
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 06 March 2025
Sec. Pediatric Oncology
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1555419
This article is part of the Research Topic The Immunosuppressive Microenvironment in Pediatric Cancers: Applications and Considerations in Immunotherapy View all 5 articles
 Chong Chen1*
Chong Chen1* Zixuan Wei2,3,4,5*
Zixuan Wei2,3,4,5*Relapsed/refractory neuroblastoma is a type of malignant solid tumor with a very poor prognosis in children. Its pathogenesis is complex, involving multiple molecular pathways and genetic alterations. Recent studies have shown that MYCN amplification, ALK mutation, TERT promoter mutation, p53 pathway inactivation, and chromosomal instability are the key mechanisms and molecular characteristics of relapsed/refractory neuroblastoma. Precision treatment strategies targeting these molecular mechanisms have shown certain prospects in preclinical studies and clinical practice. This review focuses on the relevant mechanisms and molecular characteristics of relapsed/refractory neuroblastoma, explores its relationship with treatment response and clinical prognosis, and briefly introduces the current treatment strategies to provide a theoretical basis for the development of novel and personalized therapeutic regimens to improve the prognosis of children.
Primary neuroblastoma originates from neural crest progenitor cells, cells of the sympathetic nervous system in the adrenal medulla or sympathetic chain (1–3). Neuroblastomas are remarkably heterogeneous, with unique and variable biological and clinical features, including limited disease that can spontaneously regress and widespread disseminated disease (4, 5). Based on this heterogeneity, neuroblastomas are stratified into low to intermediate risk tumors that can be surgically resected and high-risk tumors that are fatal. About half of diagnosed neuroblastomas are classified as high-risk cases (6).
Although the efficacy of multimodal therapies has been gradually improving in recent years, the 5-year survival rate for patients with high-risk neuroblastoma has only increased to approximately 50% (7). Within this group, there is also a subset of patients with a particularly poor prognosis who do not respond to initial treatment or who do not achieve complete remission, accounting for about 10-20% of children with high-risk neuroblastoma, called refractory neuroblastoma. In addition, about 40-50% of children with neuroblastoma whose disease goes into remission after treatment will eventually have the cancer relapse during or after treatment, called relapsed neuroblastoma (8–10). These tumors are usually aggressive and resistant to conventional therapies such as chemotherapy or radiation. The 5-year overall survival rate for patients with relapsed/refractory neuroblastoma is less than 20% (11). Clinically, the treatment options for these two types of tumors are usually the same, so they are generally referred to collectively as relapsed/refractory neuroblastomas.
The development of neuroblastoma reflects an abnormal developmental process of adrenal sympathetic nerve cells. Somatic variants are uncommon in neuroblastoma, whereas copy number variants across segmental chromosomal regions or even entire chromosomes are common (12). Molecular analytical studies have revealed key factors in the biology and treatment resistance of aggressive tumors, while providing important guidance for the development of new therapies. In this review, we discussed the pathogenesis of relapsed/refractory neuroblastoma and summarized some of the molecular characteristics that may be used to predict prognosis or as therapeutic targets (Table 1). In addition, we have summarized the current status of the application of drugs targeting these therapeutic targets (Table 2). In order to understand the molecular mechanisms of relapsed/refractory neuroblastoma more intuitively, we have drawn corresponding conceptual diagrams for better reading (Figures 1, 2).
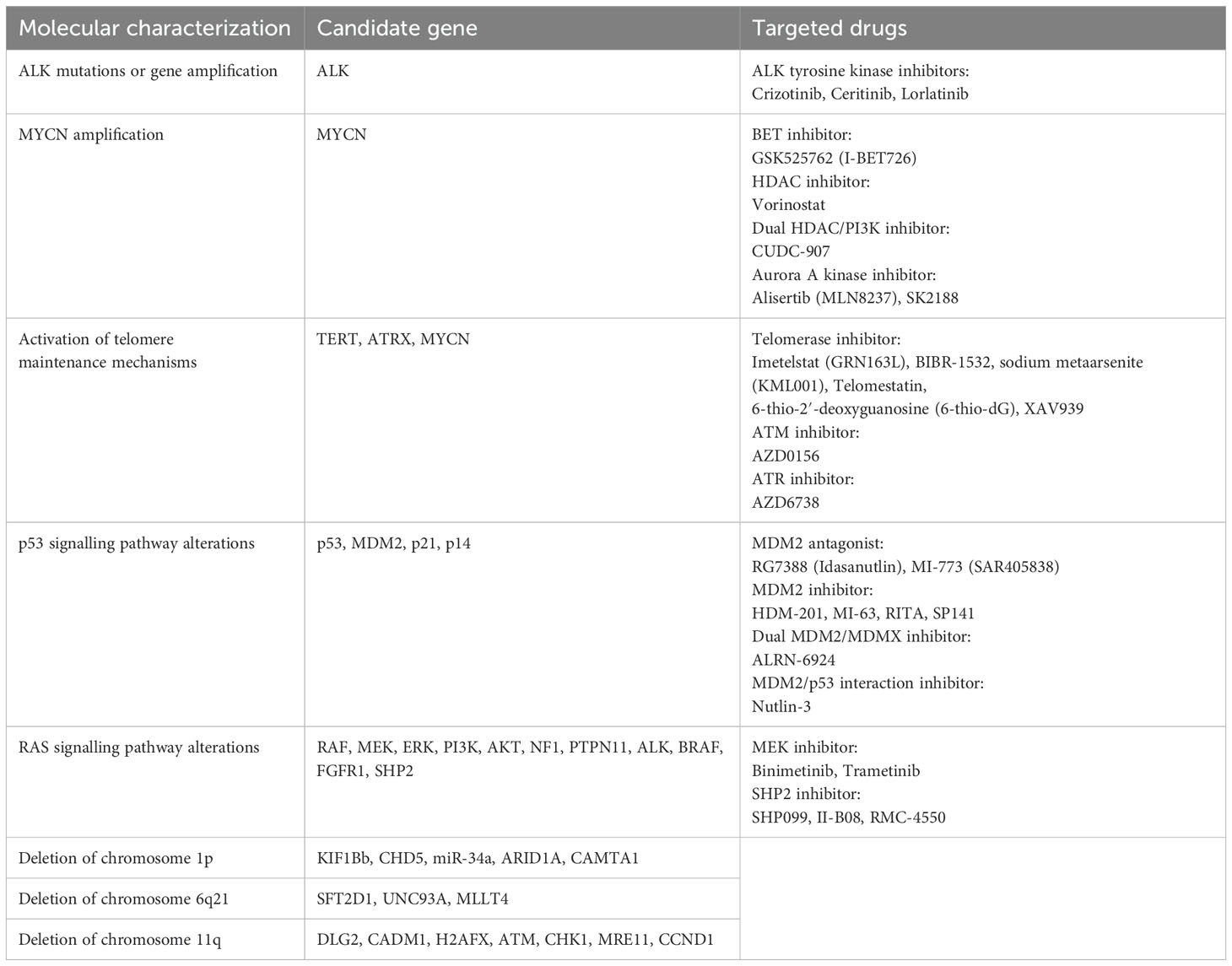
Table 1. Molecular characterization and targeted drugs in relapsed/refractory neuroblastoma.
