 Camille Zenobia
Camille Zenobia Richard P. Darveau
Richard P. Darveau
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oral. Health , 23 May 2022
Sec. Oral Infections and Microbes
Volume 3 - 2022 | https://doi.org/10.3389/froh.2022.911420
This article is part of the Research Topic Insights in Oral Infections and Microbes: 2021 View all 9 articles
The oral microbiome, with a unique emphasis on Porphyromonas gingivalis has been associated with a constellation of inflammatory diseases such as cardiovascular disease, rheumatoid arthritis, Alzheimer's disease, type II diabetes, and non-alcoholic associated fatty liver disease. Periodontal disease has also been shown to induce “leaky gut” leading to metabolic endotoxemia. Several recent studies investigating the habitants of the blood microbiome have found the majority of species appear to be derived from oral and skin bacterial communities in otherwise healthy individuals. Many of the same pathologies associated with perturbations of oral health, such as cardiovascular disease, show alterations to the composition of the blood microbiome as well as circulating neutrophil phenotypes. Gingival inflammation is associated with activated blood neutrophil phenotypes that can exacerbate a distal inflammatory insult which may explain the connection between oral and systemic inflammatory conditions. While in the oral cavity, neutrophils encounter oral microbes that are adept in manipulating neutrophil activity which can re-enter the vasculature thereafter. Endotoxin from oral microbes can differ significantly depending on bacterial community and state of oral health to alter cellular LPS tolerance mechanisms which may contribute to the primed neutrophil phenotype seen in periodontitis and provide a mechanism by which the oral-microbes can affect systemic health outcomes. This review synthesizes the studies between inflammatory diseases and oral health with emphasis on microbiome and corresponding lipopolysaccharides in immune tolerance and activation.
Despite that oral microbiota or “animalcules” were the first bacteria of the human microbiome to be described with drawings by Antoine Leeuwenhoek (inventor of the microscope) much of what we know about the human microbiome has been dictated by study of the microbial community as it pertains to gastrointestinal health. Our understanding of the human microbiome has evolved from thinking of bacteria as invaders or freeloaders to now considering them a necessary and active participant in human metabolic function. Recent investigation suggests a primary role for oral bacteria in driving gut-microbial dysbiosis leading to Inflammatory Bowel Disease, and metabolic endotoxemia [1]. Coinciding with these findings, research into characterization of the blood microbiome has shown the presence of the oral microbiome in the humoral system in otherwise healthy individuals. In dental-focused research, it has long been acknowledged that there is a link between oral health and systemic parameters of health but a direct, stepwise mechanism of action has not been elucidated that satisfies all inflammatory pathologies implicated. This review aims to synthesize the disparate study between blood microbiome, metabolic endotoxemia, and oral pathology to develop a new hypothesis and potential mechanism for the connection between oral and systemic inflammatory pathology.
The gingival tissues in the oral cavity that surround each tooth are highly microbially colonized and have evolved with our host tissues over thousands of years. Correlations have been found between oral health and cardiovascular disease [2, 3], Stroke [4], pregnancy outcomes [5], Rheumatoid Arthritis [6], Alzheimer's disease [7], type II diabetes [8], Metabolic Syndrome [9], and Non-Alcohol Associated Fatty Liver Disease [10]. Many studies have looked to specific microbial inhabitants of the oral cavity to explain the link between localized oral pathology and systemic impacts. One of the primary suspects, Porphyromonas gingivalis (P. gingivalis) has been an ideal candidate of pathological etiology with ability to induce and feed off of inflammatory mediators [11], survive intracellular conditions and travel via host cells to remote tissues [12], alter lipopolysaccharide (LPS) structures to evade host recognition [13]. More importantly, P. gingivalis has been linked to the etiology of nearly every systemic pathology that has been described as an inflammatory, systemic comorbidity of periodontitis [14, 15].
P. gingivalis has been shown to disseminate systemically either from direct release into blood through periodontal lesions or by cell to cell invasion [16–18]. Recent characterization of a human blood microbiome has shown a small but viable population of bacteria originating primarily from both oral and skin microbial communities, including P. gingivalis in otherwise healthy individuals [17, 18]. The bacteria of the blood microbiome are primarily found in the Buffy coat, implicating white blood cells as the main vehicle of dissemination. Concurrent investigation into metabolic endotoxemia has illuminated a role for oral bacteria, specific to periodontal pathology, as a driver for the induction of microbial dysbiosis of the gut and subsequent dissemination of enteric lipopolysaccharide (LPS) into the blood reviewed here [19]. The resulting scenario with blood containing oral microbes that include P. gingivalis, and enteric-derived LPS could result in very different inflammatory responses depending on the state of oral health since gingival inflammation has been shown to prime circulating neutrophils [20].
To set the stage for gingival inflammation, it is necessary to review the basics of oral biofilm succession in order to more fully discuss the nuances of the host-microbe relationship. The oral cavity is diverse in hard and soft tissue types as well as microbial biofilm diversity reflecting over 400 unique bacterial taxa [21]. The epithelium of the soft tissues, namely the junctional epithelium, that form around hard tissues (teeth) do not contain tight junctions and are proximal to the highly vascularized gingiva which allows a constant supply of innate immune cells to the gingival crevice to patrol, and control microbial load [21, 22]. The sulcus, or gingival pocket contains a very well described microbial composition that develops in a stepwise manner starting with the deposition of salivary proteins on the tooth surface, bacterial adhesins, and bacteria nutrient demands shape the development of the oral biofilm [23]. The organization of the oral biofilm is dependent on bacterial relationships that have been described and categorized into disparate complexes that, once formed, allow the biofilm to grow and form increasingly complex structures [24]. The bacterial members that comprise the early colonizers of the gingival crevice are those with the capacity to adhere to the pellicle of the tooth and are considered only moderately pathogenic and primarily Gram-positive. The resulting foundation of early biofilm allows access to a bridging community of bacteria known as the orange complex to form which have been found to be increasingly capable of causing periodontal pathology (e.g., F. nucleatum). Finally, the “Red complex” of bacteria can gain entry, these members are primarily Gram-negative, contain endotoxin, and are well described for their highly pathogenic features (e.g., P. gingivalis). The presence of “Red complex” bacteria, or keystone pathogens can cause an outgrowth of commensal microbiota that results in increased microbial burden, inflammatory pathology, and tissue destruction [25]. This classical model of microbial succession is under constant revision as we evolve our understanding of the coordinated nutritional-interplay and adherence mechanisms between microbial species.
While the periodontal-associated pathogens can remain low in abundance compared to the overall microbiome community, they can still expand in number and alter their phenotype to become increasingly pathogenic. As the microbial burden increases, so too does the inflammatory environment. The cytokine milieu and organization of the microbial community can influence the activity and expression of pathogenic features of the oral pathobionts. One example is the lipid A moiety from P. gingivalis LPS which can be altered when exposed to increase in hemin, temperature, or co-culture with F. nucleatum resulting in unique LPS phenotypes with varying capacity to inhibit Toll-Like-Receptor-4 (TLR4) activity [3, 26, 27]. P. gingivalis is considered a keystone pathogen and an etiological agent in the development of periodontitis [25]. During the progression of localized periodontal inflammation, the local gingival environment changes to potentiate a phenotype alteration in the lipid-A portion of the LPS structure. Indeed, these changes have been shown to occur in a natural periodontitis human infection; patients with periodontal disease clinically evaluated for LPS activity from plaque harvested from sites of either active disease or healthy pockets. The LPS activity was shown to be TLR4-antagonist for sites with active disease whereas otherwise healthy pockets were found to be TLR4-agonist [3, 28]. The alteration to LPS signaling in gingival pockets of periodontitis has the potential to differentially prime responding neutrophils and disrupt the LPS-tolerance mechanism [29]. While P. gingivalis has been shown to disseminate beyond the oral cavity within leukocytes or directly into vasculature, the potential for this bacteria to induce local neutrophil-specific LPS priming in the gingival crevice might be another way that the pathobiont can influence distal inflammation.
