 Lei Liao
Lei Liao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 15 April 2025
Sec. Cancer Molecular Targets and Therapeutics
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1571583
Lung cancer remains the leading cause of cancer-related mortality worldwide. Tumor-associated macrophages (TAMs) and epithelial-mesenchymal transition (EMT) are key drivers of lung cancer metastasis and drug resistance. M2-polarized TAMs dominate the immunosuppressive tumor microenvironment (TME) and promote EMT through cytokines such as TGF-β, IL-6, and CCL2. Conversely, EMT-transformed tumor cells reinforce TAM recruitment and M2 polarization through immunomodulatory factors such as CCL2 and ZEB1, thereby establishing a bidirectional interplay that fuels tumor progression. Current evidence on this interaction remains fragmented, and a comprehensive review of the TAM-EMT regulatory network and its therapeutic implications is lacking. This review systematically integrates the bidirectional regulatory mechanisms between TAMs and EMT, highlighting their roles in lung cancer progression. It also summarizes emerging therapeutic strategies targeting TAM polarization and the EMT process, emphasizing their potential for clinical translation. This study fills the gap in systematic reviews on the interaction between TAMs and EMT, providing a comprehensive theoretical foundation for future research and the development of novel lung cancer therapies.
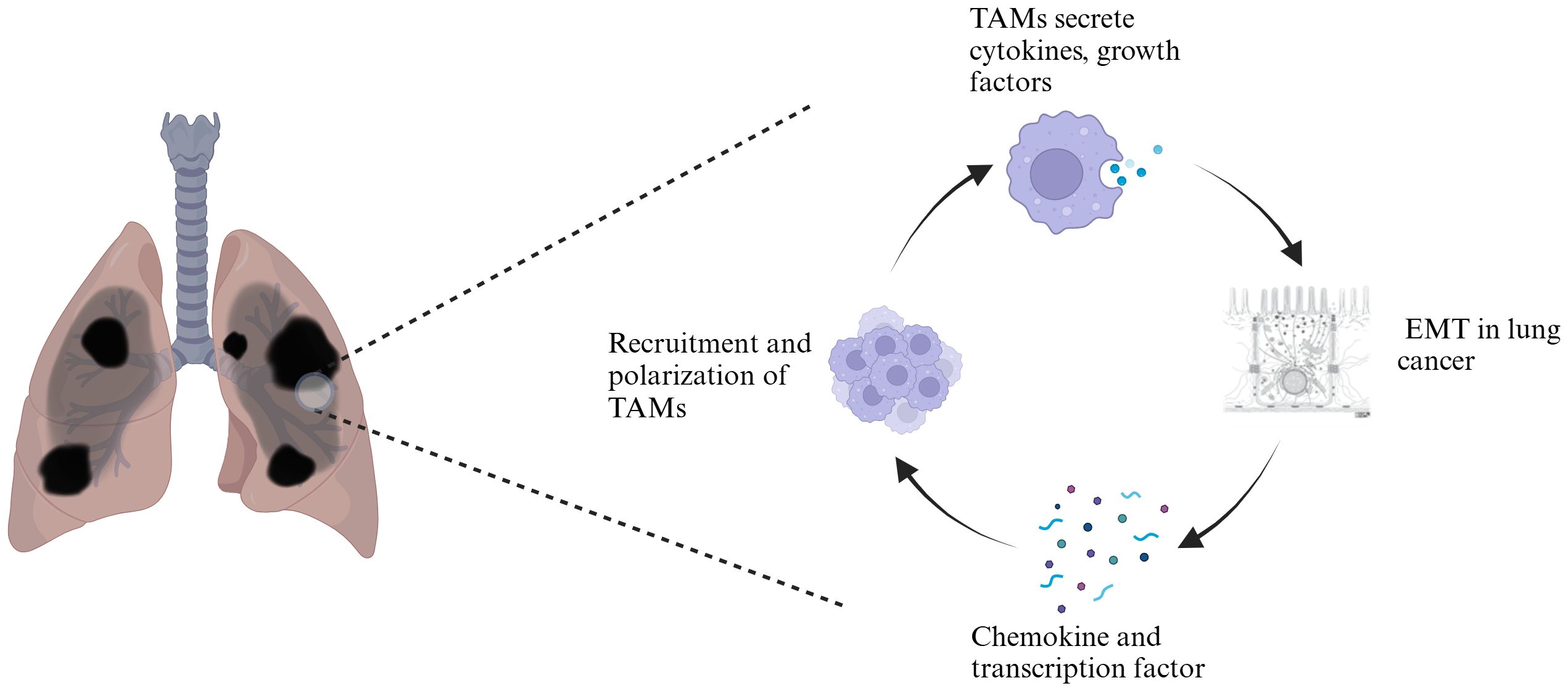
Graphical Abstract.
Primary bronchogenic carcinoma, commonly referred to as lung cancer, is the most lethal malignancy worldwide and the second most common cancer in terms of incidence (1). Lung cancer can be classified into small cell carcinoma (SCLC) and nonsmall cell carcinoma (NSCLC) on the basis of the histological characteristics of the cancer cells. NSCLC is the most common subtype, accounting for 85–90% of all lung cancer types. NSCLC comprises several histological subtypes, including lung adenocarcinoma, squamous cell carcinoma, and large cell carcinoma (2). Currently, treatment options for lung cancer include surgical resection, chemotherapy, targeted therapy, and radiotherapy. Although progress has been made in early diagnosis and treatment, the overall prognosis for patients with lung cancer remains poor.
Macrophages that infiltrate or accumulate in the tumor microenvironment are defined as tumor-associated macrophages (TAMs) and represent the predominant infiltrating immune cells in solid tumors. They exhibit high plasticity, are capable of exhibiting protumor or antitumor functions in response to various signaling stimuli (3–5), and have multiple supportive and inhibitory effects on tumor growth, progression, and metastasis (6).
Epithelial–mesenchymal transition (EMT) refers to a highly programmed process in which epithelial cells lose their original phenotypic characteristics and acquire mesenchymal cell traits (7). During EMT, epithelial-derived tumor cells lose their epithelial characteristics and acquire mesenchymal traits, resulting in reduced intercellular adhesion, loss of cell polarity, and increased cell migration, invasion, and antiapoptotic capabilities. EMT plays a crucial role not only in embryonic development and tissue repair but also in tumor progression, participating in various tumor processes, including tumor initiation, stemness, migration, oncogenic progression, vascular infiltration, malignant metastasis, and resistance to therapy (8).
In recent years, TAMs and EMT have become hot topics in cancer research. Studies have shown that the interaction between TAMs and EMT is closely related to tumor progression (9–11). Although the association between TAMs and EMT has been preliminarily reported, the bidirectional regulatory mechanisms and clinical translational potential have yet to be systematically elucidated. This review aims to explore the role of TAMs in lung cancer EMT and their clinical significance. By systematically reviewing the relevant literature, this study analyzes the interaction between TAMs and EMT, reveals their roles in lung cancer progression, and discusses the clinical potential of targeting TAMs and the EMT process, aiming to provide a theoretical basis for future research and therapeutic strategies.
Pulmonary macrophages originate from two main lineages: tissue-resident macrophages (TRMs), which can locally self-renew independently of hematopoietic stem cell pathways in adults, and monocyte-derived macrophages (MDMs), which originate from adult hematopoietic stem cells and have a shorter lifespan and tend to accumulate at sites of inflammation (12). TAMs are key immune cells in the tumor microenvironment and are derived primarily from monocytes in the bloodstream; these cells migrate to the tumor microenvironment and differentiate, with a small portion also originating from TRMs (13). The differentiation and function of TAMs are regulated by various signals in the tumor microenvironment, including cytokines and chemokines secreted by tumor cells, stromal cells, and other immune cells. Existing studies indicate that TAMs can be classified into the classical activated M1 type and the alternatively activated M2 type (4, 14–17). M1-type TAMs induced by granulocyte-macrophage colony-stimulating factor (GM-CSF), interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), lipopolysaccharides (LPS), or other pathogen-associated molecules are generally considered to exhibit pro-inflammatory and anti-tumor effects, expressing inflammatory factors such as interleukin (IL)-1β, IL-6, and TNF-α (14, 18–20); in contrast, M2-type TAMs induced by macrophage colony-stimulating factor (M-CSF), IL-4, IL-10, IL-13, transforming growth factor (TGF)-β, glucocorticoids, or immune complexes exhibit anti-inflammatory effects and promote tumor progression, expressing anti-inflammatory factors such as IL-10 and TGF-β (14, 19–21). With the advancement of research, M2-type TAMs can be further subdivided into M2a, involved in tissue fibrosis (induced by IL-4 or IL-13), M2b, which promote tumor progression (induced by immune complexes in conjunction with IL-1β or LPS), M2c, responsible for tissue remodeling (induced by IL-10, TGF-β, or glucocorticoids), and M2d, which promote angiogenesis (induced by IL-6, leukemia inhibitory factor (LIF), and adenosine) (6, 17, 18, 22) (Table 1).
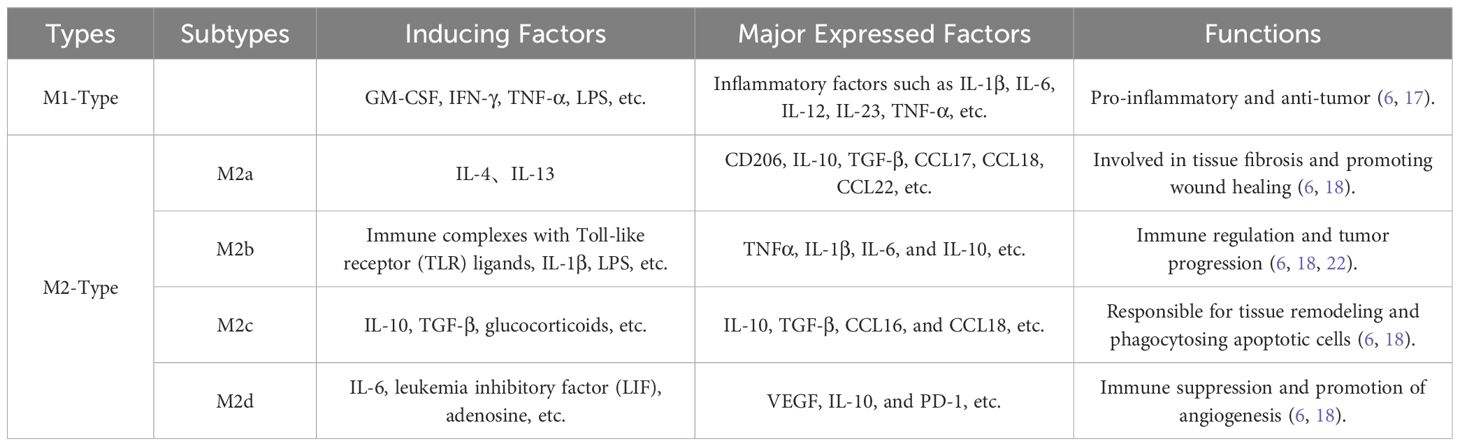
Table 1. Types and functions of TAMs.
In the tumor microenvironment, the phenotype, distribution, and density of TAMs are closely related to patient prognosis (23–25). In lung cancer tissues, the number of M2-type TAMs is usually greater than that of M1-type TAMs (26–29). Compared with tumor nodules, M1 and M2 macrophages primarily infiltrate the tumor stroma, and a higher density of M1-type macrophages is often associated with better survival rates for patients (27, 30), whereas high infiltration of M2-type macrophages generally indicates poorer prognosis (28). Although M1-type macrophages are typically associated with tumor suppression, in the initial stages of tumorigenesis, they support the initiation of tumor development by producing reactive oxygen and nitrogen intermediates. These reactive oxygen and nitrogen intermediates induce DNA damage in proliferating cells and surrounding epithelial cells, thereby increasing the risk of tumor transformation (31). Therefore, during the initial stages of tumor development, M1-type TAMs are the predominant macrophages. However, in early lung cancer, M1-type and M2-type TAMs are not mutually exclusive; some M2-type TAMs also exhibit strong M1-type characteristics. Additionally, there is a strong correlation between the density of M1-type TAMs and the density of TRMs in tumors, which is associated with better survival (26).
