 Liping Bai
Liping Bai Liang Sun
Liang Sun Juan Zou2,3*
Juan Zou2,3* Yali Chen
Yali Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Oncol., 14 March 2025
Sec. Gynecological Oncology
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1546607
Objective: Examine clinicopathological traits and differential diagnosis of ERMS in female reproductive system.
Methods: Retrospectively assess 13 patients’ data (Jan 2018 - Jun 2024, West China Second Univsity Hospital), covering clinical, histological, immunohistochemical aspects and literature review.
Results: Age 2 months - 67 years (median 21), sites in cervix (5), ovaries (3), uterus (2). Non-specific symptoms. Lesions with grape-like etc. morphologies. Immunohistochemistry: the tumor cells expressed Myogenin (11/13), Desmin (13/13), MyoD1 (12/13) and Myoglobin (5/9). 4/5 had DICER1 mutations. According to the Children’s Oncology Group Soft Tissue Sarcoma (COG-STS) risk classification, 11 low risk, 2 high risk. Treatments: 8 surgery + chemotherapy, 2 surgery + chemotherapy + radiotherapy, 2 surgery only. 4 died, 8 survived, 1 lost follow up. Follow-up 2 - 41 months (median 20).
Discussion: ERMS is rare, diagnosed by histology and immunohistochemistry, DICER1 mutation may assist. Treatment is surgery + chemo ± radiotherapy, efficacy related to multiple factors. When ERMS is diagnosed, it is mostly in the early stage, and the treatment method is mostly surgery plus chemotherapy with or without radiotherapy. However, the treatment effect is related to factors such as staging, Intergroup Rhabdomyosarcoma Study (IRS) clinical grouping, COG-STS risk, patient age, and TP53 mutation. There is no clear guideline for the treatment of adult patients.
Rhabdomyosarcoma (RMS) is a rare type of tumor that mostly occurs in children and adolescents and is less common in older adults (1). In the population under 20 years old, the overall incidence rate of RMS is approximately 4.5 cases per million patients (2). Soft tissue sarcomas account for about 3 - 7% of childhood cancers and 1% of adult cancers (1, 3, 4). Approximately half of all childhood soft tissue sarcomas are RMS, which are highly malignant tumors seen as rhabdomyoblasts with varying degrees of differentiation (3, 4). The WHO classification scheme divides rhabdomyosarcoma RMS into four different subtypes: embryonal, alveolar, spindle cell/sclerotic, and pleomorphic subtypes (5) and mentions new subtypes to be studied more, such as rhabdomyosarcoma associated with the EWSR1/FUS::TFCP2 gene fusion and the MEIS1::NCOA2 gene fusion (5, 6). Embryonal rhabdomyosarcoma (ERMS) accounts for 70 - 80% of all RMS diagnoses (5, 7). Although one-third of ERMS cases are diagnosed within 5 years after birth, they can occur at any age, including adulthood. It is worth noting that approximately half of ERMS cases originate from the head and neck region, including the orbit, while the other half occur in the genitourinary system (5, 7). Almost all cervical ERMS and nearly 67% of uterine corpus ERMS carry DICER1 mutations, while vaginal RMS are mostly DICER1 wild-type (8). This study retrospectively analyzed the clinical manifestations, treatment, and prognosis of female patients with ERMS in the female reproductive system admitted to West China Second University Hospital, Sichuan University from January 2018 to June 2024, in order to provide clinical evidence for the treatment and prognosis of this type of disease.
This retrospective, observational, single-center study was conducted at West China Second University Hospital, Sichuan University, Chengdu, China after obtaining ethical approval from the hospital’s ethics committee. The clinicopathological data of 13 female patients with ERMS in the female reproductive system admitted to West China Second University Hospital, Sichuan University from January 2018 to June 2024 were collected. The study retrospectively examined the patients’ clinical and pathological information. The pathological specimens were independently reviewed by two pathologists from West China Second University Hospital. The clinicopathological data of all 13 patients with ERMS in the reproductive system were reviewed, and a retrospective analysis was performed on their clinical and pathological characteristics, treatment methods, recurrence, post-recurrence treatment, and prognosis. The study collected basic patient information, including age, symptoms, tumor characteristics (such as location and size), surgical methods, comorbidities, adjuvant treatment, recurrence and metastasis rates, follow-up time, and prognosis. In addition, pathological characteristics and DICER1 mutation test results were also collected. The effectiveness of tumor treatment in patients was evaluated through outpatient follow-up and telephone follow-up.
Statistical analysis was performed using SPSS 27.0 software. Categorical data were presented as the number of cases and percentage (%). Normally distributed continuous data were expressed as mean ± standard deviation (xˉ ± s), while non-normally distributed continuous data were presented as median (range). Survival analysis was carried out using the Kaplan-Meier method, and the log-rank test was used for comparing survival rates. A P-value less than 0.05 was considered statistically significant.
Thirteen patients diagnosed pathologically with ERMS in the female reproductive system were included in this study. The basic characteristics of the patients are shown in Tables 1, 2. The ages of the 13 ERMS patients ranged from 2 months to 67 years, with a median age of 21 years, among which 7 patients were adults over 18 years old. The tumors originated from the cervix in 5 cases, from the ovaries in 3 cases, from the uterine body in 2 cases, from the vagina in 1 case, from the vulva in 1 case, and from the pelvic cavity in 1 case. The tumor sizes varied from 3 cm to 21 cm, with a median diameter of 8 cm. Four patients presented with vaginal bleeding (irregular vaginal bleeding and postmenopausal vaginal bleeding), accounting for 30.77%. Four patients presented with masses in the corresponding sites, accounting for 30.77%. Three patients presented with lower abdominal pain, accounting for 23.08%. One patient presented with abdominal distension, accounting for 7.70%. One patient presented with a palpable pelvic mass, accounting for 7.70%. According to the latest International Federation of Gynecology and Obstetrics (FIGO) staging: 6 cases were in stage I, 4 cases were in stage II, 2 cases were in stage III, and 1 case was in stage IV. According to the Intergroup Rhabdomyosarcoma Study Group (IRSG) clinical staging: 11 cases were in stage 1 and 2 cases were in stage 4. In terms of the IRSG clinical grouping, 7 cases were in group I, 1 case was in group II, 3 cases were in group III, and 2 cases were in group IV. According to the Children’s Oncology Group Soft Tissue Sarcoma (COG-STS) risk classification, 9 cases were at low risk, 2 cases were at medium risk, and 2 cases were at high risk. Notably, case 1 of them had a cerebellar medulloblastoma (WHO grade 4) at the age of 4 years and was treated with radiotherapy 31 times after surgical resection of the lesion, with no significant abnormality on regular review; the father of case 7 had a history of gastric cancer. The remaining patients had no family history of tumor at the cutoff of follow-up.
