 Caihong Li
Caihong Li Dongkai Cheng
Dongkai Cheng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 11 February 2025
Sec. Cancer Molecular Targets and Therapeutics
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1542811
Prostate cancer is the most common cancer among men worldwide, especially in those over 65, and is a leading cause of cancer-related mortality. The disease typically advances from an androgen-dependent state to castration-resistant prostate cancer (CRPC), which poses significant treatment challenges. The androgen receptor (AR) on the X chromosome is a central driver in this process, activating genes that govern proliferation and survival. Mutations and amplifications of the AR are closely associated with disease progression and treatment resistance. While traditional therapies such as androgen deprivation therapy (ADT) and AR antagonists like enzalutamide have been effective, resistance persists due to reactivation of AR signaling through mechanisms like ligand-independent activation. Recent research highlights the role of epigenetic modifications in enhancing AR activity and drug resistance. The tumor microenvironment, particularly interactions with cancer-associated fibroblasts (CAFs) and tumor-associated macrophages (TAMs), further complicates treatment by promoting aggressive tumor behavior and immune evasion. Future directions include developing next-generation AR antagonists, identifying AR-related biomarkers for personalized therapy, and exploring combinations with immune checkpoint inhibitors. Additionally, basal cell-lumen-derived organoids provide innovative models that can enhance understanding and treatment strategies in prostate cancer.
Prostate cancer is the second most common cancer type among men globally, accounting for 7.3% of all new cancer cases and ranking as one of the leading causes of cancer-related death in males, coming in fifth place (1). Its incidence significantly increases with age, particularly among men over 65 years old (2). Studies show that prostate cancer is a highly heterogeneous disease, with its progression typically evolving from androgen-dependent cancer to castration-resistant prostate cancer (CRPC), which is the most difficult stage of the disease to treat (3, 4).
The androgen receptor (AR) is a central driving force in the incidence and progression of prostate cancer. It activates the expression of related genes by binding to androgens (such as testosterone and dihydrotestosterone), thereby regulating cell proliferation, differentiation, and survival (5). The AR protein consists of 919 amino acids and can be structurally divided into four domains: the N-terminal domain (NTD), which is responsible for transcriptional activation; the DNA-binding domain (DBD), which mediates gene-specific binding; the hinge region (which regulates nuclear localization and stability); and the ligand-binding domain (LBD), which binds to androgens (6). The AR gene is located on the X chromosome and consists of eight exons; mutations or amplifications of this gene are often closely associated with the progression of prostate cancer and treatment resistance (7–9).
Traditional treatment strategies focus on blocking the binding of AR to androgens or inhibiting the transcriptional activity of AR, including androgen deprivation therapy (ADT) and second-generation AR antagonists (such as enzalutamide and apalutamide) (10). These treatment methods have significantly extended the progression-free survival of patients. However, the issue of resistance remains a major clinical challenge. Research indicates that the reactivation of AR signaling is a primary cause of CRPC development, with mechanisms including AR gene amplification or mutation, ligand-independent activation, bypass signaling pathway compensation, and the production of AR splice variants (such as AR-V7) (11, 12). Notably, AR-V7 is a variant that lacks the ligand-binding domain and can activate downstream signaling pathways even in the absence of androgens, making it a critical driver of CRPC resistance (13).
In recent years, studies in epigenetics have provided new perspectives for understanding the regulatory mechanisms of AR and drug resistance (14, 15). Research by Nguyen et al. found that H2AK130ac is enriched in the promoter regions of key androgen synthesis genes (such as CYP17A1) in prostate cancer cells, correlating with their high expression levels (16). Additionally, RNA modifications (such as m6A methylation) have also been shown to play a significant role in the stability and translation efficiency of AR mRNA, providing a foundation for potential therapeutic targets (17, 18). More importantly, by integrating multi-omics technologies, researchers are attempting to develop novel combination therapy strategies that target key nodes in the AR network, such as the co-inhibition of AR signaling and the PI3K/Akt pathway (19).
In CRPC, specific epigenetic changes play a crucial role in the reactivation of AR signaling (20). DNA methylation changes, particularly hypomethylation in the promoter regions of AR target genes, can lead to increased gene expression, facilitating AR reactivation even during androgen-deprivation therapy. Histone modifications, such as altered acetylation and methylation, can modify chromatin accessibility, influencing AR transcriptional activity. Increased histone acetylation at AR target sites, for instance, can enhance AR binding and transcriptional activation, promoting cancer progression despite low androgen levels. Moreover, non-coding RNAs, including microRNAs and long non-coding RNAs (lncRNAs), can regulate AR expression and activity by interacting with AR mRNA or influencing chromatin states, contributing further to AR reactivation in CRPC.
Targeting histone modifications or RNA methylation pathways holds promise but presents challenges due to the genetic heterogeneity of CRPC (21). The effectiveness of such approaches can vary across different genetic profiles, as certain modifications may be more prevalent or impactful in specific genetic contexts. While drugs targeting histone modifiers, such as Histone Deacetylase(HDAC) or Histone Methyltransferase(HMT) inhibitors, or RNA methylation processes could have broader applications, their success largely depends on identifying patient subgroups most likely to benefit from these treatments (22). This underscores the potential for employing personalized strategies that integrate genomic and epigenomic data to enhance treatment efficacy. Additionally, the possibility of resistance necessitates the exploration of combination therapies that simultaneously target multiple pathways involved in AR reactivation and CRPC progression.
In summary, as a core molecular target in prostate cancer, AR plays an indispensable role in the occurrence, progression, and treatment resistance of the disease. In-depth research into the regulatory mechanisms of AR signaling, combined with the latest findings from molecular biology and epigenetics, can enhance our understanding of the mechanisms underlying CRPC development and provide new directions and ideas for its diagnosis and treatment. This article will focus on the molecular regulatory mechanisms of AR and the latest research progress in prostate cancer treatment.
Androgens bind to the LBD of AR, inducing a conformational change that enables AR to translocate from the cytoplasm to the nucleus with the assistance of chaperone proteins, such as heat shock proteins (HSPs). These chaperones stabilize AR and facilitate its interaction with cofactors during nuclear import. Once in the nucleus, AR dimerizes and binds to androgen response elements (AREs) in the promoter regions of target genes, promoting their transcription (23).
AR enhances the expression of genes associated with cell growth, differentiation, apoptosis, and metabolism by recruiting transcription co-factors, such as members of the SRC family and CBP/p300, which form transcriptional complexes that enhance AR-mediated transcriptional activity (24, 25). Additionally, AR synergistically interacts with transcription factors like c-Jun, NF-κB, and SMAD3, alongside cofactors such as CBP and PCAF, to modulate the expression of key genes involved in sexual differentiation (e.g., SRY), gonadal function, and pubertal maturation (Figure 1) (26). For example, Cyclin D1 and Cyclin E are upregulated by AR, facilitating cell cycle progression by promoting the G1 to S phase transition. Moreover, AR regulates the expression of anti-apoptotic factors, such as Bcl-2 and Bcl-xL, while inhibiting pro-apoptotic genes like P53, thereby ensuring continuous cell survival in normal and malignant cells (27).
