 Wasef Nijim
Wasef Nijim Mohamed Moustafa2,3
Mohamed Moustafa2,3 Julia Humble
Julia Humble Mohamed Al-Shabrawey
Mohamed Al-Shabrawey
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Ophthalmol. , 07 September 2023
Sec. Retina
Volume 3 - 2023 | https://doi.org/10.3389/fopht.2023.1230581
Diabetic retinopathy (DR) is a result of neurovacular insults from hyperglycemia in diabetes mellitus (DM), and it is one of the top causes of vision loss throughout the modern world. This review article explores the role endothelial to mesenchymal transition (EndMT) has on the pathogenesis of DR. EndMT contributes to the disruption of the blood-retinal barrier, vascular leakage, neovascularization, and fibrosis observed in DR. Risk factors and biomarkers associated with DR severity are discussed, highlighting the importance of early detection and targeted therapies. Current treatments primarily focus on anti-vascular endothelial growth factor (anti-VEGF) agents, corticosteroids, and laser photocoagulation. However, emerging therapeutic strategies aimed at inhibiting EndMT and its downstream effects show promise in preventing the development and progression of DR. Understanding the molecular and cellular mechanisms underlying EndMT in DR provides valuable insights into the disease process and offers potential options for the development of potential treatments.
Diabetic Retinopathy (DR) is a microvascular complication of diabetes mellitus (DM), caused by hyperglycemic insults leading to vision-threatening damage to the retina (1). DR is the number one cause of loss of vision in adults within the United States (2). Early detection and immediate treatment are paramount in avoiding blindness from this common disease (2).
In the next ten years, the global prevalence of DM has been estimated to become 592 million (3). DR is the most common microvascular complication of diabetes (4). Ninety-three million people worldwide suffer from DR, with 25-30% of these people having vision-threatening complications (5). Of these patients, 5-8% of patients require laser treatment, and 5% require vitrectomy surgery (6).
Hyperglycemia and subsequent metabolic disorders activate inflammatory and oxidative pathways within the retina that cause damage to retinal vessels. Growth of microaneurysms and breakdown of the blood-retinal barrier (BRB) and subsequent development of diabetic macular edema (DME) and hard exudates are the typical clinical manifestations of non-proliferative diabetic retinopathy (NPDR) (1). Vasoconstriction and vascular occlusion within retinal capillaries lead to the development of ischemic pale spots ‘cotton-wool spots’ (2). Development of severe retinal ischemia eventually causes the upregulation of angiogenic factors, like Vascular Endothelial Growth Factor (VEGF) (3). The upregulation of angiogenic factors causes the characteristic neovascularization seen in proliferative diabetic retinopathy (PDR) (3).
DR affects patients with diagnosed or undiagnosed DM. The most significant risk factors for developing DR are older age, longer duration of DM, high glycemic levels, and hypertension (2). A higher HbA1C is significantly associated with advancement of DR, whereas intense glycemic control significantly correlates with a reduction in incidence and progression of DR (7, 8). Recent studies have shown high variability in glucose levels also contributes to severity of DR; therefore, controlling post-prandial glucose levels is important in reducing the progression of DR as well (9, 10). Tight blood pressure control is another important risk factor modification that has been shown to reduce the damage inflicted on the retina from DR (11). Other modifiable risk factors that are not as critical in DR but still contribute to the development and advancement of disease include: nephropathy, hyperlipidemia, smoking, and obesity (12, 13). Non-modifiable risk factors include pregnancy, age, and genetics (1, 14).
Despite the above risk factors, current studies find a variation between the development and severity of DR that cannot be fully explained by the beforementioned risk factors. Therefore, identifying newer biomarkers that can help stratify patients and determine their response to therapeutics is important (2). Systemic biomarkers that have been found to strongly correlate with severity of DR include markers for inflammation, such as C-reactive protein (CRP), homocysteine, and advanced glycation end products (AGEs) (15–17). Newer markers discovered include apolipoprotein, vitamin D, leptin, as well as various genetic markers (1). Ocular biomarkers from sampling vitreous and tears include VEGF and platelet-derived growth factor (PDGF), but these markers need more validation studies for predicting DR severity (18, 19).
DME is the most common cause of visual loss in DR patients occurring in both NPDR and PDR at various levels of disease (3, 20). DME results from leakage of fluid and proteins from retinal vessels into the macula due to breakdown of BRB (20). Hard exudates and edema can be seen on fundoscopy (21). Tractional retinal detachment (TRD) with vitreous hemorrhage is the second form of vision loss resulting from DR (20). Unlike DME, TRD only occurs in PDR due to the fibrovascular scarring generated during neovascularization being pulled on by the vitreous (3). For TRD, a ring of white scar tissue can be seen on fundoscopy (22). Fibrovascular proliferation can also cause vitreous hemorrhage, contributing to vision loss from PDR (20).
Endothelial cell to Mesenchymal Transition (EndMT) occurs during the various stages of NPDR and PDR (23). The transformation can contribute to both mechanisms of vision loss mentioned above. EndMT contributes to DME by disrupting the BRB, increasing vascular permeability due to the loss of endothelial characteristics (24). This disruption leads not only to leakage of fluid into the retina, but also of inflammatory mediators (24). EndMT contributes to fibrosis within the retina due to the gain of mesenchymal characteristics and eventually increases the likelihood of TRD during PDR (23).
There are several metabolic pathways involved in the pathogenesis of DR, such as the polyol, protein kinase c (PKC), and hexosamine pathways (25). Activation of these pathways cause upregulation of cytokines and growth factors that leads to increased vascular permeability and occlusion of blood vessels (1). Initially, the blood vessels within the retina dilate in response to the hyperglycemic insults to increase retinal metabolism (20).
Pericytes are specialized, contractile mesenchymal stem cells (MSCs) that provide vascular tone and perfusion pressure within the capillaries of the retina (26). Hyperglycemia causes apoptosis of pericyte and endothelial cells leads to microaneurysm formation, breakdown of BRB, and development of retinal ischemia (20). Insufficient circulation leads to upregulation of VEGF (27), the primary angiogenic factor that is associated with the vascular permeability and angiogenesis seen in DR (20).
The high metabolic rate of the neural retina requires the regulation of a distinct supply of specific nutrients by the BRB (28). It regulates the transport of ions, water, and nutrients and prevents immune cells and antibodies from passing into the retina tissue (29). The BRB includes the inner BRB (iBRB) and the outer BRB (oBRB) (30). The iBRB consists of endothelium, Müller cells, and pericytes (30). The capillaries of the iBRB provide oxygen, glucose, and other nutrients to neurons and prevent other molecules from entering the retina for protection (31). The oBRB is composed of retinal pigment epithelium (RPE) and functions to regulate the transport between the choriocapillaris and the retina (32). Various growth factors and cytokines that are upregulated in DR, such as VEGF, HIF-1, and IL-1 β, induce BRB breakdown through various mechanisms (33). Damage to the iBRB and oBRB leads to fluid and protein extravasation into the macula, as well as the degeneration of retinal capillaries, playing a critical role in the development of DME and severe visual loss in DR (32).
One of the advanced stages of DR, known as PDR, is characterized by neovascularization, as well as an increase in the presence of myofibroblasts (34). This process can lead to the development of fibrovascular tissue, which can result in traction on the retina, leading to TRD and eventual blindness (2). There is a switch at some point in PDR between neovascularization and fibrosis; this is known as the angio-fibrotic switch (34). As previously stated, VEGF is majorly responsible for neovascularization (20). Connective tissue growth factor (CTGF) is another factor upregulated during DR that contributes mainly to fibrosis within the eyes of patients (34). The decrease in VEGF and increase in CTGF is associated with the switch from angiogenesis to fibrosis in PDR (Figure 1) (34). CTGF is a Transforming Growth Factor-Beta (TGF-β) effector that induces tissue fibrosis during PDR (35). In addition to fibrosis during PDR, TGF-β is also a main contributor to EndMT earlier on in DR (23).