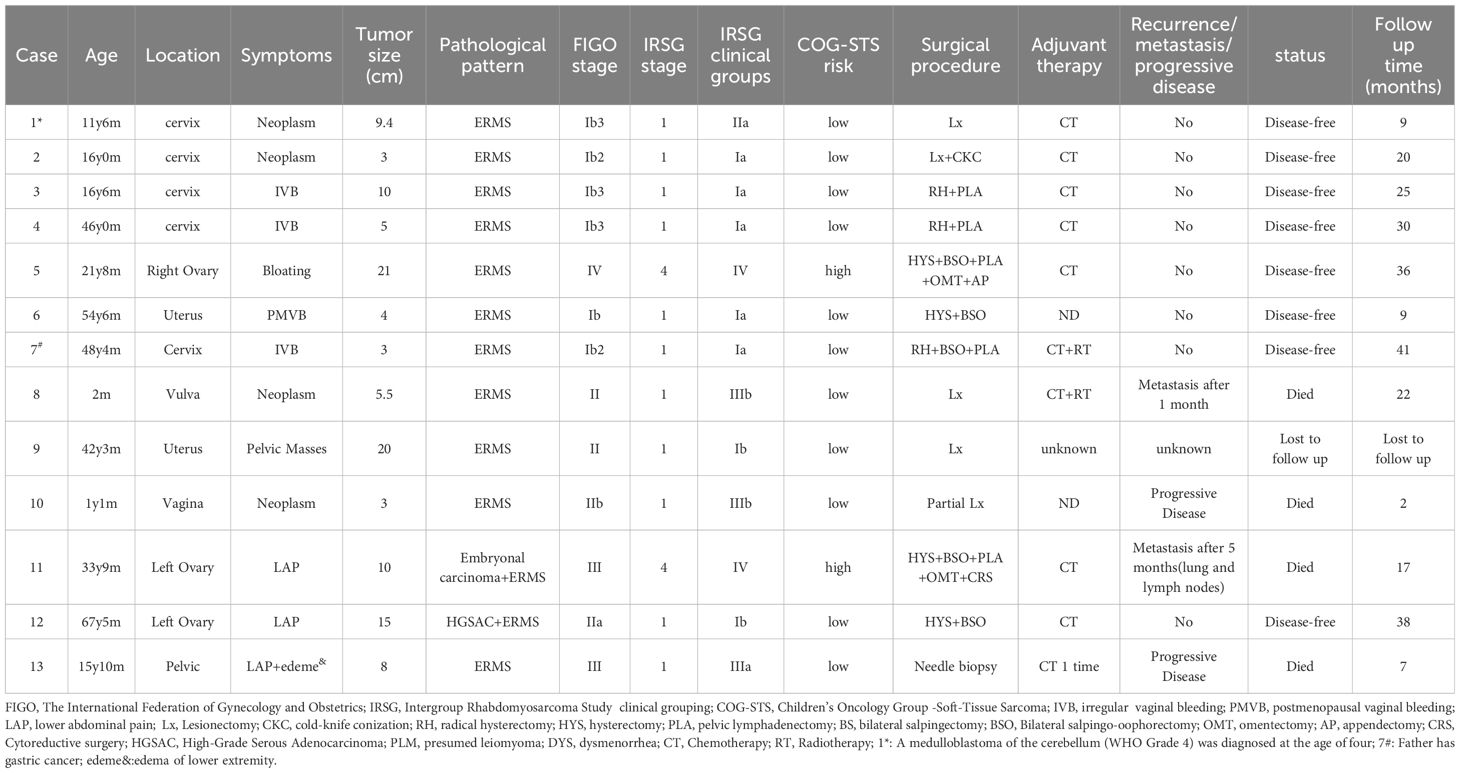
Table 1. Clinical Characteristics of ERMS.
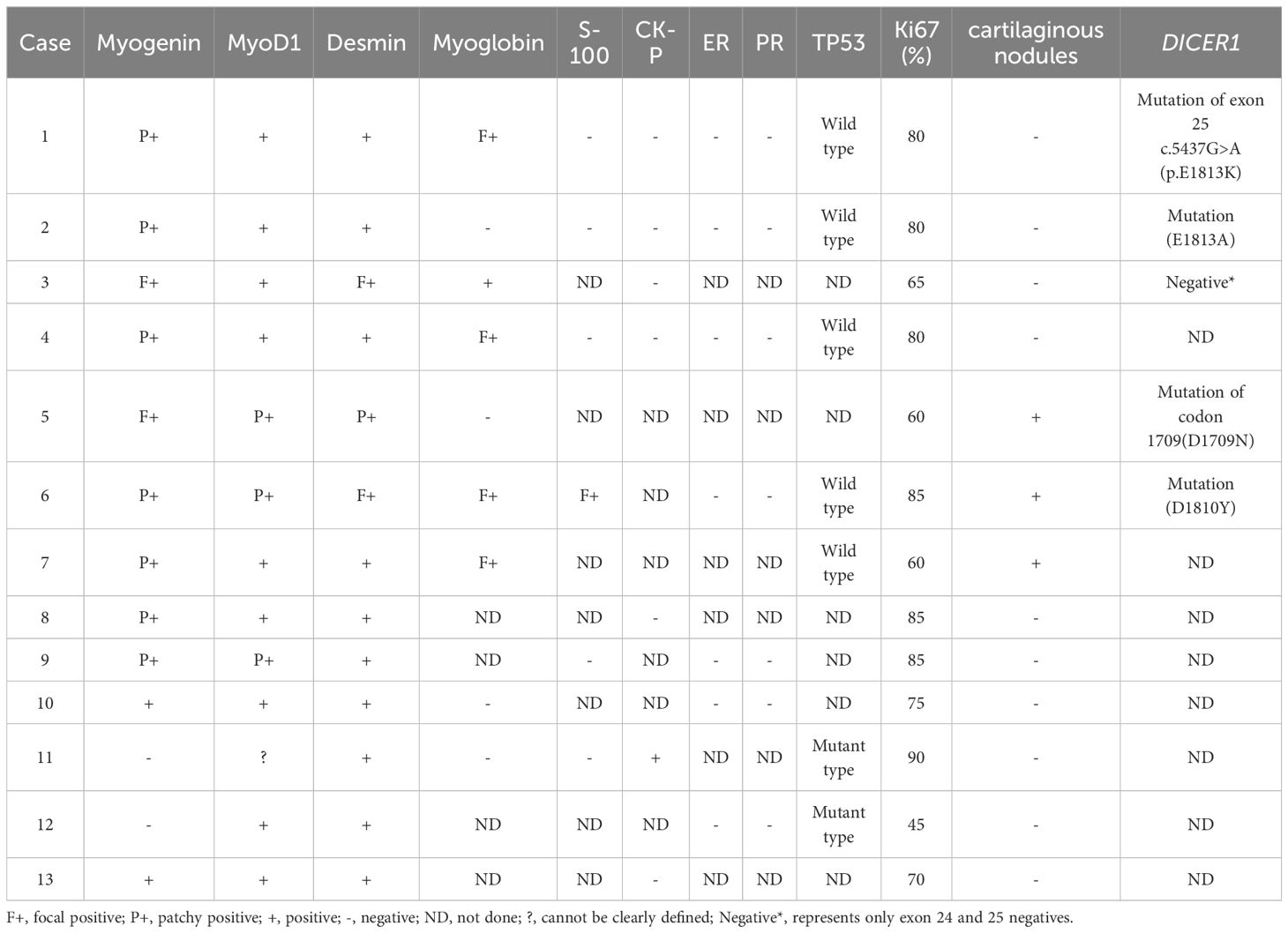
Table 2. Results of immunohistochemistry for the 13 patients with ERMS.
