
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol., 04 April 2025
Sec. Cancer Genetics
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1526044
This article is part of the Research TopicMyelodysplastic Neoplasm and Acute Myeloid Leukemia: Multi-Omics Approaches and Precision MedicineView all 3 articles
 Soumya Mikkilineni1Juan Pablo Pineda-Reyes2
Soumya Mikkilineni1Juan Pablo Pineda-Reyes2 Lindsay Wilde3Andres Ferber4
Lindsay Wilde3Andres Ferber4 Zi-Xuan Wang5Stephen Peiper2Guldeep Uppal1
Zi-Xuan Wang5Stephen Peiper2Guldeep Uppal1 Jerald Gong1
Jerald Gong1 Jinglan Liu6*
Jinglan Liu6*The MECOM (MDS1 and EVI1 complex locus) gene, located at 3q26.2, encodes an oncogenic transcription factor implicated in multiple signaling pathways. Rearrangements involving MECOM/3q26.2, including inversions, translocations, insertions and cryptic chromosomal changes, are observed in myeloid neoplasms and are associated with high-risk disease features and poor clinical outcomes. The translocation t(3;12)(q26.2;p13.1) is a rare genetic event, resulting in a fusion of the MECOM gene at 3q26.2 with the ETV6 gene at 12p13.1. To date, only 78 cases of hematologic neoplasms harboring t(3;12) have been reported in the English literature, primarily as case reports or case series. T(3;12) has been associated with abnormalities of chromosome 7, multiple hematopoietic lineage dysplasia, and poor prognosis. Given its rarity, studies on t(3;12) in myeloid neoplasms are limited. In this report, we present two additional cases exhibiting t(3;12), initially identified through routine karyotyping. The clinicopathological, cytogenetic and molecular genetic characteristics were summarized and discussed. A comprehensive review of partner genomic loci and genes mutated in myeloid neoplasms with MECOM rearrangement was conducted. The AF4 gene and the transcription elongation control pathways are proposed as potential therapeutic targets for MECOM-rearranged myeloid neoplasms.
Leukemia is a heterogeneous disease characterized by distinctive biological and clinical manifestations, driven by a variety of acquired genetic alterations. Chromosomal translocations involving the MECOM locus at 3q26.2 account for less than 1% of myeloid neoplasms. These translocations can be found in both primary and therapy-related cases and are associated with unfavorable clinical outcomes (1–3).
MECOM encodes a transcription factor, the mis-expression of which triggers leukemogenic effects through the dysregulation of its direct target genes (e.g., GATA2, PBX1, PML, etc.). Further downstream events play critical roles in upregulating protein 1, counteracting the growth-inhibitory effects of TGF-β, co-activating RAS/receptor tyrosine kinase pathway, and inhibiting differentiation and apoptosis. However, the precise functions and underlying molecular pathological mechanism by which MECOM induces leukemogenesis remain poorly understood (2, 4).
Myeloid neoplasms involving MECOM rearrangements can be classified into two groups. The first group is the classic type, which consists of inv (3)(q21q26.2) or t(3;3)(q21;q26.2), characterized by an apparent MECOM::RPN1 fusion resulting from a reciprocal chromosomal alteration. The second group is the non-classic type, represented by apparent fusions between the MECOM locus and genes other than RPN1. The involved chromosomal alterations in this group can be either balanced or unbalanced (2). The t(3;12)(q26.2;p13.1) resulting in an MECOM::ETV6 (formally named TEL) fusion, a non-classic type, is found in less than 0.1% of myeloid neoplasms (4). To date, only 78 cases have been reported in literature, and their clinical and molecular features have been reviewed and summarized by Ronaghy A. et al. (2021) (4). Herein, we describe the clinicopathological, cytogenetic, and molecular genetic features of two new cases. A comprehensive review of partner genomic loci and genes mutated in myeloid neoplasms with MECOM rearrangement was also conducted. Online data mining and pathway analysis have highlighted a possible pivotal role of the AF4 transcription elongation factor in these cases.
Case #1 is a 55-year-old female with a past medical history of early-stage melanoma, 17 years post-excision, who presented to our institution with easy bruising and fatigue. A complete blood count (CBC) revealed pancytopenia. Bone marrow biopsy showed hypocellular marrow (10-20% cellularity) with myeloid immaturity and increased CD34 and CD117-positive myeloblasts (15%). Multi-lineage dysplasia was suggested by the presence of decreased granulocytes with pseudo-Pelger-Huët anomaly (PPHA), megaloblastic erythrocytes with anisocytosis, poikilocytosis, and schistocytes, as well as immature monocytes (Table 1, Figures 1D, E). A diagnosis of myelodysplastic syndrome with excess blasts-2 (MDS-EB2) was made. The patient was initially treated with decitabine. Unfortunately, after one cycle of decitabine, a bone marrow examination revealed disease progression to hypocellular acute myeloid leukemia with 50-60% blasts. An aggressive induction approach with FLAG-Ida-venetoclax was then administered, and complete remission (CR) and negative minimal residual disease (MRD) status were achieved two months later. After completing one cycle of FLAG-Ven (without idarubicin) for consolidation, she underwent a haploidentical allogeneic hematopoietic stem cell transplant (HSCT). At the latest follow up, the patient was 834 days post-allogeneic HSCT, doing exceptionally well, and with return to baseline functional status and resolution of all symptoms. The most recent CBC reveals only mild anemia with no other concerning abnormalities. Engraftment studies have consistently shown >99% donor cells (Table 1). This patient was lost to follow-up due to a family relocation.