Oral endotoxin has been a feature of extraordinary inflammation in gingival tissues, capable of causing exacerbated inflammation in leukocyte adhesion deficiency (LAD-1) patients that lends to a distinct microbial community [30]. The neutrophils in these LAD-1 patients are unable to migrate through gingival tissues to control the microbial burden, but the localized inflammatory response is sufficient to prevent microbial infiltration into the gingiva despite the increasing microbial load and translocation of LPS into gingival tissue. The microbial community that develops in severe LAD-1 is unique from the bacterial community associated with Localized Aggressive Periodontitis (LAP) indicating a specialized relationship exists between the inflammatory etiology and resulting microbiome [30]. A similar observation has been shown in the oral microbiome from receptor knockout mice. Mice deficient in either TLR-2,−4 responsible for host-recognition of bacterial cell wall lipoproteins and LPS, were found to develop different microbial communities and decreased neutrophil recruitment [31]. Furthermore, the periodontitis that develops from P. gingivalis is TLR4 dependent unlike disease induced by ligature [31, 32]. Together, these data suggest that host-signaling and microbial communities are in lock-step as they respond to one another. Further, the etiology of inflammatory insult can induce and be shaped by specific bacterial endotoxin to impart different health outcomes.
Preserving the integrity of the gingiva is paramount for the control of microbial load in the oral cavity. Neutrophils are the primary surveillance immune cell in the gingival tissues, innervating the permeable junctional epithelium with 30,000 neutrophils passing though per minute in healthy tissues [33]. Neutrophil migration into gingival tissues still occurs in the absence of the microbiota as seen in germ-free mice, perhaps lending a resilience to the epithelium where the neutrophil has been found to rescue epithelial cells from P. gingivalis induced-cell death [34, 35]. In healthy gingival tissues, the junctional epithelium remains in a low-differentiation state with a turnover rate of about 4–6 days [36]. As the oral biofilm builds and increases complexity in the gingival crevice, the mid- and late-colonizers are adept at manipulating neutrophil antimicrobial functions [37, 38]. The dysregulation of the neutrophil can exacerbate tissue damage and lead to chronic periodontal lesions, likely due to ineffective bacterial killing [39, 40]. During periodontitis, physical disruption to the epithelium, and microbiome alterations can impact tissue-neutrophil responses to cause a local-hyperinflammatory phenotype [41]. Evaluation of circulating neutrophils confirmed that people with periodontitis have the same hyperinflammatory phenotype in the blood which does not resolve with periodontal treatment [42]. This lack of resolution is perhaps due to the development of trained immunity where LPS exposure has been found to influence memory-like responses in bone marrow neutrophils [43]. Periodontal pathology causes alterations to the connective tissue and results in the apical migration of the junctional epithelium creating an exaggerated periodontal pocket and disorganized neutrophil recruitment [44]. The increase in neutrophils recruited to the oral cavity during gingivitis and periodontitis correlates predictably with pathology [45].
In oral tissue homeostasis, the relationship between gingival epithelial cells and neutrophil recruitment is tightly regulated with a CXCR2 signaling requirement for a healthy host-response to the commensal community that develops in the interdental region of the gingival tissues [34, 46]. The CXCR2 requirement is likely due to coordinated signaling between the neutrophil and endothelial cells which has been shown to be necessary for functional host response to LPS in the lung [47]. The tissue-neutrophil appears to respond differently to different LPS structures than neutrophils in circulation. To illustrate this phenomenon, the neutrophil response to LPS was evaluated in the presence or absence of platelets or serum [48]. Seven different bacterial LPS preparations were found to induce NETosis broadly when cells were cultured in the presence of platelets, whereas neutrophils grown in platelet- and serum-free conditions produced NETs to only two of seven the LPS structures [48]. Together, a picture emerges of a protective relationship between the epithelium and neutrophil that dampens the host response to LPS type when barrier functions are intact. The progression from health to periodontal disease results in the degradation of the junctional epithelium structure around the tooth and dysregulation of neutrophil response which likely results in the platelet-rich conditions that could allow the neutrophil to respond more aggressively to unique LPS structures. Indeed, it has been shown that a subpopulation of neutrophils exist in chronic periodontitis that exhibit a pro-inflammatory phenotype with increased NETosis as measured by myeloperoxidase and histone citrullination [41]. In summary, when barrier functions are compromised, the oral neutrophil response to LPS is likely altered lending to a hyperinflammatory phenotype which propagates to the circulation.
Continued exposure to LPS typically results in tolerance and is well described for repeated LPS exposure in innate immune cells, causing a downregulation of inflammatory cytokines [49]. Much of the research in LPS tolerance is uniquely Escherichia coli (E. coli) specific but the cellular response looks very different for P. gingivalis LPS where repeated exposure in neutrophils has been shown to increase IL-8, and phosphorylated JNK production [50]. Only one P. gingivalis LPS preparation was tested in this study whereas another study found other P. gingivalis preparations impact IL-8 differentially [51]. These findings raise questions around how neutrophil tolerance might be impacted by LPS alterations, different growth conditions, or periodontal pathology. Neutrophils isolated from patients with chronic periodontitis show an exaggerated cytokine response, including IL-8 in samples isolated from both saliva and blood [42, 52]. Bone-marrow derived neutrophils have been found to develop memory-like responses to repeat LPS exposure. Neutrophils primed with either low or high dose LPS and then re-challenged after a rest-period were either sensitized or tolerized, respectively, which also affected their migration and phagocytosis functions [43]. In macrophages, priming cells with either E. coli or P. gingivalis LPS differentially impacts tolerance during subsequent cross-LPS challenge [53]. Peritoneal derived macrophages primed with P. gingivalis LPS and then challenged with E. coli LPS produce significantly more TNFα and IL-10 than those challenged first with E. coli and then with P. gingivalis. It was found that P. gingivalis LPS priming led to upregulation of TLR2 genes whereas E. coli LPS priming upregulated TLR4 genes. These mechanisms of tolerance in mouse models appear to change during the aging process where aged mice show impared tolerance to both E. coli and P. gingivalis LPS [53]. Interestingly, aged mice also naturally develop GI dysbiosis [54], periodontitis [54, 55], and altered responses to induced metabolic endotoxemia [56] which complicates the understanding of how tolerance can be disrupted with age. It is also worth noting that P. gingivalis LPS is not the only contributor to oral endotoxin (LPSo) as other oral pathobionts and their respective LPS have been found to contribute to differential host responses alone or in combination [57] and will require additional consideration when comparing microbial profiles present in health or disease. Alterations to LPS structure can clearly impact tolerance functions which are likely to affect downstream events that occur during progression of periodontitis.
