 Michael Landowski
Michael Landowski Purnima Gogoi
Purnima Gogoi Sakae Ikeda
Sakae Ikeda Akihiro Ikeda
Akihiro Ikeda- 1Department of Medical Genetics, University of Wisconsin-Madison, Madison, WI, United States
- 2McPherson Eye Research Institute, University of Wisconsin-Madison, Madison, WI, United States
Aging is the most significant risk factor for age-related diseases in general, which is true for age-related diseases in the eye including age-related macular degeneration (AMD). Therefore, in order to identify potential therapeutic targets for these diseases, it is crucial to understand the normal aging process and how its mis-regulation could cause age-related diseases at the molecular level. Recently, abnormal lipid metabolism has emerged as one major aspect of age-related symptoms in the retina. Animal models provide excellent means to identify and study factors that regulate lipid metabolism in relation to age-related symptoms. Central to this review is the role of transmembrane protein 135 (TMEM135) in the retina. TMEM135 was identified through the characterization of a mutant mouse strain exhibiting accelerated retinal aging and positional cloning of the responsible mutation within the gene, indicating the crucial role of TMEM135 in regulating the normal aging process in the retina. Over the past decade, the molecular functions of TMEM135 have been explored in various models and tissues, providing insights into the regulation of metabolism, particularly lipid metabolism, through its action in multiple organelles. Studies indicated that TMEM135 is a significant regulator of peroxisomes, mitochondria, and their interaction. Here, we provide an overview of the molecular functions of TMEM135 which is crucial for regulating mitochondria, peroxisomes, and lipids. The review also discusses the age-dependent phenotypes in mice with TMEM135 perturbations, emphasizing the importance of a balanced TMEM135 function for the health of the retina and other tissues including the heart, liver, and adipose tissue. Finally, we explore the potential roles of TMEM135 in human age-related retinal diseases, connecting its functions to the pathobiology of AMD.
1 Introduction
The retina is particularly sensitive to the effects of aging (1), allowing researchers an in vivo system to discover critical genes and pathways important in mitigating the aging process using model organisms. As wild-type mice age, it is common to observe decreases in visual function (2, 3), presence of sub-retinal pigmented epithelium (RPE) deposits (4), appearances of RPE multinucleation (5), formation of cataracts (6), development of ectopic synapses (7) and signs of neuroinflammation (7, 8) in their retinas. However, the pathways responsible for the generation of these age-dependent retinal pathologies is unknown.
Mouse genetic methodologies have been instrumental to our understanding of the molecular underpinnings of the retinal aging process. A noteworthy example of harnessing mouse forward genetics to identify a critical gene involved in retinal aging comes from the study of the FUN025 mice. The FUN025 mice originated from a N-ethyl-N-nitrosourea (ENU) mutagenesis screen and were identified as a strain showing progressive age-dependent retinal pathologies including photoreceptor cell degeneration, ectopic synapse formation, and increased retinal stress with an earlier onset (as early as two months of age) and faster rate compared to wild-type C57BL/6J mice (9–11). The progressive retinal pathologies in FUN025 mice differ from rapid retinal degeneration observed in mice with mutations in phosphodiesterase 6B (Pde6b) (12), rhodopsin (Rho) (13), and other genes linked with inherited retinal diseases in humans that is complete by two to three weeks of age (14). Importantly, the unique spatial pattern of progression for age-dependent retinal pathologies in wild-type C57BL/6J mice from the peripheral to the central retina (7) is maintained in the retina of FUN025 mice albeit with earlier onset and faster progression (11), indicating that the retinal aging process is accelerated in FUN025 mice.
Identification of the gene responsible for the age-dependent retinal phenotypes in the FUN025 strain can lead to new insights into pathways contributing to retinal aging. Positional cloning of the FUN025 line revealed a point mutation in the donor splice site of exon 12 of the transmembrane protein 135 (Tmem135) gene (11). Overexpression of Tmem135 can prolong longevity in nematodes when exposed to cold temperatures (15) but no previous studies have correlated Tmem135 with aging in mammals. The discovery of Tmem135 as a gene implicated in retinal aging of mammals lead to subsequent studies on the function of Tmem135 in cells and mice as well as associations between the pathways affected by Tmem135 and age-related retinal diseases such as age-related macular degeneration (AMD). Intriguing similarities have been observed between retinal abnormalities in Tmem135FUN025 mutant mice and retinal pathologies in AMD patients (11), as well as between gene expression profiles of the Tmem135FUN025 mutant eyecups and RPE/choroid samples from multiple stages of AMD (16).
In this review, we summarize the current literature on Tmem135. We provide an overview of the molecular functions of TMEM135 that are critical for the regulation of mitochondria, peroxisomes, and lipids. We also describe the age-dependent phenotypes of mice with perturbations in Tmem135, highlighting the concept that proper balance of Tmem135 function is vital for the health of the retina and other tissues such as the heart, liver, and adipose tissue. Lastly, we postulate about the possible roles of TMEM135 in human age-related retinal diseases by relating the roles of TMEM135 to the pathobiology of AMD.
2 Molecular functions and roles of TMEM135
Tmem135, also known as peroxisomal membrane protein 52 (Pmp52), encodes a 52 kilodalton protein with five transmembrane domains that is highly conserved across multiple species (15). Protein domains of TMEM135 also share similarities with members of the Tim17 protein family, which are central components of translocases of the mitochondrial inner membrane that are important for mitochondrial biogenesis (17). Based on this information, it is unsurprising that the TMEM135 protein shows colocalization with both mitochondria (11, 15) and peroxisomes (18–23). TMEM135 can be also found on lipid droplets, but this localization may be contingent on cellular stress such as microbial infections (24) and cold exposure (15). Further evidence indicates that TMEM135 translocates from peroxisomes to mitochondria (25), suggesting that TMEM135 is involved in functional interaction between mitochondria and peroxisomes (26). Here, we will summarize studies on the molecular roles of TMEM135, which suggest that TMEM135 is likely a multi-functional protein involved in the regulation of mitochondria and peroxisomes.
2.1 TMEM135 is a mitochondrial fission regulator
Earlier work indicates an important role of TMEM135 in “mitochondrial dynamics” (11), which is the collective term for biogenesis, fusion, fission, and mitophagy events required to preserve mitochondrial integrity within cells during times of cellular and nutritional stress (27). Fibroblasts from mice with the Tmem135FUN025 mutation that causes the loss of TMEM135 function had overly elongated mitochondrial networks that manifested in decreased number and increased size of mitochondria (11), while fibroblasts with overexpression of wild-type Tmem135 had fragmented mitochondria that were more abundant and smaller than wild-type controls (11). Colocalization between TMEM135 and a mitochondrial fission factor, dynamin-related protein 1 (DRP1), was observed at sites of mitochondrial fission (11), suggesting that TMEM135 may regulate mitochondrial fission through interaction with DRP1.
A recent study further elucidated the molecular function of TMEM135 as a regulator of DRP1, and thus, mitochondrial fission and its importance in the interaction between peroxisomes and mitochondria (25). Hu et al. observed that mitochondria appear overly fused in brown adipose tissue cultured from mice with the adipose tissue specific peroxisome deficiency, leading to impaired thermogenesis (25). Proteomic analysis of the mitochondria isolated from the peroxisome deficient brown adipocytes after cold exposure revealed TMEM135 as the most decreased protein (25), suggesting its involvement in the peroxisomal regulation of mitochondrial fission. The absence of TMEM135 on the mitochondria after cold exposure of the peroxisome deficient brown adipocytes indicated the prerequisite of TMEM135 to translocate from peroxisomes to mitochondrial membranes for the initiation of mitochondrial fission (Figure 1) (25). Investigation of the DRP1 phosphorylation state indicated that TMEM135 promotes protein kinase A (PKA)-dependent phosphorylation of DRP1 and its recruitment to mitochondria (25), defining the mechanism through which TMEM135 promotes mitochondrial fission. It was also shown that the translocation of TMEM135 from peroxisomes to mitochondria depends on plasmalogens (25), a class of glycerophospholipids containing a vinyl-ether and ester bond that are dependent on peroxisomes for their production (28). These findings add to the growing substantiation of an intimate relationship between peroxisomes and mitochondria that is needed for proper mitochondrial dynamics and homeostasis (29–33).
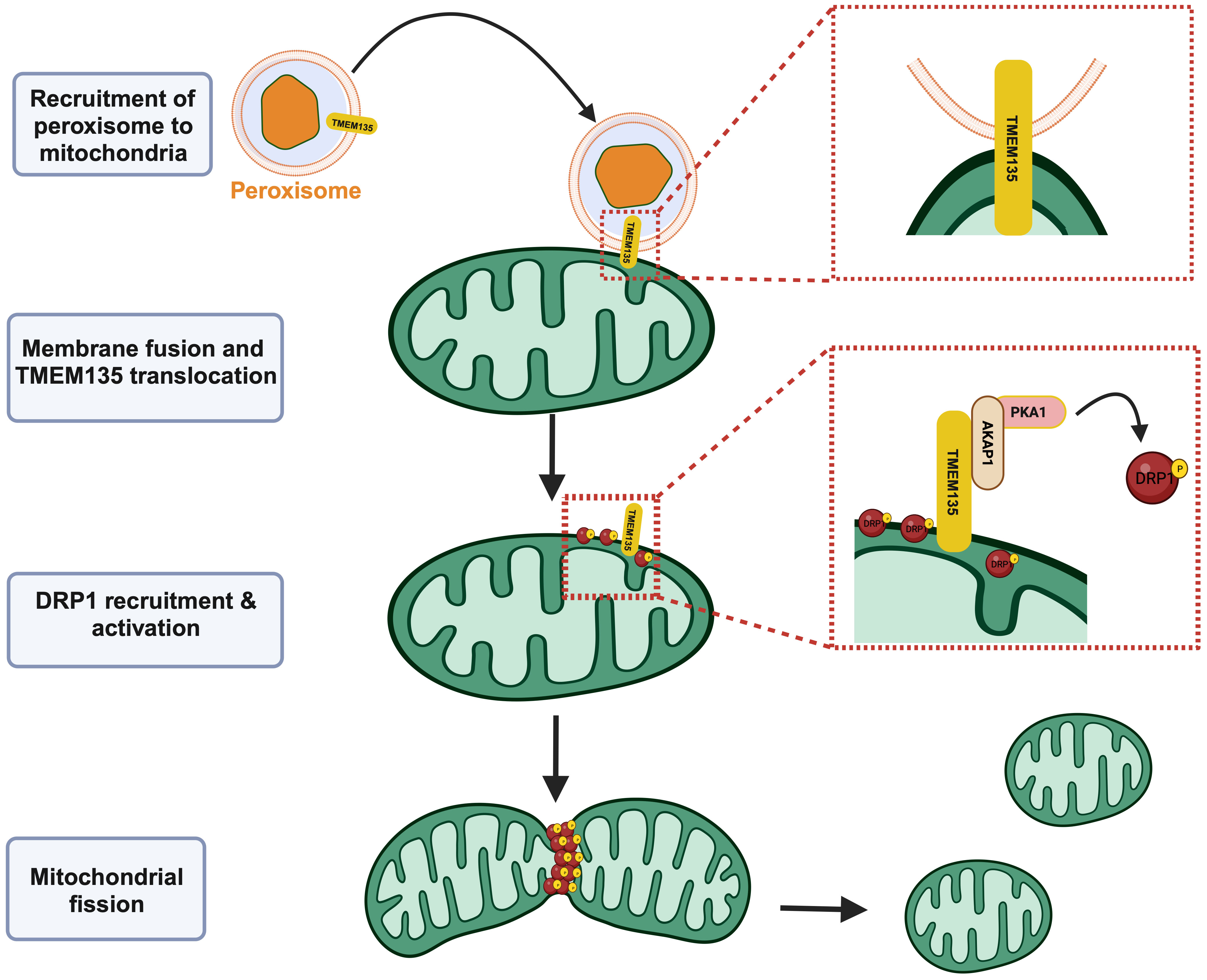
Figure 1 TMEM135 acts as a mitochondrial fission factor. Peroxisomes can interact with mitochondria, leading to the translocation of TMEM135 from the peroxisomal membrane to the mitochondrial outer membrane. The movement of TMEM135 from peroxisomes to mitochondria precipitates the recruitment of DRP1 and other proteins including AKAP1 and PKA1 that eventually leads to the activation of DRP1. Activated DRP1 causes fission of mitochondria. Image created using Biorender.com. TMEM135, transmembrane protein 135. DRP1, dynamin-related protein 1. AKAP, A-kinase anchor protein 1. PKA1, protein kinase A 1. [Figure adapted from Hu et al. (25)].
