 Aadil Sheikh
Aadil Sheikh Michael A. Curran
Michael A. Curran
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 17 March 2025
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1552750
This article is part of the Research TopicColorectal Cancer Immunotherapy and Immune MechanismsView all 3 articles
Colorectal cancer (CRC) is one of the most prevalent cancers in terms of diagnosis and mortality. Radiotherapy (RT) remains a mainstay of CRC therapy. As RT relies on DNA damage to promote tumor cell death, the activity of cellular DNA damage repair pathways can modulate cancer sensitivity to therapy. The gut microbiome has been shown to influence intestinal health and is independently associated with CRC development, treatment responses and outcomes. The microbiome can also modulate responses to CRC RT through various mechanisms such as community structure, toxins and metabolites. In this review we explore the use of RT in the treatment of CRC and the molecular factors that influence treatment outcomes. We also discuss how the microbiome can promote radiosensitivity versus radioprotection to modulate RT outcomes in CRC. Understanding the molecular interaction between the microbiome and DNA repair pathways can assist with predicting responses to RT. Once described, these connections between the microbiome and RT response can also be used to identify actionable targets for therapeutic development.
Colorectal cancer (CRC) is the third most commonly diagnosed cancer and the third leading cause of cancer-related mortality in the United States and globally (1, 2). While the incidence of CRC differs between regions, there is a strong correlation between incidence and level of socioeconomic development with a majority of cases being concentrated in North America, Western Europe and Australia (3, 4). Many CRC cases are strongly associated with modifiable risk factors including environment, lifestyle and diet (5, 6) and advancements in screening and treatment options have helped to mitigate some of this disease burden (7–9). Despite these advances increasing exposure to risk factors such as chronic health conditions including obesity and metabolic syndromes that promote long-term low-grade inflammation and the increasing adoption of the metabolically challenging “Western diet” is leading to rising case numbers and increasing incidence amongst young adults, demonstrating the major challenge that CRC poses to long-term public health and safety (7–9).
The molecular pathway of colorectal carcinogenesis is well documented with the step-wise accumulation of point mutations in critical genes that correlate to morphological changes beginning with adenoma and ending with carcinoma (10–12). Sporadic CRCs constitute a majority of the cancer cases, typically beginning with a mutation in the adenomatous polyposis coli (APC) gene, with subsequent mutations in KRAS, SMAD4, TP53 and DCC (10–12) (Figure 1). Inherited CRC form a smaller proportion of reported cases and include familial adenomatous polyposis (FAP) and Lynch syndrome (13, 14). These disease are associated with specific heterozygous inactivating mutations in APC and mismatch repair genes (i.e: MLH1, MSH2, MSH6) respectively (13–16). A sub-population of CRC cases that has been increasing in recent years is associated with chronic inflammatory states such as inflammatory bowel diseases (IBDs) (17, 18). These colitis-associated CRCs (CACs) are associated with earlier presentation, less detectable lesions, and increased genomic instability and mutational burden (17, 19–21). CACs have molecular pathogenesis that is distinct from sporadic CRCs, typically beginning with a mutation in TP53, and subsequent mutations in KRAS and SMAD4 (22) (Figure 1). An important contributor of intestinal inflammation is the microbiome and microbial community structure (23–25). Disruption of the symbiotic interactions of the microbiome with host epithelial and immune cells and/or development of dysbiosis can lead to chronic inflammation and pre-dispose patients to developing CAC (23–25).
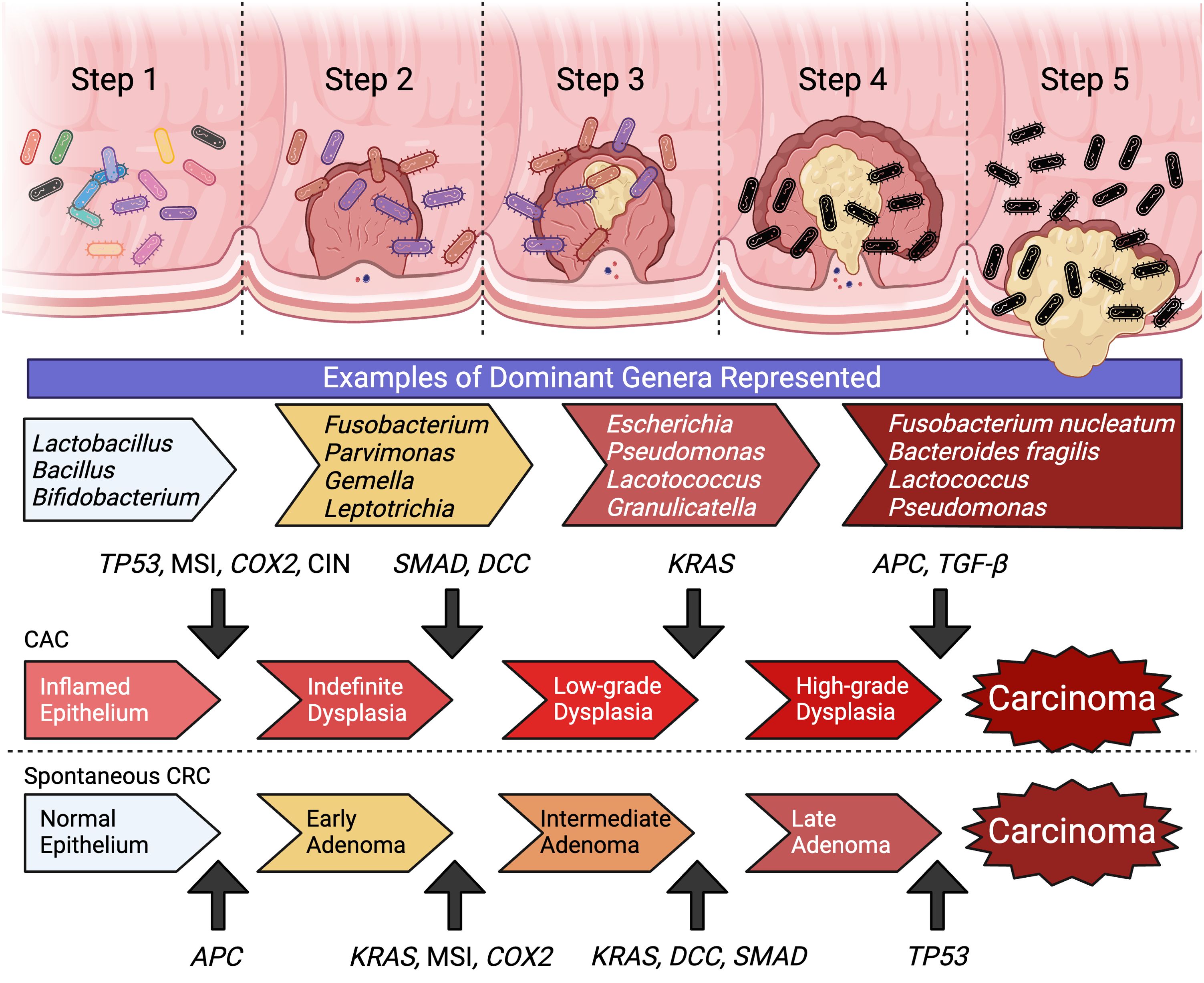
Figure 1. The molecular carcinogenesis of spontaneous CRC and CAC represent two distinct pathways of step-wise accumulation of mutations. Within the development of colon cancer there is associated with the enrichment of specific microbial genera and a concurrent loss of diversity and community structure. (Created with BioRender.com).
