 Xiaofeng He
Xiaofeng He Min Lv1
Min Lv1 Feng Wen
Feng Wen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol., 02 April 2025
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1525401
High-grade myofibroblastic sarcoma (HGMS) is exceedingly rare and highly aggressive, with a poor prognosis. Currently, there is no consensus on its definition. Wide resection is the standard of care for most patients, but clinical data on treatment outcomes remain limited. Here, we present the first reported case of HGMS originating intracranially. Surgical excision of the tumor was performed, followed by adjuvant radiotherapy with a total dose of 60 Gy in 30 fractions. As of November 2024, the patient had achieved 24 months of recurrence-free survival. This case may provide new evidence that could be useful for the treatment of rare primary intracranial HGMS.
Malignant sarcoma composed of myofibroblasts or exhibiting myofibroblastic differentiation is diagnosed as myofibroblastic sarcoma (MS) (1). It is exceedingly rare, typically low-grade, and most commonly arises in the head and neck, extremities, or trunk (1, 2). MS is graded according to the Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC) system, with recommendations to classify it at least into low (FNCLCC 1) versus high (FNCLCC 2 and 3) grades due to implications for neoadjuvant therapy (3). However, the 2020 World Health Organization (WHO) classification of soft tissue tumors continues to list only low-grade MS (LGMS) and does not establish a consensus on the definition of high-grade MS (HGMS), in contrast to the 2013 WHO classification (4). HGMS is primarily diagnosed through cytomorphological analysis combined with immunophenotyping. It is more aggressive and has a higher rate of recurrence and metastasis compared to LGMS (5). Surgical resection of the tumor and adjacent structures is the standard of care for most patients; however, clinical data on the outcomes of other treatments, such as chemotherapy, radiotherapy (RT), and targeted therapy (6), remain limited. Previous studies on HGMS are rare. Herein, we describe a patient with HGMS (FNCLCC 2) originating intracranially. To the best of our knowledge, this is the first reported case of primary intracranial HGMS.
In October 2022, a 23-year-old woman was admitted to our hospital with a 2-month history of progressive headaches and a 2-week history of diplopia. Physical examination revealed binocular diplopia but no other neurological abnormalities. Laboratory tests and systemic evaluations were unremarkable. Magnetic resonance imaging (MRI) scan of the head showed a 5.2 cm × 4.0 cm × 6.1 cm cystic mass with irregular margins located in the right fronto-parieto-occipital lobe (Figures 1A, B). The mass appeared isointense with low mixed signals on T1-weighted imaging (WI), isointense to hyperintense with low mixed signals on T2-weighted imaging (T2WI), and hyperintense on fluid-attenuated inversion recovery (FLAIR). Contrast-enhanced T1WI revealed asymmetric ring-like enhancement. Moreover, the right lateral ventricle was compressed, and the midline was slightly shifted to the left. As the lesion originated intracranially, only resection of the space-occupying mass in the right fronto-parieto-occipital lobe was performed on 27 October 2022. Upon cortical incision, a well-circumscribed, firm, vascularized parenchymatous tumor was exposed. The tumor had a gray outer surface and a red inner surface. It was large, measuring approximately 5 cm × 6 × 7 cm, and deeply located within the brain, with a distinct cyst wall in its anterior portion.