Macroscopically, the lesions were mostly solid or cystic-solid and could be grape-like, polypoid, cauliflower-like or fish-flesh-like. Microscopically, irregular bundles of immature skeletal muscle fibers are seen in a myxoid background. The cells had the characteristics of fetal myotubes, i.e., spindle-shaped outline, central oblong nucleus, and eosinophilic cytoplasm. Cartilaginous nodules were seen in 3 of 13 patients. Immunohistochemical assays, including desmin, myogenin, myoD1 (Figure 1), and the cell proliferation marker Ki67, were performed in all 13 patients; the immunohistochemical results are shown in Table 2. Among them, 5 patients underwent DICER1 gene mutation testing, and 4 of them were positive, as shown in Table 1. All 13 patients were diagnosed with ERMS, among which Case 11 and Case 12 were mixed tumors. Case 11 was combined with embryonal carcinoma; Case 12 was combined with high-grade serous adenocarcinoma of the ovary.
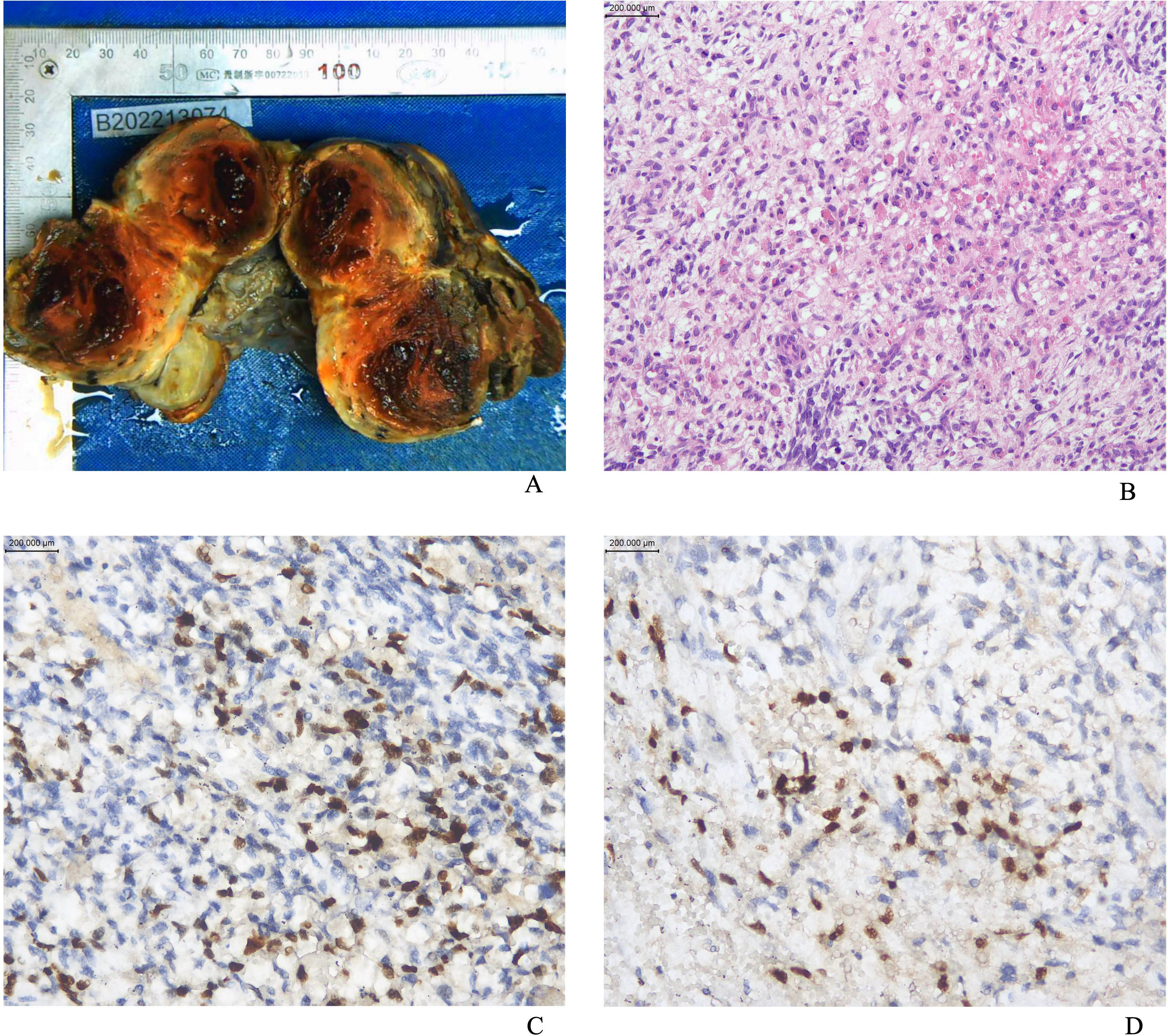
Figure 1. (A) The appearance of the gross specimen of ovarian ERMS; (B) The image of ERMS stained with hematoxylin and eosin (HE); (C) Positive staining for MyoD1; (D) Positive staining for Myogenin.
