 Peter J. Polverini
Peter J. Polverini Felipe Nör
Felipe Nör Jacques E. Nör
Jacques E. Nör
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oral. Health, 10 January 2023
Sec. Oral Cancers
Volume 3 - 2022 | https://doi.org/10.3389/froh.2022.1095842
This article is part of the Research TopicThe State of the Art in Head and Neck Cancer and Carcinogenesis Translational ResearchView all 7 articles
Cancer stem cells (CSC) are a subpopulation of cancer cells that exhibit properties of self-renewal and differentiation and have been implicated in metastasis and treatment failures. There is mounting evidence that carcinogen-initiated mucosal epithelial stem cells acquire the CSC phenotype following exposure to environmental or infectious mutagens and are responsible for promoting the malignant transformation of premalignant (dysplastic) epithelium. CSC further contribute to the progression of dysplasia by activating signaling pathways through crosstalk with various cell populations in the tumor microenvironment. Two cell types, tumor-associated macrophages (TAM) and vascular endothelial cells (EC) nurture CSC development, support CSC stemness, and contribute to tumor progression. Despite mounting evidence implicating CSC in the initiation and progression of dysplastic oral epithelium to squamous cell carcinoma (SCC), the molecular mechanisms underlying these synergistic biological processes remain unclear. This review will examine the mechanisms that underlie the transformation of normal epithelial stem cells into CSC and the mechanistic link between CSC, TAM, and EC in the growth and the malignant conversation of dysplastic oral epithelium.
Oral SCC is one of the most common malignancies worldwide (1). While there has been a modest decrease in the incidence of this disease, particularly in high-income countries, it continues to be a burden in developing countries. Additionally, in the last decade, there has been an increase in the percentage of young patients developing oropharyngeal SCC due to human papillomavirus (HPV) infection (2–5). The main risk factors for oral cancer are smoking, alcohol consumption, and DNA viruses (e.g., HPV). Unfortunately, efforts to limit the incidence of oral cancer have been met with limited success. A more effective strategy would be identifying the incipient precursor lesions, oral epithelial dysplasia (OED), at risk for malignant transformation (6–8).
While it is well known that dysplasia often precedes the development of oral SCC, the mechanism underlying the progression of epithelial dysplasias to SCC remains uncertain (6–8) CSC and the tumor microenvironment play a critical role in tumor progression. This review will highlight three cell populations that are key to tumor progression: cancer stem cells, vascular endothelium, and macrophages. The mechanism underlying the conversion of epithelial stem cells to cancer stem cells and their role in initiating crosstalk between these cell populations in driving OED to SCC will be the focus of this review.
The term “premalignant” or “precancerous” is best defined as a lesion or condition that is at increased risk of developing into an SCC (9, 10). There is considerable uncertainty when assessing the malignant potential of premalignant lesions (6). Not all clinical lesions with microscopic alterations associated with premalignancy will progress to SCC (11–13). Some of these lesions will rapidly progress to SCC or have a prolonged latency before progressing to SCC. Still, others may completely regress (6, 8, 11, 13). In addition, premalignant lesions may evolve from benign lesions that exhibit none of the recognized stages of malignant progression (8, 14). Current clinical and histopathologic criteria used to assess the risk of progression of premalignant lesions to SCC are often unable to reliably predict malignant potential (6, 11). In addition, the histologic criteria for evaluating premalignant lesions often vary from examiner to examiner (15–19).
Localized lesions and systemic conditions most commonly associated with an increased risk for progressing to SCC include leukoplakia (white patch), erythroplakia (red patch), proliferative verrucous leukoplakia, lichen planus, oral submucous fibrosis, chronic hyperplastic candidiasis, actinic cheilitis, reverse smoking, betel nut chewing, discoid lupus erythematosus, and the inherited disorders dyskeratosis congenita and Fanconi anemia (6, 19). From a clinical perspective, lesions of most concern often present as leukoplakia with or without a red component. Although these disorders have an increased statistical risk of malignant progression, it is difficult to predict the clinical outcome for any individual lesion. In addition, despite the relative ease of monitoring oral precancerous lesions, there are still no reliable biomarkers that distinguish lesions that will progress to cancer from those that will not.
Oral leukoplakia is the most common precancerous lesion. It presents as a white patch on the surface of the oral mucosa (8, 20–22). There is some variability in the risk of progression depending on its location in the oral cavity, regional geographic differences, and risk factors (tobacco smoking vs. tobacco chewing). Additionally, what is considered precancer (visual or histopathologic) varies from examiner to examiner (16). The frequency of malignant transformation of oral precancerous lesions varies with the quality and type of outcome assessed, the type of clinical studies conducted (prospective vs. retrospective), and length of follow-up (12). Also, there is considerable variation in the rate and frequency of progression of oral leukoplakia to invasive oral SCC (8, 16, 20–23). Our current understanding of the mechanism underlying malignant progression of OED remains limited. Several studies have investigated potential predictive markers of progression. The most common of these has been p53, proliferation markers Ki67 and PCNA, and a limited number of other molecular biomarkers, including cell cycle proteins, loss of heterozygosity (LOH), a range of cell surface and stromal proteins, and aberrant signaling pathways (24). To date, no single or combination of biomarkers has fulfilled the promise of predicting the onset of SCC at its earliest stages. Despite these limitations, important new information about the role of CSC and the tumor microenvironment in cancer initiation and progression has shed some light on the mechanism underlying the progression of precancerous lesions.
It is generally agreed that adult stem cells are the targets of carcinogenic agents (25, 26). There are four stages to the carcinogenic process. These include initiation, promotion, malignant transformation, and progression (27, 28). For cancer to develop, stem cells must first be exposed to an “initiating agent.” Initiation occurs following exposure to a chemical or physical carcinogen or an infectious agent. Initiation induces a reversable epigenetic modification of DNA or an irreversible molecular lesion that makes the initiated cell susceptible to the growth-promoting effect of a promoting agent (25, 26, 29, 30). Clinical and laboratory studies suggest that carcinogenesis is a process that requires multiple exposure to promoting agents (25, 31–33). Normal stem cells become immortalized, presumably due to a mutational event involving either a protooncogene or tumor suppressor gene (34–37). Once a cell is initiated, the affected cell may persist for many months or years before the second “promoting” event takes effect (38, 39).
Studies in humans and animals have provided compelling evidence that the initiated cells are de facto preneoplastic. Lesions described as leukoplakia that microscopically exhibit severe dysplasia or carcinoma in situ have taken an irreversible step toward malignant transformation (38–40). Next the initiated cell undergoes clonal expansion, during which additional changes allow it to express the malignant phenotype (25). This process, operationally, is irreversible. Several animal models have supported this concept (27, 28, 32, 39). This process is followed by transforming initiated stem cells into cancer stem cells, setting the stage for other mutational events that lead to cellular transformation and tumor progression. If malignant transformation is to occur the initiation and promotion events must occur in a specific sequence. This four-stage model of carcinogenesis has endured for over fifty years (27, 39).