The disruption of barrier function and host response in the oral cavity can allow dissemination of oral bacteria and LPS into the vasculature that coincides with alterations to systemic neutrophil phenotypes. A study of induced gingivitis in humans found the induction of gingivitis corresponded with a hyperinflammatory blood neutrophil phenotype [20, 58]. In mice with induced periodontitis, the development of a hyperinflammatory blood-neutrophil was also described and found capable of inducing exacerbated pathology when mice were subsequently challenged with peritonitis [20, 58]. The authors very nicely show that oral pathology can impact innate inflammatory responses distal to gingival insult. Additional data revealed circulatory macrophages were unaffected by induction of periodontitis. It is notable that a ligature was utilized in this mouse model of periodontitis whereas other models of induction can produce different inflammatory responses [59]. It is not yet understood if the change to circulatory neutrophil phenotype is a result of direct priming from a local gingival lesion where neutrophils are the primary responder or by indirect exposure from the release of oral bacteria, or their PAMPS (e.g., LPS) to circulation. It has been found that simple tooth-brushing can cause septic conditions [60], allowing the release of oral bacteria into circulation but sans a periodontal insult, this exposure does not appear to be a factor in driving an inflammatory neutrophil phenotype since the resolution of gingivitis, by reinstatement of oral hygiene resulted in decrease to inflammatory phenotype [20, 58]. Thus, there exists a number of ways to prime the circulating neutrophil but there seems to be a unique relationship between an inflammatory periodontal insult and the resulting circulatory neutrophil phenotype that could set the stage for chronic inflammation.
Consideration of the environmental conditions of the circulatory neutrophil that patrols the gingival tissue is key to unraveling the mystery of downstream inflammatory responses. If, for example, a high fat diet results in the outgrowth of oral bacteria which then creates gut-microbiome dysbiosis and leaky gut (as seen in models of metabolic endotoxemia discussed below), enteric endotoxin is now present in the circulation. In this scenario, the circulatory neutrophil is potentially primed with an E. coli-like LPS which could result in upregulation of TLR4 or even NETosis. If the same neutrophil is found migrating through the gingival tissue, the pathological potential for exaggerated inflammatory response could be very different if periodontal inflammation is present Figure 1. It is curious that oral hygiene can correct the circulating hyper-inflammatory neutrophil as seen during resolution of human-induced gingivitis [20, 58] whereas periodontal disease seems to crystallize the circulating neutrophil phenotype into a permanent feature [42], perhaps a function of a cross-tolerance issue from a changing LPS-environment in the gingival pocket. Figure 2 highlights the changes to neutrophil phenotypes in health and chronic periodontal disease, contrasting the neutrophil found in the oral cavity with those in circulation. It is understood that neutrophil exposure to LPS can result in tolerance or trained immunity, typically dictated by the dose which has been reviewed recently with respect to periodontal health, disease, and endotoxemia [61]. Less understood is to what extent the oral-derived LPS can participate in directing neutrophil immunity since the variable microbial load, resulting LPS dose, and LPS-phenotypes are relatively unexplored in the context of systemic health. LPS-induced trained immunity has shown an enhanced cellular response to secondary insult, similar to what the Glogauer group found in their 2-hit-animal study where mice with periodontal disease subsequently challenged with peritonitis were saddled with an exaggerated neutrophil response to the peritoneal cavity [20]. The LPS priming and tolerance mechanisms inherent to the circulating neutrophil may play a significant role in shaping trained immunity and also help explain why gingivitis is clinically reversible while periodontitis is not.
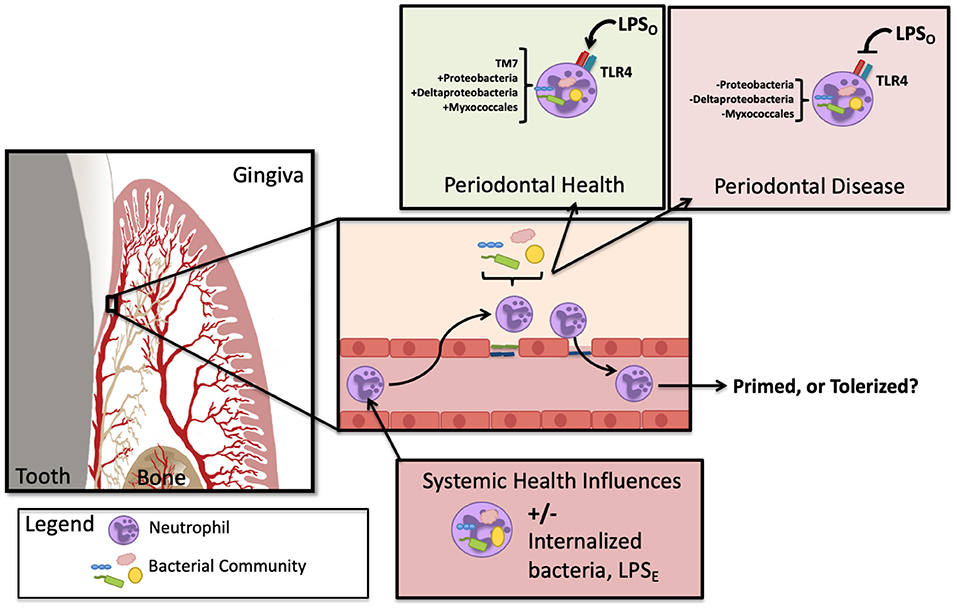
Figure 1. Oral endotoxin has the potential to prime or tolerize the infiltrating neutrophil. In circulation, the neutrophil may contain bacteria and/or be exposed to enteric LPS (LPSE) and subsequently migrate into tissues of the gingiva. Depending on the state of oral health, oral LPS (LPSO) can shift phenotypes with the changes that arise during microbial dysbiosis. Depending on the neutrophil state of activation, the subsequent agonism or antagonism of TLR4 could either prime or tolerize the migrating neutrophil. In addition, the circulating neutrophil has been found to harbor altered bacteria in periodontitis, illustrated here as described by Emery et al. [18].
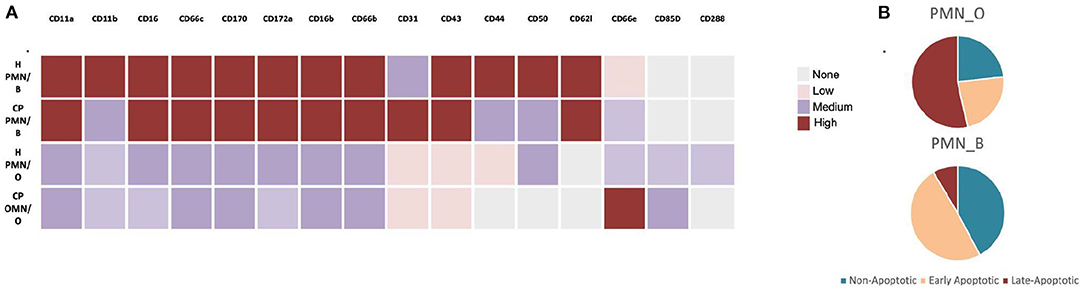
Figure 2. Oral vs. Blood Neutrophil Phenotypes. (A) Comparison of cluster differentiation expression levels of oral (O) and blood (B) neutrophils in oral- health (H) vs. chronic periodontal disease (CP). (B) Apoptotic profile of oral vs. blood neutrophils in otherwise healthy individuals. Illustrated here as described by [62, 63], respectively.
