Y, et al. CD10 As a novel marker of therapeutic resistance and cancer stem cells in head and neck squamous cell carcinoma. Br J Cancer. (2014) 111(3):506–14. doi: 10.1038/bjc.2014.289
35. Li Q, Wang Y, Xu L, Wang L, Guo Y, Guo C. High level of CD10 expression is associated with poor overall survival in patients with head and neck cancer. Int J Oral Maxillofac Surg. (2021) 50(7):857–64. doi: 10.1016/j.ijom.2020.07.037
36. Zimmerer RM, Ludwig N, Kampmann A, Bittermann G, Spalthoff S, Jungheim M, et al. CD24 + tumor-initiating Cells from oral squamous cell carcinoma induce initial angiogenesis in vivo. Microvasc Res. (2017) 112:101–8. doi: 10.1016/j.mvr.2017.03.006
37. Lin HC, Wu CL, Chen YL, Huang JS, Wong TY, Yuan K. High-level β1-integrin expression in a subpopulation of highly tumorigenic oral cancer cells. Clin Oral Investig. (2014) 18(4):1277–84. doi: 10.1007/s00784-013-1088-y
38. Martens-de Kemp SR, Brink A, Stigter-van Walsum M, Damen JM, Rustenburg F, Wu T, et al. CD98 Marks a subpopulation of head and neck squamous cell carcinoma cells with stem cell properties. Stem Cell Res. (2013) 10(3):477–88. doi: 10.1016/j.scr.2013.02.004
39. Yap TA, Olmos D, Brunetto AT, Tunariu N, Barriuso J, Riisnaes R, et al. Phase I trial of a selective c-MET inhibitor ARQ 197 incorporating proof of mechanism pharmacodynamic studies. J Clin Oncol. (2011) 29(10):1271–9. doi: 10.1200/JCO.2010.31.0367
40. Perez A, Neskey DM, Wen J, Pereira L, Reategui EP, Goodwin WJ, et al. CD44 Interacts with EGFR and promotes head and neck squamous cell carcinoma initiation and progression. Oral Oncol. (2013) 49(4):306–13. doi: 10.1016/j.oraloncology.2012.11.009
41. Rehmani HS, Issaeva N. EGFR In head and neck squamous cell carcinoma: exploring possibilities of novel drug combinations. Ann Transl Med. (2020) 8(13):813. doi: 10.21037/atm.2020.04.07
42. Adams A, Warner K, Nör JE. Salivary gland cancer stem cells. Oral Oncol. (2013) 49(9):845–53. doi: 10.1016/j.oraloncology.2013.05.013
43. Boiani M, Schöler HR. Regulatory networks in embryo-derived pluripotent stem cells. Nat Rev Mol Cell Biol. (2005) 6(11):872–81. doi: 10.1038/nrm1744
44. Chiou SH, Yu CC, Huang CY, Lin SC, Liu CJ, Tsai TH, et al. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin Cancer Res. (2008) 14(13):4085–95. doi: 10.1158/1078-0432.CCR-07-4404
45. Lee SH, Oh SY, Do SI, Lee HJ, Kang HJ, Rho YS, et al. SOX2 Regulates self-renewal and tumorigenicity of stem-like cells of head and neck squamous cell carcinoma. Br J Cancer. (2014) 111(11):2122–30. doi: 10.1038/bjc.2014.528
46. Dong Z, Liu G, Huang B, Sun J, Wu D. Prognostic significance of SOX2 in head and neck cancer: a meta-analysis. Int J Clin Exp Med. (2014) 7(12):5010.25664000
47. Chen D, Wu M, Li Y, Chang I, Yuan Q, Ekimyan-Salvo M, et al. Targeting BMI1 + cancer stem cells overcomes chemoresistance and inhibits metastases in squamous cell carcinoma. Cell Stem Cell. (2017) 20(5):621–34. doi: 10.1016/j.stem.2017.02.003
48. Chen YC, Chang CJ, Hsu HS, Chen YW, Tai LK, Tseng LM, et al. Inhibition of tumorigenicity and enhancement of radiochemosensitivity in head and neck squamous cell cancer-derived ALDH1-positive cells by knockdown of Bmi-1. Oral Oncol. (2010) 46(3):158–65. doi: 10.1016/j.oraloncology.2009.11.007
49. Destro Rodrigues MF, Sedassari BT, Esteves CM, de Andrade NP, Altemani A, de Sousa SC, et al. Embryonic stem cells markers Oct4 and Nanog correlate with perineural invasion in human salivary gland mucoepidermoid carcinoma. J Oral Pathol Med. (2017) 46(2):112–20. doi: 10.1111/jop.12449
50. Oklejas AE, Nör JE. Overcoming head and neck cancer stem cells. In: Kimple RJ, editor. Improving the therapeutic ratio in head and neck cancer. London, UK: Academic Press (2020). p. 135–58.
