 Linah A. Shahoumi
Linah A. Shahoumi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oral. Health , 21 July 2021
Sec. Oral Cancers
Volume 2 - 2021 | https://doi.org/10.3389/froh.2021.685236
This article is part of the Research Topic Therapeutic Challenges in Oral Cancer View all 6 articles
Head and neck squamous cell carcinoma (HNSCC) is currently one of the 10 most common malignancies worldwide, characterized by a biologically highly diverse group of tumors with non-specific biomarkers and poor prognosis. The incidence rate of HNSCC varies widely throughout the world, with an evident prevalence in developing countries such as those in Southeast Asia and Southern Africa. Tumor relapse and metastasis following traditional treatment remain major clinical problems in oral cancer management. Current evidence suggests that therapeutic resistance and metastasis of cancer are mainly driven by a unique subpopulation of tumor cells, termed cancer stem cells (CSCs), or cancer-initiating cells (CICs), which are characterized by their capacity for self-renewal, maintenance of stemness and increased tumorigenicity. Thus, more understanding of the molecular mechanisms of CSCs and their behavior may help in developing effective therapeutic interventions that inhibit tumor growth and progression. This review provides an overview of the main signaling cascades in CSCs that drive tumor repropagation and metastasis in oral cancer, with a focus on squamous cell carcinoma. Other oral non-SCC tumors, including melanoma and malignant salivary gland tumors, will also be considered. In addition, this review discusses some of the CSC-targeted therapeutic strategies that have been employed to combat disease progression, and the challenges of targeting CSCs, with the aim of improving the clinical outcomes for patients with oral malignancies. Targeting of CSCs in head and neck cancer (HNC) represents a promising approach to improve disease outcome. Some CSC-targeted therapies have already been proven to be successful in pre-clinical studies and they are now being tested in clinical trials, mainly in combination with conventional treatment regimens. However, some studies revealed that CSCs may not be the only players that control disease relapse and progression of HNC. Further, clinical research studying a combination of therapies targeted against head and neck CSCs may provide significant advances.
Head and neck cancer is a heterogenous group of tumors which mainly arise in the oral cavity, oropharynx, hypopharynx, salivary glands, paranasal sinuses and larynx [1]. HNC is among the most common cancers worldwide, with a high prevalence in Southeast Asia, Brazil, and Southern Africa [2]. Squamous cell carcinomas make up the majority of HNC which have an incidence of around 630,000 new cases per year worldwide, with almost 10,000 deaths in the United States annually [3, 4]. Lifestyle behaviors such as drinking alcohol, use of tobacco, and chewing betel quid are the most common risk factors associated with HNSCC [5]. Despite recent considerable advances in the therapeutic repertoire for oral cancer, the median overall survival for patients with metastatic or recurrent HNC remains <1 year [6]. Resistance to chemotherapeutic and biologic agents is responsible for the failure of many current therapeutic approaches. Accumulating evidence suggests that the molecular complexity, intratumoral heterogeneity, and presence of CSCs are responsible for local recurrence, metastatic spread, and treatment resistance in various types of cancer [7]. Intratumoral heterogeneity may be the consequence of genetic alterations, epigenetic modification, and changes in cell properties and behavior [8]. CSCs are a pluripotent subpopulation of cells in the tumor that have attributes of self-renewal, tumor initiation, differentiation, migration, and metastasis [9–11]. This subpopulation has been identified in several solid tumors, including HNC, and it shows certain characteristics that give it the ability to re-create entire heterogeneous populations of a tumor posttreatment, causing tumor recurrence and metastasis [12]. This review will discuss the characterization and molecular regulation of CSCs that drive tumor progression and metastasis in oral cancer, with a focus on HNSCC, highlighting the role of CSCs in treatment failure. Furthermore, this review will outline CSC-targeted strategies, as well as challenges of targeting CSCs, with the aim of making a potential tangible difference in clinical outcome for HNC patients.
CSCs interact with transformed cells and other stromal cells within the tumor microenvironment through adhesion molecules and paracrine factors. These microenvironmental interactions promote the differentiation of CSCs, enhance angiogenesis, recruit immune and stromal cells, and promote tumor invasion and metastasis [13] (Figure 1). CSCs were first identified in leukemia [14], and subsequently in various types of cancer, including tumors in brain [15], lung [16], colon [17], breast [18], and HNSCC [19]. CSCs are believed to have multiple unique features including the potential for differentiation and self-renewal that make them crucial for tumorigenesis [20], and potentially offer a novel area of study for developing more effective treatments for HNSCC. It was previously reported that CSCs in HNSCC play a vital role in initiation, invasion, and progression as well as resistance to chemo/radiotherapy, and they are responsible for recurrence and metastasis [21]. The literature reveals that several different markers have been used to identify CSCs in HNSCC. These markers are not only employed for selective CSC isolation but are also involved in regulating multiple biological functions of CSCs, including cell proliferation, invasion, self-renewal and survival, effectively promoting tumor progression, and recurrence.
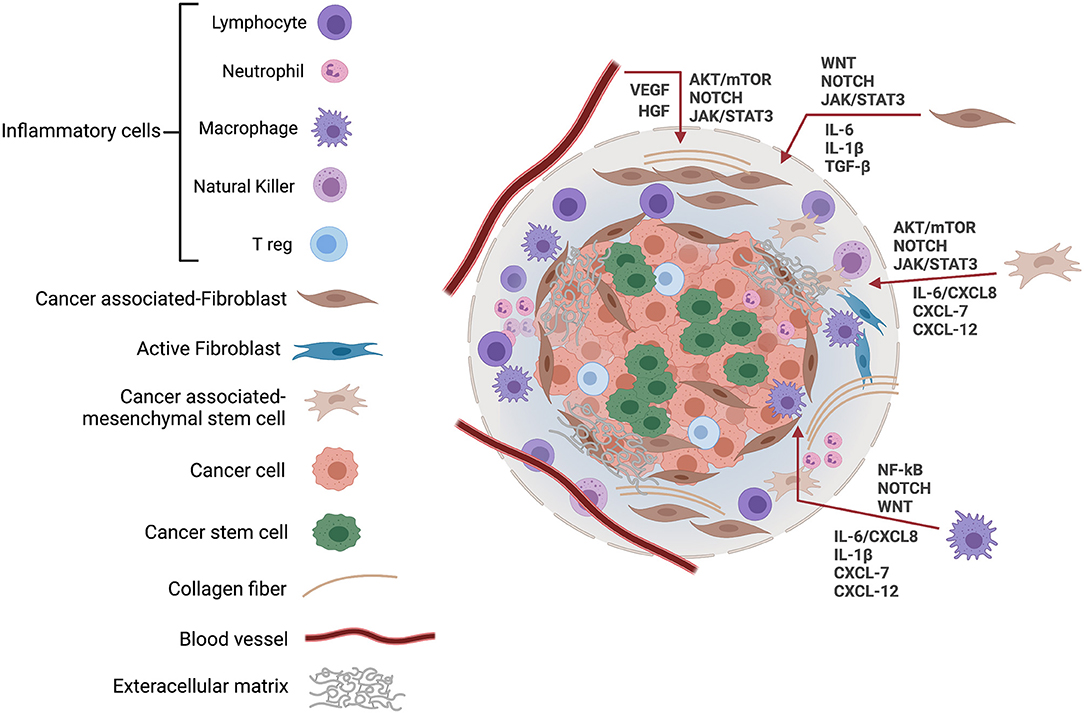
Figure 1. The cancer stem cell microenvironment. The niche or tumor microenvironment is essential for maintenance of stemness, and this also applies to CSCs where cell–cell interactions within the niche are required to support the role of CSCs in cancer initiation and progression. The CSC microenvironment also contributes to CSCs' resistance to drugs and other cancer therapies, thereby, promoting cancer recurrence. The tumor tissue microenvironment is composed of a variety of cells, including tumor cells, cancer stem cells, inflammatory cells, and cancer-associated fibroblasts, along with blood vessels and extracellular matrix. In response to hypoxic stress and matrix, CSCs induce growth factors and cytokines including IL-6, CXCL8, and VEGF to regulate their growth via EGFR, NOTCH, WNT, and other signaling cascades. JAK, Janus kinases; STAT, signal transducer, and activator of transcription; VEGF, vascular endothelial growth factor; HGF, hepatocyte growth factor; mTOR, mammalian target of rapamycin; IL, Interleukin; CXCL, CXC-motif chemokine ligand.
CSCs in HNSCC were initially identified and isolated by their high levels of expression of the hyaluronan receptor CD44 [19, 22]. CD44 is a type I transmembrane glycoprotein involved in intercellular interactions, cell migration, adhesion, and angiogenesis [23–25]. The extracellular domain of CD44 can interact with hyaluronic acid and other ligands, including cytokines, and matrix metalloproteinases [26, 27]. CD44 plays a role in cell proliferation, survival and tumorigenesis through activation of multiple tyrosine kinase-induced pathways including epidermal growth factor receptor (EGFR), Src/focal adhesion kinase (FAK), and hepatocyte growth factor receptor (MET) [28–30]. Importantly, clinical reports indicated that CD44 expression is associated with local recurrence, regional lymph node metastasis, perineural invasion, and poorer survival rate in oral squamous cell carcinoma (OSCC) [31–34], indicating its vital role in tumor recurrence and metastasis of HNSCC. In addition, Biddle et al. [35] examined the therapeutic resistance of heterogeneous CSC subpopulations that express high levels of CD44 and found that these subpopulations exhibit high plasticity and increased therapeutic resistance. CD10, a type II integral membrane protein also known as neutral endopeptidase 24.11, is another potential marker for HNSCC stem cells. Previous studies reported a role for CD10 in the differentiation and growth of neoplastic cells and that its expression was associated with tumor size, histological grade of malignancy, local recurrence, and therapeutic resistance in HNSCC [36, 37]. Moreover, HNSCC patients with high CD10 expression had significantly poorer overall survival (OS) [38]. Another well-known cell surface marker for isolation of human malignant tissue stem cells is CD133. CD133 is a transmembrane glycoprotein which has been used as a marker to identify CSCs derived from primary solid tumors [16, 39, 40]. Previously, CD133+ cell populations were found to have higher cell viability, migratory and invasive capability, colony forming ability as well as drug resistance compared with CD133− populations [41–43]. Moreover, CD133 expression has been used as a prognostic marker of survival in HNSCC and is negatively correlated with clinical outcome in these patients [44]. However, other studies reported no CD133 expression in freshly prepared HNSCC patient samples and no correlation was detected between CD133 expression and differentiation of carcinoma cells, or prognosis [45–47]. Therefore, the value of CD133 as a marker for HNSCC CSCs remains unclear and may need to be re-evaluated.
HNSCC CSCs also have elevated activity of aldehyde dehydrogenase (ALDH), an intracellular enzyme that metabolizes reactive aldehydes producedfrom alcohol and chemotherapeutic compounds into non-reactive acids [48]. Mutations and altered expression of various ALDH genes are implicated in multiple cancers including HNSCC [49–51]. ALDH1 was found to colocalize with other CSC-related markers, including Snail and MMP-9, and induce epithelial to mesenchymal transition (EMT)-related genes [49, 52]. ALDH1+ cells from HNSCC patients display a more tumorigenic phenotype, self-renewal and stemness properties, and also resistance to radiotherapy and chemotherapy [49]. Furthermore, High levels of ALDH1 in patient samples have been correlated with poor prognosis in HNSCC [53, 54]. In contrast, another clinical report investigated CSC populations in 74 locally advanced HNSCCs from an equal number of patients, treated with accelerated platinum-based radiotherapy, and revealed that high expression of ALDH1 and lack of CD44 expression in CSCs was associated with better radiotherapy outcome and favorable prognosis [55]. These contradictory results suggested that, in some tumors, terminally differentiated cancer cells may retain expression of stem cell markers. Although, tremendous efforts have been made to understand CSC-associated molecules and to find an optimal marker for CSCs in HNSCC, challenges still remain because of the heterogeneity of CSCs and the absence of one marker does not necessary mean that cells do not possess characteristics of stemness.
