 Youichi Shinozaki
Youichi Shinozaki Kazuhiko Namekata
Kazuhiko Namekata- Visual Research Project, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan
Glial cells, a type of non-neuronal cell found in the central nervous system (CNS), play a critical role in maintaining homeostasis and regulating CNS functions. Recent advancements in technology have paved the way for new therapeutic strategies in the fight against glaucoma. While intraocular pressure (IOP) is the most well-known modifiable risk factor, a significant number of glaucoma patients have normal IOP levels. Because glaucoma is a complex, multifactorial disease influenced by various factors that contribute to its onset and progression, it is imperative that we consider factors beyond IOP to effectively prevent or slow down the disease’s advancement. In the realm of CNS neurodegenerative diseases, glial cells have emerged as key players due to their pivotal roles in initiating and hastening disease progression. The inhibition of dysregulated glial function holds the potential to protect neurons and restore brain function. Consequently, glial cells represent an enticing therapeutic candidate for glaucoma, even though the majority of glaucoma research has historically concentrated solely on retinal ganglion cells (RGCs). In addition to the neuroprotection of RGCs, the proper regulation of glial cell function can also facilitate structural and functional recovery in the retina. In this review, we offer an overview of recent advancements in understanding the non-cell-autonomous mechanisms underlying the pathogenesis of glaucoma. Furthermore, state-of-the-art technologies have opened up possibilities for regenerating the optic nerve, which was previously believed to be incapable of regeneration. We will also delve into the potential roles of glial cells in the regeneration of the optic nerve and the restoration of visual function.
1 Introduction
Neurons in the mammalian central nervous system (CNS) have long been perceived as incapable of regeneration in the adult tissue (1). Similarly, retinal neurons have historically been considered non-regenerative, leading to the perception that blindness resulting from retinal neurodegenerative diseases and optic neuropathies is untreatable. Consequently, visual impairment in glaucoma, a leading global cause of blindness, has traditionally been thought of as irreversible. However, decades of extensive research have revealed that there is potential for restoring visual function even after the onset of ocular neurodegenerative diseases. Given that retinal ganglion cells (RGCs), responsible for transmitting visual information to the brain, are selectively damaged in glaucoma, therapeutic efforts have primarily focused on cell-autonomous mechanisms. These strategies have achieved significant success in preventing RGC death in glaucoma model animals and regenerating the optic nerve following optic nerve injuries. However, achieving a complete recovery of visual function remains a formidable challenge. To attain this goal, non-cell-autonomous mechanisms must also be considered, as numerous extrinsic factors play a role in regulating RGC degeneration and optic nerve regrowth. In this review, we delve into the pathogenic mechanisms of glaucoma and explore potential molecular targets for the restoration of visual function. We particularly focus on glial cells, a type of non-neuronal cell within the nervous system, as potential sources of these extrinsic factors.
2 Glaucoma
Glaucoma, progressive optic neuropathy, is the leading cause of blindness worldwide that affects more than 70 million people (2, 3). Despite the multifaceted nature of the disease, with numerous risk factors influencing its onset and progression (4), elevated intraocular pressure (IOP) is the most well-known and modifiable factor (5, 6). The scourge of blindness in glaucoma finds its genesis in the grievous impairment suffered by the optic nerve and RGCs. RGC degeneration is a hallmark of glaucoma (7), while the damage at the optic nerve head (ONH) — the part where RGC axons coalesce to form the optic nerve (8) — precedes the onset of visual field loss in glaucoma (9). Moreover, dendritic and synaptic degeneration in RGCs are also initial events that play a pivotal role in the progression of the disease (10, 11). Given the intrinsic limitations associated with the regenerative potential of both RGCs and optic nerves, extensive research endeavors have concentrated on the dual objectives of averting RGC demise and forestalling optic nerve degeneration, with the ultimate aim of reinstating visual function. A growing body of evidence has suggested that the regenerative capacity of the ocular tissue can be modifiable by various factors including the intracellular signaling molecules, extracellular factors, and environmental conditions. While interventions targeting cell-autonomous mechanisms have yielded substantial strides in optic nerve regeneration, the realization of comprehensive functional recovery remains a formidable challenge.
3 Heterogeneity in glial cells in the ocular tissue
Glial cells, non-neuronal cell types in the nervous system, are not confined to the brain or spinal cord but also exist in the ocular tissue, such as the retina and optic nerve. In the retina, three types of glial cells exist: astrocytes, Müller cells, and microglia (Figure 1A). Astrocytes localize at the innermost surface of the retina and closely attach to blood vessels with their processes. Müller cells are the retina-specific astrocyte-lineage cells which are characterized by a vertical stalk spanning through the retina. Müller cells are limited to the retina, while astrocytes are highly enriched in the ONH and optic nerve (ON). Oligodendrocytes (OLs) and their precursors (oligodendrocyte precursor cells, OPCs) are present in the ON as illustrated in Figure 1B. RGC axon remain unmyelinated in most mammalian retina, with myelination commencing behind the myelination-transition zone located behind the globe. The myelination of the optic nerve facilitates the rapid transduction of visual information from the retina to the brain. Microglia, resident immune cells in the nervous tissues, including the retina, also contribute to the heterogeneity. Prior investigations have indicated that dysregulations in Müller cells and astrocytes can lead to RGC degeneration and visual dysfunction in the absence of elevated IOP (12, 13), underscoring their pivotal roles in the pathogenesis of normal tension glaucoma (NTG). Glial cells exhibit a remarkable degree of phenotypic plasticity, demonstrating either neurotoxic or neuroprotective attributes (14, 15). While it is widely acknowledged that reactive glial cells are frequently associated with neurotoxic functions, the appropriate regulation of glial cells has the potential to mitigate neuronal damage in a variety of neurodegenerative disease and CNS injury models (16–19). For instance, microglia-derived IL-1α, TNF, and C1q can induce the transformation of astrocytes into a neurotoxic phenotype, leading to damage to RGCs (14). Blockades, such as those preventing the formation of neurotoxic astrocytes, have been shown to protect RGCs in glaucoma model mice (20). On the other hand, microglia-derived TNFα, IL-1β, and IL-6 can induce astrocytes become reactive and neuroprotective (15). The neuroprotective effects of reactive astrocytes depend on STAT3 activity. Blocking the STAT3 signal in the astrocytes exacerbates RGC damage and visual impairment in the glaucoma model (21), emphasizing the crucial role of reactive astrocytes in protecting RGCs in glaucoma. Moreover, astrocytes and microglia tend to alter their phenotype in association with the disease state, exhibiting a relatively neuroprotective phenotype during the initial stages of neurodegenerative diseases in the brain (22, 23). In the case of the DBA/2J mouse, an inherited glaucoma model, reactive astrocytes have been found to confer protective effects upon RGCs during the early stages (23). Understanding these mechanisms and the precisely modulating glial cells represent an appealing avenue for neuroprotection. Alongside the phenotypic changes of glial cells, recent advancements in single-cell RNA sequencing (scRNA-seq) have unveiled heterogeneity among glial cells, revealing distinct subclusters that exhibit neuroprotective functions even under pathological conditions in the brain (24). While the most recent scRNA-sec data from the human and mouse retinas have successfully detected several subclasses of astrocytes, Müller cells, and microglia (25–29), these data are derived from human diseases or mouse models rather than specifically from glaucoma. Furthermore, in many cases of scRNA-seq data, ocular astrocytes and microglia are found as only a minor population and single cluster (13). Obtaining their subclusters in the scRNA-seq data requires cell isolation from at least several retinae (30). Given that scRNA-seq data from brain neurodegenerative disease model animals have detected disease-associated subclusters of glial cells (i.e. disease-associated microglia or astrocytes) that dynamically affect disease progression (31, 32), ocular glial cells in the human glaucoma patients or animal models would likely exhibit similar subclusters. Technical advancements in this field will enable us to uncover glaucoma-associated subclusters of glial cells and their role in glaucoma.
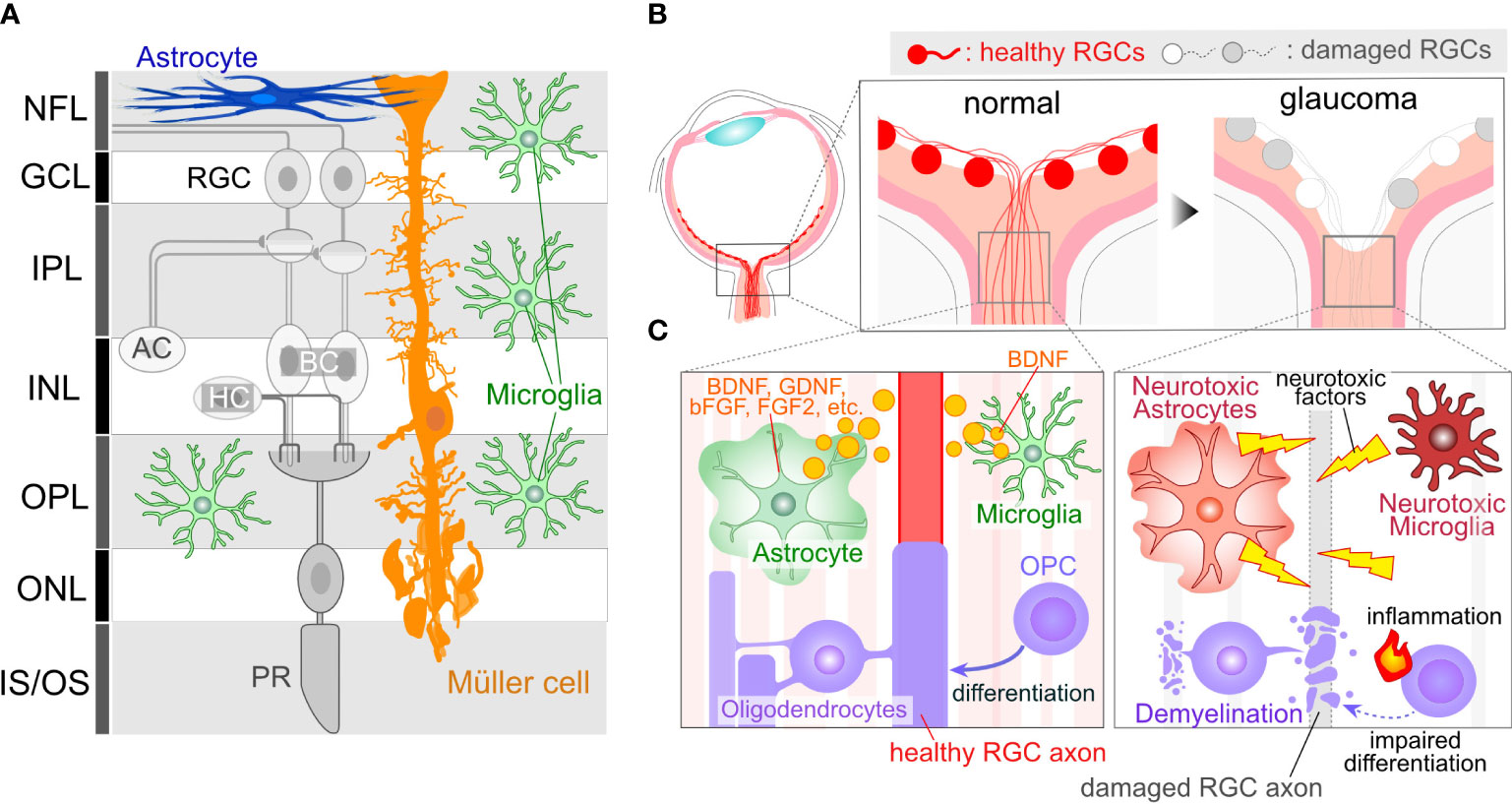
Figure 1 Glial Cells in Ocular Tissues. (A) Retinal Structure and Cellular Components: The retina comprises several neural layers. In the ganglion cell layer (GCL), one can find the cell bodies of RGCs. Additionally, some displaced amacrine cell (AC) somas are also localized within the GCL. RGC axons extend through the nerve fiber layer (NFL) and converge to form the optic nerve at the optic nerve head (ONH). Within the inner plexiform layer (IPL), RGC dendrites interact with axons from ACs or bipolar cells (BCs), forming synapses. Cell bodies for Acs and BCs reside in the inner nuclear layer (INL). Toward the outer part of the INL, horizontal cell (HC) bodies are present. In the outer plexiform layer (OPL), synapses formed by BCs, HCs, and photoreceptors (PRs) can be observed. PR cell bodies are located in the outer nuclear layer (ONL). PRs receive support from the retinal pigment epithelium, situated on the outer side of the PR inner/outer segments. Astrocytes are primarily found in the innermost retinal layer. Microglia are distributed across several retinal layers, including NFL/GCL, IPL, and OPL. Müller cells, which are retina-specific astrocyte-lineage cells, span vertically throughout the entire retinal thickness, with their cell bodies located in the INL, extending fine processes toward the synapses. (B) ONH Cupping in Glaucoma: ONH cupping represents well-characterized structural changes in the eyes of human glaucoma patients. These structural alterations may result in deformation and damage to RGC axons. Importantly, this change occurs in association with glial activations, suggesting that glial cells may contribute to the enlargement of cupping. (C) RGC axons in the ONH are unmyelinated and are directly enveloped by astrocytes and microglia. The ON is myelinated behind the optic nerve lamina region by OLs. OPCs also exist in the ON. Glial cells in normal conditions provide support for RGC axon integrity, including the production of neuroprotective factors (e.g. BDNF, GDNF, bFGF, and FGF2 from astrocytes and BDNF from microglia). However, in glaucoma, glial cells may undergo phenotypic changes, transitioning to neurotoxic states, which can lead to damage to RGC axons.