Metabolic endotoxemia is defined by chronic subclinical levels of systemic LPS. The presence of LPS release into the circulatory system is thought to stem from an inflammatory pathology resulting from “leaky gut” that has been linked to type II diabetes, non-alcoholic associated fatty liver disease, and increased cardiovascular disease; recently reviewed here [64]. Enteric LPS is considered the pathological agent that causes metabolic endotoxemia, and thus, models seeking to recapitulate what is seen in clinical pathology utilize E. coli LPS for study [65–67]. Early observations found metabolic endotoxemia can develop from a high fat, low fiber diet. As clinical and animal models were established, the mechanism of pathogenesis implicated GI microbial dysbiosis along with disruption to intestinal epithelial cells, and mucous-layer to allow enteric LPS (LPSE) to enter the circulatory system unmitigated [64, 68]. Induction of metabolic endotoxemia in clinical and animal models has been primarily through a high fat, low fiber diet, or direct injection of E. coli LPS [65–67, 69] but subsequent study of metabolic endotoxemia and periodontitis has shown that ingestion of perio-pathogens can induce similar pathology [70, 71].
P. gingivalis has been implicated in pathogenesis of metabolic endotoxemia either by release of whole, inactivated bacteria directly into blood or indirectly by ingestion and subsequent induction of intestinal microbial dysbiosis [72–74]. In an attempt to understand the capacity of different bacteria to cause metabolic endotoxemia, a recent investigation of different bacterial LPS structures found induction was limited to E. coli LPS. The other LPS structures tested were either under-acylated or mono-phosphorylated and incapable of inducing the corresponding pathology; P. gingivalis LPS was included in the study and failed to induce blood glucose levels, a requirement for induction of metabolic endotoxemia [75]. Other studies have shown that P. gingivalis LPS can in fact induce disease when combined with a high fat diet [74]. These mice also developed Non-Alcoholic Fatty Liver Disease, thus further corroborating the link between the oral pathobiont, periodontitis and fatty liver. These data implicate an etiological role for periodontitis when coupled with a high fat diet for the development of metabolic endotoxemia pathology. Intriguingly, it has also been shown that feeding a mouse a high fat diet results in natural periodontitis [71]. It is not clear if the resulting GI microbial dysbiosis that occurs with high fat diet is due to the increased ingestion of perio-pathogens or the diet itself but the developing periodontal bone loss appears to be LPS dependant [76]. A more recent study showed that removal of one of two signaling components essential for LPS recognition, TLR4 and CD14, by utilization of knockout mice were both protected from diet-induced metabolic endotoxemia despite having the same weight gain as wild type, implicating a role for the LPS recognition, not diet, as an upstream requirement for induction of GI bacterial dysbiosis and corresponding “leaky-gut” [77]. The P. gingivalis gavage model of periodontal disease is distinct from the ligature-induced model [59] with the capacity to induce GI dysbiosis, and metabolic endotoxemia [72]. Careful comparison of these two models might shed light on how periodontal priming might affect systemic inflammatory responses. It does seem plausible that the combination of high fat diet induced periodontitis (seen in the mouse model) and subsequent introduction of an under-acylated or mono-phosphorylated LPS structure, like those of P. gingivalis to the circulation could induce metabolic endotoxemia pathology and increase the risk of developing other systemic inflammatory diseases. More work is needed to identify the origins and types of circulating LPS seen in clinical metabolic endotoxemia disease.
Systemic LPS is an obvious problem with regard to circulating neutrophils. Evaluation of blood neutrophils in experimental human endotoxemia, experimental gingivitis, periodontitis, cardiovascular disease, and type II diabetes all show significant alterations to neutrophil phenotype, activation, and function [52, 58, 78–80]. Fine et al. [20] nicely demonstrated that induction of periodontitis can selectively induce blood neutrophils to cause exacerbated disease in mice subsequently challenged with peritonitis. The neutrophil appears to be the common actor on the chronic inflammatory stage. It has been shown that when the circulatory neutrophil is endotoxin-tolerized, by administration of E. coli LPS, the tolerized neutrophil is more efficient at responding to secondary infection than a non-tolerized neutrophil [81]. TLR4, the main host-cellular-receptor for endotoxin recognition, is implicated in the pathogenesis in Rheumatoid Arthritis [82], cardiovascular disease [83], and type II diabetes [84]. TLR4 inhibition is currently being explored for treatment of Rheumatoid Arthritis [85] with varying success [86]. C. Genco's lab has explored the relationship between different LPS structures of P. gingivalis in a mouse model of atherosclerosis to find that the antagonist-structure allowed for bacterial survival and disease progression while the agonist-structure did not suggesting that the TLR4-response is protective [87, 88]. In type II diabetes, the TLR4 function appears dysfunctional with neutrophils exhibiting tolerance behavior when exposed to LPS [89]. Although the neutrophil appears central to each inflammatory pathology, so too does the specific TLR4-host-response ranging from protective to pathological. Deepening our understanding around the specific neutrophil-priming events by bacterial composition, including LPS-phenotypes by tissue location will greatly improve strategies for treatment and prevention.
The blood microbiome is yet another indicator linking inflammatory pathologies where a dysbiotic microbial profile has been found in Rheumatoid Arthritis [90, 91], Diabetes [92–94], cardiovascular disease [95], non-alcohol associated fatty liver disease [93], and periodontal disease [18] when compared to healthy controls. The circulatory neutrophils have been implicated as a “Trojan Horse” [96] with capacity to allow dissemination of bacteria that remain viable and pathogenic [97]. More recently, neutrophil-specific microbiomes have been identified within the categorical study of the blood microbiome [98, 99]. Despite early evidence to the contrary, the circulatory system has long been considered a closed, sterile compartment. Recent technological advances for microbial evaluation, primarily Next Generation Sequencing have allowed scientists to re-evaluate the blood with greater sensitivity and rigor. Although methodology is still tricky and nuanced, there is clear evidence that the blood is not a sterile environment and is subject to, at minimum, a transient population of bacteria or, more likely, an ever-present bacterial community capable of impacting systemic health. It is intriguing that many of the inflammatory pathologies have in common, periodontitis, changes to neutrophil phenotypes and skewed blood microbial profiles. The present foray into characterization of the blood microbiome may shed some light on how the steady presence of circulating bacteria, and neutrophil phenotypes may contribute to a secondary insult or chronic inflammation.
Prior to broad investigation into the blood microbiome, it has long been known that viable oral pathobionts can exist within the circulatory system [100]. Studies of arterial plaques from patients with cardiovascular disease have repeatedly identified bacterial lipids as well as whole bacteria derived from the oral microbiome, including P. gingivalis [12, 101]. It is notable that gut bacteria have also been found in arterial plaques [102]. A recent study of the blood microbiome from periodontally healthy and diseased found surprisingly little difference in total bacterial DNA abundance between healthy and disease profiles but did note the presence of P. gingivalis in both study groups [18]. It would be expected that those with periodontal inflammation would also have increased bacteremia, or bacterial load but the methodology used in this study selectively evaluated whole bacteria from white blood cells. Therefore, transient bacteria that were not housed in a host cell were not considered. This is unfortunate because the cell-constrained bacterial cells are only part of a much larger picture when trying to understand the downstream circulatory effects impacted by periodontal disease. Investigation into differentiating microbial members that are cell-constrained vs. those that are transient may help uncover distinct patterns of dissemination and cell-associated microbiomes.