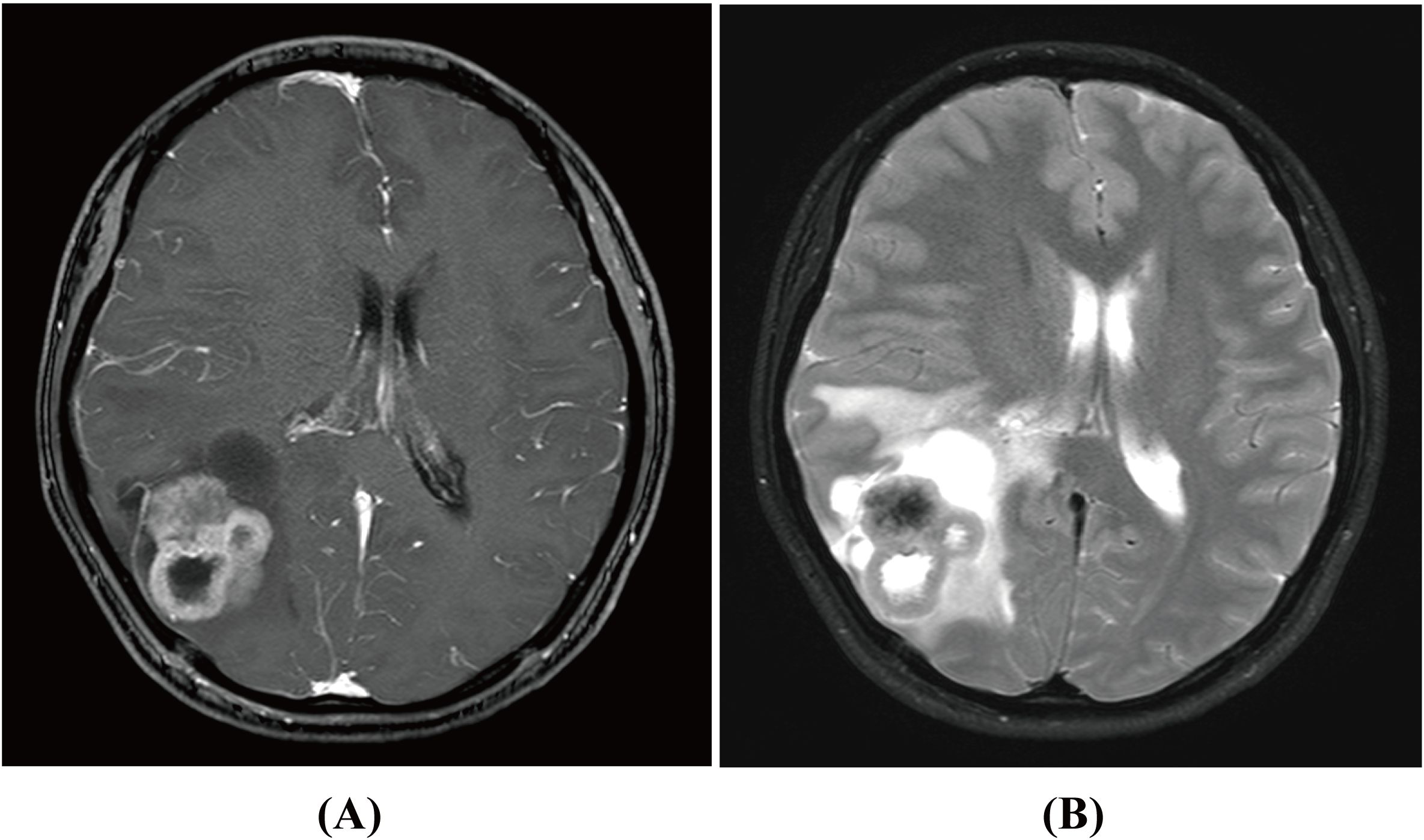
Figure 1. Magnetic resonance imaging (MRI) scan of primary intracranial myofibroblastic sarcoma in contrast-enhanced T1WI (A) and T2WI (B) before surgery.
Intraoperative frozen section analysis revealed a tumor composed of short spindle cells with hyperchromatic, distorted nuclei, an increased nuclear-to-cytoplasmic ratio, and marked nuclear atypia. Some nucleoli were also observed (Figures 2A, B). Postoperative immunohistochemical staining showed positivity for desmin, focal positivity for CD34 and somatostatin receptor 2 (SSTR2), and negativity for epithelial membrane antigen (EMA), S-100, CD99, signal transducer and activator of transcription 6 (STAT6), and smooth muscle actin (SMA). Additionally, the Ki-67 index ranged from 3% to 20%. The cytomorphology and immunophenotyping suggested a rare mesenchymal tumor. Further immunohistochemical analysis revealed that the tumor cells were positive for tri-methylation of histone H3 at lysine 27 (H3K27me3), partially positive for caldesmon, and negative for glial fibrillary acidic protein (GFAP), oligodendrocyte lineage transcription factor 2 (Olig-2), myogenin, myogenic differentiation 1 (MyoD1), and SRY-related HMG-box 10 (SOX10). Genetic testing revealed no mutation in the Dicer 1, Ribonuclease III (DICER) gene, no translocation of the Ewing Sarcoma breakpoint region 1/EWS RNA binding protein 1 (EWSR1) gene, and no amplification of the Murine Double Minute 2 (MDM2) gene in the tumor cells. Based on these findings, the tumor was ultimately diagnosed as MS and classified as grade 2 according to the FNCLCC system. Since no primary lesions were present in other organs, a diagnosis of primary intracranial HGMS (FNCLCC 2) was established.