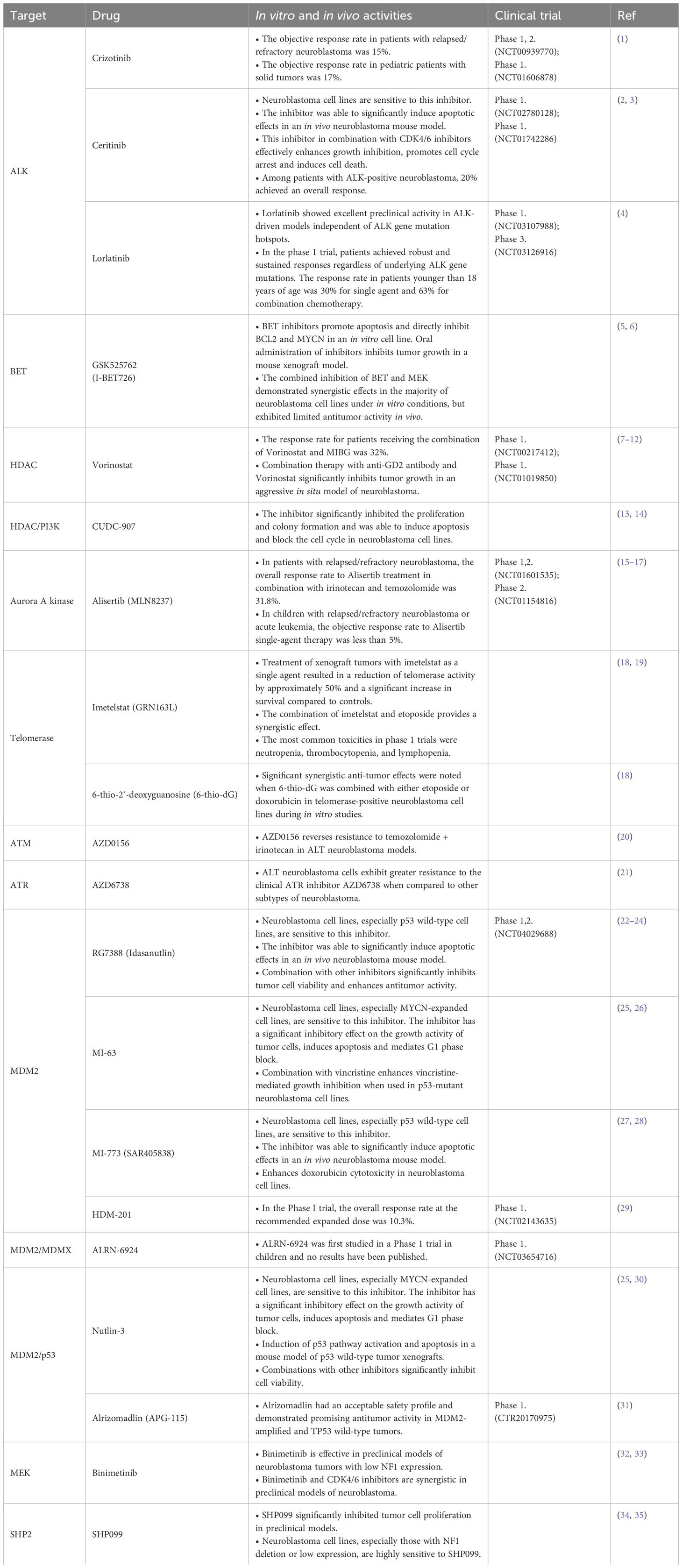
Table 2. The application of targeted agents for relapsed/refractory neuroblastoma.
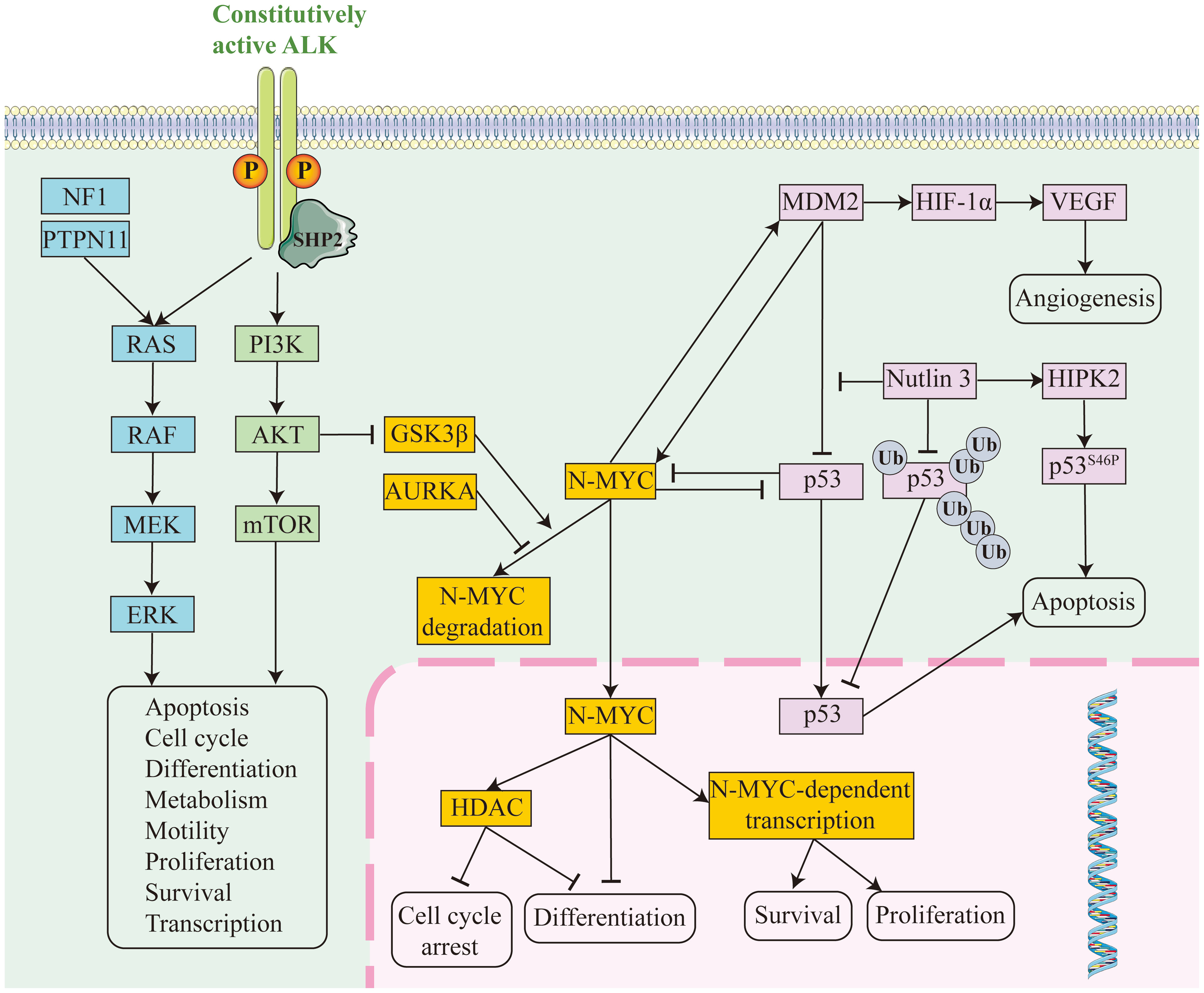
Figure 1. Conceptual diagram of molecular pathway mechanisms in relapsed/refractory neuroblastoma. RAS pathway in blue, PI3K pathway in green, MYCN pathway in orange and p53 pathway in pink.