2.2 TMEM135 is an exporter of DHA from peroxisomes
A role of TMEM135 in lipid homeostasis was first indicated by transcriptomic profiling of retinal tissues isolated from mice with the Tmem135FUN025 mutation. The retinal phenotypes of Tmem135FUN025/FUN025 mutant mice correlated with increased expression of genes involved in fatty acid metabolism, cholesterol metabolism, and steroid metabolic processes (16), suggesting that the function of TMEM135 is important for the regulation of lipid synthesis. In support of this notion, age-dependent progression of neutral lipid and cholesterol accumulation was observed in the eyecups of Tmem135FUN025/FUN025 mutant mice (16).
The relationship between TMEM135 and lipid metabolism was further defined through a high-throughput and semi-quantitative lipidomics analysis of Tmem135FUN025/FUN025 mutant tissues. Untargeted profiling of intact lipid species using liquid chromatography with tandem mass spectrometry (LC-MS/MS) in the livers, retinas, hearts, and plasmas of Tmem135FUN025/FUN025 mutant mice showed that each tissue had robust decreases in lipids containing Docosahexaenoic acid (DHA or C22:6n3) compared to wild-type control mice (34). Since all lipid classes that are known to harbor DHA were affected by the Tmem135FUN025 mutation, which was confirmed by gas chromatography mass spectrometry (GC-MS) (34), it became apparent that TMEM135 has a major task in cellular DHA homeostasis.
DHA is an omega-3 polyunsaturated fatty acid (PUFA) important for neuronal development and function as well as an important mediator of inflammation and disease (35). The concentration of DHA within tissues results from the contribution of this omega-3 PUFA from dietary sources and endogenous production within cells (36). Since the diet consumed by Tmem135FUN025/FUN025 mutant mice did not contain DHA (34), it must originate from the endogenous production from the ‘Sprecher pathway’ of DHA synthesis that takes place in the ER and completes in peroxisomes in these animals (37). The ER possesses desaturases [fatty acid desaturase 1 (Fads1) and 2 (Fads2)] and elongases [elongation of very long chain fatty acids-like 2 (Elovl2) and 5 (Elovl5)] needed for the desaturation and elongation of dietary essential fatty acid 18:3n3 to generate C24:6n3 (38). Then, C24:6n3 is imported into peroxisomes for retroconversion to C22:6n3 by their beta-oxidation enzymes (39). The ER and peroxisomal components of the ‘Sprecher pathway’ were evaluated in the livers of Tmem135FUN025/FUN025 mutant mice to determine the molecular basis of the diminished DHA concentrations due to the Tmem135FUN025 mutation (34). Remarkably, there were no decreases in any of the components, and rather there were increases in the peroxisomal beta-oxidation enzymes required to produce DHA (34) indicating that reduced DHA in Tmem135FUN025/FUN025 mutant mice does not result from a defect in the ‘Sprecher pathway’ of DHA synthesis. The remaining step where TMEM135 may play a role in the generation of DHA within cells is the export of DHA from peroxisomes (Figure 2). While the exact molecular mechanism is unknown, it is thought that there is a protein on peroxisomes capable of exporting DHA from these organelles (40). The results of the lipid and pathway investigation of Tmem135FUN025/FUN025 mutant mice strongly suggested that TMEM135 has a critical function in exporting DHA from peroxisomes to deliver DHA to the ER for esterification into lipids (Figure 2) (34). This is consistent with the postulated function of TMEM135 involving the transport of metabolites between organelles (18, 41).
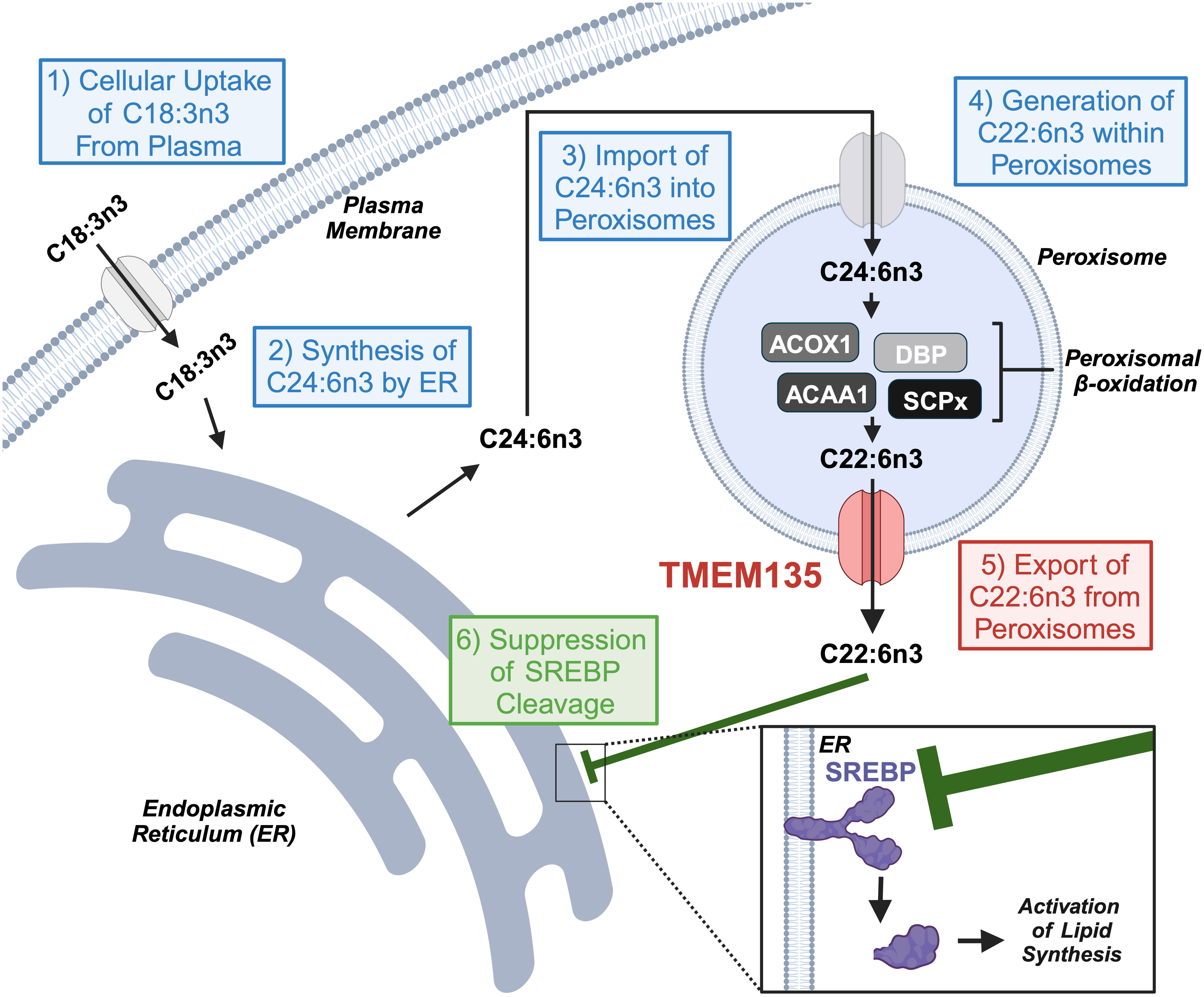
Figure 2 TMEM135 exports docosahexaenoic acid (DHA, C22:6) from peroxisomes to regulate cellular lipid synthesis. Cells utilize the ‘Sprecher pathway’ to synthesize DHA. (1) Cells uptake C18:3n3 from the blood and send it to the endoplasmic reticulum (ER). (2) The ER transforms C18:3n3 to C24:6n3 through a series of elongation and desaturation steps. (3) C24:6n3 migrates to the peroxisome for its import into its matrix. (4) Peroxisomal beta-oxidation enzymes (ACOX1, DBP, ACAA1, and SCPx) digest C24:6n3 to C22:6n3. (5) TMEM135 transports C22:6n3 from the peroxisomal matrix to the cytoplasm for its use by cells. (6) C22:6n3 accumulates within the ER to prevent the cleavage of the transcription factor SREBP and its activation of genes involved in lipid synthesis. Image created using Biorender.com. ACOX1, acyl-CoA oxidase 1. DBP, D-bifunctional protein. ACAA1, acetyl-coenzyme A acyltransferase 1. SCPx, sterol carrier protein x. SREBP, sterol response element binding protein.
2.3 TMEM135 influences peroxisome proliferation
Changes caused by Tmem135 perturbations affect the number of peroxisomes, organelles that have chief responsibilities connected with cellular metabolism through its interactions with mitochondria, lipid droplets, lysosomes, and ER (42). In cultured fibroblasts with the Tmem135FUN025 mutation, there was an increase of peroxisomes, while fibroblasts overexpressing Tmem135 showed reductions in peroxisomal number (34). It is known that peroxisome proliferation is in part mediated by the actions of the peroxisome proliferator activated receptor (PPAR) family of transcription factors (43). Peroxisomes and their protein contents were decreased in the livers of Tmem135FUN025/FUN025 mutant mice upon genetic ablation of PPAR alpha (Ppara) (34), indicating that activation of PPARa signaling is involved in increasing peroxisome proliferation in these mice. While it is unclear what drives the changes in PPAR signaling due to the changes in TMEM135 function, it is possible that impaired DHA export from peroxisomes results in the generation of peroxisome-derived metabolites that interact with PPARs such as ether phosphatidylethanolamines (EtherPEs) known to activate the PPAR signaling (44) which is increased in Tmem135FUN025/FUN025 mutant mice (34). More work is required to discern the molecular mechanism underlying the peroxisome proliferation changes observed in Tmem135 mutant and overexpressing cells, and its relationship with the TMEM135 molecular function.