4 The Roles of glial cells in the enlargement of the ONH cupping
RGCs position their cell bodies within the ganglion cell layer (GCL), with their dendrites extending into the inner plexiform layer (IPL) (Figure 1A). GC axons converge to form the optic nerve at the ONH. The optic nerve exits the eye via the lamina cribrosa (LC), a mesh-like structure through which RGC axons pass. In human patients diagnosed with glaucoma, structural alterations in the LC lead to an enlargement of the ONH cupping (Figure 1B), a characteristic feature observable through ophthalmoscopy. Given that RGC axons passing through the LC are subject to deformation and damage due to ONH cupping, it becomes imperative to elucidate the cellular and molecular mechanisms underlying the pathogenesis of glaucoma. Rodents, frequently employed as experimental or genetic models for glaucoma, have traditionally been believed not to possess an LC structure that is rich in collagen, as is the case in humans (33). Instead, the equivalent region in rodents, known as the glial lamina, is highly enriched in astrocytes expressing glial fibrillary acidic protein (GFAP) (33). Given that the human LC also consists of astrocytes, and considering the close proximity of these astrocytes to RGC axons, any changes in their function are likely to exert a significant influence on the optic nerve (Figure 1C). In addition, the LC contains microglia, which become reactive and accumulate in response to optic nerve injuries (34). In humans, the LC is enriched in collagen, a major component of the extracellular matrix (ECM) (9). The enlargement of the ONH cupping is induced by ECM remodeling, a process initiated by degradation and production of ECM. Matrix metalloproteinases (MMPs), highly expressed in astrocytes and microglia, plays a role in ECM degradation (35, 36). Single nucleotide polymorphisms (SNPs) in the MMP9 gene are associated with a higher risk of primary open-angle glaucoma (POAG) and NTG (37). Additionally, the production of ECM is crucial for tissue remodeling. Mutations or SNPs in ECM genes such as thrombospondin1 (THBS1) or fibronectin (FNDC3B) are linked to the risk of glaucoma (38, 39), and both are produced by astrocytes (40–42). Furthermore, since ONH cupping can be observed in patients irrespective of their IOP levels, including POAG and NTG, it is plausible that tissue changes and the pathogenesis of glaucoma are more closely linked to glial dysfunction than to elevated IOP.
5 The role of glial cells in promoting axonal regeneration within the ONH
Axonal injury occurring at the ONH stands as one of the pivotal events in the initiation and progression of glaucomatous pathology (Figure 2A). The optic nerve crush (ONC) model has been established as a well-recognized experimental paradigm for the assessment of axonal regeneration. Earlier investigations have unequivocally illustrated that axonal regeneration can be augmented through the grafting of peripheral nerves (43–45). This underscores the critical role played by extracellular factors and/or the microenvironment in regulating the regenerative capacity of RGC axons. In the context of spinal cord injury (SCI), the resurgence of axonal growth is contingent upon the presence of reactive astrocytes, while scar-forming astrocytes express molecules conducive to axonal growth, such as laminin (46). The effects are further potentiated by neurotrophic factors, which elicit a robust resurgence of axonal growth through the astrocytic scar and across lesion cores, exceeding control conditions by more than a hundredfold (47).
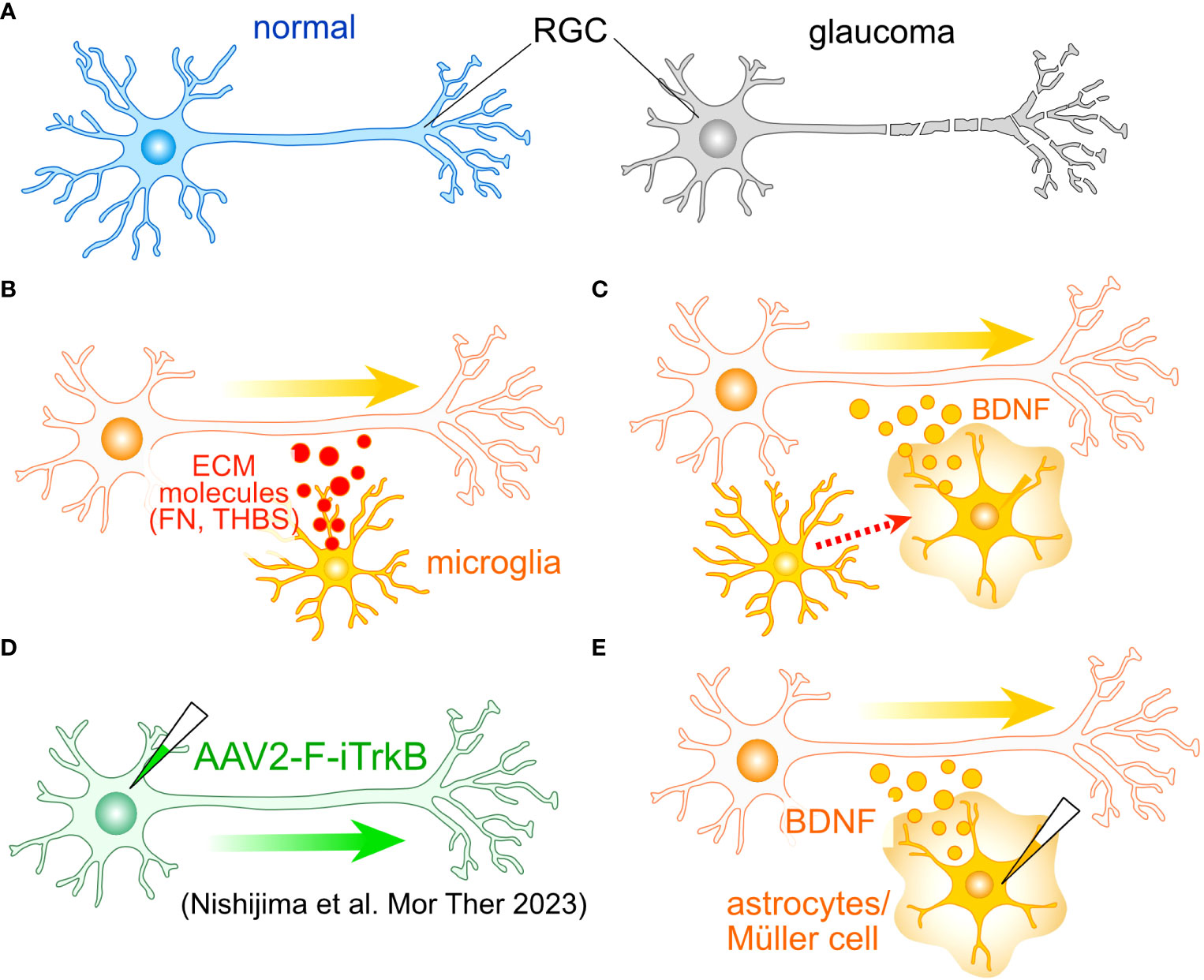
Figure 2 Potential roles of glial cells in the regeneration of RGC axon. (A) One of the most critical aspects of glaucoma is the damage to RGC axons. To identify potential molecular targets for axonal regeneration, researchers often employ the optic nerve crush (ONC) model. (B) Tissue regenerative microglia. Microglia involved in tissue regeneration express extracellular matrix (ECM) molecules like fibronectin (FN) and thrombospondin (THBS), potentially facilitating the regeneration of RGC axons. (C) Microglia-induced BDNF Expression: Microglia can induce the expression of BDNF in Müller cells, which may also accelerate axon regeneration. (D) An example of cell-autonomous enhancement of RGC axon regeneration. The induction of the farnesylated intracellular domain of TrkB (F-iTrkB) leads to a remarkable enhancement of axonal regeneration following ONC. (E) AAV-mediated expression of BDNF in astrocytes and Müller cells may stimulate axon regeneration and provide protection to RGCs, respectively.
Microglia also harbor the potential to support axonal growth. The transplantation of immature microglia has been shown to significantly enhance the recuperative process and foster axon regeneration following SCI (48). These microglial cells manifest the expression of various ECM proteins, notably including fibronectin and thrombospondin (Figure 2B). Furthermore, they exhibit the presence of endopeptidase inhibitors, which serve as crucial regulators in the resolution of inflammation. The ECM-mediated facilitation of axonal regrowth is also instigated by astrocytes (46). Moreover, microglia have been observed to elicit the expression of brain-derived neurotrophic factor (BDNF) in Müller cells (49) (Figure 2C). Concurrent administration of neurotrophic factors alongside glia-mediated support has been demonstrated to engender a robust resurgence of axon growth following SCI (47). Consequently, the amalgamation of intrinsic mechanisms with glia-mediated support holds the promise of inducing a synergistic and remarkable rejuvenation of RGC axons.
6 The role of Müller cells in the protection of RGCs
Notably, neurotrophic factor signaling in reactive astrocytes has been documented to exert a protective influence on RGCs during the early stages of glaucoma (23). Among the neurotrophic factors, BDNF and its receptor TrkB are postulated to be pivotal in upholding the integrity of RGCs in glaucoma (50, 51). Müller cells emerge as the primary source of neurotrophic factors, their induction is triggered by various stimuli and insults (52–57). The sustained expression of BDNF in Müller cells has been demonstrated to confer protection upon RGCs following optic nerve injury (58, 59). Recent research has spotlighted the adeno-associated virus (AAV)-mediated enhancement of TrkB signaling in RGCs, leading to both cryoprotection against glaucoma and a vigorous resurgence of RGC axons (60) (Figure 2D). Collectively, these findings posit that Müller cell-derived neurotrophic factors, with particular emphasis on BDNF, hold paramount importance in protecting RGCs against glaucoma. Additionally, both astrocytes and Müller cells emerge as promising candidates for promoting the regeneration of RGC axons (Figure 2E).
7 Synapse disassembly in the context of glaucoma
Glaucoma has traditionally been regarded as an optic neuropathy that results in optic nerve damage and RGC degeneration. RGC dendrites receive inputs from bipolar and amacrine cells, establishing synaptic connections in the IPL (Figure 1A). Visual information from photoreceptors is relayed to RGCs, then transmitted via the optic nerve to visual centers in the brain. Dendritic atrophy and synapse loss in RGCs can lead to visual deficits. It is well-established that RGCs undergo age-related dendritic atrophy preceding the degeneration of their cell bodies (61) (Figure 3A). Dendritic atrophy and synapse loss in RGCs represent shared structural characteristics observed in animal models of glaucoma and post-mortem human retinas (10, 11, 61–67). An accumulating body of evidence suggests that dendritic atrophy in RGCs and synapse loss within the IPL constitute early indicators of glaucomatous pathology (67–71), alongside optic nerve and RGC soma degeneration. Although achieving selective control over dendritic/synaptic atrophy poses a challenge, several studies have demonstrated that inhibiting atrophy is associated with the protection of RGC soma (11, 64, 72). Prevention of dendritic atrophy has been realized through various approaches, including blockade of the complement pathway (11), intravitreal injection of chondroitinase ABC (66) or BDNF (72). In glaucoma, synapses within the IPL are marked by complement C1q (70), and blocking the complement pathway has been shown to confer protection to RGCs (11, 73). Resident microglia eliminate the synapses with C1q and its downstream C3 (70). These extracellular signals and molecules appear to be promising targets for glaucoma treatment. Among them, ANX007, an anti-C1q monoclonal antibody, is currently undergoing clinical trials for the treatment of glaucoma (74).
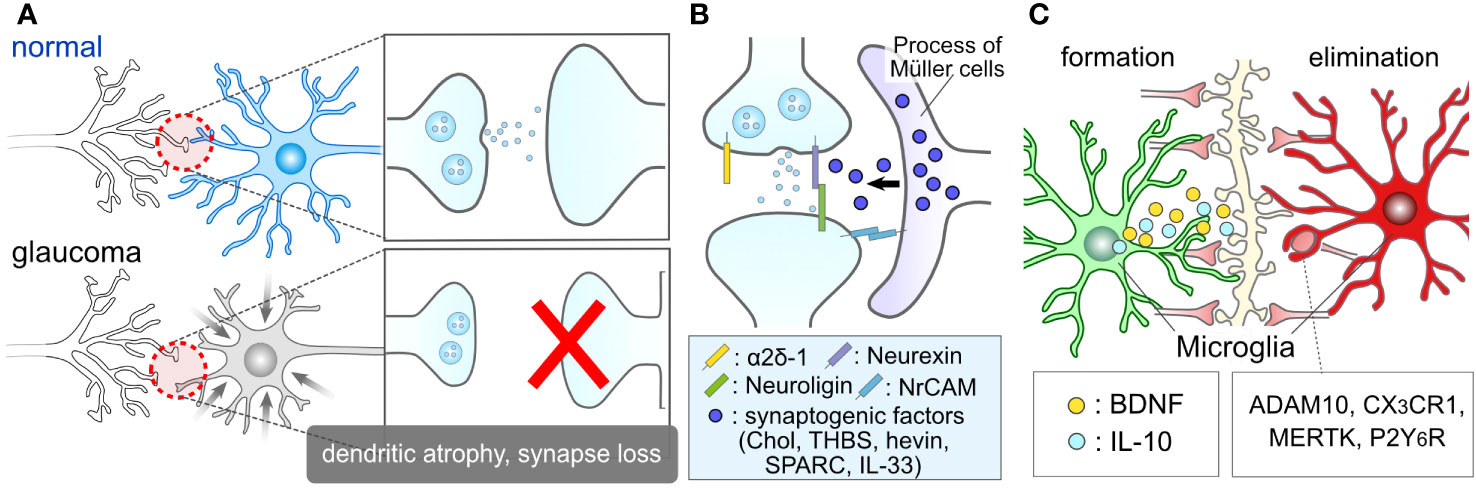
Figure 3 Glial Roles in Synapse Elimination and Formation. (A) Synaptic damage in glaucoma. Glaucoma results in the damage of RGC dendrites and retinal synapses. (B) Müller cell association with retinal synapses. The processes of Müller cells closely interact with retinal synapses. They might release synaptogenic factors, including cholesterol (Chol), thrombospondin (THBS), hevin, SPARC, interleukin-33 (IL-33), and regulate RGC synapses. (C) Microglial involvement in synapse formation and elimination. Microglia are recognized for their role in inducing synapse formation through contact and the release of factors such as BDNF and IL-10. Moreover, microglia are well-documented contributors to synapse elimination through various molecules, including ADAM10, CX3CR1, MERTK, and P2Y6R.