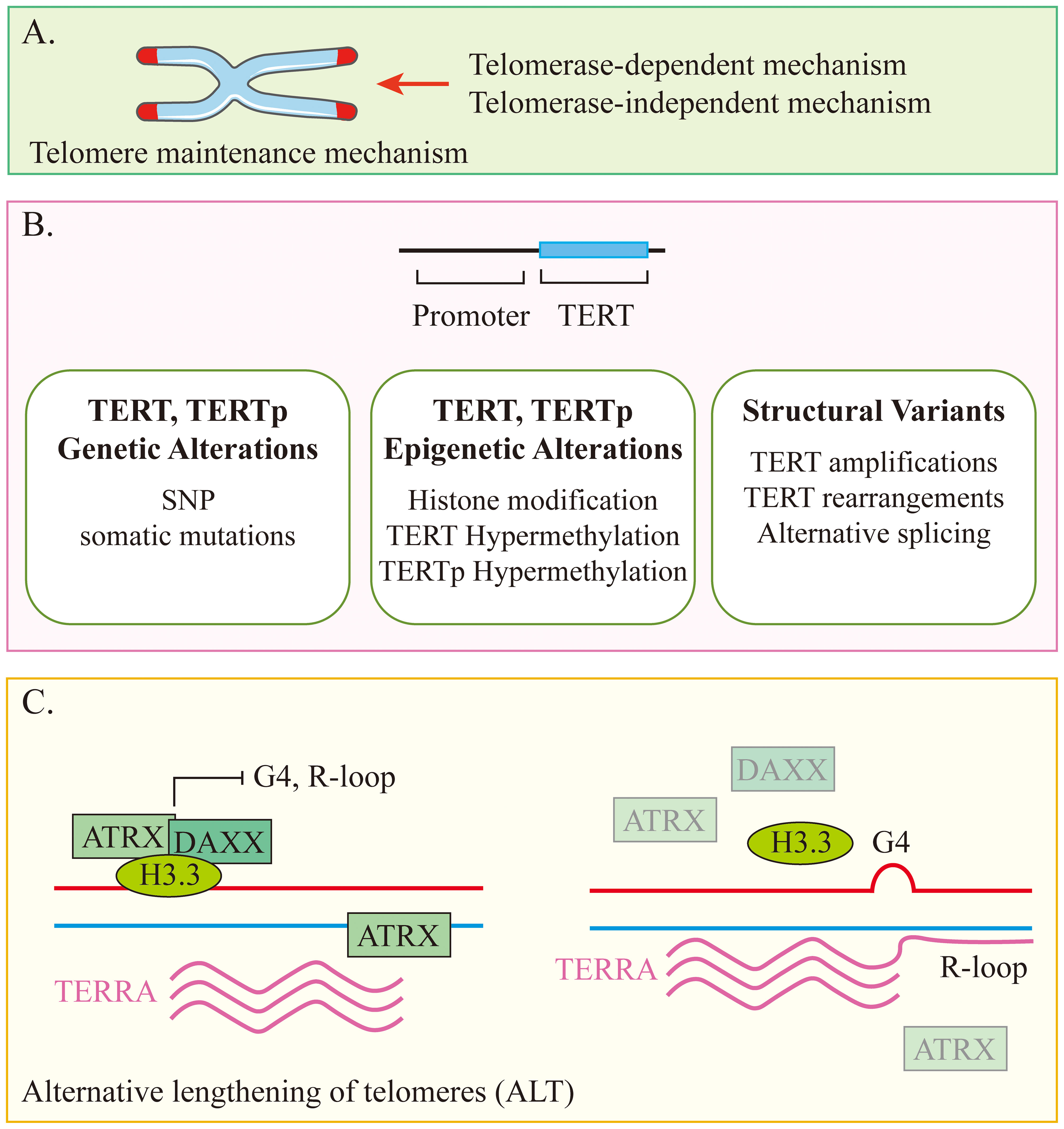
Figure 2. Conceptual diagram of the telomere maintenance mechanism. (A) Telomere maintenance mechanisms can be divided into two types based on whether or not they rely on telomerase. (B) Telomerase-dependent mechanism. (C) Telomerase-independent mechanism.
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase (RTK) whose activation generates mitogenic signaling through the RAS-MAPK and PI3K-AKT pathways (13). Point mutations, copy number amplifications, or chromosomal translocations within the structural domain of ALK tyrosine kinase can lead to oncogenic activation. ALK mutations or gene amplification are present in up to 15% of sporadic high-risk neuroblastomas, the most common somatic single-nucleotide variant in neuroblastoma and the most frequently mutated oncogene (Figure 1). The data suggest that high-risk neuroblastoma patients with ALK variants are a particularly high-risk group (14). In the SIOPEN high-risk group, ALK variants were categorized as clonal or subclonal. The difference in OS between cases with ALK amplification or clonal ALK variants and cases with subclonal ALK variants or no ALK changes was statistically significant. In multivariate models, ALK amplification and clonal ALK variants were independent predictors of poor prognosis (15). In the COG high-risk neuroblastoma population, a poorer prognosis has also been observed in cases with ALK variants or ALK amplification (14). In addition, ALK mutations can contribute to disease recurrence. Increased somatic mutations and increased ALK-activating subclonal/clonal mutations have been reported in relapsed neuroblastomas compared to tumors at diagnosis; the frequency of these mutations is higher than 20% and rises as tumors and/or plasma from relapsed patients are sequenced more routinely (10, 16, 17).
ALK tyrosine kinase structural domain variants occur mainly at three hotspot positions (positions F1174, R1275, and F1245), with 10% to 15% of the variants occurring at other kinase structural domain positions (18). The data suggest that MYCN gene amplification is often accompanied by ALK mutation activation, and the two synergistically initiate and promote the development of neuroblastoma (19, 20). At the molecular level, ALK induces MYCN transcription and stabilizes MYCN protein; conversely, MYCN increases ALK transcription (21, 22). Interestingly, a higher frequency of the F1174L mutation was observed in MYCN-amplified tumors (23). ATP-competitive ALK/Met/ROS1 tyrosine kinase inhibitors (TKIs), such as crizotinib, exhibit differential activity in preclinical models of ALK-driven neuroblastoma (24). This class of drugs primarily induces remission in patients with the ALK R1275 mutation. Primary resistance to inhibitors such as crizotinib is commonly associated with ALK hotspot mutations (e.g., F1174L and F1245C) in neuroblastomas, hence the term refractory ALK variants for such variants (14, 24). Targeting drug-resistant ALK mutations, the third-generation ALK tyrosine kinase inhibitor loratinib exerted unprecedented preclinical activity as a single agent in a xenograft model of neuroblastoma patients with three hotspot mutations (25). The first phase I study of the New Approaches to Neuroblastoma Therapy Consortium (NANT2015-02) in children (NCT03107988) explored the use of loratinib in patients with ALK-driven refractory or relapsed neuroblastoma, demonstrating the drug’s safety profile and significant clinical activity. Esther R’s team showed that the acquisition of ALK compound mutations and mutations in members of the RAS-MAPK pathway is the mechanism by which ALK-driven neuroblastoma patients develop resistance to loratinib (26). Among them, mutations in the RAS-MAPK pathway are the most common mechanism of off-target resistance to loratinib (27). Loratinib is currently enrolled in the Children’s Oncology Group (COG) (NCT03126916) and the International Society of Pediatric Oncology Europe Neuroblastoma Group (SIOPEN) phase 3 trials to further understand clinical response and drug resistance in patients (28).
MYCN, located on chromosome 2p24, is a major transcriptional regulator of cell growth, metabolism and differentiation. Approximately 50% of high-risk patients present with MYCN amplification, and these tumors tend to be the most aggressive and difficult to treat (29). High level of MYCN amplification is a driver of high-risk neuroblastoma. This is associated with reduced tumor immunogenicity and can promote tumor metastasis and recurrence (12). In addition, MYCN amplification inhibits interferon activity and chemokine expression, and its overexpression enhances tumor cell resistance to immune-mediated cytotoxicity through a variety of mechanisms, including MHC-I downregulation and inhibition of NK cell activation (30). In addition to functioning as a transcription factor, MYCN promotes disease progression and metastasis through epigenetic regulation (Figure 1) (31). In almost all multivariate regression analyses of prognostic factors, regardless of disease stage, MYCN gene amplification strongly predicted a poorer prognosis, including treatment response rate, time to tumor progression, and overall survival. Of the 4832 newly diagnosed patients enrolled in the ANBL00B1 (NCT00904241) study, patients harboring MYCN-unamplified tumors (n = 3647; 81%) had 5-year event-free survival (EFS) and overall survival (OS) rates of 77% and 87%, respectively. Patients with MYCN amplification (n = 827; 19%) had 5-year EFS and OS rates of 51% and 57%, respectively (32). In the cohort of 6,223 patients with known MYCN status in the INRG database, the hazard ratio for OS associated with MYCN amplification was 6.3. The greatest adverse prognostic impact of MYCN amplification on OS was seen in younger patients (age <18 months) (33). On this point, it has been shown that the presence of MYCN amplification appears to have a greater adverse prognostic impact in patients with other favorable characteristics (e.g., younger age and lower stage), whereas the prognostic impact of MYCN amplification is less severe in older patients with higher disease stage. However, MYCN amplification still has a negative prognostic impact even in high-risk patients, who tend to respond poorly to conventional chemotherapy and require treatment with high-dose chemotherapy and autologous stem cell transplantation (33, 34).