The microbiome is highly personalized and can influence responses to cancer treatment modalities and outcomes (26, 27). Carcinogenesis in the intestinal tract is associated with unique microbial community signatures. Early stages are associated with genera of Fusobacterium, Parvimonas, Gemella and Leptotrichia with typical lesions showing enrichment of Escherichia coli and Pseudomonas veronii, and ~40-60% of carcinomas having high carriage of Fusobacterium (Figure 1) (28, 29). The metabolism of resident microbes can improve cancer susceptibility to treatment or potentially confer resistance by modifying efficacy and toxicity of different treatment options (30). Additionally, each different class of CRC therapy can uniquely remodel the gut microbiota to influence patient outcomes (31). The effects of the microbiome on chemotherapy and immunotherapy has been studied, however data on the microbial interactions with radiotherapy (RT) is limited (30, 31). This review explores the contribution of the microbiome to the mechanism of action and responses to RT, and how this interaction may be modulated as a novel therapeutic target to improve patient outcomes.
Radiotherapy is a critical modality for cancer treatment that has been in use for decades (32). The rationale for RT is to utilize focused ionizing radiation on rapidly proliferating cancer cells leading to senescence and cell death (33, 34). RT can be delivered through several methods, either as an external beam delivered via a targeting device (external beam radiation therapy), through the placement of radioactive material near the site of the tumor/lesions (brachytherapy), or through systemic hematological dissemination of radioactive substances such as iodine or radioconjugated antibodies or pharmaceuticals (radioligand therapy) (35).
Radiation promotes several types of DNA damage in target cells including double-stranded and single-stranded breaks (DSBs and SSBs), base damage and inter-strand cross-links (36). A majority of radiation-induced cell death can be attributed to unrepaired DSBs which promote cell cycle arrest, genomic instability and mitotic catastrophe (34, 37). To ensure survival, cancer cells exploit DNA repair pathways to abrogate the damage caused by radiation. The first factor to localize to DSBs is the Mre11-Rad50-Nbs1 (MRN) complex which promotes rapid recruitment and activation of the kinase Ataxia Telangiectasia Mutated (ATM) via phosphorylation (38–40). Activation of ATM results in the downstream phosphorylation of many targets including p53, CHK2, H2AX and BRCA1 to promote cell cycle arrest and the activation of homologous and non-homologous end-joining repair mechanisms (Figure 2) (38–40). Another sentinel of DNA damage that works in conjunction with ATM is ATM- and Rad3 Related (ATR) which senses a wider range of DNA damage patterns including DSBs and SSBs. ATR is recruited to the site of lesions by the Rad9-Hus1-Rad1 complex and promotes the phosphorylation of CHK1 along with the subsequent activation of downstream repair mechanisms (Figure 2) (36, 39).
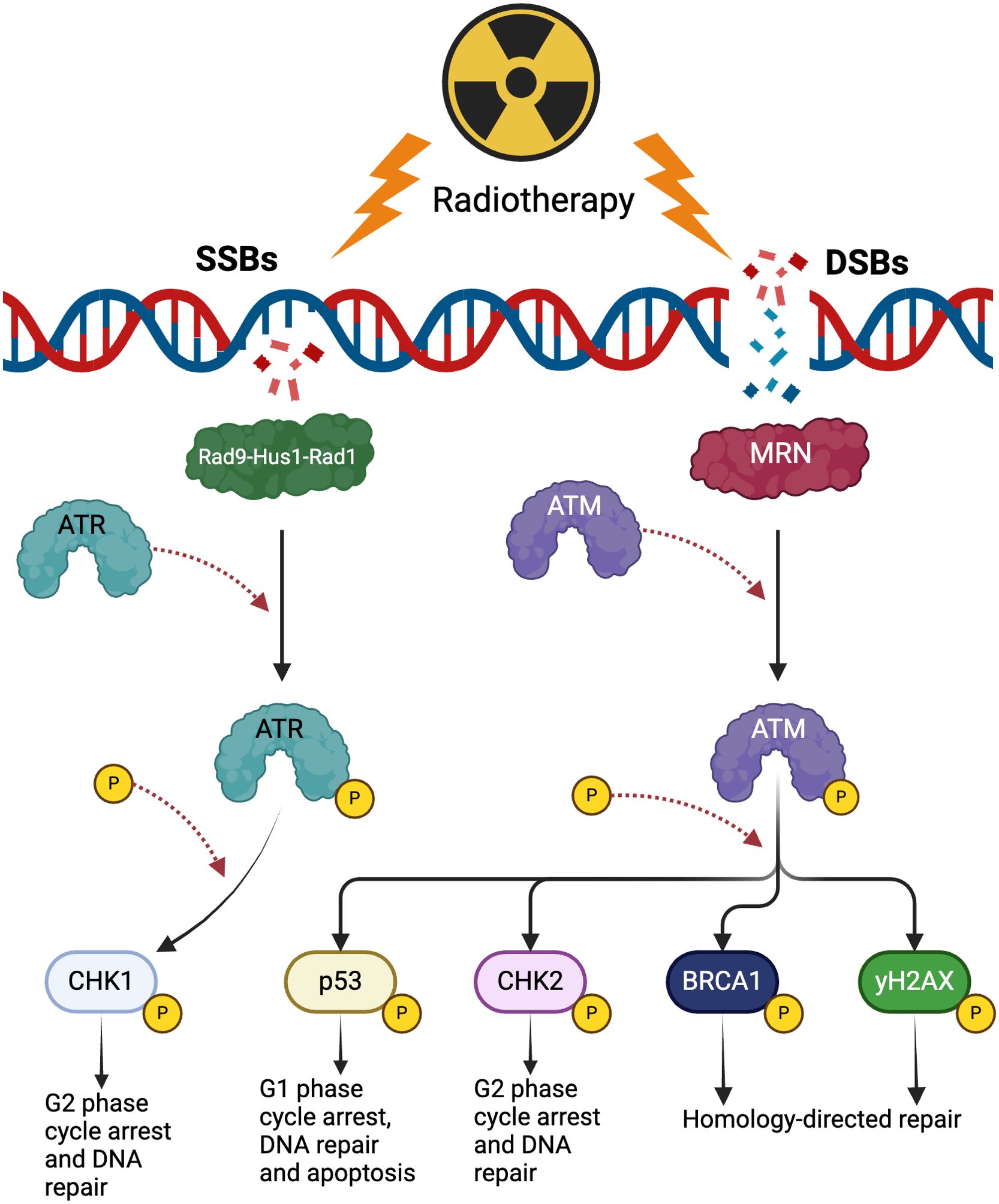
Figure 2. The effects of radiotherapy on DNA damage repair. Radiotherapy produces single-stranded breaks (SSBs) and double-stranded breaks (DSBs). SSBs are detected by the Rad9-Hus1-Rad1 complex that subsequently recruits and activates ATR. ATR phosphorylates CHK1 to promote arrest of the cell cycle in the G2 phase and activate appropriate DNA repair pathways. DSBs activate ATM which phosphorylates multiple substrates such as p53, CHK2 and BRCA1 to promote cell arrest, DNA repair and apoptosis. (Created with BioRender.com).