2.4 TMEM135 is a mediator of intracellular cholesterol trafficking
TMEM135 has been implicated to have a role in the distribution of intracellular cholesterol by two different studies (45, 46). First, Tmem135 was identified in a shRNA screen for genes involved in trafficking of cholesterol from low-density lipoprotein (LDL) to the plasma membrane of HeLa cells (45). TMEM135 was further validated as a protein involved in intracellular cholesterol trafficking by knocking it down in HeLa cells, which resulted in fewer contacts between lysosomes and peroxisomes as well as decreased cholesterol in the plasma membrane (45). These results suggested that lysosome-peroxisome trafficking of cholesterol mediated by contacts between these organelles is impaired in Tmem135 knockdown cells. This result was further confirmed in another study using RPE1 cells, an immortalized RPE cell line often utilized in cilia-focused research (46). The authors observed fewer lysosome-peroxisome contacts (46) and an increased accumulation of cholesterol in the lysosomes of cells with decreased Tmem135 expression (18). After treatment with LDL, the knockdown of Tmem135 expression impaired the ability of cholesterol from LDL particles to reach the ER. Accumulation of cholesterol was observed in the eyecups of Tmem135FUN025/FUN025 mutant mice (16), which may occur due to defective cholesterol transport in these mice. Interestingly, accumulation of cholesterol in Tmem135FUN025/FUN025 mutant eyecups coincided with upregulation of sterol regulatory element binding transcription factor 2 (SREBP2)-targeted genes that are involved in cholesterol metabolism including hydroxymethylglutaryl-CoA synthase (Hmgcs1), sterol O-acyltransferase 1 (Soat1), ATP-binding cassette subfamily A member 1 (Abca1), and ATP-binding cassette subfamily A member 1 (Abcg1) (16). It is worth exploring whether these biochemical and expression manifestations result from defective lysosome-peroxisome interactions in Tmem135FUN025/FUN025 mutant mice. Moreover, based on the TMEM135 function in DHA export (34), it would be interesting to investigate whether DHA-esterified lipids influence membrane fluidity and interactions of these organelles.
3 Tissue-specific roles of TMEM135 and its relevance to human disease
Insight into the significance of TMEM135 on metabolic functions of tissues can be gleaned by the age-related phenotypes in mice with modifications of Tmem135 function. Remarkably, all tissues from mice with a homozygous mutation in Tmem135 (Tmem135FUN025/FUN025 mutant) or overexpression of Tmem135 (Tmem135 TG) show opposing differences in their mitochondrial shape (Figure 3) and number of peroxisomes (11, 34, 47–49). However, there are specific tissues that are more sensitive to the Tmem135 mutation or overexpression (47, 50). There were also tissue-specific lipid adaptations (34). These findings indicate there is a tissue-specific reliance for TMEM135 on sustaining homeostasis through aging. Here, we will summarize the phenotypes of the Tmem135FUN025/FUN025 mutant and Tmem135 TG mice (Table 1) and the potential relevance of TMEM135 in human diseases associated with those phenotypes.
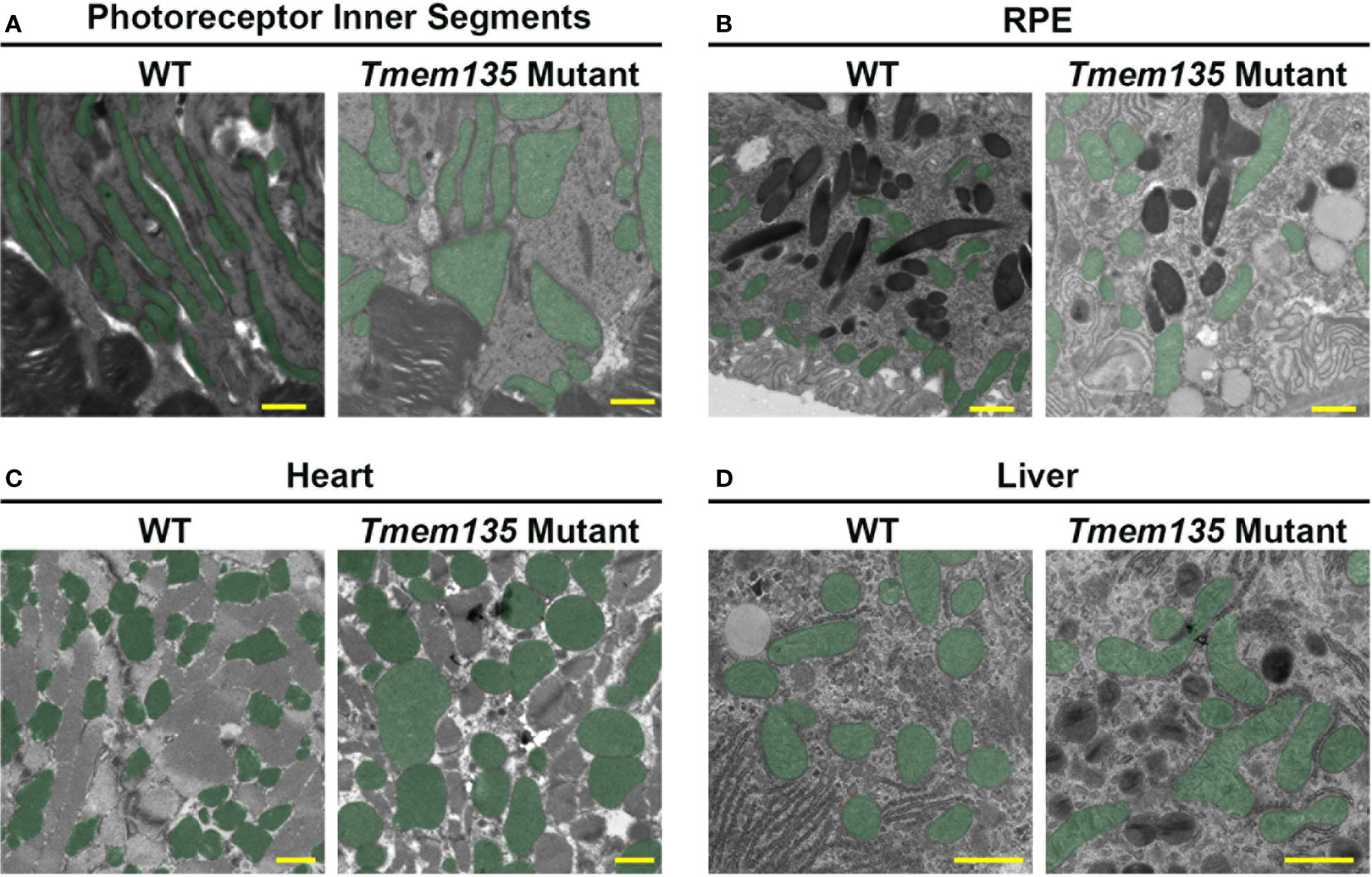
Figure 3 Mitochondrial shape in Tmem135 mutant tissues. Representative electron micrographs of mitochondria in the photoreceptor inner segments (A), retinal pigmented epithelium (RPE) (B), heart (C), and liver (D) of wild-type C57BL/6J (WT) and Tmem135 mutant mice. Mitochondria are labeled in green. Note the enlarged mitochondria in all Tmem135 mutant tissues relative to WT tissues. The magnifications for the photoreceptor inner segment and RPE micrographs are 8800X, heart micrographs are 7100X, and liver micrographs are 11500X. The scale bar in all micrographs represents 1 micron.
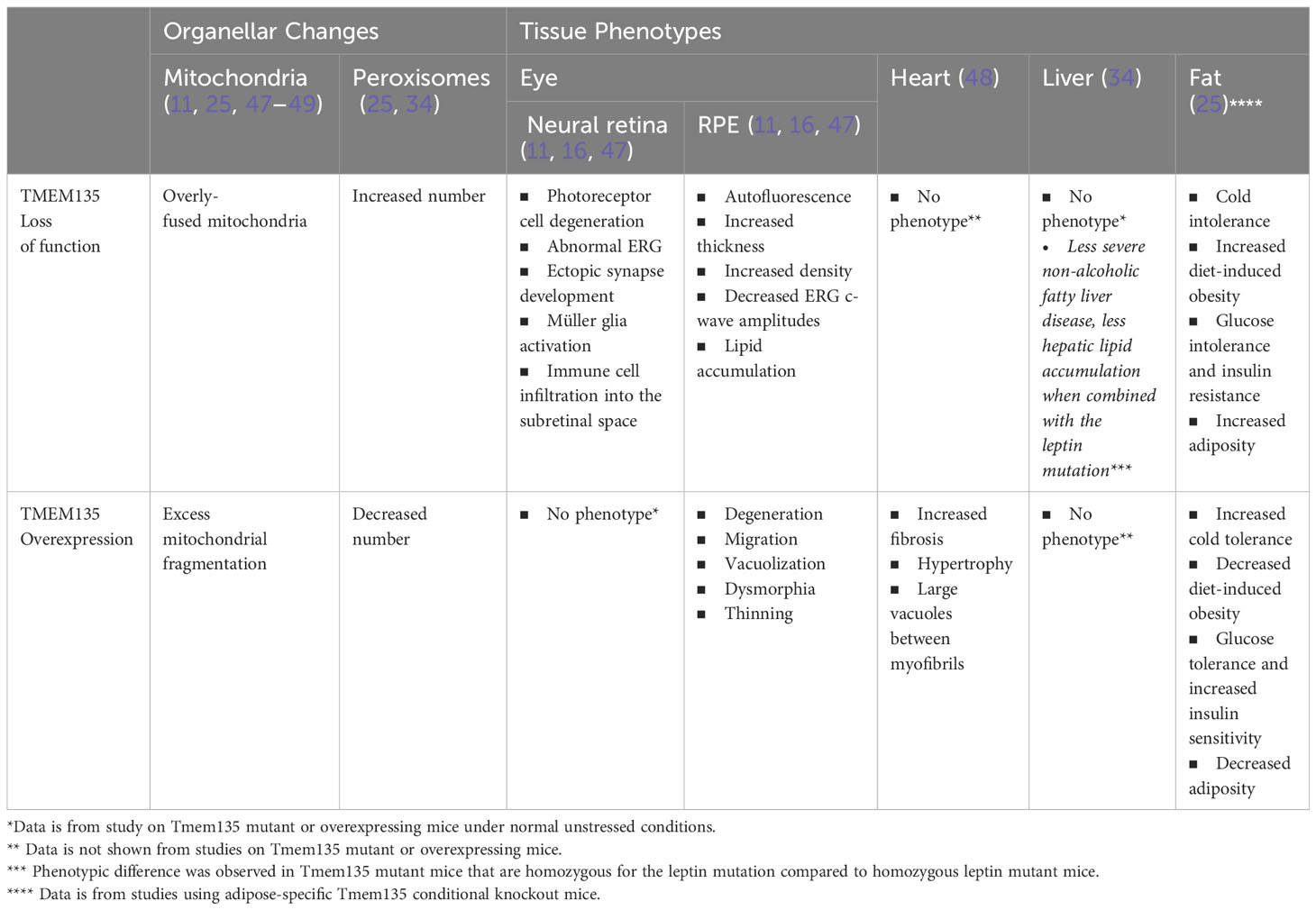
Table 1 Summary of features associated with TMEM135 loss of function and overexpression.