8 The involvement of glial cells in synaptic maintenance
Glial cells play crucial roles in the regulation of synapses, both under normal physiological conditions and in pathological contexts. For instance, glial cells serve as important regulators during the critical period, which is a developmental stage characterized by heightened synaptic plasticity within the nervous system (75, 76). Astrocytes contribute to the formation of synapses through contact-mediated signaling (77, 78) and the production of synaptogenic factors, such as cholesterol (79), thrombospondin (80), hevin, SPARC (81), IL-33 (82) and neuronal adhesion molecule (83) (Figure 3B). While these factors were initially identified in brain astrocytes, they should also be expressed by ocular astrocytes and Müller cells, as evidenced by recent single-cell RNA sequencing data in the human retina, which shows high expression of these genes in astrocytes and Müller cells (84). Microglia also play a role in synaptogenesis through direct contact with synapses (85–87) and the secretion of molecules like interleukin 10 (IL-10) and BDNF (88–90) (Figure 3C). Müller cells serve as a source of BDNF in the retina and express or produce BDNF under various conditions and stimuli (49, 54, 57, 91, 92). The BDNF signal plays a regulatory role in the formation of dendrites in RGCs (93). Given that the fine processes of Müller cells intimately associate with RGC soma (94, 95), dendrites (96), and axons (97), BDNF derived from Müller cells is likely to have a significant impact on the regulation of RGC dendrites.
Glial cells also play a pivotal role in synapse elimination. Microglia, recognized as professional phagocytes, contribute to synapse elimination through various molecular mechanisms, including complement, ADAM10, CX3CR1, MERTK, and P2Y6 receptors (70, 98–103) (Figure 3C). Microglia-mediated synapse elimination serves as a crucial regulator in both the formation and maintenance of physiological neural circuits, as well as the disruption of pathological neural circuits. Dysfunctions in purinergic signaling, such as P2Y6 receptors, have been implicated in the pathogenesis of glaucoma (104–106). Additionally, astrocytes, considered non-professional phagocytes, also participate in synapse pruning through various factors like MEGF10 and MERTK (107–110). In the absence of microglia, astrocytes adopt phagocytic capabilities via TAM receptors (111). Beyond the individual responses of these cells, astrocytes and microglia coordinate their phagocytic functions (112). Moreover, bidirectional communication between them dynamically governs their functions and exerts an influence on synaptic and neuronal conditions (14, 15, 113, 114). These findings underscore the close relationship between glial conditions and synaptic conditions, highlighting glial cells as promising therapeutic targets in the context of glaucoma.
9 Oligodendrocyte dysfunction or loss in the context of glaucoma
Glial cells surrounding the optic nerve, such as OLs and OPC, may also play an important role in the pathogenesis of glaucoma. Myelin, formed by OLs, accelerates signal transduction through axons and provides essential energetic support. Deletion of the gene encoding myelin basic protein (MBP), highly expressed in OLs, leads to axonal swelling and degeneration (115). OLs exhibit persistent turn over, continuously replenished by newly differentiated cells from OPCs. Blocking OL turnover results in reduced myelination and axonal damage (116), highlighting the indispensability of OLs for axon homeostasis and functions.
Traditionally, glaucoma is not categorized as a demyelinating disease, however, emerging evidence suggests dysfunction and potential loss of OLs in glaucoma. DBA/2J mouse model demonstrates OL loss (117). A recent human study has indicated increased radial diffusivity within the optic radiations, serving as a surrogate marker for myelin damage (118). The study also observed a delay in the conduction of multifocal visual evoked potential, indicative of slowed conduction associated with myelin loss. In both the optic nerve injury (118) and glaucoma (119) animal models, OL loss and demyelination precede RGC damage. Maintaining OPC differentiation and myelination involves cell-autonomous mechanisms, such as thyroid hormone (120). Hypothyroidism is suggested as a risk factor for glaucoma (121–123), supporting the idea that impaired OL function contributes to the pathogenesis of glaucoma.
Additionally, non-cell-autonomous mechanisms may play a role. In the DBA/2J mouse, microglia in the myelinated region express and up-regulate the expression of Galectin-3/Mac-2, a phagocytosis-related gene (117), suggesting the involvement of microglia in myelin phagocytosis and the demyelination process. OPCs also express key phagocytotic genes and engage in axon pruning during the developing stage of mouse cortex (124), though the pathological consequence of OPC phagocytotic function in glaucoma remains unclear. OPCs may contribute to neuroinflammation and demyelination via low-density lipoprotein-related receptor 1 (LRP1) (125).
Another potential mechanism involves astrocyte-mediated cholesterol support (126). In the experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis (MS), astrocytes show down-regulated cholesterol synthesis and increased immune responses. Furthermore, phagocytosis by astrocytes may contribute to the demyelination (127, 128). Beyond demyelination, altered energetic support by OLs may be a crucial factor in glaucoma. In human patients with glaucoma, OL mitochondria are small (129). In DBA/2J mouse, monocarboxylate transporter 1 (MCT1), a lactate transporter, is down-regulated in OLs (130), suggesting reduced energetic support by OLs in glaucoma.
Preserving or restoring myelin could be a promising target for glaucoma treatment. Since cholesterol synthesis is promoted during remyelination (131), expediting cholesterol synthesis in astrocytes and/or oligodendrocytes may prove beneficial for glaucoma. Activation of astrocytic ABCA1 stimulates cholesterol synthesis (126) and supports oligodendrocyte survival and myelination (132). Astrocyte-derived CXCL1 also promotes remyelination by stimulating CXCR2 in OLs (133). A ketogenic diet might present an appealing approach to enhance energy availability by reversing the decline in MCT1 (130). Considering that neuronal activity boosts myelination (134, 135), visual stimulation could also be an attractive method for restoring RGC axons and visual function (136).
10 Tools for regulating glial cells and their potential role in glaucoma treatment
As mentioned earlier, glial cells have the capacity to influence the synapses, axons, and soma of RGCs, whether in a degenerative or regenerative manner. Beyond their neuroprotective capabilities, they hold significant potential for stimulating the regeneration of ocular structures and functions. In this section, we discuss various tools for controlling glial cell functions and their potential application in future glaucoma treatments. A summary of the advantages and disadvantages of each technique is shown in Table 1.
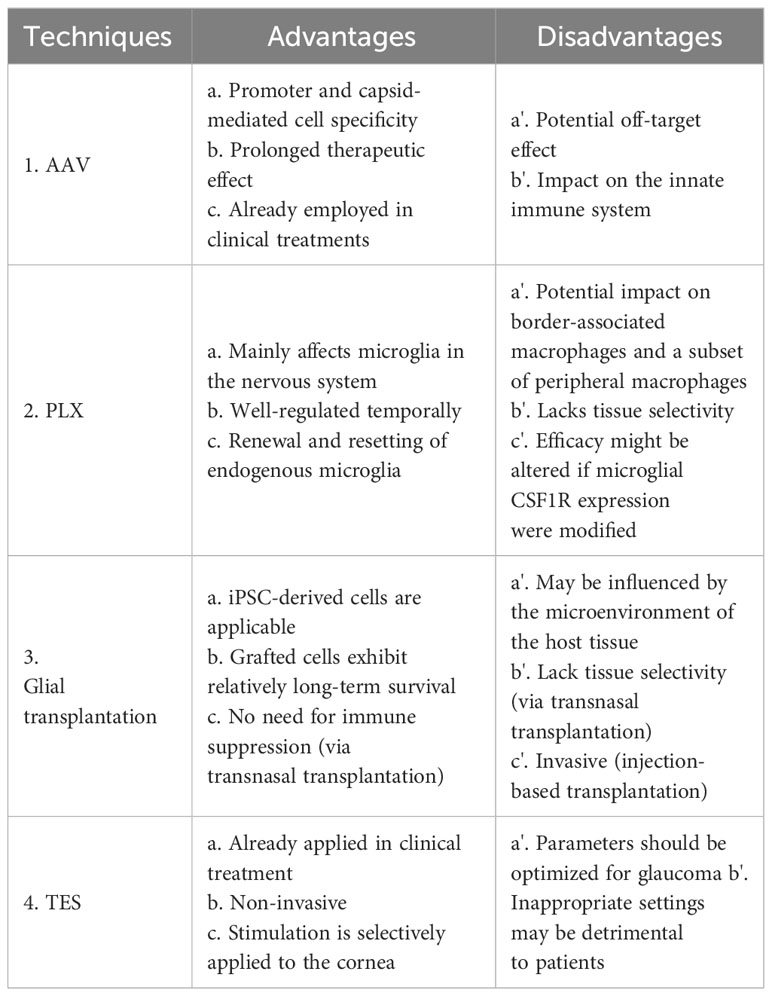
Table 1 Techniques for Glial Cell regulation: advantages and Disadvantages.
10.1 Adeno-associated virus targeting glia for gene therapy in glaucoma
The genetic approach stands as a potent method for addressing neurodegenerative diseases, including ocular conditions (137). In addition to gene therapy aimed at neurons, targeting non-neuronal cells could also prove effective in treating glaucoma. Previously approved gene therapies have operated through non-cell-autonomous mechanisms. An exemplar is Luxturna™ (voretigene neparvovec-rzyl), the inaugural gene therapy approved for treating patients afflicted with inherited retinal dystrophy, a rare genetic disorder affecting the retina (138, 139). In this disease, blindness arises from photoreceptor (PR) degeneration, yet Luxturna™ targets the retinal pigment epithelium (RPE). The restoration of RPE functions provides support and protection to PRs. Such non-cell-autonomous mechanisms could similarly be applied to glaucoma. For cell type-specific gene therapy using AAV, specific promoters tailored for each cell type are employed. The glial fibrillary acidic protein (GFAP) promoter is a key promoter for astrocytes, and the gfaABC1D promoter, exhibiting nearly 100% specificity with 2-fold greater activity (140), is now widely adopted for precise astrocyte-specific gene manipulation via AAV. For achieving Müller cell specificity, there have been developments in engineering AAV capsids. Capsid variants derived from AAV6, such as ShH10 and ShH10Y, demonstrate efficient gene expression in Müller cells upon intravitreal injection (141–143). Moreover, the retinaldehyde-binding protein 1 (RLBP1) promoter has been successful in inducing gene expression specifically in Müller cells (143). For targeting OLs, promoters for the genes encoding proteolipid protein (144), myelin basic protein (145), and the myelin-associated glycoprotein (146) are used. Presently, foundational research efforts striving to facilitate the regeneration of RGC axons and synapses are categorized into two strategies: the promotion of regenerative factors and the prevention of inhibitory factors hindering regeneration. One example of the former strategy is BDNF. As previously mentioned, glial cells serve as the primary source of neurotrophic factors, including BDNF, and they maintain close associations with RGC axons and dendrites. The AAV-mediated expression of BDNF in astrocytes and Müller cells could potentially result in efficient delivery to axons and synapses, respectively. On the other hand, an example of the latter strategy involves insulin-like growth factor (IGF). Insulin and IGF share receptors and downstream signaling pathways (147) both of which are linked to RGC protection and the regeneration of dendrites, synapses, and axons (148–151). Notably, IGF-binding protein (IGFBP), which binds to IGF and hampers its signaling, becomes upregulated in astrocytes during neurodegenerative conditions and neurodevelopmental diseases (152–154). Given that the inhibition of astrocytic IGFBP partially restores neuronal function in the brain (152), the suppression of IGFBP signaling in astrocytes and/or Müller cells may prove beneficial for safeguarding and rejuvenating RGCs by enhancing IGF signaling.
The use of targeted gene therapy in microglia presents itself as an appealing candidate for the treatment of glaucoma. Previously, inducing gene expression in microglia using AAV posed a challenge, but cutting-edge techniques now enable us to achieve such gene induction (155–157). Lin et al. initiated the evolution of the AAV capsid protein (AAV-cMG) in conjunction with the Cre-LoxP system, resulting in selective gene induction in microglia in vivo (28). Okada et al. utilized a 1.7-kb putative promoter region of the Iba1 gene for inducing gene expression in microglia/macrophage cells (156). Young et al. achieved enhanced selectivity for microglia/tissue-resident macrophages by inserting a random 21-mer into the AAV9 capsid (157). It is well-established that microglia exhibit high motility and accumulate at injury sites following ONC (34). These inherent characteristics of microglia allow us to utilize them as vectors for delivering molecules to the site of injury. Furthermore, we can employ proinflammatory gene promoters to activate the expression of the target gene (Figure 4A). For instance, promoters associated with interleukins and tumor necrosis factor α (TNFα) can be employed within this system, given that these molecules are produced by microglia at the lesion core (15). In cases of glaucoma and post-ONC, the ONH sustains damage, leading to the accumulation of microglia at the injury core. This, in turn, triggers the proinflammatory program in AAV-treated microglia, subsequently inducing the production of target genes (Figure 4B). Promoting remyelination emerges as an appealing strategy for vision recovery in glaucoma. The induction of connexin (Cx) genes, such as Cx32 and Cx47, has been associated with a protective effect against leukodystrophy (158, 159), indicating a potential impact on optic nerve remyelination. Moreover, the deletion of the Chrm1gene, which encodes muscarinic receptor 1, a negative regulator of OPC differentiation, leads to increased myelination and axon density (116). The limitation of this technique for the clinical application might lie in the efficiency of AAV delivery to the target tissue and cells. For example, achieving efficient AAV delivery to the optic nerve remains a challenging issue.