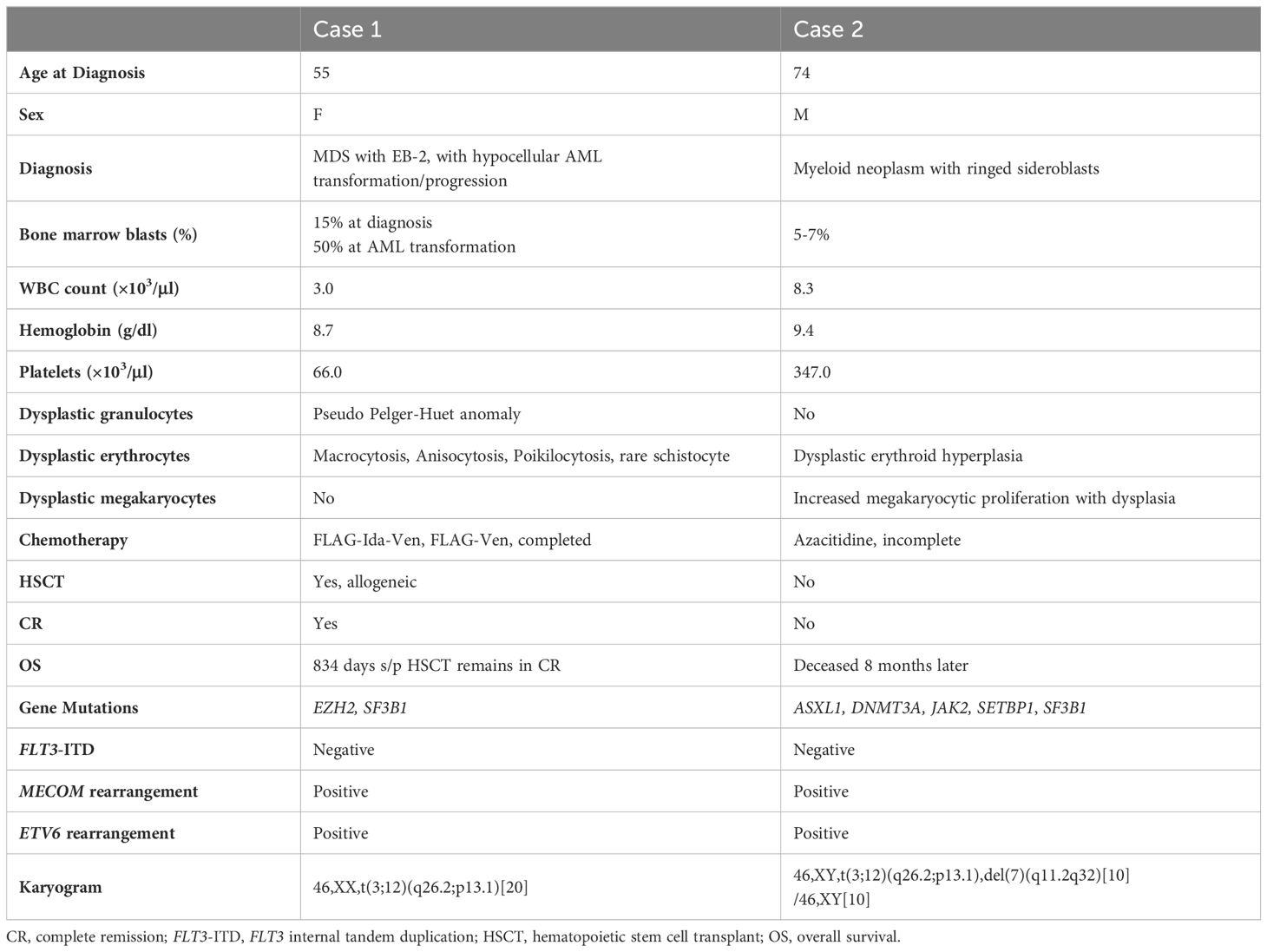
Table 1. Clinicopathologic, cytogenetic and molecular genetic features of two new cases with t(3;12)(q26.2;p13.