Once a stem cell is initiated, it is susceptible to the effects of promoting agents that relieve cells from growth constraints and expand their numbers, escaping cell death leading to an accumulation of abnormal cells (25, 41). Skin papillomas, enzyme-altered liver foci, breast nodules, colon polyps, and dysplastic oral leukoplakia harbor initiated cells that have undergone clonal expansion following exposure to a promoting agent (28, 38). Promoting agents are, for the most part, not mutagenic. Therefore, they do not induce the formation of malignant tumors. Instead, they increase the frequency of genetic and epigenetic mutations that position the abnormal cells to undergo malignant conversion. Promoting agents include hormones such as estrogen, the drug diethylstilbestrol, and various chemicals such as per- and polyfluoroalkyl substances (PFAS). Initiation and promotion must occur in sequence. When the initiation and promotion processes are reversed, i.e., when cell are first exposed to a promoting agent followed by an initiation event, cells will not undergo malignant transformation (38, 40). Animal models of skin and liver cancer have shown that initiated cells must be exposed to promoting agents repeatedly before malignant transformation results (25, 42).
Malignant conversion is when precancerous cells express the malignant phenotype. This process requires the accumulation of mutations. Malignant transformation occurs after repeated exposure to a promoter. The frequency of exposure needed to cause malignant conversion varies from tissue to tissue, but the malignant phenotype is eventually expressed. Tumor promotion contributes to the process of carcinogenesis by expanding a population of initiated cells at increased risk for malignant conversion. The relatively low probability of malignant transformation can be increased substantially by repeated exposure of precancerous cells to DNA-damaging agents. This process is mediated through activation protooncogenes and inactivation of tumor suppressor genes (34–36, 43, 44).
Once dysplastic cells have undergone malignant conversion, they enter the progression phase where they acquire more aggressive characteristics over time. A prominent feature of the malignant phenotype is increased genomic instability and unregulated growth. Further genetic and epigenetic changes occur during this process, including activating additional protooncogenes and the functional loss of tumor suppressor genes. These genetic alterations confer a growth advantage upon cells and increase their capacity for invasion and metastasis. The determining factor is the accumulation of these mutations, not the order or the stage of tumor development (25). Figure 1 depicts the relationship between the process of carcinogenesis with the progression of premalignant leukoplakia to SCC.
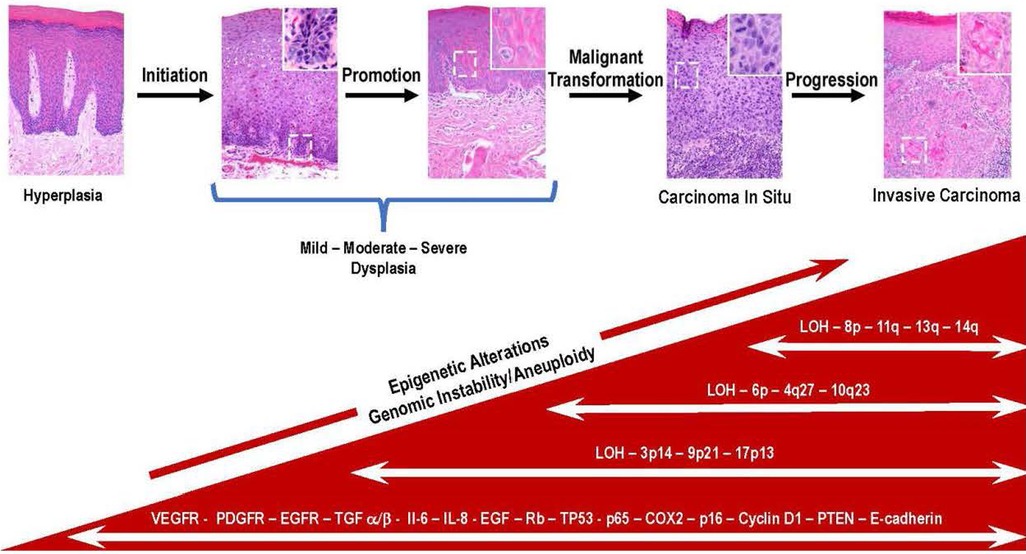
Figure 1. Key genetic and molecular events during the progression of premalignant oral epithelium to oral squamous cell carcinoma are depicted in this multistep model of histologic progression. The histological sequence of events seen in the progression of premalignant epithelium is initiated in the basal cell layer following exposure of stem cells to carcinogens. This results in irreversible damage and or epigenetic modification of DNA resulting in the emergence of a population of cancer stem cells. The subsequent exposure to promoting agents contribute to the clonal expansion of initiated cancer stem cells. With the continued exposure to promoting agents, a subset of initiated stem cells undergoes malignant conversion (i.e., carcinoma in situ). Over time cancer stem cells and bulk tumor cells acquire additional mutations many which involve loss or inactivation of tumor suppressor genes (TP53, PTEN, LOH) and overexpression of molecular mediators (Il-6, IL-8, VEGFR, PDGF). The bidirectional exchange of information between cancer and host cells contribute to acquisition of more aggressive phenotypes by the invading cancer cells (100X magnification, inset, white boarder, 400X).
Cancer stem cells (CSC) comprise a small subpopulation of cancer cells with the unique capacity to sustain tumor growth and drive tumor progression. CSC are primarily responsible for the failure of conventional and precision therapies (45–48). The properties of stemness, self-renewal, multipotency, and differentiating into heterogeneous cancer cell types define the CSC that make up the tumor mass (49–51). CSC are phenotypically and functionally heterogeneous. The phenotypic diversity gives rise to increased numbers of tumor-initiating cells (52–54). CSC have been identified in most human cancers, including breast, brain, and oral and head and neck SCC (55–58). CSC often reside at the advancing front of invasive SCC, where they are surrounded by their non-stem cell progeny and a variety of host cells that confer a survival advantage on CSC (47, 59, 60). The majority of CSC reside within a 100-mm radius of the perivascular niche (61). The perivascular niche serves as a biological nursery that helps CSC maintain their stem cell phenotype (59, 61). The perivascular niche provides the soil for CSC self-renewal and maintenance, stimulating essential signaling pathways in CSC and leading to the secretion of factors that promote angiogenesis and long-term growth of CSC (47, 62). The essential nature of the perivascular niche was demonstrated by Krishnamurthy et al. who showed that selective ablation of microvessels that comprise the perivascular niche resulted in the reduction in the population of CSC (61).