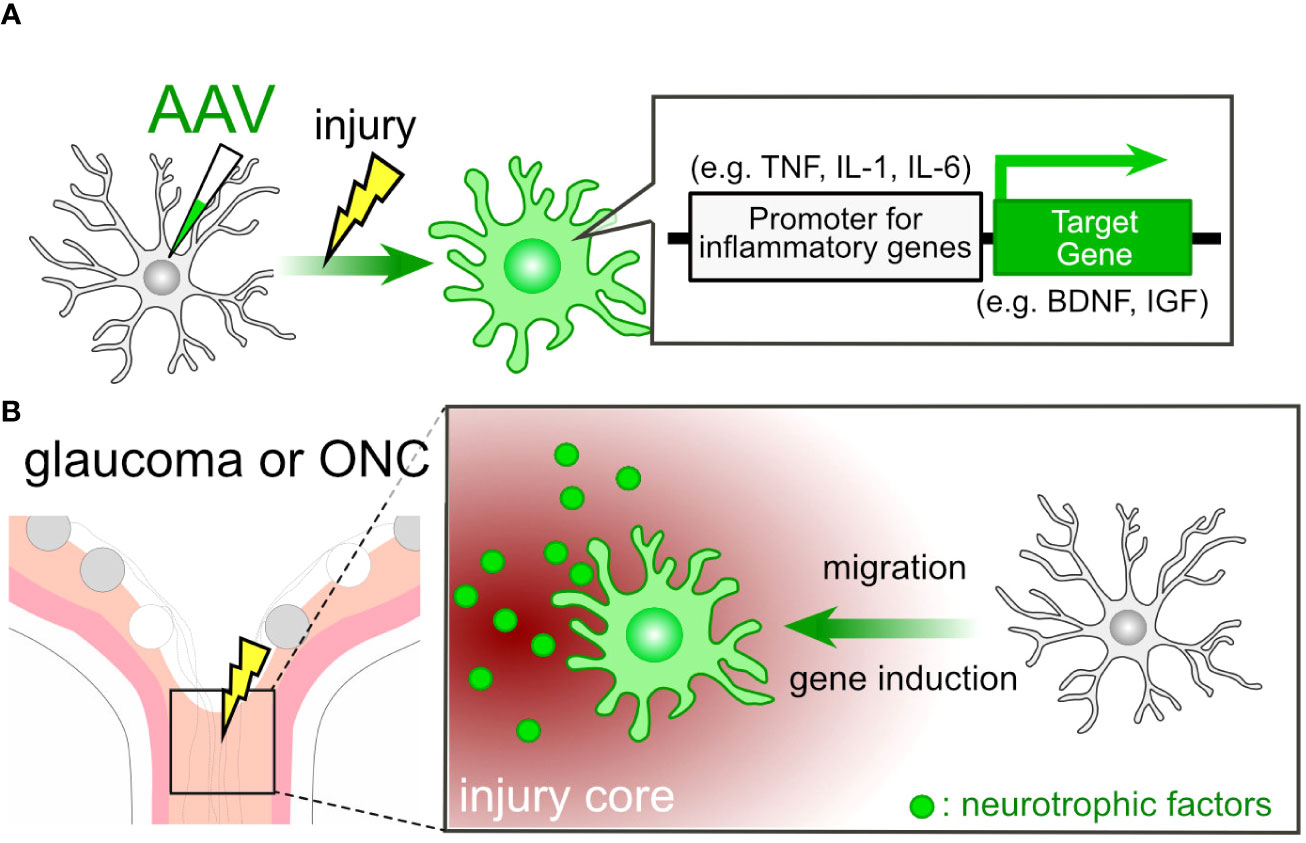
Figure 4 AAV-Mediated Cell Engineering and Target Molecule Delivery by Microglia. (A) AAV-mediated induction of neurotrophic factor genes in microglia. AAV-mediated engineering can induce the expression of neurotrophic factor genes in microglia under pathological conditions. By employing promoters associated with proinflammatory genes, microglia can produce neurotrophic factors like BDNF and IGF in response to pathological conditions. (B) Microglial Response in Glaucoma or ONC: In cases of glaucoma or ONC, the injury core triggers the microglial proinflammatory program, resulting in the induction of neurotrophic factor genes in microglia treated with AAV.
10.2 Pharmacological tools for controlling microglia: PLX compounds
PLX compounds, originally developed by Plexxikon Inc., serve as potent antagonists for the colony-stimulating factor 1 receptor (CSF1R). Oral administration of PLX3397 for either 7 or 21 days results in a reduction in brain microglia numbers by 80-90% and over 95%, respectively (160). Several analogs of PLX compounds, including PLX3397, PLX5562, and PLX647, have been developed. Oral PLX treatment also leads to a significant decrease in the number of retinal microglia (161, 162). This effect is reversible, with microglia repopulating after the discontinuation of PLX compounds (Figure 5). In the case of retinal microglia, the rate of recovery varies among retinal layers, namely the NFL/GCL, IPL, and OPL (163). Upon removal of PLX, microglia spontaneously repopulate through proliferation in both the brain and retina. The removal and repopulation of microglia induce an anti-inflammatory response and promote brain recovery following injury (164–167). Numerous studies have demonstrated that microglia alter their phenotypes to become neurotoxic, and the removal and repopulation of microglia elicit a neuroprotective effect in models of neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease (168–171). Additionally, aside from brain diseases, microglia also play a role in neurodegenerative ocular injuries and diseases (172–174). Given that PLX treatment exhibits a protective effect on RGCs against N-methyl-D-aspartate (NMDA)-mediated toxicity (162), this compound may also have potential applications in the treatment of glaucoma. Although studies have shown that axonal regeneration after ONC is unaffected by the absence of microglia (175), this condition conceals both the neurodegenerative and supportive capabilities of microglia. PLX-mediated repopulation generates ‘new’ microglia with their phenotypes and functions reset, even in pathological conditions. For example, repopulation of aged microglia converts their cellular characteristics to a more youthful state, rescuing age-associated deficits in synapses and brain functions (176). Immature microglia possess the potential for anti-inflammatory responses and tissue regeneration (48). Such a ‘microglial reset’ could also prove valuable in restoring synapses and visual function in the context of glaucoma. Of note, PLX treatment can also be detrimental in certain situations. Microglia depletion from glaucoma model mice using PLX compounds exacerbates RGC damage (177, 178). Since microglia dynamically change their phenotypes, and there might be a neuroprotective glaucoma-associated microglial subcluster, techniques for more precise control of microglia are required, which could also pose a clinical limitation.
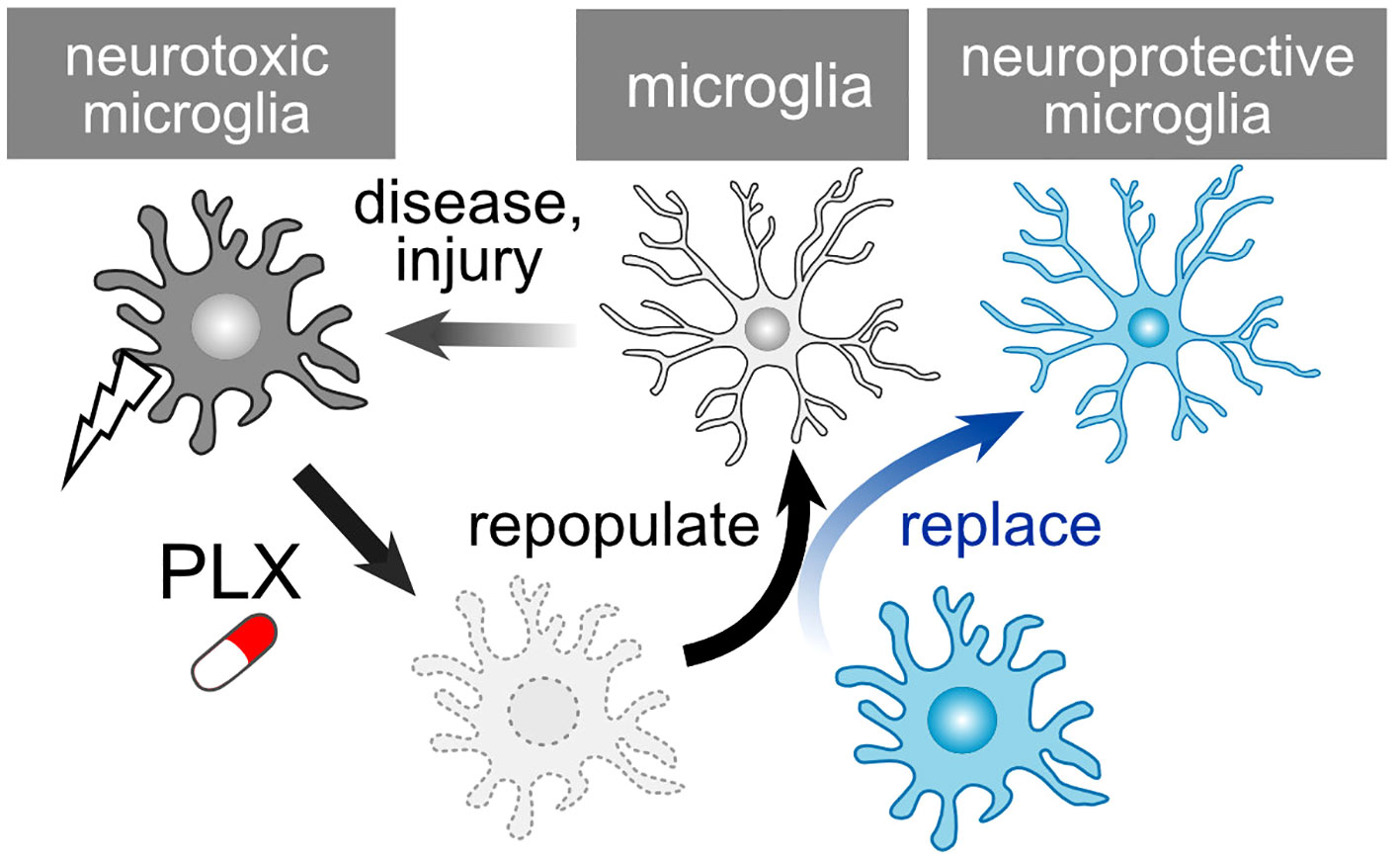
Figure 5 PLX-Mediated Repopulation and Replacement of Microglia. Microglia undergo a shift towards neurotoxic phenotypes following injury or in pathological conditions. PLX treatment depletes these microglia, and they subsequently repopulate once treatment is ceased. The newly generated microglia reset their neurotoxic characteristics. However, considering that repopulated microglia could revert to a neurotoxic phenotype, the replacement of microglia presents an additional strategy to maintain their health.
10.3 Cell transplantation
A recent study has demonstrated the feasibility of transplanting exogenous microglia into the CNS following microglial depletion induced by PLX compounds (179, 180). By combining the depletion of neurotoxic microglia with the transplantation of healthy, normal microglia, a replacement strategy can be employed (Figure 5). The depletion process appears to be crucial, as microglia extend their processes and establish their own territorial domains with an approximate diameter of 50 μm in both mouse and human brains (181). The absence of endogenous microglia permits the exogenously transplanted microglia to infiltrate and integrate into the nervous tissue. When combined with AAV-mediated functionalization of microglia (Figure 4B), this approach enables the precise delivery of specific molecules to designated sites, enhancing the efficiency of recovery while minimizing potential side effects. Transplantation can also be accomplished by introducing human iPS cell-derived microglia (iPSMG) into the mouse retina (182). Beyond microglia, intravitreal astrocyte transplantation may prove beneficial in safeguarding RGCs against damage induced by kainic acid (183). Additionally, transplantation of OPCs contributes to the neuroprotection and regeneration of the optic nerve. OPC transplantation has been shown to protect RGC in glaucoma model animal (184). Furthermore, the transplantation of OPC-rich neurospheres induces the myelination of the optic nerve (185–187). One limitation of clinical application may involve the duration for which the grafted cells survive in the host tissue. In the case of the iPSMG, transnasal transplantation to the brain maintained the grafted microglia for at least 60 days in mice (179, 180). Injected iPSMGs in the mouse retina survived over 200 days (182). However, it is not clear how long they would survive in the human tissue. Another concern is whether the grafted cells maintain their healthy phenotypes, as their phenotypes can be influenced by the microenvironment of the host tissue.
10.4 Transcorneal electrical stimulation
TES represents a non-invasive technique that administers electrical stimulation to the retina via the cornea. This approach has demonstrated therapeutic efficacy in both human patients and animal models afflicted with various injuries and diseases, including ischemic and traumatic optic neuropathies (188), axotomy (189, 190), retinal artery occlusion (191), ischemic damage (192), and photoreceptor degeneration (193, 194). TES has also exhibited a protective effect on RGCs in mouse models of glaucoma (195) and holds the potential to enhance visual function in human patients with glaucoma (196). While the precise mechanisms underlying TES are not fully elucidated, one of its neuroprotective mechanisms involves actions mediated by glial cells. For instance, TES suppresses pro-inflammatory responses by microglia (190, 197, 198) (Figure 6A). Simultaneously, TES induces the expression of various neurotrophic factors, including fibroblast growth factor 2 (FGF2), BDNF, and IGF, in Müller cells (54–56) (Figure 6B). The combination of anti-inflammatory effects and neurotrophic support is likely the primary mechanism behind RGC protection in glaucoma. As previously mentioned, these neurotrophic factors contribute not only to the protection of RGCs but also to the regeneration of dendrites, synapses, axons, and visual functions. Ongoing research aims to refine the parameters of TES for optimal neuroprotection and functional recovery (189, 199). TES is already employed in human patients with retinitis pigmentosa and has demonstrated safety and efficacy in improving visual function (200, 201). A current limitation of TES in the context of glaucoma treatment might be the absence of defined parameters. Optimized TES parameters will contribute to the development of a safe and effective treatment for glaucoma.
