
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol., 18 February 2025
Sec. Pediatric Oncology
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1514697
 Jakub Czarny1
Jakub Czarny1 Dominika Galli1
Dominika Galli1 Agnieszka Wziątek2
Agnieszka Wziątek2 Agata Pastorczak3
Agata Pastorczak3 Bartosz Szmyd3Borys Przybyszewski4
Bartosz Szmyd3Borys Przybyszewski4 Anna Raciborska4
Anna Raciborska4 Katarzyna Jończyk-Potoczna5
Katarzyna Jończyk-Potoczna5 Katarzyna Derwich2*
Katarzyna Derwich2*Germinal predisposition to malignancy is found in approximately 10% of oncological pediatric patients. As awareness of cancer risk factors associated with germline mutations increases, and with advancements in molecular techniques, more carefully selected patients are being tested. This approach enables the identification of new variants—both those that are clearly linked to tumorigenesis and candidates, which biological role needs to be functionally verified. Pathogenic variants within cancer-predisposing genes not only increase nearly eightfold the risk of secondary cancers but also may be associated with excessive toxicity of antineoplastic treatment. We present the case of a girl who developed classical Hodgkin lymphoma at the age of 8 years and secondary Ewing sarcoma at the age of 16 years. Her father was diagnosed with classical Hodgkin lymphoma at the age of 27 years. Genetic testing revealed the carriership of a germline heterozygous variant in the PALB2 gene (NM_024675.4:c.110G>A, p.Arg37His) in both the patient and her father. Since the patient was exposed to chemotherapy due to lymphoma prior to the development of secondary malignancy and the variant is classified as an aberration of unknown significance, the causative role of the PALB2 variant remains uncertain. Nevertheless, the presented case may indicate the possible interplay between inherited genetic predisposition and the exposure to cytostatic drugs, which both are involved in promoting secondary cancers in pediatric patients.
Germline predisposition to malignancies concerns approximately 10% of childhood cancer patients (1–3). However, due to rising awareness, development of molecular techniques, and their broad application to the clinical testing, the role of inborn susceptibility to malignancies in children is recognized as a significant factor promoting the development of childhood neoplasms. Pediatric patients are selected for testing of genetic predisposition to cancer using defined clinical criteria indicating on the increased genetic susceptibility to malignancy. This includes at least two malignancies in childhood (4, 5), at least one first-degree and/or two second-degree (on the same side of the family) patients with cancer under 45 years, bilateral neoplasms, parental consanguinity, specific histological types of malignancies particularly associated with germline genetic defects, excessive toxicities of oncological treatment, comorbidities (congenital anomalies, facial dysmorphisms, intellectual disability, aberrant growth, skin anomalies, hematological disorders, or immune deficiency), and secondary malignancies during childhood. Such probands require complex genetic testing using appropriate next-generation sequencing (NGS) panels, which will unravel potential causes of carcinogenesis (2, 6). The diagnosis of genetic predisposition to malignancy is beneficial both for the child with already diagnosed malignancy, as well as for their families. First, it enables introduction of the oncological surveillance and early detection of secondary neoplasms, as well as predicting potential increased toxicity and resistance to standard treatment and modifying treatment schedules. Patients’ family members may benefit from genetic counselling, oncological screening, early malignancy detection, reproductive counseling, and proper prenatal diagnosis (2, 6).
We present the case of a pediatric patient who developed Hodgkin lymphoma at the age of 8 years and secondary Ewing sarcoma at the age of 16 years with a positive family history of cancer, who carries a germline heterozygous variant of unknown significance in the PALB2 gene.
The 8-year-old female patient was referred to the pediatric oncology clinic due to cervical lymphadenopathy concurring with pharyngitis and B symptoms. The birth history was unremarkable. The patient does not present any dysmorphic features, as well as immune deficiencies, both at laboratory and clinical levels.