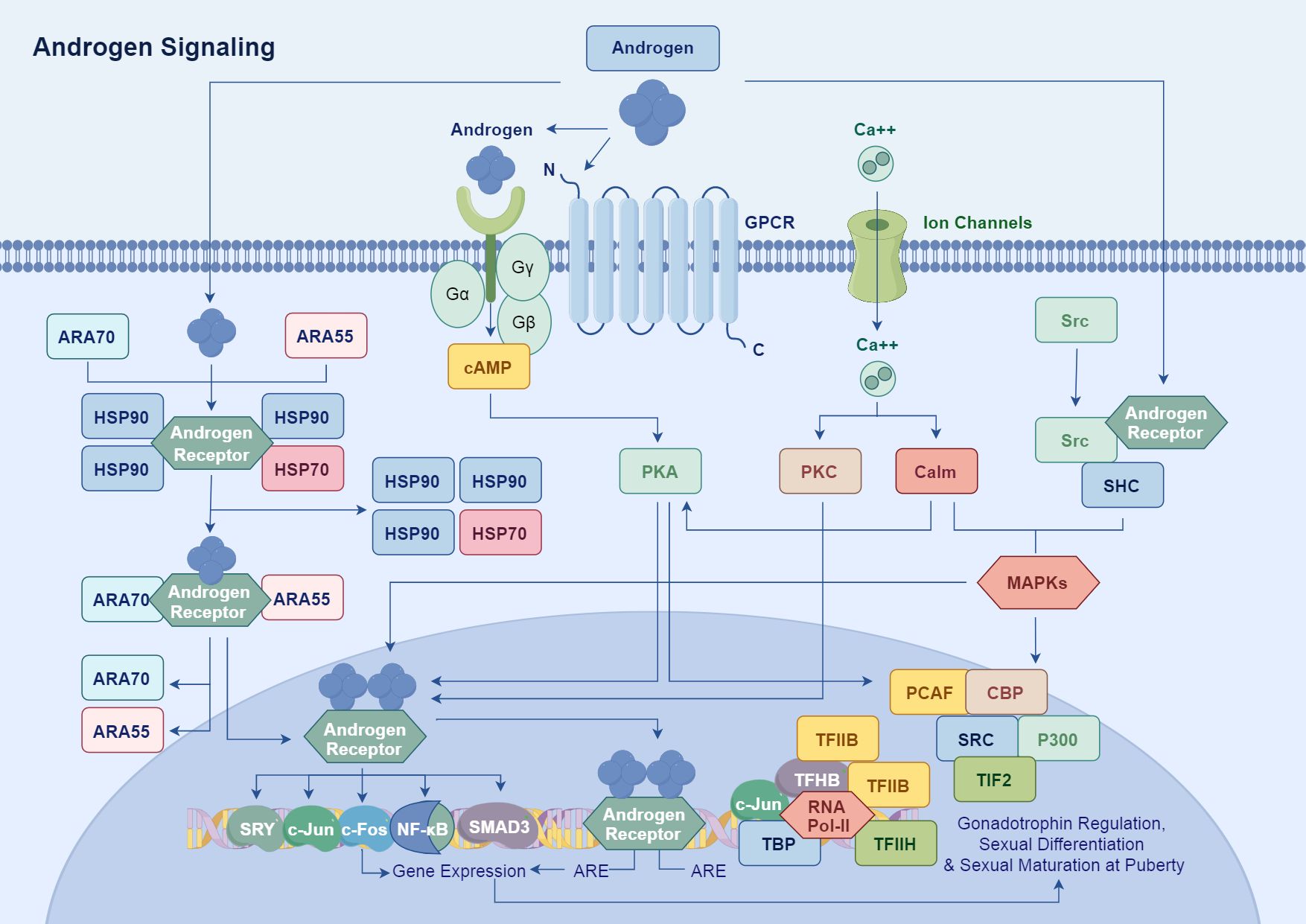
Figure 1. Androgens bind to the LBD of AR, inducing conformational changes in AR. This process allows AR to translocate from the cytoplasm to the nucleus, assisted by chaperone proteins. In the nucleus, AR forms a dimer and binds to ARE in the promoter regions of target genes, thereby promoting their transcription. AR enhances the expression of genes related to cell growth, differentiation, apoptosis, and metabolism by recruiting transcriptional co-factors such as the SRC family and CBP/p300 to form transcriptional complexes.
Androgen signaling also amplifies cellular responses through interactions with other signaling pathways, including MAPK and Src, thereby achieving comprehensive regulation of gonadal function, sexual differentiation, and pubertal maturation (28). Specifically, AR has been shown to interact with growth factor receptors like EGFR and TrkA, as well as with metalloproteases in prostate cancer, which can influence AR signaling dynamics and enhance tumor progression (29–32).
Understanding the nuances of AR signaling is critical, particularly regarding how the modulation of Cyclin D1 and Bcl-2 contributes to prostate cancer development and progression. These factors are essential, as Cyclin D1 is pivotal for cell cycle regulation, while Bcl-2 plays a crucial role in promoting cell survival, thus highlighting the importance of aberrant AR signaling in the pathology of prostate cancer.
Androgens are important factors for the growth of prostate cancer cells, and prostate cancer typically develops under the influence of androgen signaling. In the early stages, once androgens bind to AR, AR translocates to the nucleus and binds to specific enhancer elements, initiating the transcription of a series of genes related to cell proliferation and survival, including MYC and KLK3 (prostate-specific antigen) (33). The activation of this AR signaling pathway effectively promotes the growth of prostate cancer cells.
As the disease progresses, the expression levels and function of AR undergo significant changes, usually leading to a transition from an androgen-dependent state to an androgen-independent state (34). Throughout the development of prostate cancer, AR expression levels tend to gradually increase, particularly in CRPC, where the expression of AR protein significantly rises. This increase in expression is viewed as an adaptive survival strategy of tumor cells in a low-androgen environment (35).
Studies have shown that compared to hormone-sensitive prostate cancer, AR expression is notably upregulated in CRPC tissues, and this change is closely associated with enhanced proliferation capacity of tumor cells and treatment resistance (36). The elevated AR expression enables tumor cells to remain sensitive to low concentrations of androgens, maintaining the activity of the AR signaling pathway even in conditions of extremely low exogenous androgen supply. This mechanism plays a critical role in AR-mediated tumor signaling, further driving the malignant progression of the tumor.
AR not only influences the growth of tumor cells themselves but also affects tumor development and progression by regulating immune cell responses in the tumor microenvironment (37). Changes in the microenvironment are primarily reflected in several aspects:
First, tumor cells can secrete various cytokines (such as IL-6, IL-10, and TNF-α), and the changes in the expression of these cytokines not only affect the infiltration and activity of leukocytes but also promote alterations in the biochemical properties of surrounding fibroblasts. Second, tumor cells can induce an immunosuppressive microenvironment, making it difficult for the immune system to effectively attack the tumor (38). Tumor-associated macrophages (TAMs) typically exhibit characteristics that suppress T cell activity (39). Additionally, the interactions between tumor cells and the surrounding stroma enhance the invasive ability of the tumor, and changes in the stroma are also key factors in tumor metastasis.
TAMs and other immune components significantly contribute to treatment resistance in cancer by interacting with the AR (40). TAMs secrete various pro-inflammatory cytokines and growth factors, such as IL-6 and IGFs, which can activate the AR signaling pathway, promoting cancer cell growth and enhancing resistance to therapies, including antiandrogen treatment. Besides, TAMs and other immunosuppressive cells, such as myeloid-derived suppressor cells, create an immunosuppressive microenvironment that diminishes the effectiveness of antitumor immune responses, enabling tumor cells to evade control and develop resistance to treatments. Modulating cytokines like IL-6 and TNF-α could potentially shift the tumor microenvironment to support AR-targeting therapies. By regulating these cytokines, the tumor-promoting activities of TAMs might be reduced, and antitumor immune responses could be restored. Moreover, combining cytokine inhibitors with AR-targeting therapies could enhance their efficacy by mitigating the pro-inflammatory and immunosuppressive effects of the cytokines. Such modulation may also improve immune cell functionality, thus enhancing their antitumor activity and synergistically increasing the effectiveness of AR-targeting therapies.
AR also interacts with signaling pathways such as PI3K/AKT/mTOR, Wnt/β-catenin, and NF-κB, regulating cell proliferation and survival. Among these, the PI3K/AKT/mTOR signaling pathway plays a crucial role in promoting survival in prostate cancer, with its activation often accompanied by the loss of PTEN. Research indicates that there is a bidirectional regulation between the PI3K/AKT/mTOR pathway and AR signaling. Carver et al. found in mouse models that the activation of the PI3K pathway can inhibit AR expression through negative feedback, while inhibition of the PI3K pathway can upregulate AR expression (19). Furthermore, the PI3K/AKT signaling pathway can enhance the transcriptional activity of the androgen receptor, promoting tumor cell dependence on AR signaling. This reciprocal regulatory mechanism may be an important reason for treatment resistance in CRPC.
The Wnt/β-catenin signaling pathway is considered a key pro-cancer signal in prostate cancer that can interact synergistically with AR signaling to promote tumor cell proliferation and metastasis. A study by Yuan et al. demonstrated that Wnt signaling activation can enhance the interaction between β-catenin and AR by stabilizing β-catenin, thereby increasing AR’s transcriptional activity on downstream genes (41). This synergistic effect not only drives tumor cell proliferation but may also increase treatment resistance by enhancing tumor heterogeneity.
The NF-κB signaling pathway is an important regulatory factor of inflammation in the tumor microenvironment and can interact with AR signaling through various mechanisms. Thapa et al. found that NF-κB signaling activation not only directly upregulates the expression of the AR gene but can also enhance AR’s transcriptional activity by inducing the expression of co-factors that bind to AR, such as p300 (42). This interaction is particularly significant in the tumor inflammatory environment and may promote disease progression by enhancing the survival capacity of tumor cells and their immune evasion mechanisms.
Perineural invasion (PNI) is a common feature of prostate cancer, particularly evident in more aggressive tumors (43). The presence of PNI is typically associated with adverse clinical outcomes, such as an increased risk of tumor recurrence and a reduced survival rate for patients, which makes it an important prognostic marker. The microenvironment created by PNI provides favorable conditions for the survival, proliferation, and invasion of cancer cells, thereby influencing their biological behavior. Furthermore, the molecular mechanisms involved in the development of PNI primarily focus on the signaling pathways and interactions between cancer cells and nerve cells, including neurotrophic factor (NGF) and its receptors, β-adrenergic receptors, chemokines, and neural cell adhesion molecules (44). The dissemination of cancer cells around nerves is not merely a passive phenomenon; rather, it results from the complex interactions among cancer cells, nerve cells, and other components of the tumor microenvironment.