HNSCC CSCs express same proteins involved in the core network that regulates embryonic stem cells (ESCs). In particular, Octamer-binding transcription factor 4 (OCT4; POU5F1), Sex determining region Y-box 2 (SOX2) and Nanog Homeobox (NANOG) are highly enriched in both CSCs and ESCs [56, 57]. These transcription factors are considered master transcriptional regulators that orchestrate stemness properties including self-renewal, angiogenesis, migration, and resistance to apoptosis [58, 59]. OCT4 has three domains, including the POU domain (DNA binding domain) which is essential in early embryogenesis and for maintenance of pluripotent cells during embryonic development [60]. OCT4 has been reported to be overexpressed in various malignancies, including HNSCC, lung, breast, liver, and ovarian cancer [58, 61–64]. It has been suggested that OCT4 plays a crucial role in regulating EMT by increasing the expression of the mesenchymal marker N-cadherin and the transcription factor SNAI2 (Slug) to promote tumor progression and metastasis [10]. Moreover, OCT4 overexpression was observed in tumor cells metastatic to lymph nodes and also in recurrent tumors from oral squamous cell carcinoma (OSCC) patients, indicating that OCT4 may be a potential marker of recurrence and metastasis in HNSCC [10]. Furthermore, accumulating evidence indicates that OCT4 is correlated with poor survival and it has been suggested as an independent prognostic marker of HNSCC progression [58, 65]. SOX2 is a high-mobility group (HMG) domain-containing transcription factor pivotal for pluripotent cell development [66]. Indeed, SOX2 is known to complex with OCT4 which binds to important regulatory elements of POU5F1, the gene encoding OCT4, to maintain the expression of essential transcription factors in ESCs through autoregulatory and multicomponent loop network motifs [67]. In addition, SOX2 is involved in many processes important in oncogenesis including cell proliferation, stemness, migration, invasion, tumorigenesis, and chemoresistance [68]. A genomic copy number gain at the SOX2 locus results in an increase in SOX2 transcriptional activity, and this is reported to be critical for HNSCC initiation and progression [69]. Moreover, SOX2 collaborates with other oncoproteins such as signal transducer and activator of transcription 3 (STAT3) to initiate and induce SCC [70]. Overexpression of SOX2 promotes cell proliferation via cyclin B1 upregulation and enhances stem cell properties [71]. Importantly, SOX2 expression levels correlate positively with radio-chemoresistance and poor prognosis in HNSCC patients [68, 71]. On the other hand, it has been suggested that both OCT4 and SOX2 expression are associated with early tumor stage and better disease-free survival, raising the possibility that both SOX2 and OCT4 might not be the best targets to eradicate tumor relapse and progression [57]. Of interest, the reduction of SOX2 and OCT4 expression in advanced-stage HNSCC, as reported in a study by Fu et al. [57], suggests that overexpression of SOX2 seen in early lesions might decrease gradually during tumor progression. However, whether the molecular mechanisms regulating SOX2 and OCT4 are associated with a favorable prognosis in HNSCC is still unknown and was not investigated in this study. A key downstream target of OCT4 and SOX2 is NANOG, which is a variant homeobox transcription factor widely recognized as a marker for stemness [72, 73]. Functionally, NANOG is involved in blocking differentiation to maintain ESC pluripotency and has been shown to be overexpressed in various cancers including HNSCC [58, 74]. In HNSCC, NANOG can promote cell proliferation, invasion, and colony formation of the CSC population via its phosphorylation at T200 and T280 by protein kinase C epsilon (PKCε) [75]. Recently, evidence has been provided to suggest that high expression of NANOG promotes EMT, the acquisition of CSC properties, and enhances radiotherapy resistance in HNSCC [76]. Moreover, elevated NANOG expression was reported as a prognostic biomarker for OS in HNSCC and shown to correlate with poor differentiation and chemoresistance [77]. These findings indicate that NANOG may be a novel target for elimination of CSCs in HNSCC. B lymphoma Mo-MLV insertion region 1 homolog (BMI1) is a core component of the polycomb repressive complex 1 (PRC1) that mediates gene silencing via monoubiquitination of histone H2A. BMI1 is strongly linked to self-renewal and has been implicated in maintaining the stem cell pool of several tissues [78]. The N- terminal ring-finger domain of BMI1 is required for the activation of telomerase reverse transcriptase (TERT) transcription and immortalization of epithelial cells [79]. BMI1 is among the most studied CSC markers in HNSCC [80]. High expression of BMI1 in cancer was related to EMT and involved in the invasion and progression of tongue squamous cell carcinoma [81]. Several studies have reported that BMI1 is abnormally expressed in HNSCC and correlated with advanced tumor stages, drug resistance, and poor prognosis [19, 34, 82]. However, other studies could not predict survival from BMI1 expression [83, 84]. The reason of these contradictory results could be the limited scope to consider heterogeneity in CSCs as different types or states of CSCs promoting disease progression and others slowing it down.
Multiple pathways are involved in the regulation of normal stem cell differentiation and self-renewal. The NOTCH and Sonic hedgehog (SHH) pathways have been implicated in the regulation of differentiation and patterning of numerous organ systems [85]. SHH, a secreted glycoprotein, activates signaling in target cells by binding to the transmembrane receptor Patched 1 (PTCH1) which unleashes Smoothened (SMO) to trigger a series of intracellular pathways that induce the translocation of the transcription factor glioma-associated oncogene homolog 1 (GLI1) into the nucleus [86] where it induces the transcription of proliferation-associated genes including GLI1, PTCH1, WNT1, forkhead box protein M1 (FOXM1), and CCND1 [86–88]. Moreover, SHH-induced signals are positively correlated with the expression of Snail and MMP9 and negatively with E-cadherin, suggesting that SHH signaling may be an important contributor to invasion and metastasis of HNSCC [89], by driving EMT. In tumor cells, SHH signaling cascades are aberrantly activated by genetic alterations, such as loss-of-function alterations in PTCH1, gain-of-function mutations in SMO (T241M, L412F, S533N, W535L, and R562Q), and amplification of the GLI1 or GLI2 genes [90, 91]. It has been shown that SHH signaling is activated in various CSCs, including breast cancer, liver cancer, gastric cancer, and HNSCC [92–95]. Interestingly, elevated levels of GLI1 are correlated with recurrence, lymph node metastasis and the worst prognosis in HNSCC patients [89, 95]. Detection of SHH pathway components, especially GLI1 and SHH, in HNSCC might represent promising targets for future anticancer therapeutic development in HNSCC. Another important player that is linked to CSCs in HNSCC is NOTCH1. NOTCH proteins are a family of heterodimeric transmembrane receptors composed of an extracellular domain, a transmembrane domain, and an intracellular domain. The intracellular domain translocates into the nucleus, where it modulates transcription via CBF1, Suppressor of Hairless, Lag-1 (CSL) transcription factor family [96, 97]. This complex activates the transcription of target genes including HES, HERP, and HEY which are involved in cellular differentiation [97]. Other NOTCH targets include the key cell cycle regulators cyclin D1, cyclin A, p21, and p27 [97]. Abnormal expression of NOTCH receptors has been observed in different types of malignant lesions. NOTCH signaling plays an essential role in a variety of stem cell processes in HNSCC, such as cell proliferation, differentiation, survival, and self-renewal [98]. In terms of anti-cancer therapeutics, NOTCH1 Inhibition by γ-secretase inhibitors reduces tumor growth and blocks CSC function in multiple tumors including breast cancer, brain tumor, and glioma [99]. Conversely, high levels of NOTCH1 correlate with increased resistance to cisplatin in HNSCC patients [100], while high expression of both NOTCH1 and JAG1 (a NOTCH 1 ligand) is associated with poor prognosis in HNSCC [101]. In addition, inhibition of NOTCH1 delays tumorigenesis, reduces CSC self-renewal and maintenance, and improves the efficacy of cisplatin and 5-fluorouracil by targeting CSCs in HNSCC [102]. In contrast, Grilli et al. [103] have revealed that NOTCH1 expression is positively correlated with non-recurrent disease, prolonged OS rates, and better prognosis in HNSCC patients. However, these authors did not determine or further investigate the mechanisms by which NOTCH signaling functions in an anti-tumorigenic manner.
The epidermal growth factor receptor pathway is one of the signaling cascades that control CSC differentiation and maintenance in HNSCC. EGFR is a transmembrane receptor tyrosine kinase that is activated by various ligands, including epidermal growth factor (EGF) and transforming growth factor-α (TGF-α), triggering activation of downstream signaling cascades such as PI3K/AKT, MEK-ERK, and phospholipase C signaling to control cell growth, survival, differentiation, angiogenesis, and invasion [104]. Overexpression of EGFR is associated with resistance to treatment and poor clinical outcomes with HNSCC patients [105]. Accumulating evidence indicates that EGFR plays a vital role in development of HNSCC stemness. It has been shown that CD44 interacts with EGFR to promote cell proliferation, migration and induces cisplatin resistance and apoptosis inhibition in HNSCC cells [106], however, these findings should be confirmed in tissues derived from HNSCC patients. Moreover, EGFR drives HNSCC metastasis by inducing glycolysis/EMT/CSC properties through a PI3K-dependent mechanism [107]. A recent study reported that EGFR activation induces SOX2 phosphorylation at Y277, inhibiting ubiquitination, and subsequent autophagic degradation of SOX2 in a human tongue SCC cell line [108]. In addition, EGFR overexpression is positively correlated with a higher functional proportion of ALDHhigh CSCs in a human papillomavirus-16 (HPV-16)-positive cell line [109], however, the mechanism responsible for this is still unknown. WNT signaling is also critical for stem cell self-renewal and differentiation. Based on the involvement of β-catenin, WNT signaling can be divided into two pathways, namely canonical and non-canonical. WNT/β-catenin signaling pathway is activated by the binding of WNT protein to Frizzled (Fz) seven transmembrane receptor and the coreceptor lipoprotein receptor-related protein 5 or 6 (LRP 5/6) to form a functional complex [110]. Consequently, the β-catenin is uncoupled from the degradation complex and translocated into the nucleus to promote transcription of downstream targets such as cyclin D1 (CCND1), cyclooxygenase 2 (COX2), bone morphogenetic protein 4 (BMP4), matrix metalloproteinases 7 (MMP7), and C-MYC [111] (Figure 2). It has been established that WNT/β-catenin signaling plays a crucial role in maintaining the CSC phenotype in various types of cancer. A possible mechanistic explanation is that β-catenin interacts with CD44, and β-catenin inhibition by WNT decreases the expression of CD44 and OCT4 in HNSCC cells [112]. It is also reported that WNT pathway activation enhances the CSC proliferation rate and promotes stemness and sphere formation in HNSCC cell cultures, through upregulating the expression of SOX2 [113]. In addition, β-catenin plays a fundamental role in mediating cisplatin resistance by regulating DNA damage repair in HNSCC [114] (Table 1). These findings may form a basis for future studies aimed at developing novel strategies to combat drug resistance and disease recurrence in HNSCC.
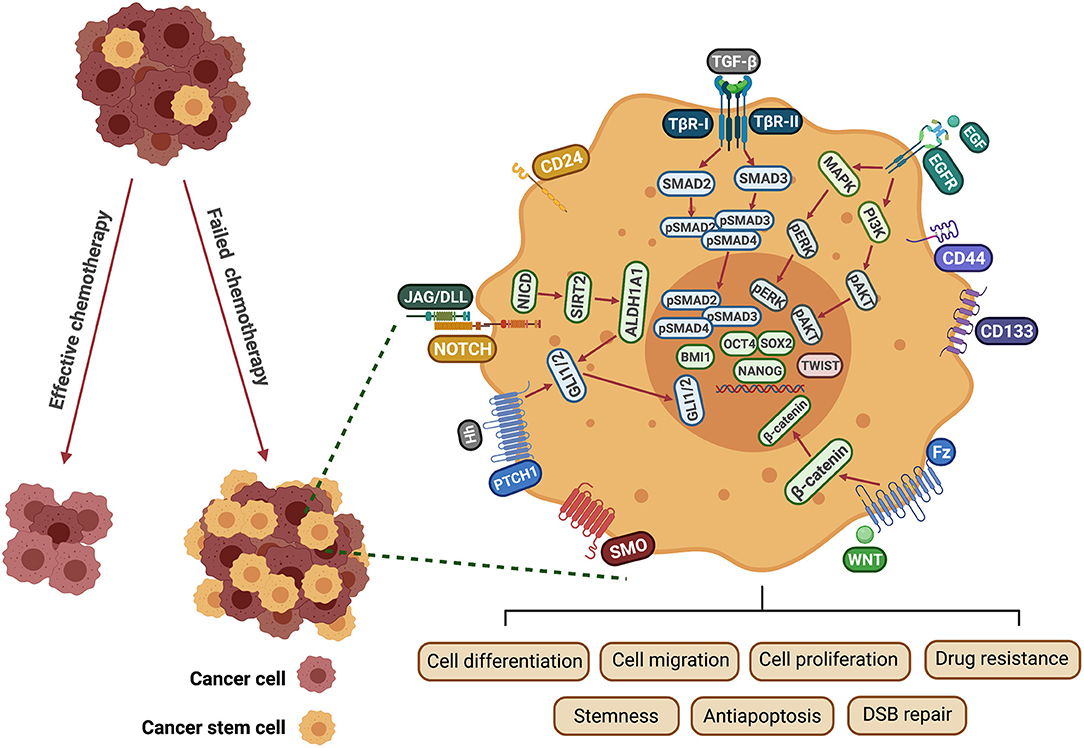
Figure 2. Mechanisms involved in CSC-induced drug resistance in HNSCC. EGF binds to the EGFR receptor tyrosine kinase resulting in activation and autophosphorylation of the receptor. This activates RAS and PI3K, triggering major signaling cascades that include MEK/ERK and PI3K/AKT. NOTCH-activated NICD1, upregulates the SIRT2/ALDH1A pathway. Hedgehog ligands (Hh) bind to Patched receptors and derepress the G-protein-coupled receptor (GPCR) SMO. Hh targets ALDH+ CSCs through GLI1 expression, which is regulated by ALDH1A1. WNT proteins bind to the Fz transmembrane receptor and the LRP 5/6 to form a functional complex. β-catenin then becomes uncoupled from the degradation complex and translocates to the nucleus to promote transcription of downstream targets. TGF-β is recognized by TβR1/2 resulting in the phosphorylation of SMAD 2/3 and formation of a SMAD 2/3/4 complex. These pathways are involved in regulating multiple biological functions of CSCs, including cell proliferation, migration, invasion, self-renewal and survival. EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; Fz, frizzled; JAG, Jagged ligand; DLL, Delta-like ligand; DSB, double-strand break; NICD, intracellular domain of NOTCH protein; SMO, smoothened; MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinases; PATCH1, Protein patched homolog 1; TGF-β, transforming growth factor; TβR, transforming growth factor β receptor; ALDH1, Aldehyde dehydrogenase 1 family; BMI1, B lymphoma Mo-MLV insertion region 1 homolog; OCT4, Octamer-binding transcription factor 4; SOX2, Sex determining region Y-box 2; NANOG, Nanog homeobox.