51. Buchwald G, van der Stoop P, Weichenrieder O, Perrakis A, van Lohuizen M, Sixma TK. Structure and E3-ligase activity of the ring–ring complex of polycomb proteins Bmi1 and Ring1b. EMBO J. (2006) 25(11):2465–74. doi: 10.1038/sj.emboj.7601144
52. Li Z, Cao R, Wang M, Myers MP, Zhang Y, Xu RM. Structure of a Bmi-1-Ring1B polycomb group ubiquitin ligase complex. J Biol Chem. (2006) 281(29):20643–9. doi: 10.1074/jbc.M602461200
53. Cao R, Tsukada YI, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell. (2005) 20(6):845–54. doi: 10.1016/j.molcel.2005.12.002
54. Gray F, Cho HJ, Shukla S, He S, Harris A, Boytsov B, et al. BMI1 Regulates PRC1 architecture and activity through homo-and hetero-oligomerization. Nat Commun. (2016) 7(1):1–2. doi: 10.1038/ncomms13343
55. Yadav AK, Sahasrabuddhe AA, Dimri M, Bommi PV, Sainger R, Dimri GP. Deletion analysis of BMI1 oncoprotein identifies its negative regulatory domain. Mol Cancer. (2010) 9(1):1–3. doi: 10.1186/1476-4598-9-158
56. Jacobs JJ, Kieboom K, Marino S, DePinho RA, Van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. (1999) 397(6715):164–8. doi: 10.1038/16476
57. Pomerantz J, Schreiber-Agus N, Liégeois NJ, Silverman A, Alland L, Chin L, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2's Inhibition of p53. Cell. (1998) 92(6):713–23. doi: 10.1016/S0092-8674(00)81400-2
58. Lowe SW, Sherr CJ. Tumor suppression by Ink4a–Arf: progress and puzzles. Curr Opin Genet Dev. (2003) 13(1):77–83. doi: 10.1016/S0959-437X(02)00013-8
59. Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. (2006) 66(12):6063–71. doi: 10.1158/0008-5472.CAN-06-0054
60. Seo E, Basu-Roy U, Zavadil J, Basilico C, Mansukhani A. Distinct functions of Sox2 control self-renewal and differentiation in the osteoblast lineage. Mol Cell Biol. (2011) 31(22):4593–608. doi: 10.1128/MCB.05798-11
61. Kim J, Hwangbo J, Wong PK. P38 MAPK-mediated Bmi-1 down-regulation and defective proliferation in ATM-deficient neural stem cells can be restored by Akt activation. PLoS One. (2011) 6(1):e16615. doi: 10.1371/journal.pone.0016615
62. Voncken JW, Niessen H, Neufeld B, Rennefahrt U, Dahlmans V, Kubben N, et al. MAPKAP Kinase 3pK phosphorylates and regulates chromatin association of the polycomb group protein Bmi1. J Biol Chem. (2005) 280(7):5178–87. doi: 10.1074/jbc.M407155200
63. Zhang Y, Xiong Y, Yarbrough WG. ARF Promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. (1998) 92(6):725–34. doi: 10.1016/S0092-8674(00)81401-4
64. Voncken JW, Schweizer D, Aagaard L, Sattler L, Jantsch MF, van Lohuizen M. Chromatin-association of the Polycomb group protein BMI1 is cell cycle-regulated and correlates with its phosphorylation status. J Cell Sci. (1999) 112(24):4627–39. doi: 10.1242/jcs.112.24.4627
65. Li SK, Smith DK, Leung WY, Cheung AM, Lam EW, Dimri GP, et al. Foxm1c counteracts oxidative stress-induced senescence and stimulates Bmi-1 expression. J Biol Chem. (2008) 283(24):16545–53. doi: 10.1074/jbc.M709604200