The progression of prostate cancer is significantly influenced by TME, particularly the role of cancer-associated fibroblasts (CAFs) (45). Recent studies highlight that CAFs express a transcriptionally inactive AR, which critically mediates their migration and invasiveness (46). Although androgen receptor signaling is essential for prostate cancer progression, its complex interplay with CAFs, including the binding of the AR to the scaffold protein filamin A, enhances CAF migration toward prostate cancer epithelial cells and increases tumor organoid size in both 2D and 3D cultures. This mechanism also results in extracellular matrix remodeling facilitated by a protease cascade activated through the AR/filamin A interaction and β1 integrin (47). Furthermore, androgen stimulation induces unique chromatin binding events in AR-expressing CAFs, altering cytokine production and promoting prostate cancer cell migration through paracrine signaling. Importantly, ADT affects AR expression in the stroma, contributing to the progression of the disease by dysregulating extracellular matrix protein levels and promoting a metastatic microenvironment. Disruption of the AR/filamin A complex using a stapled peptide effectively reduces CAF invasiveness and tumor growth in co-culture models, suggesting the potential for targeting this complex as a therapeutic strategy to enhance treatment outcomes in prostate cancer (48, 49). Overall, understanding the molecular cross-talk between AR signaling in CAFs and prostate cancer cells could pave the way for novel therapeutic approaches to combat drug resistance and metastatic disease in prostate cancer (Figure 2).
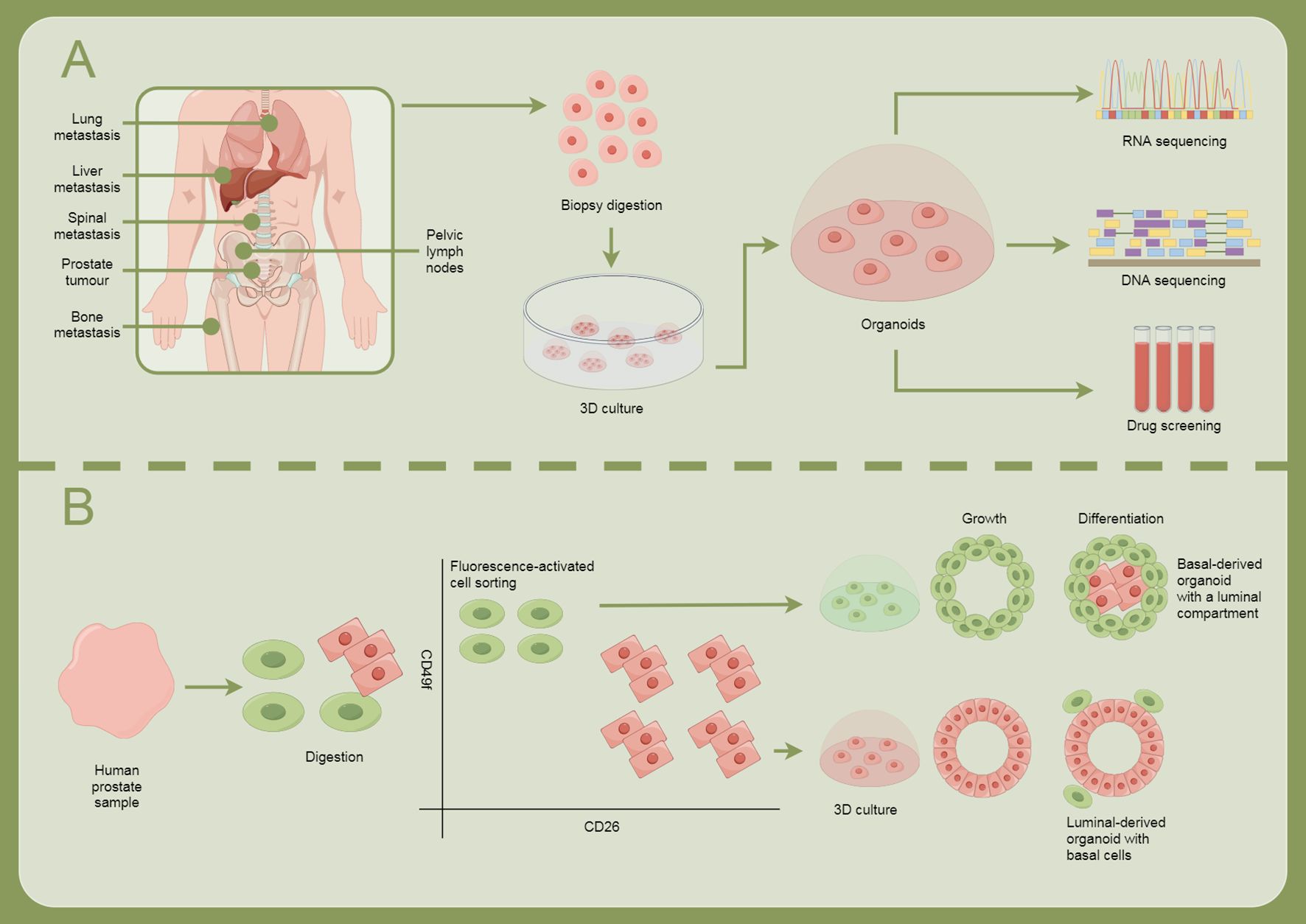
Figure 2. Illustrates how cellular models are used for research and treatment exploration in prostate cancer. (A) Prostate cancer can metastasize to sites such as the lungs, liver, spine, bones, and pelvic lymph nodes. The process involves biopsy digestion, 3D culture, organoid formation, RNA and DNA sequencing, and drug screening. (B) Human prostate samples are obtained, followed by digestion, fluorescence-activated cell sorting, and growth and differentiation, forming different types of organoids: basal-derived organoids with luminal structures, and luminal-derived organoids containing basal cells.
ADT is the mainstay treatment for metastatic prostate cancer; however, most patients eventually progress to CRPC, which exhibits poor treatment responses (50). In recent years, AR mutations have been recognized as one of the significant mechanisms underlying CRPC, primarily focusing on LBD of the androgen receptor, as this region directly participates in binding androgens or anti-androgen drugs. Various AR gene mutations are closely associated with resistance to CRPC and affect tumor cell growth and treatment response through different mechanisms (Table 1).

Table 1. The summary of androgen receptor (AR) mutation types, mutation sites, and their related therapeutic drugs.
The L701H mutation allows AR to activate downstream genes even in the absence of androgens, leading to resistance to androgen deprivation therapy (51). The T877A mutation enhances AR activity in an androgen-deprived environment, promoting the continuous growth of prostate cancer cells (51, 53). The F877L mutation alters the conformation of AR when binding with drugs, activating downstream signaling pathways and helping the tumor evade anti-androgen therapy (19). Similar to T877A, the H874Y mutation enables AR to function under androgen-deficient conditions, facilitating tumor resistance to treatment (54). The F876L mutation decreases the binding stability of anti-androgen drugs with AR, reducing the effectiveness of anti-androgen therapy (55).
Additionally, variations in the CAG repeat number in the first exon of the AR gene may also impact AR activity, with shorter CAG repeats being associated with an increased risk of prostate cancer (56). The W742C mutation is closely related to resistance to anti-androgen drugs (57). These mutation mechanisms reveal the complexity of prostate cancer treatment and provide important insights for personalized therapeutic strategies.
The existing knowledge of AR mutations, such as L701H and T877A, can be effectively utilized for precise drug design through various advanced methods. Structural biology research, including X-ray crystallography and nuclear magnetic resonance (NMR), elucidates the three-dimensional structures of AR mutants, revealing how these mutations alter the shape and charge distribution of the ligand-binding pocket, thereby affecting drug binding (58). Molecular dynamics simulations further aid in predicting optimal drug binding modes by studying the dynamic behavior of these mutants when bound to different ligands. Computer-aided drug design (CADD) is employed in virtual screening to identify candidate compounds capable of effectively targeting these mutants (59). Additionally, quantitative structure-activity relationship (QSAR) models are developed to predict the activity of new molecules on mutant ARs and optimize their chemical structures for improved efficacy (60).
High-throughput screening platforms enable the rapid identification of small molecules from large compound libraries that interact effectively with specific AR mutants. Based on screening results, directed chemical synthesis is conducted to chemically modify and optimize these molecules, enhancing their affinity and selectivity for AR mutants. Biological function validation is critical, involving testing the efficacy of candidate drugs in cell lines with specific AR mutations to assess their impact on AR activity inhibition and cancer cell proliferation. In vivo efficacy and safety assessments are conducted using mouse xenograft models harboring AR mutations, such as those found in CRPC.
In the context of personalized medicine, gene testing and drug matching utilize patients’ genomic information to determine specific AR mutation types, enabling the design of personalized treatment plans to select the most effective drugs. Furthermore, the identification of undiscovered AR mutations that are crucial for drug resistance is of utmost importance. In prostate cancer research, particularly in CRPC, there may be unknown AR mutations that play significant roles in drug resistance. High-throughput genomic sequencing broadens whole-genome or whole-exome sequencing to a larger patient population to uncover new AR mutations, which, although present in only a few patients, could be critical for drug resistance (61).
Functional validation of all identified mutations determines their impact on AR activity and drug resistance through in vitro experiments, cell line models, or animal models (62). Bioinformatics and machine learning tools are utilized to search for mutation patterns potentially associated with drug resistance from large datasets, identifying subtle yet important mutations often overlooked by traditional methods. Patient-derived models, such as organoids or xenografts, are employed for pharmacological testing to identify mutations that may lead to drug resistance, more accurately reflecting patient biological contexts. Finally, studying mutation cooperativity reveals how interactions between multiple gene mutations might also contribute to drug resistance, assisting in identifying new resistance mechanisms and addressing the challenges posed by AR mutations in prostate cancer treatment comprehensively.