During the diagnostic process, chest computed tomography (CT) revealed a well-defined, heterogeneous mediastinal tumor measuring 10.8 cm × 6.0 cm × 5.3 cm (cc × ap × ds), exhibiting contrast enhancement. The tumor adhered to the anterior chest wall, extending from the superior thoracic aperture to the level of Th8. It was in communication with the enlarged cervical lymph nodes and involved the right lung cavity without enlargement of the left hilar lymph nodes (Figure 1). Abdominal CT revealed multiple hypodense focal lesions in the spleen. Positron emission tomography (PET) CT confirmed a metabolically active conglomerate of right neck lymph nodes, a mediastinal tumor, as well as increased uptake in the spleen lesions.
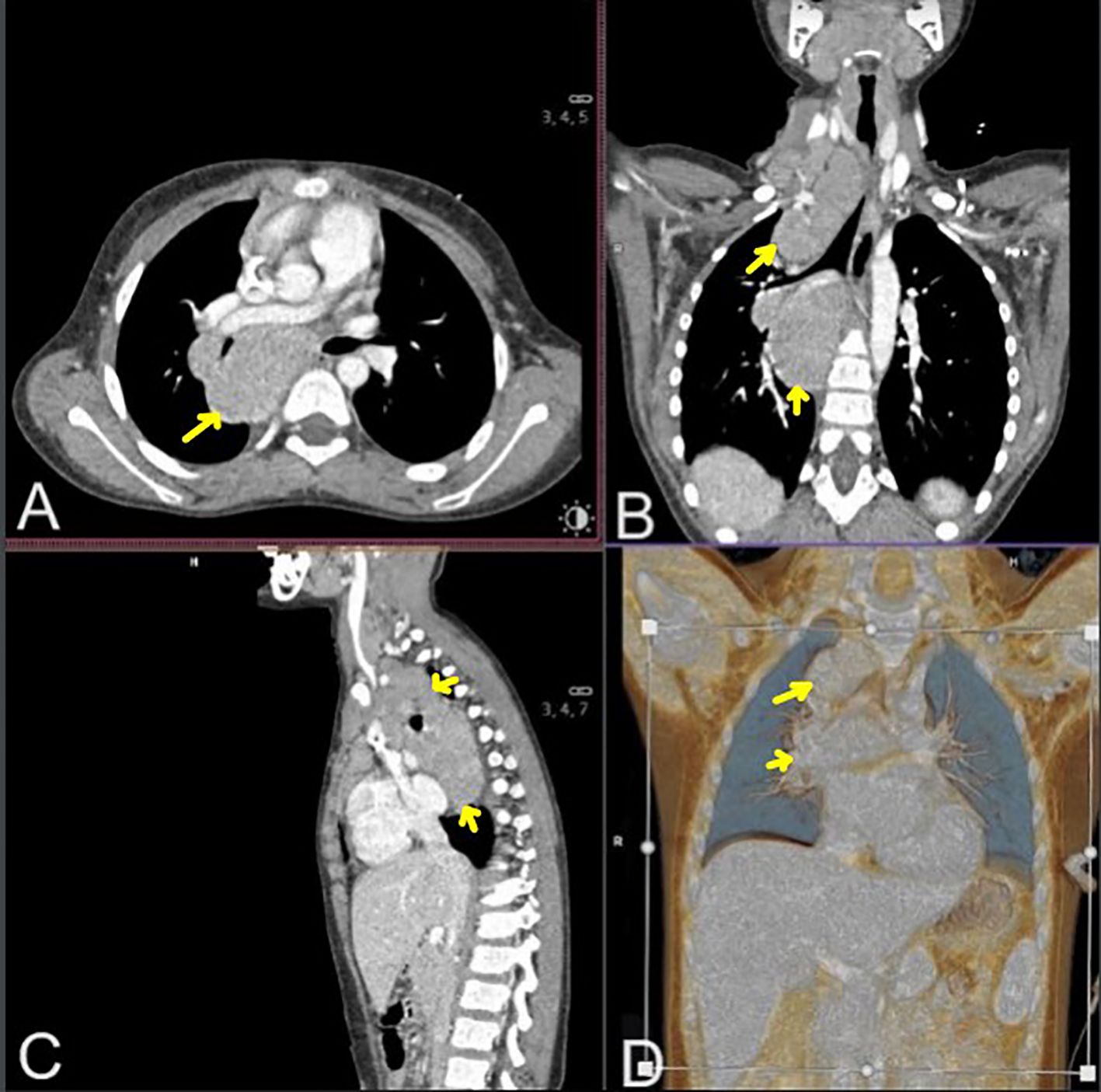
Figure 1. Chest and neck computed tomography scans in mediastinal window after contrast agent administration: transverse (A), coronal [(B) multiplanar reconstruction, (D) volume rendering technique reconstruction], and sagittal (C) section. Neck and mediastinal Hodgkin lymphoma masses are marked with yellow arrows.