Therapeutic strategies targeting MYCN through its downstream targets include bromodomain and extra-terminal domain (BET) inhibition, dual HDAC/PI3K inhibition, MDM2 inhibition and aurora A kinase inhibition. Various BET inhibitors are being used in clinical trials, such as a phase I study of GSK525762 (I-BET726) in neuroblastoma. In addition, the HDAC inhibitor vorinostat has been studied in several clinical trials in patients with NB (NCT00217412, NCT01132911, NCT02035137, NCT02559778, NCT01019850 and NCT01208454) (35–40).
Telomeres are regions of repetitive nucleotide sequences (TTAGGG) located at the ends of chromosomes. In normal dividing cells, telomeres gradually shorten with each cell replication until they reach a critical level, eventually causing the cell to be unable to replicate, and cellular senescence occurs (41). Conversely, lengthening telomeres promotes cell survival, which is known as telomere maintenance mechanism (TMM). Activation of TMM to prevent telomere shortening is necessary for the continued proliferation of cancer cells, and patients whose tumors possess TMM have a poor prognosis.
Telomerase is able to maintain telomere length by adding telomeric DNA repeats. It is a reverse transcriptase enzyme that consists of the catalytic protein subunit TERT and human telomerase RNA (hTR). Most cancers overexpress telomerase, which is usually associated with overexpression of TERT (42, 43). Alternative lengthening of telomeres (ALT) is the maintenance of telomeres in the absence of telomerase activity. In neuroblastoma, there is a strong association between ALT and loss of function (LoF) gene variants in ATRX (Figure 2) (44). Neuroblastoma cells maintain telomere length through one of several mutually exclusive mechanisms, and these tumors typically have poor response rates and poor clinical outcomes (45, 46). MYCN amplification is present in nearly 40% of high-risk neuroblastomas and is associated with upregulated expression of TERT and telomere dysfunction (47, 48). In another 23%-31% of high-risk neuroblastomas, TERT is activated through proximal chromosomal rearrangements, which induces its transcriptional upregulation (47, 49). ALT is also active in about 24% of high-risk neuroblastomas, about half of which are associated with somatic alterations in ATRX (47, 49, 50). A higher proportion of ALT-positive neuroblastoma cases were found in the relapsed patient cohort compared to the newly diagnosed cohort (10% and 48%, respectively). Patients with ALT-positive tumors also have as poor event-free survival as patients with MYCN amplification, while their long-term survival rates are very low and usually unsalvageable after progression or relapse (45, 51). In conclusion, a growing body of data supports TMM as one of the mechanisms and molecular features of most aggressive high-risk neuroblastomas that relapse or are refractory to treatment.
Drugs that target telomerase activity include imestat (GRN163L), BIBR-1532, and sodium metaarsenite (KML001), but all of these drugs suffer from excessive toxic effects. Telomestatin, 6-thio-2′-deoxyguanosine (6-thio-dG), and XAV939 have shown the ability to induce apoptosis in neuroblastoma cells in cell lines and preclinical models, but further clinical development is still needed (52). Drugs targeting ALT include ataxia-telangiectasia mutated (ATM) inhibitor combinations such as the ATM inhibitor AZD0156. Using cell lines and in vitro models derived from patients with relapsed neuroblastoma, the study by Balakrishna Koneru et al. found that constitutive ATM activation in ALT-positive cells contributes to the chemoresistant phenotype of neuroblastoma. In contrast, AZD0156 reversed resistance to temozolomide and irinotecan in ALT-positive neuroblastoma cell lines and xenograft models, providing a rationale for early clinical trials (53). Other ALT-related drugs are ataxia telangiectasia and Rad3-related (ATR) inhibitors such as AZD6738 (54). Although activation of the TMM has been shown to be a key factor in the poor prognosis of neuroblastoma, MYCN amplification and ATRX mutations, the primary drivers of the TMM, are also associated with multiple modes of transcriptional activation that drive malignant transformation. In addition, neuroblastoma patients with mutations in both TMM and the RAS/TP53 pathway have a particularly poor prognosis (55). As a result, the likelihood that aggressive relapsed/refractory neuroblastomas will be resistant to single agents targeting TMM is high, and therapeutic strategies targeting TMM will only show significant clinical efficacy in combination with agents targeting these key genes and pathways. For example, in vitro experiments targeting neuroblastoma by Janina Fischer-Mertens et al. demonstrated that 6-thio-dG and the competitive telomerase inhibitor, imestat, were more efficacious than monotherapy when combined with other agents such as etoposide (56).
The p53 is a key regulator of cell cycle checkpoints and apoptosis. Activated after cellular stress, it binds DNA in a sequence-specific manner and activates the transcription of a large number of downstream genes (including MDM2, p21, etc.), leading to apoptosis, cell cycle arrest, differentiation and DNA repair (Figure 1) (57). Mutational inactivation of the p53 gene occurs in more than half of human malignancies. Amplification of MDM2 also occurs in neuroblastoma, and even in the absence of gene amplification, MDM2 protein overexpression is often present and correlates with a poorer prognosis for patients. MDM2, located upstream of p53, is a negative regulator of p53 and acts as a ubiquitin ligase, targeting p53 for proteasome-mediated degradation, forming an autoregulatory feedback loop that tightly regulates the cellular level of p53. MDM2 also inhibits the activity of p53 by increasing the degradation of p53, contributing to tumor formation (58). MDM2 also functions independently of p53 to promote the growth, progression and development of neuroblastoma (59). For example, elevated MDM2 expression promotes multidrug resistance in neuroblastoma, leading to relapsed/refractory neuroblastoma (60). Mutations in the p53 signaling pathway occur in less than 2% of patients with a primary diagnosis of neuroblastoma (61). In contrast, in relapsed neuroblastoma after chemotherapy, the frequency of mutations in the p53/MDM2/p14ARF signaling pathway increases to almost half and leads to chemotherapy resistance (62, 63). The association of p53 mutations in neuroblastoma cells with drug resistance has been demonstrated, and these cell lines are more chemoresistant than p53 wild-type neuroblastoma cell lines (61).
Since most neuroblastomas harbor functional wild-type p53, it may be wise to target MDM2 to enhance the functional activity of p53. The MDM2 antagonist idasanutrin has shown strong antitumor effects in preclinical models (64). In addition, another MDM2 antagonist in clinical trials, MI-773 (SAR405838), has been shown to potentiate the cytotoxicity of doxorubicin in neuroblastoma cell lines (65). There are also antagonists targeting the interaction of MDM2 and p53, such as Nutlin-3, which activate the p53 pathway in chemoresistant neuroblastoma with wild-type p53, inhibit primary tumor growth, and reduce tumor metastasis in mice carrying chemoresistant neuroblastoma xenografts (66). An increasing number of targeted agents have entered early pediatric trials, such as HDM-201 (NCT02780128), an MDM2 inhibitor applied to neuroblastoma, and ALRN-6924 (NCT03654716), a dual MDM2/MDMX inhibitor applied to solid tumors in children.