In the mechanism of AR resistance, cellular metabolic reprogramming plays a crucial role in tumor cell adaptation to therapeutic stress, involving significant alterations in glucose metabolism, lipid metabolism, and amino acid metabolism (63). In glucose metabolism, resistant cells maintain energy supply by enhancing glycolysis while suppressing oxidative phosphorylation to adapt to oxidative stress (64). In terms of lipid metabolism, fatty acid synthesis and oxidation are activated to support rapid cell proliferation and improve antioxidant capacity. Research by Swinnen et al. has shown that AR-resistant tumor cells promote lipid synthesis by upregulating the expression of fatty acid synthase (FASN) and ATP-citrate lyase (ACL), thereby meeting the demands of rapid proliferation and membrane structure (65).
In amino acid metabolism, the reprogramming of glutamine metabolism and one-carbon metabolism provides energy, intermediate metabolites, and regulatory abilities for epigenetic modifications (66). These metabolic pathways work synergistically to support the growth, invasion, and immune evasion of resistant tumors. In the future, combining metabolomics with targeted metabolic inhibitors in combination therapy may help overcome AR resistance and optimize treatment strategies for prostate cancer.
Metabolic pathways, including lipid and amino acid metabolism, play a crucial role in sustaining AR-independent CRPC survival. CRPC cells can satisfy their energy requirements for growth and survival by enhancing fatty acid synthesis and β-oxidation, as well as altering cholesterol and other lipid metabolism to support membrane construction and signaling molecule production, enabling continued survival without androgen stimulation (67). Additionally, these cells reprogram amino acid metabolism by increasing glutamine utilization, which provides essential carbon and nitrogen for cell growth and supports antioxidant defenses to protect against oxidative stress. The potential for combined metabolic pathway inhibitors to enhance AR-targeting therapies lies in their ability to create therapeutic synergy. By inhibiting key metabolic pathways such as fatty acid synthesis or glutamine metabolism, the survival capabilities of CRPC cells can be weakened, increasing their sensitivity to AR-targeting therapies. This combination approach may effectively reduce the energy supply to tumors, thus inhibiting their growth. However, a significant challenge is reducing toxicity; it is crucial to develop highly selective metabolic inhibitors that target tumor metabolism without adversely affecting normal cell function. Optimizing dosing and administration regimens is essential to avoid unacceptable toxicity while ensuring the efficacy of the treatment.
The activation of bypass signaling pathways refers to tumor cells supporting their proliferation and survival through the activation of other growth factor signaling pathways, such as the PI3K/Akt/mTOR pathway, MAPK pathway, Wnt/β-catenin pathway, and IL-6/JAK/STAT3 pathway, even in the absence of AR or androgen signaling (68). These pathways can drive tumor growth independently of AR signaling. Carver et al. found that there is a reciprocal inhibitory relationship between AR and the PI3K/Akt/mTOR pathway; when AR is inhibited, the PI3K/Akt pathway is released and overly activated, promoting cell survival (19). Pencik et al. observed in PTEN-deficient mouse models that the loss of STAT3 led to decreased levels of LKB1 and phosphorylated AMPK (pAMPK), while simultaneously activating the mTOR/CREB pathway, accelerating the development of metastatic disease (69). In the context of AR resistance, the PI3K/Akt/mTOR, MAPK, Wnt/β-catenin, and IL-6/JAK/STAT3 pathways sustain tumor cell survival and progression through multi-layered regulation, highlighting the importance of the cooperation among multiple pathways as new therapeutic targets for metastatic prostate cancer and resistance.
Multi-omics technologies, which integrate genomic, transcriptomic, proteomic, and metabolomic data, enable the identification of key molecules and interactions within AR signaling pathways and compensatory pathways like PI3K/Akt (70). This comprehensive approach allows researchers to detect mutations, changes in gene expression, and metabolic reprogramming within these pathways, leading to the development of more precise drug targets. Additionally, by integrating multi-omics data, researchers can uncover how prostate cancer cells develop resistance to AR-targeted therapies by activating alternative pathways such as PI3K/Akt, providing crucial insights for designing more effective combination therapies. Multi-omics analysis also supports predicting patient responses to combination therapies: (1) Personalized Treatment Decisions: Multi-omics analysis provides information on the unique characteristics of each patient’s tumor, such as specific mutations, pathway activation statuses, and microenvironmental factors. This enables customized treatment strategies to enhance the efficacy of combination therapies. (2) Biomarker Development: By identifying biomarkers that predict treatment response, multi-omics technologies can help distinguish which patients are likely to respond well to specific combination therapies, aiding in more precise patient stratification before treatment. (3) Monitoring and Adjusting Treatment Efficacy: Multi-omics approaches allow for dynamic monitoring of molecular characteristics during treatment; for example, detected molecular changes can indicate rapid response or emerging resistance to therapy, enabling timely adjustments to treatment plans and improving long-term outcomes.
In the treatment of CRPC, AR inhibitors represent a significant therapeutic strategy (71). These drugs inhibit AR activity through various mechanisms, significantly improving patient prognosis. Enzalutamide, a non-steroidal AR antagonist, has been approved for CRPC patients and shows survival benefits by blocking the binding of androgens to AR and inhibiting its transcriptional activity (72). Abiraterone acetate lowers the systemic levels of androgens by inhibiting the CYP17 enzyme, making it an essential treatment option for CRPC patients (73). Darolutamide, a novel AR antagonist, demonstrates good efficacy in non-metastatic CRPC (nmCRPC) and exhibits a lower incidence of central nervous system side effects due to its unique molecular structure (19). The successful development and application of these drugs have provided new treatment options for CRPC patients and established new standards for clinical treatment.
Targeting AR splice variants such as AR-V7, which evade current therapies, requires innovative strategies due to the lack of the LBD that traditional AR antagonists target (74). Key approaches focus on identifying unique structural or functional domains specific to AR-V7 to develop targeted molecular inhibitors. Additionally, technologies like small molecules, antisense RNA, or siRNA could interfere with AR-V7 mRNA processing, stability, or translation, reducing its production and activity. Another promising method is utilizing PROTAC (Proteolysis Targeting Chimeras) technology to design molecules that tag AR-V7 for protein degradation, hence lowering its cellular levels (75). However, designing AR antagonists with fewer off-target effects poses several challenges. Achieving selectivity and specificity is crucial, requiring a thorough understanding of AR’s mechanisms to selectively inhibit its functions without impacting other nuclear receptor family members. Moreover, the pharmacokinetics and metabolism of compounds must be meticulously evaluated to ensure stability, proper absorption, favorable distribution, efficient metabolism, and excretion, all while maintaining effective concentrations at target tissues with a safe profile. Finally, reducing potential toxicity and adverse effects is essential to enhance patient tolerance and ensure treatment sustainability, necessitating careful consideration of the drug design and delivery processes.
Sipuleucel-T is a dendritic cell immunotherapy vaccine targeting prostate cancer. It enhances the immune system’s response to tumor cells by increasing the binding of regulatory chemokines to prostate-specific antigen (PSA) (76). As the first dendritic cell-based prostate cancer vaccine approved by the FDA, Sipuleucel-T has shown some clinical efficacy; however, it has not provided significant breakthroughs for many advanced patients, particularly those with CRPC. To overcome the limitations of dendritic cell vaccines, current research is focusing on strategies that combine them with other immunotherapeutic agents (77). For example, the combination of immune checkpoint inhibitors with dendritic cell vaccines is being explored in clinical trials. This combined treatment not only enhances the overall immune response but also effectively overcomes immune suppression in the tumor microenvironment. In addition, adjuvants such as cytokines are being used in conjunction with dendritic cell vaccines to further improve their therapeutic efficacy (78). These multi-pronged strategies suggest that the combination of dendritic cell vaccines and immunotherapy is expected to provide more effective treatment options for prostate cancer patients in the future.
For prostate cancer patients with specific biomarkers, immune checkpoint inhibitors (such as anti-PD-1/PD-L1 antibodies, like Pembrolizumab and Nivolumab) are also undergoing clinical trials (79–81). PARP inhibitors (such as Olaparib and Rucaparib) are used for prostate cancer patients with BRCA1/2 mutations or other DNA repair deficiencies, functioning by interfering with the DNA repair mechanisms of tumor cells (82, 83). Radiopharmaceuticals (such as Radium-223) target metastatic castration-resistant prostate cancer to alleviate pain from bone metastases and extend survival (84). In addition, small molecule targeted drugs against AR and its downstream signaling pathways are also in development. Meng Wu et al. discovered and identified a bifunctional small molecule, Z15, which acts as an effective AR antagonist and a selective AR degrader. Z15 is capable of directly interacting with the LBD of AR and the activation function-1 (AF-1) region, promoting AR degradation through the proteasomal pathway (85).