3.1 Ocular phenotypes of Tmem135 mutant mice
Tmem135FUN025/FUN025 mutant mice develop an age-dependent photoreceptor cell degeneration (11, 16), which coincided with visual loss, ectopic synapse development, and neuroinflammation consisting of Müller glia activation and immune cell infiltration into the subretinal space (11, 16). Tmem135FUN025/FUN025 mutant mice also showed changes in the RPE such as autofluorescence, increased thickness, increased density, decreased electroretinogram c-wave amplitudes, and lipid accumulation (11, 16, 47) (Table 1).
It is possible that the retinal phenotypes of the Tmem135FUN025/FUN025 mutant mice may be occurring due to their deficiency of DHA (34). DHA has an important role in membrane fluidity of rod photoreceptor outer segments that is required for phototransduction (51). There are reports of other mouse models with retinal DHA deficiency including elongation of very-long-chain fatty acids-like 2 (Elovl2) mutant (52), acyl-CoA synthetase 6 (Ascl6) knockout (53), major facilitator superfamily domain containing 2A (Mfsd2a) knockout (54, 55), and adiponectin receptor 1 (Adipor1) knockout mice (56, 57) that show similar retinal pathologies to those observed in Tmem135FUN025/FUN025 mutant mice (11, 16, 47).
It is also possible that the retinal phenotypes of Tmem135FUN025/FUN025 mutant mice result from enhancement of mitochondrial fusion triggered by the Tmem135 mutation (11). Boosting mitochondrial fusion in the retinas of Tmem135FUN025/FUN025 mutant mice may lead to increased nutrient intake and metabolic stress as detected in other tissues caused by excessive mitochondrial fusion (58, 59). Supporting this idea, a NMR-based metabolomics study revealed an accumulation of metabolites from glucose, amino acid and lipid metabolic pathways in primary-cultured Tmem135FUN025/FUN025 mutant RPE cells compared to wild-type RPE cells (50). In addition, mice with retinal metabolic stress such as RPE-specific vascular endothelial growth factor A (Vegfa) or superoxide dismutase 2 (Sod2) have thicker RPE like the Tmem135FUN025/FUN025 mutant mice (11, 47). Future studies will need to be undertaken to determine the roles of the reduced DHA and overly fused mitochondria in Tmem135FUN025/FUN025 mutant mice in regard to the development of their retinal pathologies.
3.2 Ocular phenotypes of Tmem135 TG mice
In contrast to Tmem135FUN025/FUN025 mutant mice, Tmem135 TG mice exhibit progressive RPE degenerative phenotypes including migration, vacuolization, dysmorphia, and thinning (47). Additionally, Tmem135 TG mice displayed thinner myelin sheaths of axons in the optic nerve (49). However, unlike Tmem135FUN025/FUN025 mutant mice, there were no signs of photoreceptor cell dysfunction or degeneration in Tmem135 TG mice at least until one year of age (47).
The retinal phenotypes of Tmem135 TG mice may result from their excess mitochondrial fragmentation (11, 47) and/or decreased peroxisome proliferation (34). It is believed that mitochondrial fission is important for the removal of damaged mitochondrial membranes in order to maintain mitochondrial function (58, 60). However, excessive mitochondrial fragmentation initiated by Tmem135 overexpression could cause mitochondrial dysfunction in the RPE and lead to degeneration of this cell type. Similar to Tmem135 TG mice, mice with RPE-specific ablation of transcription factor A, mitochondrial (Tfam) or PPARG coactivator 1 alpha (Pgc-1α) show attenuated mitochondrial function and degenerated RPE (61, 62). It is plausible that mitochondrial fragmentation in Tmem135 TG could stem from their decreased peroxisomal proliferation (34). Inhibition of proper peroxisome biogenesis by eliminating peroxisomal biogenesis factor 3 (Pex3) or peroxisomal biogenesis factor 5 (Pex5) expression promoted mitochondrial fragmentation in mouse embryonic fibroblasts (33). Recent work has shown that genetic ablation of the multifunctional protein 2 (Mfp2; also known as hydroxysteroid (17-beta) dehydrogenase 4 or Hsd17b4) gene encoding D-bifunctional protein (DBP), which is a critical enzyme required for peroxisomal beta-oxidation (34, 63), specifically in RPE cells can lead to RPE degenerative changes (64). Interestingly, expression of Mfp2 is decreased in the eyecups of Tmem135 TG mice (34). Dissecting out the roles for mitochondrial fragmentation and decreased peroxisomal proliferation will be critical to determine the contributions of these organelles to the RPE degeneration observed in Tmem135 TG mice.
3.3 Potential connection between TMEM135 and AMD
Studies from cell culture and animal experiments signal a substantial role of TMEM135 in energy homeostasis in aging. To date, there has been no direct association between TMEM135 and age-related retinal diseases including AMD. However, there are multiple levels of similarities between Tmem135 mouse models and AMD including ocular pathologies and molecular and cellular changes (11, 16). Here, we will discuss potential involvement of TMEM135 in AMD pathogenesis.
3.3.1 Mitochondrial changes in AMD
Dysfunction of mitochondria is an important pathobiological event in AMD (65–67). In the retina, there are numerous mitochondria within the photoreceptor and RPE cells (68). Mitochondria provide these retinal cells a constant supply of energy required for facilitating phototransduction and sequestering of reactive oxygen species from photons of light and other oxidative stresses (69). It is well established that aging disrupts mitochondrial homeostasis, which may predispose the retina to AMD (70). In particular, the RPE is thought to be the first tissue affected by AMD (71). Surveys of the mitochondria in the RPE of AMD-afflicted eyes uncovered robust decreases in their number and size (72). The changes in mitochondrial shape and number in the RPE of AMD donor retinas correlated with decreased mitochondrial proteins (73), increased mitochondrial DNA damage (74–76), and increased mitochondrial oxidative stress (77). To evaluate the functional consequences of these changes, RPE from human AMD donor eyes were cultured and assessed using the Seahorse Extracellular Flux Analyzer (78). These RPE cultures displayed a reduction in their glycolytic function (78) that has been validated by another group using a different method (79). The accrual of this work suggests targeting mitochondria is a viable treatment strategy for AMD as proposed by many groups in the AMD research field (80–91). It is of note that elamipretide, which targets mitochondria, recently failed in a phase 2 trial of geographic atrophy (92). However, this may still be a viable therapeutic for intermediate AMD (93), suggesting that targeting mitochondria at earlier stages of disease development may be more efficacious to treat early/intermediate AMD.
The deviations of normal mitochondria in the RPE of donor retinas hint at possible disruptions of mitochondrial dynamics in AMD. The proteins involved in mitochondrial fusion, fission, and autophagy were quantified in RPE cultures from AMD and control RPE cultures (94). Interestingly, upon treatment with the mitochondrial uncoupler FCCP, there was a disease-specific response in the RPE cultures from the AMD donor eyes including an increase of mitochondrial fission factor (MFF) (94). MFF is a protein necessary for mitochondrial fission (95), and its amplified expression correlated with the mitochondrial fragmentation typically observed in the RPE from AMD donor eyes. Since TMEM135 is a mitochondrial fission factor (11, 25, 47, 49), it may be involved in mitochondrial fragmentation in the RPE of AMD patients as well. Future work on the origins of mitochondrial fragmentation in AMD-diagnosed retinas is essential since the pharmacological inhibition of mitochondrial fission is thought to be a therapeutic target for non-exudative AMD (96).
3.3.2 Decreased DHA-containing lipids in AMD
The mammalian retina, notably the rod photoreceptor outer segments, contains the highest density of DHA than any other tissue in the body, which is important for membrane fluidity of the outer segments (97). As previously discussed, DHA can originate from dietary sources or endogenous synthesis through the ‘Sprecher pathway.’ Dietary intake of DHA has been related to decreased risk for AMD, but these findings have not been well replicated as commented in other excellent reviews (98–100). This could be due in part to the preferential uptake of DHA in different forms such as triglycerides, phosphatidylcholine, or lysophosphatidylcholine by the retina (101). Recent work suggested an important contribution of rod photoreceptor-derived DHA in AMD (102). They showed through LC-MS/MS and MALDI-molecular imaging that there was a decrease of DHA-containing phosphatidylcholines in the peripheral retinas of AMD patients (102).
Transcriptomic analysis of donor retinas also supports the claim that there is less DHA in AMD-afflicted eyes. Integrated microarray and RNA-Seq datasets (GSE29801 and GSE135092) of RPE/choroid samples from AMD patients (16, 103–105) showed increased sterol regulatory element binding transcription factor 1 (SREBP1), a transcription factor required for the synthesis of fatty acids and cholesterol, and its target genes fatty acid synthase (FASN), fatty acid desaturase 1 (FADS1), and FADS2 (16). Recent work showed that decreased DHA enhances the transcription of SREBP1 target genes (106) (Figure 2), suggesting that reduced DHA could cause increased SREBP signaling in AMD. Intriguingly, reduced DHA as well as increased expression of Srebp1 and its target genes were also observed in Tmem135FUN025/FUN025 mutant eyecup samples (16). Common molecular and pathological features between Tmem135FUN025/FUN025 mutant mice and AMD patients suggest that the role of TMEM135 in peroxisomal export of DHA within retinal cells may be important in mitigating dysregulated lipid synthesis in AMD.
3.3.3 Altered cholesterol metabolism in AMD
Cholesterol metabolism in AMD has been well investigated because there are large accumulations of esterified cholesterol within drusen, the pathological hallmark of AMD (107), and strong associations of AMD risk with genes involved in cholesterol transport (108–114). Disruptions to normal cholesterol homeostasis in the retina is thought to contribute to the onset of drusen in the sub-RPE space of the human retina (115). An understanding into retinal cholesterol metabolism comes from inquiries on the pathobiological nature of the retinal phenotype of ATP-binding cassette, subfamily A, member 4 (Abca4) knockout mice, which lead to discoveries on a vital role of dysregulated cholesterol trafficking as an important pathobiological event in the development of AMD-like pathologies in this model (116). Abca4 knockout mice are characterized by the accumulation of A2E, a major lipofuscin fluorophore, that coincides with delayed dark adaptation (117, 118). A2E can displace cholesterol from the plasma membrane of RPE cells, accumulate cholesterol within RPE cells, and impede the ability of cholesterol efflux from these cells (119). Furthermore, cholesterol accumulation can induce ceramide production in the RPE and allow for complement-mediated damage on the RPE plasma membrane in Abca4 knockout mice (120). Comparably, mice with the loss of Niemann-Pick Type C disease (NPC) intracellular cholesterol transporter 1 (NPC1), that lose the ability to transfer cholesterol from lysosomes to the cell, have impaired visual function and lipofuscin aggregation at 2 months of age (121). Given that TMEM135 has been shown to play a role in the intracellular trafficking of cholesterol between lysosomes and peroxisomes (45, 46), this function of TMEM135 may be important in sustaining cholesterol metabolism within the retina and preventing the formation of esterified cholesterol-enriched drusen in the sub-RPE space.