66. Guo WJ, Datta S, Band V, Dimri GP. Mel-18, a polycomb group protein, regulates cell proliferation and senescence via transcriptional repression of Bmi-1 and c-Myc oncoproteins. Mol Biol Cell. (2007) 18(2):536–46. doi: 10.1091/mbc.e06-05-0447
67. Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C, et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. (2009) 459(7245):387–92. doi: 10.1038/nature08040
68. Takahashi A, Ohtani N, Yamakoshi K, Iida SI, Tahara H, Nakayama K, et al. Mitogenic signalling and the p16INK4a–Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol. (2006) 8(11):1291–7. doi: 10.1038/ncb1491
69. Ismail IH, Andrin C, McDonald D, Hendzel MJ. BMI1-mediated Histone ubiquitylation promotes DNA double-strand break repair. J Cell Biol. (2010) 191(1):45–60. doi: 10.1083/jcb.201003034
70. Rodriguez-Ramirez C, Zhang Z, Warner KA, Herzog AE, Mantesso A, Zhang Z, et al. P53 inhibits Bmi-1-driven self-renewal and defines salivary gland cancer stemness. Clin Cancer Res. (2022) 28(21):4757–70. doi: 10.1158/1078-0432.CCR-22-1357
71. Silva J, García JM, Peña C, García V, Domínguez G, Suárez D, et al. Implication of polycomb members Bmi-1, Mel-18, and Hpc-2 in the regulation of p16INK4a, p14ARF, h-TERT, and c-Myc expression in primary breast carcinomas. Clin Cancer Res. (2006) 12(23):6929–36. doi: 10.1158/1078-0432.CCR-06-0788
72. Kang MK, Kim RH, Kim SJ, Yip FK, Shin KH, Dimri GP, et al. Elevated Bmi-1 expression is associated with dysplastic cell transformation during oral carcinogenesis and is required for cancer cell replication and survival. Br J Cancer. (2007) 96(1):126–33. doi: 10.1038/sj.bjc.6603529
73. Wei X, He J, Wang J, Yang X, Ma B. Bmi-1 is essential for the oncogenic potential in CD133 + human laryngeal cancer cells. Tumor Biol. (2015) 36(11):8931–42. doi: 10.1007/s13277-015-3541-9
74. Kim JH, Yoon SY, Kim CN, Joo JH, Moon SK, Choe IS, et al. The Bmi-1 oncoprotein is overexpressed in human colorectal cancer and correlates with the reduced p16INK4a/p14ARF proteins. Cancer Lett. (2004) 203(2):217–24. doi: 10.1016/j.canlet.2003.07.009
75. Wang JL, Wu JH, Hong C, Wang YN, Zhou Y, Long ZW, et al. Involvement of Bmi-1 gene in the development of gastrointestinal stromal tumor by regulating p16Ink4A/p14ARF gene expressions: an in vivo and in vitro study. Pathol Res Pract. (2017) 213(12):1542–51. doi: 10.1016/j.prp.2017.09.013
76. Vonlanthen S, Heighway J, Altermatt HJ, Gugger M, Kappeler A, Borner MM, et al. The bmi-1 oncoprotein is differentially expressed in non-small cell lung cancer and correlates with INK4A-ARF locus expression. Br J Cancer. (2001) 84(10):1372–6. doi: 10.1054/bjoc.2001.1791
77. Xu CR, Lee S, Ho C, Bommi P, Huang SA, Cheung ST, et al. Bmi1 functions as an oncogene independent of Ink4A/Arf repression in hepatic carcinogenesis Bmi1 in liver cancer development. Mol Cancer Res. (2009) 7(12):1937–45. doi: 10.1158/1541-7786.MCR-09-0333