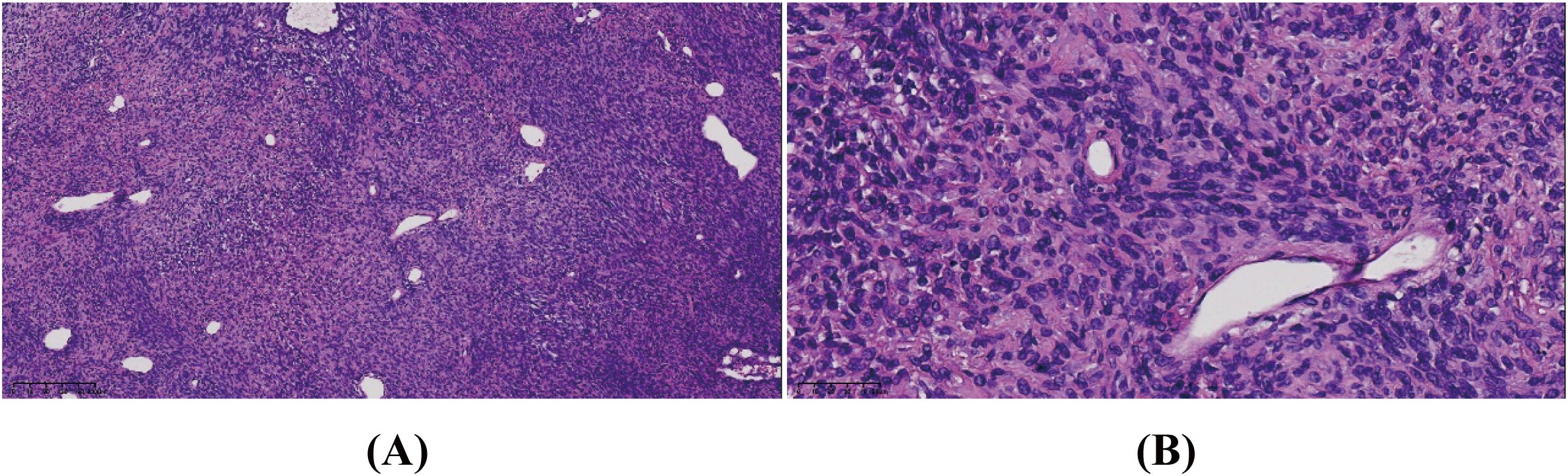
Figure 2. The tumor consisted of short spindle cells with hyperchromatic, distorted nuclei, an increased nuclear-to-cytoplasmic ratio, and marked nuclear atypia. Some nucleoli were seen. Hematoxylin–eosin stain. Original magnification: (A) × 10 and (B) × 40.
Given the rarity and aggressiveness of HGMS, the tumor’s relatively large size, and the patient’s young age, a sarcoma multidisciplinary team (MDT) discussion was conducted postoperatively. After thorough deliberation, Local RT was chosen to reduce the risk of local recurrence while minimizing systemic side effects. Chemotherapy and targeted therapy were not recommended due to their limited efficacy across the blood–brain barrier and the absence of detectable genetic mutations. Additionally, a comprehensive discussion with the patient was held before finalizing the treatment plan, considering the potential risk of cognitive dysfunction associated with local radiotherapy. The patient subsequently received adjuvant intensity-modulated radiotherapy (IMRT). The clinical target volume (CTV) encompassed the entire tumor bed, as defined by the operative record and postoperative MRI, with an additional 1.5-mm margin. The planning target volume (PTV) included the CTV plus a 5-mm margin (Figure 3A). A total dose of 60 Gy was delivered to the PTV in 30 fractions (Figure 3B) over 6 weeks, from 15 February to 28 March 2023.