A lymph node biopsy was performed. Finally, the patient was diagnosed with classical Hodgkin lymphoma, nodular sclerosis type, IIIB grade, and was qualified to the therapeutic group TL-3 according to the EuroNET-PHL-C1 treatment protocol.
The patient was administered two chemotherapy cycles of OEPA. Due to the adequate response according to the protocol, she received four cycles of COPDAC-28 and did not require radiotherapy. Complete remission was achieved. The patient was remaining under the care of the outpatient children’s oncology clinic.
Five years after the protocol completion, at the age of 16 years, she started complaining of leg pain and lower back pain. Magnetic resonance imaging (MRI) of the lumbosacral spine revealed a well-defined lesion in the S1 vertebral body, located medially on the left side, measuring 38 mm × 20 mm × 22 mm (ds × ap × cc). Within the spinal canal, compression of the cauda equina threads and segmental destruction of the cortical layer of the anterior surface of the vertebral body were visible (Figure 2). Thoracic CT revealed a well-defined, round, solid lesion measuring 10 cm × 10 cm × 11 cm (ds × ap × cc) on the left side, located medially, paravertebrally, and along the posterior-lateral chest wall. The lesion appeared to potentially originate from the sixth left rib. At the level of the lesion, the image showed destruction (osteolysis, destruction of the cortical layer, and swelling) of the posterior segment of the sixth left rib over a length of about 6 cm. The lesion penetrated the adjacent intercostal spaces at this level and intercostal muscles. Medially, the tumor adhered to the spine, causing a slight curvature of the spine to the right side with penetration into the C6/C7 intervertebral foramen. The tumor caused the displacement of mediastinal structures to the right side with forward displacement and narrowing of the left main bronchus (Figure 3). Moreover, after the second cycle of chemotherapy, three-phase scintigraphy of the skeletal system with 99mTc-MDP of activity 13 mCi revealed in the phase III whole-body and thoracic SPECT and pelvic-targeted SPECT/CT increased tracer accumulation also in the upper lateral part of the L3 vertebral body on the right side, in the head of the left femur, as well as in the projection of the distal femoral epiphyses and proximal tibial epiphysis with marked asymmetry (left over right).