Recent studies have revealed the indispensable role of the IL-6 signaling in facilitating the acquisition of cancer stem cell functions in coordination with NF-kB-dependent inflammatory signals derived from tumor cells and host cells (63–65). IL-6 is a pro-inflammatory cytokine that activates JAK/STAT3 pathway (66–68). IL-6 levels have been shown to correlate with tumor progression in a number of cancer types including oral and head and neck cancer (69–71). In addition, IL-6 is a predictive marker for recurrence rate and overall survival of head and neck SCC patients (72, 73). Studies have shown that tumor cells acquire metastatic potential through IL-6/STAT3 pathway(74). IL-6 is secreted by many different cells, including T and B cells, monocytes and macrophages, endothelial cells, fibroblasts, and CSC (47, 61). Endothelial cells secrete high levels of IL-6 and expression of IL-6R or its co-receptor gp130 at the invasive tumor front is strongly correlated with poor patient survival (47, 61). Furthermore, endothelial cell-secreted IL-6 induces epithelial mesenchymal transition and enhances migration in head and neck CSC (61). Collectively, these results demonstrate that endothelial cell-secreted IL-6 induces a migratory phenotype in head and neck CSC. It has been suggested that therapeutic blockage of the IL-6 pathway might prevent and/or delay progression of oral and head and neck SCC (47).
Several surface markers, such as CD47, CD44, CD133, and Musashi-1, a stem cell marker fond in several tissues including oral squamous epithelium, are expressed by head and neck and oral CSC along with cytoplasmic enzymes such as aldehyde dehydrogenase (ALDH) (45, 47, 61, 75, 76). CSC, together with host cells are responsible for driving tumor progression. CSC are also responsible for local recurrence, metastatic spread, and therapeutic failures (77, 78). Studies have demonstrated that initiated CSC are present in dysplastic oral epithelium (79, 80). Progression of oral cavity cancer appears to be associated with the increased presence of CSC (79, 80). CD44 is a well- studied markers of CSC in oral cancer (81, 82). It has been shown that the presence of CD44 positive cells in patients is strongly correlated with the progression of premalignant lesions to a more aggressive stage of development (severe dysplasia and carcinoma in situ). This is further evidence in support of the idea that CSC play a role in the progression of dysplasia to invasive SCC (80).
CSC exist within a complex microenvironment. Bidirectional communication between CSC and host cells plays an essential role in augmenting the tumor-promoting functions of CSC. Recent studies suggest that bidirectional communication between vascular endothelial cells, macrophages, and CSC is central in orchestrating tumor progression (83, 84).
Vascular endothelial cells and macrophages extend the tumor promoting effect of CSC in several ways (83–85). In addition to their role in establishing a stem cell niche, endothelial cells provide essential nutrients to a rapidly growing population of tumor cells through their ability to amplify the blood supply to tumors on demand(86–89). Neovascular endothelial cells and CSC have a coregulatory function(85, 90). Several differentially expressed genes in endothelial cells that are usually quiescent are upregulated during angiogenesis (86, 91). These include, among others, growth factors and growth factor receptors, matrix metalloproteinases, inhibitors of angiogenesis, and NF-kB-regulated signaling pathways (85). In addition, endothelial cells produced cytokines, including HGF, PDGF, PIGF, and IL-6, that stimulate the self-renewal and enhances the survival of CSC (92, 93).
Besides their role in self-renewal CSC from a variety of tumors have been directly implicated in activating the angiogenic switch (85, 86, 91). For example, CSC from gliomas and ovarian cancer produce elevated levels of proangiogenic mediators (85, 90). Tissue samples from patients with glioblastoma multiforme as well as CSC isolated from these tumors revealed high levels of the angiogenic mediators VEGF, VEGFR1 and VEGFR2 and the hypoxia-inducing agents HIF1α, HIF2α. Many of these proangiogenic mediators ae also produced by cells within the tumor microenvironment that communicate with CSC to promote angiogenesis (93).
It is well established that tumor angiogenesis plays a central role in tumor progression. In many cancers, tumor-associated endothelial cells have a higher proliferative rate as compared to the established microvasculature where endothelial cells are normally quiescent (94). It is well documented that the increased microvascular density (MVD) that accompanies tumor development has shown prognostic value in a variety of cancers and is an established indicator of tumor progression (80, 95, 96). A study comparing low risk (mild dysplasia) to high risk (moderate and severe) mucosal lesions showed a statistically significant correlation with MVD expression. Also increasing numbers of CD44+ CSC and CD31+ endothelial cells was positively correlated with increased MVD and high-risk dysplasias progressing to SCC (80, 97). These results further support the notion that CSC and tumor-associated endothelial cells have biologically synergistic roles in tumor progression. Furthermore, epithelial dysplasias that undergo malignant transformation are positively associated with CSC enrichment. This observation further confirms the role of vascular niche, endothelial cells, and CSC in tumor progression (80).
Tumor-associated macrophages (TAM) are a heterogeneous population of cells. In addition to phagocytic and antigen-presenting functions, they play an essential role in inflammation, resolving infections, and tissue repair. In contrast, macrophages are also responsible for tissue damage, chronic inflammatory diseases, autoimmune disorders, and tumor growth and progression (98, 99). This array of divergent functions is due in part to their ability to undergo polarization into two phenotypically distinct subpopulations designated as M1 and M2 macrophages (98–100). M1 macrophages produce an array of proinflammatory cytokines and reactive oxygen molecules that promote Th1-mediated tumoricidal responses. The M1 polarization in macrophages is mainly regulated by distinct transcriptional networks consisting the Notch, NF-κB, TGF-β, Wnt/β-catenin, and MAPK (101–105). On the other hand, M2 macrophages are considered anti-inflammatory, are involved in tissue remodeling and immune tolerance, and have protumor functions that facilitate tumor progression. Transcriptional control in M2 is mediated through STAT1 and STAT2 activation in response to type-1 α and β interferons (106). Additionally, STAT isoforms, including STAT3 and STAT6, modulate M2 polarization (104, 107–114).
The process of tumor progression is frequently associated with a phenotypic switch from M1 to M2 in tumor-associated macrophages (115). The polarization of macrophages is a labile process where the proportion of M1 and M2 TAM with tumors will vary depending on the functional status of tumor microenvironments (116, 117). Through distinct sets of autocrine and paracrine signaling molecules, transcription factors, and epigenetic modifiers, tumor cells can further differentiate TAM into subsets of tumor-promoting macrophages capable of altering the genetic and phenotypic profiles of tumor cells (118, 119).