78. Svoboda LK, Harris A, Bailey NJ, Schwentner R, Tomazou E, von Levetzow C, et al. Overexpression of HOX genes is prevalent in Ewing sarcoma and is associated with altered epigenetic regulation of developmental transcription programs. Epigenetics. (2014) 9(12):1613–25. doi: 10.4161/15592294.2014.988048
79. Andreeff M, Ruvolo V, Gadgil S, Zeng C, Coombes K, Chen W, et al. HOX Expression patterns identify a common signature for favorable AML. Leukemia. (2008) 22(11):2041–7. doi: 10.1038/leu.2008.198
80. Bhattacharya R, Kwon J, Ali B, Wang E, Patra S, Shridhar V, et al. Role of hedgehog signaling in ovarian cancer. Clin Cancer Res. (2008) 14(23):7659–66. doi: 10.1158/1078-0432.CCR-08-1414
81. Andrade NP, Warner KA, Zhang Z, Pearson AT, Mantesso A, Guimaraēs DM, et al. Survival of salivary gland cancer stem cells requires mTOR signaling. Cell Death Dis. (2021) 12(1):1–6. doi: 10.1038/s41419-021-03391-7
82. Wang MC, Li CL, Cui J, Jiao M, Wu T, Jing LI, et al. BMI-1, a promising therapeutic target for human cancer. Oncol Lett. (2015) 10(2):583–8. doi: 10.3892/ol.2015.3361
83. Zaczek A, Jóźwiak P, Ciesielski P, Forma E, Wójcik-Krowiranda K, Cwonda Ł, et al. Relationship between polycomb-group protein BMI-1 and phosphatases regulating AKT phosphorylation level in endometrial cancer. J Cell Mol Med. (2020) 24(2):1300–10. doi: 10.1111/jcmm.14782
84. You D, Wang D, Liu P, Chu Y, Zhang X, Ding X, et al. MicroRNA-498 inhibits the proliferation, migration and invasion of gastric cancer through targeting BMI-1 and suppressing AKT pathway. Hum Cell. (2020) 33(2):366–76. doi: 10.1007/s13577-019-00313-w
85. Wu Y, Tian S, Chen Y, Ji M, Qu Y, Hou P. miR-218 inhibits gastric tumorigenesis through regulating Bmi-1/Akt signaling pathway. Path Res Practice. (2019) 215(2):243–50. doi: 10.1016/j.prp.2018.10.031
86. Guo WJ, Zeng MS, Yadav A, Song LB, Guo BH, Band V, et al. Mel-18 acts as a tumor suppressor by repressing Bmi-1 expression and down-regulating Akt activity in breast cancer cells. Cancer Res. (2007) 67(11):5083–9. doi: 10.1158/0008-5472.CAN-06-4368
87. Jacobs JJ, Scheijen B, Voncken JW, Kieboom K, Berns A, van Lohuizen M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. (1999) 13(20):2678–90. doi: 10.1101/gad.13.20.2678
88. Warner KA, Nör F, Acasigua GA, Martins MD, Zhang Z, McLean SA, et al. Targeting MDM2 for treatment of adenoid cystic carcinoma MDM2 inhibition in adenoid cystic carcinoma. Clin Cancer Res. (2016) 22(14):3550–9. doi: 10.1158/1078-0432.CCR-15-1698
89. Krishnamurthy S, Warner KA, Dong Z, Imai A, Nör C, Ward BB, et al. Endothelial interleukin-6 defines the tumorigenic potential of primary human cancer stem cells. Stem Cells. (2014) 32(11):2845–57. doi: 10.1002/stem.1793
90. Song LB, Li J, Liao WT, Feng Y, Yu CP, Hu LJ, et al. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J Clin Invest. (2009) 119(12):3626–36. doi: 10.1172/JCI39374
91. Yu CC, Lo WL, Chen YW, Huang PI, Hsu HS, Tseng LM, et al. Bmi-1 regulates snail expression and promotes metastasis ability in head and neck squamous cancer-derived ALDH1 positive cells. J Oncol. (2011) 2011:609259. doi: 10.1155/2011/609259
92. Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY, Yang WH, et al. Bmi1 is essential in Twist1-induced epithelial–mesenchymal transition. Nat Cell Biol. (2010) 12(10):982–92. doi: 10.1038/ncb2099
93. Zhang Z, Dong Z, Lauxen IS, Nör JE. Endothelial cell-secreted EGF induces epithelial to mesenchymal transition and endows head and neck cancer cells with stem-like phenotype endothelial EGF and cancer stem cells. Cancer Res. (2014) 74(10):2869–81. doi: 10.1158/0008-5472.CAN-13-2032
94. Qiao B, Chen Z, Hu F, Tao Q, Lam AK. BMI-1 activation is crucial in hTERT-induced epithelial–mesenchymal transition of oral epithelial cells. Exp Mol Pathol. (2013) 95(1):57–61. doi: 10.1016/j.yexmp.2013.05.004
95. Herzog AE, Warner KA, Zhang Z, Bellile E, Bhagat MA, Castilho RM, et al. The IL-6R and Bmi-1 axis controls self-renewal and chemoresistance of head and neck cancer stem cells. Cell Death Dis. (2021) 12(11):1–2. doi: 10.1038/s41419-021-04268-5
96. Kuo SZ, Blair KJ, Rahimy E, Kiang A, Abhold E, Fan JB, et al. Salinomycin induces cell death and differentiation in head and neck squamous cell carcinoma stem cells despite activation of epithelial-mesenchymal transition and Akt. BMC Cancer. (2012) 12(1):1–4. doi: 10.1186/1471-2407-12-1
97. Magaway C, Kim E, Jacinto E. Targeting mTOR and metabolism in cancer: lessons and innovations. Cells. (2019) 8(12):1584. doi: 10.3390/cells8121584
98. Nakano T, Warner KA, Oklejas AE, Zhang Z, Rodriguez-Ramirez C, Shuman AG, et al. mTOR inhibition ablates cisplatin-resistant salivary gland cancer stem cells. J Dent Res. (2021) 100(4):377–86. doi: 10.1177/0022034520965141
99. Andrews A, Warner K, Rodriguez-Ramirez C, Pearson AT, Nör F, Zhang Z, et al. Ablation of cancer stem cells by therapeutic inhibition of the MDM2–p53 interaction in mucoepidermoid carcinoma MDM2 inhibition and cancer stem cells. Clin Cancer Res. (2019) 25(5):1588–600. doi: 10.1158/1078-0432.CCR-17-2730
100. Nör F, Warner KA, Zhang Z, Acasigua GA, Pearson AT, Kerk SA, et al. Therapeutic inhibition of the MDM2–p53 interaction prevents recurrence of adenoid cystic carcinomas MDM2 inhibition in adenoid cystic carcinoma. Clin Cancer Res. (2017) 23(4):1036–48. doi: 10.1158/1078-0432.CCR-16-1235
101. Kuo SZ, Honda CO, Li WT, Honda TK, Kim E, Altuna X, et al. Metformin results in diametrically opposed effects by targeting non-stem cancer cells but protecting cancer stem cells in head and neck squamous cell carcinoma. Int J Mol Sci. (2019) 20(1):193. doi: 10.3390/ijms20010193
102. Itoh T, Terazawa R, Kojima K, Nakane K, Deguchi T, Ando M, et al. Cisplatin induces production of reactive oxygen species via NADPH oxidase activation in human prostate cancer cells. Free Radical Res. (2011) 45(9):1033–9. doi: 10.3109/10715762.2011.591391
103. Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS, et al. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PloS One. (2013) 8(11):e81162. doi: 10.1371/journal.pone.0081162