3.4 Effects of TMEM135 modulation in other tissues
There have been published associations with TMEM135 and other human medical conditions such as osteoporosis (122–127), breast cancer (128, 129), prostate cancer (130, 131), melanoma (132, 133), non-small lung cancer (134), glioblastoma multiforme (134), non-alcoholic fatty liver disease (135), cognitive disorders (136), and metabolic disease (25). Significance of TMEM135 functions have been also indicated by phenotypes in other tissues due to Tmem135 mutation and overexpression. While readers are encouraged to refer to individual studies for details, we will summarize the phenotypes in other mouse tissues caused by modulation of Tmem135.
Overexpression of Tmem135 impacts the heart along with the RPE (48). The hearts of Tmem135 TG mice on a mixed C57BL/6J and FVB/NJ background show hypertrophy with increased fibrosis (48). Ultrastructural abnormalities such as large vacuoles co-occupying the space between myofibrils with mitochondria were observed in Tmem135 TG hearts at varying severities (48). Cardiac phenotypes of Tmem135 TG mice most likely derive from their mitochondrial fragmentation. Other mouse models with heart-specific conditional ablation of mitochondrial fusion display cardiac phenotypes comprising dilated cardiomyopathy and cardiac hypertrophy (137, 138). It remains unknown if mitochondrial fragmentation in Tmem135 TG originates from mitochondria or peroxisomes as TMEM135 can translocate from peroxisomes to mitochondria for fission events (11, 25, 34).
While livers of Tmem135FUN025/FUN025 mutant mice appear and function normally regardless of the remarkable changes in their hepatic peroxisomes and lipidome (34), physiological significance of these cellular changes could be observed when the Tmem135FUN025 mutation was combined with the leptin mutation (Lepob), which causes metabolic disease with significant hepatic lipid adjustments and dependency on functional peroxisomes in mice (139–141). Both male and female mice that are homozygous for Tmem135 and leptin mutations (Tmem135FUN025/FUN025/Lepob/ob) had lower body, liver, and gonadal fat pad weights compared to their Lepob/ob counterparts (34). The Tmem135FUN025 mutation also decreased the amount of plasma cholesterol by impairing the secretion of very low-density lipoprotein (VLDL) and LDL (142). Modifications of the classic obesity and dyslipidemia phenotype in Lepob/ob mice by the Tmem135FUN025 mutation correlated with attenuation of their non-alcoholic fatty liver disease (NAFLD) phenotypes. There were less severe NAFLD pathologies and hepatic lipid accumulation in Tmem135FUN025/FUN025/Lepob/ob mice compared to Lepob/ob mice (34). Together, these phenotypic changes suggest that impairment of TMEM135 function affects molecular pathways involved in the pathogenesis of metabolic disease with dysregulated lipid metabolism, which may include activation of PPAR signaling and increased peroxisome proliferation (34).
Most recently, adipose-specific deletion of Tmem135 was shown to result in impaired thermogenesis and increased diet-induced obesity and insulin resistance in mice, revealing significant roles of TMEM135 in the brown fat and energy homeostasis (25). Conversely, Tmem135 overexpression increased thermogenesis and prevented diet-induced obesity and insulin resistance (25). This study revealed aforementioned function of TMEM135 in the regulation of mitochondrial fission and placed TMEM135 as a critical mediator of the peroxisomal regulation of mitochondrial fission and thermogenesis (25). Additionally, the authors identified a single nucleotide polymorphism (SNP) in the human TMEM135 gene associated with increased body mass index (BMI) in a Hispanic population (25). Functional studies indicated that this specific SNP in TMEM135 reduces the function of the protein and may promote the occurrence of human metabolic diseases (25).
4 Future perspectives of TMEM135 research
There has been a wealth of knowledge on TMEM135 from new publications over the last several years. Yet, there are still many unanswered questions regarding this fascinating protein that need to be answered and its role in aging.
TMEM135 is increasingly recognized for its involvement in lipid homeostasis. Studies have shown that TMEM135 expression is elevated in conditions with lipid accumulation (15). However, the specific molecular mechanisms regulating TMEM135 expression remain to be fully elucidated. This aspect presents a potential avenue for future research to understand the role of TMEM135 in lipid homeostasis.
As detailed by the descriptions of mice with the loss-of-function mutation and overexpression of Tmem135 in this review, TMEM135 has significant roles in multiple tissues. Future studies on TMEM135 should utilize mouse genetic approaches to interrogate the role of this protein in a tissue-specific manner. These strategies could include Cre-Lox technologies or viral vectors that modulate TMEM135 expression in discrete cells. This would prevent off-target effects that may confound the interpretation of the role of TMEM135 in a particular cell-type since TMEM135 has important roles in multiple cells. Also, it is unclear the contributions of dysregulated mitochondrial dynamics, DHA concentrations, peroxisome proliferation, and intracellular trafficking on the phenotypes of the Tmem135 mutant and overexpressing mice. These pathways should be targeted to determine their contribution towards the phenotypes of these mice.
Previous studies highlight the crucial role of TMEM135 in connecting cellular organelles, particularly peroxisomes and mitochondria, as well as peroxisomes and lysosomes. The growing understanding of organelle interactions has attracted significant scientific interest. A key area of research is the role of these organelle interactions in the retina, particularly how they contribute to normal aging and their potential alteration in age-related conditions such as AMD.
Lastly, while genetic and molecular biological studies on TMEM135 have progressed, exploring the protein biochemically could yield valuable insights. For instance, identifying the structure of TMEM135, its binding partners, and small molecules that regulate its activity could pave the way for new therapeutic approaches targeting TMEM135 in metabolic and age-related diseases.
5 Conclusion
Aging is a significant stressor for tissues, but the molecular nature of aging remains a mystery. By using forward genetics, important genes and pathways involved in aging can be determined. In this review, we summarized the findings on TMEM135, an important player involved in retinal aging of mice that was uncovered through forward genetics. This unbiased phenotypic investigation led to the discovery of a critical protein involved in the regulation of mitochondrial and peroxisomal functions, as well as lipid homeostasis. Disruptions of TMEM135 function have detrimental consequences to the murine retina, but other tissues including the heart, liver, and adipose tissue can also be impacted by changes in TMEM135. Importantly, both the loss-of-function mutation in Tmem135 and overexpression of Tmem135 caused pathology development in mice, indicating the balance of TMEM135 function is required for normal healthy aging. Although no direct connection has been made between TMEM135 and age-related retinal diseases, accumulating evidence points to the involvement of TMEM135 in the molecular pathways underlying such diseases. As more information is collected on TMEM135, we will gain a better understanding of how aging contributes to disease processes, thus providing invaluable insight for the creation of novel therapies and identification of promising biomarkers for those individuals who may be affected by these conditions in the future.
Author contributions
ML: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. PG: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. SI: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. AI: Conceptualization, Funding acquisition, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors would like to acknowledge the sources of funding from the National Eye Institute (R01EY022086 to AI; P30EY016665 to UW-Madison’s Department of Ophthalmology and Visual Sciences; T32EY027721 to ML; and F32EY032766 to ML), McPherson Eye Research Institute (Timothy William Trout Chairmanship to AI), other National Institutes of Health departments for core services at UW-Madison (S10OD023526 from the NIH Office of the Director and P30CA014520 from the National Cancer Institute), and the UW-Madison’s Department of Biochemistry Endowment.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Campello L, Singh N, Advani J, Mondal AK, Corso-Diaz X, Swaroop A. Aging of the retina: molecular and metabolic turbulences and potential interventions. Annu Rev Vis Sci (2021) 7:633–64. doi: 10.1146/annurev-vision-100419-114940
2. Kolesnikov AV, Fan J, Crouch RK, Kefalov VJ. Age-related deterioration of rod vision in mice. J Neurosci (2010) 30:11222–31. doi: 10.1523/JNEUROSCI.4239-09.2010
3. Williams GA, Jacobs GH. Cone-based vision in the aging mouse. Vision Res (2007) 47:2037–46. doi: 10.1016/j.visres.2007.03.023
4. Mori H, Yamada H, Toyama K, Takahashi K, Akama T, Inoue T, et al. Developmental and age-related changes to the elastic lamina of Bruch’s membrane in mice. Graefes Arch Clin Exp Ophthalmol (2019) 257:289–301. doi: 10.1007/s00417-018-4184-5
5. Chen M, Rajapakse D, Fraczek M, Luo C, Forrester JV, Xu H. Retinal pigment epithelial cell multinucleation in the aging eye - a mechanism to repair damage and maintain homoeostasis. Aging Cell (2016) 15:436–45. doi: 10.1111/acel.12447
6. Wolf NS, Li Y, Pendergrass W, Schmeider C, Turturro A. Normal mouse and rat strains as models for age-related cataract and the effect of caloric restriction on its development. Exp Eye Res (2000) 70:683–92. doi: 10.1006/exer.2000.0835
7. Higuchi H, Macke EL, Lee WH, Miller SA, Xu JC, Ikeda S, et al. Genetic basis of age-dependent synaptic abnormalities in the retina. Mamm Genome (2015) 26:21–32. doi: 10.1007/s00335-014-9546-7
8. Xu H, Chen M, Manivannan A, Lois N, Forrester JV. Age-dependent accumulation of lipofuscin in perivascular and subretinal microglia in experimental mice. Aging Cell (2008) 7:58–68. doi: 10.1111/j.1474-9726.2007.00351.x
9. Pinto LH, Vitaterna MH, Siepka SM, Shimomura K, Lumayag S, Baker M, et al. Results from screening over 9000 mutation-bearing mice for defects in the electroretinogram and appearance of the fundus. Vision Res (2004) 44:3335–45. doi: 10.1016/j.visres.2004.07.025
10. Vitaterna MH, Pinto LH, Takahashi JS. Large-scale mutagenesis and phenotypic screens for the nervous system and behavior in mice. Trends Neurosci (2006) 29:233–40. doi: 10.1016/j.tins.2006.02.006
11. Lee WH, Higuchi H, Ikeda S, Macke EL, Takimoto T, Pattnaik BR, et al. Mouse Tmem135 mutation reveals a mechanism involving mitochondrial dynamics that leads to age-dependent retinal pathologies. Elife (2016) 5:e19264. doi: 10.7554/eLife.19264
12. Han J, Dinculescu A, Dai X, Du W, Smith WC, Pang J. Review: the history and role of naturally occurring mouse models with Pde6b mutations. Mol Vis (2013) 19:2579–89.