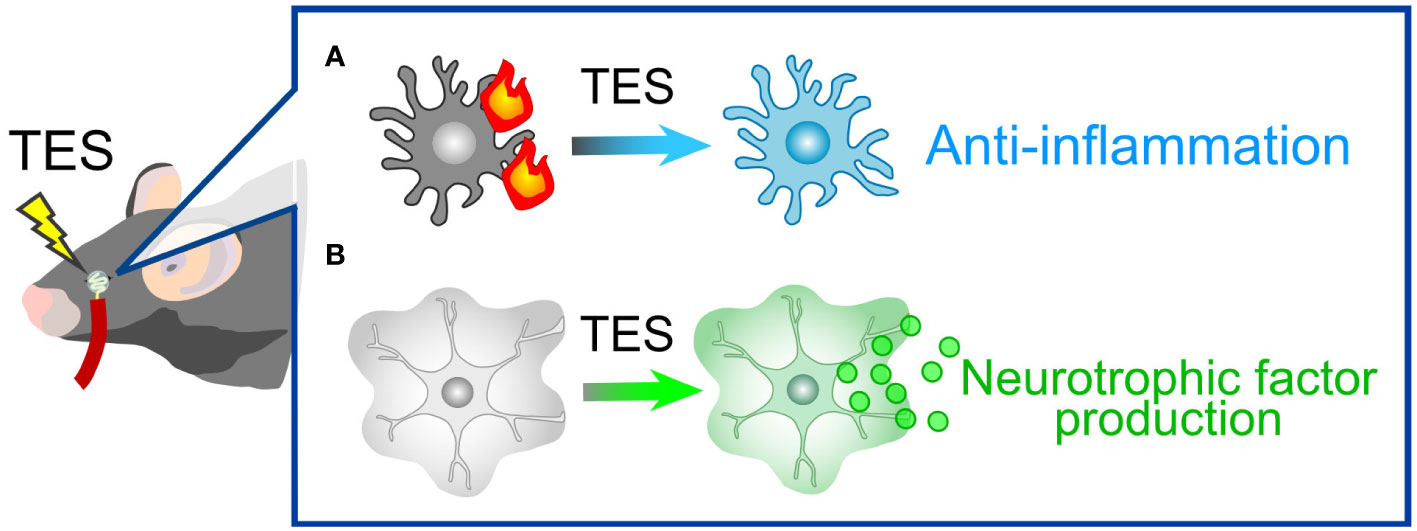
Figure 6 TES-Mediated Alterations in Glial Cells. (A) Microglial response. In pathological conditions, microglia tend to adopt pro-inflammatory phenotypes. TES effectively suppresses microglial inflammatory responses. (B) Müller Cell Expression: TES leads to an upregulation in the expression of neurotrophic factors, including FGF2, BDNF, and IGF, in Müller cells.
10.5 Extracellular vesicles
The EVs encompass membrane-derived vesicles with heterogenous groups, including exosomes and microvesicles (202). Initially described as a means to eliminate intracellular unneeded components to the extracellular space, subsequent studies have revealed their capacity for intercellular communication via transporting various molecules, including nucleic acids, lipids, and proteins. EVs are released from various cells and tissues including ocular cells and tissues. Due to their high stability and permeability to the blood-brain barrier (203), they represent attractive tools for drug delivery and hold significant potential as biomarkers for various diseases. In the ocular tissues, it has been reported that both retinal microglia and Müller cells release EVs (204–206). Considering the neuroprotective effects demonstrated by EVs from glial progenitors after traumatic brain injury (207), it is plausible that EVs from neuroprotective glial cells would similarly confer neuroprotection to RGCs. Moreover, embryonic stem cell-derived EVs, capable to delivering BDNF to other cells (208), could be employed as a therapeutic tool in glaucoma, for instance, by utilizing EVs derived from neurotrophin-overexpressing glia.
11 Concluding remarks
Glial cells are widely distributed throughout the nervous system, including ocular tissues. Pathological changes in glial cells play a pivotal role in driving RGC damage and resulting in visual deficits. Given that neuroprotective glia produce essential neurotrophic factors that impact both neuroprotection and neurodegeneration, glial cells represent an appealing therapeutic target for addressing glaucoma. With the aid of state-of-the-art techniques, we can precisely regulate glial functions, effectively suppressing neurotoxicity while enhancing neuroprotection and regeneration. Several glia-related molecules have already advanced to clinical trials, and we anticipate further advancements in drug discovery research aimed at targeting glial cells.
Author contributions
YS: Funding acquisition, Validation, Visualization, Writing – original draft, Writing – review & editing. KN: Conceptualization, Validation, Writing – review & editing. XG: Funding acquisition, Validation, Writing – review & editing. TH: Project administration, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Takeda Science Foundation (YS); Foundation for Research on Ophthalmic Disease of the Elderly (YS); Japan Society for the Promotion of Science KAKENHI grant numbers JP22K07368 (YS), JP20KK0366 (YS), JP20K07751 (KN), JP21K09688 (XG), JP21H04786 (TH), JP19KK0229 (TH), JP21H02819 (KN and TH), JP21K18279 (KN and TH); and a Grant-in-Aid for Scientific Research on Innovative Areas-Resource and Technical Support Platforms for Promoting Research “Advanced Bioimaging Support” (JP16H06280) (YS). This research was also supported by Joint Research of the Exploratory Research Center on Life and Living Systems (ExCELLS; program number 21-414) (YS).
Acknowledgments
The authors thank the members of the visual research project, Tokyo Metropolitan Institute of Medical Science for their continuous support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CNS, central nervous system; ECM, extracellular matrix; GCL, ganglion cell layer; IL, interleukin; IOP, intraocular pressure; IPL, inner plexiform layer; LC, lamina cribrosa; NMDA, N-methyl-D-aspartate; RGC, retinal ganglion cell; OL, oligodendrocyte; ONC, optic nerve crush; ONH, optic nerve head; OPC, oligodendrocyte precursor cell; PR, photoreceptor; scRNA-seq, single-cell RNA sequence.
References
1. Illis LS. Central nervous system regeneration does not occur. Spinal Cord (2012) 50:259–63. doi: 10.1038/sc.2011.132
2. Quigley HA. Number of people with glaucoma worldwide. Br J Ophthalmol (1996) 80:389–93. doi: 10.1136/bjo.80.5.389
3. Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA (2014) 311:1901–11. doi: 10.1001/jama.2014.3192
4. Weinreb RN, Leung CK, Crowston JG, Medeiros FA, Friedman DS, Wiggs JL, et al. Primary open-angle glaucoma. Nat Rev Dis Primers (2016) 2:16067. doi: 10.1038/nrdp.2016.67
5. Heijl A, Leske MC, Bengtsson B, Hyman L, Bengtsson B, Hussein M, et al. Reduction of intraocular pressure and glaucoma progression: results from the Early Manifest Glaucoma Trial. Arch Ophthalmol (2002) 120:1268–79. doi: 10.1001/archopht.120.10.1268
6. Kass MA, Heuer DK, Higginbotham EJ, Johnson CA, Keltner JL, Miller JP, et al. The Ocular Hypertension Treatment Study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch Ophthalmol (2002) 120:701–13. doi: 10.1001/archopht.120.6.701
7. Smith CA, Vianna JR, Chauhan BC. Assessing retinal ganglion cell damage. Eye (Lond) (2017) 31:209–17. doi: 10.1038/eye.2016.295
8. Quigley HA, Addicks EM. Chronic experimental glaucoma in primates. II. Effect of extended intraocular pressure elevation on optic nerve head and axonal transport. Invest Ophthalmol Vis Sci (1980) 19:137–52.
9. Sommer A, Katz J, Quigley HA, Miller NR, Robin AL, Richter RC, et al. Clinically detectable nerve fiber atrophy precedes the onset of glaucomatous field loss. Arch Ophthalmol (1991) 109:77–83. doi: 10.1001/archopht.1991.01080010079037
10. Williams PA, Howell GR, Barbay JM, Braine CE, Sousa GL, John SW, et al. Retinal ganglion cell dendritic atrophy in DBA/2J glaucoma. PloS One (2013) 8:e72282. doi: 10.1371/journal.pone.0072282
11. Williams PA, Tribble JR, Pepper KW, Cross SD, Morgan BP, Morgan JE, et al. Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma. Mol Neurodegener (2016) 11:26. doi: 10.1186/s13024-016-0091-6
12. Harada T, Harada C, Nakamura K, Quah HM, Okumura A, Namekata K, et al. The potential role of glutamate transporters in the pathogenesis of normal tension glaucoma. J Clin Invest (2007) 117:1763–70. doi: 10.1172/JCI30178
13. Shinozaki Y, Leung A, Namekata K, Saitoh S, Nguyen HB, Takeda A, et al. Astrocytic dysfunction induced by ABCA1 deficiency causes optic neuropathy. Sci Adv (2022) 8:eabq1081. doi: 10.1126/sciadv.abq1081
14. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature (2017) 541:481–7. doi: 10.1038/nature21029
15. Shinozaki Y, Shibata K, Yoshida K, Shigetomi E, Gachet C, Ikenaka K, et al. Transformation of astrocytes to a neuroprotective phenotype by microglia via P2Y(1) receptor downregulation. Cell Rep (2017) 19:1151–64. doi: 10.1016/j.celrep.2017.04.047
16. Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci (2021) 24:312–25. doi: 10.1038/s41593-020-00783-4
17. Maragakis NJ, Rothstein JD. Mechanisms of Disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol (2006) 2:679–89. doi: 10.1038/ncpneuro0355
18. Paolicelli RC, Sierra A, Stevens B, Tremblay ME, Aguzzi A, Ajami B, et al. Microglia states and nomenclature: A field at its crossroads. Neuron (2022) 110:3458–83. doi: 10.1016/j.neuron.2022.10.020
19. Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol (2010) 6:193–201. doi: 10.1038/nrneurol.2010.17
20. Guttenplan KA, Stafford BK, El-Danaf RN, Adler DI, Munch AE, Weigel MK, et al. Neurotoxic reactive astrocytes drive neuronal death after retinal injury. Cell Rep (2020) 31:107776. doi: 10.1016/j.celrep.2020.107776
21. Sun D, Moore S, Jakobs TC. Optic nerve astrocyte reactivity protects function in experimental glaucoma and other nerve injuries. J Exp Med (2017) 214:1411–30. doi: 10.1084/jem.20160412
22. Fan Z, Brooks DJ, Okello A, Edison P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain (2017) 140:792–803. doi: 10.1093/brain/aww349
23. Harder JM, Braine CE, Williams PA, Zhu X, MacNicoll KH, Sousa GL, et al. Early immune responses are independent of RGC dysfunction in glaucoma with complement component C3 being protective. Proc Natl Acad Sci U.S.A. (2017) 114:E3839–48. doi: 10.1073/pnas.1608769114
24. Choi I, Wang M, Yoo S, Xu P, Seegobin SP, Li X, et al. Autophagy enables microglia to engage amyloid plaques and prevents microglial senescence. Nat Cell Biol (2023) 25:963–74. doi: 10.1038/s41556-023-01158-0
25. Benhar I, Ding J, Yan W, Whitney IE, Jacobi A, Sud M, et al. Temporal single-cell atlas of non-neuronal retinal cells reveals dynamic, coordinated multicellular responses to central nervous system injury. Nat Immunol (2023) 24:700–13. doi: 10.1038/s41590-023-01437-w
26. Krylov A, Yu S, Veen K, Newton A, Ye A, Qin H, et al. Heterogeneity in quiescent Muller glia in the uninjured zebrafish retina drive differential responses following photoreceptor ablation. Front Mol Neurosci (2023) 16:1087136. doi: 10.3389/fnmol.2023.1087136
27. Lamas M, Martinez-Colin EJ. Muller cell molecular heterogeneity: facts and predictions. ASN Neuro (2022) 14:17590914221106903. doi: 10.1177/17590914221106903
28. Liu B, He J, Zhong L, Huang L, Gong B, Hu J, et al. Single-cell transcriptome reveals diversity of Muller cells with different metabolic-mitochondrial signatures in normal and degenerated macula. Front Neurosci (2022) 16:1079498. doi: 10.3389/fnins.2022.1079498
29. Monavarfeshani A, Yan W, Pappas C, Odenigbo KA, He Z, Segre AV, et al. Transcriptomic analysis of the ocular posterior segment completes a cell atlas of the human eye. Proc Natl Acad Sci U.S.A. (2023) 120:e2306153120. doi: 10.1073/pnas.2306153120
30. O’Koren EG, Yu C, Klingeborn M, Wong AYW, Prigge CL, Mathew R, et al. Microglial function is distinct in different anatomical locations during retinal homeostasis and degeneration. Immunity (2019) 50:723–737 e7. doi: 10.1016/j.immuni.2019.02.007
31. Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: A universal immune sensor of neurodegeneration. Cell (2018) 173:1073–81. doi: 10.1016/j.cell.2018.05.003
32. Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci (2020) 23:701–6. doi: 10.1038/s41593-020-0624-8
33. Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol (2007) 179:1523–37. doi: 10.1083/jcb.200706181
34. Qu J, Jakobs TC. The time course of gene expression during reactive gliosis in the optic nerve. PloS One (2013) 8:e67094. doi: 10.1371/journal.pone.0067094
35. Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci (2008) 28:264–78. doi: 10.1523/JNEUROSCI.4178-07.2008
36. Yin KJ, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X, et al. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J Neurosci (2006) 26:10939–48. doi: 10.1523/JNEUROSCI.2085-06.2006
37. Suh W, Won HH, Kee C. The association of single-nucleotide polymorphisms in the MMP-9 gene with normal tension glaucoma and primary open-angle glaucoma. Curr Eye Res (2018) 43:534–8. doi: 10.1080/02713683.2017.1410177
38. Fu H, Siggs OM, Knight LS, Staffieri SE, Ruddle JB, Birsner AE, et al. Thrombospondin 1 missense alleles induce extracellular matrix protein aggregation and TM dysfunction in congenital glaucoma. J Clin Invest (2022) 132:e156967. doi: 10.1172/JCI156967
39. Hysi PG, Cheng CY, Springelkamp H, Macgregor S, Bailey JNC, Wojciechowski R, et al. Genome-wide analysis of multi-ancestry cohorts identifies new loci influencing intraocular pressure and susceptibility to glaucoma. Nat Genet (2014) 46:1126–30. doi: 10.1038/ng.3087
40. Danjo Y, Shigetomi E, Hirayama YJ, Kobayashi K, Ishikawa T, Fukazawa Y, et al. Transient astrocytic mGluR5 expression drives synaptic plasticity and subsequent chronic pain in mice. J Exp Med (2022) 219:e20210989. doi: 10.1084/jem.20210989
41. Kim SK, Hayashi H, Ishikawa T, Shibata K, Shigetomi E, Shinozaki Y, et al. Cortical astrocytes rewire somatosensory cortical circuits for peripheral neuropathic pain. J Clin Invest (2016) 126:1983–97. doi: 10.1172/JCI82859
42. Tom VJ, Doller CM, Malouf AT, Silver J. Astrocyte-associated fibronectin is critical for axonal regeneration in adult white matter. J Neurosci (2004) 24:9282–90. doi: 10.1523/JNEUROSCI.2120-04.2004
43. Benowitz LI, Yin Y. Optic nerve regeneration. Arch Ophthalmol (2010) 128:1059–64. doi: 10.1001/archophthalmol.2010.152
44. Berry M, Carlile J, Hunter A. Peripheral nerve explants grafted into the vitreous body of the eye promote the regeneration of retinal ganglion cell axons severed in the optic nerve. J Neurocytol (1996) 25:147–70. doi: 10.1007/BF02284793
45. Berry M, Carlile J, Hunter A, Tsang W, Rosenstiel P, Sievers J. Optic nerve regeneration after intravitreal peripheral nerve implants: trajectories of axons regrowing through the optic chiasm into the optic tracts. J Neurocytol (1999) 28:721–41. doi: 10.1023/A:1007086004022
46. Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature (2016) 532:195–200. doi: 10.1038/nature17623
47. Anderson MA, O’Shea TM, Burda JE, Ao Y, Barlatey SL, Bernstein AM, et al. Required growth facilitators propel axon regeneration across complete spinal cord injury. Nature (2018) 561:396–400. doi: 10.1038/s41586-018-0467-6
48. Li Y, He X, Kawaguchi R, Zhang Y, Wang Q, Monavarfeshani A, et al. Microglia-organized scar-free spinal cord repair in neonatal mice. Nature (2020) 587:613–8. doi: 10.1038/s41586-020-2795-6
49. Harada T, Harada C, Kohsaka S, Wada E, Yoshida K, Ohno S, et al. Microglia-Muller glia cell interactions control neurotrophic factor production during light-induced retinal degeneration. J Neurosci (2002) 22:9228–36. doi: 10.1523/JNEUROSCI.22-21-09228.2002
50. Pease ME, McKinnon SJ, Quigley HA, Kerrigan-Baumrind LA, Zack DJ. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest Ophthalmol Vis Sci (2000) 41:764–74.