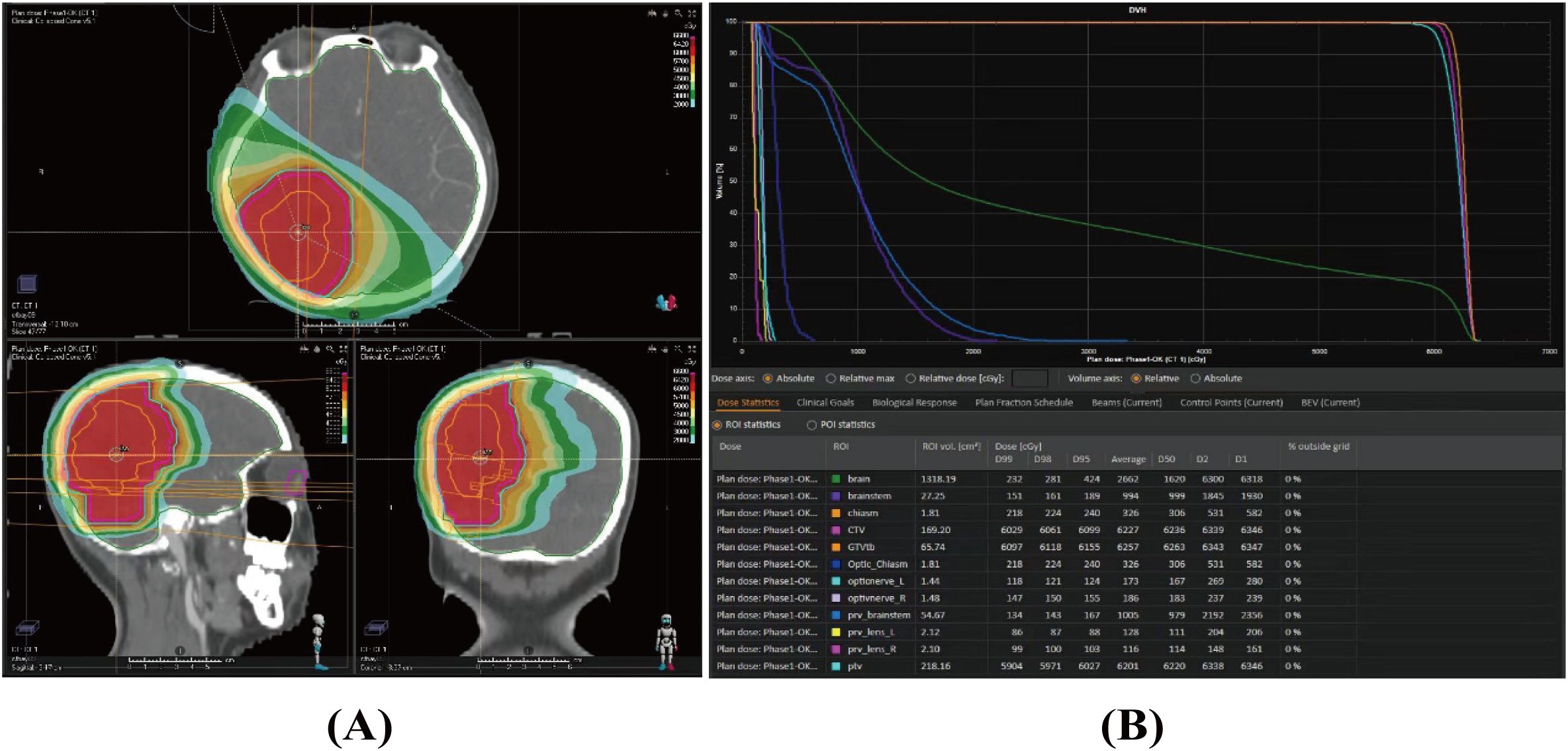
Figure 3. Dose distribution (A) and dose–volume histogram (B) of the radiotherapy plan for the case.
Since no specific serum tumor marker exists for HGMS, imaging served as the primary follow-up strategy in this case. The protocol included contrast-enhanced head MRI, along with contrast-enhanced thoracic and abdominal CT. These imaging studies were conducted 1 month after radiotherapy, then every 3 months during the first 2 years, biannually until 5 years posttreatment, and annually thereafter. Additionally, whole-body bone scans and PET/CT scans were recommended when clinically indicated. The tumor responded well to treatment, with no signs of relapse or metastasis observed during the 24-month postoperative follow-up period.
MS is an exceptionally rare malignancy, with current understanding predominantly derived from case reports and small series. First described in 1978 as a low-grade entity (LGMS) by Vasudev et al. (7), this tumor type was later refined by Mentzel et al. (8) and has undergone incremental nosological clarification in recent decades. Notably, the 2020 WHO classification retains LGMS as the only recognized myofibroblastic sarcoma subtype (4, 9), highlighting the persistent diagnostic ambiguity surrounding HGMS.