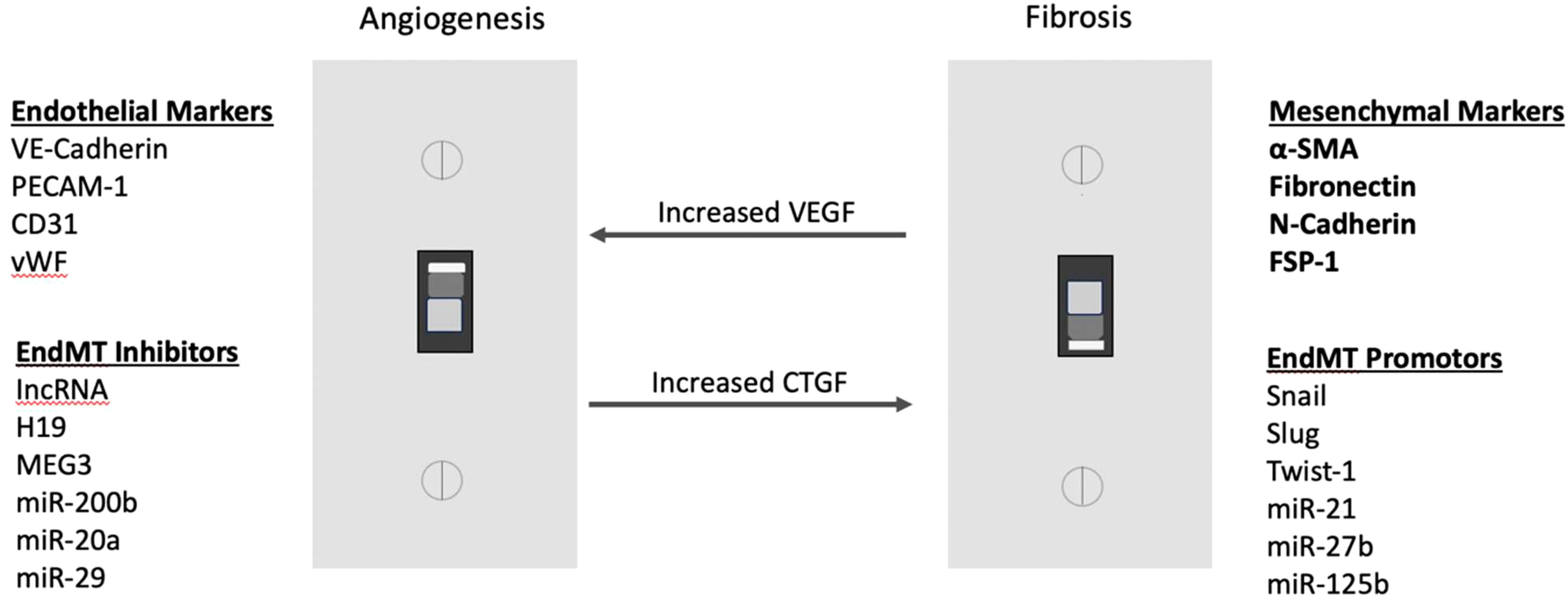
Figure 1 Angiofibrotic switch: elevated CTGF over vascular VEGF promotes fibrosis in PDR.
As previously stated, the early vascular loss in DR is a major contributor to the clinical manifestations of DR, and endothelial cell and pericyte apoptosis is only partly responsible for this vascular loss (20). Another part of the vascular loss found in DR is the transformation of endothelial cells to mesenchymal cells through EndMT (23). As a consequence of EndMT, endothelial cells undergo a shift in their cell markers and phenotype, transitioning from their original characteristics to acquire the cell markers and phenotype typically associated with mesenchymal cells (36–38). This process leads to the breakdown of the BRB and contributes to the microvascular pathogenesis of DR (23).
In addition to optimizing blood glucose, lipids, and blood pressure in diabetic patients, there are various intraocular management strategies that have become standard practice in DR patients (39, 40). For instance, the management of DME has been significantly altered with the introduction of intravitreal anti-VEGF injection therapies, such as ranibizumab, bevacizumab, and aflibercept (2). Several randomized control trials since 2010 have shown these agents significantly reduce DME and improve vision (41–45). Aflibercept has been found to be the most efficacious agent in patients with initial poor visual acuity (46). Despite all the current research on anti-VEGF treatments, the optimal frequency of injections and total time for treatment courses is still unknown (2). The current practice is to administer multiple injections within the first year of treatment and then a gradual decrease in the following years to maintain remission (39). Intravitreal anti-VEGF injections have also been shown to benefit patients with PDR (47). These medicines do, however, have some limitations and adverse effects (20). The short half-life of these agents means that initially, bimonthly injections are necessary to ensure efficacy (20). Elevation of intraocular pressure, vitreous hemorrhage, and inflammation are uncommon adverse effects (20). A rare adverse effect due to the high number of injections necessary is endophthalmitis (20, 48). Additionally, high cost and poor patient adherence are of concern when it comes to this treatment modality (20). Currently, there is research being done on other various anti-angiogenic agents that inhibit vasoconstriction and vascular occlusion within retinal capillaries multiple angiogenic factors in addition to or other than VEGF, such as Squalamine and Nesvacumab, respectively (20, 49).
In cases of refractory DME that are unresponsive to anti-VEGF treatments, intravitreal corticosteroid injections can be used (50). These refractory cases are thought to be driven by the effects of multiple cytokines (20). Corticosteroids target inflammatory mediators known to contribute to the pathogenesis of DME (20). There have been multiple clinical trials done for DME treatment using triamcinolone acetonide, dexamethasone (DEX) intravitreal implant, and fluocinolone intravitreal implant (48, 51–53). The use of intravitreal corticosteroids has been associated with a lower number of injections, lower cost, and higher compliance (20). However, given the higher incidence of adverse effects, such as cataracts, glaucoma, and vitreous hemorrhage, and no proven benefit in PDR, intravitreal corticosteroid injections are considered second-line to anti-VEGF injections (20, 48, 51–53).
On the other hand, in patients that have progressed to PDR, panretinal laser photocoagulation (PRP) is considered first-line due to its effectiveness in reducing vision loss in this patient population, especially in patients with vitreous hemorrhage complications (54–56). Aflibercept, an anti-VEGF agent previously mentioned, has been shown to result in better visual outcomes and is a safer alternative to PRP in select patients (47). Due to laser-induced retinal damage from PRP, adverse effects include mild central visual acuity loss and reduced night vision (57). However, PRP remains important as an adjuvant and rescue therapy for PDR patients with high-risk complications by reducing the rate of severe visual loss and halting the progression of retinopathy (20). In modern times, there have been efforts to develop newer laser approaches that reduce side effects (20) e.g. the pattern scanning laser (PASCAL) (58), the subthreshold micropulse diode laser (D-MPL) (59), and the navigated laser system (NAVILAS) (60).
Other treatments for DR under clinical evaluation include cardiolipin inhibitors, mitochondria-specific antioxidants, as well as other classes of antioxidants (61–63). All these treatments for DR are focused on preventing late complications of the disease. An alternate approach being investigated currently is treating the root causes and early developments within DR (23). We researched the current literature on EndMT, an early pathological event in DR and potential treatments to prevent it and subsequent complications.
EndMT is a process where endothelial cells undergo a phenotypic transition to mesenchymal cells (64). This process has been implicated in the pathogenesis of various diseases, including diabetic complications (23). EndMT in diabetes is prompted by various factors, such as elevated blood sugar levels and AGEs (23).
At the cellular level, EndMT is identified by the disappearance of endothelial markers such as CD31, while gaining mesenchymal markers such as α-smooth muscle actin (α-SMA) (65). This process is driven by various pathways, including TGF-β, Notch, and Wnt/β-catenin signaling pathways (65, 66). Furthermore, hyperglycemia can promote EndMT by triggering NF-κB signaling (67, 68). These signaling pathways ultimately lead to the increased expression of transcription factors, such as Snail, Slug, and Twist that promote EndMT (69–72). Additionally, epigenetic regulation, such as DNA methylation and histone modification, plays a role in EndMT (23, 73). One post-translational complex, methyltransferase-like 3 (METTL3), has been recently found to play an important role in EndMT. METTL3 is responsible for the N6-methyladenosine (m6a) modification, where a methyl group is added to the sixth nitrogen of adenosine in RNA. METTL3 adds the m6a modification to transient receptor potential cation channel 6 (TRPC6), which works through the calcineurin/NFAT pathway, to increase mesenchymal marker α-SMA and decrease endothelial markers CD31 and VE-cadherin (74). Another epigenetic process involved in EndMT is the methylation of maternally expressed gene 3 (MEG3) by DNA methyltransferase 1 (DMT1). Recent research has shown that MEG3 is downregulated in rat models of DR due to methylation by DMT1. Inhibition of MEG3 activates the PI3K/AKT/mTOR pathway, which is implicated in the pathogenesis of DR and EndMT (75). MicroRNAs (miRNAs) have also been shown to regulate the expression of EndMT-related genes (24). In diabetes, the expression of certain miRNAs is altered, which can promote EndMT (24, 76).
The impact of EndMT on vascular function in diabetes is multifactorial. One of the key consequences of EndMT is the loss of function of the BRB, leading to increased vascular permeability and the leakage of unwanted proteins and immune cells into the retina, causing edema and inflammation of the surrounding tissues (77, 78). EndMT also contributes to the buildup of extracellular matrix (ECM) proteins like collagen, forming fibrotic tissue (24). In DR, this excess of ECM proteins leads to the enlargement of the basement membrane in the retina. This process is part of the pathogenesis of increased vascular permeability in DR (79).