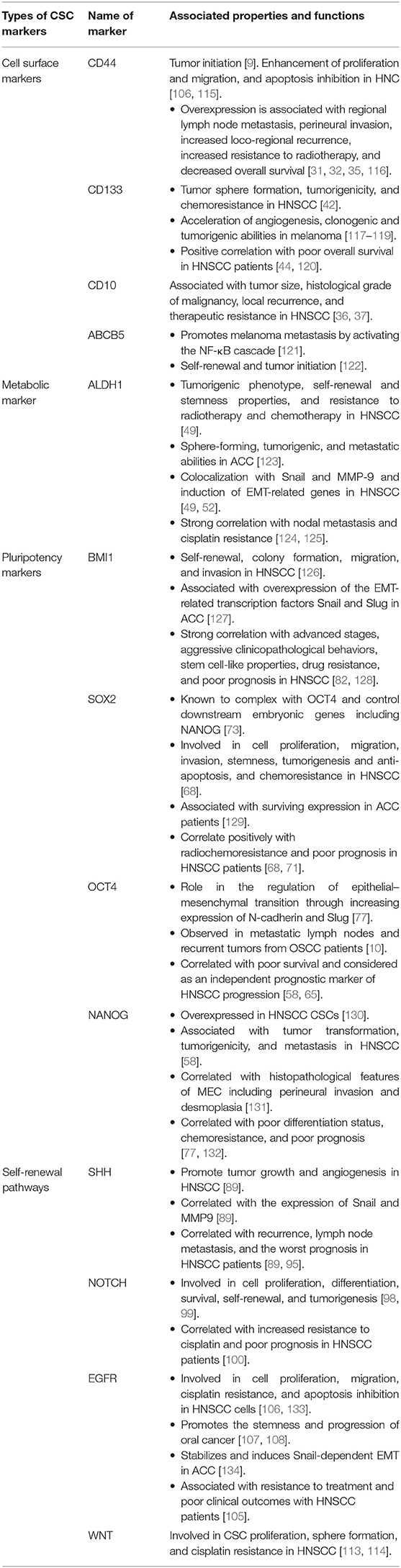
Table 1. CSC markers in HNCs. Different markers for the identification and characterization of CSCs in HNCs, including HNSCC, ACC, MEC, and oral melanoma are listed.
Salivary gland cancers are rare, accounting for only 2–6% of head and neck cancers. In spite of this, however, they constitute a significant public health issue [135]. The most two common salivary gland malignancies are mucoepidermoid carcinoma (MEC) and adenoid cystic carcinoma (ACC). MEC occurs in both the major and minor salivary glands and represents ~30% of malignant salivary gland tumors [136]. The existence of CSCs has been identified functionally in salivary gland MEC, and ALDH+ CD44+ MEC cells exhibit self-renewal and multipotency, and are highly tumorigenic [137]. In addition to these markers, BMI1, OCT4, and NANOG were highly expressed in MEC cells [131]. Importantly, the expression of OCT4 and NANOG was correlated with histopathological features of MEC including perineural invasion and desmoplasia [131]. Moreover, a combination of three cancer stem cell markers - CD44, CD133 and SOX2 - was suggested to be a powerful and practicable prognosticator for patients with MEC of minor salivary glands [138]. Additionally, there was a positive correlation between CD44 and vimentin (a marker of EMT) expression level, and the levels of both CD44 and vimentin are associated with MEC tumor grade [115]. A previous study has reported that suberoylanilide hydroxamic acid (SAHA), an inhibitor of histone deacetylases (HDACs), was capable of disrupting CSCs in MEC cell lines and sensitizing tumor cells to cisplatin treatment, emphasizing the role of MEC CSCs in the well-recognized resistance of salivary gland tumors to chemotherapy [139].
ACC is the second most common malignant salivary gland tumor, accounting for 10–25% of the total, and it is the most common histological subtype observed in patients with distant metastatic disease [140]. Previously, it has been reported that ALDH1 was expressed in stromal cells in the majority (63%) of ACCs, although, the pattern of ALDH1 expression did not affect survival of ACC patients [141]. Another study revealed that ALDH+ ACC cells generate the phenotypic diversity of the initial tumor, and have robust sphere-forming, tumorigenic, and metastatic abilities [123]. In addition, high expression of BMI1 was observed in ACC samples with distant metastases as compared to those with negative status, and there was a significant correlation with poor clinical outcome [142]. It has also been shown that BMI1 upregulation is associated with overexpression of the EMT-related transcription factors Snail and Slug (SNAI2) in ACC [127]. SOX2 is also highly expressed in ACC, being tightly associated with the clinical outcome of ACC and, therefore, it has been suggested to have utility as a prognostic marker in this tumor type [143]. Furthermore, there was a significant correlation between SOX2 and Survivin (IAP5; an anti-apoptotic protein) in ACC patients, and the levels of these were significantly higher than in the control group [129]. EGFR is another important player that is commonly upregulated in ACC and has a role in CSC self-renewal. As mentioned above, EGFR signaling holds a pivotal role in self-renewal and maintenance of stem cells. Some previous studies have reported overexpression of EGFR in a minority of ACC cases, which suggests that there is minimal involvement of this pathway in ACC pathogenesis [144, 145], although, overexpression is not necessarily indicative of pathway activity. However, other investigators have reported that 85% of ACC tumors express high levels of EGFR and that 10% of cases express mutated (active) EGFR [146, 147]. The wide variation in results could be explained, at least in part, by the difference in specificity and sensitivity of the techniques used to detect EGFR. One of the proposed mechanisms of EGFR maintenance of stemness is that it leads to stabilization of Snail, with EMT induction [134]. Therefore, analysis of downstream mediators such as Snail or other EMT regulators, or of pathway activation, may provide a more definitive answer. Another study has recently reported that blocking EGFR with erlotinib increases the activation of NOTCH1 signaling, leading to induction of stem cell-like properties [148]. Furthermore, multiple other ligands, including transforming growth factor-α (TGF-α), can activate EGFR; thus, overexpression of such ligands could be implicated in ACC pathogenesis [145]. Taken together, these results suggest that EGFR may utilize multiple mechanisms to promote CSC self-renewal and stemness in ACC (Table 1).
Oral melanoma is a rare (only 0.5% of oral malignancies) and aggressive malignancy with a very poor prognosis [149]. It presents most frequently in the hard palate and alveolar gingiva. The prognosis for patients with these tumors is poor, with a 5-year survival rate estimated at between 20 and 38% [150]. As a result of the high degree of plasticity of this cancer, and the existence of multiple mechanisms that lead to melanoma progression, the existence of a unique and specific biomarker signature for melanoma stem cells is still controversial. Among the surface markers commonly used to identify melanoma stem cells are CD133, CD271, ABCB5, and ALDH1A [151]. CD133 has been associated with CSCs in different tumors including melanoma [151, 152]. A previous study indicated that CD133+ CSCs isolated from the metastatic melanoma cell line D10 significantly induced and accelerated initial angiogenesis compared to CD133− cells [117]. Expression of CD133 has also been reported to be high in metastatic melanoma compared to primary melanoma, and CD133+ melanoma cells have higher clonogenic and tumorigenic abilities compared to CD133− cells [118, 119]. In contrast, a meta-analysis evaluated 299 melanoma cases from five studies for expression levels of CD133 and reported low power to detect a significant association between CD133 expression and melanoma progression [153], suggesting that CD133 might not be an appropriate biomarker in identifying melanoma CSCs. However, in spite of this low power, the authors found 47.9% of cases had high CD133 expression, indicating a positive correlation between CD133 expression and disease progression. High variability was observed in the expression of CD271 as a CSC marker and some controversy still exists with regard to its function in melanoma stem cells. Nevertheless, other studies have suggested that CD271 may contribute to the aggressive nature of melanoma cells and associated chemoresistance [154, 155]. Another functional driver of melanoma aggressiveness and drug resistance is ATP-binding cassette sub-family member 5 (ABCB5), which promotes drug efflux in cancer cells [156]. One study found no correlation between ABCB5 expression and tumor-initiating capacity [157]. However, in a more recent report, ABCB5 was found to enhance tumorigenic ability and promote melanoma metastasis by activating the NF-κB cascade [121]. In addition to these markers, melanoma CSCs show high ALDH activity, especially ALDH1A1 and ALDH1A3 isozymes. ALDH+ melanoma cells were found to be more tumorigenic and more resistant to chemotherapeutic agents than ALDH− cells [158]. Moreover, xenografts from ALDH+ melanoma cells displayed superior self-renewal compared to xenografts from ALDH− cells [159]. However, a recent study reported that both ALDH1A1 and ALDH1A3 correlated with favorable prognosis in metastatic BRAF wild-type and BRAF-mutant melanoma, respectively [160]. Other intracellular proteins of likely importance in melanoma stem cells are SOX2 and KLF4. As mentioned above, they are pluripotency markers and their high expression in melanoma cells promotes reprogramming toward a stem cell phenotype, inducing cell proliferation and drug resistance, and inhibiting apoptosis, [161, 162]. Recently, it has been found that the expression of OCT4, SOX2, and KLF4 was high in CSC subpopulations within the tumor nests and peritumoral stroma of head and neck metastatic malignant melanoma [163]. Another important player is c-MYC, another transcription factor regulating pluripotency and self-renewal of ESCs. For many years, c-MYC has been implicated in the development of numerous cancer types, including melanoma. It has been demonstrated that c-MYC overexpression is associated with tumor progression and is preferentially expressed in metastatic melanoma [164, 165]. Additionally, high c-MYC expression in mucocutaneus melanoma correlates with immune evasion and tumor invasiveness, hence, its association with poor prognosis and survival [164, 166]. In addition, mutations in KIT (the receptor for stem cell factor, SCF) have been found in ~15% of mucosal melanomas [167]. A previous study has indicated that KIT signaling is critical for the proliferation and migration of melanoma cells, raising the possibility that this might be a promising therapeutic target in patients with aggressive oral melanoma [168] (Table 1).
Despite the advances in targeted therapy for HNSCC, early studies from clinical trials have reported limited efficacy with monotherapy compared to conventional therapies. Thus, new therapeutic strategies targeting CSCs are under development to be used in combination with conventional non-targeted therapies to prevent tumor relapse, metastasis, and to combat chemoresistance (Figure 3).
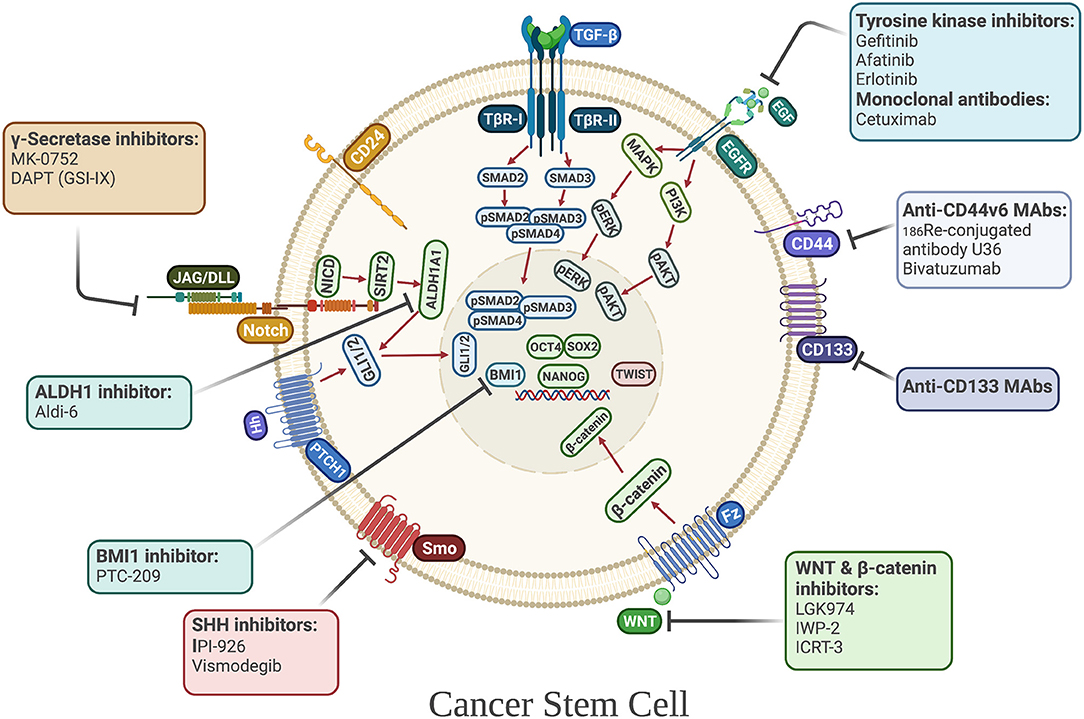
Figure 3. Cancer stem cell-directed therapies in HNSCC. Selected anti-CSC drugs currently under clinical investigation. Their mechanisms of actions include targeting CSC-associated surface markers and CSC-associated signaling pathways, including developmental pathways, that regulate the maintenance, and survival of CSCs.