51. Quigley HA, McKinnon SJ, Zack DJ, Pease ME, Kerrigan-Baumrind LA, Kerrigan DF, et al. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest Ophthalmol Vis Sci (2000) 41:3460–6.
52. Honjo M, Tanihara H, Kido N, Inatani M, Okazaki K, Honda Y. Expression of ciliary neurotrophic factor activated by retinal Muller cells in eyes with NMDA- and kainic acid-induced neuronal death. Invest Ophthalmol Vis Sci (2000) 41:552–60.
53. Morimoto T, Miyoshi T, Matsuda S, Tano Y, Fujikado T, Fukuda Y. Transcorneal electrical stimulation rescues axotomized retinal ganglion cells by activating endogenous retinal IGF-1 system. Invest Ophthalmol Vis Sci (2005) 46:2147–55. doi: 10.1167/iovs.04-1339
54. Sato T, Fujikado T, Lee TS, Tano Y. Direct effect of electrical stimulation on induction of brain-derived neurotrophic factor from cultured retinal Muller cells. Invest Ophthalmol Vis Sci (2008) 49:4641–6. doi: 10.1167/iovs.08-2049
55. Sato T, Fujikado T, Morimoto T, Matsushita K, Harada T, Tano Y. Effect of electrical stimulation on IGF-1 transcription by L-type calcium channels in cultured retinal Muller cells. Jpn J Ophthalmol (2008) 52:217–23. doi: 10.1007/s10384-008-0533-y
56. Sato T, Lee TS, Takamatsu F, Fujikado T. Induction of fibroblast growth factor-2 by electrical stimulation in cultured retinal Mueller cells. Neuroreport (2008) 19:1617–21. doi: 10.1097/WNR.0b013e3283140f25
57. Seki M, Tanaka T, Sakai Y, Fukuchi T, Abe H, Nawa H, et al. Muller Cells as a source of brain-derived neurotrophic factor in the retina: noradrenaline upregulates brain-derived neurotrophic factor levels in cultured rat Muller cells. Neurochem Res (2005) 30:1163–70. doi: 10.1007/s11064-005-7936-7
58. Di Polo A, Aigner LJ, Dunn RJ, Bray GM, Aguayo AJ. Prolonged delivery of brain-derived neurotrophic factor by adenovirus-infected Muller cells temporarily rescues injured retinal ganglion cells. Proc Natl Acad Sci U.S.A. (1998) 95:3978–83. doi: 10.1073/pnas.95.7.3978
59. Mansour-Robaey S, Clarke DB, Wang YC, Bray GM, Aguayo AJ. Effects of ocular injury and administration of brain-derived neurotrophic factor on survival and regrowth of axotomized retinal ganglion cells. Proc Natl Acad Sci U.S.A. (1994) 91:1632–6. doi: 10.1073/pnas.91.5.1632
60. Nishijima E, Honda S, Kitamura Y, Namekata K, Kimura A, Guo X, et al. Vision protection and robust axon regeneration in glaucoma models by membrane-associated Trk receptors. Mol Ther (2023) 31:810–24. doi: 10.1016/j.ymthe.2022.11.018
61. Samuel MA, Zhang Y, Meister M, Sanes JR. Age-related alterations in neurons of the mouse retina. J Neurosci (2011) 31:16033–44. doi: 10.1523/JNEUROSCI.3580-11.2011
62. Berry RH, Qu J, John SW, Howell GR, Jakobs TC. Synapse loss and dendrite remodeling in a mouse model of glaucoma. PloS One (2015) 10:e0144341. doi: 10.1371/journal.pone.0144341
63. Jakobs TC, Libby RT, Ben Y, John SW, Masland RH. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J Cell Biol (2005) 171:313–25. doi: 10.1083/jcb.200506099
64. Risner ML, Pasini S, McGrady NR, Calkins DJ. Bax contributes to retinal ganglion cell dendritic degeneration during glaucoma. Mol Neurobiol (2022) 59:1366–80. doi: 10.1007/s12035-021-02675-5
65. Tribble JR, Vasalauskaite A, Redmond T, Young RD, Hassan S, Fautsch MP, et al. Midget retinal ganglion cell dendritic and mitochondrial degeneration is an early feature of human glaucoma. Brain Commun (2019) 1:fcz035. doi: 10.1093/braincomms/fcz035
66. Tribble JR, Williams PA, Caterson B, Sengpiel F, Morgan JE. Digestion of the glycosaminoglycan extracellular matrix by chondroitinase ABC supports retinal ganglion cell dendritic preservation in a rodent model of experimental glaucoma. Mol Brain (2018) 11:69. doi: 10.1186/s13041-018-0412-5
67. Weber AJ, Kaufman PL, Hubbard WC. Morphology of single ganglion cells in the glaucomatous primate retina. Invest Ophthalmol Vis Sci (1998) 39:2304–20.
68. El-Danaf RN, Huberman AD. Characteristic patterns of dendritic remodeling in early-stage glaucoma: evidence from genetically identified retinal ganglion cell types. J Neurosci (2015) 35:2329–43. doi: 10.1523/JNEUROSCI.1419-14.2015
69. Ou Y, Jo RE, Ullian EM, Wong RO, Della Santina L. Selective vulnerability of specific retinal ganglion cell types and synapses after transient ocular hypertension. J Neurosci (2016) 36:9240–52. doi: 10.1523/JNEUROSCI.0940-16.2016
70. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
71. Weber AJ, Harman CD. Structure-function relations of parasol cells in the normal and glaucomatous primate retina. Invest Ophthalmol Vis Sci (2005) 46:3197–207. doi: 10.1167/iovs.04-0834
72. Weber AJ, Harman CD. BDNF preserves the dendritic morphology of alpha and beta ganglion cells in the cat retina after optic nerve injury. Invest Ophthalmol Vis Sci (2008) 49:2456–63. doi: 10.1167/iovs.07-1325
73. Bosco A, Anderson SR, Breen KT, Romero CO, Steele MR, Chiodo VA, et al. Complement C3-targeted gene therapy restricts onset and progression of neurodegeneration in chronic mouse glaucoma. Mol Ther (2018) 26:2379–96. doi: 10.1016/j.ymthe.2018.08.017
74. Sun Y, Wirta D, Murahashi W, Mathur V, Sankaranarayanan S, Taylor LK, et al. Safety and target engagement of complement C1q inhibitor ANX007 in neurodegenerative eye disease: results from phase I studies in glaucoma. Ophthalmol Sci (2023) 3:100290. doi: 10.1016/j.xops.2023.100290
75. Ackerman SD, Perez-Catalan NA, Freeman MR, Doe CQ. Astrocytes close a motor circuit critical period. Nature (2021) 592:414–20. doi: 10.1038/s41586-021-03441-2
76. Ribot J, Breton R, Calvo CF, Moulard J, Ezan P, Zapata J, et al. Astrocytes close the mouse critical period for visual plasticity. Science (2021) 373:77–81. doi: 10.1126/science.abf5273
77. Filosa A, Paixao S, Honsek SD, Carmona MA, Becker L, Feddersen B, et al. Neuron-glia communication via EphA4/ephrin-A3 modulates LTP through glial glutamate transport. Nat Neurosci (2009) 12:1285–92. doi: 10.1038/nn.2394
78. Murai KK, Nguyen LN, Irie F, Yamaguchi Y, Pasquale EB. Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nat Neurosci (2003) 6:153–60. doi: 10.1038/nn994
79. Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A, et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science (2001) 294:1354–7. doi: 10.1126/science.294.5545.1354
80. Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell (2005) 120:421–33. doi: 10.1016/j.cell.2004.12.020
81. Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, et al. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci U.S.A. (2011) 108:E440–9. doi: 10.1073/pnas.1104977108
82. Wang Y, Fu WY, Cheung K, Hung KW, Chen C, Geng H, et al. Astrocyte-secreted IL-33 mediates homeostatic synaptic plasticity in the adult hippocampus. Proc Natl Acad Sci U.S.A. (2021) 118:e2020810118. doi: 10.1073/pnas.2020810118
83. Takano T, Wallace JT, Baldwin KT, Purkey AM, Uezu A, Courtland JL, et al. Chemico-genetic discovery of astrocytic control of inhibition in vivo. Nature (2020) 588:296–302. doi: 10.1038/s41586-020-2926-0
84. Lukowski SW, Patel J, Andersen SB, Sim SL, Wong HY, Tay J, et al. Single-cell transcriptional profiling of aortic endothelium identifies a hierarchy from endovascular progenitors to differentiated cells. Cell Rep (2019) 27:2748–2758 e3. doi: 10.1016/j.celrep.2019.04.102
85. Gallo NB, Berisha A, Van Aelst L. Microglia regulate chandelier cell axo-axonic synaptogenesis. Proc Natl Acad Sci U.S.A. (2022) 119:e2114476119. doi: 10.1073/pnas.2114476119
86. Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, Murakoshi H, et al. Microglia contact induces synapse formation in developing somatosensory cortex. Nat Commun (2016) 7:12540. doi: 10.1038/ncomms12540
87. Weinhard L, di Bartolomei G, Bolasco G, MaChado P, Schieber NL, Neniskyte U, et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun (2018) 9:1228. doi: 10.1038/s41467-018-03566-5
88. Huang L, Jin J, Chen K, You S, Zhang H, Sideris A, et al. BDNF produced by cerebral microglia promotes cortical plasticity and pain hypersensitivity after peripheral nerve injury. PloS Biol (2021) 19:e3001337. doi: 10.1371/journal.pbio.3001337
89. Lim SH, Park E, You B, Jung Y, Park AR, Park SG, et al. Neuronal synapse formation induced by microglia and interleukin 10. PloS One (2013) 8:e81218. doi: 10.1371/journal.pone.0081218
90. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell (2013) 155:1596–609. doi: 10.1016/j.cell.2013.11.030
91. Le YZ, Xu B, Chucair-Elliott AJ, Zhang H, Zhu M, Mediates Retinal Muller Cell Viability VEGF. and neuroprotection through BDNF in diabetes. Biomolecules (2021) 11:712. doi: 10.3390/biom11050712
92. Suzumura A, Kaneko H, Funahashi Y, Takayama K, Nagaya M, Ito S, et al. n-3 fatty acid and its metabolite 18-HEPE ameliorate retinal neuronal cell dysfunction by enhancing muller BDNF in diabetic retinopathy. Diabetes (2020) 69:724–35. doi: 10.2337/db19-0550
93. Lom B, Cogen J, Sanchez AL, Vu T, Cohen-Cory S. Local and target-derived brain-derived neurotrophic factor exert opposing effects on the dendritic arborization of retinal ganglion cells in vivo. J Neurosci (2002) 22:7639–49. doi: 10.1523/JNEUROSCI.22-17-07639.2002
94. Danjo Y, Shinozaki Y, Natsubori A, Kubota Y, Kashiwagi K, Tanaka KF, et al. The mlc1 promoter directs muller cell-specific gene expression in the retina. Transl Vis Sci Technol (2022) 11:25. doi: 10.1167/tvst.11.1.25
95. Wang J, O’Sullivan ML, Mukherjee D, Punal VM, Farsiu S, Kay JN. Anatomy and spatial organization of Muller glia in mouse retina. J Comp Neurol (2017) 525:1759–77. doi: 10.1002/cne.24153
96. Tworig JM, Coate CJ, Feller MB. Excitatory neurotransmission activates compartmentalized calcium transients in Muller glia without affecting lateral process motility. Elife (2021) 10:e73202. doi: 10.7554/eLife.73202
97. Shinozaki Y, Kashiwagi K, Koizumi S. Astrocyte immune functions and glaucoma. Int J Mol Sci (2023) 24:2747. doi: 10.3390/ijms24032747
98. Dundee JM, Puigdellivol M, Butler R, Cockram TOJ, Brown GC. P2Y(6) receptor-dependent microglial phagocytosis of synapses mediates synaptic and memory loss in aging. Aging Cell (2023) 22:e13761. doi: 10.1111/acel.13761
99. Gunner G, Cheadle L, Johnson KM, Ayata P, Badimon A, Mondo E, et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat Neurosci (2019) 22:1075–88. doi: 10.1038/s41593-019-0419-y
100. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science (2011) 333:1456–8. doi: 10.1126/science.1202529
101. Park J, Choi Y, Jung E, Lee SH, Sohn JW, Chung WS. Microglial MERTK eliminates phosphatidylserine-displaying inhibitory post-synapses. EMBO J (2021) 40:e107121. doi: 10.15252/embj.2020107121
102. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
103. Wang C, Yue H, Hu Z, Shen Y, Ma J, Li J, et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science (2020) 367:688–94. doi: 10.1126/science.aaz2288
104. Hamada K, Shinozaki Y, Namekata K, Matsumoto M, Ohno N, Segawa T, et al. Loss of P2Y(1) receptors triggers glaucoma-like pathology in mice. Br J Pharmacol (2021) 178:4552–71. doi: 10.1111/bph.15637
105. Shinozaki Y, Kashiwagi K, Namekata K, Takeda A, Ohno N, Robaye B, et al. Purinergic dysregulation causes hypertensive glaucoma-like optic neuropathy. JCI Insight (2017) 2:e93456. doi: 10.1172/jci.insight.93456
106. Shinozaki Y, Saito K, Kashiwagi K, Koizumi S. Ocular P2 receptors and glaucoma. Neuropharmacology (2023) 222:109302. doi: 10.1016/j.neuropharm.2022.109302