Among the 13 patients, 1 patient underwent needle biopsy plus chemotherapy. However, due to intolerance to chemotherapy, the patient did not complete the chemotherapy treatment. As the disease progressed, the survival period was only 7 months. Twelve patients underwent surgical treatment. Among them, 5 patients had lesion resection, and 7 patients had the organ where the tumor was located removed. Among these 7 patients, 5 patients underwent radical surgeries including pelvic lymph node dissection, and 2 patients underwent hysterectomy plus bilateral adnexectomy. Among all the patients, 1 patient did not receive adjuvant treatment, 8 patients only received chemotherapy, and 2 patients received chemotherapy plus radiotherapy. Among the 10 patients who received chemotherapy, 7 patients were treated with the chemotherapy regimen of vincristine + dactinomycin + cyclophosphamide/isocyclophosphamide. One of these patients had a total survival time of 22 months due to disease progression, refractory recurrence, change of chemotherapy regimen, adjuvant radiotherapy, and secondary surgery. One patient with stage IV disease was treated with epirubicin + carboplatin chemotherapy and then albumin-bound paclitaxel + nedaplatin chemotherapy. After 36 months of follow-up, the patient is currently disease-free. One patient with combined embryonal carcinoma was treated with bleomycin + etoposide + cisplatin chemotherapy. After 17 months of follow-up, the patient has passed away. One patient with combined high-grade serous ovarian cancer was treated with paclitaxel + carboplatin chemotherapy plus bevacizumab. After 38 months of follow-up, the patient is currently disease-free. The clinical and pathological characteristics of the patients are shown in Table 3.
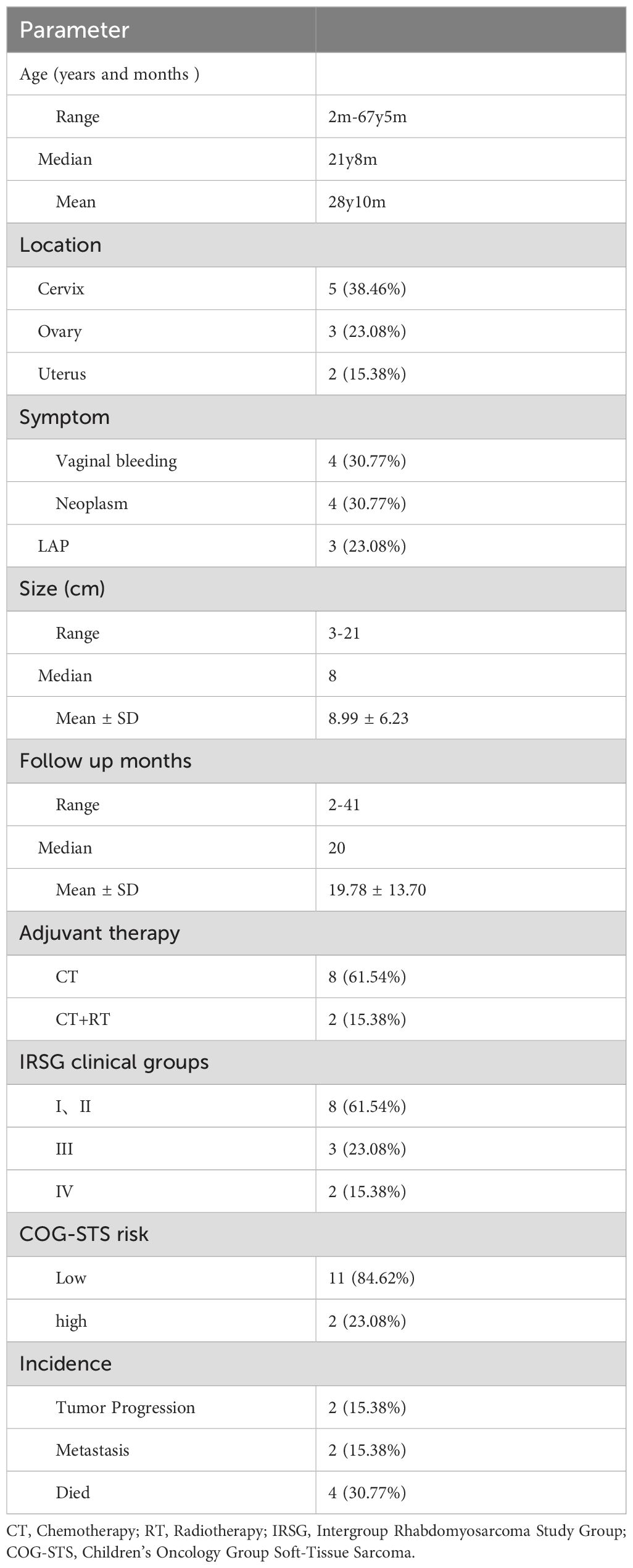
Table 3. Clinical and pathological characteristics of the patients.
During the follow-up period, of the 13 patients, 8 survived tumor-free, 4 died, and 1 was lost to follow-up. Disease progression was observed in 2 cases during the follow-up period, recurrence of metastasis in 2 cases, and death in all 4 patients. The follow-up period ranged from 2 months to 41 months, with a median follow-up period of 20 months and a 75th percentile survival time of 17 months, and the survival analysis is shown in Figure 2.