During the wound healing process, the activities of M2-type macrophages can promote the restoration of tissue homeostasis. However, in the tumor microenvironment, the polarization of M2-type TAMs plays a crucial role in tumor progression (32). In the vast majority of solid tumors, including lung cancer, M2-type TAMs are positively correlated with tumor growth and metastasis. During the development of lung cancer, M2-type TAMs drive tumor cell proliferation, survival, epithelial-to-mesenchymal transition, and immune evasion by secreting a range of molecules, including growth factors, chemokines, cytokines, and matrix metalloproteinases (MMPs), thereby promoting the invasion and metastasis of tumor cells in vivo (33–35). Among these, the expression of various molecules, such as JNK, HB-EGF, and Mincle, in M2-type macrophages promotes the growth of NSCLC cells (36, 37), whereas vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), IL-10, and exosomes containing miR-155 and miR-196a-5p are associated with lung cancer progression and metastasis (38). After lung cancer formation, TAMs in the tumor microenvironment induce M2 polarization through a series of complex molecular mechanisms, including the COX-2/PGE2/EP4 signaling axis, the AMPKα1/STING positive feedback regulatory pathways, the regulatory role of Zeb1 transcription factors, the involvement of CtBP1, the mediation by CCL2, and the secretion of circFARSA or the release of exosomes carrying PD-L1 (39–45). This leads to the gradual formation of a tumor microenvironment primarily characterized by M2 macrophages, which support and promote angiogenesis, tumor growth and survival, invasion and metastasis, and immune suppression, further driving the growth and metastasis of lung cancer.
Epithelial–mesenchymal transition (EMT) is a complex and multifaceted process that plays a crucial role in embryonic development, tissue repair, and pathological conditions such as cancer and fibrosis. During the EMT process, epithelial cancer cells undergo a series of molecular changes, including downregulation of the epithelial marker E-cadherin and upregulation of the mesenchymal markers vimentin and N-cadherin, thereby acquiring the characteristics of mesenchymal cells. During this transition, epithelial cells lose their polarity and intercellular tight junctions, acquiring enhanced migratory and invasive properties that enable them to traverse the stroma and migrate to new locations. Cancer cells undergoing EMT secrete various cytokines to remodel the tumor immune microenvironment (46).
EMT results in three main phenotypes in different biological contexts (47, 48): (1) Embryonic EMT (also known as Type 1 EMT) primarily occurs during implantation, embryogenesis, and organ development and is driven by the evolutionary need to remodel and diversify tissues to achieve proper morphogenesis and produce functional organisms. This EMT is transient and unrelated to inflammation, fibrosis, or systemic dissemination. ② Regenerative EMT (Type 2 EMT) is closely related to wound healing, tissue regeneration, and organ fibrosis. In the context of injury, regenerative EMT promotes tissue repair by producing activated mesenchymal cells, particularly myofibroblasts, which generate excessive collagen-rich extracellular matrix (ECM). ③ Cancerous EMT (Type 3 EMT) occurs in the context of tumor growth and cancer progression, particularly in tumor cells that have previously undergone genetic and epigenetic changes. During this process, tumor cells convert to a mesenchymal phenotype, thereby acquiring invasive and metastatic capabilities, which are critical steps in cancer progression and metastasis.
In lung cancer, EMT is considered a key mechanism by which tumor cells acquire migratory and invasive abilities; this phenotype is commonly observed in primary squamous cell carcinoma (SCC) and lung adenocarcinoma (LUAD) and typically occurs early in the pathogenesis of lung cancer (49). The occurrence of EMT is closely associated with metastasis, drug resistance, immune evasion, and poor prognosis in patients with lung cancer (48). The expression levels of markers, such as E-cadherin, which maintains adhesive junctions, and vimentin, which is involved in cytoskeletal remodeling, are significantly related to the prognosis of non-small cell lung cancer (NSCLC) (50, 51). As a fundamental event in the EMT process, the loss of E-cadherin expression is a key step in tumor cell infiltration and progression (52). Studies have shown that conditional deletion of E-cadherin or the expression of dominant-negative E-cadherin leads to weakened intercellular adhesion among cancer cells and induces vascular invasion and tumor growth through the upregulation of VEGF-A and VEGF-C mediated by β-catenin, thereby inducing micrometastasis in lung adenocarcinoma (53). Furthermore, EMT is also associated with tumor stem cell properties, endowing tumor cells with enhanced migratory and invasive capabilities (54, 55).
On the other hand, several genes in lung cancer, such as Twist, Snail, and TGF-β1, have been confirmed to be associated with EMT, promoting the process of EMT in lung cancer by downregulating E-cadherin and upregulating Vimentin (56–58). Additionally, lung cancer cells can induce epithelial–mesenchymal transition (EMT) through various molecules and signaling pathways. In non-small cell lung cancer, the overexpression of FoxQ1, CTEN, and EDA fibronectin (EDAFN), as well as the activation of the RAGE receptor by advanced glycation end products, can induce EMT in lung cancer cells, promoting tumor progression (59–62). H.R. et al. (63) found that NSCLC cells can induce EMT and promote proliferation, migration, and invasion by downregulating SETBP, which activates the ERK1/2 pathway.
Tumor-associated macrophages (TAMs) exhibit a multilayered and profound regulatory effect on the epithelial–mesenchymal transition (EMT) process in the tumor microenvironment, and their quantity and activation state significantly correlate with EMT regulation. In various solid tumors, including non-small cell lung cancer, the overall quantity of TAMs and the high expression of specific markers (such as CD68 and CD163) are closely related to enhanced EMT characteristics in cancer cells (such as downregulation of E-cadherin and upregulation of vimentin), indicating that EMT features are more pronounced in tumor areas rich in TAMs (33, 64–66). Furthermore, during lung cancer progression, there is a significant increase in the number of TAMs and tumor-associated fibroblasts, along with a marked increase in the expression levels of EMT markers in tumor tissues (67).
TAMs secrete various cytokines and growth factors that act on relevant signaling pathways, directly or indirectly participating in the initiation, maintenance, and progression of EMT in lung cancer, significantly promoting the loss of cell polarity and weakening intercellular adhesion, thereby disrupting the stability of intercellular connections (Figure 1). This process provides an important biological basis for the migration and invasion of tumor cells, serving as a key step in the malignant progression and metastasis of lung cancer.
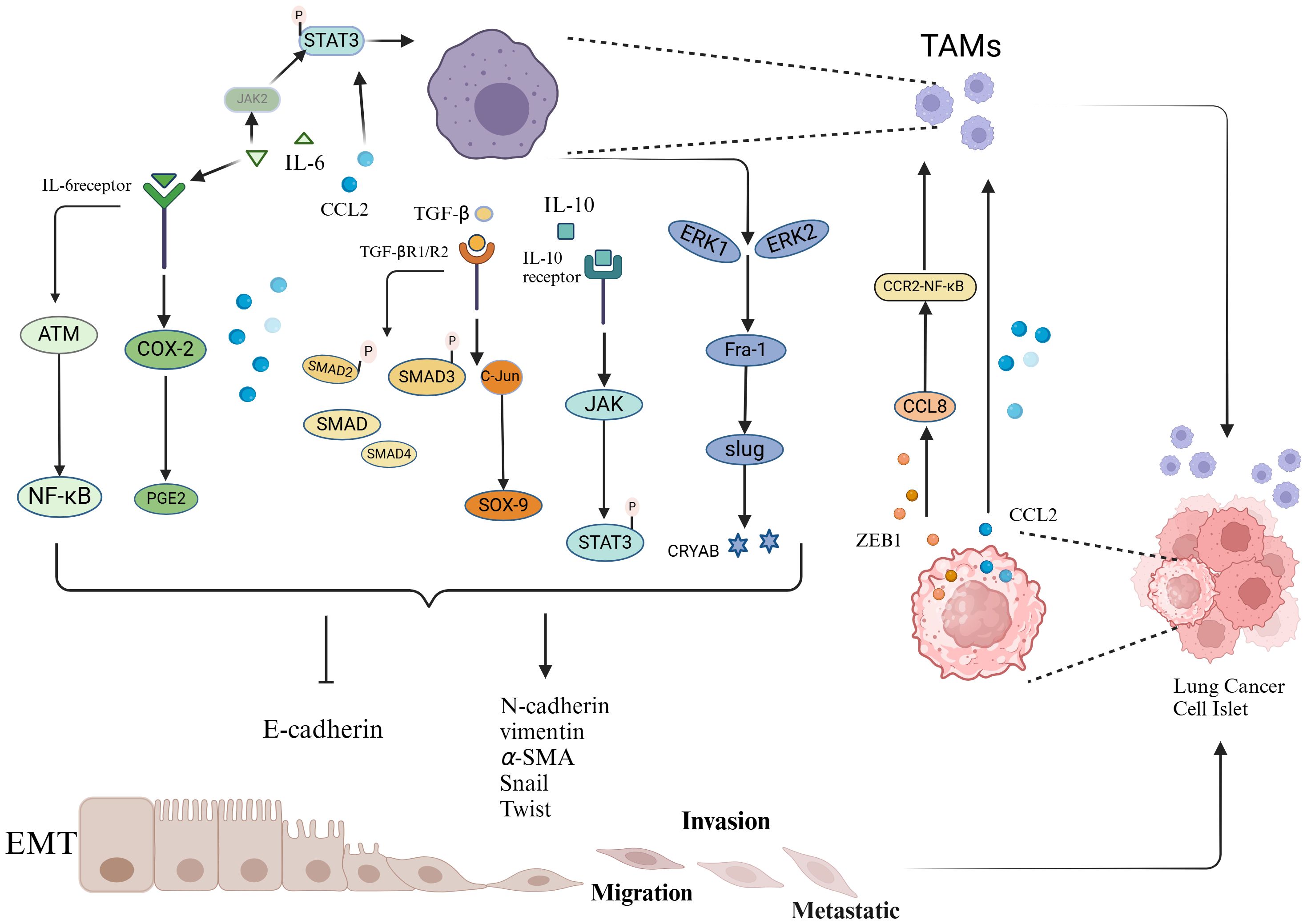
Figure 1. Schematic representation of the interaction between TAMs and EMT.
TAMs promote the EMT of tumor cells by secreting various factors, including TGF-β, IL-6, and CCL2. These factors act on intracellular signaling pathways, including COX-2/PGE2, STAT3-C/EBPβ, ATM/NF-κB, TGF-β-SMAD, SMAD/ZEB, ERK1/2/Fra-1/Slug, and JAK1-STAT3. These pathways suppress the expression of epithelial markers while promoting the expression of mesenchymal markers, thereby inducing EMT in tumor cells. Lung cancer cells undergoing EMT secrete increased levels of chemokines and transcription factors, such as the chemokine CCL2 and the transcription factor ZEB1, further enhancing the recruitment and polarization of TAMs.