In addition to direct cell killing via DNA damage, RT can promote immunogenic cell death (ICD) that results from the release of factors that stimulate the immune system to mount an anti-cancer response through various mechanisms. The innate immune system has evolved multiple receptors to sense ligands released by stressed and damaged cells collectively known as damage-associated molecular patterns (DAMPs) that are generated as a consequence of RT. There are three components of ICD that promote anti-tumor activity by the immune system: the release of ATP into the extracellular space where it acts as a chemoattractant and activator of antigen-presenting cell (APC) inflammasomes via interactions with the P2X receptors (41–43). The second component is the extracellular release of HMGB1 from the nucleus and its subsequent binding to TLR4 which promotes the activation of APCs and antigen-presentation to T cells (34, 44, 45). The final arm of ICD is the presentation of calreticulin on the outer leaflet of the cell membrane, providing an “eat me” signal to APCs and promoting phagocytosis of the dying cancer cells (34, 45). These three pillars of ICD are induced by RT and trigger the release of tumor-associated antigens (TAAs) which are taken up by APCs. This, in turn, produces an anti-tumor vaccination effect via cross-presentation, thereby promoting a robust systemic immune response utilizing both the innate and adaptive arms of anti-tumor immunity (34). While ICD can enhance radiosensitivity of cancer cells through its “tumor vaccine” immune effect, the activation of DNA repair pathways post-RT can prove detrimental to treatment outcomes by promoting radioresistance. The activity of ATM and ATR in response to RT can promote the up-regulation of “don’t eat me” signals in CRC cells such as PD-L1 and CD47 (46). The upregulation of such ligands promotes higher rates of cancer immune escape and proves to be a promising target for neo-adjuvant therapy of CRC (46).
Radiotherapy has been a standard of care for CRC for nearly thirty years (47). While it is more commonly utilized to treat rectal cancers, this modality has also been used to treat colon cancer patients at various stages of their therapeutic course, though the guidelines for utilizing RT in this context are less well defined (48, 49). For these patients, RT can be used in the neo-adjuvant setting in combination with chemotherapy, during or post-surgery (adjuvant) for removing residual cancer cells, or to treat challenging CRC cases where more conventional treatment methods are not feasible (48–53). Despite the advancements made in CRC therapeutic development, the influence of the microbiome on responses and outcomes to RT has not been fully elucidated. Understanding the interactions of the microbiome with different treatment modalities can improve their efficacy and personalize treatment options to a patient’s specific needs potentially yielding improved responses. The gut microbiome plays a crucial role in health and disease, and significant gains have been made to understand its interactions with chemotherapy (30, 31). However, its interactions with RT are still being determined and represent a promising new frontier for cancer research.
While RT has demonstrated significant efficacy in the management and treatment of CRCs, and advancements have allowed for increased precision during treatment delivery, it’s application remains limited by adverse effects on normal tissue (54). Specifically, the production of reactive-oxygen species (ROS) and DNA damage during RT effects the surrounding normal gastrointestinal epithelium, leading radiation-induced enteritis (55). The deleterious effects of RT also extend to the microbiome. RT can potentially influence microbial community structure through the elimination of commensal bacteria, leading undesirable dysbiosis due to the proliferation of pathogenic species that can further promote negative health outcomes (27, 55, 56). An exploratory analysis of 11 patients with pelvic cancers (i.e. CRC and cervical cancer) who developed adverse symptoms after RT demonstrated a lower alpha diversity index, species richness and an increased Firmicutes/Bacteroidetes ratio prior to RT when compared to healthy controls (57). Analysis of the patient microbiota post-RT demonstrated significant alterations to the differences with patients that developed diarrhea having unique microbial species profiles compared to patients who did not develop adverse symptoms (57). Further analysis of patient fecal samples over the course of treatment demonstrated a gradual decrease in species diversity in patients who experienced radiation enteropathy (54, 58). Johnson et al. observed a significant decrease in Lactobacillus, Enterobacteriaceae, and aerobic bacteria counts within 16 hours of RT (Figure 3) (59, 60). Additionally, a systematic review of 11 studies demonstrated an increase in pathogenic bacteria belonging to Proteobacteria and Fusobacteria alongside a significant decrease in commensals such as Faecalibaterium and Bifidobacterium (Figure 3) (54, 61).

Figure 3. The influence of radiation therapy on microbial community structure. Prior to RT patient fecal matter data demonstrates high species richness within (alpha diversity) and across (beta diversity) patients. Generas represented in these samples are enriched in Lactobacillus, Bacillus, Bifidobacterium, and other commensal microbes. Post-RT there is a marked decrease in community richness and structure within 16 hours. Additionally, there is a observed decrease in the Lactobacillus, Bifidobacterium and Enterobacteriaceae and an increase in pathogenic species belonging to Fusobacterium and Proteobacteria. (Created with BioRender.com).
Dysregulation of the microbiota and decreased diversity is associated with colitis and IBD. Fecal matter analysis of radiation-treated mice demonstrated an increase in beta diversity when compared to controls and an abundance of opportunistic bacteria from Akkermansia, Bacteroides, and Proteobacteria (62). Fecal matter transplantation (FMT) of the radiation-treated microbiota into germ-free (GF) mice rendered them more susceptible to radiation injury and DSS-induced colitis, and was associated with increased levels of the inflammatory cytokines IL-1β and TNF-α when compared to naïve microbiomes (62). Treatment with IL-1 receptor antagonists reduced post-radiation cytokine expression and colonic damage (62). Together the data suggests that radiation can influence the composition and diversity of the patient microbiome. The proliferation of obligate and opportunistic pathogens and the simultaneous depletion of commensal and probiotic taxa suggest that the microbiome and RT can have a synergistic effect that increases the chances of developing adverse outcomes. Concurrently the findings also highlight promising new therapeutic targets to ameliorate symptoms such as blockade of inflammatory cascades or probiotic supplementation. Patients that were provided with probiotics such as Lactobacillus and Bifidobacterium prior to radiation were found to have lower rates of diarrhea and enteritis compared to placebo groups suggesting that preventive intervention to minimize RT induced dysbiosis is feasible (63).