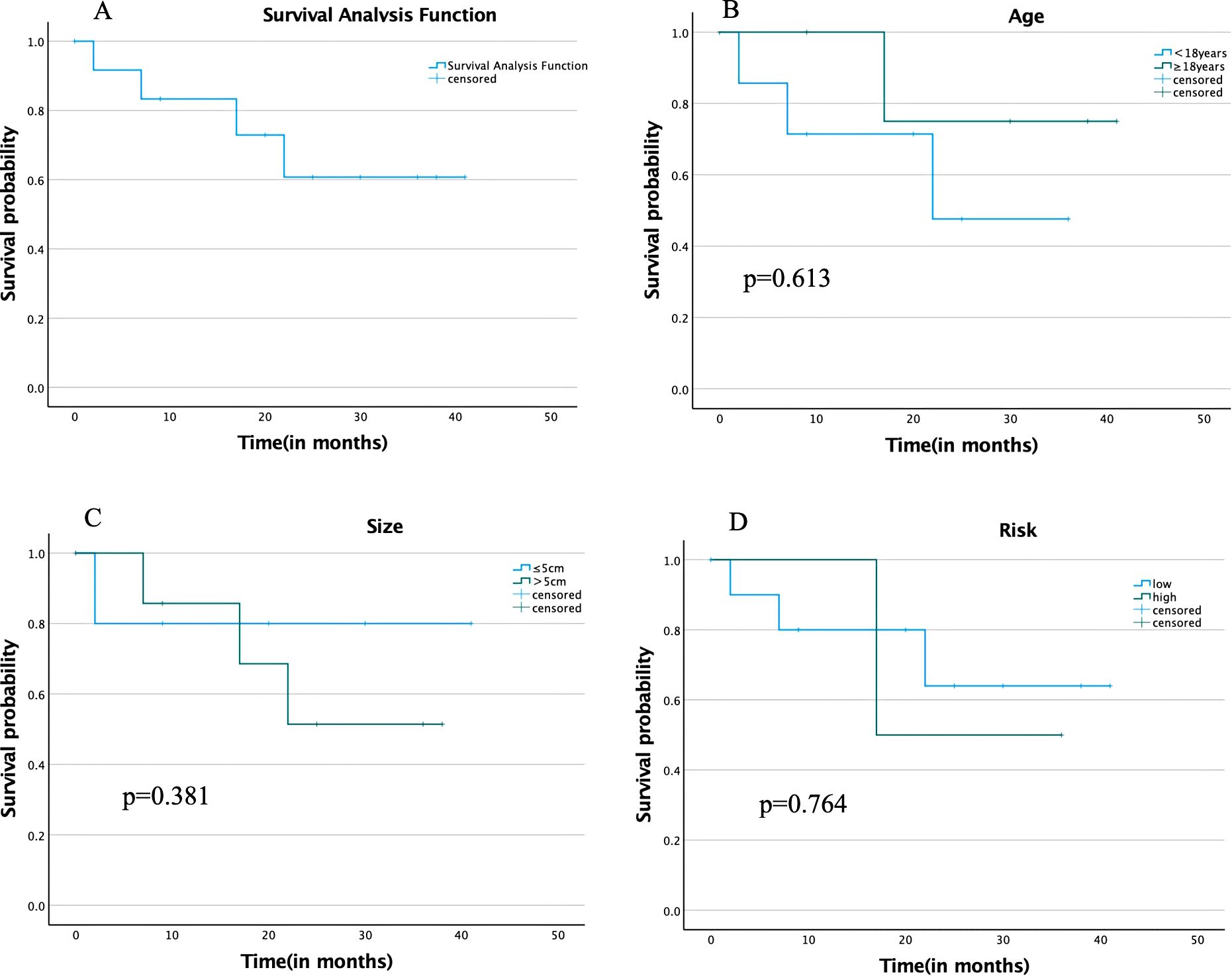
Figure 2. (A) Overall survival analysis; (B) Survival analysis by different age groups; (C) Survival analysis by different tumor size groups; (D) Survival analysis by different degrees of risk in COG-STS.
ERMS is named because it resembles skeletal muscle in embryonic development (9). ERMS is rare and difficult to diagnose. In the female reproductive system, ERMS is commonly found in the cervix and vagina. It usually presents as a polypoid mass or multiple polyps (botryoid). Histopathologically, embryonal rhabdomyosarcomas contain primitive mesenchymal cells at different stages of myogenesis. Typical embryonal rhabdomyosarcoma consists of rhabdomyoblasts with different degrees of differentiation in sparse mucus-like mesenchyme with alternating areas of dense and sparse cell density (5). The cells have small ovoid hyperchromatic nuclei and a small amount of cytoplasm, and there are often cartilaginous nodules. The diagnostic criteria for ERMS is the presence of cambium layer. Immunohistochemical markers include myogenic markers (diffuse desmin positivity, and MyoD1 and myogenin may be focally positive), and hormone receptors are often negative (8). The molecular biological characteristics are that cervical embryonal rhabdomyosarcoma and uterine corpus embryonal rhabdomyosarcoma often carry DICER1 mutations (8, 10, 11).
ERMS of the uterus usually need to be differentiated from adenosarcoma, and the morphological overlap between ERMS and adenosarcoma makes it difficult to distinguish between the two tumors. Pathological features of adenosarcomas: biphasic tumors, composed of benign epithelial and malignant mesenchymal components; usually polypoid lesions, histologically with a low-grade lobular structure, similar to lobular tumors of the breast, with glands lined by benign Müllerian epithelium, and mesenchymal stroma usually of low-grade spindle cells, which can present with differentiation of high-grade heterogeneous components (most commonly skeletal muscle) and sarcomatous overgrowth (8).On immunohistochemical testing, low-grade adenosarcomas are usually positive for CD10 and hormone receptors (8).
Heterozygous germline mutations of DICER1 were first discovered in 2009 in a series of children with pleuropulmonary blastoma (PPB) (12). The Dicer protein encoded by the DICER1 gene is an endoribonuclease that participates in the production of small RNAs such as microRNA and small interfering RNA and is crucial for the regulation of gene expression during development (10, 11, 13). The germline pathogenic variants that lead to DICER1 syndrome are usually truncating alterations (such as nonsense, frameshift, or splice - site mutations), which result in a loss of function; while the pathogenic somatic mutations are almost entirely missense substitutions that affect the five “hot spots” in the RNAse IIIb domain. These mutations change the enzyme’s ability to process microRNA, leading to an abnormal mix of microRNAs (10, 11, 13).