13. Olsson JE, Gordon JW, Pawlyk BS, Roof D, Hayes A, Molday RS, et al. Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron (1992) 9:815–30. doi: 10.1016/0896-6273(92)90236-7
14. Moshiri A. Animals models of inherited retinal disease. Int Ophthalmol Clin (2021) 61:113–30. doi: 10.1097/IIO.0000000000000368
15. Exil VJ, Silva Avila D, Benedetto A, Exil EA, Adams MR, Au C, et al. Stressed-induced TMEM135 protein is part of a conserved genetic network involved in fat storage and longevity regulation in Caenorhabditis elegans. PloS One (2010) 5:e14228. doi: 10.1371/journal.pone.0014228
16. Landowski M, Bhute VJ, Takimoto T, Grindel S, Shahi PK, Pattnaik BR, et al. A mutation in transmembrane protein 135 impairs lipid metabolism in mouse eyecups. Sci Rep (2022) 12:756. doi: 10.1038/s41598-021-04644-3
17. Zarsky V, Dolezal P. Evolution of the Tim17 protein family. Biol Direct (2016) 11:54. doi: 10.1186/s13062-016-0157-y
18. Maharjan Y, Lee JN, Kwak SA, Dutta RK, Park C, Choe SK, et al. TMEM135 regulates primary ciliogenesis through modulation of intracellular cholesterol distribution. EMBO Rep (2020) 21:e48901. doi: 10.15252/embr.201948901
19. Gronemeyer T, Wiese S, Ofman R, Bunse C, Pawlas M, Hayen H, et al. The proteome of human liver peroxisomes: identification of five new peroxisomal constituents by a label-free quantitative proteomics survey. PloS One (2013) 8:e57395. doi: 10.1371/journal.pone.0057395
20. Islinger M, Luers GH, Li KW, Loos M, Volkl A. Rat liver peroxisomes after fibrate treatment. A survey using quantitative mass spectrometry. J Biol Chem (2007) 282:23055–69. doi: 10.1074/jbc.M610910200
21. Islinger M, Li KW, Loos M, Liebler S, Angermuller S, Eckerskorn C, et al. Peroxisomes from the heavy mitochondrial fraction: isolation by zonal free flow electrophoresis and quantitative mass spectrometrical characterization. J Proteome Res (2010) 9:113–24. doi: 10.1021/pr9004663
22. Kikuchi M, Hatano N, Yokota S, Shimozawa N, Imanaka T, Taniguchi H. Proteomic analysis of rat liver peroxisome: presence of peroxisome-specific isozyme of Lon protease. J Biol Chem (2004) 279:421–8. doi: 10.1074/jbc.M305623200
23. Mi J, Kirchner E, Cristobal S. Quantitative proteomic comparison of mouse peroxisomes from liver and kidney. Proteomics (2007) 7:1916–28. doi: 10.1002/pmic.200600638
24. Bosch M, Sanchez-Alvarez M, Fajardo A, Kapetanovic R, Steiner B, Dutra F, et al. Mammalian lipid droplets are innate immune hubs integrating cell metabolism and host defense. Science (2020) 370(6514):eaay8085. doi: 10.1126/science.aay8085
25. Hu D, Tan M, Lu D, Kleiboeker B, Liu X, Park H, et al. TMEM135 links peroxisomes to the regulation of brown fat mitochondrial fission and energy homeostasis. Nat Commun (2023) 14:6099. doi: 10.1038/s41467-023-41849-8
26. He A, Dean JM, Lodhi IJ. Peroxisomes as cellular adaptors to metabolic and environmental stress. Trends Cell Biol (2021) 31:656–70. doi: 10.1016/j.tcb.2021.02.005
27. Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol (2007) 8:870–9. doi: 10.1038/nrm2275
28. Messias MCF, Mecatti GC, Priolli DG, de Oliveira Carvalho P. Plasmalogen lipids: functional mechanism and their involvement in gastrointestinal cancer. Lipids Health Dis (2018) 17:41. doi: 10.1186/s12944-018-0685-9
29. Fransen M, Lismont C, Walton P. The peroxisome-mitochondria connection: how and why? Int J Mol Sci (2017) 18(6):1126. doi: 10.3390/ijms18061126
30. Demarquoy J, Le Borgne F. Crosstalk between mitochondria and peroxisomes. World J Biol Chem (2015) 6:301–9. doi: 10.4331/wjbc.v6.i4.301
31. Lodhi IJ, Semenkovich CF. Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab (2014) 19:380–92. doi: 10.1016/j.cmet.2014.01.002
32. Wanders RJ, Waterham HR, Ferdinandusse S. Metabolic interplay between peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Front Cell Dev Biol (2015) 3:83. doi: 10.3389/fcell.2015.00083
33. Tanaka H, Okazaki T, Aoyama S, Yokota M, Koike M, Okada Y, et al. Peroxisomes control mitochondrial dynamics and the mitochondrion-dependent apoptosis pathway. J Cell Sci (2019) 132(11):jcs224766. doi: 10.1242/jcs.224766
34. Landowski M, Bhute VJ, Grindel S, Haugstad Z, Gyening YK, Tytanic M, et al. Transmembrane protein 135 regulates lipid homeostasis through its role in peroxisomal DHA metabolism. Commun Biol (2023) 6:8. doi: 10.1038/s42003-022-04404-7
35. Hishikawa D, Valentine WJ, Iizuka-Hishikawa Y, Shindou H, Shimizu T. Metabolism and functions of docosahexaenoic acid-containing membrane glycerophospholipids. FEBS Lett (2017) 591:2730–44. doi: 10.1002/1873-3468.12825
36. Li J, Pora BLR, Dong K, Hasjim J. Health benefits of docosahexaenoic acid and its bioavailability: A review. Food Sci Nutr (2021) 9:5229–43. doi: 10.1002/fsn3.2299
37. Qiu X. Biosynthesis of docosahexaenoic acid (DHA, 22:6-4, 7,10,13,16,19): two distinct pathways. Prostaglandins Leukot Essent Fatty Acids (2003) 68:181–6. doi: 10.1016/S0952-3278(02)00268-5
38. Agbaga MP, Mandal MN, Anderson RE. Retinal very long-chain PUFAs: new insights from studies on ELOVL4 protein. J Lipid Res (2010) 51:1624–42. doi: 10.1194/jlr.R005025
39. Ferdinandusse S, Denis S, Mooijer PA, Zhang Z, Reddy JK, Spector AA, et al. Identification of the peroxisomal beta-oxidation enzymes involved in the biosynthesis of docosahexaenoic acid. J Lipid Res (2001) 42:1987–95. doi: 10.1016/S0022-2275(20)31527-3
40. Wanders RJA, Waterham HR, Ferdinandusse S. Peroxisomes and their central role in metabolic interaction networks in humans. Subcell Biochem (2018) 89:345–65. doi: 10.1007/978-981-13-2233-4_15
41. Chornyi S, Lodewijk IJ, van Roermund CWT, Wanders RJA, Waterham HR. Peroxisomal metabolite and cofactor transport in humans. Front Cell Dev Biol (2020) 8:613892. doi: 10.3389/fcell.2020.613892
42. Wanders RJA, Baes M, Ribeiro D, Ferdinandusse S, Waterham HR. The physiological functions of human peroxisomes. Physiol Rev (2023) 103:957–1024. doi: 10.1152/physrev.00051.2021
43. Grygiel-Gorniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications–a review. Nutr J (2014) 13:17. doi: 10.1186/1475-2891-13-17
44. Dorninger F, Forss-Petter S, Wimmer I, Berger J. Plasmalogens, platelet-activating factor and beyond - Ether lipids in signaling and neurodegeneration. Neurobiol Dis (2020) 145:105061. doi: 10.1016/j.nbd.2020.105061
45. Chu BB, Liao YC, Qi W, Xie C, Du X, Wang J, et al. Cholesterol transport through lysosome-peroxisome membrane contacts. Cell (2015) 161:291–306. doi: 10.1016/j.cell.2015.02.019
46. Bharti K, den Hollander AI, Lakkaraju A, Sinha D, Williams DS, Finnemann SC, et al. Cell culture models to study retinal pigment epithelium-related pathogenesis in age-related macular degeneration. Exp Eye Res (2022) 222:109170. doi: 10.1016/j.exer.2022.109170
47. Landowski M, Grindel S, Shahi PK, Johnson A, Western D, Race A, et al. Modulation of tmem135 leads to retinal pigmented epithelium pathologies in mice. Invest Ophthalmol Vis Sci (2020) 61:16. doi: 10.1167/iovs.61.12.16
48. Lewis SA, Takimoto T, Mehrvar S, Higuchi H, Doebley AL, Stokes G, et al. The effect of Tmem135 overexpression on the mouse heart. PloS One (2018) 13:e0201986. doi: 10.1371/journal.pone.0201986
49. Muench NA, Patel S, Maes ME, Donahue RJ, Ikeda A, Nickells RW. The influence of mitochondrial dynamics and function on retinal ganglion cell susceptibility in optic nerve disease. Cells (2021) 10(7):1593. doi: 10.3390/cells10071593
50. Lee WH, Bhute VJ, Higuchi H, Ikeda S, Palecek SP, Ikeda A. Metabolic alterations caused by the mutation and overexpression of the Tmem135 gene. Exp Biol Med (Maywood) (2020) 245(17):1571–83. doi: 10.1177/1535370220932856
51. Lewandowski D, Sander CL, Tworak A, Gao F, Xu Q, Skowronska-Krawczyk D. Dynamic lipid turnover in photoreceptors and retinal pigment epithelium throughout life. Prog Retin Eye Res (2022) 89:101037. doi: 10.1016/j.preteyeres.2021.101037
52. Chen D, Chao DL, Rocha L, Kolar M, Nguyen Huu VA, Krawczyk M, et al. The lipid elongation enzyme ELOVL2 is a molecular regulator of aging in the retina. Aging Cell (2020) 19:e13100. doi: 10.1111/acel.13100
53. Kuroha S, Katada Y, Isobe Y, Uchino H, Shishikura K, Nirasawa T, et al. Long chain acyl-CoA synthetase 6 facilitates the local distribution of di-docosahexaenoic acid- and ultra-long-chain-PUFA-containing phospholipids in the retina to support normal visual function in mice. FASEB J (2023) 37:e23151. doi: 10.1096/fj.202300976R
54. Lobanova ES, Schuhmann K, Finkelstein S, Lewis TR, Cady MA, Hao Y, et al. Disrupted blood-retina lysophosphatidylcholine transport impairs photoreceptor health but not visual signal transduction. J Neurosci (2019) 39:9689–701. doi: 10.1523/JNEUROSCI.1142-19.2019
55. Wong BH, Chan JP, Cazenave-Gassiot A, Poh RW, Foo JC, Galam DL, et al. Mfsd2a is a transporter for the essential ω-3 fatty acid docosahexaenoic acid (DHA) in eye and is important for photoreceptor cell development. J Biol Chem (2016) 291:10501–14. doi: 10.1074/jbc.M116.721340
56. Sluch VM, Banks A, Li H, Crowley MA, Davis V, Xiang C, et al. ADIPOR1 is essential for vision and its RPE expression is lost in the Mfrp. Sci Rep (2018) 8:14339. doi: 10.1038/s41598-018-32579-9
57. Osada H, Toda E, Homma K, Guzman NA, Nagai N, Ogawa M, et al. ADIPOR1 deficiency-induced suppression of retinal ELOVL2 and docosahexaenoic acid levels during photoreceptor degeneration and visual loss. Cell Death Dis (2021) 12:458. doi: 10.1038/s41419-021-03741-5
58. Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab (2016) 27:105–17. doi: 10.1016/j.tem.2015.12.001
59. Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab (2013) 17:491–506. doi: 10.1016/j.cmet.2013.03.002
60. Zorov DB, Vorobjev IA, Popkov VA, Babenko VA, Zorova LD, Pevzner IB, et al. Lessons from the discovery of mitochondrial fragmentation (Fission): A review and update. Cells (2019) 8:1–17. doi: 10.3390/cells8020175
61. Zhao C, Yasumura D, Li X, Matthes M, Lloyd M, Nielsen G, et al. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J Clin Invest (2011) 121:369–83. doi: 10.1172/JCI44303
62. Rosales MAB, Shu DY, Iacovelli J, Saint-Geniez M. Loss of PGC-1α in RPE induces mesenchymal transition and promotes retinal degeneration. Life Sci Alliance (2019) 2:1–17. doi: 10.26508/lsa.201800212
63. Reddy JK, Hashimoto T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr (2001) 21:193–230. doi: 10.1146/annurev.nutr.21.1.193