In this study, we identified literature reports published between 2000 and 2024 using the search terms in PubMed. We searched for synonyms of “high-grade myofibroblastic sarcoma” and “myofibroblastic sarcoma” and reviewed reference lists of all included studies for additional sources. Our case was also incorporated into this review. Ultimately, only nine cases were analyzed, primarily from case reports (Table 1) (6, 10–16). We evaluated age, gender, tumor location, tumor size, tumor grade, treatment method, chemotherapy, local recurrence, and outcomes.
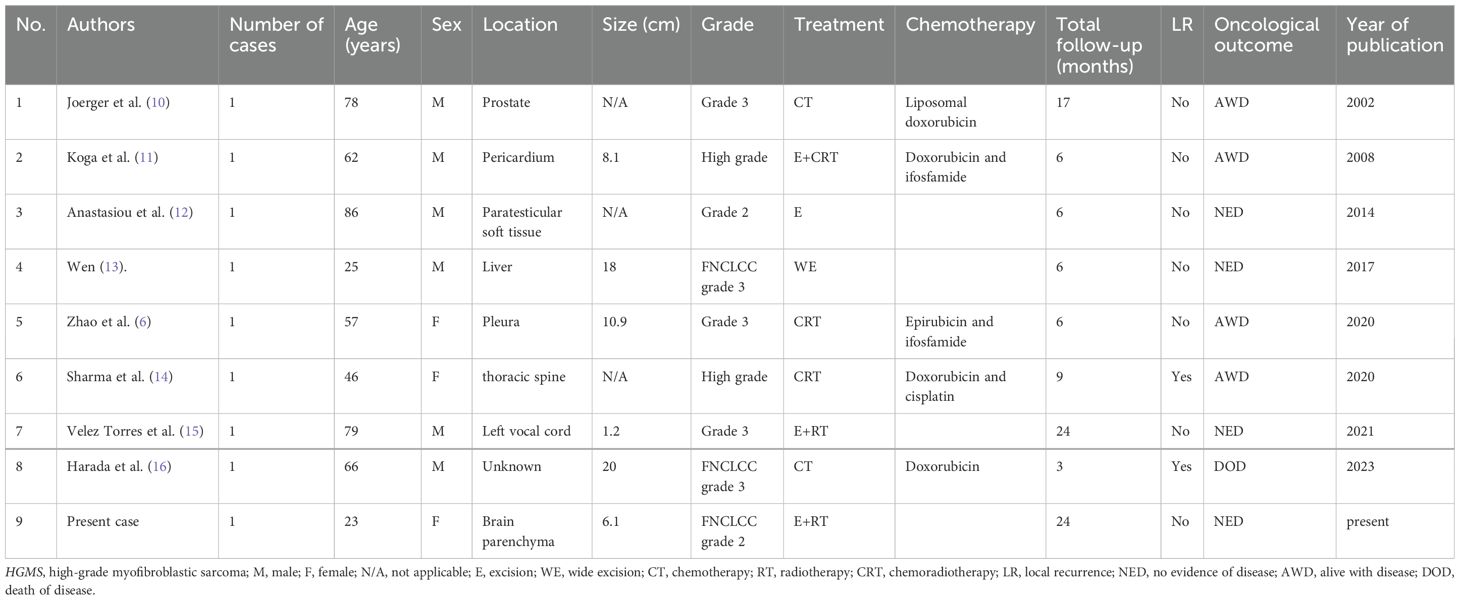
Table 1. Summary of the clinical features of eight previously reported HGMS cases and the present case.
Our systematic review revealed a male predominance (66.7%) with a median age at diagnosis of 62 years. Unlike the historical LGMS predilection for head/neck regions (1, 2), 66.7% (six out of nine) of HGMS cases arose in the trunk, while only 22.2% (two out of nine) involved head/neck. This anatomical divergence from LGMS patterns may reflect distinct biological behavior or ascertainment bias. Tumor size averaged 10.7 cm (range: 1.2–20 cm), aligning with aggressive sarcomas. The etiological analysis identified only one radiation-associated case (postprostate radiotherapy) (10), underscoring the need for larger cohorts to better delineate risk factors.