104. Alajez NM, Shi W, Hui AB, Yue S, Ng R, Lo KW, et al. Targeted depletion of BMI1 sensitizes tumor cells to P53-mediated apoptosis in response to radiation therapy. Cell Death Differ. (2009) 16(11):1469–79. doi: 10.1038/cdd.2009.85
105. Wang G, Liu L, Sharma S, Liu H, Yang W, Sun X, et al. Bmi-1 confers adaptive radioresistance to KYSE-150R esophageal carcinoma cells. Biochem Biophys Res Commun. (2012) 425(2):309–14. doi: 10.1016/j.bbrc.2012.07.087
106. Liu ZG, Liu L, Xu LH, Yi W, Tao YL, Tu ZW, et al. Bmi-1 induces radioresistance in MCF-7 mammary carcinoma cells. Oncol Rep. (2012) 27(4):1116–22. doi: 10.3892/or.2011.1615
107. Ye L, Wang C, Yu G, Jiang Y, Sun D, Zhang Z, et al. Bmi-1 induces radioresistance by suppressing senescence in human U87 glioma cells. Oncol Lett. (2014) 8(6):2601–6. doi: 10.3892/ol.2014.2606
108. Sun J, Ye L, Wang C, Li N, Wang D, Li X. MicroRNA-128 increases glioma cell radio-sensitivity by suppressing senescent evasion through oncogene Bmi-1. Int J Clin Exp Pathol. (2018) 11(3):1423.31938239
109. Sharaf K, Lechner A, Haider SP, Wiebringhaus R, Walz C, Kranz G, et al. Discrimination of cancer stem cell markers ALDH1A1, BCL11B, BMI-1, and CD44 in different tissues of HNSCC patients. Curr Oncol. (2021) 28(4):2763–74. doi: 10.3390/curroncol28040241
110. Mohamed H, Hagström J, Jouhi L, Atula T, Almangush A, Mäkitie A, et al. The expression and prognostic value of stem cell markers bmi-1, HESC5: 3, and HES77 in human papillomavirus–positive and–negative oropharyngeal squamous cell carcinoma. Tumor Biol. (2019) 41(3):1010428319840473. doi: 10.1177/1010428319840473
111. Wu TF, Li YC, Ma SR, Bing-Liu , Zhang WF, Sun ZJ. Expression and associations of TRAF1, BMI-1, ALDH1, and Lin28B in oral squamous cell carcinoma. Tumor Biol. (2017) 39(4):1010428317695930. doi: 10.1177/1010428317695930
112. Jakob M, Sharaf K, Schirmer M, Leu M, Küffer S, Bertlich M, et al. Role of cancer stem cell markers ALDH1, BCL11B, BMI-1, and CD44 in the prognosis of advanced HNSCC. Strahlenther Onkol. (2021) 197(3):231–45. doi: 10.1007/s00066-020-01653-5
113. Häyry V, Mäkinen LK, Atula T, Sariola H, Mäkitie A, Leivo I, et al. Bmi-1 expression predicts prognosis in squamous cell carcinoma of the tongue. Br J Cancer. (2010) 102(5):892–7. doi: 10.1038/sj.bjc.6605544
114. Fan Z, Li M, Chen X, Wang J, Liang X, Wang H, et al. Prognostic value of cancer stem cell markers in head and neck squamous cell carcinoma: a meta-analysis. Sci Rep. (2017) 7(1):1–8. doi: 10.1038/s41598-016-0028-x
115. Zhang X, Tian T, Sun W, Liu C, Fang X. Bmi-1 overexpression as an efficient prognostic marker in patients with nonsmall cell lung cancer. Medicine (Baltimore). (2017) 96(26):e7346. doi: 10.1097/MD.0000000000007346
116. Yuan B, Zhao H, Xue X, Zhou J, Wang X, Han Y, et al. Prognostic value and clinicopathological differences of Bmi1 in gastric cancer: a meta-analysis. Anti-Cancer Agents Med Chem. (2016) 16(4):407–13.