Almost all cervical ERMS and nearly 67% of uterine corpus ERMS carry DICER1 mutations, whereas vaginal ERMS are DICER1 wild-type (10, 14–16). Therefore, when cervical ERMS is suspected, deletion of DICER1 mutations often suggests incompatibility with this diagnosis. On the other hand, since DICER1 mutations are present in 26% to 42% of adenosarcomas (14, 17), the presence of DICER1 mutations does not differentiate ERMS from adenosarcoma. In ERMS with DICER1 mutations, genetic counseling is necessary to investigate the possibility of DICER1 syndrome. One of the 13 patients we analyzed suffered from cerebellar medulloblastoma (WHO grade 4) at the age of 4 years; in another case, the father had a history of gastric cancer. The remaining patients had no significant family history of the tumor, and none of the patients underwent genetic counseling. Five of these 13 patients underwent DICER1 genetic testing, and mutations were detected in four of them (two cervical, one ovarian, and one uterine). Another patient with cervical ERMS did not detect mutations in the DICER1 gene in exons 24 and 25, which may be related to an insufficient number of tested loci.
Currently, surgery + chemotherapy ± radiotherapy is the recommended treatment modality for ERMS (18). For children and young women, initial treatment is preferred to surgery that can completely remove the tumor (resection should be 0.5 cm beyond the tumor margins) while preserving as much organ function as possible, while reproductive adults tend to undergo surgical procedures with more complete organ removal, such as hysterectomy (19–21). Surgery that simply reduces the size of the tumor and does not completely resect the tumor is not superior to biopsy in terms of improving prognosis (19).The Children’s Oncology Group (COG) and the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG) use different chemotherapy-based regimens, the main difference being the choice of alkylating agent, with the COG using cyclophosphamide and the EpSSG using isocyclophosphamide. Comparison of these two alkylating agents shows that they do not differ significantly in therapeutic efficacy but produce different long-term side effects: isocyclophosphamide is more nephrotoxic, whereas cyclophosphamide is more gonadotoxic (22, 23).In adults, because of the rarity of RMS, chemotherapy regimens are mostly based on the selection of drugs based on experience with pediatric RMS. The response rate to chemotherapy for adult embryonal rhabdomyosarcoma is approximately 85% (1). Radiotherapy is an important part of the comprehensive treatment of RMS in children, especially for those patients with inoperable resection, microscopic residual tumor, naked eye residual tumor, or lymph node involvement, induction chemotherapy followed by simultaneous radiotherapy is the currently recommended treatment modality (19).
IRSG combines staging, clinical grouping, and pathology type to categorize RMS into low-risk, intermediate-risk, and high-risk groups. The 5-year EFS of RMS in patients in the low-risk, intermediate, and high-risk groups were 87%-90%, 65%-73%, and <30%, respectively (24), and this system was able to better predict the prognosis of patients, which is instructive for the selection of treatment options. In addition to influencing factors such as tumor stage, subgroups, and risk level grading, FOXO1 fusion positivity, age less than 1 year, age greater than 10 years, and TP53 mutations are poor prognostic factors (25–29). Higher levels of TP53 protein have been found in metastatic ERMS relative to limited ERMS (30). 95% of embryonic rhabdomyosarcomas are FOXO1 fusion-negative, which means that almost all embryonic rhabdomyosarcomas are FOXO1 fusion-negative rhabdomyosarcomas (31). ERMS is genetically free of gene fusions, but aneuploidy and multiple genetic alterations are present; ERMS has an overall favorable prognosis, but cases with diffuse interstitial changes have a poorer prognosis (6, 31, 32).
Adult patients with RMS have a lower overall survival rate than pediatric patients, and the few published series on adult patients describe a poorer prognosis, with 5-year survival rates ranging from 20-51.8% (33–36). Even with the same tissue type, site, and stage, adults still have a worse prognosis than children (35). There is an increasing number of studies related to RMS in adults (18, 26, 36–39), and the reasons for the currently reported poorer survival in adult patients may include delayed diagnosis, health system disparities, increased expression of multidrug-resistant proteins in tumors, and low tolerance to intensive therapy (18, 26, 36, 37), but also because of variations in the distribution of sarcoma subtypes and clinical behaviors in different age groups, racial differences, or biological differences (37–39). One study found that the survival of patients with embryonic and alveolar RMS in an Asian population was inferior to that previously reported in other races (39), suggesting that tumor heterogeneity may exist between different races. In another study, multiple chemoresistance genes were found to be upregulated in adult RMS patients, and pharmacological analyses showed that anthracycline-based regimens had the highest sensitivity to tumor cells in both 2D and 3D culture systems, suggesting that anthracyclines may be promising agents (37).
Four of the 13 patients reported in this article died, three of whom were minors and one an adult. Of the three minors who died, two died because they did not complete treatment and their disease progressed; one patient with vulvar ERMS who was less than 1 year old, who had a recurrence in the first month postoperatively, changed chemotherapy regimens, supplemental radiotherapy, and had a second surgery, still died at 22 months postoperatively. The other four minor patients who completed treatment survived tumor-free. The prognosis of the patients who completed treatment was consistent with the literature. Among the adult patients, one adult patient with combined embryonal carcinoma developed lung metastasis and lymph node metastasis 5 months after surgery and died 17 months after surgery. Of the remaining adult patients, 5 survived tumor-free and 1 was lost to follow-up. Of note is the case of a 21-year-old patient with ovarian ERMS, stage IV high-risk, who remained tumor-free and survived 36 months postoperatively, probably thanks to the patient’s residual-free surgical treatment and removal of metastatic lesions in the abdominopelvic cavity.
In this article, 7 of our 13 cases were adult ERMS patients, which can provide data for the study of adult rhabdomyosarcoma. However, our study has some limitations, such as a small number of cases, a low percentage of patients with genetic testing, a short follow-up period, and a lack of data on genetic counseling. For diagnostic treatment and stratified management of adults, more clinical data need to be collected and studies need to be analyzed.
ERMS of the female reproductive system is a rare malignant tumor, especially rare in adults, and is challenging to diagnose and treat. Surgery supplemented with chemotherapy, with or without radiation therapy, is the mainstay of treatment. Tumor size, tumor site, presence of metastases, and the completeness of surgery affect the patient’s prognosis. Of all rhabdomyosarcomas, embryonic rhabdomyosarcomas have a relatively good prognosis, but adult patients have a poorer prognosis. In patients with DICER1 and other gene mutations, the possibility of a tumor syndrome should be considered and genetic counseling should be done. In RMS, age-specific heterogeneity, race-specific heterogeneity, and new tissue molecular subtypes are still being studied and refined. More research is still needed on treatment strategies for adult rhabdomyosarcoma patients with poor prognosis.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
The studies involving humans were approved by Medical Research Ethics Committee of West China Second University Hospital, Sichuan University. The studies were conducted in accordance with the local legislation and institutional requirements.