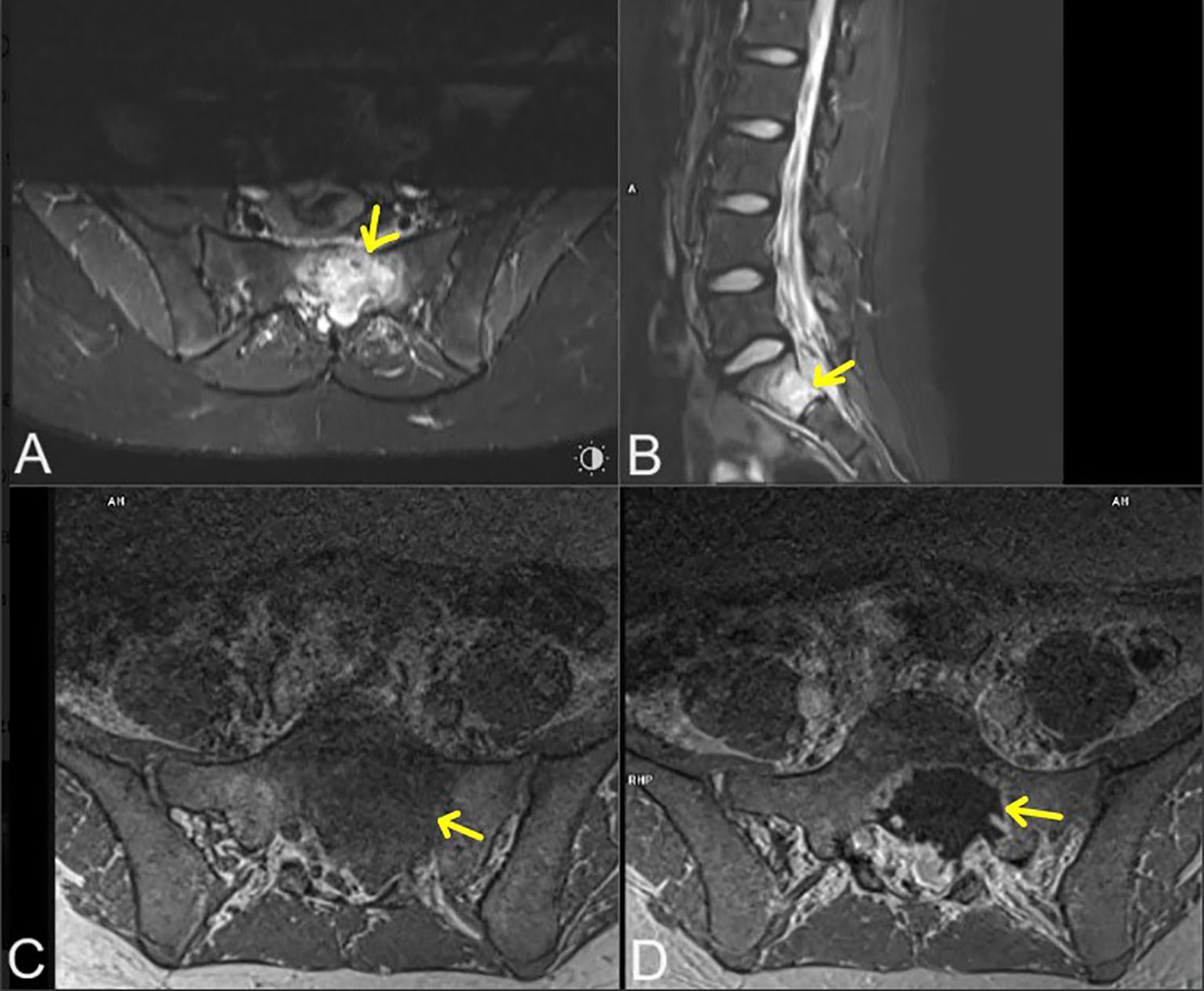
Figure 2. Lumbosacral magnetic resonance imaging in: T2-weighted (STIR) axial projection (A), T2-weighted (STIR) sagittal (B), T1-weighted (vibe) axial (C), T1-weighted (vibe) axial with contrast agent administration (D). Metastatic lesions marked with yellow arrows.

Figure 3. Chest computed tomography (mediastinal window after contrast agent administration- transverse [multiplanar reconstruction (A), volume rendering technique reconstruction (D)], coronal [volume rendering technique reconstruction (B)] and sagittal [multiplanar reconstruction (C) section]. Primary tumor mass marked with yellow arrows.
A biopsy of the chest wall tumor was performed and Ewing sarcoma was detected. Bone marrow aspiration biopsy and trepanobiopsy excluded bone marrow involvement.