Macrophages and endothelial cells influence the CSC functions through a series of reciprocal interactions between CSC, endothelial cells, and TAM (101). Neovascular endothelial cells and M1/M2 TAM produce mediators that enable CSC to maintain their stem cell phenotype while maintaining TAM proangiogenic and protumor functions (120). Cytokines produced by endothelial cells and TAM stimulate the self-renewal and survival of adjacent CSC (121, 122). The proximity of vascular niche endothelial cells to CSC provides the nurturing environment necessary to maintain the CSC phenotype and enable CSC self-renewal and long-term survival. Together, the complementary networks created by cancer stem cells, endothelial cells, and TAM contribute to tumor proliferation, growth, invasion, metastatic activity, and treatment failures. Figure 2 shows the reciprocal relationships between CSC, endothelial cells, and TAM and the mediators responsible for the protumor effects that define this relationship.
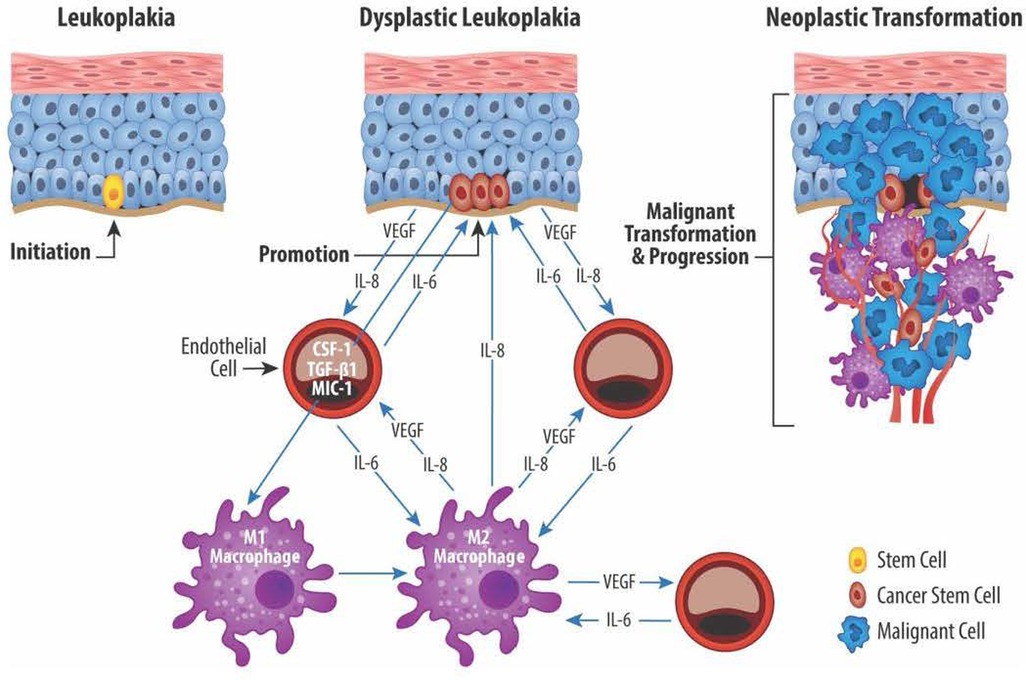
Figure 2. Reciprocal exchange of cytokine mediators between cancer stem cells (CSC), endothelial cells (EC), and macrophages (M1 and M2). CSC located in the basal layer of dysplastic epithelium produce, among others, the angiogenic mediators VEGF and IL-8. EC in turn produce IL-6 which plays an important role in maintaining the CSC stemness. EC also play a role in transitioning proinflammatory M1 macrophages to protumor M2 macrophages via production of IL-6, colony stimulating factor 1 (CSF-1), TGF-β1, macrophage inhibitory cytokine 1 (MIC-1). Lastly, M2 macrophages promote angiogenesis via production of VEGF and IL-8 and help maintain CSC stemness through IL-8 production.
The tumor microenvironment is defined by the distribution of various stromal cells and their sequential and mutually beneficial cellular interactions. Recent studies of CSC and the tumor microenvironment have revealed novel insights into the complex mechanisms that drive tumor progression and underlie treatment failures. For example, the bidirectional flow of growth factors and cytokines between CSC and host cells (EC, and TAM) promotes TAM protumor functions, sustains angiogenesis, maintains the CSC phenotype, thereby facilitating tumor growth and progression (123). CSC, like their normal stem cell counterparts, are phenotypically and functionally diverse. Studies of various human tumors have shown that phenotypically diverse subsets of CSC are present within tumors. However, their relationship with one another and their role in tumor development and progressions remain unclear. Changes in the tumor microenvironment, such as the degree of hypoxia, the composition of the inflammatory infiltrate, and their cytokine mediators, undoubtedly play a crucial role in determining the types of CSC cells that populate a tumor.
It is well established that cancer evolves from a series of random mutational events that lead to a complex series of aberrant cellular, genetic, and molecular processes. The ability of cancer cells to adapt to changes in their environment provides them with a selective growth and survival advantage. By co-opting the regenerative properties of the host, cancer cells can utilize the bidirectional flow of information between cancer cells and host cells to their advantage. It is this unique ability of cancer cells to thrive in an otherwise hostile host environment that characterizes the dynamic cancer ecosystem (124–127). Continued exploration of the mechanisms that underlie the physiologic reprogramming of CSC and the signals involved in orchestrating CSC diversity will be necessary to design therapeutic strategies that target CSC and its progeny at the incipient stages of tumor development.
PJP prepared the manuscript. FN and JEN reviewed, edited, and assisted in the preparation of the manuscript. All authors contributed to the article and approved the submitted version.