107. Byun YG, Kim NS, Kim G, Jeon YS, Choi JB, Park CW, et al. Stress induces behavioral abnormalities by increasing expression of phagocytic receptor MERTK in astrocytes to promote synapse phagocytosis. Immunity (2023) 56:2105–2120 e13. doi: 10.1016/j.immuni.2023.07.005
108. Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature (2013) 504:394–400. doi: 10.1038/nature12776
109. Lee JH, Kim JY, Noh S, Lee H, Lee SY, Mun JY, et al. Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature (2021) 590:612–7. doi: 10.1038/s41586-020-03060-3
110. Shi X, Luo L, Wang J, Shen H, Li Y, Mamtilahun M, et al. Stroke subtype-dependent synapse elimination by reactive gliosis in mice. Nat Commun (2021) 12:6943. doi: 10.1038/s41467-021-27248-x
111. Konishi H, Okamoto T, Hara Y, Komine O, Tamada H, Maeda M, et al. Astrocytic phagocytosis is a compensatory mechanism for microglial dysfunction. EMBO J (2020) 39:e104464. doi: 10.15252/embj.2020104464
112. Damisah EC, Hill RA, Rai A, Chen F, Rothlin CV, Ghosh S, et al. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci Adv (2020) 6:eaba3239. doi: 10.1126/sciadv.aba3239
113. Shinozaki Y, Nomura M, Iwatsuki K, Moriyama Y, Gachet C, Koizumi S. Microglia trigger astrocyte-mediated neuroprotection via purinergic gliotransmission. Sci Rep (2014) 4:4329. doi: 10.1038/srep04329
114. Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science (2018) 359:1269–73. doi: 10.1126/science.aal3589
115. Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science (1998) 280:1610–3. doi: 10.1126/science.280.5369.1610
116. Zhi JJ, Wu SL, Wu HQ, Ran Q, Gao X, Chen JF, et al. Insufficient oligodendrocyte turnover in optic nerve contributes to age-related axon loss and visual deficits. J Neurosci (2023) 43:1859–70. doi: 10.1523/JNEUROSCI.2130-22.2023
117. Son JL, Soto I, Oglesby E, Lopez-Roca T, Pease ME, Quigley HA, et al. Glaucomatous optic nerve injury involves early astrocyte reactivity and late oligodendrocyte loss. Glia (2010) 58:780–9. doi: 10.1002/glia.20962
118. You Y, Joseph C, Wang C, Gupta V, Liu S, Yiannikas C, et al. Demyelination precedes axonal loss in the transneuronal spread of human neurodegenerative disease. Brain (2019) 142:426–42. doi: 10.1093/brain/awy338
119. Nakazawa T, Nakazawa C, Matsubara A, Noda K, Hisatomi T, She H, et al. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J Neurosci (2006) 26:12633–41. doi: 10.1523/JNEUROSCI.2801-06.2006
120. Baas D, Legrand C, Samarut J, Flamant F. Persistence of oligodendrocyte precursor cells and altered myelination in optic nerve associated to retina degeneration in mice devoid of all thyroid hormone receptors. Proc Natl Acad Sci U.S.A. (2002) 99:2907–11. doi: 10.1073/pnas.052482299
121. Gillow JT, Shah P, O’Neill EC. Primary open angle glaucoma and hypothyroidism: chance or true association? Eye (Lond) (1997) 11(Pt 1):113–4. doi: 10.1038/eye.1997.22
122. McLenachan J, Davies DM. Glaucoma and the thyroid. Br J Ophthalmol (1965) 49:441–4. doi: 10.1136/bjo.49.8.441
123. Thvilum M, Brandt F, Brix TH, Hegedus L. The interrelation between hypothyroidism and glaucoma: a critical review and meta-analyses. Acta Ophthalmol (2017) 95:759–67. doi: 10.1111/aos.13412
124. Buchanan J, Elabbady L, Collman F, Jorstad NL, Bakken TE, Ott C, et al. Oligodendrocyte precursor cells ingest axons in the mouse neocortex. Proc Natl Acad Sci U.S.A. (2022) 119:e2202580119. doi: 10.1073/pnas.2202580119
125. Fernandez-Castaneda A, Chappell MS, Rosen DA, Seki SM, Beiter RM, Johanson DM, et al. The active contribution of OPCs to neuroinflammation is mediated by LRP1. Acta Neuropathol (2020) 139:365–82. doi: 10.1007/s00401-019-02073-1
126. Itoh N, Itoh Y, Tassoni A, Ren E, Kaito M, Ohno A, et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc Natl Acad Sci U.S.A. (2018) 115:E302–9. doi: 10.1073/pnas.1716032115
127. Ponath G, Ramanan S, Mubarak M, Housley W, Lee S, Sahinkaya FR, et al. Myelin phagocytosis by astrocytes after myelin damage promotes lesion pathology. Brain (2017) 140:399–413. doi: 10.1093/brain/aww298
128. Wan T, Zhu W, Zhao Y, Zhang X, Ye R, Zuo M, et al. Astrocytic phagocytosis contributes to demyelination after focal cortical ischemia in mice. Nat Commun (2022) 13:1134. doi: 10.1038/s41467-022-28777-9
129. Rosko L, Smith VN, Yamazaki R, Huang JK. Oligodendrocyte bioenergetics in health and disease. Neuroscientist (2019) 25:334–43. doi: 10.1177/1073858418793077
130. Harun-Or-Rashid M, Pappenhagen N, Palmer PG, Smith MA, Gevorgyan V, Wilson GN, et al. Structural and functional rescue of chronic metabolically stressed optic nerves through respiration. J Neurosci (2018) 38:5122–39. doi: 10.1523/JNEUROSCI.3652-17.2018
131. Voskuhl RR, Itoh N, Tassoni A, Matsukawa MA, Ren E, Tse V, et al. Gene expression in oligodendrocytes during remyelination reveals cholesterol homeostasis as a therapeutic target in multiple sclerosis. Proc Natl Acad Sci U.S.A. (2019) 116:10130–9. doi: 10.1073/pnas.1821306116
132. Molina-Gonzalez I, Holloway RK, Jiwaji Z, Dando O, Kent SA, Emelianova K, et al. Astrocyte-oligodendrocyte interaction regulates central nervous system regeneration. Nat Commun (2023) 14:3372. doi: 10.1038/s41467-023-39046-8
133. Karim H, Kim SH, Lapato AS, Yasui N, Katzenellenbogen JA, Tiwari-Woodruff SK. Increase in chemokine CXCL1 by ERbeta ligand treatment is a key mediator in promoting axon myelination. Proc Natl Acad Sci U.S.A. (2018) 115:6291–6. doi: 10.1073/pnas.1721732115
134. Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science (2014) 344:1252304. doi: 10.1126/science.1252304
135. Noori R, Park D, Griffiths JD, Bells S, Frankland PW, Mabbott D, et al. Activity-dependent myelination: A glial mechanism of oscillatory self-organization in large-scale brain networks. Proc Natl Acad Sci U.S.A. (2020) 117:13227–37. doi: 10.1073/pnas.1916646117
136. Lim JH, Stafford BK, Nguyen PL, Lien BV, Wang C, Zukor K, et al. Neural activity promotes long-distance, target-specific regeneration of adult retinal axons. Nat Neurosci (2016) 19:1073–84. doi: 10.1038/nn.4340
137. Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discovery (2019) 18:358–78. doi: 10.1038/s41573-019-0012-9
138. High KA, Roncarolo MG. Gene therapy. N Engl J Med (2019) 381:455–64. doi: 10.1056/NEJMra1706910
139. Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (2017) 390:849–60. doi: 10.1016/S0140-6736(17)31868-8
140. Lee Y, Messing A, Su M, Brenner M. GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia (2008) 56:481–93. doi: 10.1002/glia.20622
141. Klimczak RR, Koerber JT, Dalkara D, Flannery JG, Schaffer DV. A novel adeno-associated viral variant for efficient and selective intravitreal transduction of rat Muller cells. PloS One (2009) 4:e7467. doi: 10.1371/journal.pone.0007467
142. Koerber JT, Klimczak R, Jang JH, Dalkara D, Flannery JG, Schaffer DV. Molecular evolution of adeno-associated virus for enhanced glial gene delivery. Mol Ther (2009) 17:2088–95. doi: 10.1038/mt.2009.184
143. Pellissier LP, Hoek RM, Vos RM, Aartsen WM, Klimczak RR, Hoyng SA, et al. Specific tools for targeting and expression in Muller glial cells. Mol Ther Methods Clin Dev (2014) 1:14009. doi: 10.1038/mtm.2014.9
144. Tanaka KF, Matsui K, Sasaki T, Sano H, Sugio S, Fan K, et al. Expanding the repertoire of optogenetically targeted cells with an enhanced gene expression system. Cell Rep (2012) 2:397–406. doi: 10.1016/j.celrep.2012.06.011
145. Chen H, McCarty DM, Bruce AT, Suzuki K, Suzuki K. Gene transfer and expression in oligodendrocytes under the control of myelin basic protein transcriptional control region mediated by adeno-associated virus. Gene Ther (1998) 5:50–8. doi: 10.1038/sj.gt.3300547
146. von Jonquieres G, Frohlich D, Klugmann CB, Wen X, Harasta AE, Ramkumar R, et al. Recombinant human myelin-associated glycoprotein promoter drives selective AAV-mediated transgene expression in oligodendrocytes. Front Mol Neurosci (2016) 9:13. doi: 10.3389/fnmol.2016.00013
147. Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer (2012) 12:159–69. doi: 10.1038/nrc3215
148. Agostinone J, Alarcon-Martinez L, Gamlin C, Yu WQ, Wong ROL, Di Polo A. Insulin signalling promotes dendrite and synapse regeneration and restores circuit function after axonal injury. Brain (2018) 141:1963–80. doi: 10.1093/brain/awy142
149. Duan X, Qiao M, Bei F, Kim IJ, He Z, Sanes JR. Subtype-specific regeneration of retinal ganglion cells following axotomy: effects of osteopontin and mTOR signaling. Neuron (2015) 85:1244–56. doi: 10.1016/j.neuron.2015.02.017
150. Kermer P, Klocker N, Labes M, Bahr M. Insulin-like growth factor-I protects axotomized rat retinal ganglion cells from secondary death via PI3-K-dependent Akt phosphorylation and inhibition of caspase-3 In vivo. J Neurosci (2000) 20:2–8. doi: 10.1523/JNEUROSCI.20-02-00722.2000
151. Zhang Y, Williams PR, Jacobi A, Wang C, Goel A, Hirano AA, et al. Elevating growth factor responsiveness and axon regeneration by modulating presynaptic inputs. Neuron (2019) 103:39–51 e5. doi: 10.1016/j.neuron.2019.04.033
152. Caldwell ALM, Sancho L, Deng J, Bosworth A, Miglietta A, Diedrich JK, et al. Aberrant astrocyte protein secretion contributes to altered neuronal development in multiple models of neurodevelopmental disorders. Nat Neurosci (2022) 25:1163–78. doi: 10.1038/s41593-022-01150-1
153. Liu X, Yao DL, Bondy CA, Brenner M, Hudson LD, Zhou J, et al. Astrocytes express insulin-like growth factor-I (IGF-I) and its binding protein, IGFBP-2, during demyelination induced by experimental autoimmune encephalomyelitis. Mol Cell Neurosci (1994) 5:418–30. doi: 10.1006/mcne.1994.1052
154. Watanabe K, Uemura K, Asada M, Maesako M, Akiyama H, Shimohama S, et al. The participation of insulin-like growth factor-binding protein 3 released by astrocytes in the pathology of Alzheimer’s disease. Mol Brain (2015) 8:82. doi: 10.1186/s13041-015-0174-2
155. Lin R, Zhou Y, Yan T, Wang R, Li H, Wu Z, et al. Directed evolution of adeno-associated virus for efficient gene delivery to microglia. Nat Methods (2022) 19:976–85. doi: 10.1038/s41592-022-01547-7
156. Okada Y, Hosoi N, Matsuzaki Y, Fukai Y, Hiraga A, Nakai J, et al. Development of microglia-targeting adeno-associated viral vectors as tools to study microglial behavior in vivo. Commun Biol (2022) 5:1224. doi: 10.1038/s42003-022-04200-3
157. Young A, Neumann B, Segel M, Chen CZ, Tourlomousis P, Franklin RJM. Targeted evolution of adeno-associated virus capsids for systemic transgene delivery to microglia and tissue-resident macrophages. Proc Natl Acad Sci U.S.A. (2023) 120:e2302997120. doi: 10.1073/pnas.2302997120
158. Georgiou E, Sidiropoulou K, Richter J, Papaneophytou C, Sargiannidou I, Kagiava A, et al. Gene therapy targeting oligodendrocytes provides therapeutic benefit in a leukodystrophy model. Brain (2017) 140:599–616. doi: 10.1093/brain/aww351
159. Schiza N, Sargiannidou I, Kagiava A, Karaiskos C, Nearchou M, Kleopa KA. Transgenic replacement of Cx32 in gap junction-deficient oligodendrocytes rescues the phenotype of a hypomyelinating leukodystrophy model. Hum Mol Genet (2015) 24:2049–64. doi: 10.1093/hmg/ddu725
160. Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron (2014) 82:380–97. doi: 10.1016/j.neuron.2014.02.040
161. Ebneter A, Kokona D, Jovanovic J, Zinkernagel MS. Dramatic effect of oral CSF-1R kinase inhibitor on retinal microglia revealed by in vivo scanning laser ophthalmoscopy. Transl Vis Sci Technol (2017) 6:10. doi: 10.1167/tvst.6.2.10
162. Takeda A, Shinozaki Y, Kashiwagi K, Ohno N, Eto K, Wake H, et al. Microglia mediate non-cell-autonomous cell death of retinal ganglion cells. Glia (2018) 66:2366–84. doi: 10.1002/glia.23475