LB: Data curation, Formal analysis, Investigation, Methodology, Resources, Software, Writing – original draft, Writing – review & editing. LH: Supervision, Conceptualization, Project administration, Validation, Investigation, Visualization, Writing – original draft, Writing – review & editing. LS: Conceptualization, Investigation, Project administration, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. JZ: Data curation, Formal analysis, Resources, Supervision, Writing – original draft, Writing – review & editing. YC: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Project administration, Resources, Software, Supervision, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research and/or publication of this article.
Sincere gratitude is extended to the Department of Pathology, West China Second University Hospital, Sichuan University, for their provision of pathological images and associated pathological data.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ferrari A, Dileo P, Casanova M, Bertulli R, Meazza C, Gandola L, et al. Rhabdomyosarcoma in adults. A retrospective analysis of 171 patients treated at a single institution. Cancer. (2003) 98:571–80. doi: 10.1002/cncr.11550
2. Ognjanovic S, Linabery AM, Charbonneau B, Ross JA. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975-2005. Cancer. (2009) 115:4218–26. doi: 10.1002/cncr.24465
3. Hawkins DS, Spunt SL, Skapek SX. COG Soft Tissue Sarcoma Committee. Children's Oncology Group's 2013 blueprint for research: Soft tissue sarcomas. Pediatr Blood Cancer. (2013) 60:1001–8. doi: 10.1002/pbc.24435
4. Egas-Bejar D, Huh WW. Rhabdomyosarcoma in adolescent and young adult patients: current perspectives. Adolesc Health Med Ther. (2014) 5:115–25. doi: 10.2147/AHMT.S44582
5. Parham DM, Rudzinski ER. Rhabdomyosarcoma & Embryonal Rhabdomyosarcoma]In WHO Classification of Tumours Editorial Board. (Ed.), WHO Classification of Tumours: Soft Tissue and Bone Tumours (2020) (5th ed, pp198 – 204). World Health Organization.
6. Agaram NP. Evolving classification of rhabdomyosarcoma. Histopathology. (2022) 80:98–108. doi: 10.1111/his.14449
7. Rudzinski ER, Kelsey A, Vokuhl C, Linardic CM, Shipley J, Hettmer S, et al. Pathology of childhood rhabdomyosarcoma: A consensus opinion document from the Children's Oncology Group, European Pediatric Soft Tissue Sarcoma Study Group, and the Cooperative Weichteilsarkom Studiengruppe. Pediatr Blood Cancer. (2021) 68:e28798. doi: 10.1002/pbc.28798
8. Croce S, Devouassoux-Shisheboran M, Pautier P, Ray-Coquard I, Treilleux I, Neuville A, et al. Uterine sarcomas and rare uterine mesenchymal tumors with Malignant potential. Diagnostic guidelines of the French Sarcoma Group and the Rare Gynecological Tumors Group. Gynecol Oncol. (2022) 167:373–89. doi: 10.1016/j.ygyno.2022.07.031
9. Gerharz CD, Gabbert H, Moll R, Mellin W, Engers R, Gabbiani G. The intraclonal and interclonal phenotypic heterogeneity in a rhabdomyosarcoma cell line with abortive imitation of embryonic myogenesis. Virchows Arch B Cell Pathol Incl Mol Pathol. (1988) 55:193–206. doi: 10.1007/BF02896576
10. Apellaniz-Ruiz M, McCluggage WG, Foulkes WD. DICER1-associated embryonal rhabdomyosarcoma and adenosarcoma of the gynecologic tract: Pathology, molecular genetics, and indications for molecular testing. Genes Chromosomes Cancer. (2021) 60:217–33. doi: 10.1002/gcc.22913
11. Han LM, Weiel JJ, Longacre TA, Folkins AK. DICER1-associated tumors in the female genital tract: molecular basis, clinicopathologic features, and differential diagnosis. Adv Anat Pathol. (2022) 29:297–308. doi: 10.1097/PAP.0000000000000351
12. Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. (2009) 325:965. doi: 10.1126/science.1174334
13. Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer. (2014) 14:662–72. doi: 10.1038/nrc3802
14. de Kock L, Yoon JY, Apellaniz-Ruiz M, Pelletier D, McCluggage WG, Stewart CJR, et al. Significantly greater prevalence of DICER1 alterations in uterine embryonal rhabdomyosarcoma compared to adenosarcoma. Mod Pathol. (2020) 33:1207–19. doi: 10.1038/s41379-019-0436-0
15. Kommoss FKF, Stichel D, Mora J, Esteller M, Jones DTW, Pfister SM, et al. Clinicopathologic and molecular analysis of embryonal rhabdomyosarcoma of the genitourinary tract: evidence for a distinct DICER1-associated subgroup. Mod Pathol. (2021) 34:1558–69. doi: 10.1038/s41379-021-00804-y
16. Bennett JA, Ordulu Z, Young RH, Pinto A, Van de Vijver K, Burandt E, et al. Embryonal rhabdomyosarcoma of the uterine corpus: a clinicopathological and molecular analysis of 21 cases highlighting a frequent association with DICER1 mutations. Mod Pathol. (2021) 34:1750–62. doi: 10.1038/s41379-021-00821-x
17. Bean GR, Anderson J, Sangoi AR, Krings G, Garg K. DICER1 mutations are frequent in müllerian adenosarcomas and are independent of rhabdomyosarcomatous differentiation. Mod Pathol. (2019) 32:280–9. doi: 10.1038/s41379-018-0132-5
18. Bisogno G, Compostella A, Ferrari A, Pastore G, Cecchetto G, Garaventa A, et al. Rhabdomyosarcoma in adolescents: a report from the AIEOP Soft Tissue Sarcoma Committee. Cancer. (2012) 118:821–7. doi: 10.1002/cncr.26355
19. Rogers TN, Dasgupta R. Management of rhabdomyosarcoma in pediatric patients. Surg Oncol Clin N Am. (2021) 30:339–53. doi: 10.1016/j.soc.2020.11.003