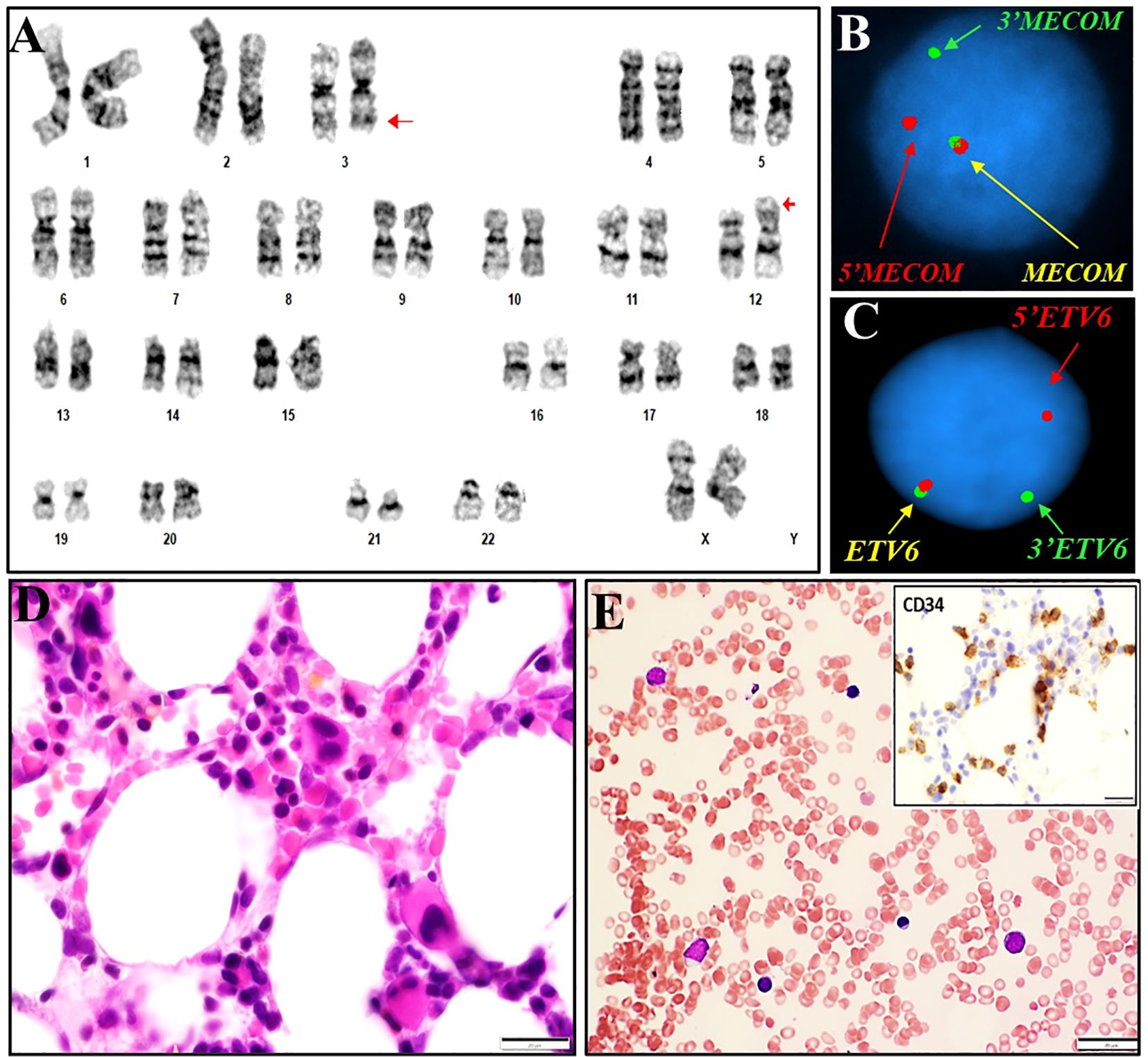
Figure 1. Cytogenetics, FISH and histomorphological findings of case #1. (A) Representative karyogram of a translocation 46,XX,t(3;12)(q26.2;p13) observed in 20/20 metaphases. Arrows indicate aberrant chromosomes. (B) FISH using dual-color break-apart probes for MECOM 3q26.2 demonstrating one normal fusion signal, one split signal for 5’ end region of MECOM (green), and one split signal for the 3’ end region of MECOM (red), consistent with rearrangement at MECOM locus. (C) FISH using dual-color break-apart probes for ETV6 at 12p13.1 demonstrating one normal fusion signal, one split signal for 5’ end region of ETV6 (red), and one split signal for the 3’ end region of ETV6 (green), consistent with rearrangement at ETV6 locus. (D, E): Bone marrow biopsy and touch prep showing dysplasia in erythroid precursors.
Case #2 is a 74-year-old male who presented to the clinic with progressive fatigue and dyspnea. The patient had a history of a long-standing anemia and was diagnosed with a myelodysplastic/myeloproliferative neoplasm (MDS/MPN) with a JAK-2 mutation at an outside hospital approximately three years prior. He had been closely monitored for 34 months with no remarkable events until he developed progressive anemia, high ferritin and iron saturation, and negative HFE mutations. His platelet count and white blood cell count remained normal. The bone marrow examination revealed a hypercellular bone marrow (90%) with trilineage hematopoiesis, dysplastic erythroid hyperplasia, increased megakaryocytic proliferation with dysplasia, increased ringed sideroblasts (34% of bone marrow erythroid precursors), mild increased blasts (5-7%), and mild diffuse reticulin fibrosis, consistent with the patient’s known history (Table 1, Figures 2D, E). The patient was started on treatment with azacytidine; the hemoglobin level increased but remained low. By cycle 4, the patient continued to feel fatigued and showed no improvement in anemia. He declined a repeat bone marrow biopsy and was transferred to hospice care. The patient was deceased a month later (Table 1).