Members of the RAS protein family are GDP-GTP-regulated switches that regulate the cytoplasmic-nuclear signaling network that controls normal cellular processes. They send signals through a series of molecular pathways (such as RAF/MEK/ERK and PI3K/AKT, etc.) to regulate cell survival, proliferation, and differentiation (Figure 1). With constitutively activating mutations in the RAS family of genes in up to 30% of cancers, dysregulation of RAS-dependent signaling is essential for tumorigenesis (67). Mutations in the RAS pathway occur frequently in neuroblastoma, not only in the RAS gene itself, but also through mutations in regulatory proteins and downstream signaling components (e.g., NF1 and PTPN11) or by triggering constitutive activation of the receptor kinases of the pathway (e.g., ALK) (68). RAS pathway mutations in neuroblastoma, especially in high-risk patients, are strongly associated with poor prognosis. Patients harboring RAS pathway mutations have a worse prognosis than those harboring ALK mutations. Patients with both RAS pathway mutations and telomere maintenance mechanisms have an extremely poor prognosis (55). It was found that the frequency of mutations in the RAS-MAPK signaling pathway was significantly increased in relapsed neuroblastoma tumors, including mutations in ALK, NF1, BRAF, PTPN11, FGFR1, and the three RAS genes, and that the mutations in this pathway were mutually exclusive (69).
Targeting the RAS signaling pathway has been a challenge, but effective therapies applied to inhibit RAS-driven neuroblastoma have not been developed for more than three decades (70). Therefore, attempts to target the RAS pathway have focused on inhibiting its upstream or downstream effectors.MEK is an effector molecule of RAF, and in vitro experiments have found that neuroblastoma cell lines with mutations in the RAS family are sensitive to MEK inhibitors, but the presence of MYCN amplification leads to resistance to this class of drugs. The tyrosine phosphatase SHP2 encoded by PTPN11 is an activator of RAS. SHP2 inhibitors alone are susceptible to resistance in neuroblastoma, however, dual inhibition of SHP2 and the RAS effectors RAF, MEK, or ERK demonstrated synergistic effects (71). Therefore, combinations of drugs targeting this pathway may be an effective strategy for the treatment of relapsed/refractory neuroblastoma. A single-agent clinical trial of trametinib as a treatment for patients with RAS-mutated relapsed neuroblastoma is currently underway (NCT02780128).
Chromosomal alterations are very common in neuroblastoma, occurring in about 90% of patients, and there is a clear correlation between the type of alteration and prognosis (72). In particular, large segmental chromosome imbalances and localized aberrations are common in high-risk tumors, and any type of segmental chromosome aberrations is associated with a poorer prognosis, perhaps because loss of segmental chromosomes leads to inactivation of tumor suppressor genes. Relapsed neuroblastoma often occurs with deletions of chromosome 1p or 6q21 (73). Tumor suppressor genes located at 1p36 may include KIF1Bb, CHD5, miR-34a, ARID1A and CAMTA1 (74, 75). Among them, it has been shown that deletion of ARID1A promotes cell invasion and migration and causes neuroblastoma cells to exhibit enriched mesenchymal-type gene features (73). Pauline Depuydt et al. showed that neuroblastomas in the presence of a deletion in the distal region of chromosome 6q are highly aggressive and highly susceptible to developing into relapsed/refractory tumors (76). They also identified a number of candidate genes located in this region that were strongly associated with prognosis, such as SFT2D1, UNC93A, and MLLT4. In children younger than 18 months, only segmental chromosome aberrations led to relapse and death, and 11q deletion was the strongest prognostic marker (77). In addition, patients with heterozygous deletions of 11q are less likely to respond to induction therapy, thus promoting the development of refractory neuroblastoma (78). Many studies have been conducted on genes within the deletion region of chromosome 11q, including DLG2, CADM1, H2AFX, ATM, CHK1, MRE11, and CCND1, which are tumor suppressor genes. Many of these belong to cell growth control regulatory genes or DNA repair genes, which favor tumor development in the presence of haploinsufficiency (79–81).
The state of neuroblastoma cells significantly influences treatment response and prognosis. Based on RNA sequencing and epigenomic analyses, a method has been proposed to describe cell states, classifying neuroblastoma cells into a differentiated adrenergic cell population and a less differentiated mesenchymal cell population (82–86). These two cancer cell states have different gene expression profiles and are dynamic and programmable to transform into each other (73, 87, 88). For example, deletion of chromosome 1p, leading to loss of the tumor suppressor gene ARID1A, promotes the development of the mesenchymal state (73). Adrenergic cells are generally more sensitive to initial treatments, whereas mesenchymal cells exhibit stronger resistance to cytotoxic chemotherapy (82). Enrichment of mesenchymal gene expression signatures has been observed in relapsed samples. These data suggest that cellular states influence treatment sensitivity (82, 86, 87, 89, 90). Under therapeutic pressures such as chemotherapy or radiation, adrenergic cells may transition to the mesenchymal state, enabling tumor cells to acquire enhanced survival capabilities and drug resistance (91). This phenotypic plasticity plays a critical role in the development of relapsed/refractory neuroblastoma.
Relapsed/refractory neuroblastoma occurs by complex mechanisms involving multiple genetic, epigenetic, and tumor microenvironmental changes. Currently, despite our initial understanding of their molecular characterization, there is a lack of effective therapeutic tools to deal with their relapse and drug resistance. These tumors often exhibit genetic mutations, chromosomal instability, and aberrant activation of key pathways, leading to poor therapeutic response and poor clinical prognosis. In-depth study of these molecular mechanisms can help reveal new therapeutic targets and provide patients with more precise and personalized treatment options. Future research should focus on the discovery of new biomarkers, improvement of existing therapeutic strategies, and exploration of the clinical application of innovative therapeutic tools such as immunotherapy and targeted therapy, in the hope of bringing new breakthroughs in the treatment of relapsed/refractory neuroblastoma.
Our understanding of relapsed/refractory neuroblastoma will continue to deepen as molecular biology and genomics technologies continue to evolve. Future studies will rely more on advanced methods such as high-throughput sequencing technology, single-cell genomics, and tumor microenvironmental analysis to fully resolve the interactions between tumor cells and the surrounding environment. In addition, the development of gene editing technologies and immunotherapy offers new possibilities for neuroblastoma treatment, especially in targeting tumor-associated gene mutations, immune escape mechanisms, and cellular drug resistance interventions. Through the combined application of these innovative strategies, we expect to achieve more effective personalized treatment in the future and improve the survival and quality of life of patients with relapsed/refractory neuroblastoma.
CC: Conceptualization, Project administration, Writing – review & editing. ZW: Writing – original draft.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was sponsored by the Tianjin health research project (No. TJWJ2021MS010 to C Chen).