Support by the University of Michigan Distinguished University Professor funds to Peter J. Polverini
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bouvard V, Wentzensen N, Mackie A, Berkhof J, Brotherton J, Giorgi-Rossi P, et al. The IARC perspective on cervical cancer screening. N Engl J Med. (2021) 385(20):1908–18. doi: 10.1056/NEJMsr2030640
2. Gillison ML, D'Souza G, Westra W, Sugar E, Xiao W, Begum S, et al. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J Natl Cancer Inst. (2008) 100(6):407–20. doi: 10.1093/jnci/djn025
3. Chen SY, Massa S, Mazul AL, Kallogjeri D, Yaeger L, Jackson RS, et al. The association of smoking and outcomes in HPV-positive oropharyngeal cancer: a systematic review. Am J Otolaryngol. (2020) 41(5):102592. doi: 10.1016/j.amjoto.2020.102592
4. Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. (2010) 363(1):24–35. doi: 10.1056/NEJMoa0912217
5. Lechner M, Liu J, Masterson L, Fenton TR. HPV-associated oropharyngeal cancer: epidemiology, molecular biology and clinical management. Nat Rev Clin Oncol. (2022) 19(5):306–27. doi: 10.1038/s41571-022-00603-7
6. Speight PM, Khurram SA, Kujan O. Oral potentially malignant disorders: risk of progression to malignancy. Oral Surg Oral Med Oral Pathol Oral Radiol. (2018) 125(6):612–27. doi: 10.1016/j.oooo.2017.12.011
7. Arduino PG, Surace A, Carbone M, Elia A, Massolini G, Gandolfo S, et al. Outcome of oral dysplasia: a retrospective hospital-based study of 207 patients with a long follow-up. J Oral Pathol Med. (2009) 38(6):540–4. doi: 10.1111/j.1600-0714.2009.00782.x
8. Chaturvedi AK, Udaltsova N, Engels EA, Katzel JA, Yanik EL, Katki HA, et al. Oral leukoplakia and risk of progression to oral cancer: a population-based cohort study. J Natl Cancer Inst. (2020) 112(10):1047–54. doi: 10.1093/jnci/djz238
9. Curtius K, Wright NA, Graham TA. Evolution of premalignant disease. Cold Spring Harb Perspect Med. (2017) 7(12):1–19. doi: 10.1101/cshperspect.a026542
10. Curtius K, Wright NA, Graham TA. An evolutionary perspective on field cancerization. Nat Rev Cancer. (2018) 18(1):19–32. doi: 10.1038/nrc.2017.102
11. Lingen MW, Kalmar JR, Karrison T, Speight PM. Critical evaluation of diagnostic aids for the detection of oral cancer. Oral Oncol. (2008) 44(1):10–22. doi: 10.1016/j.oraloncology.2007.06.011
12. National Cancer Institute Clinical Trials. [updated 2014]. Available from: http://www.cancer.gov/clinicaltrials
13. Lingen MW, Abt E, Agrawal N, Chaturvedi AK, Cohen E, D'Souza G, et al. Evidence-based clinical practice guideline for the evaluation of potentially malignant disorders in the oral cavity: a report of the American dental association. JADA. (2017) 148(10):712–27. e10. doi: 10.1016/j.adaj.2017.07.032
14. Abrahams E, Ginsburg GS, Silver M. The personalized medicine coalition: goals and strategies. Am J Pharmacogen. (2005) 5(6):345–55. doi: 10.2165/00129785-200505060-00002
15. Warnakulasuriya S. Histological grading of oral epithelial dysplasia: revisited. J Pathol. (2001) 194(3):294–7. doi: 10.1002/1096-9896(200107)194:3%3C294::AID-PATH911%3E3.0.CO;2-Q
16. Warnakulasuriya S, Reibel J, Bouquot J, Dabelsteen E. Oral epithelial dysplasia classification systems: predictive value, utility, weaknesses and scope for improvement. J Oral Pathol Med. (2008) 37(3):127–33. doi: 10.1111/j.1600-0714.2007.00584.x
17. Katz HC, Shear M, Altini M. A critical evaluation of epithelial dysplasia in oral mucosal lesions using the smith-pindborg method of standardization. J Oral Pathol. (1985) 14(6):476–82. doi: 10.1111/j.1600-0714.1985.tb00519.x
18. Speight PM, Abram TJ, Floriano PN, James R, Vick J, Thornhill MH, et al. Interobserver agreement in dysplasia grading: toward an enhanced gold standard for clinical pathology trials. Oral Surg Oral Med Oral Pathol Oral Radiol. (2015) 120(4):474–82. e2. doi: 10.1016/j.oooo.2015.05.023
19. Warnakulasuriya S, Kujan O, Aguirre-Urizar JM, Bagan JV, Gonzalez-Moles MA, Kerr AR, et al. Oral potentially malignant disorders: a consensus report from an international seminar on nomenclature and classification, convened by the WHO collaborating centre for oral cancer. Oral Dis. (2021) 27(8):1862–80. doi: 10.1111/odi.13704
20. Mehanna HM, Rattay T, Smith J, McConkey CC. Treatment and follow-up of oral dysplasia - a systematic review and meta-analysis. Head Neck. (2009) 31(12):1600–9. doi: 10.1002/hed.21131
21. Yanik EL, Katki HA, Silverberg MJ, Manos MM, Engels EA, Chaturvedi AK. Leukoplakia, oral cavity cancer risk, and cancer survival in the U.S. Elderly. Cancer Prev Res (Phila). (2015) 8(9):857–63. doi: 10.1158/1940-6207.CAPR-15-0091
22. William WN J, Papadimitrakopoulou V, Lee JJ, Mao L, Cohen EE, Lin HY, et al. Erlotinib and the risk of oral cancer: the erlotinib prevention of oral cancer (EPOC) randomized clinical trial. JAMA Oncol. (2016) 2(2):209–16. doi: 10.1001/jamaoncol.2015.4364
23. Lee JJ, Hong WK, Hittelman WN, Mao L, Lotan R, Shin DM, et al. Predicting cancer development in oral leukoplakia: ten years of translational research. Clin Cancer Res. (2000) 6(5):1702–10.10815888
24. Nikitakis NG, Pentenero M, Georgaki M, Poh CF, Peterson DE, Edwards P, et al. Molecular markers associated with development and progression of potentially premalignant oral epithelial lesions: current knowledge and future implications. Oral Surg Oral Med Oral Pathol Oral Radiol. (2018) 125(6):650–69. doi: 10.1016/j.oooo.2018.03.012
25. Scott RE, Wille JJ Jr., Wier ML. Mechanisms for the initiation and promotion of carcinogenesis: a review and a new concept. Mayo Clin Proc. (1984) 59(2):107–17. doi: 10.1016/S0025-6196(12)60244-4
26. Trosko JE. Human adult stem cells as the target cells for the initiation of carcinogenesis and for the generation of “cancer stem cells”. Int J Stem Cells. (2008) 1(1):8–26. doi: 10.15283/ijsc.2008.1.1.8
27. Berenblum I. Quantitative carcinogenesis in relation to human exposures. Cancer Detect Prev. (1988) 11(3-6):135–7.3390839