To date, one of the most promising strategies for targeting HNSCC CSCs is blocking the key self-renewal signaling cascades, such as those regulated by EGFR, NOTCH, WNT, and SHH. Inhibition of EGFR is being employed in advanced and recurrent HNSCC treatment. It has been determined that gefitinib (a tyrosine kinase inhibitor) preferentially targets CSCs, eliminating tumor regrowth, and increasing sensitivity to cisplatin in nasopharyngeal carcinoma [169]. Moreover, blocking EGFR with gefitinib reduces the expression of c-MYC and NANOG, essential factors for reprogramming of induced pluripotent stem cells [169]. Interestingly, treatment with the anti-EGFR antibody, cetuximab, induces CSC differentiation, and inhibits radiochemoresistance of CSCs in HNSCC [170]. Another study has indicated that afatinib (a second-generation tyrosine kinase inhibitor) reduces the self-renewal and invasive properties of HNSCC CSCs in culture by downregulating CD44 and OCT4, inhibiting tumor sphere formation and growth, and inducing radiosensitization [171]. In contrast, in a phase III study, monotherapy with gefitinib did not improve the OS in patients with recurrent and/or metastatic HNSCC [172]. However, the combination of EGFR inhibitors with cisplatin or radiotherapy shows improved response compared to monotherapy with erlotinib or cetuximab, raising the possibility that resistance of tumors to EGFR inhibitor monotherapy could be related to the functional heterogeneity of CSCs in advanced HNSCC [170, 173]. As NOTCH, WNT, and SHH signaling play essential roles in CSC maintenance in HNSCC [174], these pathways are considered as attractive targets for treatment of recurrent/metastatic HNSCC. A preclinical study found that blocking NOTCH1 with the γ-secretase inhibitor, DAPT, modifies the HNSCC CSC phenotype, reducing chemoresistance in tissues post chemotherapy, and also lymph node metastasis [102]. In a non-randomized phase I trial, the combination of a NOTCH inhibitor, MK-0752, with an mTOR inhibitor, ridaforolimus, showed clinical activity in metastatic HNSCC; however, a high number of adverse effects were reported [175]. For WNT signaling, the inhibitor of β-catenin responsive transcription (ICRT-3) arrests the cell cycle and decreases the motility of HNSCC cells [176]. Other preclinical studies have focused on inhibition of Porcupine, a membrane-bound acyltransferase, and found that Porcupine inhibitors such as LGK974 and IWP-2, induce apoptosis, inhibit cell migration and reduce the expression of CSC markers in HNSCC [177, 178]. A similar pattern is also seen with SHH inhibitors, as one study has shown that treating EGFR inhibitor-resistant HNSCC cells with the SHH inhibitor IPI-926 reduces tumor growth and blocks tumor recurrence in patient-derived HNSCC xenografts [179]. Moreover, vismodegib, a SHH pathway inhibitor, decreases expression of GLI1 and Survivin, and promotes radiation-induced DNA damage in HNSCC cells [180]. Another study investigated the dual targeting of EGFR and SHH pathways and found a reduction of cell proliferation and colony forming ability of HNSCC cells [181]. Furthermore, a pilot study testing cetuximab and IPI-926 in patients with recurrent and/or metastatic HNSCC has revealed good tolerability and anti-tumor efficacy [182], indicating that combinational therapy blocking EGFR and SHH might improve the clinical outcomes for HNSCC patients.
The markers used to identify and enrich CSCs may have potential as targets for HNSCC therapy. Among the first reports involving therapeutic targeting of CD44, one study investigated the effect of 186Re-conjugated U36 antibody against the splice variant CD44v6, which was well-tolerated and showed initial promise. However, long-term disease stabilization was only observed in one of 13 patients [183]. Another clinical study indicated that the anti-CD44v6 monoclonal antibody BIWA 4 (bivatuzumab) has antitumor effects and disease stabilization was observed in patients with recurrent locoregional and/or metastatic HNSCC [184]. Labeling BIWA 4 with both a near-infrared fluorescent dye (IRDye800CW) and a radioactive label (Indium-111) was used recently to detect xenografted HNSCC cells, raising the possibility that targeting CD44v6 might be useful for fluorescence-guided surgery [185]. Anti-CD133 agents have also been investigated as targeted anti-CSC therapy in HNSCC. A preclinical study revealed that CD133 knockdown decreases the percentage of side-population CSCs, diminishes tumor growth in vivo, and overcomes chemoresistance [43]. Another study in which a bacterial toxin (cytolethal distending toxin) was conjugated to an anti-human CD133 monoclonal antibody demonstrated inhibition of CD133+-induced cell proliferation in cultures of established HNSCC cell lines [186]. Another approach has favored targeting the drug-detoxification enzyme ALDH1A1, and a strong correlation was found between high levels of ALDH1A1 expression and cisplatin resistance that were reversible by an ALDH1A1 inhibitor which blocks CSC-related activity [124]. In addition, treatment with a novel small molecule inhibitor of ALDH3A1 (Aldi-6) decreased cell viability, and the combination of Aldi-6 and cisplatin caused a profound reduction of cell viability and a greater reduction in tumor size in vivo [187]. Taken together, it seems that combinational therapy using CSC inhibitors together with standard chemotherapy agents could hold significant promise as a future therapeutic strategy for HNSCC.
Another potential therapeutic target to eradicate CSCs is the transcription factor NANOG. Targeting NANOG in combination with cisplatin suppressed stem cell properties of HNSCC cells and enhanced apoptosis and chemosensitivity [188]. Another recent preclinical study found that the combination of NANOG inhibition and radiotherapy produced an additive effect with a decrease in cell viability, stemness properties, and radiotherapy resistance of CD44+ HNSCC cells [76]. In the same study, the authors investigated the involvement of ERK1/2-NANOG signaling on tumor growth and metastasis and found that ERK1/2-NANOG inhibition may reverse CSC phenotypes and have potential to reduce tumor progression and metastasis in HNSCC patients. Additionally, combined treatment with 4SC-202, a novel selective class I histone deacetylase (HDAC) inhibitor, and INK128, a selective mTORC1/C2 inhibitor, synergistically inhibits SOX2 expression and cell growth, and reduces ALDH1+ CSCs and sphere-forming ability of HNSCC cells [189], suggesting that combined HDAC and mTORC1/C2 inhibition selectively targets the self-renewing capacity of CSCs and is more effective and promising than conventional chemotherapy. Furthermore, knockdown of BMI1 was shown to increase the sensitivity of HNSCC cells to radio/chemotherapy in HNSCC-ALDH1+ cells [190]. Other preclinical studies have revealed that the BMI1 inhibitor, PTC-209, inhibits cell proliferation and migration, eliminates lymph node metastases, and reduces colony formation as well as the percentage of ALDH+ cells [82, 191]. Recently, it has been demonstrated that combination therapy with PTC-209 augmented PD1 immune checkpoint blockade and eliminated BMI1+ CSCs by inducing tumor cell-intrinsic immunity, resulting in the inhibition of metastasis and relapse of HNSCC in vivo [192]. Although, monotherapy with the BMI1 inhibitor suppressed HNSCC growth and metastasis, it was not as effective as combination therapy with anti-PD1.
Malignant salivary gland tumors are characterized by frequent local recurrence and distant metastasis, and no satisfactory method has been determined to treat these lesions. Surgery and radiotherapy are reserved to treat localized disease, while chemotherapy is necessary to manage recurrent and/or metastatic tumors, although, no chemotherapy regimen has yet been proven to improve OS [193–197]. Due to these unfavorable outcomes, various targeted agents have been investigated as potential new treatments for salivary gland carcinomas. As mentioned above, EGFR overexpression occurs in a high proportion of ACC cases, and varies markedly among different histotypes, making it a potentially attractive therapeutic target. A phase II study revealed no correlation between EGFR expression/status and response to cetuximab, although, the majority of ACC patients obtained disease stabilization [198]. Consistent with this study, another clinical trial observed a response with lapatinib treatment with prolonged tumor stabilization of more than 6 months in recurrent or metastatic salivary gland carcinoma patients, suggesting that targeted therapy with anti-EGFR therapeutics may improve the clinical outcomes of patients suffering from malignant salivary gland tumors [199]. However, another phase II study demonstrated no objective responses with gefitinib as a monotherapy in patients with advanced salivary gland cancer [200]. Continued work toward the development of predictive biomarkers to inform better treatment options for patients is needed to improve clinical outcome in ACC. A case report indicated that the combination of radiotherapy and cetuximab was well-tolerated and showed a complete remission in a patient with recurrent high grade MEC [201], suggesting that, as for SCC, the combination of anti-EGFR and conventional therapies could be more effective than anti-EGFR monotherapy to manage salivary gland carcinomas. However, further clinical investigation is needed to determine the impact of combinational therapy of anti-EGFR and conventional therapies on patient with MEC. NOTCH1 has also been tested as a therapeutic target for salivary gland CSCs. Genetic reduction of NOTCH1 using RNA interference suppresses proliferation, migration, and clonogenic growth of ACC cells in culture, and reduces the number of metastatic nodules in the lungs of immunodeficient mice bearing ACC xenografts [202]. In an open-label phase I trial, LY3039478, a selective oral NOTCH inhibitor, was well-tolerated; however, no partial or complete responses were observed, although, the majority of the patients (58%) with ACC achieved stable disease [203]. Additionally, Brontictuzumab, a humanized monoclonal antibody that binds to the NOTCH1 juxtamembrane negative regulatory region, was found to have antitumor efficacy in a subcohort of patients with NOTCH-activated ACC [204]. Notably, cisplatin treatment increased the proportion of ALDHhigh/CD44high cells, the number and size of spheroids and BMI1 expression in MEC cell lines. However, the combination of cisplatin and the mTOR inhibitor, temsirolimus, decreased the expression of BMI1, inhibited mTOR signaling, reduced spheroid formation, and decreased the CSC fraction in MEC patient-derived xenograft tumors [205]. Moreover, the combination of cisplatin and the histone acetyltransferase inhibitor, vorinostat, reduced ALDH expression, and CSC numbers, as well as increasing chemosensitivity of MEC cells to cisplatin [139]. Overall, it is likely that combination treatment of targeted therapy with conventional agents might hold promise as a path to improve clinical outcomes. Currently, several clinical trials of targeted therapy for salivary gland malignancies are ongoing.
Previous studies have demonstrated that melanoma cells that have attained a chemoresistant phenotype express multiple stem cell markers, as mentioned above, suggesting that melanoma CSCs may drive the phenotype of these tumor and, thus, represent a very attractive target for novel treatment. One potential therapeutic target is the receptor tyrosine kinase KIT. In phase II clinical trials, the KIT inhibitor imatinib has been evaluated in patients with mucosal melanoma harboring KIT mutations, with an overall disease control rate of ~50%. Importantly, around half of patients with KIT mutations responded to imatinib treatment while none harboring KIT amplification showed a similar response [206]. Another study tested the impact of blocking the ABCB5 transporter on recurrent melanoma, but found that knockdown of ABCB5 did not re-sensitize BRAF inhibitor-resistant melanoma cell lines, in spite of the fact that ABCB5 is highly expressed in malignant melanoma-initiating cells [207]. This suggests that ABCB5 may not be a useful therapeutic target for patients with BRAF inhibitor-resistant melanoma. Another important player that mediates stemness properties in mucosal melanoma, and therefore, might be a promising target, is ALDH. In a recent preclinical study, activated nifuroxazide, which belongs to the family of 5-nitrofuranes, specifically targets ALDH1high melanoma cells by reducing the survival of melanoma CSCs [208]. In the same study, it was shown that combination of a BRAF inhibitor and a MEK inhibitor led to an adaptive increase in ALDHhigh subpopulations in a subset of melanoma cell lines. However, in phase III trials, combination of the BRAF inhibitor, dabrafenib, and the MEK inhibitor, trametinib, resulted in improved progression-free survival, and overall response and survival rates compared to dabrafenib monotherapy in untreated patients who had metastatic melanoma with BRAF V600E or V600K mutations [209]. Furthermore, the combination of the BRAF inhibitor, vemurafenib, and the MEK inhibitor, cobimetinib, also provided a significant benefit compared to vemurafenib alone [210], suggesting that dual targeting may provide better clinical outcomes for metastatic melanoma. Moreover, it also indicates that caution is advised when translating preclinical study results, carried out with cell lines, to patient populations. Additionally, dual targeting with the Bcl-2 inhibitor, ABT-737, and the γ-secretase Inhibitor, GSI, revealed a high efficiency in reducing the cell viability, disrupting colony formation, decreasing ALDH+ cells, and inhibiting the self-renewal of melanoma CSCs [211], indicating that this combinational therapy might be another promising strategy to address treatment relapse of malignant melanoma.
Even though, substantial progress has been made recently in the development of different therapeutic strategies to treat HNC, including targeted therapy, unfavorable clinical response is still a common problem due to therapeutic resistance. Conventional therapies have limited therapeutic effects against the CSC subpopulation. CSC-targeted therapy has been found to be a promising strategy to sensitize resistant tumor cells, eliminate residual tumor-initiating cells, and prevent disease relapse, resulting in improved OS. Multiple CSC biomarkers have been identified that are correlated with poor prognosis, and are attractive targets for therapy. CSC-targeted monotherapies are frequently associated with development of resistant phenotypes or lack of clinical response. Combinatorial therapies address different facets of the CSC phenotype, and hold promise to improve HNC patient outcomes. However, there is a pressing need to discover more specific CSC properties and their druggable targets, as well as distinguishing patient-specific CSC subsets, in order to increase specificity of treatments and reduce serious side-effects experienced by cancer patients. As another point of note, the utility of CSC-targeted therapy could be complicated as a result of serious adverse effects due to targeting pathways that are critical for normal tissue stem cells. Thus, further clinical research to determine the therapeutic value of targeting CSC markers, as well as a more comprehensive understanding of the nuances of cancer stem cells, would help to improve the clinical outcomes of HNC patients.
LS conceived the idea, wrote and revised the manuscript, and is solely responsible for its content.
LS acknowledges start-up funding provided by Augusta University.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author is grateful for guidance and mentorship of Dr. Andrew Yeudall.