64. Kocherlakota S, Das Y, Swinkels D, Vanmunster M, Callens M, Vinckier S, et al. The murine retinal pigment epithelium requires peroxisomal beta-oxidation to maintain lysosomal function and prevent dedifferentiation. Proc Natl Acad Sci U.S.A. (2023) 120:e2301733120. doi: 10.1073/pnas.2301733120
65. Lenin RR, Koh YH, Zhang Z, Yeo YZ, Parikh BH, Seah I, et al. Dysfunctional autophagy, proteostasis, and mitochondria as a prelude to age-related macular degeneration. Int J Mol Sci (2023) 24(10):8763. doi: 10.3390/ijms24108763
66. Jimenez-Loygorri JI, Benitez-Fernandez R, Viedma-Poyatos A, Zapata-Munoz J, Villarejo-Zori B, Gomez-Sintes R, et al. Mitophagy in the retina: Viewing mitochondrial homeostasis through a new lens. Prog Retin Eye Res (2023) 96:101205. doi: 10.1016/j.preteyeres.2023.101205
67. Barot M, Gokulgandhi MR, Mitra AK. Mitochondrial dysfunction in retinal diseases. Curr Eye Res (2011) 36:1069–77. doi: 10.3109/02713683.2011.607536
68. Stone J, van Driel D, Valter K, Rees S, Provis J. The locations of mitochondria in mammalian photoreceptors: relation to retinal vasculature. Brain Res (2008) 1189:58–69. doi: 10.1016/j.brainres.2007.10.083
69. Eells JT. Mitochondrial dysfunction in the aging retina. Biol (Basel) (2019) 8(2):31. doi: 10.3390/biology8020031
70. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell (2023) 186:243–78. doi: 10.1016/j.cell.2022.11.001
71. Somasundaran S, Constable IJ, Mellough CB, Carvalho LS. Retinal pigment epithelium and age-related macular degeneration: A review of major disease mechanisms. Clin Exp Ophthalmol (2020) 48:1043–56. doi: 10.1111/ceo.13834
72. Feher J, Kovacs I, Artico M, Cavallotti C, Papale A, Balacco Gabrieli C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol Aging (2006) 27:983–93. doi: 10.1016/j.neurobiolaging.2005.05.012
73. Nordgaard CL, Karunadharma PP, Feng X, Olsen TW, Ferrington DA. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci (2008) 49:2848–55. doi: 10.1167/iovs.07-1352
74. Kenney MC, Atilano SR, Boyer D, Chwa M, Chak G, Chinichian S, et al. Characterization of retinal and blood mitochondrial DNA from age-related macular degeneration patients. Invest Ophthalmol Vis Sci (2010) 51:4289–97. doi: 10.1167/iovs.09-4778
75. Terluk MR, Kapphahn RJ, Soukup LM, Gong H, Gallardo C, Montezuma SR, et al. Investigating mitochondria as a target for treating age-related macular degeneration. J Neurosci (2015) 35:7304–11. doi: 10.1523/JNEUROSCI.0190-15.2015
76. Ferrington DA, Kapphahn RJ, Leary MM, Atilano SR, Terluk MR, Karunadharma P, et al. Increased retinal mtDNA damage in the CFH variant associated with age-related macular degeneration. Exp Eye Res (2016) 145:269–77. doi: 10.1016/j.exer.2016.01.018
77. Muste JC, Russell MW, Chen AX, Seth K, Iyer AI, Valentim CCS, et al. Functional imaging of mitochondria in age-related macular degeneration using flavoprotein fluorescence. Ophthalmic Surg Lasers Imaging Retina (2023) 54:24–31. doi: 10.3928/23258160-20221214-03
78. Ferrington DA, Ebeling MC, Kapphahn RJ, Terluk MR, Fisher CR, Polanco JR, et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol (2017) 13:255–65. doi: 10.1016/j.redox.2017.05.015
79. Zhang M, Jiang N, Chu Y, Postnikova O, Varghese R, Horvath A, et al. Dysregulated metabolic pathways in age-related macular degeneration. Sci Rep (2020) 10:2464. doi: 10.1038/s41598-020-59244-4
80. Dabravolski SA. Mitochondria-derived peptides in healthy ageing and therapy of age-related diseases. Adv Protein Chem Struct Biol (2023) 136:197–215. doi: 10.1016/bs.apcsb.2023.02.015
81. Voloboueva LA, Killilea DW, Atamna H, Ames BN. N-tert-butyl hydroxylamine, a mitochondrial antioxidant, protects human retinal pigment epithelial cells from iron overload: relevance to macular degeneration. FASEB J (2007) 21:4077–86. doi: 10.1096/fj.07-8396com
82. Millington-Ward S, Chadderton N, Finnegan LK, Post IJM, Carrigan M, Nixon R, et al. RPE-directed gene therapy improves mitochondrial function in murine dry AMD models. Int J Mol Sci (2023) 24(4):3847. doi: 10.3390/ijms24043847
83. Sreekumar PG, Ishikawa K, Spee C, Mehta HH, Wan J, Yen K, et al. The mitochondrial-derived peptide humanin protects RPE cells from oxidative stress, senescence, and mitochondrial dysfunction. Invest Ophthalmol Vis Sci (2016) 57:1238–53. doi: 10.1167/iovs.15-17053
84. Rajapakse D, Curtis T, Chen M, Xu H. Zinc protects oxidative stress-induced RPE death by reducing mitochondrial damage and preventing lysosome rupture. Oxid Med Cell Longev (2017) 2017:6926485. doi: 10.1155/2017/6926485
85. Nashine S, Cohen P, Nesburn AB, Kuppermann BD, Kenney MC. Characterizing the protective effects of SHLP2, a mitochondrial-derived peptide, in macular degeneration. Sci Rep (2018) 8:15175. doi: 10.1038/s41598-018-33290-5
86. Brown EE, Lewin AS, Ash JD. Mitochondria: potential targets for protection in age-related macular degeneration. Adv Exp Med Biol (2018) 1074:11–7. doi: 10.1007/978-3-319-75402-4_2
87. Kaarniranta K, Kajdanek J, Morawiec J, Pawlowska E, Blasiak J. PGC-1alpha Protects RPE Cells of the Aging Retina against Oxidative Stress-Induced Degeneration through the Regulation of Senescence and Mitochondrial Quality Control. The Significance for AMD Pathogenesis. Int J Mol Sci (2018) 19(8):2317. doi: 10.3390/ijms19082317
88. Shu DY, Frank SI, Fitch TC, Karg MM, Butcher ER, Nnuji-John E, et al. Dimethyl fumarate blocks tumor necrosis factor-alpha-driven inflammation and metabolic rewiring in the retinal pigment epithelium. Front Mol Neurosci (2022) 15:896786. doi: 10.3389/fnmol.2022.896786
89. Ebeling MC, Polanco JR, Qu J, Tu C, Montezuma SR, Ferrington DA. Improving retinal mitochondrial function as a treatment for age-related macular degeneration. Redox Biol (2020) 34:101552. doi: 10.1016/j.redox.2020.101552
90. Mehrzadi S, Hemati K, Reiter RJ, Hosseinzadeh A. Mitochondrial dysfunction in age-related macular degeneration: melatonin as a potential treatment. Expert Opin Ther Targets (2020) 24:359–78. doi: 10.1080/14728222.2020.1737015
91. Beeson C, Peterson YK, Perron N, Bandyopadhyay M, Nasarre C, Beeson G, et al. Newly identified chemicals preserve mitochondrial capacity and decelerate loss of photoreceptor cells in murine retinal degeneration models. J Ocul Pharmacol Ther (2021) 37:367–78. doi: 10.1089/jop.2020.0140
92. Gasparian SA, Skrehot HC, Weng CY. Therapies for geographic atrophy. Int Ophthalmol Clin (2024) 64:5–20. doi: 10.1097/IIO.0000000000000515
93. Allingham MJ, Mettu PS, Cousins SW. Phase 1 clinical trial of elamipretide in intermediate age-related macular degeneration and high-risk drusen: ReCLAIM high-risk drusen study. Ophthalmol Sci (2022) 2:100095. doi: 10.1016/j.xops.2021.100095
94. Fisher CR, Shaaeli AA, Ebeling MC, Montezuma SR, Ferrington DA. Investigating mitochondrial fission, fusion, and autophagy in retinal pigment epithelium from donors with age-related macular degeneration. Sci Rep (2022) 12:21725. doi: 10.1038/s41598-022-26012-5
95. Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol (2010) 191:1141–58. doi: 10.1083/jcb.201007152
96. Yako T, Nakamura M, Nakamura S, Hara H, Shimazawa M. Pharmacological inhibition of mitochondrial fission attenuates oxidative stress-induced damage of retinal pigmented epithelial cells. J Pharmacol Sci (2021) 146:149–59. doi: 10.1016/j.jphs.2021.03.012
97. Fliesler SJ, Anderson RE. Chemistry and metabolism of lipids in the vertebrate retina. Prog Lipid Res (1983) 22:79–131. doi: 10.1016/0163-7827(83)90004-8
98. Zhong Y, Wang K, Jiang L, Wang J, Zhang X, Xu J, et al. Dietary fatty acid intake, plasma fatty acid levels, and the risk of age-related macular degeneration (AMD): a dose-response meta-analysis of prospective cohort studies. Eur J Nutr (2021) 60:3013–27. doi: 10.1007/s00394-020-02445-4
99. van Leeuwen EM, Emri E, Merle BMJ, Colijn JM, Kersten E, Cougnard-Gregoire A, et al. A new perspective on lipid research in age-related macular degeneration. Prog Retin Eye Res (2018) 67:56–86. doi: 10.1016/j.preteyeres.2018.04.006
100. Walchuk C, Suh M. Nutrition and the aging retina: A comprehensive review of the relationship between nutrients and their role in age-related macular degeneration and retina disease prevention. Adv Food Nutr Res (2020) 93:293–332. doi: 10.1016/bs.afnr.2020.04.003
101. Sugasini D, Yalagala PCR, Subbaiah PV. Efficient enrichment of retinal DHA with dietary lysophosphatidylcholine-DHA: potential application for retinopathies. Nutrients (2020) 12(10):3114. doi: 10.3390/nu12103114
102. Gordon WC, Kautzmann MI, Jun B, Cothern ML, Fang Z, Bazan NG. Rod-specific downregulation of omega-3 very-long-chain polyunsaturated fatty acid pathway in age-related macular degeneration. Exp Eye Res (2023) 235:109639. doi: 10.1016/j.exer.2023.109639
103. Newman AM, Gallo NB, Hancox LS, Miller NJ, Radeke CM, Maloney MA, et al. Systems-level analysis of age-related macular degeneration reveals global biomarkers and phenotype-specific functional networks. Genome Med (2012) 4:16. doi: 10.1186/gm315
104. Orozco LD, Chen HH, Cox C, Katschke KJ Jr., Arceo R, Espiritu C, et al. Integration of eQTL and a single-cell atlas in the human eye identifies causal genes for age-related macular degeneration. Cell Rep (2020) 30:1246–1259 e6. doi: 10.1016/j.celrep.2019.12.082
105. Dhirachaikulpanich D, Li X, Porter LF, Paraoan L. Integrated microarray and RNAseq transcriptomic analysis of retinal pigment epithelium/choroid in age-related macular degeneration. Front Cell Dev Biol (2020) 8:808. doi: 10.3389/fcell.2020.00808
106. Hishikawa D, Yanagida K, Nagata K, Kanatani A, Iizuka Y, Hamano F, et al. Hepatic levels of DHA-containing phospholipids instruct SREBP1-mediated synthesis and systemic delivery of polyunsaturated fatty acids. iScience (2020) 23:101495. doi: 10.1016/j.isci.2020.101495
107. Wang L, Clark ME, Crossman DK, Kojima K, Messinger JD, Mobley JA, et al. Abundant lipid and protein components of drusen. PloS One (2010) 5:e10329. doi: 10.1371/journal.pone.0010329
108. Klaver CC, Kliffen M, van Duijn CM, Hofman A, Cruts M, Grobbee DE, et al. Genetic association of apolipoprotein E with age-related macular degeneration. Am J Hum Genet (1998) 63:200–6. doi: 10.1086/301901
109. Schmidt S, Saunders AM, de la Paz MA, Postel EA, Heinis RM, . Agarwal A, et al. Pericak-Vance, Association of the apolipoprotein E gene with age-related macular degeneration: possible effect modification by family history, age, and gender. Mol Vis (2000) 6:287–93.