We acknowledge Tianjin Union Medical Center, The First Affiliated Hospital of Nankai University and Tianjin Medical University Cancer Institute and Hospital for support of excellent conditions. They provide us with a good platform for this review. This work was supported by the Tianjin health research project, under Grant [number TJWJ2021MS010]. All authors have read the journal’s authorship agreement and policy on disclosure of potential conflicts of interest.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Dong R, Yang R, Zhan Y, Lai HD, Ye CJ, Yao XY, et al. Single-cell characterization of Malignant phenotypes and developmental trajectories of adrenal neuroblastoma. Cancer Cell. (2020) 38:716–733.e6. doi: 10.1016/j.ccell.2020.08.014
2. Ponzoni M, Bachetti T, Corrias MV, Brignole C, Pastorino F, Calarco E, et al. Recent advances in the developmental origin of neuroblastoma: an overview. J Exp Clin Cancer Res. (2022) 41:92. doi: 10.1186/s13046-022-02281-w
3. Jansky S, Sharma AK, Körber V, Quintero A, Toprak UH, Wecht EM, et al. Single-cell transcriptomic analyses provide insights into the developmental origins of neuroblastoma. Nat Genet. (2021) 53:683–93. doi: 10.1038/s41588-021-00806-1
4. Lundberg KI, Treis D, Johnsen JI. Neuroblastoma heterogeneity, plasticity, and emerging therapies. Curr Oncol Rep. (2022) 24:1053–62. doi: 10.1007/s11912-022-01270-8
5. Gomez RL, Ibragimova S, Ramachandran R, Philpott A, Ali FR. Tumoral heterogeneity in neuroblastoma. Biochim Biophys Acta Rev Cancer. (2022) 1877:188805. doi: 10.1016/j.bbcan.2022.188805
6. Segura MF, Soriano A, Roma J, Piskareva O, Jiménez C, Boloix A, et al. Methodological advances in the discovery of novel neuroblastoma therapeutics. Expert Opin Drug Discovery. (2022) 17:167–79. doi: 10.1080/17460441.2022.2002297
7. Pinto NR, Applebaum MA, Volchenboum SL, Matthay KK, London WB, Ambros PF, et al. Advances in risk classification and treatment strategies for neuroblastoma. J Clin Oncol. (2015) 33:3008–17. doi: 10.1200/JCO.2014.59.4648
8. Park JR, Villablanca JG, Hero B, Kushner BH, Wheatley K, Beiske KH, et al. Early-phase clinical trial eligibility and response evaluation criteria for refractory, relapsed, or progressive neuroblastoma: A consensus statement from the National Cancer Institute Clinical Trials Planning Meeting. Cancer. (2022) 128:3775–83. doi: 10.1002/cncr.v128.21
9. Grossmann LD, Chen CH, Uzun Y, Thadi A, Wolpaw AJ, Louault K, et al. Identification and characterization of chemotherapy-resistant high-risk neuroblastoma persister cells. Cancer Discovery. (2024) 14:2387–406. doi: 10.1158/2159-8290.CD-24-0046
10. Rosswog C, Fassunke J, Ernst A, Schömig-Markiefka B, Merkelbach-Bruse S, Bartenhagen C, et al. Genomic ALK alterations in primary and relapsed neuroblastoma. Br J Cancer. (2023) 128:1559–71. doi: 10.1038/s41416-023-02208-y
11. Zhou MJ, Doral MY, DuBois SG, Villablanca JG, Yanik GA, Matthay KK. Different outcomes for relapsed versus refractory neuroblastoma after therapy with (131)I-metaiodobenzylguanidine ((131)I-MIBG). Eur J Cancer. (2015) 51:2465–72. doi: 10.1016/j.ejca.2015.07.023
12. Qiu B, Matthay KK. Advancing therapy for neuroblastoma. Nat Rev Clin Oncol. (2022) 19:515–33. doi: 10.1038/s41571-022-00643-z
13. Guan J, Borenäs M, Xiong J, Lai WY, Palmer RH, Hallberg B. IGF1R contributes to cell proliferation in ALK-mutated neuroblastoma with preference for activating the PI3K-AKT signaling pathway. Cancers (Basel). (2023) 15:4252. doi: 10.3390/cancers15174252
14. Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell. (2014) 26:682–94. doi: 10.1016/j.ccell.2014.09.019
15. Bellini A, Pötschger U, Bernard V, Lapouble E, Baulande S, Ambros PF, et al. Frequency and prognostic impact of ALK amplifications and mutations in the european neuroblastoma study group (SIOPEN) high-risk neuroblastoma trial (HR-NBL1). J Clin Oncol. (2021) 39:3377–90. doi: 10.1200/JCO.21.00086
16. Schleiermacher G, Javanmardi N, Bernard V, Leroy Q, Cappo J, Rio Frio T, et al. Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol. (2014) 32:2727–34. doi: 10.1200/JCO.2013.54.0674
17. Padovan-Merhar OM, Raman P, Ostrovnaya I, Kalletla K, Rubnitz KR, Sanford EM, et al. Enrichment of targetable mutations in the relapsed neuroblastoma genome. PloS Genet. (2016) 12:e1006501. doi: 10.1371/journal.pgen.1006501
18. Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. (2008) 455:930–5. doi: 10.1038/nature07261
19. Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell. (2012) 22:117–30. doi: 10.1016/j.ccr.2012.06.001
20. Heukamp LC, Thor T, Schramm A, De Preter K, Kumps C, De Wilde B, et al. Targeted expression of mutated ALK induces neuroblastoma in transgenic mice. Sci Transl Med. (2012) 4:141ra91. doi: 10.1126/scitranslmed.3003967
21. Schönherr C, Ruuth K, Kamaraj S, Wang CL, Yang HL, Combaret V, et al. Anaplastic Lymphoma Kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene. (2012) 31:5193–200. doi: 10.1038/onc.2012.12
22. Hasan MK, Nafady A, Takatori A, Kishida S, Ohira M, Suenaga Y, et al. ALK is a MYCN target gene and regulates cell migration and invasion in neuroblastoma. Sci Rep. (2013) 3:3450. doi: 10.1038/srep03450
23. De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res. (2010) 16:4353–62. doi: 10.1158/1078-0432.CCR-09-2660
24. Foster JH, Voss SD, Hall DC, Minard CG, Balis FM, Wilner K, et al. Activity of crizotinib in patients with ALK-aberrant relapsed/refractory neuroblastoma: A children’s oncology group study (ADVL0912). Clin Cancer Res. (2021) 27:3543–8. doi: 10.1158/1078-0432.CCR-20-4224
25. Infarinato NR, Park JH, Krytska K, Ryles HT, Sano R, Szigety KM, et al. The ALK/ROS1 inhibitor PF-06463922 overcomes primary resistance to crizotinib in ALK-driven neuroblastoma. Cancer Discovery. (2016) 6:96–107. doi: 10.1158/2159-8290.CD-15-1056
26. Berko ER, Witek GM, Matkar S, Petrova ZO, Wu MA, Smith CM, et al. Circulating tumor DNA reveals mechanisms of lorlatinib resistance in patients with relapsed/refractory ALK-driven neuroblastoma. Nat Commun. (2023) 14:2601. doi: 10.1038/s41467-023-38195-0
27. Berlak M, Tucker E, Dorel M, Winkler A, McGearey A, Rodriguez-Fos E, et al. Mutations in ALK signaling pathways conferring resistance to ALK inhibitor treatment lead to collateral vulnerabilities in neuroblastoma cells. Mol Cancer. (2022) 21:126. doi: 10.1186/s12943-022-01583-z
28. Goldsmith KC, Park JR, Kayser K, Malvar J, Chi YY, Groshen SG, et al. Lorlatinib with or without chemotherapy in ALK-driven refractory/relapsed neuroblastoma: phase 1 trial results. Nat Med. (2023) 29:1092–102. doi: 10.1038/s41591-023-02297-5
29. Mercatelli D, Balboni N, Palma A, Aleo E, Sanna PP, Perini G, et al. Single-cell gene network analysis and transcriptional landscape of MYCN-amplified neuroblastoma cell lines. Biomolecules. (2021) 11:177. doi: 10.3390/biom11020177
30. Kennedy PT, Zannoupa D, Son MH, Dahal LN, Woolley JF. Neuroblastoma: an ongoing cold front for cancer immunotherapy. J Immunother Cancer. (2023) 11:e007798. doi: 10.1136/jitc-2023-007798