Deepening the understanding of the relationships between periodontitis, metabolic endotoxemia, and blood microbiome will require additional study. The host-microbiome has been shown to impact neutrophil aging [103] and is required for steady-state hematopoiesis [103–105], implicating an intimately coordinated relationship exists between tissue and bone compartments. Both MyD88-dependent-TLR and Nod-Like-Receptors are involved in steady-state-hematopoiesis further underscoring the bacterial requirement, and quite likely LPS, for driving the regulation [105]. Another investigation into the microbial regulation of osteo-immunomodulatory effects suggest the oral microbiome, by selective use of an antibiotic mouthwash, specifically induces the pro-osteoclastic activity in the alveolar bone compartment [105, 106]. Together, these studies indicate that specific microbial-niches can modulate disparate immune-functions. The microbiomes that contain Gram negative bacteria and LPS content are capable of inducing and maintaining the populations of circulating neutrophil activity; careful evaluation is needed to improve our understanding of neutrophil activation, tolerance, and trained immunity in the context of systemic health. The circulating neutrophil, which we now understand can harbor bacteria acquired by patrolling local tissue sites that are colonized by pathogenic bacteria, can be primed or tolerized by exposure to different or subsequent LPS-phenotypes (defined by tissue-specific bacterial communities) and then traffic to the lymph or distal tissues with muted or exaggerated responses. It will be imperative to understand which tissue-specific-bacteria/communities impart the most meaningful systemic benefit or pathology to inform and improve treatment strategies for the constellation of inflammatory pathologies associated with periodontal disease.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
CZ was employed by Os Salutem LLC.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. She Y-Y, Kong X-B, Ge Y-P, Liu Z-Y, Chen J-Y, Jiang J-W, et al. Periodontitis and inflammatory bowel disease: a meta-analysis. BMC Oral Health. (2020) 20:67. doi: 10.1186/s12903-020-1053-5
2. Scannapieco FA, Bush RB, Paju S. Associations between periodontal disease and risk for atherosclerosis, cardiovascular disease, and stroke. a systematic review. Ann Periodontol. (2003) 8:38–53. doi: 10.1902/annals.2003.8.1.38
3. Al-Qutub MN, Braham PH, Karimi-Naser LM, Liu X, Genco CA, Darveau RP. Hemin-dependent modulation of the lipid A structure of Porphyromonas gingivalis lipopolysaccharide. Infect Immun. (2006) 74:4474–85. doi: 10.1128/IAI.01924-05
4. Beck J, Garcia R, Heiss G, Vokonas PS, Offenbacher S. Periodontal disease and cardiovascular disease. J Periodontol. (1996) 67:1123–37. doi: 10.1902/jop.1996.67.10s.1123
5. Scannapieco FA, Bush RB, Paju S. Periodontal disease as a risk factor for adverse pregnancy outcomes. a systematic review. Ann Periodontol. (2003) 8:70–8. doi: 10.1902/annals.2003.8.1.70
6. Renvert S, Berglund JS, Persson GR, Söderlin MK. The association between rheumatoid arthritis and periodontal disease in a population-based cross-sectional case-control study. BMC Rheumatology. (2020) 4:1–8. doi: 10.1186/s41927-020-00129-4
7. Hu X, Zhang J, Qiu Y, Liu Z. Periodontal disease and the risk of Alzheimer's disease and mild cognitive impairment: a systematic review and meta-analysis. Psychogeriatrics. (2021) 21:813–25. doi: 10.1111/psyg.12743
8. Genco RJ, Graziani F, Hasturk H. Effects of periodontal disease on glycemic control, complications, and incidence of diabetes mellitus. Periodontol. (2020) 2000:59–65. doi: 10.1111/prd.12271
9. Gobin R, Tian D, Liu Q, Wang J. Periodontal diseases and the risk of metabolic syndrome: an updated systematic review and meta-analysis. Front Endocrinol. (2020) 11:336. doi: 10.3389/fendo.2020.00336
10. Alakhali MS, Al-Maweri SA, Al-Shamiri HM, Al-Haddad K, Halboub E. The potential association between periodontitis and non-alcoholic fatty liver disease: a systematic review. Clin Oral Investig. (2018) 22:2965–74. doi: 10.1007/s00784-018-2726-1
11. Darveau RP, Belton CM, Reife RA, Lamont RJ. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun. (1998) 66:1660–5. doi: 10.1128/IAI.66.4.1660-1665.1998
12. Olsen I, Progulske-Fox A. Invasion of porphyromonas gingivalis strains into vascular cells and tissue. J Oral Microbiol. (2015) 7:28788. doi: 10.3402/jom.v7.28788
13. Reife RA, Coats SR, Al-Qutub M, Dixon DM, Braham PA, Billharz RJ, et al. Porphyromonas gingivalis lipopolysaccharide lipid A heterogeneity: differential activities of tetra- and penta-acylated lipid A structures on E-selectin expression and TLR4 recognition. Cell Microbiol. (2006) 8:857–68. doi: 10.1111/j.1462-5822.2005.00672.x
14. Johansson A. Periodontitis: From Dysbiotic Microbial Immune Response to Systemic Inflammation. MDPI (2020).
15. Mei F, Xie M, Huang X, Long Y, Lu X, Wang X, et al. Porphyromonas gingivalis and its systemic impact: current status. Pathogens. (2020) 9:944. doi: 10.3390/pathogens9110944
16. Li L, Michel R, Cohen J, Decarlo A, Kozarov E. Intracellular survival and vascular cell-to-cell transmission of Porphyromonas gingivalis. BMC Microbiol. (2008) 8:26. doi: 10.1186/1471-2180-8-26
17. Whittle E, Leonard MO, Harrison R, Gant TW, Tonge DP. Multi-method characterization of the human circulating microbiome. Front Microbiol. (2018) 9:3266. doi: 10.3389/fmicb.2018.03266
18. Emery DC, Cerajewska TL, Seong J, Davies M, Paterson A, Allen-Birt SJ, et al. Comparison of blood bacterial communities in periodontal health and periodontal disease. Front Cell Infect Microbiol. (2020) 10:577485. doi: 10.3389/fcimb.2020.577485
19. Olsen I, Yamazaki K. Can oral bacteria affect the microbiome of the gut? J Oral Microbiol. (2019) 11:1586422. doi: 10.1080/20002297.2019.1586422