(1) IL-6
Interleukin-6 (IL-6) is a multifunctional cytokine that plays a significant role in regulating the immune system (68). Research by Dehai et al. (69) found that THP-1-derived macrophages promote the invasiveness and EMT process of lung adenocarcinoma cells by secreting IL-6. In another study, IL-6 was shown to promote the nuclear translocation of β-catenin via the COX-2/PGE2 pathway, thereby inducing EMT and enhancing tumor cell invasion (70). Furthermore, a study by Hu et al. (71) reported that TAMs promote IL-6 expression and activate the EMT pathway by forming an IL6-STAT3-C/EBPβ-IL6 positive feedback loop. Notably, IL-6 is also associated with chemotherapy resistance and prognosis in lung cancer patients; studies have shown that IL-6 can activate and enhance chemotherapy resistance in lung cancer by activating the ATM/NF-κB pathway (72). Additionally, high levels of IL-6 in plasma are regarded as important markers for poor prognosis in chemotherapy patients (73).
(2) TGF-β and related signaling pathways
Transforming growth factor-beta (TGF-β) is a prototypical member of a structurally and functionally related family of proteins that regulate cell proliferation, migration, and the differentiation of a variety of cell types (74). In the tumor microenvironment, TGF-β has a dual role in tumor progression. In the early stages of cancer, it acts as an effective tumor suppressor by inhibiting the cell cycle and promoting apoptosis, thereby suppressing tumor initiation and progression. However, in the middle to late stages of cancer, tumor cells may develop resistance to TGF-β or be reprogrammed by it (75), transforming TGF-β into a tumor promoter that induces epithelial–mesenchymal transition (EMT) and enhances the invasive and metastatic capabilities of tumor cells, as well as their resistance to chemotherapy. Furthermore, it supports cancer growth and progression by activating tumor angiogenesis and cancer-associated fibroblasts, as well as enabling tumors to evade immune responses (74, 76).
TGF-β is a key factor released by TAMs and has been shown to significantly promote the transition of epithelial cells to mesenchymal cells by activating various intrinsic pathways, such as the AKT, SMAD, and β-catenin pathways, thereby increasing cell migration and invasion (77–81). In the lung cancer microenvironment, TAMs can promote EMT in tumor cells by releasing TGF-β, which acts on signaling pathways such as the Smad/ZEB and C-jun/SMAD3 pathways, thereby increasing the metastatic potential and proliferation ability of tumors (82, 83). Research by J. A et al. (84) further revealed that B7-H4-expressing macrophages may regulate the EMT process by secreting TGF-β1, thereby promoting pleural metastasis in lung cancer. Additionally, a study by Bonde et al. (64) reached similar conclusions, indicating that TAMs induce EMT in cancer cells within tumors through TGF-β signaling and the β-catenin pathway.
(3) CCL2
CCL2 is a key chemokine that regulates the migration of monocytes/macrophages into the TME (85, 86). In lung cancer tissues, CCL2 secreted by TAMs promotes EMT through a dual regulatory mechanism: on one hand, it significantly downregulates the expression of the epithelial marker E-cadherin, while on the other, it simultaneously upregulates the expression levels of the mesenchymal marker vimentin, as well as matrix metalloproteinases MMP-2 and MMP-9, thereby enhancing the invasion and migration capabilities of NSCLC cells (87). Notably, CCL2 and IL-6 exhibit a significant synergistic effect in EMT induction. They mutually induce each other to activate STAT3 phosphorylation. In turn, activated STAT3 can regulate the expression of IL-6 and CCL2, forming a positive feedback loop that not only amplifies STAT3 signaling but also drives a cascade reaction in the EMT process. Additionally, CCL2 can significantly enhance IL-6-induced EMT by upregulating the expression of the transcription factor Twist (88).
(4) Other Pathways
In addition to the aforementioned classical cytokine pathways, several studies have revealed non-classical mechanisms by which TAMs regulate EMT in lung cancer: Li et al. (38) found that exosomal miR-155/miR-196a-5p can promote EMT in NSCLC cells through epigenetic modifications. Cao et al. (89) identified the IL-10/STAT3 phosphorylation cascade as a critical activator of EMT. Guo et al. (90) revealed that M2 macrophages enhance EMT by upregulating CRYAB expression and activating the ERK1/2/Fra-1/Slug signaling axis.
EMT, while promoting tumor invasion and metastasis, is often accompanied by the attraction and infiltration of TAMs (91, 92). After undergoing EMT, tumor cells typically release increased levels of chemokines (such as CCL2), further promoting the recruitment and polarization of TAMs. This interaction forms a positive feedback loop that collectively drives tumor progression (Figure 1). Currently, the regulatory mechanisms by which EMT influences TAMs primarily involve the following pathways:
(1) TWIST1/CCL2-Mediated Macrophage Recruitment and M2 Polarization
TWIST1 is a key regulatory factor in the interplay between TAMs and EMT. On one hand, TWIST1 directly participates in the EMT process by regulating E-cadherin and other related proteins, thereby promoting EMT in lung cancer cells. On the other hand, activated TWIST1 upregulates the expression of CCL2, which acts as a chemokine to attract macrophages and induce their polarization toward the M2 phenotype. The polarized M2 macrophages, in turn, promote EMT in lung cancer cells, forming a positive feedback loop that continuously drives tumor progression and metastasis (93). Additionally, Wang et al. (42) found that elevated CtBP1 protein expression can induce EMT in NSCLC cells and regulate the activation of the NF-κB signaling pathway, leading to increased CCL2 secretion, which in turn promotes TAM recruitment and polarization.
(2) ZEB1 Promotes TAM Accumulation and Infiltration in the Hypoxic TME
Hypoxia is a critical microenvironmental stressor that regulates various potent immunosuppressive phenomena associated with tumor progression (94). Hypoxia triggers EMT in various cancers, including breast, prostate, and oral cancers (95). The accumulation of TAMs in the hypoxic TME is closely associated with malignant tumor progression. Moreover, hypoxic regions exhibit significant spatial overlap with EMT invasion fronts and TAM distribution (95, 96). ZEB1 is a key transcription factor in EMT, endowing cancer cells with an invasive, mesenchymal-like phenotype and serving as a predictor of poor clinical prognosis in most cancers. In the hypoxic TME, ZEB1 promotes macrophage infiltration by activating the transcription of CCL8, which subsequently attracts macrophages via the CCR2-NF-κB pathway, enhancing TAM accumulation in the tumor microenvironment (97).
In addition to the aforementioned factors, various inflammatory cytokines induced during the EMT process, such as the pro-inflammatory and immunoregulatory TNF-α (98), neutrophil-recruiting GROs (99), and angiogenesis-promoting IL-8 (100), also play crucial roles in the interaction between EMT and TAMs. These factors may collectively contribute to creating a favorable environment for tumor development.
The interaction between TAMs and EMT is also significant in other types of cancer. In pancreatic ductal adenocarcinoma, Xiong et al. (101) demonstrated that TAMs drive EMT by activating the Snail transcription factor via the TGF-β/Smad2/3/4 signaling axis, thereby promoting stromal invasion and tumor microenvironment remodeling. In hepatocellular carcinoma, Zhang et al. (102) discovered a HIF-1α/IL-1β signaling loop between cancer cells and TAMs in the hypoxic microenvironment, leading to epithelial-mesenchymal transition and metastasis. In rectal cancer, Zheng et al. (103) found that the long non-coding RNA LINC00543 enhances EMT through the pre-miR-506-3p/FOXQ1 axis, leading to the upregulation of CCL2, which in turn promotes CCL2-mediated macrophage recruitment and M2-like polarization. Further research by Wei et al. (104) confirmed that TAMs can enhance EMT progression via the STAT3/miR-506-3p/FoxQ1 pathway, leading to increased CCL2 production, which facilitates macrophage recruitment. This establishes an “EMT-CCL2-TAM” positive feedback loop that drives rectal cancer progression. In breast cancer, Su et al. (105) found that EMT-processed cancer cells secrete GM-CSF, which activates macrophages into a TAM-like phenotype. The activated macrophages, in turn, secrete CCL18, inducing EMT in cancer cells and establishing a positive feedback loop. This loop is crucial for promoting breast cancer cell metastasis and is associated with poor prognosis in breast cancer patients.
In summary, the interaction between TAMs and EMT creates a positive feedback mechanism that not only enhances the migratory capacity of tumor cells but also leads to sustained immune suppression within the tumor microenvironment, facilitating tumor cell escape from host immune surveillance and clearance.
Therapeutic strategies targeting TAMs and EMT are being progressively developed, showing promising preclinical prospects. By inhibiting the recruitment or polarization of TAMs, their supportive role in tumors can be diminished, thereby enhancing the efficacy of conventional treatments such as chemotherapy and radiotherapy. Additionally, blocking the EMT process to reduce the migration, invasion, and drug resistance of tumor cells is also considered a highly promising strategy for lung cancer treatment. Here, we summarize the current therapeutic strategies targeting TAMs and EMT as key points (Table 2) and outline the progress of lung cancer clinical trials related to TAMs and EMT (Table 3).
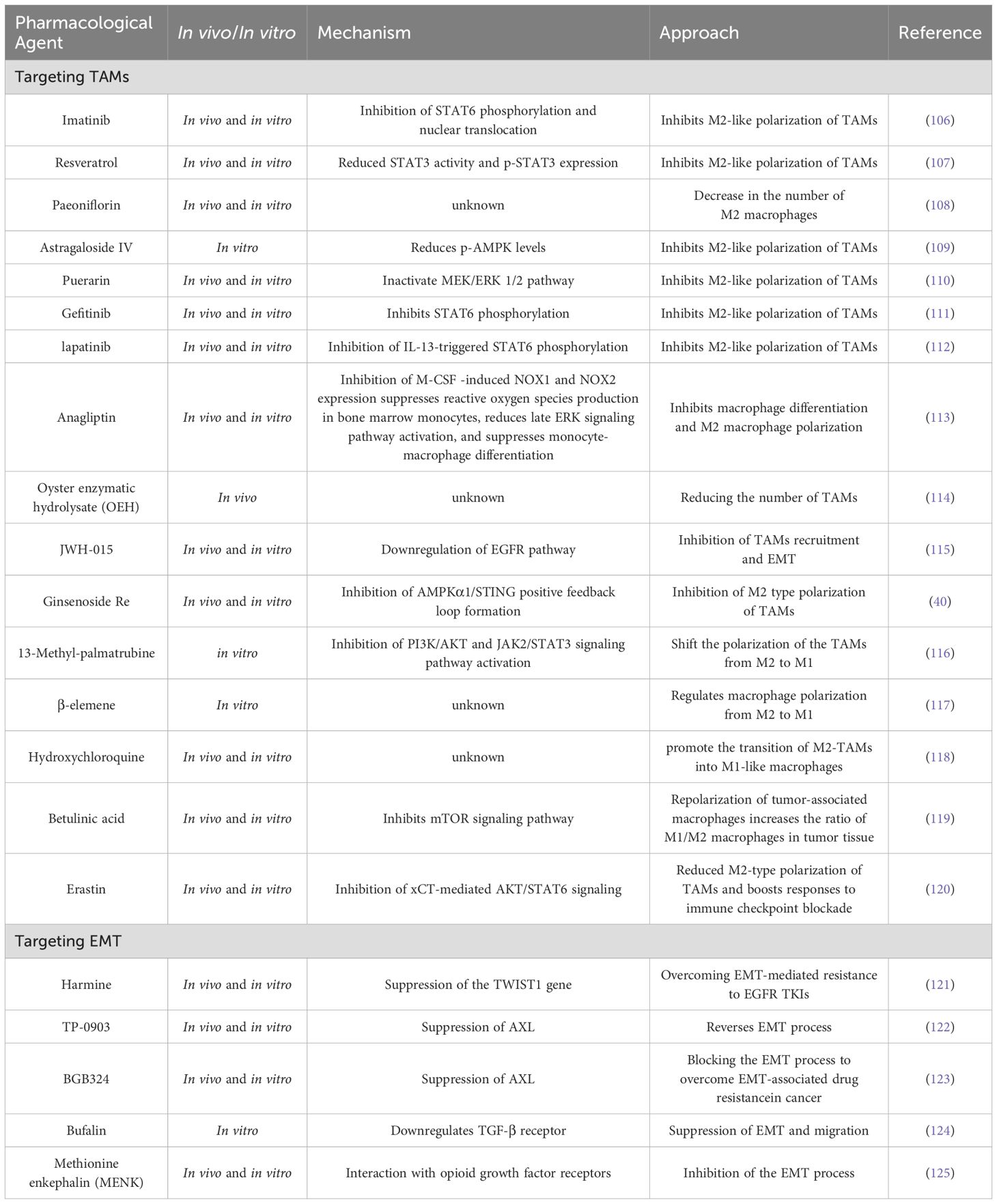
Table 2. Therapeutic approaches targeting TAMs and EMT in lung cancer.