DR, a leading cause of visual impairment in adults, can be categorized into NPDR and PDR stages (20). EndMT occurs throughout these stages, contributing to increased vascular permeability and fibrovascular scarring (23). Endothelial cells within retinal capillaries help maintain the BRB, which consists of cells joined together to prevent certain substances from entering the retina from the circulatory system (80). EndMT disrupts the integrity of the BRB, permitting plasma and proteins to enter the retina, which can give rise to complications like DME (80, 81).
In DR, EndMT leads to thickening of the basement membrane and increased deposition of matrix proteins, contributing to the breakdown of the BRB (24, 82). Additionally, EndMT increases the number of myofibroblasts, which can lead to TRD because of an increase in fibrosis within the retina (79). Hyperglycemia, suppression of long non-coding RNAs (lncRNAs), upregulation of Notch2, and various circRNAs and miRNAs are all implicated in EndMT induction through TGF-β signaling in DR (76, 81). Overexpression of H19 and MEG3, effectively prevents EndMT (24, 75, 81). Inhibition of Notch2 and suppression of various circRNAs are also potential therapeutic targets for preventing EndMT in DR (76).
We conducted our own study on EndMT within DR. The goal of our study was to evaluate if there is a significant difference in the amount of EndMT at different stages of the disease. EndMT and fibroproliferative transformation were characterized by de novo cellular expression of α-SMA. We used Ins2Akita mice as an experimental model of type 1 diabetes. The mice eyes were dissected to retrieve their retinas. We then performed immunofluorescence (IF) on the retinas of 23 diabetic mice at 4-weeks (8 mice), 12-weeks (8 mice), and 32-weeks (7 mice) old. We also performed IF on the retinas of 9 non-diabetic mice that were 12-weeks-old to use as a control. The antibodies used to stain the retina slides were for α-SMA, CD31, and DAPI. These were used as markers for mesenchymal tissue, endothelial cells, and nuclear markers, respectively. Images of these slides were taken at 20x using fluorescence microscopy. The intensity of α-SMA was analyzed using Image J software, and statistical analysis was performed using One-way ANOVA Multiple Comparisons Test with a p-value <0.05 considered significant. An increase in α-SMA intensity correlated with an increase in EndMT. Our results indicated that there was a significant increase in EndMT in the retina of diabetic mice compared to the control group (Figures 2A, C). Moreover, EndMT was significantly increased in the 12-week-old diabetic mice when compared to the 4-week-old and 32-week-old diabetic mice (Figures 2B, C). . There was no significant difference between the amount of EndMT when comparing the 4-week-old diabetic mice with the 32-week-old diabetic mice (Figure 2C). The significant increase of EndMT, at 12-weeks in comparison to 4-weeks may be due to a temporal requirement for diabetes to initiate the process of EndMT. The significant decline of EndMT, at 32-weeks in comparison to 12-weeks, could be attributed to the loss of pericytes and endothelial cells by 24 weeks in DR (83). Pericytes are mesenchymal stem cells, so they contribute to the increase in mesenchymal cell population found with EndMT (83).
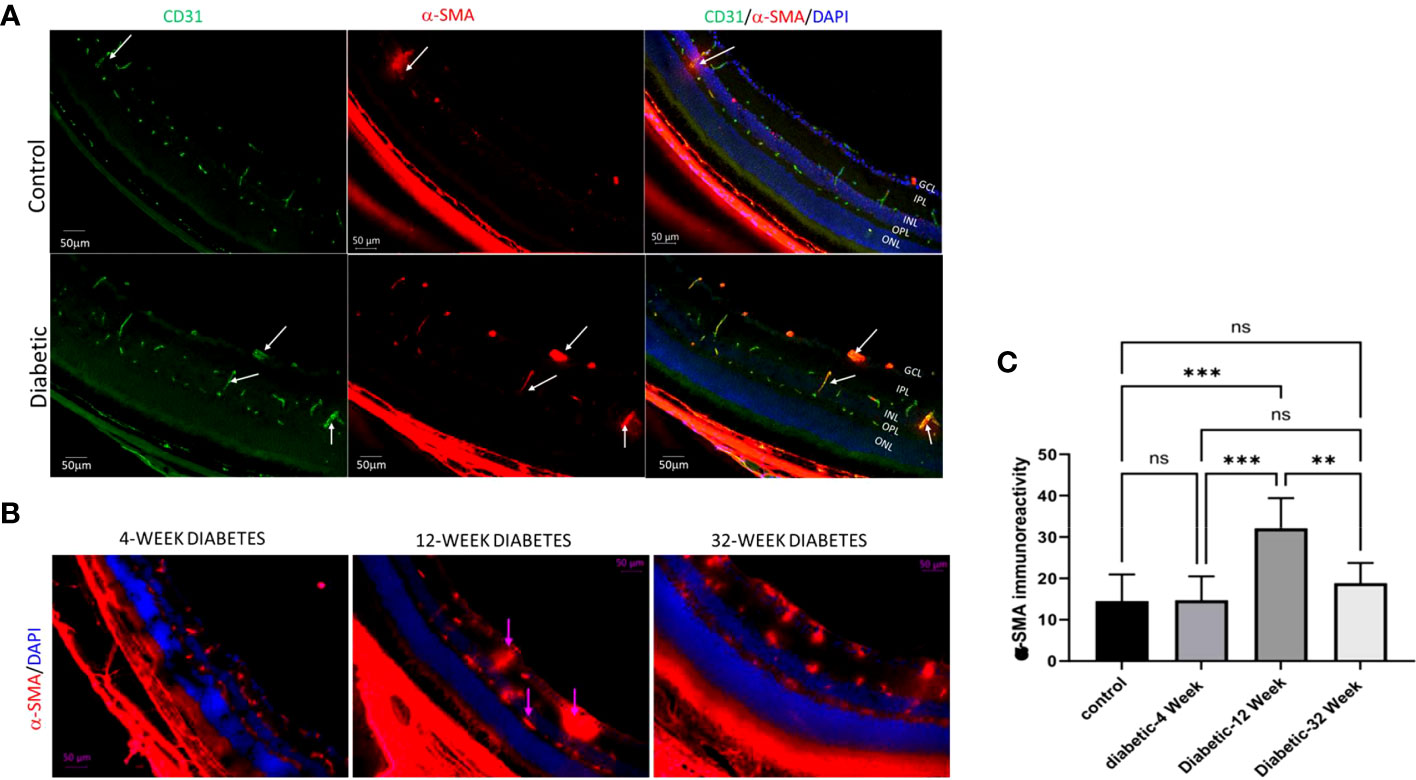
Figure 2 (A) Immunofluorescence of α-SMA in retinal cryosections of 12-Week diabetic vs. Control (10X magnification). (B) 4 Week Diabetic vs. 12 Week Diabetic vs. 32 Week Diabetic α-SMA Immunoreactivity in retinas of diabetic mice (20x magnification). (C) One-way ANOVA multiple comparison test of α-SMA immunoreactivity intensity for 12 Week Control Group, 4 Week Diabetic, 12 Week Diabetic, and 32 Week diabetic mice. (Scale bar= 50μm). **P<.005, ***P<.0005, and ns, not significant.
Our findings are consistent with the suggestion that EndMT plays a crucial role in the pathogenesis of microvascular dysfunction in DR. This transformation could lead to loss of endothelial cells, capillary degeneration, vascular leakage, and retinal hyperpermeability, leading to complications like DME and TRD. Our study provides valuable insight into the temporal changes of EndMT in the context of diabetic retinopathy and highlights the importance of further research in this area. Our research group has started investigating EndMT in postmortem human retina that were obtained from Georgia Eye Bank and processed to prepare paraffin-embedded sections. Slides from 3 diabetic human samples and 3 non-diabetic human samples were used in an IF experiment, with α-SMA as a mesenchymal marker, DAPI as a marker for the nucleus, and VEGFR2 marking endothelial cells. Our preliminary findings suggest that diabetic human retinas exhibit a higher amount of mesenchymal tissue compared to control groups as shown by a marked increase in α-SMA immunoreactivity in retinal vessels of diabetic human subjects compared to the non-diabetic group (Figure 3). Further research is needed to confirm these results and to explore the temporal changes of EndMT in diabetic human retinas.