Researchers are working to develop more efficient and selective AR antagonists to overcome the limitations of existing drugs (86). Precision-targeted therapies against AR-V7 are anticipated to significantly improve treatment outcomes for patients with CRPC, especially for those who have developed resistance to current therapies (87). Non-coding RNAs regulate the activity of the AR signaling pathway through various mechanisms, influencing the onset and progression of prostate cancer (88–92). Firstly, non-coding RNAs can directly bind to AR or regulate its downstream target genes, thereby affecting tumor cell proliferation, invasion, and resistance. Additionally, certain non-coding RNAs can mediate the expression or activity of AR variants (such as AR-V7) in CRPC, resulting in resistance to traditional AR-targeted therapies. Non-coding RNAs also indirectly influence the activity of AR signaling in the tumor microenvironment by regulating immune cell infiltration and inflammatory responses, further promoting tumor progression and resistance (93).
Research by Yaru Xu et al. indicates that Zinc Finger Protein 397 (ZNF397) is a true co-activator of AR, and Ten-eleven translocation 2(TET2) inhibitors can eliminate the resistance of ZNF397-deficient tumors to AR-targeted therapies. This mechanism reveals how prostate cancer can acquire lineage plasticity through epigenetic regulation and offers new clinical intervention strategies to overcome resistance caused by this lineage plasticity (94).
Developing personalized treatment plans based on patients’ genomic and tumor characteristics is expected to enhance treatment efficacy. In prostate cancer research, basal cell-derived organoids serve as an innovative in vitro culture model that effectively simulates the tissue structure and function of the prostate (95). These organoids are not only used to study tumor biology but also provide a new platform for personalized treatment and drug screening. Basal cell-derived organoids offer a highly promising experimental tool for prostate cancer research, helping scientists to better understand the biological characteristics of prostate cancer and facilitating the development and implementation of personalized treatment approaches.
Lumen-forming basal-derived organoids represent an important three-dimensional cell culture model that better mimics the biological features of glands such as the prostate (Figure 3) (96). These organoids can reflect cell growth and differentiation while reproducing complex interactions within tissue structures and the microenvironment, making them particularly suitable for studying diseases like prostate cancer. As a preclinical model, basal-derived organoids have broad applications in drug screening and disease mechanism research (97). For instance, studies on personalized treatment using patient-derived organoids are continuously advancing (98). Additionally, the application of gene editing technologies like CRISPR provides convenience for functional gene studies in organoids, aiding in the in-depth understanding of tumor initiation and progression mechanisms.
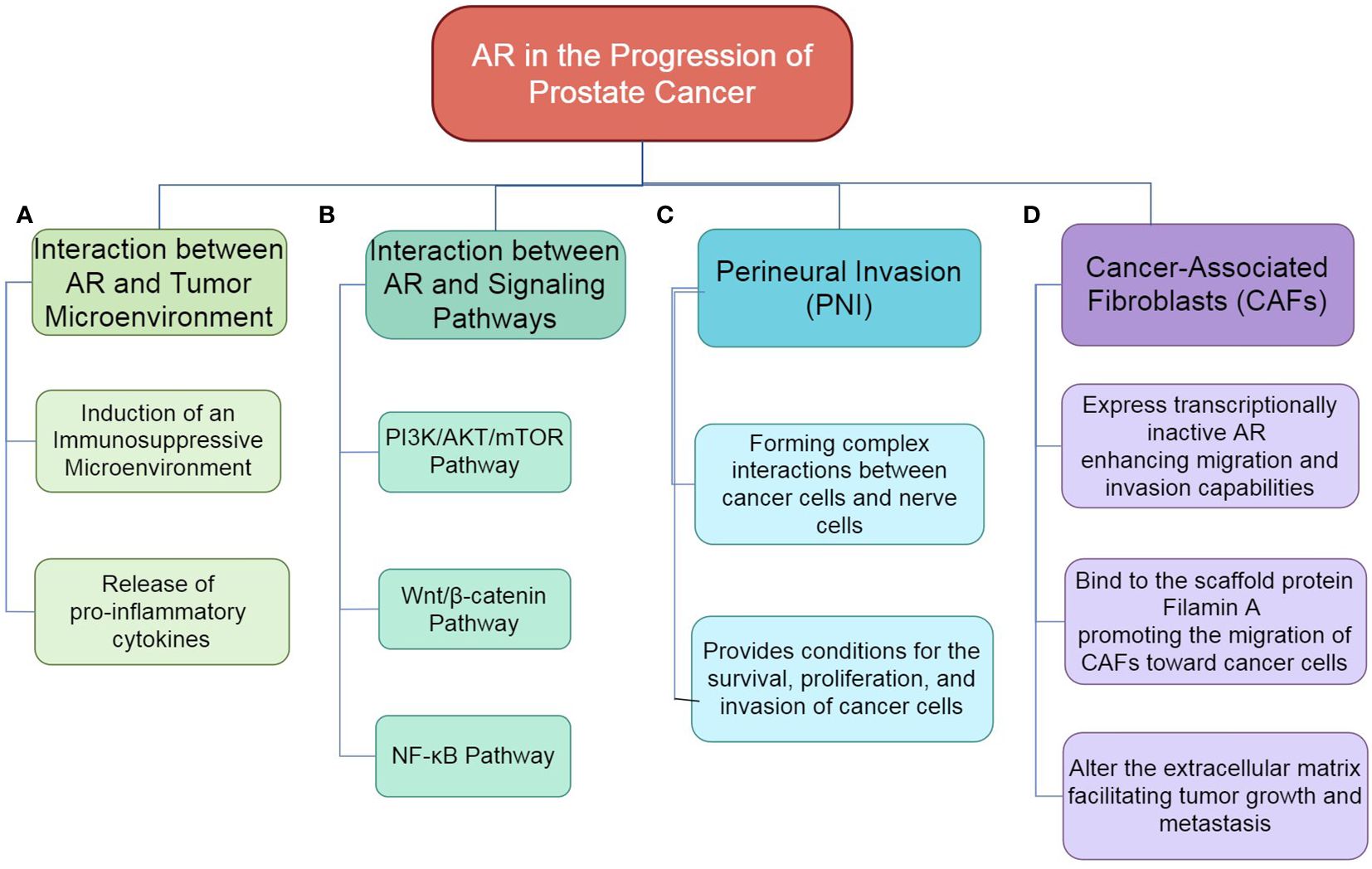
Figure 3. The role of AR in the progression of prostate cancer. (A) Interaction between AR and Tumor Microenvironment: AR influences the tumor microenvironment by mediating interactions with various stromal components, which can promote tumor growth and progression. (B) Interaction between AR and Signaling Pathways: AR interacts with multiple signaling pathways, including PI3K/AKT/mTOR, Wnt/β-catenin and, NF-κB, thereby modulating cellular processes such as proliferation, survival, and invasion. (C) PNI: AR is implicated in the process of perineural invasion, where cancer cells invade surrounding nerve tissues, contributing to tumor spread and associated pain. (D) Role of CAFs: AR within CAFs enhances their migratory and invasive properties, facilitating crosstalk with cancer cells and promoting a supportive microenvironment for tumor progression.
Patient-derived organoids, particularly basal-derived organoids, demonstrate high accuracy in replicating patient-specific tumor responses in drug screening due to their structural and functional similarity to the original tumor tissue, preserving the genomic characteristics and heterogeneity of the patient’s tumor (99). These organoids often provide more accurate predictions of tumor responses because they can replicate complex cancer biology features, such as cell differentiation states and the influence of the tumor microenvironment. However, it is essential to evaluate the stability of these models during long-term culture and understand their differences from the in vivo environment. To integrate organoids into routine clinical practice for therapy decisions in a cost-effective manner, several strategies can be employed: optimizing production processes by standardizing organoid cultivation and analysis to reduce costs and time; applying automation technologies to increase yield and consistency, making organoids more suitable for clinical settings; and utilizing high-throughput screening platforms, which can significantly reduce the time and cost of drug testing, enabling large-scale application. Furthermore, combining organoids with genomic sequencing and other molecular diagnostic tools can quickly identify potential patient responses to specific therapies, offering more cost-effective personalized treatment recommendations.
Non-coding RNAs, particularly long non-coding RNAs (lncRNAs), have emerged as potential therapeutic targets due to their role in regulating the AR signaling pathway and their significant contribution to prostate cancer progression and drug resistance (100). Targeting these molecules could thus represent an innovative therapeutic strategy. To ensure the efficacy and safety of therapies targeting non-coding RNAs, comprehensive studies on biological mechanisms are essential for identifying key targets and developing effective interfering molecules. Additionally, optimized delivery systems need to be established to ensure the effectiveness and specificity of these molecules. in vivo, minimize off-target effects, and enhance treatment safety. For advanced CRPC patients, optimizing immune therapies like checkpoint inhibitors and vaccines such as Sipuleucel-T involves several strategies. The effectiveness of checkpoint inhibitors can be enhanced through combination therapies that amplify immune responses, such as pairing them with conventional therapies or drugs targeting metabolic pathways to counteract the immunosuppressive tumor microenvironment. Vaccines can be improved by selecting more immunogenic target antigens or using immune adjuvants to boost the immune system’s recognition capabilities. Personalized vaccine strategies, which tailor treatments based on each patient’s unique tumor antigen profile, also hold promise. The role of biomarkers is critical, as identifying and validating those predicting treatment efficacy can help in selecting the appropriate patient groups, thereby improving the overall effectiveness of immunotherapy.