1. Daraei P, Moore CE. Racial disparity among the head and neck cancer population. J Cancer Educ. (2015) 30:546–51. doi: 10.1007/s13187-014-0753-4
2. Simard EP, Torre LA, Jemal A. International trends in head and neck cancer incidence rates: differences by country, sex and anatomic site. Oral Oncol. (2014) 50:387–403. doi: 10.1016/j.oraloncology.2014.01.016
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. (2017) 67:7–30. doi: 10.3322/caac.21387
4. Jou A, Hess J. Epidemiology and molecular biology of head and neck cancer. Oncol Res Treat. (2017) 40:328–32. doi: 10.1159/000477127
5. Petti S. Lifestyle risk factors for oral cancer. Oral Oncol. (2009) 45:340–50. doi: 10.1016/j.oraloncology.2008.05.018
6. Price KA, Cohen EE. Current treatment options for metastatic head and neck cancer. Curr Treat Options Oncol. (2012) 13:35–46. doi: 10.1007/s11864-011-0176-y
7. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
8. Mazor T, Pankov A, Song JS, Costello JF. Intratumoral heterogeneity of the epigenome. Cancer Cell. (2016) 29:440–51. doi: 10.1016/j.ccell.2016.03.009
9. Bhaijee F, Pepper DJ, Pitman KT, Bell D. Cancer stem cells in head and neck squamous cell carcinoma: a review of current knowledge and future applications. Head Neck. (2012) 34:894–9. doi: 10.1002/hed.21801
10. Tsai LL, Hu FW, Lee SS, Yu CH, Yu CC, Chang YC. Oct4 mediates tumor initiating properties in oral squamous cell carcinomas through the regulation of epithelial-mesenchymal transition. PLoS ONE. (2014) 9:e87207. doi: 10.1371/journal.pone.0087207
11. Masui T, Ota I, Yook JI, Mikami S, Yane K, Yamanaka T, et al. Snail-induced epithelial-mesenchymal transition promotes cancer stem cell-like phenotype in head and neck cancer cells. Int J Oncol. (2014) 44:693–9. doi: 10.3892/ijo.2013.2225
12. Abbaszadegan MR, Bagheri V, Razavi MS, Momtazi AA, Sahebkar A, Gholamin M. Isolation, identification, and characterization of cancer stem cells: a review. J Cell Physiol. (2017) 232:2008–18. doi: 10.1002/jcp.25759
13. Lacina L, Plzak J, Kodet O, Szabo P, Chovanec M, Dvorankova B, et al. Cancer microenvironment: what can we learn from the stem cell niche. Int J Mol Sci. (2015) 16:24094–110. doi: 10.3390/ijms161024094
14. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. (1997) 3:730–7. doi: 10.1038/nm0797-730
15. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. (2004) 432:396–401. doi: 10.1038/nature03128
16. Wang P, Gao Q, Suo Z, Munthe E, Solberg S, Ma L, et al. Identification and characterization of cells with cancer stem cell properties in human primary lung cancer cell lines. PLoS ONE. (2013) 8:e57020. doi: 10.1371/journal.pone.0057020
17. Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. (2007) 445:111–5. doi: 10.1038/nature05384
18. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. (2003) 100:3983–8. doi: 10.1073/pnas.0530291100
19. Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. (2007) 104:973–8. doi: 10.1073/pnas.0610117104
20. Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. (2007) 58:267–84. doi: 10.1146/annurev.med.58.062105.204854
21. Albers AE, Chen C, Köberle B, Qian X, Klussmann JP, Wollenberg B, et al. Stem cells in squamous head and neck cancer. Crit Rev Oncol Hematol. (2012) 81:224–40. doi: 10.1016/j.critrevonc.2011.03.004
22. Locke M, Heywood M, Fawell S, Mackenzie IC. Retention of intrinsic stem cell hierarchies in carcinoma-derived cell lines. Cancer Res. (2005) 65:8944–50. doi: 10.1158/0008-5472.CAN-05-0931
23. Ludwig N, Szczepanski MJ, Gluszko A, Szafarowski T, Azambuja JH, Dolg L, et al. CD44(+) tumor cells promote early angiogenesis in head and neck squamous cell carcinoma. Cancer Lett. (2019) 467:85–95. doi: 10.1016/j.canlet.2019.10.010
24. Mărgăritescu C, Pirici D, Simionescu C, Stepan A. The utility of CD44, CD117 and CD133 in identification of cancer stem cells (CSC) in oral squamous cell carcinomas (OSCC). Rom J Morphol Embryol. (2011) 52(Suppl. 3):985–93.
25. Dzwonek J, Wilczynski GM. CD44: molecular interactions, signaling and functions in the nervous system. Front Cell Neurosci. (2015) 9:175. doi: 10.3389/fncel.2015.00175
26. Kajita M, Itoh Y, Chiba T, Mori H, Okada A, Kinoh H, et al. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J Cell Biol. (2001) 153:893–904. doi: 10.1083/jcb.153.5.893
27. Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. (2003) 4:33–45. doi: 10.1038/nrm1004
28. Skupien A, Konopka A, Trzaskoma P, Labus J, Gorlewicz A, Swiech L, et al. CD44 regulates dendrite morphogenesis through Src tyrosine kinase-dependent positioning of the Golgi. J Cell Sci. (2014) 127(Pt. 23):5038–51. doi: 10.1242/jcs.154542
29. Matzke A, Sargsyan V, Holtmann B, Aramuni G, Asan E, Sendtner M, et al. Haploinsufficiency of c-Met in cd44-/- mice identifies a collaboration of CD44 and c-Met in vivo. Mol Cell Biol. (2007) 27:8797–806. doi: 10.1128/MCB.01355-07
30. Wang SJ, Bourguignon LY. Hyaluronan and the interaction between CD44 and epidermal growth factor receptor in oncogenic signaling and chemotherapy resistance in head and neck cancer. Arch Otolaryngol Head Neck Surg. (2006) 132:771–8. doi: 10.1001/archotol.132.7.771
31. Yanamoto S, Yamada S, Takahashi H, Naruse T, Matsushita Y, Ikeda H, et al. Expression of the cancer stem cell markers CD44v6 and ABCG2 in tongue cancer: effect of neoadjuvant chemotherapy on local recurrence. Int J Oncol. (2014) 44:1153–62. doi: 10.3892/ijo.2014.2289
32. Sawant S, Gokulan R, Dongre H, Vaidya M, Chaukar D, Prabhash K, et al. Prognostic role of Oct4, CD44 and c-Myc in radio-chemo-resistant oral cancer patients and their tumourigenic potential in immunodeficient mice. Clin Oral Investig. (2016) 20:43–56. doi: 10.1007/s00784-015-1476-6
33. Saghravanian N, Anvari K, Ghazi N, Memar B, Shahsavari M, Aghaee MA. Expression of p63 and CD44 in oral squamous cell carcinoma and correlation with clinicopathological parameters. Arch Oral Biol. (2017) 82:160–5. doi: 10.1016/j.archoralbio.2017.06.011
34. Jakob M, Sharaf K, Schirmer M, Leu M, Küffer S, Bertlich M, et al. Role of cancer stem cell markers ALDH1, BCL11B, BMI-1, and CD44 in the prognosis of advanced HNSCC. Strahlenther Onkol. (2020) 197:231–45. doi: 10.1055/s-0040-1710997
35. Biddle A, Gammon L, Liang X, Costea DE, Mackenzie IC. Phenotypic plasticity determines cancer stem cell therapeutic resistance in oral squamous cell carcinoma. EBioMedicine. (2016) 4:138–45. doi: 10.1016/j.ebiom.2016.01.007
36. Fukusumi T, Ishii H, Konno M, Yasui T, Nakahara S, Takenaka Y, et al. CD10 as a novel marker of therapeutic resistance and cancer stem cells in head and neck squamous cell carcinoma. Br J Cancer. (2014) 111:506–14. doi: 10.1038/bjc.2014.289
37. Piattelli A, Fioroni M, Iezzi G, Perrotti V, Stellini E, Piattelli M, et al. CD10 expression in stromal cells of oral cavity squamous cell carcinoma: a clinic and pathologic correlation. Oral Dis. (2006) 12:301–4. doi: 10.1111/j.1601-0825.2005.01196.x
38. Li Q, Wang Y, Xu L, Wang L, Guo Y, Guo C. High level of CD10 expression is associated with poor overall survival in patients with head and neck cancer. Int J Oral Maxillofac Surg. (2020) doi: 10.1016/j.ijom.2020.07.037
39. Li B, McCrudden CM, Yuen HF, Xi X, Lyu P, Chan KW, et al. CD133 in brain tumor: the prognostic factor. Oncotarget. (2017) 8:11144–59. doi: 10.18632/oncotarget.14406
40. Lazarova P, Wu Q, Kvalheim G, Suo Z, Haakenstad KW, Metodiev K, et al. Growth factor receptors in hematopoietic stem cells: EPH family expression in CD34+ and CD133+ cell populations from mobilized peripheral blood. Int J Immunopathol Pharmacol. (2006) 19:49–56. doi: 10.1177/205873920601900105
41. Wang J, Wu Y, Gao W, Li F, Bo Y, Zhu M, et al. Identification and characterization of CD133(+)CD44(+) cancer stem cells from human laryngeal squamous cell carcinoma cell lines. J Cancer. (2017) 8:497–506. doi: 10.7150/jca.17444
42. Zhang Q, Shi S, Yen Y, Brown J, Ta JQ, Le AD. A subpopulation of CD133(+) cancer stem-like cells characterized in human oral squamous cell carcinoma confer resistance to chemotherapy. Cancer Lett. (2010) 289:151–60. doi: 10.1016/j.canlet.2009.08.010
43. Yu CC, Hu FW, Yu CH, Chou MY. Targeting CD133 in the enhancement of chemosensitivity in oral squamous cell carcinoma-derived side population cancer stem cells. Head Neck. (2016) 38(Suppl. 1):E231–8. doi: 10.1002/hed.23975
44. Yu L, Zhou L, Wu S, Gong X, Feng Z, Ma L, et al. Clinicopathological significance of cancer stem cells marked by CD133 and KAI1/CD82 expression in laryngeal squamous cell carcinoma. World J Surg Oncol. (2014) 12:118. doi: 10.1186/1477-7819-12-118
45. de Moraes FP, Lourenço SV, Ianez RC, de Sousa EA, Silva MM, Damascena AS, et al. Expression of stem cell markers in oral cavity and oropharynx squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol. (2017) 123:113–22. doi: 10.1016/j.oooo.2016.09.009
46. Okamoto H, Fujishima F, Nakamura Y, Zuguchi M, Ozawa Y, Takahashi Y, et al. Significance of CD133 expression in esophageal squamous cell carcinoma. World J Surg Oncol. (2013) 11:51. doi: 10.1186/1477-7819-11-51
47. Hang D, Dong HC, Ning T, Dong B, Hou DL, Xu WG. Prognostic value of the stem cell markers CD133 and ABCG2 expression in esophageal squamous cell carcinoma. Dis Esophagus. (2012) 25:638–44. doi: 10.1111/j.1442-2050.2011.01298.x
48. Ma I, Allan AL. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev Rep. (2011) 7:292–306. doi: 10.1007/s12015-010-9208-4
49. Chen YC, Chen YW, Hsu HS, Tseng LM, Huang PI, Lu KH, et al. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem Biophys Res Commun. (2009) 385:307–13. doi: 10.1016/j.bbrc.2009.05.048
50. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. (2007) 1:555–67. doi: 10.1016/j.stem.2007.08.014
51. Rasper M, Schäfer A, Piontek G, Teufel J, Brockhoff G, Ringel F, et al. Aldehyde dehydrogenase 1 positive glioblastoma cells show brain tumor stem cell capacity. Neuro Oncol. (2010) 12:1024–33. doi: 10.1093/neuonc/noq070
52. Sterz CM, Kulle C, Dakic B, Makarova G, Böttcher MC, Bette M, et al. A basal-cell-like compartment in head and neck squamous cell carcinomas represents the invasive front of the tumor and is expressing MMP-9. Oral Oncol. (2010) 46:116–22. doi: 10.1016/j.oraloncology.2009.11.011
53. Yu CC, Lo WL, Chen YW, Huang PI, Hsu HS, Tseng LM, et al. Bmi-1 regulates snail expression and promotes metastasis ability in head and neck squamous cancer-derived ALDH1 positive cells. J Oncol. (2011) 2011:609259. doi: 10.1155/2011/609259
54. Wang Y, Zhe H, Gao P, Zhang N, Li G, Qin J. Cancer stem cell marker ALDH1 expression is associated with lymph node metastasis and poor survival in esophageal squamous cell carcinoma: a study from high incidence area of northern China. Dis Esophagus. (2012) 25:560–5. doi: 10.1111/j.1442-2050.2011.01279.x
55. Koukourakis MI, Giatromanolaki A, Tsakmaki V, Danielidis V, Sivridis E. Cancer stem cell phenotype relates to radio-chemotherapy outcome in locally advanced squamous cell head-neck cancer. Br J Cancer. (2012) 106:846–53. doi: 10.1038/bjc.2012.33
56. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. (2007) 318:1917–20. doi: 10.1126/science.1151526
57. Fu TY, Hsieh IC, Cheng JT, Tsai MH, Hou YY, Lee JH, et al. Association of OCT4, SOX2, and NANOG expression with oral squamous cell carcinoma progression. J Oral Pathol Med. (2016) 45:89–95. doi: 10.1111/jop.12335