117. Yu J, Chen L, Bao Z, Liu Y, Liu G, Li F, et al. BMI-1 promotes invasion and metastasis in endometrial adenocarcinoma and is a poor prognostic factor. Oncol Rep. (2020) 43(5):1630–40. doi: 10.3892/or.2020.7539
118. Kreso A, Van Galen P, Pedley NM, Lima-Fernandes E, Frelin C, Davis T, et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat Med. (2014) 20(1):29–36. doi: 10.1038/nm.3418
119. Jia L, Zhang W, Wang CY. BMI1 Inhibition eliminates residual cancer stem cells after PD1 blockade and activates antitumor immunity to prevent metastasis and relapse. Cell Stem Cell. (2020) 27(2):238–53. doi: 10.1016/j.stem.2020.06.022
120. Di Pietro A, Polmear J, Cooper L, Damelang T, Hussain T, Hailes L, et al. Targeting BMI-1 in B cells restores effective humoral immune responses and controls chronic viral infection. Nat Immunol. (2022) 23(1):86–98. doi: 10.1038/s41590-021-01077-y
121. Duan Q, Li H, Gao C, Zhao H, Wu S, Wu H, et al. High glucose promotes pancreatic cancer cells to escape from immune surveillance via AMPK-Bmi1-GATA2-MICA/B pathway. J Exp Clin Cancer Res. (2019) 38(1):1–3. doi: 10.1186/s13046-019-1209-9
122. Zhang D, Huang J, Wang F, Ding H, Cui Y, Yang Y, et al. BMI1 Regulates multiple myeloma-associated macrophage's pro-myeloma functions. Cell Death Dis. (2021) 12(5):1–6. doi: 10.1038/s41419-021-03748-y
123. Infante JR, Bedard PL, Shapiro G, Bauer TM, Prawira A, Laskin O, et al. Phase 1 results of PTC596, a novel small molecule targeting cancer stem cells (CSCs) by reducing levels of BMI1 protein.
124. Seipel K, Kopp B, Bacher U, Pabst T. BMI1-Inhibitor PTC596 in combination with MCL1 inhibitor S63845 or MEK inhibitor trametinib in the treatment of acute leukemia. Cancers (Basel). (2021) 13(3):581. doi: 10.3390/cancers13030581
125. Ismail IH, McDonald D, Strickfaden H, Xu Z, Hendzel MJ. A small molecule inhibitor of polycomb repressive complex 1 inhibits ubiquitin signaling at DNA double-strand breaks. J Biol Chem. (2013) 288(37):26944–54. doi: 10.1074/jbc.M113.461699
Keywords: head and neck cancer, salivary gland cancer (SGC), cancer stem cell (CSC), self-renewal, bmi-1, therapeutic resistance, head and neck squamos cell carcinoma, targeted therapy
Citation: Herzog AE, Somayaji R and Nör JE (2023) Bmi-1: A master regulator of head and neck cancer stemness. Front. Oral. Health 4:1080255. doi: 10.3389/froh.2023.1080255
Received: 26 October 2022; Accepted: 2 January 2023;
Published: 16 January 2023.
Edited by:
Fernanda Visioli, Federal University of Rio Grande do Sul, BrazilReviewed by:
Hope M Amm, University of Alabama at Birmingham, United StatesVui King Vincent-Chong, University at Buffalo, United States
© 2023 Herzog, Somayaji and Nör. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacques E. Nör amVub3JAdW1pY2guZWR1
Specialty Section: This article was submitted to Oral Cancers, a section of the journal Frontiers in Oral Health