28. Berenblum I, Shubik P. A new, quantitative, approach to the study of the stages of chemical cartinogenesis in the mouse's Skin. Br J Cancer. (1947) 1(4):383–91. doi: 10.1038/bjc.1947.36
29. Gazdzicka J, Golabek K, Strzelczyk JK, Ostrowska Z. Epigenetic modifications in head and neck cancer. Biochem Genet. (2020) 58(2):213–44. doi: 10.1007/s10528-019-09941-1
30. Bajaj J, Diaz E, Reya T. Stem cells in cancer initiation and progression. J Cell Biol. (2020) 219(1):1–12. doi: 10.1083/jcb.201911053
31. Yuspa SH. Overview of carcinogenesis: past, present and future. Carcinogenesis. (2000) 21(3):341–4. doi: 10.1093/carcin/21.3.341
32. Yuspa SH, Poirier MC. Chemical carcinogenesis: from animal models to molecular models in one decade. Adv Cancer Res. (1988) 50:25–70. doi: 10.1016/S0065-230X(08)60434-0
33. Hussain SP, Harris CC. Molecular epidemiology and carcinogenesis: endogenous and exogenous carcinogens. Mutat Res. (2000) 462(2-3):311–22. doi: 10.1016/S1383-5742(00)00015-6
34. Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res. (2001) 477(1-2):7–21. doi: 10.1016/S0027-5107(01)00091-4
36. Sidransky D. Molecular genetics of head and neck cancer. Curr Opin Oncol. (1995) 7(3):229–33. doi: 10.1097/00001622-199505000-00007
37. Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. (1994) 54(18):4855–78.8069852
38. Hennings H, Devor D, Wenk ML, Slaga TJ, Former B, Colburn NH, et al. Comparison of two-stage epidermal carcinogenesis initiated by 7,12-dimethylbenz(a)anthracene or N-methyl-n'-nitro-N-nitrosoguanidine in newborn and adult SENCAR and BALB/c mice. Cancer Res. (1981) 41(3):773–9.6780187
39. Slaga TJ, Fischer SM, Nelson K, Gleason GL. Studies on the mechanism of skin tumor promotion: evidence for several stages in promotion. Proc Natl Acad Sci. (1980) 77(6):3659–63. doi: 10.1073/pnas.77.6.3659
40. Becker FF. Presidential address. Recent concepts of initiation and promotion in carcinogenesis. Am J Pathol. (1981) 105(1):3–9.7294157
41. Scott RE, Maercklein PB. An initiator of carcinogenesis selectively and stably inhibits stem cell differentiation: a concept that initiation of carcinogenesis involves multiple phases. Proc Natl Acad Sci. (1985) 82(9):2995–9. doi: 10.1073/pnas.82.9.2995
42. Boutwell RK, Verma AK, Ashendel CL, Astrup E. Mouse skin: a useful model system for studying the mechanism of chemical carcinogenesis. Carcinog Compr Surv. (1982) 7:1–12.6802487
43. Califano J, Westra WH, Meininger G, Corio R, Koch WM, Sidransky D. Genetic progression and clonal relationship of recurrent premalignant head and neck lesions. Clin Cancer Res. (2000) 6(2):347–52.10690509
44. Ha PK, Chang SS, Glazer CA, Califano JA, Sidransky D. Molecular techniques and genetic alterations in head and neck cancer. Oral Oncol. (2009) 45(4-5):335–9. doi: 10.1016/j.oraloncology.2008.05.015
45. Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci. (2007) 104(3):973–8. doi: 10.1073/pnas.0610117104
46. Clarke MF. Clinical and therapeutic implications of cancer stem cells. Reply. N Engl J Med. (2019) 381(10):e19. doi: 10.1056/NEJMc1908886
47. Krishnamurthy S, Nor JE. Head and neck cancer stem cells. J Dent Res. (2012) 91(4):334–40. doi: 10.1177/0022034511423393
48. Albers AE, Chen C, Koberle B, Qian X, Klussmann JP, Wollenberg B, et al. Stem cells in squamous head and neck cancer. Crit Rev Oncol Hematol. (2012) 81(3):224–40. doi: 10.1016/j.critrevonc.2011.03.004
49. Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell. (2004) 118(5):635–48. doi: 10.1016/j.cell.2004.08.012
50. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. (2001) 414(6859):105–11. doi: 10.1038/35102167
51. Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer. (2016) 114(12):1305–12. doi: 10.1038/bjc.2016.152
52. Antoniou A, Hebrant A, Dom G, Dumont JE, Maenhaut C. Cancer stem cells, a fuzzy evolving concept: a cell population or a cell property? Cell Cycle. (2013) 12(24):3743–8. doi: 10.4161/cc.27305
53. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 Is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. (2007) 1(5):555–67. doi: 10.1016/j.stem.2007.08.014
54. Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. (2010) 140(1):62–73. doi: 10.1016/j.cell.2009.12.007
55. Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS. Stem cells in normal breast development and breast cancer. Cell Prolif. (2003) 36(Suppl 1):59–72. doi: 10.1046/j.1365-2184.36.s.1.6.x
56. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci. (2003) 100(7):3983–8. doi: 10.1073/pnas.0530291100
57. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. (2003) 63(18):5821–8.14522905
58. Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res. (2007) 67(3):1030–7. doi: 10.1158/0008-5472.CAN-06-2030
59. Krishnamurthy S, Warner KA, Dong Z, Imai A, Nor C, Ward BB, et al. Endothelial interleukin-6 defines the tumorigenic potential of primary human cancer stem cells. Stem Cells. (2014) 32(11):2845–57. doi: 10.1002/stem.1793
60. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. (2006) 444(7120):756–60. doi: 10.1038/nature05236
61. Krishnamurthy S, Dong Z, Vodopyanov D, Imai A, Helman JI, Prince ME, et al. Endothelial cell-initiated signaling promotes the survival and self-renewal of cancer stem cells. Cancer Res. (2010) 70(23):9969–78. doi: 10.1158/0008-5472.CAN-10-1712
62. Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. (2006) 66(16):7843–8. doi: 10.1158/0008-5472.CAN-06-1010
63. Hirano T. IL-6 in inflammation, autoimmunity and cancer. Int Immunol. (2021) 33(3):127–48. doi: 10.1093/intimm/dxaa078
64. Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. (1990) 10(5):2327–34. doi: 10.1128/mcb.10.5.2327-2334.1990
65. Akira S, Isshiki H, Nakajima T, Kinoshita S, Nishio Y, Hashimoto S, et al. A nuclear factor for the IL-6 gene (NF-IL6). Chem Immunol. (1992) 51:299–322.1567545
66. Hashemi V, Ahmadi A, Malakotikhah F, Chaleshtari MG, Baghi Moornani M, Masjedi A, et al. Silencing of p68 and STAT3 synergistically diminishes cancer progression. Life Sci. (2020) 249:117499. doi: 10.1016/j.lfs.2020.117499