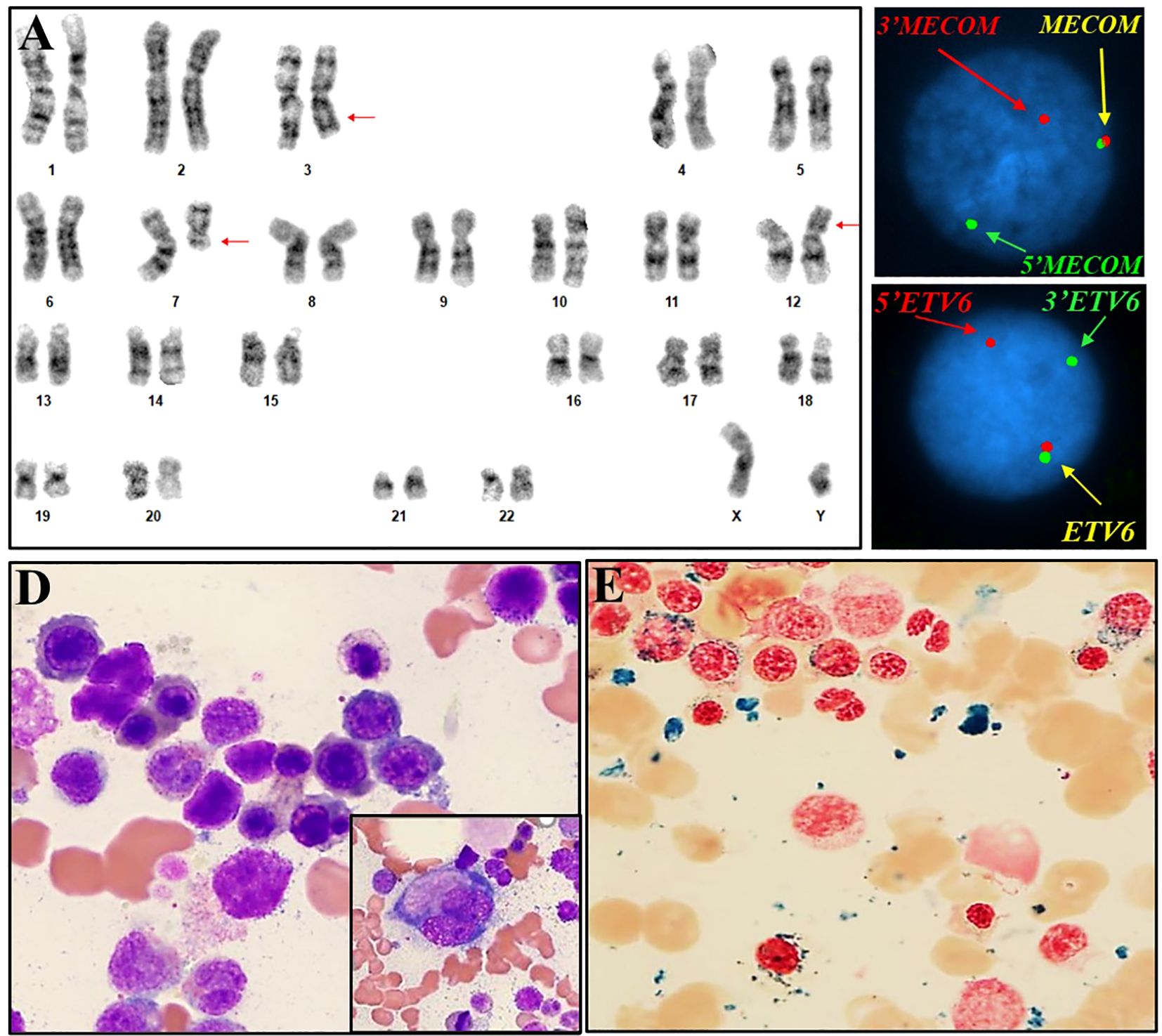
Figure 2. Cytogenetic, FISH and histomorphological findings of case #2. (A) Representative karyogram of translocation 46,XY,t(3;12)(q26.2;p13),del(7)(q11.2q32) in 10/20 metaphases. Arrows indicate aberrant chromosomes. (B) FISH using dual-color break-apart probes for MECOM 3q26.2 demonstrating one normal fusion signal, one split signal for 5’ end region of MECOM (green), and one split signal for the 3’ end region of MECOM (red), consistent with rearrangement at MECOM locus. (C) FISH using dual-color break-apart probes for ETV6 at 12p13.1 demonstrating one normal fusion signal, one split signal for 5’ end region of ETV6 (red), and one split signal for 3’ end region of ETV6 (green), consistent with rearrangement at ETV6 locus. (D) Bone marrow aspirate showing dysplasia in erythroid precursors with irregular nuclear contours, budding, basophilic stippling and megaloblastoid features, megakaryocyte with dysplasia and hypolobated morphology (inlet). (E) Ringed sideroblasts (iron stain).
Flow cytometric immunophenotyping was performed on cell suspensions prepared from fresh bone marrow specimens using standard methods. A 7-color staining procedure was conducted with the following commercially available monoclonal antibodies: CD2, CD3, CD4, CD5, CD7, CD8, CD10, CD13, CD14, CD16, CD19, CD20, CD22, CD23, CD33, CD34, CD38, CD45, CD56, CD57, CD64, CD71, CD117, HLA-DR, kappa, lambda, IgG1 (FITC), IgG1 (PE), MPO, TdT, cCD3, cCD22, cCD79a, CD11b, CD11c and CD235a (Becton Dickinson Immunocytometry System, Beckman Coulter Inc., Dako Inc.). Bone marrow smears, core biopsies and immunohistochemistry staining were processed and analyzed with standard protocols or following the manufacturer’s instructions for hematological neoplasms (Ventana Medical Systems).
Chromosome analyses were performed on metaphases harvested from 24- and 48-hour cultured, unstimulated bone marrow cells. Standard GTW-banding procedures were followed. Interphase FISH was conducted using dual color RPN1/MECOM translocation, MECOM break-apart, and ETV6 break-apart probe sets (CytoTest Inc., Rockville, MD, USA). Karyotyping and scoring of metaphases as well as FISH images capturing and analysis, were performed with the ASI system (Applied Spectrum Imaging, Carlsbad, CA, USA). The karyograms and FISH signals were described according to the International System for Human Cytogenetic Nomenclature (ISCN) 2020.
A next-generation sequencing (NGS)-based analysis was performed to detect somatic mutations in the coding sequences of 73 genes that are recurrently mutated in hematological neoplasms, as described previously (5). The cutoff for common hotspot variants was set at 5%, while for other variants it was 10%. Pathological sequence variants were described according to guidelines by the Human Genome Variation Society (http://www.hgvs.org/mutnomen) based on the genomic reference sequence of genome build hg19.