Immunohistochemical profiling revealed variable expression of desmin (11–13, 15), SMA (6, 11–13, 15, 16), and vimentin (13, 16), with no pathognomonic markers identified. The molecular mechanisms underlying HGMS remain incompletely understood due to its rarity; however, emerging evidence implicates several key pathways: (1) Anaplastic lymphoma kinase (ALK) dynamics—while ALK fusions characterize inflammatory myofibroblastic tumors (IMT) (17), HGMS typically lacks rearrangements. However, rare cases of ALK overexpression suggest a potential progression from precursors with ALK alterations. (2) TP53 inactivation: studies have shown that TP53 mutations or deletions are common in undifferentiated pleomorphic sarcoma (UPS) (18, 19), leiomyosarcoma (LMS) (19, 20), and osteosarcoma (21). These genetic alterations correlate with both shorter disease-free survival and increased response to anthracyclines in sarcomas (19). (3) MDM2 amplification: although this TP53 antagonist is prevalent in liposarcoma and osteosarcoma (22), no detectable amplification was found in our cohort; however, systematic screening remains warranted. (4) Phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR) activation: while direct mechanistic evidence is lacking, dysregulation of this pathway in sarcomas (23, 24) may contribute to HGMS progression.
Notably, our case excluded DICER1 mutations (associated with pediatric central nervous system tumors) (25–27), EWSR1 rearrangements (a hallmark of Ewing sarcoma) (28), and MDM2 amplification (common in liposarcoma and osteosarcoma) (22), highlighting the molecular distinctiveness of HGMS.
In our study, complete surgical resection with negative margins (achieved in 55.6% of cases) was the cornerstone of management, with three out of five resected patients remaining disease-free at 6–24 months. Adjuvant RT and chemotherapy (doxorubicin-based regimens in 55.6%) appeared beneficial, with only one reported mortality. These findings align with sarcoma management principles, where wide excision and multimodal therapy optimize outcomes.
HGMS resides within the fibroblastic or myofibroblastic spectrum, which encompasses a group of rare tumor types with often-overlapping clinicopathologic features, ranging in biological potential from benign to overtly malignant (4, 29). Among these, IMT is a common type of intermediate (locally aggressive) tumor composed of myofibroblasts, frequently harboring ALK or ROS1 fusions (17). Local recurrence is common, and ALK inhibitors (e.g., crizotinib) have shown promise in advanced cases. Adult-type fibrosarcoma is a high-grade spindle cell sarcoma characterized by a herringbone architecture, poor prognosis, and a lack of targetable drivers (4). Myxofibrosarcoma, a malignancy with curvilinear vessels and myxoid stroma, often exhibits complex genomic alterations and a high propensity for local recurrence (30). Unlike LGMS, which lacks recurrent cytogenetic alterations (29), HGMS demonstrates TP53/MDM2/PI3K axis perturbations, suggesting a divergent pathogenesis.
Although this study represents the largest aggregation of HGMS cases, it inherits limitations inherent to retrospective case synthesis, including heterogeneous reporting, selection bias, and insufficient molecular profiling. Tumor heterogeneity and therapeutic variability hinder definitive conclusions regarding prognostic factors. Therefore, prospective registries integrating next-generation sequencing (e.g., for TP53 and PI3K pathway alterations) and standardized treatment protocols are essential. These approaches are crucial for transforming HGMS from a poorly understood condition to a malignancy with evidence-based management strategies.
In conclusion, our analysis advances the characterization of HGMS as a trunk-predominant sarcoma affecting older men, necessitating aggressive resection and multimodal therapy. The molecular overlaps with TP53-driven sarcomas and PI3K pathway activation provide a rationale for targeted therapy trials. The presented intracranial HGMS case—potentially the first report—further illustrates the tumor’s anatomical versatility. Only through international collaboration and molecular profiling can this enigmatic entity transition from histopathological curiosity to a biologically defined therapeutic target.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
XH: Conceptualization, Validation, Visualization, Writing – original draft, Writing – review & editing, Data curation, Methodology. ML: Data curation, Formal Analysis, Methodology, Supervision, Writing – review & editing. JY: Data curation, Investigation, Software, Writing – original draft. JH: Data curation, Formal Analysis, Project administration, Software, Writing – review & editing. XD: Methodology, Project administration, Resources, Supervision, Writing – original draft. YY: Data curation, Formal Analysis, Resources, Writing – review & editing. HZ: Conceptualization, Investigation, Methodology, Resources, Validation, Visualization, Writing – review & editing. FW: Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Coffin CM, Alaggio R. Fibroblastic and myofibroblastic tumors in children and adolescents. Pediatr Dev Pathol. (2012) 15:127–80. doi: 10.2350/10-12-0944-PB.1
2. Yonezawa H, Yamamoto N, Hayashi K, Takeuchi A, Miwa S, Igarashi K, et al. Low-grade myofibroblastic sarcoma of the levator scapulae muscle: a case report and literature review. BMC Musculoskelet Disord. (2020) 21:836. doi: 10.1186/s12891-020-03857-3
3. Obeidin F. Grading. PathologyOutlines.com . Available online at: https://www.pathologyoutlines.com/topic/softtissuegrading.html (Accessed October 17, 2024).