67. Masjedi A, Hashemi V, Hojjat-Farsangi M, Ghalamfarsa G, Azizi G, Yousefi M, et al. The significant role of interleukin-6 and its signaling pathway in the immunopathogenesis and treatment of breast cancer. Biomed Pharmacother. (2018) 108:1415–24. doi: 10.1016/j.biopha.2018.09.177
68. Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. (2018) 15(4):234–48. doi: 10.1038/nrclinonc.2018.8
69. Qin X, Yan M, Wang X, Xu Q, Wang X, Zhu X, et al. Cancer-associated fibroblast-derived IL-6 promotes head and neck cancer progression via the osteopontin-NF-kappa B signaling pathway. Theranostics. (2018) 8(4):921–40. doi: 10.7150/thno.22182
70. Li M, Jin S, Zhang Z, Ma H, Yang X. Interleukin-6 facilitates tumor progression by inducing ferroptosis resistance in head and neck squamous cell carcinoma. Cancer Lett. (2022) 527:28–40. doi: 10.1016/j.canlet.2021.12.011
71. Pencik J, Wiebringhaus R, Susani M, Culig Z, Kenner L. IL-6/STAT3/ARF: the guardians of senescence, cancer progression and metastasis in prostate cancer. Swiss Med Wkly. (2015) 145:w14215. doi: 10.4414/smw.2015.14215
72. Weng YS, Tseng HY, Chen YA, Shen PC, Al Haq AT, Chen LM, et al. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Molec Cancer. (2019) 18(1):42. doi: 10.1186/s12943-019-0988-0
73. Wu J, Gao FX, Wang C, Qin M, Han F, Xu T, et al. IL-6 and IL-8 secreted by tumour cells impair the function of NK cells via the STAT3 pathway in oesophageal squamous cell carcinoma. J Exp Clin Cancer Res. (2019) 38(1):321. doi: 10.1186/s13046-019-1310-0
74. Saini U, Naidu S, ElNaggar AC, Bid HK, Wallbillich JJ, Bixel K, et al. Elevated STAT3 expression in ovarian cancer ascites promotes invasion and metastasis: a potential therapeutic target. Oncogene. (2017) 36(2):168–81. doi: 10.1038/onc.2016.197
75. Ye X, Wang X, Lu R, Zhang J, Chen X, Zhou G. CD47 As a potential prognostic marker for oral leukoplakia and oral squamous cell carcinoma. Oncol Lett. (2018) 15(6):9075–80. doi: 10.3892/ol.2018.8520
76. Ravindran G, Devaraj H. Aberrant expression of CD133 and musashi-1 in preneoplastic and neoplastic human oral squamous epithelium and their correlation with clinicopathological factors. Head Neck. (2012) 34(8):1129–35. doi: 10.1002/hed.21896
77. Das PK, Islam F, Lam AK. The roles of cancer stem cells and therapy resistance in colorectal carcinoma. Cells. (2020) 9(6):1–21. doi: 10.3390/cells9061392
78. Dawood S, Austin L, Cristofanilli M. Cancer stem cells: implications for cancer therapy. Oncology (2014) 28(12):1101–7. 10.25510809
79. Chen ZG. The cancer stem cell concept in progression of head and neck cancer. J Oncol. (2009) 2009:894064. doi: 10.1155/2009/894064
80. Surendran S, Siddappa G, Mohan A, Hicks W Jr., Jayaprakash V, Mimikos C, et al. Cancer stem cell and its niche in malignant progression of oral potentially malignant disorders. Oral Oncol. (2017) 75:140–7. doi: 10.1016/j.oraloncology.2017.11.003
81. Ghuwalewala S, Ghatak D, Das P, Dey S, Sarkar S, Alam N, et al. CD44(high)CD24(low) Molecular signature determines the cancer stem cell and EMT phenotype in oral squamous cell carcinoma. Stem Cell Res. (2016) 16(2):405–17. doi: 10.1016/j.scr.2016.02.028
82. Keysar SB, Jimeno A. More than markers: biological significance of cancer stem cell-defining molecules. Molec Cancer Therapeu. (2010) 9(9):2450–7. doi: 10.1158/1535-7163.MCT-10-0530
83. Kumar S, Bar-Lev L, Sharife H, Grunewald M, Mogilevsky M, Licht T, et al. Identification of vascular cues contributing to cancer cell stemness and function. Angiogenesis. (2022) 25(3):355–71. doi: 10.1007/s10456-022-09830-z
84. Muller L, Tunger A, Plesca I, Wehner R, Temme A, Westphal D, et al. Bidirectional crosstalk between cancer stem cells and immune cell subsets. Front Immunol. (2020) 11:140. doi: 10.3389/fimmu.2020.00140
85. D'Alessio A, Proietti G, Lama G, Biamonte F, Lauriola L, Moscato U, et al. Analysis of angiogenesis related factors in glioblastoma, peritumoral tissue and their derived cancer stem cells. Oncotarget. (2016) 7(48):78541–56. doi: 10.18632/oncotarget.12398
86. Ribatti D, Nico B, Crivellato E, Roccaro AM, Vacca A. The history of the angiogenic switch concept. Leukemia. (2007) 21(1):44–52. doi: 10.1038/sj.leu.2404402
87. Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. (1989) 339(6219):58–61. doi: 10.1038/339058a0
88. Gimbrone MA Jr., Gullino PM. Angiogenic capacity of preneoplastic lesions of the murine mammary gland as a marker of neoplastic transformation. Cancer Res. (1976) 36(7 PT 2):2611–20.1277168
89. Gimbrone MA Jr., Gullino PM. Neovascularization induced by intraocular xenografts of normal, preneoplastic, and neoplastic mouse mammary tissues. J Natl Cancer Inst. (1976) 56(2):305–18. doi: 10.1093/jnci/56.2.305
90. Markowska A, Sajdak S, Markowska J, Huczynski A. Angiogenesis and cancer stem cells: new perspectives on therapy of ovarian cancer. Eur J Med Chem. (2017) 142:87–94. doi: 10.1016/j.ejmech.2017.06.030
91. Ribatti D. Cancer stem cells and tumor angiogenesis. Cancer Lett. (2012) 321(1):13–7. doi: 10.1016/j.canlet.2012.02.024
92. Kim HS, Chen YC, Nor F, Warner KA, Andrews A, Wagner VP, et al. Endothelial-derived interleukin-6 induces cancer stem cell motility by generating a chemotactic gradient towards blood vessels. Oncotarget. (2017) 8(59):100339–52. doi: 10.18632/oncotarget.22225
93. Jiang X, Wang J, Deng X, Xiong F, Zhang S, Gong Z, et al. The role of microenvironment in tumor angiogenesis. J Exp Clin Cancer Res. (2020) 39(1):204. doi: 10.1186/s13046-020-01709-5
94. Hobson B, Denekamp J. Endothelial proliferation in tumours and normal tissues: continuous labelling studies. Br J Cancer. (1984) 49(4):405–13. doi: 10.1038/bjc.1984.66
95. Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. J Clin Invest. (2010) 120(1):41–50. doi: 10.1172/JCI41004
96. Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. (2007) 11(1):69–82. doi: 10.1016/j.ccr.2006.11.020