58. Chiou SH, Yu CC, Huang CY, Lin SC, Liu CJ, Tsai TH, et al. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin Cancer Res. (2008) 14:4085–95. doi: 10.1158/1078-0432.CCR-07-4404
59. Schröck A, Bode M, Göke FJ, Bareiss PM, Schairer R, Wang H, et al. Expression and role of the embryonic protein SOX2 in head and neck squamous cell carcinoma. Carcinogenesis. (2014) 35:1636–42. doi: 10.1093/carcin/bgu094
60. Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. (1998) 95:379–91. doi: 10.1016/S0092-8674(00)81769-9
61. Ezeh UI, Turek PJ, Reijo RA, Clark AT. Human embryonic stem cell genes OCT4, NANOG, STELLAR, and GDF3 are expressed in both seminoma and breast carcinoma. Cancer. (2005) 104:2255–65. doi: 10.1002/cncr.21432
62. Chen YC, Hsu HS, Chen YW, Tsai TH, How CK, Wang CY, et al. Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS ONE. (2008) 3:e2637. doi: 10.1371/journal.pone.0002637
63. Wang XQ, Ongkeko WM, Chen L, Yang ZF, Lu P, Chen KK, et al. Octamer 4 (Oct4) mediates chemotherapeutic drug resistance in liver cancer cells through a potential Oct4-AKT-ATP-binding cassette G2 pathway. Hepatology. (2010) 52:528–39. doi: 10.1002/hep.23692
64. Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. (2008) 68:4311–20. doi: 10.1158/0008-5472.CAN-08-0364
65. Sinha N, Mukhopadhyay S, Das DN, Panda PK, Bhutia SK. Relevance of cancer initiating/stem cells in carcinogenesis and therapy resistance in oral cancer. Oral Oncol. (2013) 49:854–62. doi: 10.1016/j.oraloncology.2013.06.010
66. Boiani M, Schöler HR. Regulatory networks in embryo-derived pluripotent stem cells. Nat Rev Mol Cell Biol. (2005) 6:872–84. doi: 10.1038/nrm1744
67. Chew JL, Loh YH, Zhang W, Chen X, Tam WL, Yeap LS, et al. Reciprocal transcriptional regulation of Pou5f1 and Sox2 via the Oct4/Sox2 complex in embryonic stem cells. Mol Cell Biol. (2005) 25:6031–46. doi: 10.1128/MCB.25.14.6031-6046.2005
68. Chou MY, Hu FW, Yu CH, Yu CC. Sox2 expression involvement in the oncogenicity and radiochemoresistance of oral cancer stem cells. Oral Oncol. (2015) 51:31–9. doi: 10.1016/j.oraloncology.2014.10.002
69. Freier K, Knoepfle K, Flechtenmacher C, Pungs S, Devens F, Toedt G, et al. Recurrent copy number gain of transcription factor SOX2 and corresponding high protein expression in oral squamous cell carcinoma. Genes Chromosomes Cancer. (2010) 49:9–16. doi: 10.1002/gcc.20714
70. Bass AJ, Wang TC. An inflammatory situation: SOX2 and STAT3 cooperate in squamous cell carcinoma initiation. Cell Stem Cell. (2013) 12:266–8. doi: 10.1016/j.stem.2013.02.004
71. Lee SH, Oh SY, Do SI, Lee HJ, Kang HJ, Rho YS, et al. SOX2 regulates self-renewal and tumorigenicity of stem-like cells of head and neck squamous cell carcinoma. Br J Cancer. (2014) 111:2122–30. doi: 10.1038/bjc.2014.528
72. Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. (2006) 38:431–40. doi: 10.1038/ng1760
73. Rodda DJ, Chew JL, Lim LH, Loh YH, Wang B, Ng HH, et al. Transcriptional regulation of nanog by OCT4 and SOX2. J Biol Chem. (2005) 280:24731–7. doi: 10.1074/jbc.M502573200
74. Han J, Zhang F, Yu M, Zhao P, Ji W, Zhang H, et al. RNA interference-mediated silencing of NANOG reduces cell proliferation and induces G0/G1 cell cycle arrest in breast cancer cells. Cancer Lett. (2012) 321:80–8. doi: 10.1016/j.canlet.2012.02.021
75. Xie X, Piao L, Cavey GS, Old M, Teknos TN, Mapp AK, et al. Phosphorylation of Nanog is essential to regulate Bmi1 and promote tumorigenesis. Oncogene. (2014) 33:2040–52. doi: 10.1038/onc.2013.173
76. Huang C, Yoon C, Zhou XH, Zhou YC, Zhou WW, Liu H, et al. ERK1/2-Nanog signaling pathway enhances CD44(+) cancer stem-like cell phenotypes and epithelial-to-mesenchymal transition in head and neck squamous cell carcinomas. Cell Death Dis. (2020) 11:266. doi: 10.1038/s41419-020-2448-6
77. Tsai LL, Yu CC, Chang YC, Yu CH, Chou MY. Markedly increased Oct4 and Nanog expression correlates with cisplatin resistance in oral squamous cell carcinoma. J Oral Pathol Med. (2011) 40:621–8. doi: 10.1111/j.1600-0714.2011.01015.x
78. Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. (2003) 423:302–5. doi: 10.1038/nature01587
79. Allegra E, Trapasso S, Pisani D, Puzzo L. The role of BMI1 as a biomarker of cancer stem cells in head and neck cancer: a review. Oncology. (2014) 86:199–205. doi: 10.1159/000358598
80. Curtarelli RB, Gonçalves JM, Dos Santos LGP, Savi MG, Nör JE, Mezzomo LAM, et al. Expression of cancer stem cell biomarkers in human head and neck carcinomas: a systematic review. Stem Cell Rev Rep. (2018) 14:769–84. doi: 10.1007/s12015-018-9839-4
81. Kurihara K, Isobe T, Yamamoto G, Tanaka Y, Katakura A, Tachikawa T. Expression of BMI1 and ZEB1 in epithelial-mesenchymal transition of tongue squamous cell carcinoma. Oncol Rep. (2015) 34:771–8. doi: 10.3892/or.2015.4032
82. Chen D, Wu M, Li Y, Chang I, Yuan Q, Ekimyan-Salvo M, et al. Targeting BMI1(+) cancer stem cells overcomes chemoresistance and inhibits metastases in squamous cell carcinoma. Cell Stem Cell. (2017) 20:621–34.e6. doi: 10.1016/j.stem.2017.02.003
83. Fan Z, Li M, Chen X, Wang J, Liang X, Wang H, et al. Prognostic value of cancer stem cell markers in head and neck squamous cell carcinoma: a meta-analysis. Sci Rep. (2017) 7:43008. doi: 10.1038/srep43008
84. Lundberg M, Renkonen S, Haglund C, Mattila PS, Leivo I, Hagström J, et al. Association of BMI-1 and p16 as prognostic factors for head and neck carcinomas. Acta Otolaryngol. (2016) 136:501–5. doi: 10.3109/00016489.2015.1122227
85. Goetz SC, Ocbina PJ, Anderson KV. The primary cilium as a Hedgehog signal transduction machine. Methods Cell Biol. (2009) 94:199–222. doi: 10.1016/S0091-679X(08)94010-3
86. Chari NS, McDonnell TJ. The sonic hedgehog signaling network in development and neoplasia. Adv Anat Pathol. (2007) 14:344–52. doi: 10.1097/PAP.0b013e3180ca8a1d
87. Katoh M. Genomic testing, tumor microenvironment and targeted therapy of Hedgehog-related human cancers. Clin Sci. (2019) 133:953–70. doi: 10.1042/CS20180845
88. Chinchilla P, Xiao L, Kazanietz MG, Riobo NA. Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle. (2010) 9:570–79. doi: 10.4161/cc.9.3.10591
89. Fan HX, Wang S, Zhao H, Liu N, Chen D, Sun M, et al. Sonic hedgehog signaling may promote invasion and metastasis of oral squamous cell carcinoma by activating MMP-9 and E-cadherin expression. Med Oncol. (2014) 31:41. doi: 10.1007/s12032-014-0041-5
90. Reinders MG, van Hout AF, Cosgun B, Paulussen AD, Leter EM, Steijlen PM, et al. New mutations and an updated database for the patched-1 (PTCH1) gene. Mol Genet Genomic Med. (2018) 6:409–15. doi: 10.1002/mgg3.380
91. Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell. (2015) 27:327–41. doi: 10.1016/j.ccell.2015.02.001
92. Zhou M, Hou Y, Yang G, Zhang H, Tu G, Du YE, et al. LncRNA-Hh strengthen cancer stem cells generation in twist-positive breast cancer via activation of hedgehog signaling pathway. Stem Cells. (2016) 34:55–66. doi: 10.1002/stem.2219
93. Zhang C, Li C, He F, Cai Y, Yang H. Identification of CD44+CD24+ gastric cancer stem cells. J Cancer Res Clin Oncol. (2011) 137:1679–86. doi: 10.1007/s00432-011-1038-5
94. Zhang K, Che S, Pan C, Su Z, Zheng S, Yang S, et al. The SHH/Gli axis regulates CD90-mediated liver cancer stem cell function by activating the IL6/JAK2 pathway. J Cell Mol Med. (2018) 22:3679–90. doi: 10.1111/jcmm.13651
95. Cavicchioli Buim ME, Gurgel CA, Gonçalves Ramos EA, Lourenço SV, Soares FA. Activation of sonic hedgehog signaling in oral squamous cell carcinomas: a preliminary study. Hum Pathol. (2011) 42:1484–90. doi: 10.1016/j.humpath.2010.12.015
96. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. (2009) 137:216–33. doi: 10.1016/j.cell.2009.03.045
98. Lee SH, Do SI, Lee HJ, Kang HJ, Koo BS, Lim YC. Notch1 signaling contributes to stemness in head and neck squamous cell carcinoma. Lab Invest. (2016) 96:508–16. doi: 10.1038/labinvest.2015.163
99. Pannuti A, Foreman K, Rizzo P, Osipo C, Golde T, Osborne B, et al. Targeting Notch to target cancer stem cells. Clin Cancer Res. (2010) 16:3141–52. doi: 10.1158/1078-0432.CCR-09-2823
100. Gu F, Ma Y, Zhang Z, Zhao J, Kobayashi H, Zhang L, et al. Expression of Stat3 and Notch1 is associated with cisplatin resistance in head and neck squamous cell carcinoma. Oncol Rep. (2010) 23:671–6. doi: 10.3892/or_00000683
101. Lin JT, Chen MK, Yeh KT, Chang CS, Chang TH, Lin CY, et al. Association of high levels of Jagged-1 and Notch-1 expression with poor prognosis in head and neck cancer. Ann Surg Oncol. (2010) 17:2976–83. doi: 10.1245/s10434-010-1118-9
102. Zhao ZL, Zhang L, Huang CF, Ma SR, Bu LL, Liu JF, et al. NOTCH1 inhibition enhances the efficacy of conventional chemotherapeutic agents by targeting head neck cancer stem cell. Sci Rep. (2016) 6:24704. doi: 10.1038/srep24704
103. Grilli G, Hermida-Prado F, Álvarez-Fernández M, Allonca E, Álvarez-González M, Astudillo A, et al. Impact of Notch signaling on the prognosis of patients with head and neck squamous cell carcinoma. Oral Oncol. (2020) 110:105003. doi: 10.1016/j.oraloncology.2020.105003
104. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. (2005) 5:341–54. doi: 10.1038/nrc1609
105. Argiris A, Harrington KJ, Tahara M, Schulten J, Chomette P, Ferreira Castro A, et al. Evidence-based treatment options in recurrent and/or metastatic squamous cell carcinoma of the head and neck. Front Oncol. (2017) 7:72. doi: 10.3389/fonc.2017.00072
106. Perez A, Neskey DM, Wen J, Pereira L, Reategui EP, Goodwin WJ, et al. CD44 interacts with EGFR and promotes head and neck squamous cell carcinoma initiation and progression. Oral Oncol. (2013) 49:306–13. doi: 10.1016/j.oraloncology.2012.11.009
107. Xu Q, Zhang Q, Ishida Y, Hajjar S, Tang X, Shi H, et al. EGF induces epithelial-mesenchymal transition and cancer stem-like cell properties in human oral cancer cells via promoting Warburg effect. Oncotarget. (2017) 8:9557–71. doi: 10.18632/oncotarget.13771
108. Lv XX, Zheng XY, Yu JJ, Ma HR, Hua C, Gao RT. EGFR enhances the stemness and progression of oral cancer through inhibiting autophagic degradation of SOX2. Cancer Med. (2020) 9:1131–40. doi: 10.1002/cam4.2772
109. Tang AL, Hauff SJ, Owen JH, Graham MP, Czerwinski MJ, Park JJ, et al. UM-SCC-104: a new human papillomavirus-16-positive cancer stem cell-containing head and neck squamous cell carcinoma cell line. Head Neck. (2012) 34:1480–91. doi: 10.1002/hed.21962
110. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. (2009) 17:9–26. doi: 10.1016/j.devcel.2009.06.016
111. Kléber M, Sommer L. Wnt signaling and the regulation of stem cell function. Curr Opin Cell Biol. (2004) 16:681–7. doi: 10.1016/j.ceb.2004.08.006
112. Roy S, Kar M, Roy S, Padhi S, Kumar A, Thakur S, et al. Inhibition of CD44 sensitizes cisplatin-resistance and affects Wnt/β-catenin signaling in HNSCC cells. Int J Biol Macromol. (2020) 149:501–12. doi: 10.1016/j.ijbiomac.2020.01.131
113. Le PN, Keysar SB, Miller B, Eagles JR, Chimed TS, Reisinger J, et al. Wnt signaling dynamics in head and neck squamous cell cancer tumor-stroma interactions. Mol Carcinog. (2019) 58:398–410. doi: 10.1002/mc.22937
114. Roy S, Kar M, Roy S, Saha A, Padhi S, Banerjee B. Role of β-catenin in cisplatin resistance, relapse and prognosis of head and neck squamous cell carcinoma. Cell Oncol. (2018) 41:185–200. doi: 10.1007/s13402-017-0365-1