A literature review was performed by searching published articles in PubMed using keywords “MECOM”, “3q”, “t(3;12)” and “acute leukemia”. The search was conducted exclusively in English and included studies published until November 2024. No specific inclusion or exclusion criteria were applied, except for the exclusion of non-English articles. Fusion partner genes of MECOM were identified through this literature review, as well as by searching the Atlas of Genetics and Cytogenetics in Oncology and Hematology (https://atlasgeneticsoncology.org) and the Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer (https://mitelmandatabase.isb-cgc.org). Functional annotation and enrichment analysis were conducted using online bioinformatics tools, including the Gene Ontology (GO) knowledgebase (6, 7) and Enrichr-KG (https://maayanlab.cloud/enrichr-kg) (8). Notably, since most of the selected cases in the literature were diagnosed before the recently published WHO 5th classification of myeloid neoplasms, their original diagnoses and classifications are retained in this report.
In case #1, a conventional cytogenetics study observed a karyogram of 46,XX,t(3;12)(q26.2;p13.1)[20] (Figure 1A). FISH testing with the AML panel probes was negative, except for the presence of three copies of the MECOM probes with the RPN1/MECOM translocation probe set (data not shown), suggesting a MECOM rearrangement with an unknown partner. Reflex testing using gene-specific break-apart probes revealed a signal pattern indicative of rearrangement of the MECOM gene at 3q26.2 and the ETV6 gene at 12p13.1, consistent with a reciprocal translocation between MECOM and ETV6 and the t(3;12) observed in the conventional cytogenetics study (Figures 1A-C). NGS-based gene panel analysis revealed pathogenic mutations in genes EZH2 and SF3B1 (Table 1). In case #2, a conventional cytogenetics study observed a karyogram of 46,XX,t(3;12)(q26.2;p13.1),del (7)(q11.2q32)[10/20] (Figure 2A). Rearrangements of the MECOM gene and the ETV6 gene were confirmed by FISH studies (Figures 2B, C). NGS-based gene panel analysis revealed pathogenic mutations in the genes ASXL1, DNMT3A, JAK2, SETBP1 and SF3B1 (Table 1).
Myeloid neoplasms with structural alterations involving the chromosomal locus 3q26.2, as reported in the Atlas of Genetics and Cytogenetics in Oncology and Hematology, the Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer, and various literature reports (2, 9–11) were reviewed.
Thirty-five known genes located on 13 different chromosomes at 30 genomic loci involved in structural rearrangements with the MECOM locus at the 3q26.2 were documented, including translocations, inversions and insertions, either balanced or unbalanced (Table 2). Clinico-pathological and molecular genetics data are summarized, showing an overall aggressive presentation, poor treatment response and shorter survival time (Table 2). Gene ontology analysis, functional annotation, and pathway hunting were further attempted on the two groups of genes with online bioinformatic tools, the Gene Ontology (GO) knowledgebase (6, 7) and Enrichr-KG (https://maayanlab.cloud/enrichr-kg) (8, 12, 13). GO analysis identified “regulation of DNA-templated transcription” (GO: 00006355; https://www.ebi.ac.uk/QuickGO/term/GO:0006355) and “negative regulation of DNA-templated transcription” (GO:0045892; https://www.ebi.ac.uk/QuickGO/term/GO:0045892) as the top annotated biological processes, each comprising 13 and 10 genes, respectively (Supplementary Table 1, Supplementary Figures 1A, B).
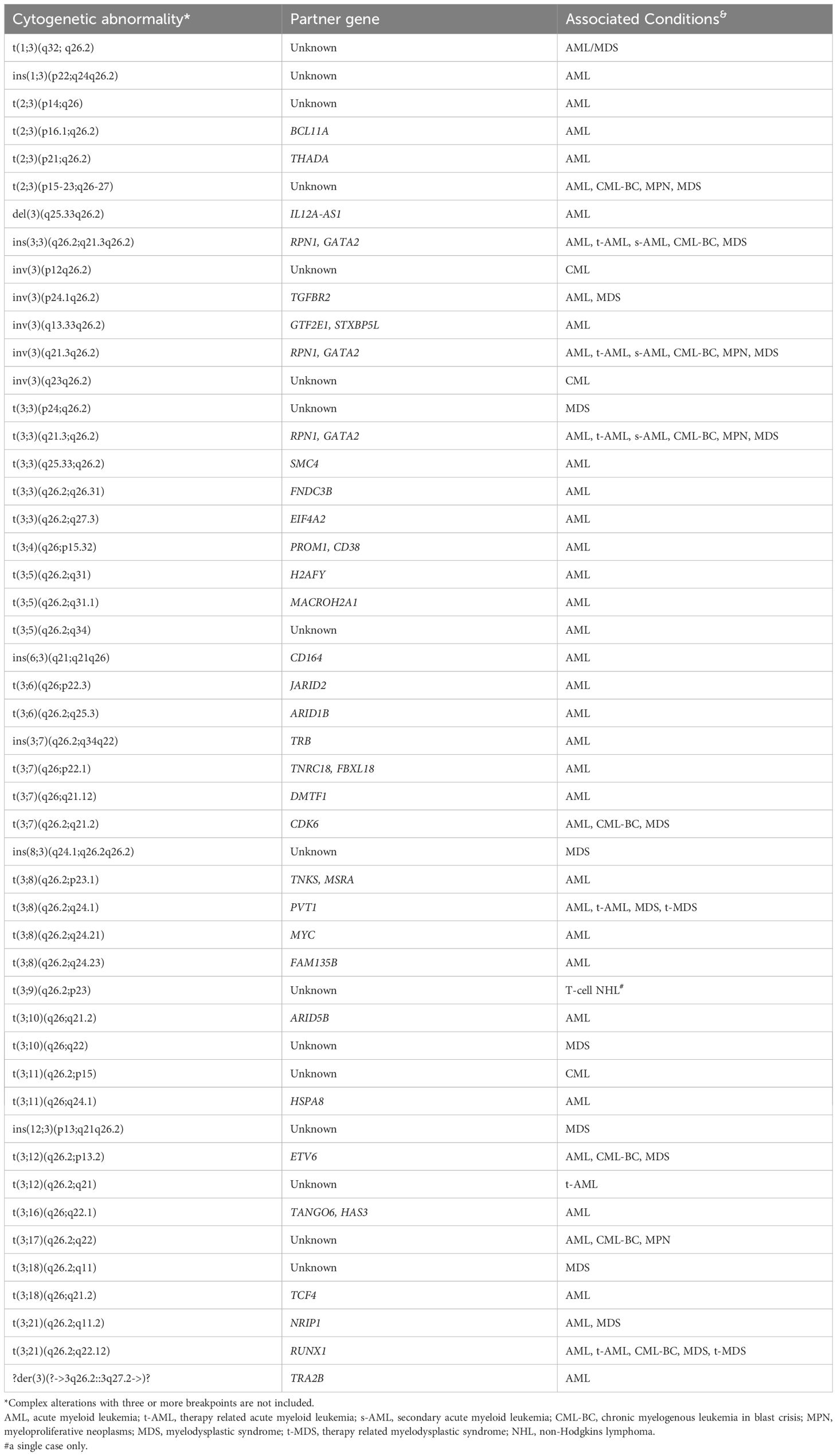
Table 2. Structural abnormalities resulting in chimeric gene fusion between MECOM locus at 3q26.2 and various partner genomic loci, with either known or unknown gene content in hematologic neoplasms, as documented in the public databases and literature.