163. Zhang Y, Zhao L, Wang X, Ma W, Lazere A, Qian HH, et al. Repopulating retinal microglia restore endogenous organization and function under CX3CL1-CX3CR1 regulation. Sci Adv (2018) 4:eaap8492. doi: 10.1126/sciadv.aap8492
164. Henry RJ, Ritzel RM, Barrett JP, Doran SJ, Jiao Y, Leach JB, et al. Microglial depletion with CSF1R inhibitor during chronic phase of experimental traumatic brain injury reduces neurodegeneration and neurological deficits. J Neurosci (2020) 40:2960–74. doi: 10.1523/JNEUROSCI.2402-19.2020
165. Rice RA, Pham J, Lee RJ, Najafi AR, West BL, Green KN. Microglial repopulation resolves inflammation and promotes brain recovery after injury. Glia (2017) 65:931–44. doi: 10.1002/glia.23135
166. Rice RA, Spangenberg EE, Yamate-Morgan H, Lee RJ, Arora RP, Hernandez MX, et al. Elimination of microglia improves functional outcomes following extensive neuronal loss in the hippocampus. J Neurosci (2015) 35:9977–89. doi: 10.1523/JNEUROSCI.0336-15.2015
167. Wang Y, Wernersbach I, Strehle J, Li S, Appel D, Klein M, et al. Early posttraumatic CSF1R inhibition via PLX3397 leads to time- and sex-dependent effects on inflammation and neuronal maintenance after traumatic brain injury in mice. Brain Behav Immun (2022) 106:49–66. doi: 10.1016/j.bbi.2022.07.164
168. George S, Rey NL, Tyson T, Esquibel C, Meyerdirk L, Schulz E, et al. Microglia affect alpha-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol Neurodegener (2019) 14:34. doi: 10.1186/s13024-019-0335-3
169. Olmos-Alonso A, Schetters ST, Sri S, Askew K, Mancuso R, Vargas-Caballero M, et al. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain (2016) 139:891–907. doi: 10.1093/brain/awv379
170. Sosna J, Philipp S, Albay R 3rd, Reyes-Ruiz JM, Baglietto-Vargas D, LaFerla FM, et al. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol Neurodegener (2018) 13:11. doi: 10.1186/s13024-018-0244-x
171. Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M, et al. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain (2016) 139:1265–81. doi: 10.1093/brain/aww016
172. Okunuki Y, Mukai R, Nakao T, Tabor SJ, Butovsky O, Dana R, et al. Retinal microglia initiate neuroinflammation in ocular autoimmunity. Proc Natl Acad Sci U.S.A. (2019) 116:9989–98. doi: 10.1073/pnas.1820387116
173. Okunuki Y, Mukai R, Pearsall EA, Klokman G, Husain D, Park DH, et al. Microglia inhibit photoreceptor cell death and regulate immune cell infiltration in response to retinal detachment. Proc Natl Acad Sci U.S.A. (2018) 115:E6264–73. doi: 10.1073/pnas.1719601115
174. Yang X, Zhao L, Campos MM, Abu-Asab M, Ortolan D, Hotaling N, et al. CSF1R blockade induces macrophage ablation and results in mouse choroidal vascular atrophy and RPE disorganization. Elife (2020) 9:e55564. doi: 10.7554/eLife.55564
175. Hilla AM, Diekmann H, Fischer D. Microglia are irrelevant for neuronal degeneration and axon regeneration after acute injury. J Neurosci (2017) 37:6113–24. doi: 10.1523/JNEUROSCI.0584-17.2017
176. Elmore MRP, Hohsfield LA, Kramar EA, Soreq L, Lee RJ, Pham ST, et al. Replacement of microglia in the aged brain reverses cognitive, synaptic, and neuronal deficits in mice. Aging Cell (2018) 17:e12832. doi: 10.1111/acel.12832
177. Diemler CA, Cossete T, Heuer S, Goodrich MM, MacLean M, Marola O, et al. Microglia depletion increases susceptibility for glaucomatous neurodegeneration in ocular hypertensive mice. ARVO Annu Meeting Abstract (2023) 64:1595.
178. Tan Z, Guo Y, Shrestha M, Sun D, Gregory-Ksander M, Jakobs TC. Microglia depletion exacerbates retinal ganglion cell loss in a mouse model of glaucoma. Exp Eye Res (2022) 225:109273. doi: 10.1016/j.exer.2022.109273
179. Parajuli B, Saito H, Shinozaki Y, Shigetomi E, Miwa H, Yoneda S, et al. Transnasal transplantation of human induced pluripotent stem cell-derived microglia to the brain of immunocompetent mice. Glia (2021) 69:2332–48. doi: 10.1002/glia.23985
180. Parajuli B, Shinozaki Y, Shigetomi E, Koizumi S. Transplantation of human induced pluripotent stem cell-derived microglia in immunocompetent mice brain via non-invasive transnasal route. J Vis Exp (2022) 183. doi: 10.3791/63574-v
181. Tvrdik P, Kalani MYS. In vivo imaging of microglial calcium signaling in brain inflammation and injury. Int J Mol Sci (2017) 18:2366. doi: 10.3390/ijms18112366
182. Ma W, Xhao L, Xu B, Fariss RN, Redmond TM, Zou J, et al. Human iPSC-derived Microglia Cells Integrated into Mouse Retina and Recapitulated Features of Endogenous Microglia Cells. bioRxiv (2023). doi: 10.7554/eLife.90695.1
183. Livne-Bar I, Wei J, Liu HH, Alqawlaq S, Won GJ, Tuccitto A, et al. Astrocyte-derived lipoxins A4 and B4 promote neuroprotection from acute and chronic injury. J Clin Invest (2017) 127:4403–14. doi: 10.1172/JCI77398
184. Bull ND, Irvine KA, Franklin RJ, Martin KR. Transplanted oligodendrocyte precursor cells reduce neurodegeneration in a model of glaucoma. Invest Ophthalmol Vis Sci (2009) 50:4244–53. doi: 10.1167/iovs.08-3239
185. Ader M, Meng J, Schachner M, Bartsch U. Formation of myelin after transplantation of neural precursor cells into the retina of young postnatal mice. Glia (2000) 30:301–10. doi: 10.1002/(SICI)1098-1136(200005)30:3<301::AID-GLIA9>3.0.CO;2-S
186. Laeng P, Molthagen M, Yu EG, Bartsch U. Transplantation of oligodendrocyte progenitor cells into the rat retina: extensive myelination of retinal ganglion cell axons. Glia (1996) 18:200–10. doi: 10.1002/(SICI)1098-1136(199611)18:3<200::AID-GLIA4>3.0.CO;2-2
187. Yang X, Zou H, Jung G, Richard G, Linke SJ, Ader M, et al. Nonneuronal control of the differential distribution of myelin along retinal ganglion cell axons in the mouse. Invest Ophthalmol Vis Sci (2013) 54:7819–27. doi: 10.1167/iovs.13-12596
188. Fujikado T, Morimoto T, Matsushita K, Shimojo H, Okawa Y, Tano Y. Effect of transcorneal electrical stimulation in patients with nonarteritic ischemic optic neuropathy or traumatic optic neuropathy. Jpn J Ophthalmol (2006) 50:266–73. doi: 10.1007/s10384-005-0304-y
189. Morimoto T, Miyoshi T, Sawai H, Fujikado T. Optimal parameters of transcorneal electrical stimulation (TES) to be neuroprotective of axotomized RGCs in adult rats. Exp Eye Res (2010) 90:285–91. doi: 10.1016/j.exer.2009.11.002
190. Yin H, Yin H, Zhang W, Miao Q, Qin Z, Guo S, et al. Transcorneal electrical stimulation promotes survival of retinal ganglion cells after optic nerve transection in rats accompanied by reduced microglial activation and TNF-alpha expression. Brain Res (2016) 1650:10–20. doi: 10.1016/j.brainres.2016.08.034
191. Inomata K, Shinoda K, Ohde H, Tsunoda K, Hanazono G, Kimura I, et al. Transcorneal electrical stimulation of retina to treat longstanding retinal artery occlusion. Graefes Arch Clin Exp Ophthalmol (2007) 245:1773–80. doi: 10.1007/s00417-007-0610-9
192. Wang X, Mo X, Li D, Wang Y, Fang Y, Rong X, et al. Neuroprotective effect of transcorneal electrical stimulation on ischemic damage in the rat retina. Exp Eye Res (2011) 93:753–60. doi: 10.1016/j.exer.2011.09.022
193. Ni YQ, Gan DK, Xu HD, Xu GZ, Da CD. Neuroprotective effect of transcorneal electrical stimulation on light-induced photoreceptor degeneration. Exp Neurol (2009) 219:439–52. doi: 10.1016/j.expneurol.2009.06.016
194. Yu H, Enayati S, Chang K, Cho K, Lee SW, Talib M, et al. Noninvasive electrical stimulation improves photoreceptor survival and retinal function in mice with inherited photoreceptor degeneration. Invest Ophthalmol Vis Sci (2020) 61:5. doi: 10.1167/iovs.61.4.5
195. Jassim AH, Cavanaugh M, Shah JS, Willits R, Inman DM. Transcorneal electrical stimulation reduces neurodegenerative process in a mouse model of glaucoma. Ann BioMed Eng (2021) 49:858–70. doi: 10.1007/s10439-020-02608-8
196. Ota Y, Ozeki N, Yuki K, Shiba D, Kimura I, Tsunoda K, et al. The efficacy of transcorneal electrical stimulation for the treatment of primary open-angle glaucoma: A pilot study. Keio J Med (2018) 67:45–53. doi: 10.2302/kjm.2017-0015-OA
197. Fu L, Fung FK, Lo AC, Chan YK, So KF, Wong IY, et al. Transcorneal electrical stimulation inhibits retinal microglial activation and enhances retinal ganglion cell survival after acute ocular hypertensive injury. Transl Vis Sci Technol (2018) 7:7. doi: 10.1167/tvst.7.3.7
198. Zhou WT, Ni YQ, Jin ZB, Zhang M, Wu JH, Zhu Y, et al. Electrical stimulation ameliorates light-induced photoreceptor degeneration in vitro via suppressing the proinflammatory effect of microglia and enhancing the neurotrophic potential of Muller cells. Exp Neurol (2012) 238:192–208. doi: 10.1016/j.expneurol.2012.08.029
199. Stett A, Schatz A, Gekeler F, Franklin J. Transcorneal electrical stimulation dose-dependently slows the visual field loss in retinitis pigmentosa. Transl Vis Sci Technol (2023) 12:29. doi: 10.1167/tvst.12.2.29
200. Miura G, Sugawara T, Kawasaki Y, Tatsumi T, Nizawa T, Baba T, et al. Clinical trial to evaluate safety and efficacy of transdermal electrical stimulation on visual functions of patients with retinitis pigmentosa. Sci Rep (2019) 9:11668. doi: 10.1038/s41598-019-48158-5
201. Schatz A, Rock T, Naycheva L, Willmann G, Wilhelm B, Peters T, et al. Transcorneal electrical stimulation for patients with retinitis pigmentosa: a prospective, randomized, sham-controlled exploratory study. Invest Ophthalmol Vis Sci (2011) 52:4485–96. doi: 10.1167/iovs.10-6932
202. van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol (2018) 19:213–28. doi: 10.1038/nrm.2017.125
203. Elliott RO, He M. Unlocking the power of exosomes for crossing biological barriers in drug delivery. Pharmaceutics (2021) 13:122. doi: 10.3390/pharmaceutics13010122
204. Aires ID, Ribeiro-Rodrigues T, Boia R, Catarino S, Girao H, Ambrosio AF, et al. Exosomes derived from microglia exposed to elevated pressure amplify the neuroinflammatory response in retinal cells. Glia (2020) 68:2705–24. doi: 10.1002/glia.23880
205. Carapia AK, Martinez-Colin EJ, Segura-Villalobos D, Victoria-Chavez RY, Lezama I, Martinez-Martinez E, et al. Muller glia to muller glia extracellular vesicle-dependent signaling induces multipotency genes nestin and lin28 expression in response to N-methyl-D-aspartate (NMDA) exposure. ASN Neuro (2023) 15:17590914231183272. doi: 10.1177/17590914231183272
206. Demais V, Pohl A, Wunderlich KA, Pfaller AM, Kaplan L, Barthelemy A, et al. Release of VAMP5-positive extracellular vesicles by retinal Muller glia in vivo. J Extracell Vesicles (2022) 11:e12254. doi: 10.1002/jev2.12254
207. Salikhova DI, Timofeeva AV, Golovicheva VV, Fatkhudinov TK, Shevtsova YA, Soboleva AG, et al. Extracellular vesicles of human glial cells exert neuroprotective effects via brain miRNA modulation in a rat model of traumatic brain injury. Sci Rep (2023) 13:20388. doi: 10.1038/s41598-023-47627-2
Keywords: adeno-associated virus, astrocytes, cell transplantation, glaucoma, intraocular pressure, microglia, Müller cells
Citation: Shinozaki Y, Namekata K, Guo X and Harada T (2024) Glial cells as a promising therapeutic target of glaucoma: beyond the IOP. Front. Ophthalmol. 3:1310226. doi: 10.3389/fopht.2023.1310226
Received: 09 October 2023; Accepted: 18 December 2023;
Published: 08 January 2024.
Edited by:
Kenji Matsushita, Osaka University, JapanReviewed by:
Stephanie Louise Grillo, Penn State Milton S. Hershey Medical Center, United StatesYukihiro Shiga, University of Montreal Hospital Research Centre (CRCHUM), Canada
Copyright © 2024 Shinozaki, Namekata, Guo and Harada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youichi Shinozaki, c2hpbm96YWtpLXlpQGlnYWt1a2VuLm9yLmpw