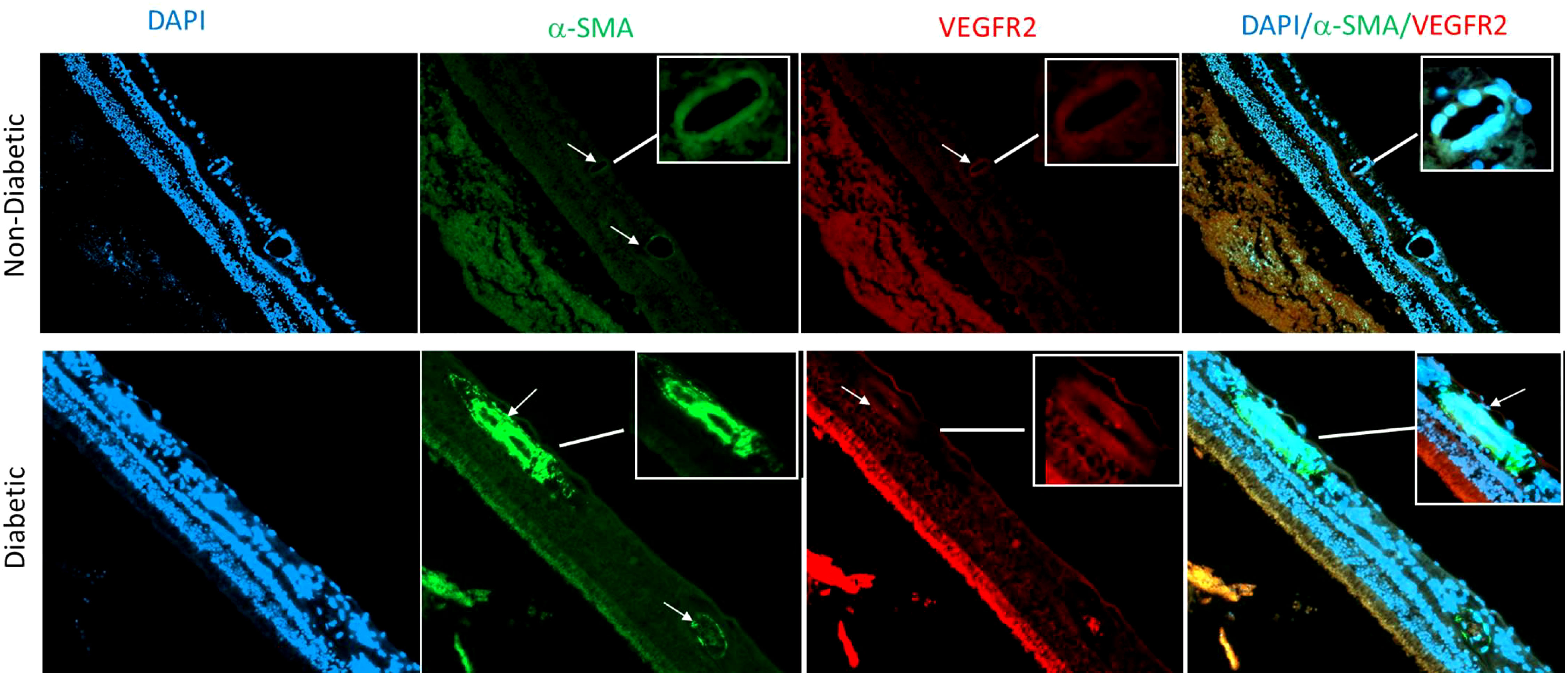
Figure 3 Immunofluorescence of α-SMA in retina sections of human subjects using anti- α SMA (green), vascular marker VEGFR2 (red) and nuclear marker DAPI (blue). There is an obvious increase in α-SMA immunoreactivity in retinal vessels (arrows) of diabetic human donors compared to the non-diabetic donors.
Exploring therapeutic targets for preventing EndMT in diabetic patients is important, and it involves researching various molecules and pathways. While the exact interplay and regulation between these pathways are not yet fully understood, several potential targets have emerged based on extensive research.
Among the pathways implicated in hyperglycemia-induced EndMT, inhibition of Notch, canonical TGF-β, or non-canonical TGF-β signaling alone has shown promise in preventing EndMT in the context of DR (23). However, understanding the extent of pathway overlap and co-regulation remains a subject of ongoing investigation. Intriguingly, a few medications usually used for glucose control in diabetics, such as sodium-glucose co-transporter 2 (SGLT2) inhibitors and GLP-1 agonists, have also shown effects in suppressing EndMT in different organs (84–86). These medications activate AMP-activated protein kinase (AMPK), which inhibits intracellular TGF-β signaling (85–87).
Sprouty-related proteins with EVH1 domain (SPRED2), a member of the Sprouty/SPRED family, areis known for theirits negative regulation of the Ras/Raf/ERK/MAPK signaling pathway. SPRED 2 is another emerging target under investigation as demonstrated by a study exploring its role on EndMT in DR. The researchers used diabetic rat models and human retinal endothelial cells (HRECs) treated with high glucose to simulate DR. The results showed that SPRED2 expression was reduced in the retinal tissues of diabetic rats and high glucose-treated HRECs. By increasing the levels of SPRED2, the researchers observed a suppression of endothelial injury, inhibition of EndMT by regulating specific markers, improvement of tight junction components, and downregulation of the MAPK signaling pathway. These findings suggest that SPRED2 could be a promising therapeutic target for managing the progression of DR (88).
Other proteins, such as Raf kinase inhibitor protein (RKIP), also play a role in EndMT within DR. Increased RKIP levels have been found to exhibit inhibitory effects on the cellular processes associated with EndMT. Moreover, RKIP downregulation has been found to reduce expression of endothelial markers CD31 and vWF in HRCECs under glucose-induced conditions, while RKIP overexpression resulted in their upregulation. These results suggest that RKIP exerts its action by negatively regulating glucose-induced EndMT and associated cellular events in HRCECs, thereby indicating finding ways to increase RKIP can be a potential therapy for managing EndMT in DR (89).
Additionally, the involvement of LPA-1, a receptor for lysophosphatidic acid (LPA), is being studied as a therapeutic target for EndMT in DR. One study demonstrated that the downregulation of acylglycerol kinase (AGK), an enzyme involved in LPA production, suppressed EndMT in HRECs by modulating the LPA-1/TGF-β/Notch signaling pathway. These results suggest that targeting LPA-1 and its associated signaling pathways may hold therapeutic potential for managing EndMT in the context of DR (90).
Furthermore, recent research has established the role of formyl peptide receptor 2 (FRPR2) in DR, contributing to both pathological neovascularization and EndMT. FRPR2 is a G-protein coupled receptor expressed in a variety of cells, including endothelial cells and glial cells. An in vitro study showed that high glucose upregulates FRPR2 in human endothelial cells. They also found that in FRPR2 knock-out diabetic mice, there was a significant decrease in mesenchymal markers on retinal endothelial cells compared to wild-type diabetic mice. This shows that FRPR2 plays an important role in EndMT and may have potential as a novel therapeutic target for DR (91).
Select dietary supplements have displayed potential in inhibiting EndMT. Notably, supplements like resveratrol and eicosapentaenoic acid have shown inhibitory effects on the PKC pathway, effectively impeding the induction of TGF-β and endothelin-1 (ET-1), and subsequently suppressing EndMT in retinal and glomerular endothelial cells, respectively (92, 93). In addition to small molecule compounds, nucleic acid-based approaches have also been explored. Short interfering RNAs (siRNAs) targeting pro-EndMT genes and lncRNAs that mimic anti-EndMT microRNAs have shown promise in preventing EndMT. Experimental silencing of lncRNAs ZFAS1 and MALAT1, as well as induction of various miRNAs, have proven to be effective in suppressing hyperglycemia-induced EndMT through different molecular pathways (76, 94–98). Moreover, synthetic lncRNAs have emerged as a potential strategy to suppress glucose-induced EndMT (99).
Experimental upregulation of lncRNAs H19 and MEG3, which are inhibited by glucose, has shown promise in preventing EndMT in specifically DR (81, 100). Moreover, emerging evidence suggests that specific members of the bone morphogenetic protein (BMP) family (BMP2, BMP4, and BMP9), a subgroup of the TGF-β superfamily, play crucial roles in the regulation of the EndMT process in DR (101, 102). Targeting the inhibitory modulation of these BMPs represents a promising therapeutic approach for preventing or attenuating EndMT in DR.
In summary, EndMT exerts its influence on both the non-proliferative and proliferative stages of DR, contributing to the mechanisms underlying blindness in DR (Figure 4). The critical role played by EndMT in the development of microvascular dysfunction in DR (Figure 5) suggests the need for further investigations to understand the underlying molecular and cellular mechanisms. This should lead to the identification of new therapeutic strategies to mitigate the early pathological changes in DR and preserve vision of diabetic patients.