31. Epp S, Chuah SM, Halasz M. Epigenetic dysregulation in MYCN-amplified neuroblastoma. Int J Mol Sci. (2023) 24:17085. doi: 10.3390/ijms242317085
32. Irwin MS, Naranjo A, Zhang FF, Cohn SL, London WB, Gastier-Foster JM, et al. Revised neuroblastoma risk classification system: A report from the children’s oncology group. J Clin Oncol. (2021) 39:3229–41. doi: 10.1200/JCO.21.00278
33. Campbell K, Shyr D, Bagatell R, Fischer M, Nakagawara A, Nieto AC, et al. Comprehensive evaluation of context dependence of the prognostic impact of MYCN amplification in neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer. (2019) 66:e27819. doi: 10.1002/pbc.27819
34. Morgenstern DA, Pötschger U, Moreno L, Papadakis V, Owens C, Ash S, et al. Risk stratification of high-risk metastatic neuroblastoma: A report from the HR-NBL-1/SIOPEN study. Pediatr Blood Cancer. (2018) 65:e27363. doi: 10.1002/pbc.v65.11
35. Gadde S, Kleynhans A, Holien JK, Bhadbhade M, Nguyen PLD, Mittra R, et al. Pyrimido[1,2-a]benzimidazoles as inhibitors of oncoproteins ubiquitin specific protease 5 and MYCN in the childhood cancer neuroblastoma. Bioorg Chem. (2023) 136:106462. doi: 10.1016/j.bioorg.2023.106462
36. Li M, Hu Y, Wang J, Xu Y, Hong Y, Zhang L, et al. The dual HDAC and PI3K inhibitor, CUDC−907, inhibits tumor growth and stem−like properties by suppressing PTX3 in neuroblastoma. Int J Oncol. (2024) 64:14. doi: 10.3892/ijo.2023.5602
37. Rishfi M, Krols S, Martens F, Bekaert SL, Sanders E, Eggermont A, et al. Targeted AURKA degradation: Towards new therapeutic agents for neuroblastoma. Eur J Med Chem. (2023) 247:115033. doi: 10.1016/j.ejmech.2022.115033
38. Zafar A, Wang W, Liu G, Xian W, McKeon F, Zhou J, et al. Targeting the p53-MDM2 pathway for neuroblastoma therapy: Rays of hope. Cancer Lett. (2021) 496:16–29. doi: 10.1016/j.canlet.2020.09.023
39. Shi X, Wang Y, Zhang L, Zhao W, Dai X, Yang YG, et al. Targeting bromodomain and extra-terminal proteins to inhibit neuroblastoma tumorigenesis through regulating MYCN. Front Cell Dev Biol. (2022) 10:1021820. doi: 10.3389/fcell.2022.1021820
40. Bishayee K, Nazim UM, Kumar V, Kang J, Kim J, Huh SO, et al. Reversing the HDAC-inhibitor mediated metabolic escape in MYCN-amplified neuroblastoma. BioMed Pharmacother. (2022) 150:113032. doi: 10.1016/j.biopha.2022.113032
41. Aubert G, Lansdorp PM. Telomeres and aging. Physiol Rev. (2008) 88:557–79. doi: 10.1152/physrev.00026.2007
42. Liu M, Zhang Y, Jian Y, Gu L, Zhang D, Zhou H, et al. The regulations of telomerase reverse transcriptase (TERT) in cancer. Cell Death Dis. (2024) 15:90. doi: 10.1038/s41419-024-06454-7
43. Barthel FP, Wei W, Tang M, Martinez-Ledesma E, Hu X, Amin SB, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet. (2017) 49:349–57. doi: 10.1038/ng.3781
44. Hoang SM, O’Sullivan RJ. Alternative lengthening of telomeres: building bridges to connect chromosome ends. Trends Cancer. (2020) 6:247–60. doi: 10.1016/j.trecan.2019.12.009
45. Koneru B, Lopez G, Farooqi A, Conkrite KL, Nguyen TH, Macha SJ, et al. Telomere maintenance mechanisms define clinical outcome in high-risk neuroblastoma. Cancer Res. (2020) 80:2663–75. doi: 10.1158/0008-5472.CAN-19-3068
46. Roderwieser A, Sand F, Walter E, Fischer J, Gecht J, Bartenhagen C, et al. Telomerase is a prognostic marker of poor outcome and a therapeutic target in neuroblastoma. JCO Precis Oncol. (2019) 3:1–20. doi: 10.1200/PO.19.00072
47. Valentijn LJ, Koster J, Zwijnenburg DA, Hasselt NE, van Sluis P, Volckmann R, et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet. (2015) 47:1411–4. doi: 10.1038/ng.3438
48. Lee JW, Son MH, Cho HW, Ma YE, Yoo KH, Sung KW, et al. Clinical significance of MYCN amplification in patients with high-risk neuroblastoma. Pediatr Blood Cancer. (2018) 65:e27257. doi: 10.1002/pbc.v65.10
49. Peifer M, Hertwig F, Roels F, Dreidax D, Gartlgruber M, Menon R, et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature. (2015) 526:700–4. doi: 10.1038/nature14980
50. Dagg RA, Pickett HA, Neumann AA, Napier CE, Henson JD, Teber ET, et al. Extensive proliferation of human cancer cells with ever-shorter telomeres. Cell Rep. (2017) 19:2544–56. doi: 10.1016/j.celrep.2017.05.087
51. Hartlieb SA, Sieverling L, Nadler-Holly M, Ziehm M, Toprak UH, Herrmann C, et al. Alternative lengthening of telomeres in childhood neuroblastoma from genome to proteome. Nat Commun. (2021) 12:1269. doi: 10.1038/s41467-021-21247-8
52. Siteni S, Grichuk A, Shay JW. Telomerase in cancer therapeutics. Cold Spring Harb Perspect Biol. (2024) 16(12):a041703. doi: 10.1101/cshperspect.a041703
53. Koneru B, Farooqi A, Nguyen TH, Chen WH, Hindle A, Eslinger C, et al. ALT neuroblastoma chemoresistance due to telomere dysfunction-induced ATM activation is reversible with ATM inhibitor AZD0156. Sci Transl Med. (2021) 13:eabd5750. doi: 10.1126/scitranslmed.abd5750
54. Foote KM, Nissink JWM, McGuire T, Turner P, Guichard S, Yates JWT, et al. Discovery and characterization of AZD6738, a potent inhibitor of ataxia telangiectasia mutated and rad3 related (ATR) kinase with application as an anticancer agent. J Med Chem. (2018) 61:9889–907. doi: 10.1021/acs.jmedchem.8b01187
55. Ackermann S, Cartolano M, Hero B, Welte A, Kahlert Y, Roderwieser A, et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science. (2018) 362:1165–70. doi: 10.1126/science.aat6768
56. Fischer-Mertens J, Otte F, Roderwieser A, Rosswog C, Kahlert Y, Werr L, et al. Telomerase-targeting compounds Imetelstat and 6-thio-dG act synergistically with chemotherapy in high-risk neuroblastoma models. Cell Oncol (Dordr). (2022) 45:991–1003. doi: 10.1007/s13402-022-00702-8
57. Hassin O, Oren M. Drugging p53 in cancer: one protein, many targets. Nat Rev Drug Discovery. (2023) 22:127–44. doi: 10.1038/s41573-022-00571-8
58. Koo N, Sharma AK, Narayan S. Therapeutics targeting p53-MDM2 interaction to induce cancer cell death. Int J Mol Sci. (2022) 23:5005. doi: 10.3390/ijms23095005
59. Gu L, Zhang H, He J, Li J, Huang M, Zhou M. MDM2 regulates MYCN mRNA stabilization and translation in human neuroblastoma cells. Oncogene. (2012) 31:1342–53. doi: 10.1038/onc.2011.343
60. Gundem G, Levine MF, Roberts SS, Cheung IY, Medina-Martínez JS, Feng Y, et al. Clonal evolution during metastatic spread in high-risk neuroblastoma. Nat Genet. (2023) 55(6):1022–33. doi: 10.1038/s41588-023-01395-x
61. Tweddle DA, Pearson ADJ, Haber M, Norris MD, Xue C, Flemming C, et al. The p53 pathway and its inactivation in neuroblastoma. Cancer Lett. (2003) 197:93–8. doi: 10.1016/S0304-3835(03)00088-0
62. Carr-Wilkinson J, O’Toole K, Wood KM, Challen CC, Baker AG, Board JR, et al. High frequency of p53/MDM2/p14ARF pathway abnormalities in relapsed neuroblastoma. Clin Cancer Res. (2010) 16:1108–18. doi: 10.1158/1078-0432.CCR-09-1865
63. Carr J, Bell E, Pearson ADJ, Kees UR, Beris H, Lunec J, et al. Increased frequency of aberrations in the p53/MDM2/p14(ARF) pathway in neuroblastoma cell lines established at relapse. Cancer Res. (2006) 66:2138–45. doi: 10.1158/0008-5472.CAN-05-2623