Given the impacts of RT-induced dysbiosis on patient outcomes, there has been a growing interest in exploring how to manipulate the microbiome to improve the efficacy of therapy and to reduce adverse consequences. Studies demonstrate that the microbiome can mitigate the negative effects of RT and even promote cellular radiosensitivity to improve clinical results (55). A seminal study reported finding a significant radioprotective potential of the microbiome and microbial-derived metabolites such as short-chain fatty acids (SCFAs) and indoles. Researchers found a small percentage of mice that could survive high doses of radiation and maintain a normal lifespan (55, 64). The mice were designated as “elite survivors” and were found to have a gut microbiome enriched with Lachnospiraceae and Enterococcaceae. FMT of elite survivors into GF and specific-pathogen free (SPF) mice demonstrated an increase in survival rate of recipients post-radiation (64). Analysis of fecal samples from patients with leukemia corroborated the findings of the murine study; patients who did not experience diarrhea post-radiation had an abundance of Lachnospiraceae and Enerococcaceae (64). Further investigation to understand the correlation between radioprotection and Lachnospiraceae revealed that SCFAs like acetate, butyrate and propionate produced by the bacteria can mediate radioprotection through inhibition of inflammatory pathways, reduction of ROS generation in cells, and decreased DNA damage (64).
In addition to protecting patients from the negative consequences of RT, the microbiome has demonstrated a capacity to improve the efficacy of radiation. Treatment of murine cancer models with antibiotics to deplete the microbiome prior to radiation has been shown to reduce its efficacy while simultaneously expanding commensal fungi populations (65). Depletion of the commensal fungi and re-establishment of bacteria resulted in a decrease in the count of pro-tumor macrophages and an increase in anti-tumor CD8+ T cell mediated responses (65). Analysis of the microbiota of patients that responded to RT showed an enrichment of Faecalibaterium when compared to patients that did not respond to treatment (66). Further metabolomics revealed that cyclic-di-AMP, a second messenger produced by the bacteria, is upregulated in responsive patients and activates the cGAS-STING pathway to regulate immune sensitivity to RT (66). Microbial metabolites such butyrate and methylglyoxal can also promote radiosensitivity through activation of innate immune receptors, and autophagy/apoptosis (67, 68). The emerging data suggests that the manipulation of the microbiome through targeted antibiotics, supplementation with probiotics, or FMT can improve RT outcomes (63–65). While the exact mechanisms governing the interactions of the microbiome and radiosensitivity have yet to be revealed, there are promising results that point to activation of the immune system and DNA damage responses that correlate with the mechanism of action of RT.
With increasing evidence demonstrating the interplay of the microbiome with outcomes from RT, it has become imperative to understand the mechanisms that govern these interactions. While the microbiome affects many physiological processes, in the context of RT it is critical to elucidate the influence of the microbiome on DNA repair pathways. As RT relies on DNA damage to induce cell death, the subsequent activation of DNA repair can promote cancer cell survival and lead to poor therapeutic outcomes (32, 46).
Multiple studies have demonstrated a correlation between the microbiome and cancer-inducing DNA damage in CRC, particularly with in relation to specific species such as Bacteroides fragilis and Fusobacterium nucleatum (69–72). However, limited data exists on the specific pathways bridging the microbiome and alterations in DNA repair, though there is compelling evidence suggesting a potential link through SCFAs and other microbial-derived metabolites in influencing DNA repair activity (26). SCFAs originate from the metabolism of non-dietary carbohydrates by the microbiome and have many documented effects on health and homeostasis (73). Butyrate has been shown to have cytotoxic effects on CRC cells through the inhibition of histone deacetylases (HDACs) which can promote apoptosis (Figure 4) (26, 74, 75). HDACs regulate epigenetic modification of histones through removal acetyl groups resulting in chromatin condensation and transcriptional repression. This HDAC-mediated modulation of gene expression affects carcinogenesis and radioresistance by influencing DNA repair, apoptosis and cell cycle pathways (76, 77). HDACs are upregulated in CRC and are a promising therapeutic target to improve treatment efficacy and outcomes (78–80). HDAC1 and HDAC2 are enriched in CRC cells and have promote radioresistance by localizing to DSBs and activate DNA repair through non-homologous end-joining (NHEJ) (81). Inhibition of HDACs in CRC has demonstrated increased apoptosis and reduced cell growth; further investigations show that HDACi improves CRC radiosensitivity through induced expression of Caspase-3 and stimulation of autophagy (82, 83). Other metabolites originating from commensal bacteria have been found to have radioprotective effects on cells through several DNA repair pathways (84). Indoles produced by commensal E. coli have been shown to prevent DNA damage and aneuploidy in Caenorhabditis elegans by interacting with hus-1 and cku-70, components of SSB and DSB repair pathways respectively (Figure 4) (84). Within the context of cancer, it has been found that indole-3-carbinol (I3C), a natural compound from vegetables, has significant anti-tumor properties in breast cancer (85). I3C has been shown to down-regulate the expression of estrogen-responsive genes and upregulate and work synergistically with BRCA1 to suppress estrogen receptor signaling (Figure 4) (85). Though the evidence demonstrates the influence of indoles on DNA damage detection and repair, the precise mechanism by which these molecules dynamically influence radioprotection or radiosensitivity have yet to be fully elucidated.

Figure 4. Potential mechanisms by which the microbiome can influence RT responses through interactions with DNA repair pathways. Butyrate and other SCFA products from fermentation of fiber in the intestinal lumen can promote DNA repair through multiple pathways. Butyrate-mediated HDAC inhibition can prompt DSB repair through NHEJ and contribute to radioresistance. Simultaneously it can promote activation of caspase-3 via HDACi which leads to autophagy and radiosenstivity. Butryrate also upregulates the expression of isocitrate dehydrogenase, pyruvate dehydrogenase, ACoA and α-Ketoglutarate. Interaction of butyrate with α-Ketoglutarate leads to epigenetic remodeling of the MLH1 and MSH2 promoters which suppress CRC development and can potentially radiosensitize cells. Butryrate can interact with β-catenin through an unknown mechanism independent of APC which in turn has an unknown interaction with MMR proteins to drive proliferation in dMMR CRC. Indole metabolites from the microbiome can induce similar opposing effects as SCFAs. Indoles can interact with repair proteins like hus-1 to promote homologous repair and cku-70 to promote NHEJ. Activation of both pathways have been shown to have radioprotective effects in C. elegans embryos. Indoles can also work with BRCA1 to inhibit expression of estrogen-responsive genes in breast cancer. Further investigation into the role of indole-DNA repair interactions in CRC can further understanding of RT outcomes and responses. (Created with BioRender.com).