Prostate cancer is the second most common cancer among men worldwide, with incidence rates significantly increasing with age. Although androgen deprivation therapy and second-generation AR antagonists have made some progress in improving patient prognosis, the development of CRPC and its resistance remain significant challenges. AR plays a central role in prostate cancer, with the activity of its signaling pathway directly influencing the proliferation and survival of tumor cells. The resistance of CRPC is primarily associated with AR mutations, activation of downstream signaling pathways, and metabolic reprogramming, enabling tumor cells to survive in a low-androgen environment. In recent years, new therapeutic strategies targeting AR have emerged, including novel AR antagonists, immunotherapy, and targeted therapies, providing new options. Future research should focus on developing antagonists against AR-V7, exploring AR-related biomarkers, and achieving personalized treatment plans to improve the early diagnosis and prognosis of CRPC. In summary, in-depth research into the regulatory mechanisms of AR and metabolic reprogramming will bring new hope for the treatment of prostate cancer.
CL: Methodology, Writing – original draft. DC: Formal Analysis, Validation, Writing – review & editing. PL: Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We thank Figdraw (www.figdraw.com) for expert assistance in the pattern drawing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
CRPC, Castration-Resistant Prostate Cancer; AR, Androgen Receptor; ADT, Androgen Deprivation Therapy; LBD, Ligand-binding domain; NTD, N-terminal domain; DBD, DNA-binding domain; AREs, Androgen response elements; TAMs, Tumor-associated macrophages; PSA, Prostate-specific antigen; FASN, Fatty acid synthase; ACL, ATP-citrate lyase; AF-1, Activation function-1; CDKs, cyclin-dependent kinases; TET2, Ten-eleven translocation 2; TME, Tumor Microenvironment; CAFs, Cancer-associated fibroblasts; PNI, Perineural invasion; NGF, Neurotrophic factor.
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. (2020) 70:7–30. doi: 10.3322/caac.21590
3. Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. (2015) 15:701–11. doi: 10.1038/nrc4016
4. Nurul Azwa Abd Wahab N, Lajis NH, Abas F, Othman I, Naidu R. Mechanism of anti-cancer activity of curcumin on androgen-dependent and androgen-independent prostate cancer. Nutrients. (2020) 12:679. doi: 10.3390/nu12030679
5. Wang G, Zhao D, Spring DJ, DePinho RA. Genetics and biology of prostate cancer. Genes Dev. (2018) 32:1105–40. doi: 10.1101/gad.315739.118
6. Shafi AA, Yen AE, Weigel NL. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol Ther. (2013) 140:223–38. doi: 10.1016/j.pharmthera.2013.07.003
7. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. (2014) 371:1028–38. doi: 10.1056/NEJMoa1315815
8. Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. The androgen receptor gene mutations database: 2012 update. Hum Mutat. (2012) 33:887–94. doi: 10.1002/humu.22046
9. Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. (2002) 20:3001–15. doi: 10.1200/JCO.2002.10.018
10. Zheng Z, Li J, Liu Y, Shi Z, Xuan Z, Yang K, et al. The crucial role of AR-V7 in enzalutamide-resistance of castration-resistant prostate cancer. Cancers (Basel). (2022) 14:4877. doi: 10.3390/cancers14194877
11. Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ, et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell. (2018) 174:758–69. doi: 10.1016/j.cell.2018.06.039
12. Cucchiara V, Yang JC, Mirone V, Gao AC, Rosenfeld MG, Evans CP. Epigenomic regulation of androgen receptor signaling: potential role in prostate cancer therapy. Cancers (Basel). (2017) 9:9. doi: 10.3390/cancers9010009
13. Csizmarik A, Hadaschik B, Kramer G, Nyirady P, Szarvas T. Mechanisms and markers of resistance to androgen signaling inhibitors in patients with metastatic castration-resistant prostate cancer. Urol Oncol. (2021) 39:728.e13–728.e24. doi: 10.1016/j.urolonc.2021.01.030
14. Beltran H, Prandi D, Mosquera JM. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. (2016) 22:298–305. doi: 10.1038/nm.4045
15. Gujrati H, Ha S, Wang B-D. Deregulated microRNAs involved in prostate cancer aggressiveness and treatment resistance mechanisms. Cancers (Basel). (2023) 15:3140. doi: 10.3390/cancers15123140
16. Nguyen T, Sridaran D, Chouhan S, Weimholt C, Wilson A, Luo J, et al. Histone H2A Lys130 acetylation epigenetically regulates androgen production in prostate cancer. Nat Commun. (2023) 14:3357. doi: 10.1038/s41467-023-38887-7
17. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. (2004) 10:33–9. doi: 10.1038/nm972
18. Barbieri I, Kouzarides T. Role of RNA modifications in cancer. Nat Rev Cancer. (2020) 20:303–22. doi: 10.1038/s41568-020-0253-2
19. Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. (2011) 19:575–86. doi: 10.1016/j.ccr.2011.04.008
20. Imamura J, Ganguly S, Muskara A, Liao RS, Nguyen JK, Weight C, et al. Lineage plasticity and treatment resistance in prostate cancer: the intersection of genetics, epigenetics, and evolution. Front Endocrinol (Lausanne). (2023) 30:1191311. doi: 10.3389/fendo.2023.1191311
21. Sadeesh N, Scaravilli M, Latonen L. Proteomic landscape of prostate cancer: the view provided by quantitative proteomics, integrative analyses, and protein interactomes. Cancers (Basel). (2021) 13:4829. doi: 10.3390/cancers13194829
22. Parveen R, Harihar D, Chatterji BP. Recent histone deacetylase inhibitors in cancer therapy. Cancer. (2023) 129:3372–80. doi: 10.1002/cncr.34974
23. Olson BM, Bradley ES, Sawicki T, Zhong W, Ranheim EA. Safety and immunological efficacy of a DNA vaccine encoding the androgen receptor ligand-binding domain (AR-LBD). Prostate. (2017) 77:812–21. doi: 10.1002/pros.23321
24. Sardar S, McNair CM, Ravindranath L, Chand SN. AR coactivators, CBP/p300, are critical mediators of DNA repair in prostate cancer. Oncogene. (2024) 43:3197–213. doi: 10.1038/s41388-024-03148-4
25. Welti J, Sharp A, Brooks N, Yuan W, McNair C, Chand SN, et al. Targeting the p300/CBP axis in lethal prostate cancer. Cancer Discovery. (2021) 11:1118–37. doi: 10.1158/2159-8290.CD-20-0751
26. Hsu CC, Hu CD. Transcriptional activity of c-Jun is critical for the suppression of AR function. Mol Cell Endocrinol. (2013) 372:12–22. doi: 10.1016/j.mce.2013.03.004
27. Kim SH, Nam KH, Hwang KA, Choi KC. Influence of hexabromocyclododecane and 4-nonylphenol on the regulation of cell growth, apoptosis and migration in prostatic cancer cells. Toxicol In Vitro. (2016) 32:240–7. doi: 10.1016/j.tiv.2016.01.008
28. Shorning BY, Dass MS, Smalley MJ, Pearson HB. The PI3K-AKT-mTOR pathway and prostate cancer: at the crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci. (2020) 21:4507. doi: 10.3390/ijms21124507
29. Zhu M-L, Kyprianou N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr Relat Cancer. (2008) 15:841–9. doi: 10.1677/ERC-08-0084
30. Léotoing L, Manin M, Monté D, Baron S, Communal Y, Lours C, et al. Crosstalk between androgen receptor and epidermal growth factor receptor-signalling pathways: a molecular switch for epithelial cell differentiation. J Mol Endocrinol. (2007) 39:151–62. doi: 10.1677/JME-07-0021
31. Donato MD, Cernera G, Auricchio F, Migliaccio A, Castoria G. Cross-talk between androgen receptor and nerve growth factor receptor in prostate cancer cells: implications for a new therapeutic approach. Cell Death Discovery. (2018) 4:5. doi: 10.1038/s41420-017-0024-3
32. Donato MD, Cernera G, Migliaccio A, Castoria G. Nerve growth factor induces proliferation and aggressiveness in prostate cancer cells. Cancers (Basel). (2019) 11:784. doi: 10.3390/cancers11060784