Given the suggested role in transcription for genes involved in the MECOM rearrangement, Enrichr-KG was utilized to search for transcription binding sites and the corresponding factors bound to those sites enriched from ChEA2022 libraries across the aforementioned 35 genes. Chromatin immunoprecipitation sequencing (ChIP-seq) studies on a human leukemia cell line SEM documented from ChEA2022 study (14), identified 18 of the 35 genes as putative targets of the transcription elongation factor AF4 (Supplementary Table 2, Supplementary Figure 2).
Furthermore, although NGS-based cancer mutational panel testing data were available in literature for only a limited number of cases with MECOM rearrangement, 38 genes carrying pathogenic mutations co-existing with MECOM rearrangement were identified, including those in the current two cases (Supplementary Table 3). Enrichr-KG gene set enrichment analysis of these 38 genes showed that 20 of them are also targets of AF4 in SEM cells based on the ChIP-seq study (14) (Supplementary Figure 3).
MECOM/3q26 abnormalities result in overexpression of MECOM rather than generating a chimeric gene. These abnormalities mainly involve the juxtaposition of regulatory sequences near MECOM, functioning as a super enhancer or a promoter (15). In myeloid neoplasms, t(3;12)(q26.2;p13.1) resulting in an apparent MECOM::ETV6 fusion was found in less than 0.1% of these cases. The corresponding neoplastic transformation is believed to be triggered by MECOM overexpression driven by the ETV6 promoter (4, 10, 15). To date, only 78 cases have been reported in literature. Through routine conventional cytogenetics study, we have identified two new cases with t(3;12).
In case #1, t(3;12) was found at the diagnosis, indicative of a driver event, while in case #2, it appeared as an acquired event during disease progression. Deletion 7q was observed in case #2, consistent with -7/7q being the most common secondary chromosomal abnormality in patients with MECOM rearrangements (Tang et al., 2019a, Tang et al., 2019b). Reports also suggest that t(3;12) is associated with the transformation of MDS to AML (2, 16), which was found in case #1. However, in contrast to the significantly high failure rates of therapy in literature (2, 16) and along with a quick AML transformation, case #1 is doing exceptionally well at the latest follow-up more than two years post-HSCT, suggesting that aggressive induction and consolidation therapy along with transplant are an effective approach treating patients with t(3;12). The poor prognosis of case #2 is most likely due to the presence of an additional 7q deletion, which by itself is a high-risk genetic marker in myeloid neoplasms. Despite the poor prognosis of case #2, his blast count was only 5-7%, which is consistent with findings that the prognosis of MDS or AML cases with MECOM rearrangements is not dependent on blast count (11). Although the expression level of MECOM was not directly measured, it is hypothesized to be elevated in both cases. Intriguingly, a recent study demonstrated that the hypomethylating agent 5-aza-2-deoxycytidine can induce widespread apoptosis in MECOM-high leukemia cells, highlighting its potential as a therapeutic agent for leukemia with MECOM overexpression (17). However, in case #1, the patient was initially treated with decitabine, a hypomethylating agent, upon diagnosis of MDS-EB2, but still progressed to AML. In case #2, the patient was treated with Azacitidine, a similar hypomethylating agent, but did not respond well, either. Although interesting, given that the current report includes only two cases, the clinical outcomes observed cannot be considered statistically conclusive. Both cases showed dysplasia affecting at least two lineages, consistent with dysplasia being a distinct morphological feature associated with MECOM rearrangements (2, 16).
To date, more than 30 genomic loci have been reported to fuse with the MECOM locus in human myeloid neoplasms. This diversity raises the question of whether any common traits are shared among these the fusion partners? Specifically, are there links among different MECOM rearrangement partners? Through a review of the literature and online database searches, 35 known genes at 30 genomic loci located at the fusion breakpoints with MECOM were identified. We demonstrate here that AF4 target genes are overrepresented among these fusion partners.