20. Lawrence W Jr, Hays DM, Heyn R, Beltangady M, Maurer HM. Surgical lessons from the Intergroup Rhabdomyosarcoma Study (IRS) pertaining to extremity tumors. World J Surg. (1988) 12:676–84. doi: 10.1007/BF01655884
21. Dasgupta R, Rodeberg DA. Update on rhabdomyosarcoma. Semin Pediatr Surg. (2012) 21:68–78. doi: 10.1053/j.sempedsurg.2011.10.007
22. Arndt CA, Stoner JA, Hawkins DS, Rodeberg DA, Hayes-Jordan AA, Paidas CN, et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: children's oncology group study D9803. J Clin Oncol. (2009) 27:5182–8. doi: 10.1200/JCO.2009.22.3768
23. Dixon SB, Bjornard KL, Alberts NM, Armstrong GT, Brinkman TM, Chemaitilly W, et al. Factors influencing risk-based care of the childhood cancer survivor in the 21st century. CA Cancer J Clin. (2018) 68:133–52. doi: 10.3322/caac.21445
24. Malempati S, Hawkins DS. Rhabdomyosarcoma: review of the Children's Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Blood Cancer. (2012) 59:5–10. doi: 10.1002/pbc.24118
25. Bradley JA, Kayton ML, Chi YY, Hawkins DS, Tian J, Breneman J, et al. Treatment approach and outcomes in infants with localized rhabdomyosarcoma: A report from the soft tissue sarcoma committee of the children's oncology group. Int J Radiat Oncol Biol Phys. (2019) 103:19–27. doi: 10.1016/j.ijrobp.2018.08.017
26. van der Graaf WTA, Orbach D, Judson IR, Ferrari A. Soft tissue sarcomas in adolescents and young adults: a comparison with their pediatric and adult counterparts. Lancet Oncol. (2017) 18:e166–75. doi: 10.1016/S1470-2045(17)30099-2
27. Hibbitts E, Chi YY, Hawkins DS, Barr FG, Bradley JA, Dasgupta R, et al. Refinement of risk stratification for childhood rhabdomyosarcoma using FOXO1 fusion status in addition to established clinical outcome predictors: A report from the Children's Oncology Group. Cancer Med. (2019) 8:6437–48. doi: 10.1002/cam4.2504
28. Shern JF, Selfe J, Izquierdo E, Patidar R, Chou HC, Song YK, et al. Genomic classification and clinical outcome in rhabdomyosarcoma: A report from an international consortium. J Clin Oncol. (2021) 39:2859–71. doi: 10.1200/JCO.20.03060
29. De Salvo GL, Del Bianco P, Minard-Colin V, Chisholm J, Jenney M, Guillen G, et al. Reappraisal of prognostic factors used in the European Pediatric Soft Tissue Sarcoma Study Group RMS 2005 study for localized rhabdomyosarcoma to optimize risk stratification and generate a prognostic nomogram. Cancer. (2024) 130:2351–60. doi: 10.1002/cncr.35258
30. Leuschner I, Langhans I, Schmitz R, Harms D, Mattke A, Treuner J, et al. p53 and mdm-2 expression in Rhabdomyosarcoma of childhood and adolescence: clinicopathologic study by the Kiel Pediatric Tumor Registry and the German Cooperative Soft Tissue Sarcoma Study. Pediatr Dev Pathol. (2003) 6:128–36. doi: 10.1007/s10024-001-0097-z
31. Parham DM, Barr FG. Classification of rhabdomyosarcoma and its molecular basis. Adv Anat Pathol. (2013) 20:387–97. doi: 10.1097/PAP.0b013e3182a92d0d
32. Lawrence W Jr, Anderson JR, Gehan EA, Maurer H. Pretreatment TNM staging of childhood rhabdomyosarcoma: a report of the Intergroup Rhabdomyosarcoma Study Group. Children's Cancer Study Group. Pediatric Oncology Group. Cancer. (1997) 80:1165–70. doi: 10.1002/(SICI)1097-0142(19970915)80:6<1165::AID-CNCR21>3.0.CO;2-5
33. Esnaola NF, Rubin BP, Baldini EH, Vasudevan N, Demetri GD, Fletcher CD, et al. Response to chemotherapy and predictors of survival in adult rhabdomyosarcoma. Ann Surg. (2001) 234:215–23. doi: 10.1097/00000658-200108000-00012
34. Little DJ, Ballo MT, Zagars GK, Pisters PW, Patel SR, El-Naggar AK, et al. Adult rhabdomyosarcoma: outcome following multimodality treatment. Cancer. (2002) 95:377–88. doi: 10.1002/cncr.10669
35. Sultan I, Qaddoumi I, Yaser S, Rodriguez-Galindo C, Ferrari A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol. (2009) 27:3391–7. doi: 10.1200/JCO.2008.19.7483
36. Bergamaschi L, Bertulli R, Casanova M, Provenzano S, Chiaravalli S, Gasparini P, et al. Rhabdomyosarcoma in adults: analysis of treatment modalities in a prospective single-center series. Med Oncol. (2019) 36:59. doi: 10.1007/s12032-019-1282-0
37. De Vita A, Ferrari A, Miserocchi G, Vanni S, Domizio C, Fonzi E, et al. Identification of a novel RAB3IP-HMGA2 fusion transcript in an adult head and neck rhabdomyosarcoma. Oral Dis. (2022) 28:2052–4. doi: 10.1111/odi.14036
38. De Vita A, Vanni S, Fausti V, Cocchi C, Recine F, Miserocchi G, et al. Deciphering the genomic landscape and pharmacological profile of uncommon entities of adult rhabdomyosarcomas. Int J Mol Sci. (2021) 22:11564. doi: 10.3390/ijms222111564
Keywords: embryonal rhabdomyosarcoma, DICER1, female reproductive system, clinicopathology, immunohistochemistry
Citation: Bai L, Han L, Sun L, Zou J and Chen Y (2025) Clinicopathological analysis of 13 patients with embryonal rhabdomyosarcoma of the female reproductive system in the Chinese population. Front. Oncol. 15:1546607. doi: 10.3389/fonc.2025.1546607
Received: 17 December 2024; Accepted: 17 February 2025;
Published: 14 March 2025.
Edited by:
Jian-Jun Wei, Northwestern University, United StatesReviewed by:
Alessandro De Vita, Scientific Institute of Romagna for the Study and Treatment of Tumors (IRCCS), ItalyCopyright © 2025 Bai, Han, Sun, Zou and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Zou, aHVheGlwYXRoempAMTYzLmNvbQ==; Yali Chen, WWFsaWNoZW4xODJAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.