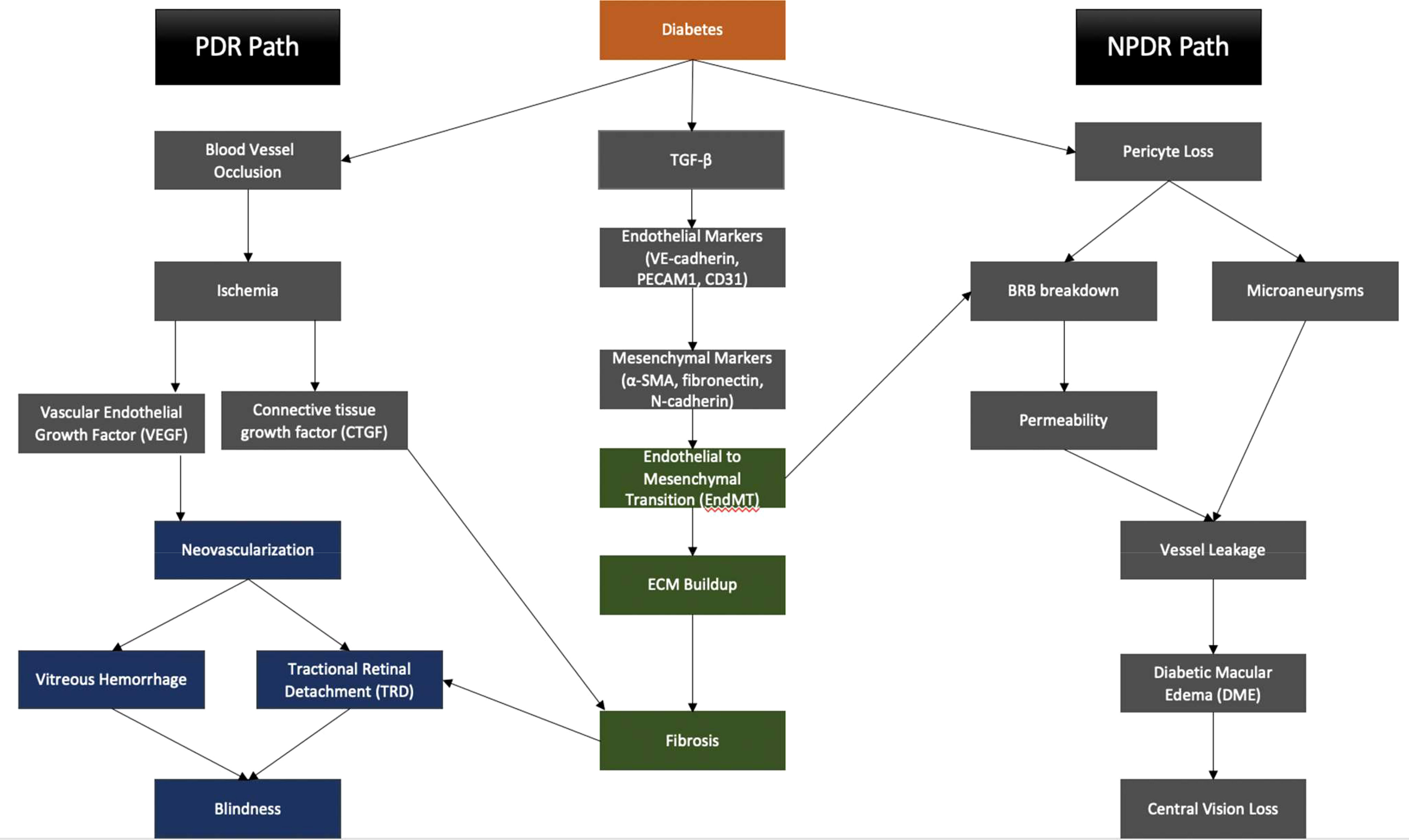
Figure 4 EndMT contributes to the pathogenesis of PDR and NPDR, eventually leading to central vision loss and blindness.
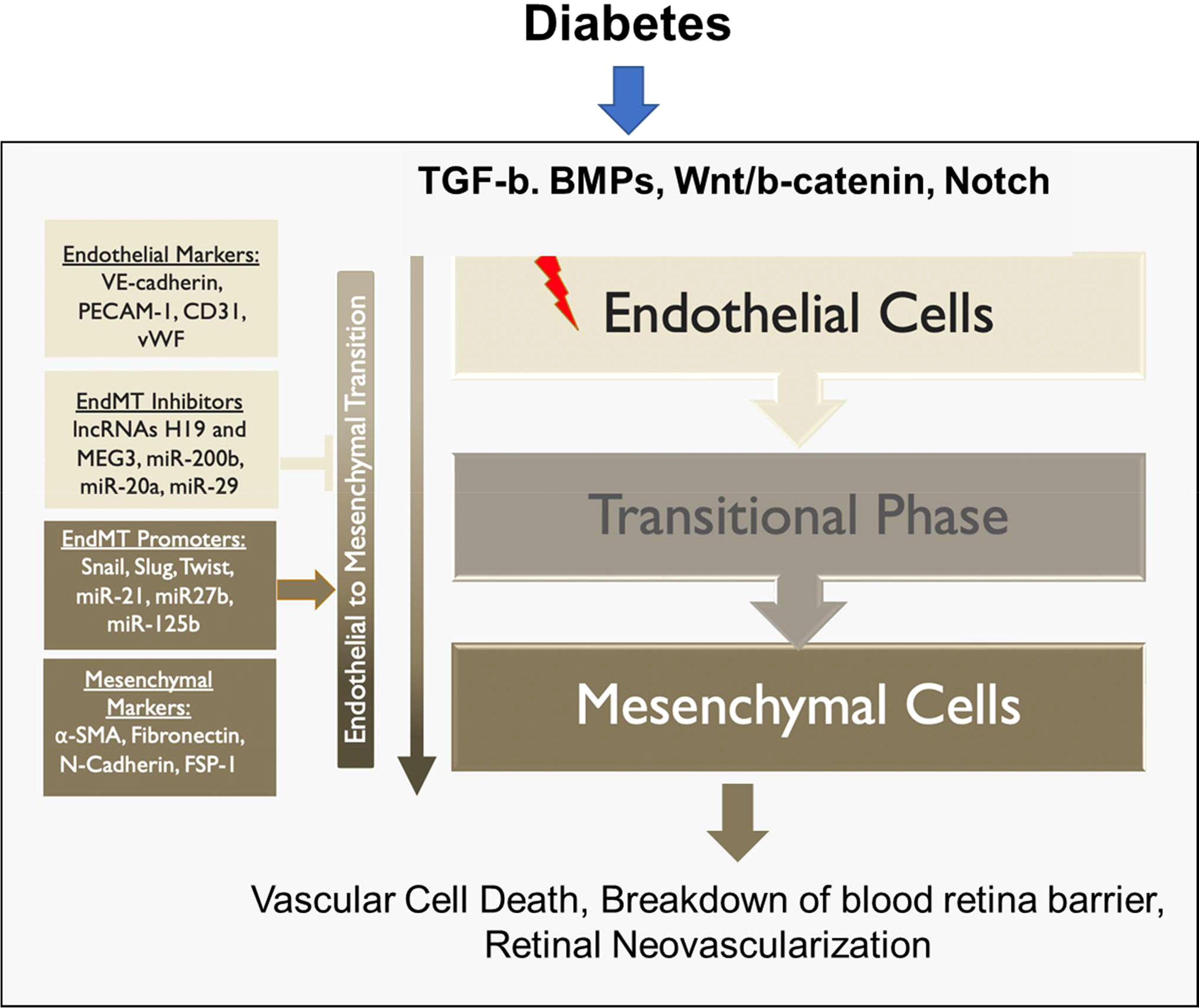
Figure 5 Role of EndMT in the pathogenesis of diabetes-induced retinal microvascular dysfunction.
WN – Conducted lab experiments mentioned in manuscript, wrote full initial draft and created figures of manuscript, revised and edited manuscript. JH – Revised and edited manuscript, conducted lab experiments mentioned in manuscript. MM – Conducted lab experiments mentioned in manuscript. MA-S – Conception of lab experiments mentioned in manuscript, conducted lab experiments, revised, and edited manuscript. All authors contributed to the article and approved thesubmitted version.
Start-up Fund from Oakland University (OU) and Oakland University William Beaumont School of Medicine (OUWB-SOM) to MA-S, NIH/NEI 1) R01 EY030054 (MA-S). Eye Research Center and Eye Research Institute Core Facility (OUWB and OU)
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author MA-S declared that they were an editorial board member of Frontiers at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Shukla UV, Tripathy K. Diabetic retinopathy, in statPearls. Treasure Island (FL): StatPearls Publishing (2023).