AF4 is primarily known to be the fusion partner of the KMT2A/MLL gene in infant acute lymphoblastic leukemia with chromosomal translocation t(5;12)(q31;q23). The AF4 gene (also known as ALF transcription elongation factor 4 or AFF4) is located on 5q31.1, and encodes a protein that belongs to the AF4/LAF4/FMR2 (ALF) family. AF4 directly interacts with ELL proteins (ELL, ELL2 or ELL3) and the P-TEFb complex to form the super elongation complex (SEC). SEC then stimulates transcript elongation by converting the RNA polymerase II (Pol II) into its elongating form with an enhanced catalytic rate at multiple sites along the DNA. Additionally, the AF4 family/ENL family/P-TEFb complex (AEP) is capable of activating transcription by binding to acetylated H3K9/18/27. Further functional verification of AF4 involvement in MECOM rearrangement-based myeloid neoplasms will help elucidate the corresponding molecular etiology and identify therapeutic targets (18–21).
The limitations of this study include:
1. Reported data in literature primarily focus on clinical observations. We analyzed and predicted biological functions using multiple database searches and online bioinformatics tools. To advance in vivo proof-of-concept investigations, initial steps could include isolating endogenously occurring MECOM::ETV6 fusion products from the primary leukemia patient cells, mapping the nucleotide sequences of the breakpoints, characterizing potential functional elements, and generating immortalized cell lines using the chimeric fusion genes.
2. There is a lack of genome-wide analyses of MECOM-rearranged leukemia. In fact, very limited data is available in literature. Only recently has NGS-based cancer panel sequencing begun to be tested on these patients, and the data is not available for the majority of reported cases. Given the complex biological nature of human cancer, genome-wide analyses, including epigenomics, genomics, proteomics and metabolomics studies, are warranted to further investigate and characterize key signaling pathways and networks and critical elements associated with leukemia containing the MECOM rearrangement.
3. Due to limited resources, we were unable to but propose to conduct the following studies: isolation of the full-length MECOM::ETV6 fusion product, identification of the breakpoints, characterization of the functional domains of both MECOM and ETV6 included in the chimeric gene, and in vivo functional studies accessing MECOM expression and the responses of predicted signaling pathways. Additionally, future in vitro and in vivo studies are needed to provide experimental evidence evaluating the functional role of AF4 in MECOM-mediated leukemogenesis.
4. This study is based on only two cases, which limits the general applicability of the conclusions. Future research with larger sample sizes and potential collaborations is warranted to enhance the robustness and applicability of the findings.
5. The potential complex interplay between co-existing genetic mutations and MECOM rearrangements in disease progression, clonal evolution, and treatment response requires further exploration. Molecular classification of these mutations could guide the selection of targeted therapies, such as inhibitors of specific pathways. By incorporating clinical data, such as electronic medical records, the impact of these mutations on the efficacy of standard treatments like chemotherapy or stem cell transplantation can be further studied. Due to the limited availability of such data for the cases studied in this manuscript, the genetic analyses currently require further refinement.
Several questions remain to be answered. For example, is t(3;12) sufficient to transform myeloid progenitors? Is AF4 a potential link between MECOM-rearranged leukemia and CTCF pathways, given that both bind to acetylated H3K9/18/27? Is there any molecular mechanistic overlap between MECOM and KMT2A rearranged leukemia? Since AF4 is one of the partner genes for KMT2A, could therapeutic approaches currently utilized or studied for leukemia with KMT2 rearrangement be applicable to those with MECOM rearrangement?
In summary, we have described the clinical, histomorphological and genetic features of two new patients with myeloid neoplasms carrying a t(3;12). We demonstrate that AF4 is likely an upstream regulator in physiologic MECOM-dependent transcriptional pathways, and propose that it might be one of the major mechanisms of MECOM-mediated leukemic transformation. Our study provides further insight into the molecular etiology of MECOM translocation-based myeloid neoplasms. We propose that the AF4 gene and the transcription elongation control pathway could be potential targets for the treatment of MECOM-associated myeloid neoplasms. Additionally, our results underscore the significance of conducting routine conventional cytogenetics assessment in myeloid neoplasms. For future clinical and research studies of acute myeloid leukemia with t(3;12), we suggest grouping all patients with various chromosomal rearrangements involving the MECOM locus at 3q26.2 to address the issue of small sample sizes.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Written informed consent was obtained from the participants for the publication of this case report.