64. Chen L, Pastorino F, Berry P, Bonner J, Kirk C, Wood KM, et al. Preclinical evaluation of the first intravenous small molecule MDM2 antagonist alone and in combination with temozolomide in neuroblastoma. Int J Cancer. (2019) 144:3146–59. doi: 10.1002/ijc.v144.12
65. Lu J, Guan S, Zhao Y, Yu Y, Wang Y, Shi Y, et al. Novel MDM2 inhibitor SAR405838 (MI-773) induces p53-mediated apoptosis in neuroblastoma. Oncotarget. (2016) 7:82757–69. doi: 10.18632/oncotarget.12634
66. Van Maerken T, Ferdinande L, Taildeman J, Lambertz I, Yigit N, Vercruysse L, et al. Antitumor activity of the selective MDM2 antagonist nutlin-3 against chemoresistant neuroblastoma with wild-type p53. J Natl Cancer Inst. (2009) 101:1562–74. doi: 10.1093/jnci/djp355
67. Weiss RA. A perspective on the early days of RAS research. Cancer Metastasis Rev. (2020) 39:1023–8. doi: 10.1007/s10555-020-09919-1
68. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. (2013) 45:279–84. doi: 10.1038/ng.2529
69. Eleveld TF, Oldridge DA, Bernard V, Koster J, Colmet Daage L, Diskin SJ, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet. (2015) 47:864–71. doi: 10.1038/ng.3333
70. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discovery. (2014) 13:828–51. doi: 10.1038/nrd4389
71. Valencia-Sama I, Ladumor Y, Kee L, Adderley T, Christopher G, Robinson CM, et al. NRAS status determines sensitivity to SHP2 inhibitor combination therapies targeting the RAS-MAPK pathway in neuroblastoma. Cancer Res. (2020) 80:3413–23. doi: 10.1158/0008-5472.CAN-19-3822
72. Janoueix-Lerosey I, Schleiermacher G, Michels E, Mosseri V, Ribeiro A, Lequin D, et al. Overall genomic pattern is a predictor of outcome in neuroblastoma. J Clin Oncol. (2009) 27:1026–33. doi: 10.1200/JCO.2008.16.0630
73. Shi H, Tao T, Abraham BJ, Durbin AD, Zimmerman MW, Kadoch C, et al. ARID1A loss in neuroblastoma promotes the adrenergic-to-mesenchymal transition by regulating enhancer-mediated gene expression. Sci Adv. (2020) 6:eaaz3440. doi: 10.1126/sciadv.aaz3440
74. Schlisio S, Kenchappa RS, Vredeveld LCW, George RE, Stewart R, Greulich H, et al. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. (2008) 22:884–93. doi: 10.1101/gad.1648608
75. Guan J, Hallberg B, Palmer RH. Chromosome imbalances in neuroblastoma-recent molecular insight into chromosome 1p-deletion, 2p-gain, and 11q-deletion identifies new friends and foes for the future. Cancers (Basel). (2021) 13:5897. doi: 10.3390/cancers13235897
76. Depuydt P, Boeva V, Hocking TD, Cannoodt R, Ambros IM, Ambros PF, et al. Genomic amplifications and distal 6q loss: novel markers for poor survival in high-risk neuroblastoma patients. J Natl Cancer Inst. (2018) 110:1084–93. doi: 10.1093/jnci/djy022
77. Ambros IM, Tonini GP, Pötschger U, Gross N, Mosseri V, Beiske K, et al. Age dependency of the prognostic impact of tumor genomics in localized resectable MYCN-nonamplified neuroblastomas. Report from the SIOPEN biology group on the LNESG trials and a COG validation group. J Clin Oncol. (2020) 38:3685–97. doi: 10.1200/JCO.18.02132
78. Pinto N, Naranjo A, Hibbitts E, Kreissman SG, Granger MM, Irwin MS, et al. Predictors of differential response to induction therapy in high-risk neuroblastoma: A report from the Children’s Oncology Group (COG). Eur J Cancer. (2019) 112:66–79. doi: 10.1016/j.ejca.2019.02.003
79. Mlakar V, Jurkovic Mlakar S, Lopez G, Maris JM, Ansari M, Gumy-Pause F. 11q deletion in neuroblastoma: a review of biological and clinical implications. Mol Cancer. (2017) 16:114. doi: 10.1186/s12943-017-0686-8
80. Johnson AF, Nguyen HT, Veitia RA. Causes and effects of haploinsufficiency. Biol Rev Camb Philos Soc. (2019) 94:1774–85. doi: 10.1111/brv.12527
81. Siaw JT, Javanmardi N, Van den Eynden J, Lind DE, Fransson S, Martinez-Monleon A, et al. 11q deletion or ALK activity curbs DLG2 expression to maintain an undifferentiated state in neuroblastoma. Cell Rep. (2020) 32:108171. doi: 10.1016/j.celrep.2020.108171
82. van Groningen T, Koster J, Valentijn LJ, Zwijnenburg DA, Akogul N, Hasselt NE, et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat Genet. (2017) 49:1261–6. doi: 10.1038/ng.3899
83. Jahangiri L, Pucci P, Ishola T, Pereira J, Cavanagh ML, Turner SD. Deep analysis of neuroblastoma core regulatory circuitries using online databases and integrated bioinformatics shows their pan-cancer roles as prognostic predictors. Discovery Oncol. (2021) 12:56. doi: 10.1007/s12672-021-00452-3
84. Westerhout EM, Hamdi M, Stroeken P, Nowakowska NE, Lakeman A, van Arkel J, et al. Mesenchymal-type neuroblastoma cells escape ALK inhibitors. Cancer Res. (2022) 82:484–96. doi: 10.1158/0008-5472.CAN-21-1621
85. van Wezel EM, van Zogchel LMJ, van Wijk J, Timmerman I, Vo NK, Zappeij-Kannegieter L, et al. Mesenchymal neuroblastoma cells are undetected by current mRNA marker panels: the development of a specific neuroblastoma mesenchymal minimal residual disease panel. JCO Precis Oncol. (2019) 3:PO.18.00413. doi: 10.1200/PO.18.00413
86. Boeva V, Louis-Brennetot C, Peltier A, Durand S, Pierre-Eugène C, Raynal V, et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat Genet. (2017) 49:1408–13. doi: 10.1038/ng.3921
87. van Groningen T, Akogul N, Westerhout EM, Chan A, Hasselt NE, Zwijnenburg DA, et al. A NOTCH feed-forward loop drives reprogramming from adrenergic to mesenchymal state in neuroblastoma. Nat Commun. (2019) 10:1530. doi: 10.1038/s41467-019-09470-w
88. Thirant C, Peltier A, Durand S, Kramdi A, Louis-Brennetot C, Pierre-Eugène C, et al. Reversible transitions between noradrenergic and mesenchymal tumor identities define cell plasticity in neuroblastoma. Nat Commun. (2023) 14:2575. doi: 10.1038/s41467-023-38239-5
89. Decaesteker B, Denecker G, Van Neste C, Dolman EM, Van Loocke W, Gartlgruber M, et al. TBX2 is a neuroblastoma core regulatory circuitry component enhancing MYCN/FOXM1 reactivation of DREAM targets. Nat Commun. (2018) 9:4866. doi: 10.1038/s41467-018-06699-9
90. Durbin AD, Zimmerman MW, Dharia NV, Abraham BJ, Iniguez AB, Weichert-Leahey N, et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat Genet. (2018) 50:1240–6. doi: 10.1038/s41588-018-0191-z
Keywords: neuroblastoma, relapsed/refractory, molecular characterization, mechanism, targeted therapy, precision oncology
Citation: Chen C and Wei Z (2025) Mechanisms and molecular characterization of relapsed/refractory neuroblastomas. Front. Oncol. 15:1555419. doi: 10.3389/fonc.2025.1555419
Received: 04 January 2025; Accepted: 18 February 2025;
Published: 06 March 2025.
Edited by:
Ling Xu, Jinan University, ChinaReviewed by:
Janith Ananda Seneviratne, Peter MacCallum Cancer Centre, AustraliaCopyright © 2025 Chen and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chong Chen, Y2hvbmdjaGVuQHRtdS5lZHUuY24=; Zixuan Wei, eml4dWFud2VpNzdAZm94bWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.