20. Fine N, Chadwick JW, Sun C, Parbhakar KK, Khoury N, Barbour A, et al. Periodontal inflammation primes the systemic innate immune response. J Dent Res. (2021) 100:318–25. doi: 10.1177/0022034520963710
21. Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner ACR, Yu W-H, et al. The human oral microbiome. J Bacteriol. (2010) 192:5002. doi: 10.1128/JB.00542-10
22. Bosshardt DD, Lang NP. The junctional epithelium: from health to disease. J Dent Res. (2005) 84. doi: 10.1177/154405910508400102
23. Kolenbrander PE, Palmer RJJr, Periasamy S, Jakubovics NS. Oral multispecies biofilm development and the key role of cell-cell distance. Nat Rev Microbiol. (2010) 8:471–80. doi: 10.1038/nrmicro2381
24. Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL. Microbial complexes in subgingival plaque. J Clin Periodontol. (1998) 25. doi: 10.1111/j.1600-051X.1998.tb02419.x
25. Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. (2012) 10:717–25. doi: 10.1038/nrmicro2873
26. Curtis MA, Percival RS, Devine D, Darveau RP, Coats SR, Rangarajan M, et al. Temperature-dependent modulation of Porphyromonas gingivalis lipid A structure and interaction with the innate host defenses. Infect Immun. (2011) 79:1187–93. doi: 10.1128/IAI.00900-10
27. Lee SH, Baek DH. Characteristics of porphyromonas gingivalis lipopolysaccharide in co-culture with fusobacterium nucleatum. Mol Oral Microbiol. (2013) 28:230–8. doi: 10.1111/omi.12020
28. To TT, Gümüş P, Nizam N, Buduneli N, Darveau RP. Subgingival plaque in periodontal health antagonizes at toll-like receptor 4 and inhibits E-Selectin expression on endothelial cells. Infect Immun. (2016) 84:120–6. doi: 10.1128/IAI.00693-15
29. Zaric S, Shelburne C, Darveau R, Quinn DJ, Weldon S, Taggart CC, et al. Impaired immune tolerance to Porphyromonas gingivalis lipopolysaccharide promotes neutrophil migration and decreased apoptosis. Infect Immun. (2010) 78:4151–6. doi: 10.1128/IAI.00600-10
30. Moutsopoulos NM, Chalmers NI, Barb JJ, Abusleme L, Greenwell-Wild T, Dutzan N, et al. Subgingival microbial communities in leukocyte adhesion deficiency and their relationship with local immunopathology. PLoS Pathog. (2015) 11:e1004698. doi: 10.1371/journal.ppat.1004698
31. Chang AM, Liu Q, Hajjar AM, Greer A, McLean JS, Darveau RP. Toll-like receptor-2 and−4 responses regulate neutrophil infiltration into the junctional epithelium and significantly contribute to the composition of the oral microbiota. J Periodontol. (2019) 90:1202-12. doi: 10.1002/JPER.18-0719
32. Lin M, Hu Y, Wang Y, Kawai T, Wang Z, Han X. Different engagement of TLR2 and TLR4 in Porphyromonas gingivalis vs. ligature-induced periodontal bone loss Braz. Oral Res. (2017) 31:e63. doi: 10.1590/1807-3107bor-2017.vol31.0063
33. Schiött CR, Löe H. The origin and variation in number of leukocytes in the human saliva. J Periodontal Res. (1970) 5:36–41. doi: 10.1111/j.1600-0765.1970.tb01835.x
34. Zenobia C, Luo XL, Hashim A, Abe T, Jin L, Chang Y, et al. Commensal bacteria-dependent select expression of CXCL2 contributes to periodontal tissue homeostasis. Cell Microbiol. (2013) 15:1419–26. doi: 10.1111/cmi.12127
35. Galicia JC, Benakanakere MR, Stathopoulou PG, Kinane DF. Neutrophils rescue gingival epithelial cells from bacterial-induced apoptosis. J Leukoc Biol. (2009) 86:181–6. doi: 10.1189/jlb.0109003
36. Jiang Q, Yu Y, Ruan H, Luo Y, Guo X. Morphological and functional characteristics of human gingival junctional epithelium. BMC Oral Health. (2014) 14:30. doi: 10.1186/1472-6831-14-30
37. Kurgan S, Kansal S, Nguyen D, Stephens D, Koroneos Y, Hasturk H, et al. Strain-specific impact of fusobacterium nucleatum on neutrophil function. J Periodontol. (2017) 88:380–9. doi: 10.1902/jop.2016.160212
38. Sochalska M, Potempa J. Manipulation of neutrophils by porphyromonas gingivalis in the development of periodontitis. Front Cell Infect Microbiol. (2017) 7:197. doi: 10.3389/fcimb.2017.00197
39. Sima C, Glogauer M. Neutrophil dysfunction and host susceptibility to periodontal inflammation: current state of knowledge. Curr Oral Health Rep. (2014) 1:95–103. doi: 10.1007/s40496-014-0015-x
40. Hirschfeld J. Neutrophil subsets in periodontal health and disease: a mini review. Front Immunol. (2019) 10:3001. doi: 10.3389/fimmu.2019.03001
41. Fine N, Hassanpour S, Borenstein A, Sima C, Oveisi M, Scholey J, et al. Distinct oral neutrophil subsets define health and periodontal disease states. J Dent Res. (2016) 95:931–8. doi: 10.1177/0022034516645564
42. Ling MR, Chapple ILC, Matthews JB. Peripheral blood neutrophil cytokine hyper-reactivity in chronic periodontitis. Innate Immun. (2015) 21:714–25. doi: 10.1177/1753425915589387
43. Lajqi T, Braun M, Kranig SA, Frommhold D, Pöschl J, Hudalla H. LPS induces opposing memory-like inflammatory responses in mouse bone marrow neutrophils. Int J Mol Sci. (2021) 22:9803. doi: 10.3390/ijms22189803
44. Nanci A, Bosshardt DD. Structure of periodontal tissues in health and disease. Periodontol 2000. (2006) 40:11–28. doi: 10.1111/j.1600-0757.2005.00141.x
45. Khoury W, Glogauer J, Tenenbaum HC, Glogauer M. Oral inflammatory load: Neutrophils as oral health biomarkers. J Periodontal Res. (2020) 55:594–601. doi: 10.1111/jre.12758
46. Greer A, Irie K, Hashim A, Leroux BG, Chang AM, Curtis MA, et al. Site-specific neutrophil migration and CXCL2 expression in periodontal tissue. J Dent Res. (2016) 95:946. doi: 10.1177/0022034516641036
47. Reutershan J, Morris MA, Burcin TL, Smith DF, Chang D, Saprito MS, et al. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J Clin Invest. (2006) 116:695–702. doi: 10.1172/JCI27009
48. Pieterse E, Rother N, Yanginlar C, Hilbrands LB, van der Vlag J. Neutrophils discriminate between lipopolysaccharides of different bacterial sources and selectively release neutrophil extracellular traps. Front Immunol. (2016) 7:484. doi: 10.3389/fimmu.2016.00484