SM: Conceptualization, Data curation, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. JPP-R: Conceptualization, Data curation, Methodology, Writing – original draft, Writing – review & editing. LW: Data curation, Formal analysis, Investigation, Resources, Supervision, Writing – review & editing. AF: Data curation, Formal analysis, Investigation, Resources, Supervision, Writing – review & editing. Z-XW: Data curation, Investigation, Methodology, Resources, Writing – review & editing. SP: Data curation, Resources, Validation, Writing – review & editing. GU: Data curation, Formal analysis, Methodology, Writing – review & editing. JG: Data curation, Methodology, Resources, Writing – review & editing. JL: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1526044/full#supplementary-material
1. Hinai AA, Valk PJ. Review: Aberrant EVI1 expression in acute myeloid leukaemia. Br J Haematol. (2016) 172:870–8. doi: 10.1111/bjh.2016.172.issue-6
2. Tang Z, Tang G, Hu S, Patel KP, Yin CC, Wang W, et al. Deciphering the complexities of MECOM rearrangement-driven chromosomal aberrations. Cancer Genet. (2019) 233-234:21–31. doi: 10.1016/j.cancergen.2019.03.002
3. Lugthart S, Groschel S, Beverloo HB, Kayser S, Valk PJ, van-Zelderen-Bhola SL, et al. Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J Clin Oncol. (2010) 28:3890–8. doi: 10.1200/JCO.2010.29.2771
4. Ronaghy A, Hu S, Tang Z, Wang W, Tang G, Loghavi S, et al. Myeloid neoplasms associated with t(3;12)(q26.2;p13) are clinically aggressive, show myelodysplasia, and frequently harbor chromosome 7 abnormalities. Mod Pathol. (2021) 34:300–13. doi: 10.1038/s41379-020-00663-z
5. Simen BB, Yin L, Goswami CP, Davis KO, Bajaj R, Gong JZ, et al. Validation of a next-generation-sequencing cancer panel for use in the clinical laboratory. Arch Pathol Lab Med. (2015) 139:508–17. doi: 10.5858/arpa.2013-0710-OA
6. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Gene Ontol Consortium Nat Genet. (2000) 25:25–9. doi: 10.1038/75556
7. Gene Ontology C, Aleksander SA, Balhoff J, Carbon S, Cherry JM, Drabkin HJ, et al. The gene ontology knowledgebase in 2023. Genetics. (2023) 224:14. doi: 10.1093/genetics/iyad031
8. Evangelista JE, Xie Z, Marino GB, Nguyen N, Clarke DJB, Ma’ayan A. Enrichr-KG: bridging enrichment analysis across multiple libraries. Nucleic Acids Res. (2023) 51:W168–W79. doi: 10.1093/nar/gkad393
9. Gao J, Gurbuxani S, Zak T, Kocherginsky M, Ji P, Wehbe F, et al. Comparison of myeloid neoplasms with nonclassic 3q26.2/MECOM versus classic inv(3)/t(3;3) rearrangements reveals diverse clinicopathologic features, genetic profiles, and molecular mechanisms of MECOM activation. Genes Chromosomes Cancer. (2022) 61:71–80. doi: 10.1002/gcc.23004
10. Ottema S, Mulet-Lazaro R, Beverloo HB, Erpelinck C, van Herk S, van der Helm R, et al. Atypical 3q26/MECOM rearrangements genocopy inv(3)/t(3;3) in acute myeloid leukemia. Blood. (2020) 136:224–34. doi: 10.1182/blood.2019003701
11. Summerer I, Haferlach C, Meggendorfer M, Kern W, Haferlach T, Stengel A. Prognosis of MECOM (EVI1)-rearranged MDS and AML patients rather depends on accompanying molecular mutations than on blast count. Leuk Lymphoma. (2020) 61:1756–9. doi: 10.1080/10428194.2020.1737689
12. Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma’ayan A. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics. (2010) 26:2438–44. doi: 10.1093/bioinformatics/btq466
13. Keenan AB, Torre D, Lachmann A, Leong AK, Wojciechowicz ML, Utti V, et al. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. (2019) 47:W212–W24. doi: 10.1093/nar/gkz446
14. Benito JM, Godfrey L, Kojima K, Hogdal L, Wunderlich M, Geng H, et al. MLL-rearranged acute lymphoblastic leukemias activate BCL-2 through H3K79 methylation and are sensitive to the BCL-2-specific antagonist ABT-199. Cell Rep. (2015) 13:2715–27. doi: 10.1016/j.celrep.2015.12.003
15. Li Q, Wang F, Zhang X, Liu S, Sun MZ, Yan J. The ETV6-MECOM fusion protein promotes EMT-related properties by repressing the transactivation activity of E-cadherin promoter in K562 leukemia cells. Biochem Biophys Rep. (2024) 38:101667. doi: 10.1016/j.bbrep.2024.101667
16. Tang G, Hu S, Wang SA, Xie W, Lin P, Xu J, et al. t(3;8)(q26.2;q24) often leads to MECOM/MYC rearrangement and is commonly associated with therapy-related myeloid neoplasms and/or disease progression. J Mol Diagn. (2019) 21:343–51. doi: 10.1016/j.jmoldx.2018.10.005
17. Li F, He W, Geng R, Xie X. Myeloid leukemia with high EVI1 expression is sensitive to 5-aza-2’-deoxycytidine by targeting miR-9. Clin Transl Oncol. (2020) 22:137–43. doi: 10.1007/s12094-019-02121-y
18. Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, et al. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. (2010) 37:429–37. doi: 10.1016/j.molcel.2010.01.026
19. Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell. (2010) 17:198–212. doi: 10.1016/j.ccr.2009.12.040
20. Yokoyama A. Leukemogenesis via aberrant self-renewal by the MLL/AEP-mediated transcriptional activation system. Cancer Sci. (2021) 112:3935–44. doi: 10.1111/cas.v112.10
Keywords: t(3;12), MECOM, ETV6, cytogenetics, myeloid, neoplasms, AF4
Citation: Mikkilineni S, Pineda-Reyes JP, Wilde L, Ferber A, Wang Z-X, Peiper S, Uppal G, Gong J and Liu J (2025) Case Report: Myeloid neoplasms with the t(3;12)(q26.2;p13.1)/MECOM-ETV6 translocation: report of two new cases and review of the literature. Front. Oncol. 15:1526044. doi: 10.3389/fonc.2025.1526044
Received: 11 November 2024; Accepted: 04 February 2025;
Published: 04 April 2025.
Edited by:
Teresa de Souza Fernandez, National Cancer Institute (INCA), BrazilReviewed by:
Nathalia Lopez Duarte, Rio de Janeiro Municipal Health Secretariat (SMS-RJ), BrazilCopyright © 2025 Mikkilineni, Pineda-Reyes, Wilde, Ferber, Wang, Peiper, Uppal, Gong and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinglan Liu, amluZ2xhbi5saXVAamVmZmVyc29uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.