A prominent subset of CRC is defined by mismatch repair (MMR) deficiencies, characterized by a high frequency of microsatellite instability (86). This characteristic is high penetrant in hereditary syndromes such as Lynch syndrome (hereditary nonpolyposis CRC; HNPCC) due to germline mutation in MMR genes such as MLH1, MSH2, and MSH6 (86). Patients with CRCs that are MMR deficient (dMMR) are found to have higher tumor mutational burden (TMB) and respond to immunotherapy better than patients MMR-proficient (pMMR) CRC (87). The differences in clinical and biological characteristics between dMMR and pMMR cancers extend to the microbiome (87). Studies have demonstrated an enrichment of species that have a high association with CRC such as Bacteroides fragilis and Fusobacterium nucleatum in patients with dMMR CRC (88). Additionally, it has been found that patients with pMMR CRC tended to have higher alpha diversity and distinct beta diversity when compared to dMMR CRC patients, with the latter having enrichment in genera such as Fusobacterium, Streptococcus, Akkermansia, and Prevotella (89). A study analyzing the development of CRC in MSH2-deficient mice showed that antibiotic depletion of the microbiome prevented the development of polyps (90). Further metabolic analysis implicated butyrate in promoting higher cell turnover and increased differentiation in transit-amplifying cells in the colonic crypts (75, 90). The authors of the study hypothesized that butyrate drives proliferation and increased β-catenin activity in dMMR colon cells (90). They found that that β-catenin is increased MSH2-deficient cells in an APC-independent manner and postulated that MMR proteins can directly affect β-catenin signaling, though the exact mechanism remains to be elucidated (Figure 4) (90). Paradoxically it has also been found that butyrate can suppress CRC development through MMR via epigenetic remodeling (75). In HT-29 and Caco-2 cells, butyrate has been found to upregulate expression of isocitrate dehydrogenase and pyruvate dehydrogenase, as well as increase levels of acetyl-CoA and α-ketoglutarate (75). Furthermore, butyrate was found to interact with α-ketoglutarate to promote histone acetylation and DNA demethylation of the promoters of MLH1 and MSH2 (Figure 4) (75). Consideration of MMR-status is becoming more important in CRC treatments due to its role as a molecular biomarker and prognostic indicator (91). While dMMR patients have been shown to have improved disease-free survival with neo-adjuvant chemotherapy, it has also been shown that dMMR tumors respond poorly to RT while pMMR patients respond well (91).
While the mechanistic associations remain to be described, the sum of the existing data suggests a compelling association between the microbiome and microbial metabolites and susceptibility to RT that at least partially depends on modulation of DNA damage repair pathways. Future studies which elucidate the pathways connecting specific microbial elements to their cognate mechanisms of modulation of RT sensitivity may open new avenues for therapeutic intervention to increase RT efficacy while diminishing adverse effects on normal tissue via targeted microbiome modulation. While some therapeutically actionable microbial pathways are likely widely shared, other interventions may require more personalized microbiome profiling and customized therapeutic interventions.
The microbiome has well-documented effects on host physiology, carcinogenesis and treatment outcomes (25, 27, 28, 56, 88). While the interactions of the microbiome with chemotherapy in CRC are well-appreciated, less work has been done to understand the interplay of the microbiome with RT (26, 31). Though RT is not as common a treatment modality for colon cancer as it is for rectal cancer, it is becoming increasingly used for CRC as evidence emerges for its efficacy in treating locally advanced tumors (32, 51). As RT utilizes DNA damage to promote cell death and anti-tumor responses, it is evident that factors enhancing or dampening responses need to be considered to maximize the efficacy of treatment (30, 32, 33, 43, 51). Microbial community structure, colonization of specific genera, and metabolites have demonstrated a capacity to promote radiosensitivity or, in some cases, radioresistance in CRC (27, 31, 54). Given the microbiome’s capacity to manipulate multiple molecular pathways within host cells, an important question arises as to how it influences DNA repair pathways in CRC and, as a result, modulates RT effectiveness (58, 67, 75). Several studies have investigated the mechanisms involving the microbiome and DNA repair. SCFAs such as butyrate have been demonstrated to have contradictory effects on DNA repair. On one hand it has been shown to upregulate NHEJ by inhibiting HDACs, and can inhibit cancer progression by promoting activation of silenced MMR genes through DNA demethylation and histone acetylation in an α-ketoglutarate dependent manner (26, 75). Conversely, butyrate can promote oncogenic progression and exacerbate dMMR CRC by interacting with β-catenin independent of APC (90). Other metabolites such as indoles can promote radioprotection by interacting with proteins in SSB and DSB repair pathways such as BRCA1 (84, 85). Investigating the impact of the microbiome on DNA repair pathways directly informs on the downstream sensitivity to RT and warrants further investigation.
Several future research directions may be pursued in the future and will help us understand the molecular mechanisms that influence the microbiome-DNA repair-radiotherapy axis. The first will be to investigate the association of microbial community structure and specific species with the expression of various DNA repair proteins involved in NHEJ, homologous end-joining (HEJ), base excision repair (BER), and nucleotide excision repair (NER) in CRC cells. Additionally, it will be critical to define the role of specific microbial metabolites, proteins, or even toxins to influence the different repair pathways either through direct or epigenetic interactions. Furthermore, understanding how these DNA repair proteins will be modified in the presence of a combination of microbial products and RT will assist with understanding key therapeutic windows for dosage and length of treatment to maximize efficiency when moving these strategies to the clinic. Also considering the influence of different methods that impact the microbiome such as probiotics, prebiotics, antibiotic and FMT will provide evidence that can inform standard-of-care guidelines. Such advances will enable us to establish a direct mechanistic link between the microbiome and radiotherapy and promises to serve as both biomarker and therapeutic target in combatting CRC.
AS: Conceptualization, Writing – original draft, Writing – review & editing. MC: Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The study received R25 grant: CA265800-01A1.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. SEER Cancer Stat Facts: Colorectal Cancer. Bethesda, MD: National Cancer Institute. Available online at: https://seer.cancer.gov/statfacts/html/colorect.html (Accessed June 30, 2024).
2. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA. Cancer J Clin. (2024) 74:12–49. doi: 10.3322/caac.21820
3. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
4. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. (2017) 66:683–91. doi: 10.1136/gutjnl-2015-310912
5. Sharma R, Karki R, Sundaram B, Wang Y, Vogel P, Kanneganti T-D. Global, regional, and national burden of colorectal cancer and its risk factors, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Gastroenterol Hepatol. (2022) 7:627–47. doi: 10.1016/S2468-1253(22)00044-9
6. Sawicki T, Ruszkowska M, Danielewicz A, Niedźwiedzka E, Arłukowicz T, Przybyłowicz KE. A review of colorectal cancer in terms of epidemiology, risk factors, development, symptoms and diagnosis. Cancers. (2021) 13:2025. doi: 10.3390/cancers13092025
7. Guren MG. The global challenge of colorectal cancer. Lancet Gastroenterol Hepatol. (2019) 4:894–5. doi: 10.1016/S2468-1253(19)30329-2
8. Xi Y, Xu P. Global colorectal cancer burden in 2020 and projections to 2040. Transl Oncol. (2021) 14:101174. doi: 10.1016/j.tranon.2021.101174
9. Haggar FA, Boushey RP. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg. (2009) 22:191–7. doi: 10.1055/s-0029-1242458
10. Mármol I, Sánchez-de-Diego C, Pradilla Dieste A, Cerrada E, Rodriguez Yoldi MJ. Colorectal carcinoma: A general overview and future perspectives in colorectal cancer. Int J Mol Sci. (2017) 18:197. doi: 10.3390/ijms18010197
11. Ionescu VA, Gheorghe G, Bacalbasa N, Chiotoroiu AL, Diaconu C. Colorectal cancer: from risk factors to oncogenesis. Medicina (Mex.). (2023) 59:1646. doi: 10.3390/medicina59091646
12. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. (1990) 61:759–67. doi: 10.1016/0092-8674(90)90186-I
13. Leclerc J, Vermaut C, Buisine M-P. Diagnosis of lynch syndrome and strategies to distinguish lynch-related tumors from sporadic MSI/dMMR tumors. Cancers. (2021) 13:467. doi: 10.3390/cancers13030467
14. Preisler L, Habib A, Shapira G, Kuznitsov-Yanovsky L, Mayshar Y, Carmel-Gross I, et al. Heterozygous APC germline mutations impart predisposition to colorectal cancer. Sci Rep. (2021) 11:5113. doi: 10.1038/s41598-021-84564-4
15. Testa U, Pelosi E, Castelli G. Colorectal cancer: genetic abnormalities, tumor progression, tumor heterogeneity, clonal evolution and tumor-initiating cells. Med Sci. (2018) 6:31. doi: 10.3390/medsci6020031
16. Abildgaard AB, Nielsen SV, Bernstein I, Stein A, Lindorff-Larsen K, Hartmann-Petersen R. Lynch syndrome, molecular mechanisms and variant classification. Br J Cancer. (2023) 128:726–34. doi: 10.1038/s41416-022-02059-z
17. Yashiro M. Ulcerative colitis-associated colorectal cancer. World J Gastroenterol. (2014) 20:16389–97. doi: 10.3748/wjg.v20.i44.16389
18. Stidham RW, Higgins PDR. Colorectal cancer in inflammatory bowel disease. Clin Colon Rectal Surg. (2018) 31:168–78. doi: 10.1055/s-0037-1602237
19. Munkholm P. Review article: the incidence and prevalence of colorectal cancer in inflammatory bowel disease. Aliment. Pharmacol Ther. (2003) 18:1–5. doi: 10.1046/j.1365-2036.18.s2.2.x
20. Choi C-HR, Bakir IA, Hart AL, Graham TA. Clonal evolution of colorectal cancer in IBD. Nat Rev Gastroenterol Hepatol. (2017) 14:218–29. doi: 10.1038/nrgastro.2017.1
21. Beaugerie L, Itzkowitz SH. Cancers complicating inflammatory bowel disease. N Eng J Med. (2015) 372:1441–52. doi: 10.1056/NEJMra1403718
22. Zhou RW, Harpaz N, Itzkowitz SH, Parsons RE. Molecular mechanisms in colitis-associated colorectal cancer. Oncogenesis. (2023) 12:1–11. doi: 10.1038/s41389-023-00492-0
23. Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. (2012) 13:260–70. doi: 10.1038/nrg3182
24. Young VB. The role of the microbiome in human health and disease: an introduction for clinicians. BMJ. (2017) 356:j831. doi: 10.1136/bmj.j831
25. Glassner KL, Abraham BP, Quigley EMM. The microbiome and inflammatory bowel disease. J Allergy Clin Immunol. (2020) 145:16–27. doi: 10.1016/j.jaci.2019.11.003
26. Eaton SE, Kaczmarek J, Mahmood D, McDiarmid AM, Norarfan AN, Scott EG, et al. Exploiting dietary fibre and the gut microbiota in pelvic radiotherapy patients. Br J Cancer. (2022) 127:2087–98. doi: 10.1038/s41416-022-01980-7
27. Yi Y, Lu W, Shen L, Wu Y, Zhang Z. The gut microbiota as a booster for radiotherapy: novel insights into radio-protection and radiation injury. Exp Hematol Oncol. (2023) 12:48. doi: 10.1186/s40164-023-00410-5
28. Nakatsu G, Li X, Zhou H, Sheng J, Wong SH, Wu WKK, et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun. (2015) 6:8727. doi: 10.1038/ncomms9727
29. Villéger R, Lopès A, Veziant J, Gagnière J, Barnich N, Billard E, et al. Microbial markers in colorectal cancer detection and/or prognosis. World J Gastroenterol. (2018) 24:2327–47. doi: 10.3748/wjg.v24.i22.2327
30. Alexander JL, Wilson ID, Teare J, Marchesi JR, Nicholson JK, Kinross JM. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat Rev Gastroenterol Hepatol. (2017) 14:356–65. doi: 10.1038/nrgastro.2017.20
31. Liu J, Liu C, Yue J. Radiotherapy and the gut microbiome: facts and fiction. Radiat. Oncol. (2021) 16:9. doi: 10.1186/s13014-020-01735-9
32. Delaney G, Jacob S, Featherstone C, Barton M. The role of radiotherapy in cancer treatment. Cancer. (2005) 104:1129–37. doi: 10.1002/cncr.v104:6
33. Verheij M. Clinical biomarkers and imaging for radiotherapy-induced cell death. Cancer Metastasis Rev. (2008) 27:471–80. doi: 10.1007/s10555-008-9131-1
34. Chan Wah Hak CML, Rullan A, Patin EC, Pedersen M, Melcher AA, Harrington KJ. Enhancing anti-tumour innate immunity by targeting the DNA damage response and pattern recognition receptors in combination with radiotherapy. Front Oncol. (2022) 12. doi: 10.3389/fonc.2022.971959
35. Baskar R, Dai J, Wenlong N, Yeo R, Yeoh K-W. Biological response of cancer cells to radiation treatment. Front Mol Biosci. (2014) 1. doi: 10.3389/fmolb.2014.00024