2. Lin KY, Hsih W-H, Lin Y-B, Wen C-Y, Chang T-J. Update in the epidemiology, risk factors, screening, and treatment of diabetic retinopathy. J Diabetes Investig (2021) 12(8):1322–5. doi: 10.1111/jdi.13480
3. Duh EJ, Sun JK, Stitt AW. Diabetic retinopathy: current understanding, mechanisms, and treatment strategies. JCI Insight (2017) 2(14). doi: 10.1172/jci.insight.93751
4. Antonetti DA, Klein R, Gardner TW. Diabetic retinopathy. N Engl J Med (2012) 366(13):1227–39. doi: 10.1056/NEJMra1005073
5. Wilkinson-Berka JL, Miller AG. Update on the treatment of diabetic retinopathy. ScientificWorldJournal (2008) 8:98–120. doi: 10.1100/tsw.2008.25
6. Moutray T, Evans JR, Lois N, Armstrong DJ, Peto T, Azuara-Blanco A, et al. Different lasers and techniques for proliferative diabetic retinopathy. Cochrane Database Syst Rev (2018) 3(3):CD012314. doi: 10.1002/14651858.CD012314.pub2
7. Song KH, Jeong J-S, Kim MK, Kwon H-S, Baek K-H, Ko S-H, et al. Discordance in risk factors for the progression of diabetic retinopathy and diabetic nephropathy in patients with type 2 diabetes mellitus. J Diabetes Investig (2019) 10(3):745–52. doi: 10.1111/jdi.12953
8. Hainsworth DP, Bebu I, Aiello LP, Sivitz W, Gubitosi-Klug R, Malone J, et al. Risk factors for retinopathy in type 1 diabetes: The DCCT/EDIC study. Diabetes Care (2019) 42(5):875–82. doi: 10.2337/dc18-2308
9. Lu J, Ma X, Zhang L, Mo Y, Ying L, Lu W, et al. Glycemic variability assessed by continuous glucose monitoring and the risk of diabetic retinopathy in latent autoimmune diabetes of the adult and type 2 diabetes. J Diabetes Investig (2019) 10(3):753–9. doi: 10.1111/jdi.12957
10. Takao T, Takahashi K, Yoshida Y, Kushiyama A, Onishi Y, Tahara T, et al. Effect of postprandial hyperglycemia at clinic visits on the incidence of retinopathy in patients with type 2 diabetes: An analysis using real-world long-term follow-up data. J Diabetes Investig (2020) 11(4):930–7. doi: 10.1111/jdi.13194
11. Group U.K.P.D.S. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. UK Prospective Diabetes Study Group. BMJ (1998) 317(7160):703–13.
12. Estacio RO, McFarling E, Biggerstaff S, Jeffers BW, Johnson D, Schrier RW, et al. Overt albuminuria predicts diabetic retinopathy in Hispanics with NIDDM. Am J Kidney Dis (1998) 31(6):947–53. doi: 10.1053/ajkd.1998.v31.pm9631838
13. Chew EY, Davis MD, Danis RP, Lovato JF, Perdue LH, Greven C, et al. The effects of medical management on the progression of diabetic retinopathy in persons with type 2 diabetes: the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Eye Study. Ophthalmology (2014) 121(12):2443–51. doi: 10.1016/j.ophtha.2014.07.019
14. Schreur V, Asten Fv, Ng H, Weeda J, Groenewoud JMM, Tack CJ, et al. Risk factors for development and progression of diabetic retinopathy in Dutch patients with type 1 diabetes mellitus. Acta Ophthalmol (2018) 96(5):459–64. doi: 10.1111/aos.13815
15. Song J, Liu SCX, Duan H, Kong J, Li Z. Relationship between C-reactive protein level and diabetic retinopathy: A systematic review and meta-analysis. PLoS One (2015) 10(12):e0144406. doi: 10.1371/journal.pone.0144406
16. Tawfik A, Mohamed R, Elsherbiny NM, DeAngelis MM, Bartoli M, Al-Shabrawey M. Homocysteine: A potential biomarker for diabetic retinopathy. J Clin Med (2019) 8(1). doi: 10.3390/jcm8010121
17. Xu J, Chen L-J, Yu J, Wang H-J, Zhang F, Liu Q, et al. Involvement of advanced glycation end products in the pathogenesis of diabetic retinopathy. Cell Physiol Biochem (2018) 48(2):705–17. doi: 10.1159/000491897
18. Han L, Zhang L, Xing W, Zhuo R, Lin X, Hao Y, et al. The associations between VEGF gene polymorphisms and diabetic retinopathy susceptibility: a meta-analysis of 11 case-control studies. J Diabetes Res (2014) 2014:805801. doi: 10.1155/2014/805801
19. Praidou A, Klangas I, Papakonstantinou E, Androudi S, Georgiadis N, Karakiulakis G, et al. Vitreous and serum levels of platelet-derived growth factor and their correlation in patients with proliferative diabetic retinopathy. Curr Eye Res (2009) 34(2):152–61. doi: 10.1080/02713680802585920
20. Wang W, Lo ACY. Diabetic retinopathy: Pathophysiology and treatments. Int J Mol Sci (2018) 19(6). doi: 10.3390/ijms19061816
21. Litvin TV, Ozawa GY, George H. Bresnick M, Cuadros JA, Muller MS, Elsner AE, et al. Utility of hard exudates for the screening of macular edema. Optom Vis Sci (2014) 91(4):370–5. doi: 10.1097/OPX.0000000000000205
22. Mishra C, Tripathy K. Retinal traction detachment, in statPearls. Treasure Island (FL): StatPearls Publishing (2023).
23. Wang E, Wang H, Chakrabarti S. Endothelial-to-mesenchymal transition: An underappreciated mediator of diabetic complications. Front Endocrinol (2023) 14. doi: 10.3389/fendo.2023.1050540
24. Cao Y, Feng B, Chen S, Chu Y, Chakrabarti S. Mechanisms of endothelial to mesenchymal transition in the retina in diabetes. Invest Ophthalmol Vis Sci (2014) 55(11):7321–31. doi: 10.1167/iovs.14-15167
25. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes (2005) 54(6):1615–25. doi: 10.2337/diabetes.54.6.1615
26. Beltramo E, Porta M. Pericyte loss in diabetic retinopathy: mechanisms and consequences. Curr Med Chem (2013) 20(26):3218–25. doi: 10.2174/09298673113209990022
27. Huang H, He J, Johnson DK, Wei Y, Liu Y, Wang S, et al. Deletion of placental growth factor prevents diabetic retinopathy and is associated with Akt activation and HIF1alpha-VEGF pathway inhibition. Diabetes (2015) 64(1):200–12. doi: 10.2337/db14-0016
28. Runkle EA, Antonetti DA. The blood-retinal barrier: structure and functional significance. Methods Mol Biol (2011) 686:133–48. doi: 10.1007/978-1-60761-938-3_5
29. Cunha-Vaz J, Bernardes R, Lobo C. Blood-retinal barrier. Eur J Ophthalmol (2011) 21 Suppl 6:S3–9. doi: 10.5301/EJO.2010.6049
30. Hosoya K, Tachikawa M. The inner blood-retinal barrier: molecular structure and transport biology. Adv Exp Med Biol (2012) 763:85–104. doi: 10.1007/978-1-4614-4711-5_4
31. Diaz-Coranguez M, Ramos C, Antonetti DA. The inner blood-retinal barrier: Cellular basis and development. Vision Res (2017) 139:123–37. doi: 10.1016/j.visres.2017.05.009
32. Rudraraju M, Narayanan SP, SOmanath PR. Regulation of blood-retinal barrier cell-junctions in diabetic retinopathy. Pharmacol Res (2020) 161:105115. doi: 10.1016/j.phrs.2020.105115
33. Vinores SA. Breakdown of the blood–retinal barrier. Encyclopedia Eye (2010) 2010:12. doi: 10.1016/B978-0-12-374203-2.00137-8
34. Kuiper EJ, Nieuwenhoven FAV, de Smet MD, Meurs JCv, Tanck MW, Oliver N, et al. The angio-fibrotic switch of VEGF and CTGF in proliferative diabetic retinopathy. PLoS One (2008) 3(7):e2675. doi: 10.1371/journal.pone.0002675
35. Gerhardinger C, Dagher Z, Sebastiani P, Park YS, Lorenzi M. The transforming growth factor-beta pathway is a common target of drugs that prevent experimental diabetic retinopathy. Diabetes (2009) 58(7):1659–67. doi: 10.2337/db08-1008
36. Piera-Velazquez S, Jimenez SA. Molecular mechanisms of endothelial to mesenchymal cell transition (EndoMT) in experimentally induced fibrotic diseases. Fibrogenesis Tissue Repair (2012) 5(Suppl 1):S7. doi: 10.1186/1755-1536-5-S1-S7
37. Yoshimatsu Y, Watabe T. Roles of TGF-beta signals in endothelial-mesenchymal transition during cardiac fibrosis. Int J Inflam 2011 (2011) p:724080.