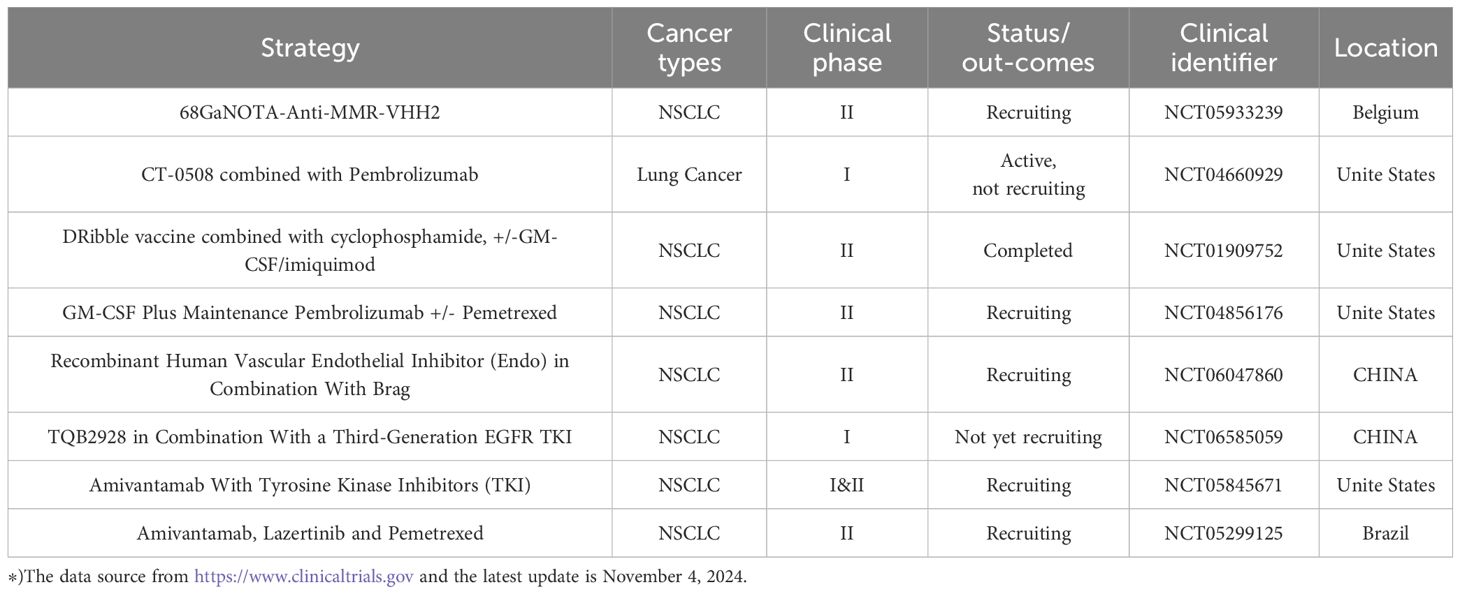
Table 3. Clinical trials related to TAMs and EMT in lung cancer therapy.
In the lung cancer microenvironment, TAMs constitute a major component of immune cells. They not only act as immunosuppressive cells, enabling lung cancer cells to escape immune surveillance but also directly promote cancer cell proliferation, survival, invasion, and metastasis (126). Therefore, the precise regulation and development of therapeutic strategies targeting TAMs have become important research directions in the field of lung cancer treatment.
(1) Modulating the number of TAMs
As mentioned earlier, M2-type TAMs dominate the lung cancer microenvironment, and their high infiltration at tumor sites is a crucial factor in tumor progression. Therefore, reducing the density of TAMs, particularly the number of M2-type TAMs, can effectively inhibit lung cancer progression. Blocking various chemokines produced by tumor and stromal cells, such as monocyte chemoattractant protein-1 (MCP-1), prostaglandin E2 (PGE2), colony-stimulating factor 1, and CCN3, can inhibit the recruitment of TAMs to tumor sites, thereby suppressing tumor progression and preventing metastasis (127, 128). Several drugs, such as ginsenoside (40), imatinib (106), resveratrol (107), paeoniflorin (108), astragaloside IV (109), puerarin (110), gefitinib (111), apatinib (112), and anagliptin (113), have been shown to inhibit M2 polarization of macrophages and reduce TAM infiltration, thereby suppressing lung cancer progression. Additionally, two studies indicated that oyster hydrolysate (OEH) and cannabinoid receptor-2 agonists could reduce the recruitment of macrophages to tumor sites and inhibit M2-type macrophage-induced EMT, thereby decreasing the migration and invasion capabilities of lung cancer cells (114, 115). On the other hand, increasing the number of M1-type TAMs is also considered an effective approach to suppress tumors. Liu et al. (129) found that the overexpression of the transcription factor TCF21 promotes the polarization of TAMs to M1 macrophages and enhances the impact of macrophages on T-cell activity, thereby strengthening the body’s antitumor immune response.
(2) Altering the function of TAMs
Given that TAMs exhibit significant plasticity and can polarize into different phenotypes in response to various stimuli in the tumor microenvironment, “reprogramming” TAMs to adopt an antitumor phenotype represents a promising therapeutic strategy. A series of drugs, such as 13-methyl-palmatrubine (13MP) derived from the flower of Crotalaria juncea (116), β-caryophyllene (117), hydroxychloroquine (118), and birch bark acid (119), have been found to regulate the polarization state of TAMs by modulating signaling pathways such as the AMPKα1/STING, mTOR, PI3K/AKT, and JAK/STAT3 pathways, facilitating the transition from the tumor-promoting M2 phenotype to the antitumor M1 phenotype and thereby inhibiting lung cancer growth and progression. Additionally, Lee et al. (130) found that inhibiting the Wnt/β-catenin signaling pathway can also reprogram TAMs into an M1-like phenotype that suppresses tumors. Lin et al. (131) achieved combined administration of osimertinib and panobinostat (Pan) via a liposome codelivery system modified with lactoferrin; this strategy not only reversed EMT-related resistance by repolarizing TAMs from the M2 phenotype to the M1 phenotype but also inhibited tumor metabolism and angiogenesis, providing a new therapeutic approach to overcoming resistance in NSCLC. Notably, the application of nanotechnology, such as ultrasound-mediated PLGA-PEI nanobubbles carrying STAT6 siRNA and intraperitoneal injection of HA-PEI nanoparticles, has also demonstrated the ability to repolarize TAMs from the M2 phenotype to the M1 phenotype, thereby inhibiting the progression of non-small cell lung cancer (132, 133).
(3) TAMs and immunotherapy in lung cancer
TAMs play a crucial role in immunotherapy for lung cancer. They inhibit phagocytosis and tumor immunity by expressing PD-1, and blocking PD-1/PD-L1 in vivo can enhance macrophage phagocytosis and reduce tumor growth. Monoclonal antibodies that block PD-1/PD-L1 have demonstrated significant clinical efficacy in various cancer patients, including those with non-small cell lung cancer (NSCLC) (134). In NSCLC, MARCO-expressing TAMs exhibit an M2 phenotype that promotes tumor growth, and this phenotype is positively correlated with immune response pathways (135). The polarization state of TAMs can affect the activity of NF-κB, which participates in immune evasion by regulating PD-L1 expression in tumor cells. Thus, inhibiting the NF-κB signaling pathway may help reduce PD-L1 expression and enhance the efficacy of immunotherapy (136). Furthermore, xCT derived from TAMs is closely associated with poor prognosis in lung cancer patients. xCT deficiency downregulates the AKT/STAT6 signaling pathway, inhibits M2 polarization of macrophages, increases T-cell infiltration, and activates inflammatory and immune responses. The xCT inhibitor erastin enhances sensitivity to PD-L1 and effectively suppresses lung cancer progression (120). Additionally, several other molecules and signaling pathways that target TAMs have been identified in lung cancer, such as TCF21 (129), ADPGK-AS1 (137), TLR4 (138) and JMJD6/STAT3/IL-10 (139), which are also considered potential targets for immunotherapy. Notably, the recently introduced chimeric antigen receptor macrophage (CAR-M) therapy has opened new possibilities for the immunotherapy of solid tumors (140). Although CAR-M has not yet been reported for the treatment of lung cancer, several CAR-M therapies, such as CT-0508 (NCT04660929) and MCY-M11 (NCT03608618), have been approved by the U.S. Food and Drug Administration (FDA) to enter clinical trials for the treatment of recurrent or metastatic solid tumors overexpressing HER2 as well as recurrent/refractory ovarian cancer and peritoneal mesothelioma (141, 142). With continuous advancements in gene editing technologies, synthetic biology, and biomaterials science, CAR-M therapy is expected to become a crucial component of cancer immunotherapy, providing a powerful tool for overcoming solid tumors, including lung cancer.
The EMT process plays a crucial role in the metastasis, immune evasion, and chemotherapy resistance of lung cancer. Therefore, therapeutic strategies targeting EMT are particularly important.
(1) EGFR-AKT:
Epidermal growth factor receptor (EGFR)-mutant tumors have become key targets in the study of lung cancer resistance since their discovery in 2004. Currently, the third-generation EGFR inhibitor osimertinib is widely used in first-line treatment (143). However, in recent years, several previously uncommon acquired resistance mechanisms have emerged in lung cancer patients treated with osimertinib, with increasing frequency. These mechanisms include acquired EGFR mutations (e.g., C797S), amplification of MET and HER2, and small-cell transformation (144–146). Notably, epithelial–mesenchymal transition (EMT) transcription factors such as ZEB1, Slug, and TWIST1 have been identified as drivers of EGFR TKI resistance mediated by EMT, suggesting that these transcription factors could be potential targets for treating EMT-related resistance in lung cancer (121, 147–149).
(2) Regulation of microRNAs and circRNAs:
Research on EMT-related microRNAs and circRNAs has provided new therapeutic approaches and strategies for lung cancer patients. Specifically, studies have shown that in cancer cells undergoing Snail-induced epithelial–mesenchymal transition (EMT), tumor cells can deliver miR-21 via exosomes to inhibit the activity of the NLRP3 inflammasome in TAMs, thereby enhancing the resistance of tumor cells (150). Another study (151) revealed that miR-138-5p inhibits the epithelial–mesenchymal transition, growth, and metastasis of lung adenocarcinoma cells by targeting ZEB2. These findings suggest that modulating specific microRNAs could be an effective strategy for targeting EMT. Additionally, Jiang et al. (152) discussed the roles of EMT-inducing and EMT-suppressing circRNAs in lung cancer. They reported that EMT-inducing circRNAs primarily promote EMT-mediated metastasis by affecting key members of EMT-related signaling pathways in NSCLC (e.g., Wnt/β-catenin), whereas EMT-suppressing circRNAs play a significant role in inhibiting EMT-mediated metastasis in NSCLC by acting as sponges for microRNAs, influencing the expression of EMT transcription factors (EMT-TFs), EMT-related signaling, and EMT markers.