115. Irani S, Jafari B. Expression of vimentin and CD44 in mucoepidermoid carcinoma: a role in tumor growth. Indian J Dent Res. (2018) 29:333–40. doi: 10.4103/ijdr.IJDR_184_17
116. Mannelli G, Magnelli L, Deganello A, Busoni M, Meccariello G, Parrinello G, et al. Detection of putative stem cell markers, CD44/CD133, in primary and lymph node metastases in head and neck squamous cell carcinomas. A preliminary immunohistochemical and in vitro study. Clin Otolaryngol. (2015) 40:312–20. doi: 10.1111/coa.12368
117. Zimmerer RM, Matthiesen P, Kreher F, Kampmann A, Spalthoff S, Jehn P, et al. Putative CD133+ melanoma cancer stem cells induce initial angiogenesis in vivo. Microvasc Res. (2016) 104:46–54. doi: 10.1016/j.mvr.2015.12.001
118. Klein WM, Wu BP, Zhao S, Wu H, Klein-Szanto AJ, Tahan SR. Increased expression of stem cell markers in malignant melanoma. Mod Pathol. (2007) 20:102–7. doi: 10.1038/modpathol.3800720
119. Zimmerer RM, Korn P, Demougin P, Kampmann A, Kokemüller H, Eckardt AM, et al. Functional features of cancer stem cells in melanoma cell lines. Cancer Cell Int. (2013) 13:78. doi: 10.1186/1475-2867-13-78
120. Oliveira LR, Castilho-Fernandes A, Oliveira-Costa JP, Soares FA, Zucoloto S, Ribeiro-Silva A. CD44+/CD133+ immunophenotype and matrix metalloproteinase-9: influence on prognosis in early-stage oral squamous cell carcinoma. Head Neck. (2014) 36:1718–26. doi: 10.1002/hed.23527
121. Wang S, Tang L, Lin J, Shen Z, Yao Y, Wang W, et al. ABCB5 promotes melanoma metastasis through enhancing NF-κB p65 protein stability. Biochem Biophys Res Commun. (2017) 492:18–26. doi: 10.1016/j.bbrc.2017.08.052
122. Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, et al. Identification of cells initiating human melanomas. Nature. (2008) 451:345–9. doi: 10.1038/nature06489
123. Sun S, Wang Z. ALDH high adenoid cystic carcinoma cells display cancer stem cell properties and are responsible for mediating metastasis. Biochem Biophys Res Commun. (2010) 396:843–8. doi: 10.1016/j.bbrc.2010.04.170
124. Kulsum S, Sudheendra HV, Pandian R, Ravindra DR, Siddappa G, R N, et al. Cancer stem cell mediated acquired chemoresistance in head and neck cancer can be abrogated by aldehyde dehydrogenase 1 A1 inhibition. Mol Carcinog. (2017) 56:694–711. doi: 10.1002/mc.22526
125. Michifuri Y, Hirohashi Y, Torigoe T, Miyazaki A, Kobayashi J, Sasaki T, et al. High expression of ALDH1 and SOX2 diffuse staining pattern of oral squamous cell carcinomas correlates to lymph node metastasis. Pathol Int. (2012) 62:684–9. doi: 10.1111/j.1440-1827.2012.02851.x
126. He Q, Liu Z, Zhao T, Zhao L, Zhou X, Wang A. Bmi1 drives stem-like properties and is associated with migration, invasion, and poor prognosis in tongue squamous cell carcinoma. Int J Biol Sci. (2015) 11:1–10. doi: 10.7150/ijbs.10405
127. Yi C, Li BB, Zhou CX. Bmi-1 expression predicts prognosis in salivary adenoid cystic carcinoma and correlates with epithelial-mesenchymal transition-related factors. Ann Diagn Pathol. (2016) 22:38–44. doi: 10.1016/j.anndiagpath.2015.10.015
128. Nör C, Zhang Z, Warner KA, Bernardi L, Visioli F, Helman JI, et al. Cisplatin induces Bmi-1 and enhances the stem cell fraction in head and neck cancer. Neoplasia. (2014) 16:137–46. doi: 10.1593/neo.131744
129. Luo Y, Liu T, Fei W, Yue XG. Correlation between SOX2 and Survivin clinical features in patients with salivary adenoid cystic carcinoma. J Infect Public Health. (2019) 12:847–53. doi: 10.1016/j.jiph.2019.03.015
130. Pozzi V, Sartini D, Rocchetti R, Santarelli A, Rubini C, Morganti S, et al. Identification and characterization of cancer stem cells from head and neck squamous cell carcinoma cell lines. Cell Physiol Biochem. (2015) 36:784–98. doi: 10.1159/000430138
131. Destro Rodrigues MF, Sedassari BT, Esteves CM, de Andrade NP, Altemani A, de Sousa SC, et al. Embryonic stem cells markers Oct4 and Nanog correlate with perineural invasion in human salivary gland mucoepidermoid carcinoma. J Oral Pathol Med. (2017) 46:112–20. doi: 10.1111/jop.12449
132. Lee HJ, Kang YH, Lee JS, Byun JH, Kim UK, Jang SJ, et al. Positive expression of NANOG, mutant p53, and CD44 is directly associated with clinicopathological features and poor prognosis of oral squamous cell carcinoma. BMC Oral Health. (2015) 15:153. doi: 10.1186/s12903-015-0120-9
133. Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. (2006) 24:2666–72. doi: 10.1200/JCO.2005.04.8306
134. Wang Y, Hu J, Wang Y, Ye W, Zhang X, Ju H, et al. EGFR activation induced snail-dependent EMT and myc-dependent PD-L1 in human salivary adenoid cystic carcinoma cells. Cell Cycle. (2018) 17:1457–70. doi: 10.1080/15384101.2018.1489177
135. Speight PM, Barrett AW. Salivary gland tumours. Oral Dis. (2002) 8:229–40. doi: 10.1034/j.1601-0825.2002.02870.x
136. Luna MA. Salivary mucoepidermoid carcinoma: revisited. Adv Anat Pathol. (2006) 13:293–307. doi: 10.1097/01.pap.0000213058.74509.d3
137. Adams A, Warner K, Pearson AT, Zhang Z, Kim HS, Mochizuki D, et al. ALDH/CD44 identifies uniquely tumorigenic cancer stem cells in salivary gland mucoepidermoid carcinomas. Oncotarget. (2015) 6:26633–50. doi: 10.18632/oncotarget.5782
138. Xu W, Wang Y, Qi X, Xie J, Wei Z, Yin X, et al. Prognostic factors of palatal mucoepidermoid carcinoma: a retrospective analysis based on a double-center study. Sci Rep. (2017) 7:43907. doi: 10.1038/srep43907
139. Guimarães DM, Almeida LO, Martins MD, Warner KA, Silva AR, Vargas PA, et al. Sensitizing mucoepidermoid carcinomas to chemotherapy by targeted disruption of cancer stem cells. Oncotarget. (2016) 7:42447–60. doi: 10.18632/oncotarget.9884
140. Alfieri S, Granata R, Bergamini C, Resteghini C, Bossi P, Licitra LF, et al. Systemic therapy in metastatic salivary gland carcinomas: a pathology-driven paradigm? Oral Oncol. (2017) 66:58–63. doi: 10.1016/j.oraloncology.2016.12.016
141. Zhou JH, Hanna EY, Roberts D, Weber RS, Bell D. ALDH1 immunohistochemical expression and its significance in salivary adenoid cystic carcinoma. Head Neck. (2013) 35:575–8. doi: 10.1002/hed.23003
142. Chang B, Li S, He Q, Liu Z, Zhao L, Zhao T, et al. Deregulation of Bmi-1 is associated with enhanced migration, invasion and poor prognosis in salivary adenoid cystic carcinoma. Biochim Biophys Acta. (2014) 1840:3285–91. doi: 10.1016/j.bbagen.2014.08.005
143. Dai W, Tan X, Sun C, Zhou Q. High expression of SOX2 is associated with poor prognosis in patients with salivary gland adenoid cystic carcinoma. Int J Mol Sci. (2014) 15:8393–406. doi: 10.3390/ijms15058393
144. Chen CH, Li BY, Wan JT, Sun A, Leu JS, Chiang CP. Expression of epidermal growth factor in salivary adenoid cystic carcinoma. Proc Natl Sci Counc Repub China B. (2001) 25:90–6.
145. Gibbons MD, Manne U, Carroll WR, Peters GE, Weiss HL, Grizzle WE. Molecular differences in mucoepidermoid carcinoma and adenoid cystic carcinoma of the major salivary glands. Laryngoscope. (2001) 111:1373–8. doi: 10.1097/00005537-200108000-00011
146. Williams MD, Roberts DB, Kies MS, Mao L, Weber RS, El-Naggar AK. Genetic and expression analysis of HER-2 and EGFR genes in salivary duct carcinoma: empirical and therapeutic significance. Clin Cancer Res. (2010) 16:2266–74. doi: 10.1158/1078-0432.CCR-09-0238
147. Vered M, Braunstein E, Buchner A. Immunohistochemical study of epidermal growth factor receptor in adenoid cystic carcinoma of salivary gland origin. Head Neck. (2002) 24:632–6. doi: 10.1002/hed.10104
148. Wang Y, Han Y, Xu S, Zhang L, Zhang X, Deng J, et al. Targeting EGFR enriches stem cell-like properties in salivary adenoid cystic carcinoma by activating the notch1 pathway. Cancer Manag Res. (2020) 12:6655–63. doi: 10.2147/CMAR.S253500
149. Meleti M, Leemans CR, Mooi WJ, Vescovi P, van der Waal I. Oral malignant melanoma: a review of the literature. Oral Oncol. (2007) 43:116–21. doi: 10.1016/j.oraloncology.2006.04.001
150. Ebenezer J. Malignant melanoma of the oral cavity. Indian J Dent Res. (2006) 17:94–6. doi: 10.4103/0970-9290.29882
151. Grasso C, Anaka M, Hofmann O, Sompallae R, Broadley K, Hide W, et al. Iterative sorting reveals CD133+ and CD133- melanoma cells as phenotypically distinct populations. BMC Cancer. (2016) 16:726. doi: 10.1186/s12885-016-2759-2
152. Roudi R, Ebrahimi M, Sabet MN, Najafi A, Nourani MR, Fomeshi MR, et al. Comparative gene-expression profiling of CD133(+) and CD133(-) D10 melanoma cells. Future Oncol. (2015) 11:2383–93. doi: 10.2217/fon.15.174
153. Madjd Z, Erfani E, Gheytanchi E, Moradi-Lakeh M, Shariftabrizi A, Asadi-Lari M. Expression of CD133 cancer stem cell marker in melanoma: a systematic review and meta-analysis. Int J Biol Markers. (2016) 31:e118–25. doi: 10.5301/jbm.5000209
154. Redmer T, Walz I, Klinger B, Khouja S, Welte Y, Schäfer R, et al. The role of the cancer stem cell marker CD271 in DNA damage response and drug resistance of melanoma cells. Oncogenesis. (2017) 6:e291. doi: 10.1038/oncsis.2016.88
155. Vidal A, Redmer T. Decoding the role of CD271 in melanoma. Cancers. (2020) 12:2460. doi: 10.3390/cancers12092460
156. Begicevic RR, Falasca M. ABC transporters in cancer stem cells: beyond chemoresistance. Int J Mol Sci. (2017) 18:2362. doi: 10.3390/ijms18112362
157. Quintana E, Shackleton M, Foster HR, Fullen DR, Sabel MS, Johnson TM, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell. (2010) 18:510–23. doi: 10.1016/j.ccr.2010.10.012
158. Luo Y, Dallaglio K, Chen Y, Robinson WA, Robinson SE, McCarter MD, et al. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cells. (2012) 30:2100–13. doi: 10.1002/stem.1193
159. Boonyaratanakornkit JB, Yue L, Strachan LR, Scalapino KJ, LeBoit PE, Lu Y, et al. Selection of tumorigenic melanoma cells using ALDH. J Invest Dermatol. (2010) 130:2799–808. doi: 10.1038/jid.2010.237
160. Samson JM, Ravindran Menon D, Smith DE, Baird E, Kitano T, Gao D, et al. Clinical implications of ALDH1A1 and ALDH1A3 mRNA expression in melanoma subtypes. Chem Biol Interact. (2019) 314:108822. doi: 10.1016/j.cbi.2019.108822
161. Wang Y, Sun HJ, Li RG, Wang XM, Cheng ZQ, Lou N. Reprogramming factors induce proliferation and inhibit apoptosis of melanoma cells by changing the expression of particular genes. Mol Med Rep. (2019) 19:967–73. doi: 10.3892/mmr.2018.9753
162. Hüser L, Sachindra S, Granados K, Federico A, Larribère L, Novak D, et al. SOX2-mediated upregulation of CD24 promotes adaptive resistance toward targeted therapy in melanoma. Int J Cancer. (2018) 143:3131–42. doi: 10.1002/ijc.31609
163. Yoganandarajah V, Patel J, van Schaijik B, Bockett N, Brasch HD, Paterson E, et al. Identification of cancer stem cell subpopulations in head and neck metastatic malignant melanoma. Cells. (2020) 9:324. doi: 10.3390/cells9020324
164. Kfoury A, Armaro M, Collodet C, Sordet-Dessimoz J, Giner MP, Christen S, et al. AMPK promotes survival of c-Myc-positive melanoma cells by suppressing oxidative stress. EMBO J. (2018) 37:e97673. doi: 10.15252/embj.201797673
165. Kraehn GM, Utikal J, Udart M, Greulich KM, Bezold G, Kaskel P, et al. Extra c-myc oncogene copies in high risk cutaneous malignant melanoma and melanoma metastases. Br J Cancer. (2001) 84:72–9. doi: 10.1054/bjoc.2000.1535