36. Deng S, Vlatkovic T, Li M, Zhan T, Veldwijk MR, Herskind C. Targeting the DNA damage response and DNA repair pathways to enhance radiosensitivity in colorectal cancer. Cancers. (2022) 14:4874. doi: 10.3390/cancers14194874
37. Huang R-X, Zhou P-K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduction Targeting Ther. (2020) 5:1–27. doi: 10.1038/s41392-020-0150-x
38. Lee J-H, Paull TT. Direct activation of the ATM protein kinase by the mre11/rad50/nbs1 complex. Science. (2004) 304:93–6. doi: 10.1126/science.1091496
39. Maréchal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect Biol. (2013) 5:a012716. doi: 10.1101/cshperspect.a012716
40. Lee J-H, Paull TT. ATM activation by DNA double-strand breaks through the mre11-rad50-nbs1 complex. Science. (2005) 308:551–4. doi: 10.1126/science.1108297
41. Silva-Vilches C, Ring S, Mahnke K. ATP and its metabolite adenosine as regulators of dendritic cell activity. Front Immunol. (2018) 9:2581. doi: 10.3389/fimmu.2018.02581
42. Ma Y, Kepp O, Ghiringhelli F, Apetoh L, Aymeric L, Locher C, et al. Chemotherapy and radiotherapy: Cryptic anticancer vaccines. Semin Immunol. (2010) 22:113–24. doi: 10.1016/j.smim.2010.03.001
43. Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat. Oncol. (2015) 25:11–7. doi: 10.1016/j.semradonc.2014.07.005
44. Kono K, Mimura K. Immunogenic tumor cell death induced by chemoradiotherapy in a clinical setting. OncoImmunology. (2013) 2:e22197. doi: 10.4161/onci.22197
45. Gameiro SR, Jammed ML, Wattenberg MM, Tsang KY, Ferrone S, Hodge JW. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget. (2013) 5:403–16. doi: 10.18632/oncotarget.1719
46. Hsieh RC-E, Krishnan S, Wu R-C, Boda AR, Liu A, Winkler M, et al. ATR-mediated CD47 and PD-L1 upregulation restricts radiotherapy-induced immune priming and abscopal responses in colorectal cancer. Sci Immunol. (2022) 7:eabl9330. doi: 10.1126/sciimmunol.abl9330
47. Steele GD Jr, Augenlicht LH, Begg CB, Gelber RD, Green MR, Kinsella TJ, et al. Adjuvant therapy for patients with colon and rectal cancer. JAMA. (1990) 264:1444–50. doi: 10.1001/jama.1990.03450110090034
48. Colon cancer treatment (PDQ®) - NCI(2024). Available online at: https://www.cancer.gov/types/colorectal/hp/colon-treatment-pdq (Accessed July 01, 2024).
49. Radiation therapy for colorectal cancer | Radiation for colorectal cancer. Available online at: https://www.cancer.org/cancer/types/colon-rectal-cancer/treating/radiation-therapy.html (Accessed July 01, 2024).
50. Roeder F, Meldolesi E, Gerum S, Valentini V, Rödel C. Recent advances in (chemo-)radiation therapy for rectal cancer: a comprehensive review. Radiat. Oncol. (2020) 15:262. doi: 10.1186/s13014-020-01695-0
51. Häfner MF, Debus J. Radiotherapy for colorectal cancer: current standards and future perspectives. Visc. Med. (2016) 32:172–7. doi: 10.1159/000446486
52. Willett CG, Fung CY, Kaufman DS, Efird J, Shellito PC. Postoperative radiation therapy for high-risk colon carcinoma. J Clin Oncol Off J Am Soc Clin Oncol. (1993) 11:1112–7. doi: 10.1200/JCO.1993.11.6.1112
53. Willett CG, Goldberg S, Shellito PC, Grossbard M, Clark J, Fung C, et al. Does postoperative irradiation play a role in the adjuvant therapy of stage T4 colon cancer? Cancer J Sci Am. (1999) 5:242–7.
54. Fernandes A, Oliveira A, Soares R, Barata P. The effects of ionizing radiation on gut microbiota, a systematic review. Nutrients. (2021) 13:3025. doi: 10.3390/nu13093025
55. Moraitis I, Guiu J, Rubert J. Gut microbiota controlling radiation-induced enteritis and intestinal regeneration. Trends Endocrinol Metab. (2023) 34:489–501. doi: 10.1016/j.tem.2023.05.006
56. Wang W, Cui B, Nie Y, Sun L, Zhang F. Radiation injury and gut microbiota-based treatment. Protein Cell. (2024) 15:83–97. doi: 10.1093/procel/pwad044
57. Wang A, Ling Z, Yang Z, Kiela PR, Wang T, Wang C, et al. Gut microbial dysbiosis may predict diarrhea and fatigue in patients undergoing pelvic cancer radiotherapy: A pilot study. PloS One. (2015) 10:e0126312. doi: 10.1371/journal.pone.0126312
58. Reis Ferreira M, Andreyev HJN, Mohammed K, Truelove L, Gowan SM, Li J, et al. Microbiota- and radiotherapy-induced gastrointestinal side-effects (MARS) study: A large pilot study of the microbiome in acute and late-radiation enteropathy. Clin Cancer Res. (2019) 25:6487–500. doi: 10.1158/1078-0432.CCR-19-0960
59. Johnson LB, Riaz AA, Adawi D, Wittgren L, Bäck S, Thornberg C, et al. Radiation enteropathy and leucocyte-endothelial cell reactions in a refined small bowel model. BMC Surg. (2004) 4:10. doi: 10.1186/1471-2482-4-10
60. Jian Y, Zhang D, Liu M, Wang Y, Xu Z-X. The impact of gut microbiota on radiation-induced enteritis. Front Cell Infect Microbiol. (2021) 11:586392. doi: 10.3389/fcimb.2021.586392
61. Wang Z, Wang Q, Wang X, Zhu L, Chen J, Zhang B, et al. Gut microbial dysbiosis is associated with development and progression of radiation enteritis during pelvic radiotherapy. J Cell Mol Med. (2019) 23:3747–56. doi: 10.1111/jcmm.2019.23.issue-5
62. Gerassy-Vainberg S, Blatt A, Danin-Poleg Y, Gershovich K, Sabo E, Nevelsky A, et al. Radiation induces proinflammatory dysbiosis: transmission of inflammatory susceptibility by host cytokine induction. Gut. (2018) 67:97–107. doi: 10.1136/gutjnl-2017-313789
63. Demers M, Dagnault A, Desjardins J. A randomized double-blind controlled trial: Impact of probiotics on diarrhea in patients treated with pelvic radiation. Clin Nutr. (2014) 33:761–7. doi: 10.1016/j.clnu.2013.10.015
64. Guo H, Chou W-C, Lai Y, Liang K, Tam JW, Brickey WJ, et al. Multi-omics analyses of radiation survivors identify radioprotective microbes and metabolites. Science. (2020) 370:eaay9097. doi: 10.1126/science.aay9097
65. Shiao SL, Kershaw KM, Limon JJ, You S, Yoon J, Ko EY, et al. Commensal bacteria and fungi differentially regulate tumor responses to radiation therapy. Cancer Cell. (2021) 39:1202–1213.e6. doi: 10.1016/j.ccell.2021.07.002