4. Sbaraglia M, Bellan E, Dei Tos AP. The 2020 WHO Classification of Soft Tissue Tumours: news and perspectives. Pathologica. (2020) 113:70–84. doi: 10.32074/1591-951X-213
5. Fisher C. Myofibroblastic Malignancies. Adv Anatomic Pathol. (2004) 11:190–201. doi: 10.1097/01.pap.0000131773.16130.aa
6. Zhao R, Wang J, Zhang H, Chi Y, Bi N. High-grade myofibroblastic sarcoma of the pleura: A case report and literature review. Thorac Cancer. (2020) 11:3011–4. doi: 10.1111/1759-7714.13613
7. Vasudev KS, Harris M. A sarcoma of myofibroblasts: an ultrastructural study. Arch Pathol Lab Med. (1978) 102:185–8.
8. Mentzel T, Dry S, Katenkamp D, Fletcher CDM. Low-grade myofibroblastic sarcoma: analysis of 18 cases in the spectrum of myofibroblastic tumors. Am J Surg Pathol. (1998) 22:1228–38. doi: 10.1097/00000478-199810000-00008
9. Anderson WJ, Doyle LA. Updates from the 2020 world health organization classification of soft tissue and bone tumours. Histopathology. (2021) 78:644–57. doi: 10.1111/his.14265
10. Joerger M, Oehlschlegel C, Cerny T, Gillessen S. Postradiation high-grade myofibroblastic sarcoma of the prostate – a rare entity of prostatic tumors – responding to liposomal doxorubicin. Oncol Res Treat. (2002) 25:558–61. doi: 10.1159/000068628
11. Koga S, Ikeda S, Urata J, Chijiwa R, Abe K, Hayashi T, et al. Primary high-grade myofibroblastic sarcoma arising from the pericardium. Circ J. (2008) 72:337–9. doi: 10.1253/circj.72.337
12. Anastasiou I, Levis PK, Katafigiotis I, Karaolanis G, Palla V-V, Felekouras E, et al. High grade myofibroblastic sarcoma of paratesticular soft tissues. Case Rep Oncol Med. (2014) 2014:1–3. doi: 10.1155/2014/768379
13. Wen J, Zhao W, Li C, Shen J-Y, Wen T-F. High-grade myofibroblastic sarcoma in the liver: A case report. World J Gastroenterol. (2017) 23:7054–8. doi: 10.3748/wjg.v23.i38.7054
14. Sharma M, Lee H, Madrigal FC, Terry MJ, Hattab EM, Altstadt T. High-grade myofibroblastic sarcoma involving the thoracic spine: A rare entity. Clin Neurol Neurosurg. (2020) 199:106307. doi: 10.1016/j.clineuro.2020.106307
15. Velez Torres JM, Martinez Duarte E, Diaz-Perez JA, Leibowitz J, Weed DT, Thomas G, et al. Primary sarcomas of the larynx: A clinicopathologic study of 27 cases. Head Neck Pathol. (2021) 15:905–16. doi: 10.1007/s12105-021-01314-8
16. Harada T, Togawa T, Miyamoto H, Matsushita Y, Hatachi Y. A unique case of high-grade myofibroblastic sarcoma initially presenting with oral symptoms. Clin Case Rep. (2023) 11:e7218. doi: 10.1002/ccr3.7218
17. Hunt AL, Nutcharoen A, Randall J, Papazian A, Deeken J, Maxwell GL, et al. Integration of multi-omic data in a molecular tumor board reveals EGFR-associated ALK-inhibitor resistance in a patient with inflammatory myofibroblastic cancer. Oncologist. (2023) 28:730–6. doi: 10.1093/oncolo/oyad129
18. Cui Y, Han L, Shang J, Fang W, Zhao M, Chen D, et al. Primary cardiac undifferentiated pleomorphic sarcoma is associated with TP53 mutation during lack of MDM2 amplification, and targeted sequencing analysis reveals potentially actionable targets. Hum Pathol. (2022) 123:113–22. doi: 10.1016/j.humpath.2022.02.006
19. Nassif EF, Auclin E, Bahleda R, Honoré C, Mir O, Dumont S, et al. TP53 mutation as a prognostic and predictive marker in sarcoma: Pooled analysis of MOSCATO and ProfiLER precision medicine trials. Cancers (Basel). (2021) 13:3362. doi: 10.3390/cancers13133362
20. Yang C-Y, Liau J-Y, Huang W-J, Chang Y-T, Chang M-C, Lee J-C, et al. Targeted next-generation sequencing of cancer genes identified frequent TP53 and ATRX mutations in leiomyosarcoma. Am J Transl Res. (2015) 7:2072–81.
21. Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. (2014) 7:104–12. doi: 10.1016/j.celrep.2014.03.003
22. Sciot R. MDM2 amplified sarcomas: A literature review. Diagn (Basel). (2021) 11:496. doi: 10.3390/diagnostics11030496
23. Wani AK, Singh R, Akhtar N, Prakash A, Nepovimova E, Oleksak P, et al. Targeted inhibition of the PI3K/akt/mTOR signaling axis: Potential for sarcoma therapy. Mini Rev Med Chem. (2024) 24:1496–520. doi: 10.2174/0113895575270904231129062137
24. Hiraki-Hotokebuchi Y, Yamada Y, Kohashi K, Yamamoto H, Endo M, Setsu N, et al. Alteration of PDGFRβ-akt-mTOR pathway signaling in fibrosarcomatous transformation of dermatofibrosarcoma protuberans. Hum Pathol. (2017) 67:60–8. doi: 10.1016/j.humpath.2017.07.001
25. Cardona AF, Chamorro Ortiz DF, Ruíz-Patiño A, Gomez D, Muñoz Á, Ardila DV, et al. DICER1-associated central nervous system sarcoma: A comprehensive clinical and genomic characterization of case series of young adult patients. Neurooncol Pract. (2023) 10:381–90. doi: 10.1093/nop/npad014
26. Kamihara J, Paulson V, Breen MA, Laetsch TW, Rakheja D, Shulman DS, et al. DICER1-associated central nervous system sarcoma in children: Comprehensive clinicopathologic and genetic analysis of a newly described rare tumor. Mod Pathol. (2020) 33:1910–21. doi: 10.1038/s41379-020-0516-1
27. Kosteniuk SE, Michaiel G, Dunham C. A case of primary intracranial sarcoma, DICER1-mutant, in a child with a germline DICER1 mutation. Brain Sci. (2023) 13:1040. doi: 10.3390/brainsci13071040
28. Flucke U, van Noesel MM, Siozopoulou V, Creytens D, Tops BBJ, van Gorp JM, et al. EWSR1-the most common rearranged gene in soft tissue lesions, which also occurs in different bone lesions: An updated review. Diagn (Basel). (2021) 11:1093. doi: 10.3390/diagnostics11061093
29. Baranov E, Hornick JL. Soft tissue special issue: Fibroblastic and myofibroblastic neoplasms of the head and neck. Head Neck Pathol. (2020) 14:43–58. doi: 10.1007/s12105-019-01104-3
Keywords: primary intracranial, high-grade myofibroblastic sarcoma, resection, radiotherapy, case report
Citation: He X, Lv M, Yuan J, He J, Du X, Yang Y, Zhang H and Wen F (2025) Case Report: Primary intracranial high-grade myofibroblastic sarcoma and literature review. Front. Oncol. 15:1525401. doi: 10.3389/fonc.2025.1525401
Received: 20 December 2024; Accepted: 11 March 2025;
Published: 02 April 2025.
Edited by:
Vinay Kumar, The Pennsylvania State University, United StatesReviewed by:
Vishakha Anand Pawar, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2025 He, Lv, Yuan, He, Du, Yang, Zhang and Wen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Wen, d2VuZmVuZzA2NTVAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.