49. Ziegler-Heitbrock HW. Molecular mechanism in tolerance to lipopolysaccharide. J Inflamm. (1995) 45:13–26.
50. Gu J-Y, Liu Y-J, Zhu X-Q, Qiu J-Y, Sun Y. Effects of endotoxin tolerance induced by porphyromonas gingivalis lipopolysaccharide on inflammatory responses in neutrophils. Inflammation. (2020) 43:1692–706. doi: 10.1007/s10753-020-01243-8
51. Herath TDK, Darveau RP, Seneviratne CJ, Wang C-Y, Wang Y, Jin L. Tetra- and penta-acylated lipid A structures of porphyromonas gingivalis LPS differentially activate TLR4-Mediated NF-κB signal transduction cascade and immuno-inflammatory response in human gingival fibroblasts. PLoS ONE. (2013) 8:e58496. doi: 10.1371/journal.pone.0058496
52. Lakschevitz FS, Aboodi GM, Glogauer M. Oral neutrophil transcriptome changes result in a pro-survival phenotype in periodontal diseases. PLoS ONE. (2013) 8:e0068983. doi: 10.1371/journal.pone.0068983
53. Sun Y, Li H, Yang M-F, Shu W, Sun M-J, Xu Y. Effects of aging on endotoxin tolerance induced by lipopolysaccharides derived from porphyromonas gingivalis and escherichia coli. PLoS ONE. (2012) 7:e39224. doi: 10.1371/journal.pone.0039224
54. Alam MS, Gangiredla J, Hasan NA, Barnaba T, Tartera C. Aging-induced dysbiosis of gut microbiota as a risk factor for increased infection. Front Immunol. (2021) 12:672353. doi: 10.3389/fimmu.2021.672353
55. Liang S, Hosur KB, Domon H, Hajishengallis G. Periodontal inflammation and bone loss in aged mice. J Periodontal Res. (2010) 45:574–8. doi: 10.1111/j.1600-0765.2009.01245.x
56. Saito H, Sherwood ER, Varma TK, Evers BM. Effects of aging on mortality, hypothermia, and cytokine induction in mice with endotoxemia or sepsis. Mech Ageing Dev. (2003) 124:1047–58. doi: 10.1016/j.mad.2003.08.002
57. Bodet C, Chandad F, Grenier D. Inflammatory responses of a macrophage/epithelial cell co-culture model to mono and mixed infections with Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia. Microbes Infect. (2006) 8:27–35. doi: 10.1016/j.micinf.2005.05.015
58. Wellappuli NC, Fine N, Lawrence HP, Goldberg M, Tenenbaum HC, Glogauer M. Oral and blood neutrophil activation states during experimental gingivitis. JDR Clin Transl Res. (2018) 3:65–75. doi: 10.1177/2380084417742120
59. Barel O, Aizenbud Y, Tabib Y, Jaber Y, Leibovich A, Horev Y, et al. γδ T Cells differentially regulate bone loss in periodontitis models. J Dent Res. (2021) 101:428–36. doi: 10.1177/00220345211042830
60. Lockhart PB, Brennan MT, Sasser HC, Fox PC, Paster BJ, Bahrani-Mougeot FK. Bacteremia associated with toothbrushing and dental extraction. Circulation. (2008) 117:3118–25. doi: 10.1161/CIRCULATIONAHA.107.758524
61. Vitkov L, Muñoz LE, Knopf J, Schauer C, Oberthaler H, Minnich B, et al. Connection between periodontitis-induced low-grade endotoxemia and systemic diseases: neutrophils as protagonists and targets. Int J Mol Sci. (2021) 22:4647. doi: 10.3390/ijms22094647
62. Lakschevitz FS, Hassanpour S, Rubin A, Fine N, Sun C, Glogauer M. Identification of neutrophil surface marker changes in health and inflammation using high-throughput screening flow cytometry. Exp Cell Res. (2016) 342:200–9. doi: 10.1016/j.yexcr.2016.03.007
63. Rijkschroeff P, Jansen IDC, van der Weijden FA, Keijser BJF, Loos BG, Nicu EA. Oral polymorphonuclear neutrophil characteristics in relation to oral health: a cross-sectional, observational clinical study. Int J Oral Sci. (2016) 8:191–8. doi: 10.1038/ijos.2016.23
64. Mohammad S, Thiemermann C. Role of metabolic endotoxemia in systemic inflammation and potential interventions. Front Immunol. (2020) 11:594150. doi: 10.3389/fimmu.2020.594150
65. Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, et al. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia. (2007) 50:2374–83. doi: 10.1007/s00125-007-0791-0
66. Mehta NN, McGillicuddy FC, Anderson PD, Hinkle CC, Shah R, Pruscino L, et al. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes. (2010) 59:172–81. doi: 10.2337/db09-0367
67. Patel PN, Shah RY, Ferguson JF, Reilly MP. Human experimental endotoxemia in modeling the pathophysiology, genomics, and therapeutics of innate immunity in complex cardiometabolic diseases. Arterioscler Thromb Vasc Biol. (2015) 35:525–34. doi: 10.1161/ATVBAHA.114.304455
68. Ghosh SS, Wang J, Yannie PJ, Ghosh S. Intestinal barrier dysfunction, LPS translocation, and disease development. J Endocr Soc. (2020) 4:bvz039. doi: 10.1210/jendso/bvz039
69. Zhou X, Han D, Xu R, Li S, Wu H, Qu C, et al. A model of metabolic syndrome and related diseases with intestinal endotoxemia in rats fed a high fat and high sucrose diet. PLoS ONE. (2014) 9:e115148. doi: 10.1371/journal.pone.0115148
70. Arimatsu K, Yamada H, Miyazawa H, Minagawa T, Nakajima M, Ryder MI, et al. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci Rep. (2014) 4:4828. doi: 10.1038/srep04828
71. Fujita Y, Maki K. High-fat diet-induced obesity triggers alveolar bone loss and spontaneous periodontal disease in growing mice. BMC Obes. (2015) 3:1. doi: 10.1186/s40608-016-0082-8
72. Nakajima M, Arimatsu K, Kato T, Matsuda Y, Minagawa T, Takahashi N, et al. Oral administration of P. gingivalis induces dysbiosis of gut microbiota and impaired barrier function leading to dissemination of enterobacteria to the liver. PLOS ONE. (2015) 10:e0134234. doi: 10.1371/journal.pone.0134234
73. Nibali L, Henderson B. The Human Microbiota and Chronic Disease: Dysbiosis as a Cause of Human Pathology. London: John Wiley and Sons (2016). doi: 10.1002/9781118982907
74. Sasaki N, Katagiri S, Komazaki R, Watanabe K, Maekawa S, Shiba T, et al. Endotoxemia by porphyromonas gingivalis injection aggravates non-alcoholic fatty liver disease, disrupts glucose/lipid metabolism, and alters gut microbiota in mice. Front Microbiol. (2018) 9:2470. doi: 10.3389/fmicb.2018.02470
75. Anhê FF, Barra NG, Cavallari JF, Henriksbo BD, Schertzer JD. Metabolic endotoxemia is dictated by the type of lipopolysaccharide. Cell Rep. (2021) 36:109691. doi: 10.1016/j.celrep.2021.109691
76. Blasco-Baque V, Serino M, Vergnes J-N, Riant E, Loubieres P, Arnal J-F, et al. High-fat diet induces periodontitis in mice through lipopolysaccharides (LPS) receptor signaling: protective action of estrogens. PLoS ONE. (2012) 7:e48220. doi: 10.1371/journal.pone.0048220
77. Dalby MJ, Aviello G, Ross AW, Walker AW, Barrett P, Morgan PJ. Diet induced obesity is independent of metabolic endotoxemia and TLR4 signalling, but markedly increases hypothalamic expression of the acute phase protein, SerpinA3N. Sci Rep. (2018) 8:15648. doi: 10.1038/s41598-018-33928-4
78. Visser T, Pillay J, Pickkers P, Leenen LPH, Koenderman L. Homology in systemic neutrophil response induced by human experimental endotoxemia and by trauma. Shock. (2012) 37:145–51. doi: 10.1097/SHK.0b013e31823f14a4
79. Maréchal P, Tridetti J, Nguyen M-L, Wéra O, Jiang Z, Gustin M, et al. Neutrophil phenotypes in coronary artery disease. J Clin Med Res. (2020) 9:1602. doi: 10.3390/jcm9051602
80. Kleinstein SE, McCorrison J, Ahmed A, Hasturk H, Van Dyke TE, Freire M. Transcriptomics of type 2 diabetic and healthy human neutrophils. BMC Immunol. (2021) 22:37. doi: 10.1186/s12865-021-00428-6
81. Ariga SK, Abatepaulo FB, Melo ESA, Velasco IT, Pinheiro da Silva F, de Lima TM, et al. Endotoxin tolerance drives neutrophil to infectious site. Shock. (2014) 42:168–73. doi: 10.1097/SHK.0000000000000175
82. Elshabrawy HA, Essani AE, Szekanecz Z, Fox DA, Shahrara S. TLRs future potential therapeutic targets for RA. Autoimmun Rev. (2017) 16:103–13. doi: 10.1016/j.autrev.2016.12.003
83. Yang Y, Lv J, Jiang S, Ma Z, Wang D, Hu W, et al. The emerging role of Toll-like receptor 4 in myocardial inflammation. Cell Death Dis. (2016) 7:e2234. doi: 10.1038/cddis.2016.140
84. Taha IM, Abdu Allah AM, Abd El Gayed EM. Expression of toll-like receptor 4 and its connection with type 2 diabetes mellitus. Cell Mol Biol. (2018) 64:15–20. doi: 10.14715/cmb/2018.64.13.4
85. Samarpita S, Kim JY, Rasool MK, Kim KS. Investigation of toll-like receptor (TLR) 4 inhibitor TAK-242 as a new potential anti-rheumatoid arthritis drug. Arthritis Res Ther. (2020) 22:16. doi: 10.1186/s13075-020-2097-2
86. Claire Barnard SMR. TLR4 “Not a Relevant Target” for RA. rheumatology.medicinematters.com. (2020). Available online at: https://rheumatology.medicinematters.com/rheumatoid-arthritis-/treatment/tlr4-not-a-relevant-target-for-ra/17545280 (accessed March 14, 2022).