166. Lin X, Sun R, Zhao X, Zhu D, Zhao X, Gu Q, et al. C-myc overexpression drives melanoma metastasis by promoting vasculogenic mimicry via c-myc/snail/Bax signaling. J Mol Med. (2017) 95:53–67. doi: 10.1007/s00109-016-1452-x
167. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. (2006) 24:4340–6. doi: 10.1200/JCO.2006.06.2984
168. Posch C, Moslehi H, Sanlorenzo M, Green G, Vujic I, Panzer-Grümayer R, et al. Pharmacological inhibitors of c-KIT block mutant c-KIT mediated migration of melanocytes and melanoma cells in vitro and in vivo. Oncotarget. (2016) 7:45916–25. doi: 10.18632/oncotarget.10001
169. Ma L, Zhang G, Miao XB, Deng XB, Wu Y, Liu Y, et al. Cancer stem-like cell properties are regulated by EGFR/AKT/β-catenin signaling and preferentially inhibited by gefitinib in nasopharyngeal carcinoma. FEBS J. (2013) 280:2027–41. doi: 10.1111/febs.12226
170. Setúbal Destro Rodrigues MF, Gammon L, Rahman MM, Biddle A, Nunes FD, Mackenzie IC. Effects of Cetuximab and Erlotinib on the behaviour of cancer stem cells in head and neck squamous cell carcinoma. Oncotarget. (2018) 9:13488–500. doi: 10.18632/oncotarget.24416
171. Macha MA, Rachagani S, Qazi AK, Jahan R, Gupta S, Patel A, et al. Afatinib radiosensitizes head and neck squamous cell carcinoma cells by targeting cancer stem cells. Oncotarget. (2017) 8:20961–73. doi: 10.18632/oncotarget.15468
172. Stewart JS, Cohen EE, Licitra L, Van Herpen CM, Khorprasert C, Soulieres D, et al. Phase III study of gefitinib compared with intravenous methotrexate for recurrent squamous cell carcinoma of the head and neck [corrected]. J Clin Oncol. (2009) 27:1864–71. doi: 10.1200/JCO.2008.17.0530
173. Herchenhorn D, Dias FL, Viegas CM, Federico MH, Araújo CM, Small I, et al. Phase I/II study of erlotinib combined with cisplatin and radiotherapy in patients with locally advanced squamous cell carcinoma of the head and neck. Int J Radiat Oncol Biol Phys. (2010) 78:696–702. doi: 10.1016/j.ijrobp.2009.08.079
174. Chen D, Wang CY. Targeting cancer stem cells in squamous cell carcinoma. Precis Clin Med. (2019) 2:152–65. doi: 10.1093/pcmedi/pbz016
175. Piha-Paul SA, Munster PN, Hollebecque A, Argilés G, Dajani O, Cheng JD, et al. Results of a phase 1 trial combining ridaforolimus and MK-0752 in patients with advanced solid tumours. Eur J Cancer. (2015) 51:1865–73. doi: 10.1016/j.ejca.2015.06.115
176. Sogutlu F, Kayabasi C, Ozmen Yelken B, Asik A, Gasimli R, Dogan F, et al. The effect of ICRT-3 on Wnt signaling pathway in head and neck cancer. J Cell Biochem. (2019) 120:380–95. doi: 10.1002/jcb.27393
177. Kleszcz R, Szymańska A, Krajka-Kuzniak V, Baer-Dubowska W, Paluszczak J. Inhibition of CBP/β-catenin and porcupine attenuates Wnt signaling and induces apoptosis in head and neck carcinoma cells. Cell Oncol. (2019) 42:505–20. doi: 10.1007/s13402-019-00440-4
178. Zimmerli D, Cecconi V, Valenta T, Hausmann G, Cantù C, Restivo G, et al. WNT ligands control initiation and progression of human papillomavirus-driven squamous cell carcinoma. Oncogene. (2018) 37:3753–62. doi: 10.1038/s41388-018-0244-x
179. Keysar SB, Le PN, Anderson RT, Morton JJ, Bowles DW, Paylor JJ, et al. Hedgehog signaling alters reliance on EGF receptor signaling and mediates anti-EGFR therapeutic resistance in head and neck cancer. Cancer Res. (2013) 73:3381–92. doi: 10.1158/0008-5472.CAN-12-4047
180. Hehlgans S, Booms P, Güllülü Ö, Sader R, Rödel C, Balermpas P, et al. Radiation sensitization of basal cell and head and neck squamous cell carcinoma by the hedgehog pathway inhibitor vismodegib. Int J Mol Sci. (2018) 19:2485. doi: 10.3390/ijms19092485
181. Liebig H, Günther G, Kolb M, Mozet C, Boehm A, Dietz A, et al. Reduced proliferation and colony formation of head and neck squamous cell carcinoma (HNSCC) after dual targeting of EGFR and hedgehog pathways. Cancer Chemother Pharmacol. (2017) 79:411–20. doi: 10.1007/s00280-017-3239-3
182. Bowles DW, Keysar SB, Eagles JR, Wang G, Glogowska MJ, McDermott JD, et al. A pilot study of cetuximab and the hedgehog inhibitor IPI-926 in recurrent/metastatic head and neck squamous cell carcinoma. Oral Oncol. (2016) 53:74–9. doi: 10.1016/j.oraloncology.2015.11.014
183. Colnot DR, Quak JJ, Roos JC, van Lingen A, Wilhelm AJ, van Kamp GJ, et al. Phase I therapy study of 186Re-labeled chimeric monoclonal antibody U36 in patients with squamous cell carcinoma of the head and neck. J Nucl Med. (2000) 41:1999–2010.
184. Börjesson PK, Postema EJ, Roos JC, Colnot DR, Marres HA, van Schie MH, et al. Phase I therapy study with (186)Re-labeled humanized monoclonal antibody BIWA 4 (bivatuzumab) in patients with head and neck squamous cell carcinoma. Clin Cancer Res. (2003) 9(10 Pt. 2):3961–72s.
185. Odenthal J, Rijpkema M, Bos D, Wagena E, Croes H, Grenman R, et al. Targeting CD44v6 for fluorescence-guided surgery in head and neck squamous cell carcinoma. Sci Rep. (2018) 8:10467. doi: 10.1038/s41598-018-28059-9
186. Damek-Poprawa M, Volgina A, Korostoff J, Sollecito TP, Brose MS, O'Malley BW, et alet al. Targeted inhibition of CD133+ cells in oral cancer cell lines. J Dent Res. (2011) 90:638–45. doi: 10.1177/0022034510393511
187. Kim J, Shin JH, Chen CH, Cruz L, Farnebo L, Yang J, et al. Targeting aldehyde dehydrogenase activity in head and neck squamous cell carcinoma with a novel small molecule inhibitor. Oncotarget. (2017) 8:52345–56. doi: 10.18632/oncotarget.17017
188. Huang CE, Yu CC, Hu FW, Chou MY, Tsai LL. Enhanced chemosensitivity by targeting Nanog in head and neck squamous cell carcinomas. Int J Mol Sci. (2014) 15:14935–48. doi: 10.3390/ijms150914935
189. Liang X, Deng M, Zhang C, Ping F, Wang H, Wang Y, et al. Combined class I histone deacetylase and mTORC1/C2 inhibition suppresses the initiation and recurrence of oral squamous cell carcinomas by repressing SOX2. Cancer Lett. (2019) 454:108–19. doi: 10.1016/j.canlet.2019.04.010
190. Chen YC, Chang CJ, Hsu HS, Chen YW, Tai LK, Tseng LM, et al. Inhibition of tumorigenicity and enhancement of radiochemosensitivity in head and neck squamous cell cancer-derived ALDH1-positive cells by knockdown of Bmi-1. Oral Oncol. (2010) 46:158–65. doi: 10.1016/j.oraloncology.2009.11.007
191. Wang Q, Li Z, Wu Y, Huang R, Zhu Y, Zhang W, et al. Pharmacological inhibition of Bmi1 by PTC-209 impaired tumor growth in head neck squamous cell carcinoma. Cancer Cell Int. (2017) 17:107. doi: 10.1186/s12935-017-0481-z
192. Jia L, Zhang W, Wang CY. BMI1 inhibition eliminates residual cancer stem cells after PD1 blockade and activates antitumor immunity to prevent metastasis and relapse. Cell Stem Cell. (2020) 27:238–53.e6. doi: 10.1016/j.stem.2020.06.022
193. Hsieh CE, Lin CY, Lee LY, Yang LY, Wang CC, Wang HM, et al. Adding concurrent chemotherapy to postoperative radiotherapy improves locoregional control but not overall survival in patients with salivary gland adenoid cystic carcinoma-a propensity score matched study. Radiat Oncol. (2016) 11:47. doi: 10.1186/s13014-016-0617-7
194. Mifsud MJ, Tanvetyanon T, McCaffrey JC, Otto KJ, Padhya TA, Kish J, et al. Adjuvant radiotherapy versus concurrent chemoradiotherapy for the management of high-risk salivary gland carcinomas. Head Neck. (2016) 38:1628–33. doi: 10.1002/hed.24484
195. Gebhardt BJ, Ohr JP, Ferris RL, Duvvuri U, Kim S, Johnson JT, et al. Concurrent chemoradiotherapy in the adjuvant treatment of high-risk primary salivary gland malignancies. Am J Clin Oncol. (2018) 41:888–93. doi: 10.1097/COC.0000000000000386
196. Wang X, Luo Y, Li M, Yan H, Sun M, Fan T. Management of salivary gland carcinomas - a review. Oncotarget. (2017) 8:3946–56. doi: 10.18632/oncotarget.13952
197. Lassche G, van Boxtel W, Ligtenberg MJL, van Engen-van Grunsven ACH, van Herpen CML. Advances and challenges in precision medicine in salivary gland cancer. Cancer Treat Rev. (2019) 80:101906. doi: 10.1016/j.ctrv.2019.101906
198. Locati LD, Bossi P, Perrone F, Potepan P, Crippa F, Mariani L, et al. Cetuximab in recurrent and/or metastatic salivary gland carcinomas: a phase II study. Oral Oncol. (2009) 45:574–8. doi: 10.1016/j.oraloncology.2008.07.010
199. Agulnik M, Cohen EW, Cohen RB, Chen EX, Vokes EE, Hotte SJ, et al. Phase II study of lapatinib in recurrent or metastatic epidermal growth factor receptor and/or erbB2 expressing adenoid cystic carcinoma and non adenoid cystic carcinoma malignant tumors of the salivary glands. J Clin Oncol. (2007) 25:3978–84. doi: 10.1200/JCO.2007.11.8612
200. Jakob JA, Kies MS, Glisson BS, Kupferman ME, Liu DD, Lee JJ, et al. Phase II study of gefitinib in patients with advanced salivary gland cancers. Head Neck. (2015) 37:644–9. doi: 10.1002/hed.23647
201. Milanovic D, Jeremic B, Kayser G, Rischke HC, Pfeiffer J, Henke A. Relapsing high grade mucoepidermoid carcinoma. Long-lasting complete response following reirradiation and EGFR blockade. Strahlenther Onkol. (2012) 188:518–22. doi: 10.1007/s00066-012-0096-1
202. Chen W, Cao G, Yuan X, Zhang X, Zhang Q, Zhu Y, et al. Notch-1 knockdown suppresses proliferation, migration and metastasis of salivary adenoid cystic carcinoma cells. J Transl Med. (2015) 13:167. doi: 10.1186/s12967-015-0520-2
203. Even C, Lassen U, Merchan J, Le Tourneau C, Soria JC, Ferte C, et al. Safety and clinical activity of the Notch inhibitor, crenigacestat (LY3039478), in an open-label phase I trial expansion cohort of advanced or metastatic adenoid cystic carcinoma. Invest New Drugs. (2020) 38:402–9. doi: 10.1007/s10637-019-00739-x
204. Ferrarotto R, Eckhardt G, Patnaik A, LoRusso P, Faoro L, Heymach JV, et al. A phase I dose-escalation and dose-expansion study of brontictuzumab in subjects with selected solid tumors. Ann Oncol. (2018) 29:1561–8. doi: 10.1093/annonc/mdy171
205. Nakano T, Warner KA, Oklejas AE, Zhang Z, Rodriguez-Ramirez C, Shuman AG, et al. mTOR inhibition ablates cisplatin-resistant salivary gland cancer stem cells. J Dent Res. (2020) 100:377–86. doi: 10.1177/0022034520965141
206. Hodi FS, Corless CL, Giobbie-Hurder A, Fletcher JA, Zhu M, Marino-Enriquez A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol. (2013) 31:3182–90. doi: 10.1200/JCO.2012.47.7836
207. Xiao J, Egger ME, McMasters KM, Hao H. Differential expression of ABCB5 in BRAF inhibitor-resistant melanoma cell lines. BMC Cancer. (2018) 18:675. doi: 10.1186/s12885-018-4583-3
208. Sarvi S, Crispin R, Lu Y, Zeng L, Hurley TD, Houston DR, et al. ALDH1 bio-activates nifuroxazide to eradicate ALDH(High) melanoma-initiating cells. Cell Chem Biol. (2018) 25:1456–69.e6. doi: 10.1016/j.chembiol.2018.09.005
209. Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. (2014) 371:1877–88. doi: 10.1056/NEJMoa1406037
210. Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. (2014) 371:1867–76. doi: 10.1056/NEJMoa1408868
Keywords: cancer stem cells, head and neck, squamous cell carcinoma, self-renewal, tumor relapse, targeted therapy
Citation: Shahoumi LA (2021) Oral Cancer Stem Cells: Therapeutic Implications and Challenges. Front. Oral. Health 2:685236. doi: 10.3389/froh.2021.685236
Received: 24 March 2021; Accepted: 25 June 2021;
Published: 21 July 2021.
Edited by:
Ahmad Waseem, Queen Mary University of London, United KingdomReviewed by:
Ian Mackenzie, Queen Mary University of London, United KingdomCopyright © 2021 Shahoumi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linah A. Shahoumi, bHNoYWhvdW1pQGF1Z3VzdGEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.