110. Zareparsi S, Reddick AC, Branham KE, Moore KB, Jessup L, Thoms S, et al. Association of apolipoprotein E alleles with susceptibility to age-related macular degeneration in a large cohort from a single center. Invest Ophthalmol Vis Sci (2004) 45:1306–10. doi: 10.1167/iovs.03-1253
111. McKay GJ, Patterson CC, Chakravarthy U, Dasari S, Klaver CC, Vingerling JR, et al. Evidence of association of APOE with age-related macular degeneration: a pooled analysis of 15 studies. Hum Mutat (2011) 32:1407–16. doi: 10.1002/humu.21577
112. Chen W, Stambolian D, Edwards AO, Branham KE, Othman M, Jakobsdottir J, et al. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc Natl Acad Sci U.S.A. (2010) 107:7401–6. doi: 10.1073/pnas.0912702107
113. Neale BM, Fagerness J, Reynolds R, Sobrin L, Parker M, Raychaudhuri S, et al. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc Natl Acad Sci U.S.A. (2010) 107:7395–400. doi: 10.1073/pnas.0912019107
114. Wang YF, Han Y, Zhang R, Qin L, Wang MX, Ma L. CETP/LPL/LIPC gene polymorphisms and susceptibility to age-related macular degeneration. Sci Rep (2015) 5:15711. doi: 10.1038/srep15711
115. Pikuleva IA, Curcio CA. Cholesterol in the retina: the best is yet to come. Prog Retin Eye Res (2014) 41:64–89. doi: 10.1016/j.preteyeres.2014.03.002
116. Landowski M, Bowes Rickman C. Targeting lipid metabolism for the treatment of age-related macular degeneration: insights from preclinical mouse models. J Ocul Pharmacol Ther (2022) 38:3–32. doi: 10.1089/jop.2021.0067
117. Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell (1999) 98:13–23. doi: 10.1016/S0092-8674(00)80602-9
118. Charbel Issa P, Barnard AR, Singh MS, Carter E, Jiang Z, Radu RA, et al. Fundus autofluorescence in the Abca4(-/-) mouse model of Stargardt disease–correlation with accumulation of A2E, retinal function, and histology. Invest Ophthalmol Vis Sci (2013) 54:5602–12. doi: 10.1167/iovs.13-11688
119. Lakkaraju A, Finnemann SC, Rodriguez-Boulan E. The lipofuscin fluorophore A2E perturbs cholesterol metabolism in retinal pigment epithelial cells. Proc Natl Acad Sci U.S.A. (2007) 104:11026–31. doi: 10.1073/pnas.0702504104
120. Tan LX, Toops KA, Lakkaraju A. Protective responses to sublytic complement in the retinal pigment epithelium. Proc Natl Acad Sci U.S.A. (2016) 113:8789–94. doi: 10.1073/pnas.1523061113
121. Claudepierre T, Paques M, Simonutti M, Buard I, Sahel J, Maue RA, et al. Lack of Niemann-Pick type C1 induces age-related degeneration in the mouse retina. Mol Cell Neurosci (2010) 43:164–76. doi: 10.1016/j.mcn.2009.10.007
122. Moayyeri A, Hsu YH, Karasik D, Estrada K, Xiao SM, Nielson C, et al. Genetic determinants of heel bone properties: genome-wide association meta-analysis and replication in the GEFOS/GENOMOS consortium. Hum Mol Genet (2014) 23:3054–68. doi: 10.1093/hmg/ddt675
123. Scheideler M, Elabd C, Zaragosi LE, Chiellini C, Hackl H, Sanchez-Cabo F, et al. Comparative transcriptomics of human multipotent stem cells during adipogenesis and osteoblastogenesis. BMC Genomics (2008) 9:340. doi: 10.1186/1471-2164-9-340
124. Correa-Rodriguez M, Viatte S, Massey J, Schmidt-RioValle J, Rueda-Medina B, Orozco G. Analysis of SNP-SNP interactions and bone quantitative ultrasound parameter in early adulthood. BMC Med Genet (2017) 18:107. doi: 10.1186/s12881-017-0468-6
125. Lu HF, Hung KS, Chu HW, Wong HS, Kim J, Kim MK, et al. Meta-analysis of genome-wide association studies identifies three loci associated with stiffness index of the calcaneus. J Bone Miner Res (2019) 34:1275–83. doi: 10.1002/jbmr.3703
126. Zhang N, Jiang H, Bai Y, Lu X, Feng M, Guo Y, et al. The molecular mechanism study of insulin on proliferation and differentiation of osteoblasts under high glucose conditions. Cell Biochem Funct (2019) 37:385–94. doi: 10.1002/cbf.3415
127. Wang R, Zhang M, Hu Y, He J, Lin Q, Peng N. MiR-100-5p inhibits osteogenic differentiation of human bone mesenchymal stromal cells by targeting TMEM135. Hum Cell (2022) 35:1671–83. doi: 10.1007/s13577-022-00764-8
128. Teraoka SN, Bernstein JL, Reiner AS, Haile RW, Bernstein L, Lynch CF, et al. Single nucleotide polymorphisms associated with risk for contralateral breast cancer in the Women’s Environment, Cancer, and Radiation Epidemiology (WECARE) Study. Breast Cancer Res (2011) 13:R114. doi: 10.1186/bcr3057
129. Natrajan R, Mackay A, Lambros MB, Weigelt B, Wilkerson PM, Manie E, et al. A whole-genome massively parallel sequencing analysis of BRCA1 mutant oestrogen receptor-negative and -positive breast cancers. J Pathol (2012) 227:29–41. doi: 10.1002/path.4003
130. Yu YP, Ding Y, Chen Z, Liu S, Michalopoulos A, Chen R, et al. Novel fusion transcripts associate with progressive prostate cancer. Am J Pathol (2014) 184:2840–9. doi: 10.1016/j.ajpath.2014.06.025
131. Yu YP, Liu S, Nelson J, Luo JH. Detection of fusion gene transcripts in the blood samples of prostate cancer patients. Sci Rep (2021) 11:16995. doi: 10.1038/s41598-021-96528-9
132. Potrony M, Puig-Butille JA, Farnham JM, Gimenez-Xavier P, Badenas C, Tell-Marti G, et al. Genome-wide linkage analysis in Spanish melanoma-prone families identifies a new familial melanoma susceptibility locus at 11q. Eur J Hum Genet (2018) 26:1188–93. doi: 10.1038/s41431-018-0149-8
133. Wang H, Liu H, Dai W, Luo S, Amos CI, Lee JE, et al. Association of genetic variants of TMEM135 and PEX5 in the peroxisome pathway with cutaneous melanoma-specific survival. Ann Transl Med (2021) 9:396. doi: 10.21037/atm-20-2117
134. Yu YP, Liu P, Nelson J, Hamilton RL, Bhargava R, Michalopoulos G, et al. Identification of recurrent fusion genes across multiple cancer types. Sci Rep (2019) 9:1074. doi: 10.1038/s41598-019-38550-6
135. Aguayo-Orozco A, Bois FY, Brunak S, Taboureau O. Analysis of time-series gene expression data to explore mechanisms of chemical-induced hepatic steatosis toxicity. Front Genet (2018) 9:396. doi: 10.3389/fgene.2018.00396
136. Franic S, Groen-Blokhuis MM, Dolan CV, Kattenberg MV, Pool R, Xiao X, et al. Intelligence: shared genetic basis between Mendelian disorders and a polygenic trait. Eur J Hum Genet (2015) 23:1378–83. doi: 10.1038/ejhg.2015.3
137. Wai T, Garcia-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, et al. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science (2015) 350:aad0116. doi: 10.1126/science.aad0116
138. Song M, Mihara K, Chen Y, Scorrano L, Dorn GW 2nd. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab (2015) 21:273–86. doi: 10.1016/j.cmet.2014.12.011
139. Gao Q, Jia Y, Yang G, Zhang X, Boddu PC, Petersen B, et al. PPARalpha-deficient ob/ob obese mice become more obese and manifest severe hepatic steatosis due to decreased fatty acid oxidation. Am J Pathol (2015) 185:1396–408. doi: 10.1016/j.ajpath.2015.01.018
140. Carmona MC, Louche K, Nibbelink M, Prunet B, Bross A, Desbazeille M, et al. Fenofibrate prevents Rosiglitazone-induced body weight gain in ob/ob mice. Int J Obes (Lond) (2005) 29:864–71. doi: 10.1038/sj.ijo.0802943
141. Stocks B, Gonzalez-Franquesa A, Borg ML, Bjornholm M, Niu L, Zierath JR, et al. Integrated liver and plasma proteomics in obese mice reveals complex metabolic regulation. Mol Cell Proteomics (2022) 21:100207. doi: 10.1016/j.mcpro.2022.100207
Keywords: TMEM135, AMD, mitochondria, peroxisomes, lipid, DHA, RPE, photoreceptors
Citation: Landowski M, Gogoi P, Ikeda S and Ikeda A (2024) Roles of transmembrane protein 135 in mitochondrial and peroxisomal functions - implications for age-related retinal disease. Front. Ophthalmol. 4:1355379. doi: 10.3389/fopht.2024.1355379
Received: 13 December 2023; Accepted: 12 January 2024;
Published: 31 January 2024.
Edited by:
James Hurley, University of Washington, United StatesReviewed by:
Daniel Taran Hass, University of Washington, United StatesThomas Wubben, University of Michigan, United States
Copyright © 2024 Landowski, Gogoi, Ikeda and Ikeda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Akihiro Ikeda, YWlrZWRhQHdpc2MuZWR1
†These authors share first authorship