38. van Meeteren LA, ten Dijke P. Regulation of endothelial cell plasticity by TGF-beta. Cell Tissue Res (2012) 347(1):177–86. doi: 10.1007/s00441-011-1222-6
39. Wong TY, Sun J, Kawasaki R, Ruamviboonsuk P, Gupta N, Lansingh VC, et al. Guidelines on diabetic eye care: The international council of ophthalmology recommendations for screening, follow-up, referral, and treatment based on resource settings. Ophthalmology (2018) 125(10):1608–22. doi: 10.1016/j.ophtha.2018.04.007
40. Solomon SD, Chew E, Duh EJ, Sobrin L, Sun JK, VanderBeek BL, et al. Diabetic retinopathy: A position statement by the American Diabetes Association. Diabetes Care (2017) 40(3):412–8. doi: 10.2337/dc16-2641
41. Michaelides M, Kaines A, Hamilton RD, Fraser-Bell S, Rajendram R, Quhill F, et al. A prospective randomized trial of intravitreal bevacizumab or laser therapy in the management of diabetic macular edema (BOLT study) 12-month data: report 2. Ophthalmology (2010) 117(6):1078–1086.e2. doi: 10.1016/j.ophtha.2010.03.045
42. Mitchell P, Bandello F, Schmidt-Erfurth U, Lang GE, Massin P, Schlingemann RO, et al. The RESTORE study: ranibizumab monotherapy or combined with laser versus laser monotherapy for diabetic macular edema. Ophthalmology (2011) 118(4):615–25. doi: 10.1016/j.ophtha.2011.01.031
43. Nguyen QD, Brown DM, Marcus DM, Boyer DS, Patel S, Feiner L, et al. Ranibizumab for diabetic macular edema: results from 2 phase III randomized trials: RISE and RIDE. Ophthalmology (2012) 119(4):789–801. doi: 10.1016/j.ophtha.2011.12.039
44. Brown DM, Schmidt-Erfurth U, Do DV, Holz FG, Boyer DS, Midena E, et al. Intravitreal aflibercept for diabetic macular edema: 100-week results from the VISTA and VIVID studies. Ophthalmology (2015) 122(10):2044–52. doi: 10.1016/j.ophtha.2015.06.017
45. Wells JA, Glassman AR, Ayala AR, Jampol LM, Aiello LP, Antoszyk AN, et al. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema. N Engl J Med (2015) 372(13):1193–203.
46. Wells JA, Glassman AR, Ayala AR, Jampol LM, Bressler NM, Bressler SB, et al. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema: Two-year results from a comparative effectiveness randomized clinical trial. Ophthalmology (2016) 123(6):1351–9. doi: 10.1016/j.ophtha.2016.02.022
47. Sivaprasad S, Prevost AT, Vasconcelos JC, Riddell A, Murphy C, Kelly J, et al. Clinical efficacy of intravitreal aflibercept versus panretinal photocoagulation for best corrected visual acuity in patients with proliferative diabetic retinopathy at 52 weeks (CLARITY): a multicentre, single-blinded, randomised, controlled, phase 2b, non-inferiority trial. Lancet (2017) 389(10085):2193–203. doi: 10.1016/S0140-6736(17)31193-5
48. Elman MJ, Aiello LP, Beck RW, Bressler NM, Bressler SB, Edwards AR, et al. Randomized trial evaluating ranibizumab plus prompt or deferred laser or triamcinolone plus prompt laser for diabetic macular edema. Ophthalmology (2010) 117(6):1064–1077.e35.
49. Wroblewski JJ, Hu AY. Topical squalamine 0.2% and intravitreal ranibizumab 0.5 mg as combination therapy for macular edema due to branch and central retinal vein occlusion: An open-label, randomized study. Ophthalmic Surg Lasers Imaging Retina (2016) 47(10):914–23.
50. Lattanzio R, Cicinelli MV, Bandello F. Intravitreal steroids in diabetic macular edema. Dev Ophthalmol (2017) 60:78–90.
51. Boyer DS, Yoon YH, Belfort R Jr, Bandello F, Maturi RK, Augustin AJ, et al. Three-year, randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with diabetic macular edema. Ophthalmology (2014) 121(10):1904–14.
52. Pacella F, Romano MR, Turchetti P, Tarquini G, Carnovale A, Mollicone A, et al. An eighteen-month follow-up study on the effects of Intravitreal Dexamethasone Implant in diabetic macular edema refractory to anti-VEGF therapy. Int J Ophthalmol (2016) 9(10):1427–32.
53. Campochiaro PA, Brown DM, Pearson A, Ciulla T, Boyer D, Holz FG, et al. Long-term benefit of sustained-delivery fluocinolone acetonide vitreous inserts for diabetic macular edema. Ophthalmology (2011) 118(4):626–635.e2. doi: 10.1016/j.ophtha.2010.12.028
54. Bressler NM, Beck RW, Ferris FL. Panretinal photocoagulation for proliferative diabetic retinopathy. N Engl J Med (2011) 365(16):1520–6. doi: 10.1056/NEJMct0908432
55. Ogden TE, Callahan F, Riekhof FT. The electroretinogram after peripheral retinal ablation in diabetic retinopathy. Am J Ophthalmol (1976) 81(4):397–402. doi: 10.1016/0002-9394(76)90293-2
56. Patz A, Fine S, Finkelstein D, Prout T, Aiello L, Bradley R. Photocoagulation treatment of proliferative diabetic retinopathy: The second report of diabetic retinopathy study findings. Ophthalmology (1978) 85(1):82–106.
57. Fong DS, Girach A, Boney A. Visual side effects of successful scatter laser photocoagulation surgery for proliferative diabetic retinopathy: a literature review. Retina (2007) 27(7):816–24. doi: 10.1097/IAE.0b013e318042d32c
58. Blumenkranz MS, Yellachich D, Andersen DE, Wiltberger MW, Mordaunt D, Marcellino GR, et al. Semiautomated patterned scanning laser for retinal photocoagulation. Retina (2006) 26(3):370–6. doi: 10.1097/00006982-200603000-00024
59. Vujosevic S, Martini F, Convento E, Longhin E, Kotsafti O, Parrozzani R, et al. Subthreshold laser therapy for diabetic macular edema: metabolic and safety issues. Curr Med Chem (2013) 20(26):3267–71. doi: 10.2174/09298673113209990030
60. Neubauer AS, Langer J, Liegl R, Haritoglou C, Wolf A, Kozak I, et al. Navigated macular laser decreases retreatment rate for diabetic macular edema: a comparison with conventional macular laser. Clin Ophthalmol (2013) 7:121–8.
61. Alam NM, Mills WC, Wong AA, Douglas RM, Szeto HH, Prusky GT, et al. A mitochondrial therapeutic reverses visual decline in mouse models of diabetes. Dis Model Mech (2015) 8(7):701–10.
62. Gębka A, Serkies-Minuth E, Raczyńska D. Effect of the administration of alpha-lipoic acid on contrast sensitivity in patients with type 1 and type 2 diabetes. Mediators Inflamm 2014 (2014) p:131538.