87. Hayashi C, Papadopoulos G, Gudino CV, Weinberg EO, Barth KR, Madrigal AG, et al. Protective role for TLR4 signaling in atherosclerosis progression as revealed by infection with a common oral pathogen. J Immunol. (2012) 189:3681–8. doi: 10.4049/jimmunol.1201541
88. Slocum C, Coats SR, Hua N, Kramer C, Papadopoulos G, Weinberg EO, et al. Distinct lipid a moieties contribute to pathogen-induced site-specific vascular inflammation. PLoS Pathog. (2014) 10:e1004215. doi: 10.1371/journal.ppat.1004215
89. Kuwabara WMT, Yokota CNF, Curi R, Alba-Loureiro TC. Obesity and Type 2 Diabetes mellitus induce lipopolysaccharide tolerance in rat neutrophils. Sci Rep. (2018) 8:17534. doi: 10.1038/s41598-018-35809-2
90. Hammad DBM, Hider SL, Liyanapathirana VC, Tonge DP. Molecular characterization of circulating microbiome signatures in rheumatoid arthritis. Front Cell Infect Microbiol. (2019) 9:440. doi: 10.3389/fcimb.2019.00440
91. Mo X-B, Dong C-Y, He P, Wu L-F, Lu X, Zhang Y-H, et al. Alteration of circulating microbiome and its associated regulation role in rheumatoid arthritis: evidence from integration of multiomics data. Clin Transl Med. (2020) 10:e229. doi: 10.1002/ctm2.229
92. Qiu J, Zhou H, Jing Y, Dong C. Association between blood microbiome and type 2 diabetes mellitus: a nested case-control study. J Clin Lab Anal. (2019) 33:e22842. doi: 10.1002/jcla.22842
93. Yun Y, Kim H-N, Lee E-J, Ryu S, Chang Y, Shin H, et al. Fecal and blood microbiota profiles and presence of nonalcoholic fatty liver disease in obese versus lean subjects. PLoS ONE. (2019) 14:e0213692. doi: 10.1371/journal.pone.0213692
94. Velmurugan G, Dinakaran V, Rajendhran J, Swaminathan K. Blood microbiota and circulating microbial metabolites in diabetes and cardiovascular disease. Trends Endocrinol Metab. (2020) 31:835–47. doi: 10.1016/j.tem.2020.01.013
95. Amar J, Lange C, Payros G, Garret C, Chabo C, Lantieri O, et al. Blood microbiota dysbiosis is associated with the onset of cardiovascular events in a large general population: the DESIR study. PLoS ONE. (2013) 8:e54461. doi: 10.1371/journal.pone.0054461
96. Thwaites GE, Gant V. Are bloodstream leukocytes trojan horses for the metastasis of staphylococcus aureus? Nat Rev Microbiol. (2011) 9:215–22. doi: 10.1038/nrmicro2508
97. Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J Immunol. (2000) 164:3713–22. doi: 10.4049/jimmunol.164.7.3713
98. Li Q, Wang C, Tang C, Zhao X, He Q, Li J. Identification and characterization of blood and neutrophil-associated microbiomes in patients with severe acute pancreatitis using next-generation sequencing. Front Cell Infect Microbiol. (2018) 8:5. doi: 10.3389/fcimb.2018.00005
99. Wang C, Li Q, Tang C, Zhao X, He Q, Tang X, et al. Characterization of the blood and neutrophil-specific microbiomes and exploration of potential bacterial biomarkers for sepsis in surgical patients. Immun Inflamm Dis. (2021) 9:1343–57. doi: 10.1002/iid3.483
100. Kozarov EV, Dorn BR, Shelburne CE, Dunn WA, Progulske-Fox A. Human atherosclerotic plaque contains viable invasive actinobacillus actinomycetemcomitans and porphyromonas gingivalis. Arterioscler Thromb Vasc Biol. (2005) 25:e17–8. doi: 10.1161/01.ATV.0000155018.67835.1a
101. Stelzel M, Conrads G, Pankuweit S, Maisch B, Vogt S, Moosdorf R, et al. Detection of Porphyromonas gingivalis DNA in aortic tissue by PCR. J Periodontol. (2002) 73:868–70. doi: 10.1902/jop.2002.73.8.868
102. Koren O, Spor A, Felin J, Fåk F, Stombaugh J, Tremaroli V, et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U S A. (2011) 108:4592–8. doi: 10.1073/pnas.1011383107
103. Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, et al. Neutrophil ageing is regulated by the microbiome. Nature. (2015) 525:528–32. doi: 10.1038/nature15367
104. Balmer ML, Schürch CM, Saito Y, Geuking MB, Li H, Cuenca M, et al. Microbiota-derived compounds drive steady-state granulopoiesis via MyD88/TICAM signaling. J Immunol. (2014) 193:5273–83. doi: 10.4049/jimmunol.1400762
105. Yan H, Baldridge MT, King KY. Hematopoiesis and the bacterial microbiome. Blood. (2018) 132:559–64. doi: 10.1182/blood-2018-02-832519
Keywords: metabolic endotoxemia, Porphyromonas gingivalis, periodontal disease, gut dysbiosis, systemic inflammation
Citation: Zenobia C and Darveau RP (2022) Does Oral Endotoxin Contribute to Systemic Inflammation? Front. Oral. Health 3:911420. doi: 10.3389/froh.2022.911420
Received: 02 April 2022; Accepted: 29 April 2022;
Published: 23 May 2022.
Edited by:
Hatice Hasturk, The Forsyth Institute, United StatesReviewed by:
Tomoki Maekawa, Niigata University, JapanCopyright © 2022 Zenobia and Darveau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Camille Zenobia, Y2FtaWxsZS56ZW5vYmlhQG9zc2FpbnN0aXR1dGUuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.