33. Ito S, Kayukawa N, Ueda T, Taniguchi H, Morioka Y, Hongo F, et al. MRGBP promotes AR-mediated transactivation of KLK3 and TMPRSS2 via acetylation of histone H2A.Z in prostate cancer cells. Biochim Biophys Acta Gene Regul Mech. (2018) 1874:9399. doi: 10.1016/j.bbagrm.2018.07.014
34. Chandrasekaran B, Tapadar S, Wu B, Saran U, Tyagi A, Johnston A, et al. Antiandrogen-equipped histone deacetylase inhibitors selectively inhibit androgen receptor (AR) and AR-splice variant (AR-SV) in castration-resistant prostate cancer (CRPC). Cancers (Basel). (2023) 15:1769. doi: 10.3390/cancers15061769
35. Whitton B, Okamoto H, Rose-Zerilli M, Packham G, Crabb SJ. V-ATPase inhibition decreases mutant androgen receptor activity in castrate-resistant prostate cancer. Mol Cancer Ther. (2021) 20:739–48. doi: 10.1158/1535-7163.MCT-20-0662
36. Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. (2005) 23:32. doi: 10.1200/JCO.2005.03.4777
37. Park J, Hsueh P-C, Li Z, Ho P-C. Microenvironment-driven metabolic adaptations guiding CD8+ T cell anti-tumor immunity. Immunity. (2023) 56:32–42. doi: 10.1016/j.immuni.2022.12.008
38. Gholiha AR, Hollander P, Glimelius I, Hedstrom G, Molin D. Revisiting IL-6 expression in the tumor microenvironment of classical Hodgkin lymphoma. Blood Adv. (2021) 5:1671–81. doi: 10.1182/bloodadvances.2020003664
39. Wang D, Cheng C, Chen X, Wang J, Liu K, Jing N. IL-1β is an androgen-responsive target in macrophages for immunotherapy of prostate cancer. Adv Sci (Weinh). (2023) 10:e2206889. doi: 10.1002/advs.202206889
40. Hatano K, Fujita K, Nonomura N. Application of anti-inflammatory agents in prostate cancer. J Clin Med. (2020) 9:2680. doi: 10.3390/jcm9082680
41. Yuan X, Cai C, Chen S, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. (2014) 33:2815–25. doi: 10.1038/onc.2013.235
42. Thapa D, Meng P, Bedolla RG, Reddick RL, Kumar AP, Ghosh R. NQO1 suppresses NF-κB-p300 interaction to regulate inflammatory mediators associated with prostate tumorigenesis. Cancer Res. (2014) 74:5644–55. doi: 10.1158/0008-5472.CAN-14-0562
43. Niu Y, Förster S, Muders M. The role of perineural invasion in prostate cancer and its prognostic significance. Cancers (Basel). (2022) 14:4065. doi: 10.3390/cancers14174065
44. Bakst RL, Wong RJ. Mechanisms of perineural invasion. J Neurol Surg B Skull Base. (2016) 77:96–106. doi: 10.1055/s-0036-1571835
45. Cioni B, Nevedomskaya E, Melis MHM, Burgsteden Jv, Stelloo S, Hodel E, et al. Loss of androgen receptor signaling in prostate cancer-associated fibroblasts (CAFs) promotes CCL2- and CXCL8-mediated cancer cell migration. Mol Oncol. (2018) 12:1308–23. doi: 10.1002/1878-0261.12327
46. Tang Q, Cheng B, Dai R, Wang R. The role of androgen receptor in cross talk between stromal cells and prostate cancer epithelial cells. Front Cell Dev Biol. (2021) 9:729498. doi: 10.3389/fcell.2021.729498
47. Donato MD, Zamagni A, Galasso G, Zazzo ED, Giovannelli P, Barone MV, et al. The androgen receptor/filamin A complex as a target in prostate cancer microenvironment. Cell Death Dis. (2021) 12:127. doi: 10.1038/s41419-021-03402-7
48. Chen H, Fang S, Zhu X, Liu H. Cancer-associated fibroblasts and prostate cancer stem cells: crosstalk mechanisms and implications for disease progression. Front Cell Dev Biol. (2024) 12:1412337. doi: 10.3389/fcell.2024.1412337
49. Clark KC, Wu Y, Taylor RA, Daly RJ. Novel therapeutic targets and biomarkers associated with prostate cancer-associated fibroblasts (CAFs). Crit Rev Oncog. (2022) 27:1–24. doi: 10.1615/CritRevOncog.2022043478
50. Antonarakis ES, Zhang N, Saha J, Nevalaita L, Ikonen T, Tsai LJ, et al. Prevalence and spectrum of AR ligand-binding domain mutations detected in circulating-tumor DNA across disease states in men with metastatic castration-resistant prostate cancer. JCO Precis Oncol. (2024) 8:e2300330. doi: 10.1200/PO.23.00330
51. van de Wijngaart DJ, Molier M, Lusher SJ, Hersmus R, Jenster G, Trapman J, et al. Systematic structure-function analysis of androgen receptor Leu701 mutants explains the properties of the prostate cancer mutant L701H. J Biol Chem. (2010) 285:5097–105. doi: 10.1074/jbc.M109.039958
52. Steketee K, Timmerman L, Ziel-van-der-Made ACJ, Doesburg P, Brinkmann AO, Trapman J. Broadened ligand responsiveness of androgen receptor mutants obtained by random amino acid substitution of H874 and mutation hot spot T877 in prostate cancer. Int J Cancer. (2002) 100:309–17. doi: 10.1002/ijc.10495
53. Bertolini R, Goepfert C, Andrieu T, Nichols S, Walter MA, Frey FJ, et al. 18F-RB390: innovative ligand for imaging the T877A androgen receptor mutant in prostate cancer via positron emission tomography (PET). Prostate. (2015) 75:348–59. doi: 10.1002/pros.22919
54. Southwell J, Chowdhury SF, Gottlieb B, Beitel LK, Lumbroso R, Purisima EO, et al. An investigation into CAG repeat length variation and N/C terminal interactions in the T877A mutant androgen receptor found in prostate cancer. J Steroid Biochem Mol Biol. (2008) 111:138–46. doi: 10.1016/j.jsbmb.2008.04.009
55. Wu Z, Wang K, Yang Z, Pascal LE, Nelson JB, Takubo K, et al. A novel androgen receptor antagonist JJ-450 inhibits enzalutamide-resistant mutant ARF876L nuclear import and function. Prostate. (2020) 80:319–28. doi: 10.1002/pros.23945
56. Ledet EM, Lilly MB, Sonpavde G, Lin E, Nussenzveig RH, Barata PC, et al. Comprehensive analysis of AR alterations in circulating tumor DNA from patients with advanced prostate cancer. Oncologist. (2020) 25:327–33. doi: 10.1634/theoncologist.2019-0115
57. Doamekpor SK, Peng P, Xu R, Ma L, Tong Y, Tong L. A partially open conformation of an androgen receptor ligand-binding domain with drug-resistance mutations. Acta Crystallogr F Struct Biol Commun. (2023) 79:95–104. doi: 10.1107/S2053230X23002224
58. Asangani I, Blair IA, Duyne GV, Hilser VJ, Moiseenkova-Bell V, Plymate S, et al. Using biochemistry and biophysics to extinguish androgen receptor signaling in prostate cancer. J Biol Chem. (2021) 296:100240. doi: 10.1074/jbc.REV120.012411
59. Batista-Silva JP, Gomes D, Sousa SF, Sousa Â, Passarinha LA. Advances in structure-based drug design targeting membrane protein markers in prostate cancer. Drug Discovery Today. (2024) 29:104130. doi: 10.1016/j.drudis.2024.104130
60. Popa SL, Pop C, Dita MO, Brata VD, Bolchis R, Czako Z, et al. Deep learning and antibiotic resistance. Antibiotics (Basel). (2022) 11:1674. doi: 10.3390/antibiotics11111674
61. Ramanand SG, Mani RS. Genetic, environmental, and nuclear factors governing genomic rearrangements. Adv Exp Med Biol. (2019) 1210:57–66. doi: 10.1007/978-3-030-32656-2_3
62. Dai C, Dehm SM, Sharifi N. Targeting the androgen signaling axis in prostate cancer. J Clin Oncol. (2023) 41:4267–78. doi: 10.1200/JCO.23.00433
63. Kocak A, Yildiz M. Molecular dynamics simulations reveal the plausible agonism/antagonism mechanism by steroids on androgen receptor mutations. J Mol Graph Model. (2022) 111:108081. doi: 10.1016/j.jmgm.2021.108081
64. Bader DA, McGuire SE. Tumour metabolism and its unique properties in prostate adenocarcinoma. Nat Rev Urol. (2020) 17:214–31. doi: 10.1038/s41585-020-0288-x
65. Swinnen JV, Heemers H, van de Sande T, de Schrijver E, Brusselmans K, Heyns W, et al. Androgens, lipogenesis and prostate cancer. J Steroid Biochem Mol Biol. (2004) 92:273–9. doi: 10.1016/j.jsbmb.2004.10.013
66. Corbin JM, Ruiz-Echevarría MJ. One-carbon metabolism in prostate cancer: the role of androgen signaling. Int J Mol Sci. (2016) 17:1208. doi: 10.3390/ijms17081208
67. Uo T, Sprenger CC, Plymate SR. Androgen receptor signaling and metabolic and cellular plasticity during progression to castration resistant prostate cancer. Front Oncol. (2020) 10:580617. doi: 10.3389/fonc.2020.580617