97. Ludwig N, Szczepanski MJ, Gluszko A, Szafarowski T, Azambuja JH, Dolg L, et al. CD44(+) Tumor cells promote early angiogenesis in head and neck squamous cell carcinoma. Cancer Lett. (2019) 467:85–95. doi: 10.1016/j.canlet.2019.10.010
98. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23(11):549–55. doi: 10.1016/S1471-4906(02)02302-5
99. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. (2010) 11(10):889–96. doi: 10.1038/ni.1937
100. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8(12):958–69. doi: 10.1038/nri2448
101. Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. (2007) 67(11):5064–6. doi: 10.1158/0008-5472.CAN-07-0912
102. Huang R, Wang S, Wang N, Zheng Y, Zhou J, Yang B, et al. CCL5 Derived from tumor-associated macrophages promotes prostate cancer stem cells and metastasis via activating beta-catenin/STAT3 signaling. Cell Death Dis. (2020) 11(4):234. doi: 10.1038/s41419-020-2435-y
103. Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. (2013) 12:86. doi: 10.1186/1476-4598-12-86
104. Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. (2008) 205(6):1261–8. doi: 10.1084/jem.20080108
105. Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C, et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal. (2014) 26(2):192–7. doi: 10.1016/j.cellsig.2013.11.004
106. Radharani NNV, Yadav AS, Nimma R, Kumar TVS, Bulbule A, Chanukuppa V, et al. Tumor-associated macrophage derived IL-6 enriches cancer stem cell population and promotes breast tumor progression via stat-3 pathway. Cancer Cell Int. (2022) 22(1):122. doi: 10.1186/s12935-022-02527-9
107. Noben-Trauth N, Shultz LD, Brombacher F, Urban JF Jr., Gu H, Paul WE. An interleukin 4 (IL-4)-independent pathway for CD4+ T cell IL-4 production is revealed in IL-4 receptor-deficient mice. Proc Natl Acad Sci. (1997) 94(20):10838–43. doi: 10.1073/pnas.94.20.10838
108. Fujioka S, Niu J, Schmidt C, Sclabas GM, Peng B, Uwagawa T, et al. NF-kappaB and AP-1 connection: mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol Cell Biol. (2004) 24(17):7806–19. doi: 10.1128/MCB.24.17.7806-7819.2004
109. Tugal D, Liao X, Jain MK. Transcriptional control of macrophage polarization. Arterioscler Thromb Vasc Biol. (2013) 33(6):1135–44. doi: 10.1161/ATVBAHA.113.301453
110. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. IRF5 Promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. (2011) 12(3):231–8. doi: 10.1038/ni.1990
111. Hagemann T, Biswas SK, Lawrence T, Sica A, Lewis CE. Regulation of macrophage function in tumors: the multifaceted role of NF-kappaB. Blood. (2009) 113(14):3139–46. doi: 10.1182/blood-2008-12-172825
112. Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, et al. STAT6 Transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity. (2010) 33(5):699–712. doi: 10.1016/j.immuni.2010.11.009
113. Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, et al. Kruppel-like factor 4 regulates macrophage polarization. J Clin Invest. (2011) 121(7):2736–49. doi: 10.1172/JCI45444
114. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. (2010) 11(10):936–44. doi: 10.1038/ni.1920
115. Zaynagetdinov R, Sherrill TP, Polosukhin VV, Han W, Ausborn JA, McLoed AG, et al. A critical role for macrophages in promotion of urethane-induced lung carcinogenesis. J Immunol. (2011) 187(11):5703–11. doi: 10.4049/jimmunol.1100558
116. Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. (2005) 65(8):3437–46. doi: 10.1158/0008-5472.CAN-04-4262
117. Ojalvo LS, King W, Cox D, Pollard JW. High-density gene expression analysis of tumor-associated macrophages from mouse mammary tumors. Am J Pathol. (2009) 174(3):1048–64. doi: 10.2353/ajpath.2009.080676
118. Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. (2010) 70(14):5728–39. doi: 10.1158/0008-5472.CAN-09-4672
119. Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin's Lymphoma. N Eng J Med. (2010) 362(10):875–85. doi: 10.1056/NEJMoa0905680
120. Raggi C, Mousa HS, Correnti M, Sica A, Invernizzi P. Cancer stem cells and tumor-associated macrophages: a roadmap for multitargeting strategies. Oncogene. (2016) 35(6):671–82. doi: 10.1038/onc.2015.132
121. Hamerlik P, Lathia JD, Rasmussen R, Wu Q, Bartkova J, Lee M, et al. Autocrine VEGF-VEGFR2-neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med. (2012) 209(3):507–20. doi: 10.1084/jem.20111424
122. Okamoto R, Ueno M, Yamada Y, Takahashi N, Sano H, Suda T, et al. Hematopoietic cells regulate the angiogenic switch during tumorigenesis. Blood. (2005) 105(7):2757–63. doi: 10.1182/blood-2004-08-3317
123. Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L, et al. Tumor self-seeding by circulating cancer cells. Cell. (2009) 139(7):1315–26. doi: 10.1016/j.cell.2009.11.025
124. Schwab ED, Pienta KJ. Cancer as a complex adaptive system. Med Hypotheses. (1996) 47(3):235–41. doi: 10.1016/S0306-9877(96)90086-9
125. Massague J, Ganesh K. Metastasis-Initiating cells and ecosystems. Cancer Discov. (2021) 11(4):971–94. doi: 10.1158/2159-8290.CD-21-0010
126. Martinez-Outschoorn UE, Bartrons M, Bartrons R. Editorial: cancer ecosystems. Front Oncol. (2019) 9(718):1–4. doi: 10.3389/fonc.2019.00718
Keywords: angiogenesis, cancer stem cells, carcinogenesis, endothelial cells, epithelial dysplasia, macrophages, premalignant, tumor progression
Citation: Polverini PJ, Nör F and Nör JE (2023) Crosstalk between cancer stem cells and the tumor microenvironment drives progression of premalignant oral epithelium. Front. Oral. Health 3:1095842. doi: 10.3389/froh.2022.1095842
Received: 11 November 2022; Accepted: 19 December 2022;
Published: 10 January 2023.
Edited by:
Fernanda Visioli, Federal University of Rio Grande do Sul, BrazilReviewed by:
Samapika Routray, All India Institute of Medical Sciences Bhubaneswar, India© 2023 Polverini, Nör and Nör. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter J. Polverini bmVvdmFzQHVtaWNoLmVkdQ==
Specialty Section: This article was submitted to Oral Cancers, a section of the journal Frontiers in Oral Health
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.