(3) Targeting the TAM Family Receptor Tyrosine Kinase AXL:
AXL is a receptor tyrosine kinase of the TAM family that has become an important factor influencing the resistance of NSCLC and other cancers to chemotherapy, radiotherapy, and targeted therapy because of its key role in mediating EMT and immune evasion (153, 154). Studies have indicated that AXL expression is significantly upregulated in EGFR-mutant non-small cell lung cancer cells that have developed resistance and that its degradation rate is inhibited. Therefore, regulating the degradation rate of AXL is expected to be a new strategy to overcome gefitinib resistance (155). Several studies have shown that Axl inhibitors can increase the sensitivity of tumors to chemotherapy and radiotherapy and may also help overcome tumor immune evasion (122, 123, 156, 157). Furthermore, the combination of AXL with ATR inhibitors has been shown to be an effective strategy for treating lung cancer. This approach not only increases the sensitivity of non-small cell lung cancer (NSCLC) cells to ATR inhibitors but can also be used to treat SLFN11-low tumors resistant to platinum-based and PARP inhibitors while inhibiting the progression of small cell lung cancer (SCLC) by modulating tumor-associated macrophages (TAMs) and the epithelial–mesenchymal transition (EMT) (158, 159).
In addition to the aforementioned pathways, current strategies targeting the EMT process for lung cancer treatment involve various molecules and mechanisms that play significant roles in inhibiting epithelial–mesenchymal transition (EMT) in lung cancer, opening new perspectives and providing new ideas for its treatment. Among these factors, TGF-β, a key factor in inducing EMT, is also regarded as an important target for inhibiting the EMT process in lung cancer. For example, research has indicated that bufalin can inhibit the EMT process induced by TGF-β in human lung cancer A549 cells by downregulating the expression of TGF-β receptors (124). Additionally, Zhang et al. (125) reported that methionine enkephalin (MENK) can inhibit the growth, migration, invasion, and EMT of lung cancer cells by interacting with opioid growth factor receptors, thereby combating lung cancer. Research by Kim et al. (160) indicated that apoptotic cancer cells treated with ultraviolet irradiation can secrete PTEN (phosphatase and tensin homolog) and PPARγ (peroxisome proliferator-activated receptor gamma) ligands, which inhibit EMT and metastasis in lung cancer cells.
Tumor-associated macrophages (TAMs) play crucial roles in the process of epithelial–mesenchymal transition (EMT) in lung cancer. TAMs promote EMT in tumor cells by releasing cytokines and growth factors, significantly enhancing the migration and invasion capabilities of tumor cells. Moreover, tumor cells that have undergone EMT alter the microenvironment, impacting the function of TAMs and thereby creating a positive feedback loop. This complex interaction not only deepens our understanding of the biology of lung cancer but also provides new targets for the development of therapeutic strategies.
With further research into the mechanisms of TAMs and EMT, we hope to discover more effective biomarkers and explore treatment strategies based on these mechanisms. Future research should focus on elucidating the specific roles of TAMs and EMT in different tumor microenvironments, as well as how to translate these findings into clinical applications to increase treatment efficacy and improve patient outcomes.
In summary, TAMs play an important role in the EMT process in lung cancer, and a deeper understanding of their interactions will help us better comprehend the complexities of lung cancer and provide more effective treatment options for patients.
LL: Data curation, Visualization, Writing – original draft, Writing – review & editing, Conceptualization, Validation. YW: Funding acquisition, Resources, Writing – review & editing. SF: Data curation, Project administration, Writing – review & editing. XW: Investigation, Resources, Software, Writing – review & editing. XZ: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82260727), the Innovation Team Construction Project of Kunming Medical University (CXTD202203), the Yunnan Provincial Department of Education (2024J0196), and the First-Class Discipline Team of Kunming Medical University (2024XKTDTS13).
We are grateful to the members of our laboratory for critical discussion of this work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
2. Abdelaziz HM, Gaber M, Abd-Elwakil MM, Mabrouk MT, Elgohary MM, Kamel NM, et al. Inhalable particulate drug delivery systems for lung cancer therapy: Nanoparticles, microparticles, nanocomposites and nanoaggregates. J Control Release. (2018) 269:374–92. doi: 10.1016/j.jconrel.2017.11.036
3. Cacho-Díaz B, García-Botello DR, Wegman-Ostrosky T, Reyes-soto G, Ortiz-sánchez E, Herrera-montalvo LA. Tumor microenvironment differences between primary tumor and brain metastases. J Transl Med. (2020) 18:1. doi: 10.1186/s12967-019-02189-8
4. Wang C, Gao Q, Wu J, Lu M, Wang J, Ma T. The biological role of macrophage in lung and its implications in lung cancer immunotherapy. Adv Biol (Weinh). (2024) 8:e2400119. doi: 10.1002/adbi.202400119
5. Guo H, Shu J, Hu G, Liu B, Li J, Sun J, et al. Downregulation of RCN1 inhibits esophageal squamous cell carcinoma progression and M2 macrophage polarization. PloS One. (2024) 19:e0302780. doi: 10.1371/journal.pone.0302780
6. Sedighzadeh SS, Khoshbin AP, Razi S, Keshavarz-fathi M, Rezaei N. A narrative review of tumor-associated macrophages in lung cancer: regulation of macrophage polarization and therapeutic implications. Transl Lung Cancer Res. (2021) 10:1889–916. doi: 10.21037/tlcr-20-1241
7. Manfioletti G, Fedele M. Epithelial-mesenchymal transition (EMT) 2021. Int J Mol Sci. (2022) 23:5848. doi: 10.3390/ijms23105848
8. Pastushenko I, Blanpain C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. (2019) 29:212–26. doi: 10.1016/j.tcb.2018.12.001
9. Lin CY, Lin CJ, Chen KH, Wu JC, Huang SH, Wang SM. Macrophage activation increases the invasive properties of hepatoma cells by destabilization of the adherens junction. FEBS Lett. (2006) 580:3042–50. doi: 10.1016/j.febslet.2006.04.049
10. Vlaicu P, Mertins P, Mayr T, Widschwender P, Ataseven B, Högel B, et al. Monocytes/macrophages support mammary tumor invasivity by co-secreting lineage-specific EGFR ligands and a STAT3 activator. BMC Cancer. (2013) 13:197. doi: 10.1186/1471-2407-13-197
11. Liu CY, Xu JY, Shi XY, Huang W, Ruan TY, Xie P, et al. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab Invest. (2013) 93:844–54. doi: 10.1038/labinvest.2013.69
12. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
13. Lahmar Q, Keirsse J, Laoui D, Movahedi K, Van Overmeire E, Van Ginderachter JA. Tissue-resident versus monocyte-derived macrophages in the tumor microenvironment. Biochim Biophys Acta. (2016) 1865:23–34. doi: 10.1016/j.bbcan.2015.06.009
14. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci. (2021) 22:6995. doi: 10.3390/ijms22136995
15. Gordon S, Plüddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev. (2014) 262:36–55. doi: 10.1111/imr.2014.262.issue-1
16. Murray PJ. Macrophage polarization. Annu Rev Physiol. (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
17. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). (2014) 6:1670–90. doi: 10.3390/cancers6031670
18. Anders CB, Lawton TMW, Smith HL, Garret J, Doucette MM, Ammons MCB. Use of integrated metabolomics, transcriptomics, and signal protein profile to characterize the effector function and associated metabotype of polarized macrophage phenotypes. J Leukoc Biol. (2022) 111:667–93. doi: 10.1002/JLB.6A1120-744R
19. Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-based approaches for cancer immunotherapy. Cancer Res. (2021) 81:1201–8. doi: 10.1158/0008-5472.CAN-20-2990
20. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.v233.9
21. Mantovani A, Sica A, Locati M. New vistas on macrophage differentiation and activation. Eur J Immunol. (2007) 37:14–6. doi: 10.1002/eji.200636910
22. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol. (2019) 106:345–58. doi: 10.1002/JLB.3RU1018-378RR
23. Becker M, Müller CB, De Bastiani MA, Klamt F. The prognostic impact of tumor-associated macrophages and intra-tumoral apoptosis in non-small cell lung cancer. Histol Histopathol. (2014) 29:21–31. doi: 10.14670/HH-29.21
24. Ryder M, Ghossein RA, Ricarte-Filho JC, Knauf JA, Fagin JA. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr Relat Cancer. (2008) 15:1069–74. doi: 10.1677/ERC-08-0036
25. Welsh TJ, Green RH, Richardson D, Waller DA, O'byrne KJ, Bradding P. Macrophage and mast-cell invasion of tumor cell islets confers a marked survival advantage in non-small-cell lung cancer. J Clin Oncol. (2005) 23:8959–67. doi: 10.1200/JCO.2005.01.4910
26. Garrido-Martin EM, Mellows TWP, Clarke J, Ganesan AP, Wood O, Cazaly A, et al. M1(hot) tumor-associated macrophages boost tissue-resident memory T cells infiltration and survival in human lung cancer. J Immunother Cancer. (2020) 8:e000778–e000778. doi: 10.1136/jitc-2020-000778
27. Ohri CM, Shikotra A, Green RH, Waller DA, Bradding P. Macrophages within NSCLC tumour islets are predominantly of a cytotoxic M1 phenotype associated with extended survival. Eur Respir J. (2009) 33:118–26. doi: 10.1183/09031936.00065708
28. Jackute J, Zemaitis M, Pranys D, Sitkauskiene B, Miliauskas S, Vaitkiene S, et al. Distribution of M1 and M2 macrophages in tumor islets and stroma in relation to prognosis of non-small cell lung cancer. BMC Immunol. (2018) 19:3. doi: 10.1186/s12865-018-0241-4
29. Aldawsari HM, Gorain B, Alhakamy NA, Md S. Role of therapeutic agents on repolarisation of tumour-associated macrophage to halt lung cancer progression. J Drug Target. (2020) 28:166–75. doi: 10.1080/1061186X.2019.1648478
30. Ma J, Liu L, Che G, Yu N, Dai F, You Z. The M1 form of tumor-associated macrophages in non-small cell lung cancer is positively associated with survival time. BMC Cancer. (2010) 10:112. doi: 10.1186/1471-2407-10-112
31. Maeda H, Akaike T. Nitric oxide and oxygen radicals in infection, inflammation, and cancer. Biochem Biokhimiia. (1998) 63:854–65.
32. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. (2011) 11:723–37. doi: 10.1038/nri3073
33. Liu S, Zhang S, Dong H, Jin X, Sun J, Zhou H, et al. CD63 + tumor-associated macrophages drive the progression of hepatocellular carcinoma through the induction of epithelial-mesenchymal transition and lipid reprogramming. BMC Cancer. (2024) 24:698. doi: 10.1186/s12885-024-12472-7
34. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. (2010) 141:39–51. doi: 10.1016/j.cell.2010.03.014
35. Allavena P, Mantovani A. Immunology in the clinic review series; focus on cancer: tumour-associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol. (2012) 167:195–205. doi: 10.1111/j.1365-2249.2011.04515.x
36. Zhang J, Li H, Wu Q, Chen Y, Deng Y, Yang Z, et al. Tumoral NOX4 recruits M2 tumor-associated macrophages via ROS/PI3K signaling-dependent various cytokine production to promote NSCLC growth. Redox Biol. (2019) 22:101116. doi: 10.1016/j.redox.2019.101116
37. Li C, Xue VW, Wang QM, Lian GY, Huang XR, Lee TL, et al. The mincle/syk/NF-κB signaling circuit is essential for maintaining the protumoral activities of tumor-associated macrophages. Cancer Immunol Res. (2020) 8:1004–17. doi: 10.1158/2326-6066.CIR-19-0782
38. Erratum to tumor-associated macrophages secret exosomal miR-155 and miR-196a-5p to promote metastasis of non-small-cell lung cancer. Transl Lung Cancer Res. (2021) 10:4047–8. doi: 10.21037/tlcr-2021-4
39. Zhao J, Zhu Q, Zhang Y, Li G, Zhang Y, Li F, et al. Role of COX-2/PGE2/EP4 axis-induced macrophage functional activation in NSCLC development. Chin J Lung Cancer. (2024) 27:245–56. doi: 10.3779/j.issn.1009-3419.2024.101.05
40. Tang X, Zhu M, Zhu Z, Tang W, Zhang H, Chen Y, et al. Ginsenoside Re inhibits non-small cell lung cancer progression by suppressing macrophage M2 polarization induced by AMPKα1/STING positive feedback loop. Phytother Res. (2024) 38(11):5088–106. doi: 10.1002/ptr.v38.11
41. Guo Y, Lu X, Chen Y, Rendon B, Mitchell RA, Cuatrecasas M, et al. Zeb1 induces immune checkpoints to form an immunosuppressive envelope around invading cancer cells. Sci Adv. (2021) 7:7455–7455. doi: 10.1126/sciadv.abd7455
42. Wang Z, Zhao Y, Xu H, Liang F, Zou Q, Wang C, et al. CtBP1 promotes tumour-associated macrophage infiltration and progression in non-small-cell lung cancer. J Cell Mol Med. (2020) 24:11445–56. doi: 10.1111/jcmm.15751
43. Fridlender ZG, Kapoor V, Buchlis G, Cheng G, Sun J, Wang LC, et al. Monocyte chemoattractant protein-1 blockade inhibits lung cancer tumor growth by altering macrophage phenotype and activating CD8+ cells. Am J Respir Cell Mol Biol. (2011) 44:230–7. doi: 10.1165/rcmb.2010-0080OC
44. Lu X, Shen J, Huang S, Liu D, Wang H. Tumor cells-derived exosomal PD-L1 promotes the growth and invasion of lung cancer cells in vitro via mediating macrophages M2 polarization. Eur J Histochem. (2023) 67:3784–3784. doi: 10.4081/ejh.2023.3784
45. Chen T, Liu Y, Li C, Xu C, Ding C, Chen J, et al. Tumor-derived exosomal circFARSA mediates M2 macrophage polarization via the PTEN/PI3K/AKT pathway to promote non-small cell lung cancer metastasis. Cancer Treat Res Commun. (2021) 28:100412. doi: 10.1016/j.ctarc.2021.100412
46. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. (2019) 20:69–84. doi: 10.1038/s41580-018-0080-4
47. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. (2009) 119:1420–8. doi: 10.1172/JCI39104
48. Menju T, Date H. Lung cancer and epithelial-mesenchymal transition. Gen Thorac Cardiovasc Surg. (2021) 69:781–9. doi: 10.1007/s11748-021-01595-4
49. Prudkin L, Liu DD, Ozburn NC, Sun M, Behrens C, Tang X, et al. Epithelial-to-mesenchymal transition in the development and progression of adenocarcinoma and squamous cell carcinoma of the lung. Mod Pathol. (2009) 22:668–78. doi: 10.1038/modpathol.2009.19
50. Sulzer MA, Leers MP, Van Noord JA, Bollen EC, Theunissen PH. Reduced E-cadherin expression is associated with increased lymph node metastasis and unfavorable prognosis in non-small cell lung cancer. Am J Respir Crit Care Med. (1998) 157:1319–23. doi: 10.1164/ajrccm.157.4.9703099
51. Soltermann A, Tischler V, Arbogast S, Braun J, Probst-Hensch N, Weder W, et al. Prognostic significance of epithelial-mesenchymal and mesenchymal-epithelial transition protein expression in non-small cell lung cancer. Clin Cancer Res. (2008) 14:7430–7. doi: 10.1158/1078-0432.CCR-08-0935
52. Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. (1998) 392:190–3. doi: 10.1038/32433
53. Ceteci F, Ceteci S, Karreman C, Kramer BW, Asan E, Götz R, et al. Disruption of tumor cell adhesion promotes angiogenic switch and progression to micrometastasis in RAF-driven murine lung cancer. Cancer Cell. (2007) 12:145–59. doi: 10.1016/j.ccr.2007.06.014
54. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
55. Tirino V, Camerlingo R, Bifulco K, Irollo E, Montella R, Paino F, et al. TGF-β1 exposure induces epithelial to mesenchymal transition both in CSCs and non-CSCs of the A549 cell line, leading to an increase of migration ability in the CD133+ A549 cell fraction. Cell Death Dis. (2013) 4:e620. doi: 10.1038/cddis.2013.144
56. Pallier K, Cessot A, Côté JF, Just PA, Cazes A, Fabre E, et al. TWIST1 a new determinant of epithelial to mesenchymal transition in EGFR mutated lung adenocarcinoma. PloS One. (2012) 7:e29954. doi: 10.1371/journal.pone.0029954
57. Pirozzi G, Tirino V, Camerlingo R, Franco R, La Rocca A, Liguori E, et al. Epithelial to mesenchymal transition by TGFβ-1 induction increases stemness characteristics in primary non small cell lung cancer cell line. PloS One. (2011) 6:e21548. doi: 10.1371/journal.pone.0021548
58. Blaukovitsch M, Halbwedl I, Kothmaier H, Gogg-Kammerer M, Popper HH. Sarcomatoid carcinomas of the lung–are these histogenetically heterogeneous tumors? Virchows Arch. (2006) 449:455–61. doi: 10.1007/s00428-006-0256-8
59. Feng J, Zhang X, Zhu H, Wang X, Ni S, Huang J. FoxQ1 overexpression influences poor prognosis in non-small cell lung cancer, associates with the phenomenon of EMT. PloS One. (2012) 7:e39937. doi: 10.1371/journal.pone.0039937
60. Lu X, Gao J, Zhang Y, Zhao T, Cai H, Zhang T. CTEN induces epithelial-mesenchymal transition (EMT) and metastasis in non small cell lung cancer cells. PloS One. (2018) 13:e0198823. doi: 10.1371/journal.pone.0198823
61. Amin A, Mokhdomi TA, Bukhari S, Wani Z, Chikan NA, Shah BA, et al. Lung cancer cell-derived EDA-containing fibronectin induces an inflammatory response from monocytes and promotes metastatic tumor microenvironment. J Cell Biochem. (2021) 122:562–76. doi: 10.1002/jcb.v122.5
62. Chen MC, Chen KC, Chang GC, Lin H, Wu CC, Kao WH, et al. RAGE acts as an oncogenic role and promotes the metastasis of human lung cancer. Cell Death Dis. (2020) 11:1–13. doi: 10.1038/s41419-020-2432-1
63. Li HR, Gao J, Jin C, Jiang JH, Ding JY. Downregulation of SETBP1 promoted non-small cell lung cancer progression by inducing cellular EMT and disordered immune status. Am J Transl Res. (2020) 12:447–62.
64. Bonde AK, Tischler V, Kumar S, Soltermann A, Schwendener RA. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer. (2012) 12:35. doi: 10.1186/1471-2407-12-35
65. Hu Y, He MY, Zhu LF, Yang CC, Zhou ML, Wang Q, et al. Tumor-associated macrophages correlate with the clinicopathological features and poor outcomes via inducing epithelial to mesenchymal transition in oral squamous cell carcinoma. J Exp Clin Cancer Res. (2016) 35:12. doi: 10.1186/s13046-015-0281-z
66. Gao L, Zhang W, Zhong WQ, Liu ZJ, Li HM, Yu ZL, et al. Tumor associated macrophages induce epithelial to mesenchymal transition via the EGFR/ERK1/2 pathway in head and neck squamous cell carcinoma. Oncol Rep. (2018) 40:2558–72. doi: 10.3892/or.2018.6657
67. Naito M, Satoh Y, Ishii G. Microenvironmental changes in the progression from adenocarcinoma in situ to early-invasive adenocarcinoma of the lung. Cancer Res. (2017) 77:Abstract 5921. doi: 10.1158/1538-7445.AM2017-5921
68. Yao X, Huang J, Zhong H, Shen N, Faggioni R, Fung M, et al. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol Ther. (2014) 141:125–39. doi: 10.1016/j.pharmthera.2013.09.004
69. Dehai C, Bo P, Qiang T, Lihua S, Fang L, Shi J, et al. Enhanced invasion of lung adenocarcinoma cells after co-culture with THP-1-derived macrophages via the induction of EMT by IL-6. Immunol Lett. (2014) 160:1–10. doi: 10.1016/j.imlet.2014.03.004
70. Che D, Zhang S, Jing Z, Shang L, Jin S, Liu F, et al. Macrophages induce EMT to promote invasion of lung cancer cells through the IL-6-mediated COX-2/PGE(2)/β-catenin signalling pathway. Mol Immunol. (2017) 90:197–210. doi: 10.1016/j.molimm.2017.06.018
71. Hu Z, Sui Q, Jin X, Shan G, Huang Y, Yi Y, et al. IL6-STAT3-C/EBPβ-IL6 positive feedback loop in tumor-associated macrophages promotes the EMT and metastasis of lung adenocarcinoma. J Exp Clin Cancer Res. (2024) 43:63. doi: 10.1186/s13046-024-02989-x
72. Yan HQ, Huang XB, Ke SZ, Jiang YN, Zhang YH, Wang YN, et al. Interleukin 6 augments lung cancer chemotherapeutic resistance via ataxia-telangiectasia mutated/NF-kappaB pathway activation. Cancer Sci. (2014) 105:1220–7. doi: 10.1111/cas.2014.105.issue-9
73. Chang CH, Hsiao CF, Yeh YM, Chang GC, Tsai YH, Chen YM, et al. Circulating interleukin-6 level is a prognostic marker for survival in advanced nonsmall cell lung cancer patients treated with chemotherapy. Int J Cancer. (2013) 132:1977–85. doi: 10.1002/ijc.v132.9
74. Hao Y, Baker D, Ten Dijke P. TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. Int J Mol Sci. (2019) 20:2767–2767. doi: 10.3390/ijms20112767
75. Batlle E, Massagué J. Transforming growth factor-β Signaling in immunity and cancer. Immunity. (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
76. Liu ZW, Zhang YM, Zhang LY, Zhou T, Li YY, Zhou GC, et al. Duality of interactions between TGF-β and TNF-α During tumor formation. Front Immunol. (2021) 12:810286. doi: 10.3389/fimmu.2021.810286
77. Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. (2001) 12:27–36. doi: 10.1091/mbc.12.1.27
78. Gal A, Sjöblom T, Fedorova L, Imreh S, Beug H, Moustakas A. Sustained TGF beta exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene. (2008) 27:1218–30. doi: 10.1038/sj.onc.1210741
79. Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. (2000) 275:36803–10. doi: 10.1074/jbc.M005912200
80. Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol. (2009) 11:943–50. doi: 10.1038/ncb1905
81. Nawshad A, Lagamba D, Polad A, Hay ED. Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: implications for embryogenesis and tumor metastasis. Cells Tissues Organs. (2005) 179:11–23. doi: 10.1159/000084505
82. Zhang S, Che D, Yang F, Chi C, Meng H, Shen J, et al. Tumor-associated macrophages promote tumor metastasis via the TGF-β/SOX9 axis in non-small cell lung cancer. Oncotarget. (2017) 8:99801–15. doi: 10.18632/oncotarget.21068
83. Sumitomo R, Menju T, Shimazu Y, Toyazaki T, Chiba N, Miyamoto H, et al. M2-like tumor-associated macrophages promote epithelial-mesenchymal transition through the transforming growth factor β/Smad/zinc finger e-box binding homeobox pathway with increased metastatic potential and tumor cell proliferation in lung squamous cell carcinoma. Cancer Sci. (2023) 114:4521–34. doi: 10.1111/cas.v114.12
84. Huang JA, Cheng C, Zhang XH. The role of B7-H4-expressing macrophage in Malignant pleural effusion. J Thorac Oncol. (2015) 10:S603.
85. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. (2011) 475:222–5. doi: 10.1038/nature10138
86. Ding M, He SJ, Yang J. MCP-1/CCL2 mediated by autocrine loop of PDGF-BB promotes invasion of lung cancer cell by recruitment of macrophages via CCL2-CCR2 axis. J Interferon Cytokine Res. (2019) 39:224–32. doi: 10.1089/jir.2018.0113
87. Xu H, Wang J, Al-Nusaif M, Ma H, Le W. CCL2 promotes metastasis and epithelial-mesenchymal transition of non-small cell lung cancer via PI3K/Akt/mTOR and autophagy pathways. Cell Prolif. (2024) 57:e13560. doi: 10.1111/cpr.13560
88. Chen W, Gao Q, Han S, Pan F, Fan W. The CCL2/CCR2 axis enhances IL-6-induced epithelial-mesenchymal transition by cooperatively activating STAT3-Twist signaling. Tumour Biol. (2015) 36:973–81. doi: 10.1007/s13277-014-2717-z
89. Cao Y, Wu Y, Tu H, Gu Z, Yu F, Huang W, et al. (-)-Guaiol inhibit epithelial-mesenchymal transition in lung cancer via suppressing M2 macrophages mediated STAT3 signaling pathway. Heliyon. (2023) 9:e19817. doi: 10.1016/j.heliyon.2023.e19817
90. Guo Z, Song J, Hao J, Zhao H, Du X, Li E, et al. M2 macrophages promote NSCLC metastasis by upregulating CRYAB. Cell Death Dis. (2019) 10:377. doi: 10.1038/s41419-019-1618-x
91. Xia L, Huang W, Tian D, Zhang L, Qi X, Chen Z, et al. Forkhead box Q1 promotes hepatocellular carcinoma metastasis by transactivating ZEB2 and VersicanV1 expression. Hepatology. (2014) 59:958–73. doi: 10.1002/hep.26735
92. Liu S, Yan B, Tao Y. Radioresistance is linked with stem-like properties via activation of aryl hydrocarbon receptor. Cancer Res. (2017) 77:4757. doi: 10.1158/1538-7445.AM2017-4757
93. Wang J, Wang X, Wang Y, Li S, Wang X. Krüppel like factor 6 splice variant 1 (KLF6-SV1) overexpression recruits macrophages to participate in lung cancer metastasis by up-regulating TWIST1. Cancer Biol Ther. (2019) 20:680–91. doi: 10.1080/15384047.2018.1550570
94. Nobre AR, Entenberg D, Wang Y, Condeelis J, Aguirre-Ghiso JA. The different routes to metastasis via hypoxia-regulated programs. Trends Cell Biol. (2018) 28:941–56. doi: 10.1016/j.tcb.2018.06.008
95. Joseph JP, Harishankar MK, Pillai AA, Devi A. Hypoxia induced EMT: A review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. (2018) 80:23–32. doi: 10.1016/j.oraloncology.2018.03.004
96. Henze AT, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest. (2016) 126:3672–9. doi: 10.1172/JCI84427
97. Chen XJ, Deng YR, Wang ZC, Wei WF, Zhou CF, Zhang YM, et al. Hypoxia-induced ZEB1 promotes cervical cancer progression via CCL8-dependent tumour-associated macrophage recruitment. Cell Death Dis. (2019) 10:508. doi: 10.1038/s41419-019-1748-1
98. Cruceriu D, Baldasici O, Balacescu O, Berindan-Neagoe I. The dual role of tumor necrosis factor-alpha (TNF-α) in breast cancer: molecular insights and therapeutic approaches. Cell Oncol (Dordr). (2020) 43:1–18. doi: 10.1007/s13402-019-00489-1
99. Jinquan T, Frydenberg J, Mukaida N, Bonde J, Larsen CG, Matsushima K, et al. Recombinant human growth-regulated oncogene-alpha induces T lymphocyte chemotaxis. A process regulated via IL-8 receptors by IFN-gamma, TNF-alpha, IL-4, IL-10, and IL-13. J Immunol. (1995) 155:5359–68. doi: 10.4049/jimmunol.155.11.5359
100. Lin Y, Huang R, Chen L, Li S, Shi Q, Jordan C, et al. Identification of interleukin-8 as estrogen receptor-regulated factor involved in breast cancer invasion and angiogenesis by protein arrays. Int J Cancer. (2004) 109:507–15. doi: 10.1002/ijc.v109:4
101. Xiong C, Zhu Y, Xue M, Jiang Y, Zhong Y, Jiang L, et al. Tumor-associated macrophages promote pancreatic ductal adenocarcinoma progression by inducing epithelial-to-mesenchymal transition. Aging (Albany NY). (2021) 13:3386–404. doi: 10.18632/aging.202264
102. Zhang J, Zhang Q, Lou Y, Fu Q, Chen Q, Wei T, et al. Hypoxia-inducible factor-1α/interleukin-1β signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology. (2018) 67:1872–89. doi: 10.1002/hep.29681
103. Zheng J, Dou R, Zhang X, Zhong B, Fang C, Xu Q, et al. LINC00543 promotes colorectal cancer metastasis by driving EMT and inducing the M2 polarization of tumor associated macrophages. J Transl Med. (2023) 21:153. doi: 10.1186/s12967-023-04009-6
104. Wei C, Yang C, Wang S, Shi D, Zhang C, Lin X, et al. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol Cancer. (2019) 18:64. doi: 10.1186/s12943-019-0976-4
105. Su S, Liu Q, Chen J, Chen J, Chen F, He C, et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell. (2014) 25:605–20. doi: 10.1016/j.ccr.2014.03.021
106. Yao Z, Zhang J, Zhang B, Liang G, Chen X, Yao F, et al. Imatinib prevents lung cancer metastasis by inhibiting M2-like polarization of macrophages. Pharmacol Res. (2018) 133:121–31. doi: 10.1016/j.phrs.2018.05.002
107. Sun L, Chen B, Jiang R, Li J, Wang B. Resveratrol inhibits lung cancer growth by suppressing M2-like polarization of tumor associated macrophages. Cell Immunol. (2017) 311:86–93. doi: 10.1016/j.cellimm.2016.11.002
108. Wu Q, Chen GL, Li YJ, Chen Y, Lin FZ. Paeoniflorin inhibits macrophage-mediated lung cancer metastasis. Chin J Nat Med. (2015) 13:925–32. doi: 10.1016/S1875-5364(15)30098-4
109. Xu F, Cui WQ, Wei Y, Cui J, Qiu J, Hu LL, et al. Astragaloside IV inhibits lung cancer progression and metastasis by modulating macrophage polarization through AMPK signaling. J Exp Clin Cancer Res. (2018) 37:207. doi: 10.1186/s13046-018-0878-0
110. Kang H, Zhang J, Wang B, Liu M, Zhao J, Yang M, et al. Puerarin inhibits M2 polarization and metastasis of tumor-associated macrophages from NSCLC xenograft model via inactivating MEK/ERK 1/2 pathway. Int J Oncol. (2017) 50:545–54. doi: 10.3892/ijo.2017.3841
111. Tariq M, Zhang JQ, Liang GK, He QJ, Ding L, Yang B. Gefitinib inhibits M2-like polarization of tumor-associated macrophages in Lewis lung cancer by targeting the STAT6 signaling pathway. Acta Pharmacol Sin. (2017) 38:1501–11. doi: 10.1038/aps.2017.124
112. Tariq M, Hussain N, Rehman K, Akash MSH, Al Haddad AHI, Said ASA, et al. Macrophages M2 polarization is involved in lapatinib-mediated chemopreventive effects in the lung cancer. BioMed Pharmacother. (2023) 161:114527. doi: 10.1016/j.biopha.2023.114527
113. Zuo B, Li T, Liu X, Wang S, Cheng J, Liu X, et al. Dipeptidyl peptidase 4 inhibitor reduces tumor-associated macrophages and enhances anti-PD-L1-mediated tumor suppression in non-small cell lung cancer. Clin Transl Oncol. (2023) 25:3188–202. doi: 10.1007/s12094-023-03187-5
114. Jin QG, Zhou M, Hu YL, Chen L, Liu Y, Wang YQ, et al. Inhibitory effect and mechanism of oyster enzymatic hydrolysate on lung metastasis in the subcutaneous lewis lung cancer model in mice. Kafkas Universitesi Veteriner Fakultesi Dergisi. (2021) 27:73–82. doi: 10.9775/kvfd.2020.24776
115. Ravi J, Elbaz M, Wani NA, Nasser MW, Ganju RK. Cannabinoid receptor-2 agonist inhibits macrophage induced EMT in non-small cell lung cancer by downregulation of EGFR pathway. Mol Carcinog. (2016) 55:2063–76. doi: 10.1002/mc.v55.12
116. Wu Z, Zhou J, Chen F, Yu J, Li H, Li Q, et al. 13-Methyl-palmatrubine shows an anti-tumor role in non-small cell lung cancer via shifting M2 to M1 polarization of tumor macrophages. Int Immunopharmacol. (2022) 104:108468. doi: 10.1016/j.intimp.2021.108468
117. Yu X, Xu M, Li N, Li Z, Li H, Shao S, et al. β-elemene inhibits tumor-promoting effect of M2 macrophages in lung cancer. Biochem Biophys Res Commun. (2017) 490:514–20. doi: 10.1016/j.bbrc.2017.06.071
118. Li Y, Cao F, Li M, Li P, Yu Y, Xiang L, et al. Hydroxychloroquine induced lung cancer suppression by enhancing chemo-sensitization and promoting the transition of M2-TAMs to M1-like macrophages. J Exp Clin Cancer Res. (2018) 37:259. doi: 10.1186/s13046-018-0938-5
119. Zeng AQ, Chen X, Dai Y, Zhao JN. Betulinic acid inhibits non-small cell lung cancer by repolarizing tumor-associated macrophages via mTOR signaling pathway. Zhongguo Zhong Yao Za Zhi. (2024) 49:2376–84. doi: 10.19540/j.cnki.cjcmm.20240122.401
120. Tang B, Wang Y, Xu W, Zhu J, Weng Q, Chen W, et al. Macrophage xCT deficiency drives immune activation and boosts responses to immune checkpoint blockade in lung cancer. Cancer Lett. (2023) 554:216021. doi: 10.1016/j.canlet.2022.216021
121. Yochum ZA, Cades J, Wang H, Chatterjee S, Simons BW, O'brien JP, et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene. (2019) 38:656–70. doi: 10.1038/s41388-018-0482-y
122. Taverna JA, Hung CN, Lin CL, Osmulski PA, Gaczynska ME, Wang CM, et al. AXL Inhibitor TP-0903 attenuates TGFβ-Hippo signaling in lung adenocarcinoma cells. Mol Cancer Res. (2020) 18:55–55. doi: 10.1158/1557-3125.HIPPO19-B29
123. Wnuk-Lipinska K, Tiron C, Gausdal G, Sandal T, Frink R, Hinz S, et al. BGB324, a selective small molecule Axl kinase inhibitor to overcome EMT-associated drug resistance in carcinomas: Therapeutic rationale and early clinical studies. Cancer Res. (2014) 74:1747. doi: 10.1158/1538-7445.AM2014-1747
124. Zhao L, Liu S, Che X, Hou K, Ma Y, Li C, et al. Bufalin inhibits TGF-β-induced epithelial-to-mesenchymal transition and migration in human lung cancer A549 cells by downregulating TGF-β receptors. Int J Mol Med. (2015) 36:645–52. doi: 10.3892/ijmm.2015.2268