68. Labrecque MP, Brown LG, Coleman IM, Nguyen HM, Dalrymple S, Brennen WN. Targeting the fibroblast growth factor pathway in molecular subtypes of castration-resistant prostate cancer. Prostate. (2024) 84:100–10. doi: 10.1002/pros.24630
69. Pencik J, Philippe C, Schlederer M, Atas E, Pecoraro M. STAT3/LKB1 controls metastatic prostate cancer by regulating mTORC1/CREB pathway. Mol Cancer. (2023) 22:133. doi: 10.1186/s12943-023-01825-8
70. Nevedomskaya E, Haendler B. From omics to multi-omics approaches for in-depth analysis of the molecular mechanisms of prostate cancer. Int J Mol Sci. (2022) 23:6281. doi: 10.3390/ijms23116281
71. Zamagni A, Cortesi M, Zanoni M, Tesei A. Non-nuclear AR signaling in prostate cancer. Front Chem. (2019) 7:651. doi: 10.3389/fchem.2019.00651
72. Freedland SJ, de Almeida Luz M, De Giorgi U, Gleave M. Improved outcomes with enzalutamide in biochemically recurrent prostate cancer. N Engl J Med. (2023) 389:1453–65. doi: 10.1056/NEJMoa2303974
73. Szmulewitz RZ, Peer CJ, Ibraheem A, Martinez E, Kozloff MF. Prospective international randomized phase II study of low-dose abiraterone with food versus standard dose abiraterone in castration-resistant prostate cancer. J Clin Oncol. (2018) 36:1389–95. doi: 10.1200/JCO.2017.76.4381
74. Jacob A, Raj R, Allison DB, Myint ZW. Androgen receptor signaling in prostate cancer and therapeutic strategies. Cancers (Basel). (2021) 13:5417. doi: 10.3390/cancers13215417
75. Wang L-Y, Hung C-L, Wang T-C, Hsu H-C, Kung H-J, Lin K-H. PROTACs as therapeutic modalities for drug discovery in castration-resistant prostate cancer. Annu Rev Pharmacol Toxicol. (2025) 65:375–96. doi: 10.1146/annurev-pharmtox-030624-110238
76. Cao B, Kim M, Reizine NM, Moreira DM. Adverse events and androgen receptor signaling inhibitors in the treatment of prostate cancer: a systematic review and multivariate network meta-analysis. Eur Urol Oncol. (2023) 6:237–50. doi: 10.1016/j.euo.2023.01.001
77. Handy CE, Antonarakis ES. Sipuleucel-T for the treatment of prostate cancer: novel insights and future directions. Future Oncol. (2018) 14:907–17. doi: 10.2217/fon-2017-0531
78. Sutherland SIM, Ju X, Horvath LG, Clark GJ. Moving on from Sipuleucel-T: new dendritic cell vaccine strategies for prostate cancer. Front Immunol. (2021) 12:641307. doi: 10.3389/fimmu.2021.641307
79. McNeel DG, Eickhoff JC, Wargowski E, Zahm C, Staab MJ, Straus J, et al. Concurrent, but not sequential, PD-1 blockade with a DNA vaccine elicits anti-tumor responses in patients with metastatic, castration-resistant prostate cancer. Oncotarget. (2018) 9:25586–96. doi: 10.18632/oncotarget.25387
80. Tsai AK, Kagalwalla S, Langer J, Le-Kumar T, Le-Kumar V, Antonarakis ES. Pembrolizumab for metastatic castration-resistant prostate cancer: trials and tribulations. Expert Opin Biol Ther. (2024) 24:51–62. doi: 10.1080/14712598.2024.2311750
81. McNeel DG, Emamekhoo H, Eickhoff JC, Kyriakopoulos CE, Wargowski E, Tonelli TP, et al. Phase 2 trial of a DNA vaccine (pTVG-HP) and nivolumab in patients with castration-sensitive non-metastatic (M0) prostate cancer. J Immunother Cancer. (2023) 11:e008067. doi: 10.1136/jitc-2023-008067
82. Saad F, Clarke NW, Oya M, Shore N, Procopio G, Guedes JD, et al. Olaparib plus abiraterone versus placebo plus abiraterone in metastatic castration-resistant prostate cancer (PROpel): final prespecified overall survival results of a randomised, double-blind, phase 3 trial. Lancet Oncol. (2023) 24:1094–108. doi: 10.1016/S1470-2045(23)00382-0
83. Wu MS, Goldberg H. Role of Rucaparib in the treatment of prostate cancer: clinical perspectives and considerations. Cancer Manag Res. (2022) 14:3159–74. doi: 10.2147/CMAR.S353411
84. Ko JJ, Mbuagbaw L, Tyldesley S, Lowther J, Sunderland K, Royer C, et al. Real-world evaluation of access-driven Canadian treatment sequences in progressive prostate cancer (REACTIVATE). Can Urol Assoc J. (2024) 1:E194–E203. doi: 10.5489/cuaj.8620
85. Wu M, Zhang R, Zhang Z, Zhang N, Li C, Xie Y, et al. Selective androgen receptor degrader (SARD) to overcome antiandrogen resistance in castration-resistant prostate cancer. eLife. (2023) 12:e70700. doi: 10.7554/eLife.70700
86. Gebrael G, Galarza Fortuna G, Sayegh N, Swami U, Agarwal N. Advances in the treatment of metastatic prostate cancer. Trends Cancer. (2023) 9:840–54. doi: 10.1016/j.trecan.2023.06.009
87. Uo T, Plymate SR, Sprenger CC. The potential of AR-V7 as a therapeutic target. Expert Opin Ther Targets. (2018) 22:201–16. doi: 10.1080/14728222.2018.1439016
88. Zhang A, Zhao JC, Kim J, Fong KW, Yang YA, Chakravarti D, et al. LncRNA HOTAIR enhances the androgen-receptor-mediated transcriptional program and drives castration-resistant prostate cancer. Cell Rep. (2015) 13:209–21. doi: 10.1016/j.celrep.2015.08.069
89. Guo H, Ahmed M, Zhang F, Yao CQ, Li S, Liang Y, et al. Modulation of long noncoding RNAs by risk SNPs underlying genetic predispositions to prostate cancer. Nat Genet. (2016) 48:1142–50. doi: 10.1038/ng.3637
90. Zhang Y, Pitchiaya S, Cieslik M, Niknafs YS, Tien JC, Hosono Y, et al. Analysis of the androgen receptor-regulated lncRNA landscape identifies a role for ARLNC1 in prostate cancer progression. Nat Genet. (2018) 50:814–24. doi: 10.1038/s41588-018-0120-1
91. Kroiss A, Vincent S, Decaussin-Petrucci M, Meugnier E, Viallet J, Ruffion A, et al. Androgen-regulated microRNA-135a decreases prostate cancer cell migration and invasion through downregulating ROCK1 and ROCK2. Oncogene. (2015) 34:2846–55. doi: 10.1038/onc.2014.222
92. Leite KR, Morais DR, Florez MG, Reis ST, Iscaife A, Viana N, et al. The role of microRNAs 371 and 34a in androgen receptor control influencing prostate cancer behavior. Urol Oncol. (2015) 33:e15–22. doi: 10.1016/j.urolonc.2015.03.002
93. Yang Y, Liu KY, Liu Q, Cao Q. Androgen receptor-related non-coding RNAs in prostate cancer. Front Cell Dev Biol. (2021) 9:660853. doi: 10.3389/fcell.2021.660853
94. Xu Y, Yang Y, Wang Z, Sjöström M, Jiang Y, Tang Y. ZNF397 deficiency triggers TET2-driven lineage plasticity and AR-targeted therapy resistance in prostate cancer. Cancer Discovery. (2024) 14:1496–521. doi: 10.1158/2159-8290.CD-23-0539
95. Crowley L, Shen MM. Heterogeneity and complexity of the prostate epithelium: new findings from single-cell RNA sequencing studies. Cancer Lett. (2022) 525:108–14. doi: 10.1016/j.canlet.2021.10.035
96. Crowell PD, Giafaglione JM, Hashimoto T, Diaz JA, Goldstein AS. Evaluating the differentiation capacity of mouse prostate epithelial cells using organoid culture. J Vis Exp. (2019) 153):60223. doi: 10.3791/60223
97. Sachs N, Papaspyropoulos A, Zomer-van-Ommen DD, Heo I. Long-term expanding human airway organoids for disease modeling. EMBO J. (2019) 38:e100300. doi: 10.15252/embj.2018100300
98. Driehuis E, Kretzschmar K, Clevers H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat Protoc. (2020) 15:3380–409. doi: 10.1038/s41596-020-0379-4
99. Yang R, Yu Y. Patient-derived organoids in translational oncology and drug screening. Cancer Lett. (2023) 562:216180. doi: 10.1016/j.canlet.2023.216180
Keywords: prostate cancer, androgen receptor (AR), epigenetics, drug resistance, personalized therapy
Citation: Li C, Cheng D and Li P (2025) Androgen receptor dynamics in prostate cancer: from disease progression to treatment resistance. Front. Oncol. 15:1542811. doi: 10.3389/fonc.2025.1542811
Received: 10 December 2024; Accepted: 23 January 2025;
Published: 11 February 2025.
Edited by:
Bekir Cinar, Clark Atlanta University, United StatesReviewed by:
Rajdeep Das, University Hospitals Cleveland Medical Center, United StatesCopyright © 2025 Li, Cheng and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Li, bHB3eHFsempAc29odS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.