66. Li Z, Zhang Y, Hong W, Wang B, Chen Y, Yang P, et al. Gut microbiota modulate radiotherapy-associated antitumor immune responses against hepatocellular carcinoma Via STING signaling. Gut Microbes. (2022) 14:2119055. doi: 10.1080/19490976.2022.2119055
67. Dong J, Wang B, Xiao Y, Liu J, Wang Q, Xiao H, et al. Roseburia intestinalis sensitizes colorectal cancer to radiotherapy through the butyrate/OR51E1/RALB axis. Cell Rep. (2024) 43:113846. doi: 10.1016/j.celrep.2024.113846
68. Zhou H, Wang L, Lin Z, Jiang C, Chen X, Wang K, et al. Methylglyoxal from gut microbes boosts radiosensitivity and radioimmunotherapy in rectal cancer by triggering endoplasmic reticulum stress and cGAS-STING activation. J Immunother. Cancer. (2023) 11:e007840. doi: 10.1136/jitc-2023-007840
69. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor immune microenvironment. Cell Host Microbe. (2013) 14:207–15. doi: 10.1016/j.chom.2013.07.007
70. Chung L, Thiele Orberg E, Geis AL, Chan JL, Fu K, DeStefano Shields CE, et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe. (2018) 23:203–214.e5. doi: 10.1016/j.chom.2018.01.007
71. Haghi F, Goli E, Mirzaei B, Zeighami H. The association between fecal enterotoxigenic B. fragilis with colorectal cancer. BMC Cancer. (2019) 19:879. doi: 10.1186/s12885-019-6115-1
72. Chen Y, Peng Y, Yu J, Chen T, Wu Y, Shi L, et al. Invasive Fusobacterium nucleatum activates beta-catenin signaling in colorectal cancer via a TLR4/P-PAK1 cascade. Oncotarget. (2017) 8:31802–14. doi: 10.18632/oncotarget.15992
73. Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. (2016) 7:189–200. doi: 10.1080/19490976.2015.1134082
74. Vinolo MAR, Rodrigues HG, Hatanaka E, Sato FT, Sampaio SC, Curi R. Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J Nutr Biochem. (2011) 22:849–55. doi: 10.1016/j.jnutbio.2010.07.009
75. Sun X, Zhu M-J. Butyrate inhibits indices of colorectal carcinogenesis via enhancing α-ketoglutarate-dependent DNA demethylation of mismatch repair genes. Mol Nutr Food Res. (2018) 62:e1700932.
76. Salek Farrokhi A, Mohammadlou M, Abdollahi M, Eslami M, Yousefi B. Histone deacetylase modifications by probiotics in colorectal cancer. J Gastrointest. Cancer. (2020) 51:754–64. doi: 10.1007/s12029-019-00338-2
77. Ling R, Wang J, Fang Y, Yu Y, Su Y, Sun W, et al. HDAC-an important target for improving tumor radiotherapy resistance. Front Oncol. (2023) 13:1193637. doi: 10.3389/fonc.2023.1193637
78. Chou C-W, Wu M-S, Huang W-C, Chen C-C. HDAC inhibition decreases the expression of EGFR in colorectal cancer cells. PloS One. (2011) 6:e18087. doi: 10.1371/journal.pone.0018087
79. Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect Med. (2016) 6:a026831. doi: 10.1101/cshperspect.a026831
80. Rajendran P, Delage B, Dashwood WM, Yu T-W, Wuth B, Williams DE, et al. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol Cancer. (2011) 10:68. doi: 10.1186/1476-4598-10-68
81. Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. (2010) 17:1144–51. doi: 10.1038/nsmb.1899
82. Sanaei M, Kavoosi F. Effect of 5-Aza-2’-Deoxycytidine in Comparison to Valproic Acid and Trichostatin A on Histone Deacetylase 1, DNA Methyltransferase 1, and CIP/KIP Family (p21, p27, and p57) Genes Expression, Cell Growth Inhibition, and Apoptosis Induction in Colon Cancer SW480 Cell Line. Adv Biomed Res. (2019) 8:52. doi: 10.4103/abr.abr_91_19
83. He G, Wang Y, Pang X, Zhang B. Inhibition of autophagy induced by TSA sensitizes colon cancer cell to radiation. Tumor Biol. (2014) 35:1003–11. doi: 10.1007/s13277-013-1134-z
84. Sonowal R, Swimm AI, Cingolani F, Parulekar N, Cleverley TL, Sahoo A, et al. A microbiota and dietary metabolite integrates DNA repair and cell death to regulate embryo viability and aneuploidy during aging. Sci Adv. (2023) 9:eade8653. doi: 10.1126/sciadv.ade8653
85. Meng Q, Rosen EM, Fan S, Yuan F, Goldberg ID, Auborn K. Indole-3-carbinol is a negative regulator of estrogen receptor-α Signaling in human tumor cells. J Nutr. (2000) 130:2927–31. doi: 10.1093/jn/130.12.2927
86. Sinicrope FA. DNA mismatch repair and adjuvant chemotherapy in sporadic colon cancer. Nat Rev Clin Oncol. (2010) 7:174–7. doi: 10.1038/nrclinonc.2009.235
87. Sillo TO, Beggs AD, Middleton G, Akingboye A. The gut microbiome, microsatellite status and the response to immunotherapy in colorectal cancer. Int J Mol Sci. (2023) 24:5767. doi: 10.3390/ijms24065767
88. Hale VL, Jeraldo P, Chen J, Mundy M, Yao J, Priya S, et al. Distinct microbes, metabolites, and ecologies define the microbiome in deficient and proficient mismatch repair colorectal cancers. Genome Med. (2018) 10:78. doi: 10.1186/s13073-018-0586-6
89. Jin M, Wu J, Shi L, Zhou B, Shang F, Chang X, et al. Gut microbiota distinct between colorectal cancers with deficient and proficient mismatch repair: A study of 230 CRC patients. Front Microbiol. (2022) 13. doi: 10.3389/fmicb.2022.993285
90. Belcheva A, Irrazabal T, Robertson SJ, Streutker C, Maughan H, Rubino S, et al. Gut microbial metabolism drives transformation of msh2-deficient colon epithelial cells. Cell. (2014) 158:288–99. doi: 10.1016/j.cell.2014.04.051
Keywords: radiotherapy, microbiome, colorectal cancer, DNA damage, DNA repair
Citation: Sheikh A and Curran MA (2025) The influence of the microbiome on radiotherapy and DNA damage responses. Front. Oncol. 15:1552750. doi: 10.3389/fonc.2025.1552750
Received: 29 December 2024; Accepted: 24 February 2025;
Published: 17 March 2025.
Edited by:
Zheng Liu, National Cancer Center of China, ChinaReviewed by:
Daming Zhang, First Affiliated Hospital of Harbin Medical University, ChinaCopyright © 2025 Sheikh and Curran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael A. Curran, bWN1cnJhbkBtZGFuZGVyc29uLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.