125. Zhang S, Geng J, Shan F, Shan Y, Griffin N, Wu B, et al. Methionine enkephalin suppresses lung cancer metastasis by regulating the polarization of tumor-associated macrophages and the distribution of myeloid-derived suppressor cells in the tumor microenvironment and inhibiting epithelial-mesenchymal transition. Int Immunopharmacol. (2023) 118:110064. doi: 10.1016/j.intimp.2023.110064
126. Conway EM, Pikor LA, Kung SH, Hamilton MJ, Lam S, Lam WL, et al. Macrophages, inflammation, and lung cancer. Am J Respir Crit Care Med. (2016) 193:116–30. doi: 10.1164/rccm.201508-1545CI
127. Ruytinx P, Proost P, Van Damme J, Struyf S. Chemokine-induced macrophage polarization in inflammatory conditions. Front Immunol. (2018) 9. doi: 10.3389/fimmu.2018.01930
128. Kawaguchi Y, Ohshio Y, Watanabe A, Shiratori T, Okamoto K, Ueda K, et al. Depletion of tumor-associated macrophages inhibits lung cancer growth and enhances the antitumor effect of cisplatin. Cancer Sci. (2023) 114:750–63. doi: 10.1111/cas.v114.3
129. Liu H, He R, Yang X, Huang B, Liu H. Mechanism of TCF21 downregulation leading to immunosuppression of tumor-associated macrophages in non-small cell lung cancer. Pharmaceutics. (2023) 15:2295–2295. doi: 10.3390/pharmaceutics15092295
130. Sarode P, Zheng X, Giotopoulou GA, Weigert A, Kuenne C, Günther S, et al. Reprogramming of tumor-associated macrophages by targeting β-catenin/FOSL2/ARID5A signaling: A potential treatment of lung cancer. Sci Adv. (2020) 6:eaaz6105. doi: 10.1126/sciadv.aaz6105
131. Lin TT, Xiong W, Chen GH, He Y, Long L, Gao XF, et al. Epigenetic-based combination therapy and liposomal codelivery overcomes osimertinib-resistant NSCLC via repolarizing tumor-associated macrophages. Acta Pharmacol Sin. (2024) 45:867–78. doi: 10.1038/s41401-023-01205-4
132. Shu H, Lv W, Ren ZJ, Li H, Dong T, Zhang Y, et al. Ultrasound-mediated PLGA-PEI nanobubbles carrying STAT6 siRNA enhances NSCLC treatment via repolarizing tumor-associated macrophages from M2 to M1 phenotypes. Curr Drug Delivery. (2024) 21:1114–27. doi: 10.2174/1567201820666230724151545
133. Parayath NN, Parikh A, Amiji MM. Repolarization of tumor-associated macrophages in a genetically engineered nonsmall cell lung cancer model by intraperitoneal administration of hyaluronic acid-based nanoparticles encapsulating microRNA-125b. Nano Lett. (2018) 18:3571–9. doi: 10.1021/acs.nanolett.8b00689
134. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, Mccracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. (2017) 545:495–9. doi: 10.1038/nature22396
135. La Fleur L, Boura VF, Alexeyenko A, Berglund A, Pontén V, Mattsson JSM, et al. Expression of scavenger receptor MARCO defines a targetable tumor-associated macrophage subset in non-small cell lung cancer. Int J Cancer. (2018) 143:1741–52. doi: 10.1002/ijc.v143.7
136. Antonangeli F, Natalini A, Garassino MC, Sica A, Santoni A, Di Rosa F. Regulation of PD-L1 expression by NF-κB in cancer. Front Immunol. (2020) 11:584626. doi: 10.3389/fimmu.2020.584626
137. Karger A, Mansouri S, Leisegang MS, Weigert A, Günther S, Kuenne C, et al. ADPGK-AS1 long noncoding RNA switches macrophage metabolic and phenotypic state to promote lung cancer growth. EMBO J. (2023) 42:e111620. doi: 10.15252/embj.2022111620
138. Wang S, Hu Q, Chang Z, Liu Y, Gao Y, Luo X, et al. Moringa oleifera leaf polysaccharides exert anti-lung cancer effects upon targeting TLR4 to reverse the tumor-associated macrophage phenotype and promote T-cell infiltration. Food Funct. (2023) 14:4607–20. doi: 10.1039/D2FO03685A
139. Chen S, Wang M, Lu T, Liu Y, Hong W, He X, et al. JMJD6 in tumor-associated macrophage regulates macrophage polarization and cancer progression via STAT3/IL-10 axis. Oncogene. (2023) 42:2737–50. doi: 10.1038/s41388-023-02781-9
140. Hao J, Zhao X, Wang C, Cao X, Liu Y. Recent advances in nanoimmunotherapy by modulating tumor-associated macrophages for cancer therapy. Bioconjug Chem. (2024) 35:867–82. doi: 10.1021/acs.bioconjchem.4c00242
141. Klichinsky M, Gabrusiewicz K, Anderson N, Schmierer M, Best A, Zeeman M, et al. CT-0508 is an anti-HER2 chimeric antigen receptor (CAR) macrophage with targeted anti-tumor activity that promotes a pro-inflammatory solid tumor microenvironment. Cancer Res. (2020) 80:3242. doi: 10.1158/1538-7445.AM2020-3242
142. Annunziata CM, Ghobadi A, Pennella EJ, Vanas J, Powell C, Pavelova M, et al. Feasibility and preliminary safety and efficacy of first-in-human intraperitoneal delivery of MCYM11, anti-human-mesothelin CAR mRNA transfected into peripheral blood mononuclear cells, for ovarian cancer and Malignant peritoneal mesothelioma. J Clin Oncol. (2020) 38:Abstract 3014. doi: 10.1200/JCO.2020.38.15_suppl.3014
143. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. (2018) 378:113–25. doi: 10.1056/NEJMoa1713137
144. Ricordel C, Llamas-Gutierrez F, Chiforeanu D, Lena H, Corre R. Large cell neuroendocrine lung carcinoma transformation as an acquired resistance mechanism to osimertinib. J Thorac Oncol. (2017) 12:e184–e6. doi: 10.1016/j.jtho.2017.07.019
145. Ou SI, Agarwal N, Ali SM. High MET amplification level as a resistance mechanism to osimertinib (AZD9291) in a patient that symptomatically responded to crizotinib treatment post-osimertinib progression. Lung Cancer. (2016) 98:59–61. doi: 10.1016/j.lungcan.2016.05.015
146. Planchard D, Loriot Y, André F, Gobert A, Auger N, Lacroix L, et al. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann Oncol. (2015) 26:2073–8. doi: 10.1093/annonc/mdv319
147. Yoshida T, Song L, Bai Y, Kinose F, Li J, Ohaegbulam KC, et al. ZEB1 mediates acquired resistance to the epidermal growth factor receptor-tyrosine kinase inhibitors in non-small cell lung cancer. PloS One. (2016) 11:e0147344. doi: 10.1371/journal.pone.0147344
148. Zhang T, Guo L, Creighton CJ, Lu Q, Gibbons DL, Yi ES, et al. A genetic cell context-dependent role for ZEB1 in lung cancer. Nat Commun. (2016) 7:12231. doi: 10.1038/ncomms12231
149. Chang TH, Tsai MF, Su KY, Wu SG, Huang CP, Yu SL, et al. Slug confers resistance to the epidermal growth factor receptor tyrosine kinase inhibitor. Am J Respir Crit Care Med. (2011) 183:1071–9. doi: 10.1164/rccm.201009-1440OC
150. Cheng HY, Hsieh CH, Lin PH, Chen YT, Hsu DSS, Tai SK, et al. Snail-regulated exosomal microRNA-21 suppresses NLRP3 inflammasome activity to enhance cisplatin resistance. J ImmunoTherapy Cancer. (2022) 10:e004832. doi: 10.1136/jitc-2022-004832
151. Zhu D, Gu L, Li Z, Jin W, Lu Q, Ren T. MiR-138-5p suppresses lung adenocarcinoma cell epithelial-mesenchymal transition, proliferation and metastasis by targeting ZEB2. Pathol Res Pract. (2019) 215:861–72. doi: 10.1016/j.prp.2019.01.029
152. Jiang M, Fang S, Zhao X, Zhou C, Gong Z. Epithelial-mesenchymal transition-related circular RNAs in lung carcinoma. Cancer Biol Med. (2021) 18:411–20. doi: 10.20892/j.issn.2095-3941.2020.0238
153. Ramkumar K, Della Corte CM, Diao L, Cardnell RJ, Papadimitrakopoulou VA, Heymach JV, et al. Clinical, genomic, and immune landscape of the receptor tyrosine kinase AXL in non-small cell lung cancer. Mol Cancer Ther. (2019) 18:Abstract B024. doi: 10.1158/1535-7163.TARG-19-B024
154. Zhang YP, Huang HY, Yang YJ, Xu DD, Zhang KR, Zhu L. Role of receptor tyrosine kinase AXL in tumor drug resistance. J Shanghai Jiaotong Univ (Medical Science). (2018) 38:819–24. doi: 10.3969/j.issn.1674-8115.2018.07.020
155. Kim D, Bach DH, Hong JY, Park HJ, Lee SK. Overexpression and extended degradation rate of Axl in acquired gefitinib-resistant non-small cell lung cancer cells. Cancer Res. (2018) 78:Abstract 894. doi: 10.1158/1538-7445.AM2018-894
156. Wium M, Ajayi-Smith AF, Paccez JD, Zerbini LF. The role of the receptor tyrosine kinase axl in carcinogenesis and development of therapeutic resistance: An overview of molecular mechanisms and future applications. Cancers. (2021) 13:1521–1521. doi: 10.3390/cancers13071521
157. Soh KK, Kim W, Lee YS, Peterson P, Siddiqui-Jain A, Warner SL, et al. AXL inhibition leads to a reversal of a mesenchymal phenotype sensitizing cancer cells to targeted agents and immuno-oncology therapies. Cancer Res. (2016) 76:Abstract 235. doi: 10.1158/1538-7445.AM2016-235
158. Ramkumar K, Tong P, Fan Y, Peng D, Heymach JV, Gibbons DL, et al. Targeting AXL sensitizes nonsmall cell lung cancer to ATR inhibitors by enhancing replication stress. Cancer Res. (2019) 79:Abstract 276. doi: 10.1158/1538-7445.AM2019-276
159. Ramkumar K, Stewart CA, Tanimoto A, Wang Q, Xi Y, Morris BB, et al. Combined inhibition of AXL and ATR enhances replication stress, cell death and immune response in small cell lung cancer. Cancer Res. (2023) 83:Abstract 6206. doi: 10.1158/1538-7445.AM2023-6206
Keywords: tumor-associated macrophages, epithelial-mesenchymal transformation, lung cancer, tumor microenvironment, tumor therapy
Citation: Liao L, Wang Y-X, Fan S-S, Hu Y-Y, Wang X-C and Zhang X (2025) The role and clinical significance of tumor-associated macrophages in the epithelial–mesenchymal transition of lung cancer. Front. Oncol. 15:1571583. doi: 10.3389/fonc.2025.1571583
Received: 05 February 2025; Accepted: 25 March 2025;
Published: 15 April 2025.
Edited by:
Rui Vitorino, University of Aveiro, PortugalReviewed by:
Noha M. Mesbah, Suez Canal University, EgyptCopyright © 2025 Liao, Wang, Fan, Hu, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xue-Chang Wang, eW53YW5neGNAMTYzLmNvbQ==; Xuan Zhang, c25vb3B5a21AMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.