63. Moschos MM, Dettoraki M, Tsatsos M, Kitsos G, Kalogeropoulos C. Effect of carotenoids dietary supplementation on macular function in diabetic patients. Eye Vis (Lond) (2017) 4:23. doi: 10.1186/s40662-017-0088-4
64. Bischoff J. Endothelial-to-mesenchymal transition. Circ Res (2019) 124(8):1163–5. doi: 10.1161/CIRCRESAHA.119.314813
65. Cho JG, Lee A, Chang W, Lee M-S, Kim J. Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front Immunol (2018) 9:294. doi: 10.3389/fimmu.2018.00294
66. Piera-Velazquez S, Jimenez SA. Endothelial to mesenchymal transition: Role in physiology and in the pathogenesis of human diseases. Physiol Rev (2019) 99(2):1281–324. doi: 10.1152/physrev.00021.2018
67. Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, et al. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest (2015) 125(12):4514–28. doi: 10.1172/JCI82719
68. Giordo R, Ahmed YMA, Allam H, Abusnana S, Pappalardo L, Nasrallah GK, et al. EndMT regulation by small RNAs in diabetes-associated fibrotic conditions: Potential link with oxidative stress. Front Cell Dev Biol (2021) 9:683594. doi: 10.3389/fcell.2021.683594
69. Medici D, Potenta S, Kalluri R. Transforming growth factor-β2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem J (2011) 437(3):515–20. doi: 10.1042/BJ20101500
70. Kokudo T, Suzuki Y, Yoshimatsu Y, Yamazaki T, Watabe T, Miyazono K, et al. Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci (2008) 121(Pt 20):3317–24. doi: 10.1242/jcs.028282
71. ROmano LA, Runyan RB. Slug is an essential target of TGFbeta2 signaling in the developing chicken heart. Dev Biol (2000) 223(1):91–102. doi: 10.1006/dbio.2000.9750
72. Chakraborty S, Wirrig EE, Hinton RB, Merrill WH, Spicer DB, Yutzey KE, et al. Twist1 promotes heart valve cell proliferation and extracellular matrix gene expression during development in vivo and is expressed in human diseased aortic valves. Dev Biol (2010) 347(1):167–79. doi: 10.1016/j.ydbio.2010.08.021
73. Zeng L, Wang G, Ummarino D, Margariti A, Xu Q, Xiao Q, et al. Histone deacetylase 3 unconventional splicing mediates endothelial-to-mesenchymal transition through transforming growth factor β2. J Biol Chem (2013) 288(44):31853–66. doi: 10.1074/jbc.M113.463745
74. Kong C, Zhang F, Hu R, Wang L. METTL3 promotes endothelium-mesenchymal transition of pulmonary artery endothelial cells by regulating TRPC6/calcineurin/NFAT signaling pathways. Evid Based Complement Alternat Med (2023) 2023:8269356. doi: 10.1155/2023/8269356
75. He Y, Dan Y, Gao X, Huang L, Lv H, Chen J, et al. DNMT1-mediated lncRNA MEG3 methylation accelerates endothelial-mesenchymal transition in diabetic retinopathy through the PI3K/Akt/mTOR signaling pathway. Am J Physiol Endocrinol Metab (2021) 320(3):E598–e608. doi: 10.1152/ajpendo.00089.2020
76. Zhang J, Zeng Y, Chen J, Cai D, Chen C, Zhang S, et al. miR-29a/b cluster suppresses high glucose-induced endothelial-mesenchymal transition in human retinal microvascular endothelial cells by targeting Notch2. Exp Ther Med (2019) 17(4):3108–16. doi: 10.3892/etm.2019.7323
77. Wautier JL, Wautier MP. Vascular permeability in diseases. Int J Mol Sci (2022) 23(7). doi: 10.3390/ijms23073645
78. Alvandi Z, Bischoff J. Endothelial-mesenchymal transition in cardiovascular disease. Arterioscler Thromb Vasc Biol (2021) 41(9):2357–69. doi: 10.1161/ATVBAHA.121.313788
79. Abu El-Asrar AM, De Hertogh G, Eynde Kvd, Alam K, Raemdonck KV, Opdenakker G, et al. Myofibroblasts in proliferative diabetic retinopathy can originate from infiltrating fibrocytes and through endothelial-to-mesenchymal transition (EndoMT). Exp Eye Res (2015) 132:179–89. doi: 10.1016/j.exer.2015.01.023
80. Eshaq RS, Aldalati AMZ, Alexander JS, Harris NR. Diabetic retinopathy: Breaking the barrier. Pathophysiology (2017) 24(4):229–41. doi: 10.1016/j.pathophys.2017.07.001
81. Thomas AA, Biswas S, Feng B, Chen S, Gonde J, Chakrabarti S, et al. lncRNA H19 prevents endothelial-mesenchymal transition in diabetic retinopathy. Diabetologia (2019) 62(3):517–30. doi: 10.1007/s00125-018-4797-6
82. Roy S, Kim D. Retinal capillary basement membrane thickening: Role in the pathogenesis of diabetic retinopathy. Prog Retin Eye Res (2021) 82:100903. doi: 10.1016/j.preteyeres.2020.100903
83. Pfister F, Feng Y, Hagen Fv, Hoffmann S, Molema G, Hillebrands J-L, et al. Pericyte migration: a novel mechanism of pericyte loss in experimental diabetic retinopathy. Diabetes (2008) 57(9):2495–502. doi: 10.2337/db08-0325
84. Kanasaki K, Shi S, Kanasaki M, He J, Nagai T, Nakamura Y, et al. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes (2014) 63(6):2120–31. doi: 10.2337/db13-1029
85. Tsai TH, Lee C-H, Cheng C-I, Fang Y-N, Chung S-Y, Chen S-M, et al. Liraglutide inhibits endothelial-to-mesenchymal transition and attenuates neointima formation after endovascular injury in streptozotocin-induced diabetic mice. Cells (2019) 8(6). doi: 10.3390/cells8060589
86. Tian J, Zhang M, Suo M, Liu D, Wang X, Liu M, et al. Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J Cell Mol Med (2021) 25(16):7642–59. doi: 10.1111/jcmm.16601
87. Lin H, Li N, He H, Ying Y, Sunkara S, Luo L, et al. AMPK inhibits the stimulatory effects of TGF-β on smad2/3 activity, cell migration, and epithelial-to-mesenchymal transition. Mol Pharmacol (2015) 88(6):1062–71. doi: 10.1124/mol.115.099549
88. Liu T, Zhao J, Lin C. Sprouty-related proteins with EVH1 domain (SPRED2) prevents high-glucose induced endothelial-mesenchymal transition and endothelial injury by suppressing MAPK activation. Bioengineered (2022) 13(5):13882–92. doi: 10.1080/21655979.2022.2086351
89. Feng L, Zhang C, Liu G, Wang F L, Zhang C, Liu G, Wang F. RKIP negatively regulates the glucose induced angiogenesis and endothelial-mesenchymal transition in retinal endothelial cells. Exp Eye Res (2019) 189: 107851. doi: 10.1016/j.exer.2019.107851
90. Wang H, Feng Z, Han X, Xing Y, Zhang X. Downregulation of acylglycerol kinase suppresses high-glucose-induced endothelial-mesenchymal transition in human retinal microvascular endothelial cells through regulating the LPAR1/TGF-β/Notch signaling pathway. Can J Physiol Pharmacol (2022) 100(2):142–50. doi: 10.1139/cjpp-2021-0265
91. Lou X, Liu S, Shi J, Chen H, Wang Z, Le Y, et al. The G protein coupled formyl-peptide receptor 2 (FPR2) promotes endothelial-mesenchymal transition in diabetic retinopathy. Ophthalmic Res (2023) 66(1):681–91. doi: 10.1159/000529578
92. Giordo R, Nasrallah GK, Posadino AM, Galimi F, Capobianco G, Eid AH, et al. Resveratrol-elicited PKC inhibition counteracts NOX-mediated endothelial to mesenchymal transition in human retinal endothelial cells exposed to high glucose. Antioxidants (Basel) (2021) 10(2). doi: 10.3390/antiox10020224
93. Yasuzawa T, Nakamura T, Ueshima S, Mima A. Protective effects of eicosapentaenoic acid on the glomerular endothelium via inhibition of endMT in diabetes. J Diabetes Res (2021) 2021:2182225. doi: 10.1155/2021/2182225
94. Feng B, Cao Y, Chen S, Chu X, Chu Y, Chakrabarti S, et al. miR-200b mediates endothelial-to-mesenchymal transition in diabetic cardiomyopathy. Diabetes (2016) 65(3):768–79. doi: 10.2337/db15-1033
95. Wang Z, Wang Z, Gao L, Xiao L, Yao R, Du B, et al. miR-222 inhibits cardiac fibrosis in diabetic mice heart via regulating Wnt/β-catenin-mediated endothelium to mesenchymal transition. J Cell Physiol (2020) 235(3):2149–60. doi: 10.1002/jcp.29119
96. Jordan NP, Tingle SJ, Shuttleworth VG, Cooke K, Redgrave RE, Singh E, et al. MiR-126-3p is dynamically regulated in endothelial-to-mesenchymal transition during fibrosis. Int J Mol Sci (2021) 22(16). doi: 10.3390/ijms22168629
97. Feng B, Liu J, Wang E, Su Z, Chakrabarti S. Endothelial derived miRNA-9 mediated cardiac fibrosis in diabetes and its regulation by ZFAS1. PLoS One (2022) 17(10):e0276076. doi: 10.1371/journal.pone.0276076
98. Liu B, Qiang L, Wang G-D, Duan Q, Liu J. LncRNA MALAT1 facilities high glucose induced endothelial to mesenchymal transition and fibrosis via targeting miR-145/ZEB2 axis. Eur Rev Med Pharmacol Sci (2019) 23(8):3478–86.
99. Mercer TR, Munro T, Mattick JS. The potential of long noncoding RNA therapies. Trends Pharmacol Sci (2022) 43(4):269–80. doi: 10.1016/j.tips.2022.01.008
100. Yin Q, He M, Huang L, Zhang X, Zhan J, Hu J, et al. lncRNA ZFAS1 promotes ox-LDL induced EndMT through miR-150-5p/Notch3 signaling axis. Microvasc Res (2021) 134:104118. doi: 10.1016/j.mvr.2020.104118
101. Perera N, Ritchie RH, Tate M. The role of bone morphogenetic proteins in diabetic complications. ACS Pharmacol Transl Sci (2020) 3(1):11–20. doi: 10.1021/acsptsci.9b00064
Keywords: diabetic retinopathy, endothelial-mesenchymal transition, PDR, diabetic macular edema, retinal detachment, therapeutic targets, NPDR
Citation: Nijim W, Moustafa M, Humble J and Al-Shabrawey M (2023) Endothelial to mesenchymal cell transition in diabetic retinopathy: targets and therapeutics. Front. Ophthalmol. 3:1230581. doi: 10.3389/fopht.2023.1230581
Received: 29 May 2023; Accepted: 11 August 2023;
Published: 07 September 2023.
Edited by:
Mohammad Shamsul Ola, King Saud University, Saudi ArabiaCopyright © 2023 Nijim, Moustafa, Humble and Al-Shabrawey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohamed Al-Shabrawey, bWFsc2hhYnJhd2V5QG9ha2xhbmQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.