She underwent preoperative chemotherapy cycles of vincristine-doxorubicin-cyclophosphamide (the fourth adjuvant cycle did not include doxorubicin due to the planned radiotherapy) and ifosfamid-etopophos (due to an allergic reaction to etoposide). After the fourth cycle of chemotherapy, VMAT radiotherapy was administered to the area of metastatic lesions in the sacrum, L3 vertebra, and left femur at a total dose of 3,600 cGy. Preoperative imaging tests (MRI, PET-CT) showed a good response to the induction therapy. Subsequently, a resection of the tumor was performed (the sixth rib and part of the seventh rib on the left side were removed). Next, she continued the adjuvant chemotherapy, as well as VMAT breast-preserving radiotherapy, which was distributed to the lodge of the tumor with the margin at a dose of 4,500 cGy. Currently, the patient remains in complete remission under the care of the pediatric oncology outpatient clinic.
In the biopsy material, FoundationOne® CDx NGS panel detected EWSR1::FLI1 fusion and the following sequence variants: in PALB2 gene (p.Arg37His, in SGK1 gene (p.Ala40fs*21), in TSC2 gene (p.Ala357Val), in MRE11A gene (p.Ile120Val), in PIM1 gene (p.Glu124Gln), in RPTOR gene (p.Ala862Thr). No tumor mutational burden and microsatellite instability were revealed.
Given the family’s positive history of lymphoma, the patient was offered genetic testing for genetic predisposition to malignancy. We performed trio exome sequencing using germinal DNA isolated from peripheral blood collected at remission from the patient and her parents. The analysis was performed using the hg19 reference genome and the DRAGEN Germline Pipeline (basespace.com). The variants, which are shared only by the child and the father, were firstly filtered based on a list of genes related to childhood cancer risk and associated with immunological and/or hematological abnormalities. We analyzed only variants observed in more than seven, which were rarer than 5% in the obtained databases. We identified two possibly causative heterozygous alterations in proband and father: PALB2 (NM_024675.4:c.110G>A; p.Arg37His) and DDX41 (NM_016222.4:c.299-3C>Tp.)? (Figure 4, Supplementary Table S1). As the variant in DDX41 could probably lead to lymphoproliferation (7) through affecting splice junctions, we assessed splicing based on patients’ mRNA, but the effect on splicing was not proved. Therefore, in the further analysis, we focused on the PALB2 variant. Bioinformatic analysis showed its rarity—it was observed that only 10 alleles were observed among 251,458 total alleles, according to gnomAD Exomes Version: 2.1.1 encompassing the following subpopulations comprise: African/African-American, Remaining, Admixed American, European (non-Finnish), South Asian, European (Finnish), Ashkenazi Jewish, and East Asian. Remarkably, there were no reported homozygous cases for this variant, further underscoring its uncommon nature (GnomAD and ExAC databases). Furthermore, the variant’s conservation score, as assessed by both PhastCons100way (1.000) and PhyloP100way (4.318), points to its biological significance. Clinical relevance is underscored by the analysis of ClinVar submissions, where 11 of 12 entries related to hereditary cancer syndromes classified the variant as a Variant of Uncertain Significance (VUS), while only one submission suggested a likely benign nature. The majority of these reports are grounded in clinical assessments of germline DNA. In-silico predictions showed a nuanced view. Two predictors (DANN and SIFT) support a pathogenic interpretation, while a significant number indicate uncertain significance, and 10 suggest benign or moderately supportive evidence of benignity (see Supplementary Table S1 for further information). Conversely, meta-analyses of in-silico predictors, which integrate evidence across various computational tools, lean toward a benign classification, with all assessed meta-scores offering moderate or supportive evidence for a benign interpretation (see Supplementary Table S1). Familial segregation analysis revealed that both variants were present not only in the patient with lymphoma but also in two of the proband’s brothers (currently at the ages 6 and 9 years), who have no history of neoplasms to date after the evaluation of the pediatric oncologist. This could be attributed to the variable degree of penetrance and the relatively young age of these boys. Considering these diverse and sometimes conflicting findings, we conducted a comprehensive literature review to further explore the implications of these variants, which is discussed in detail in the subsequent sections of this paper.
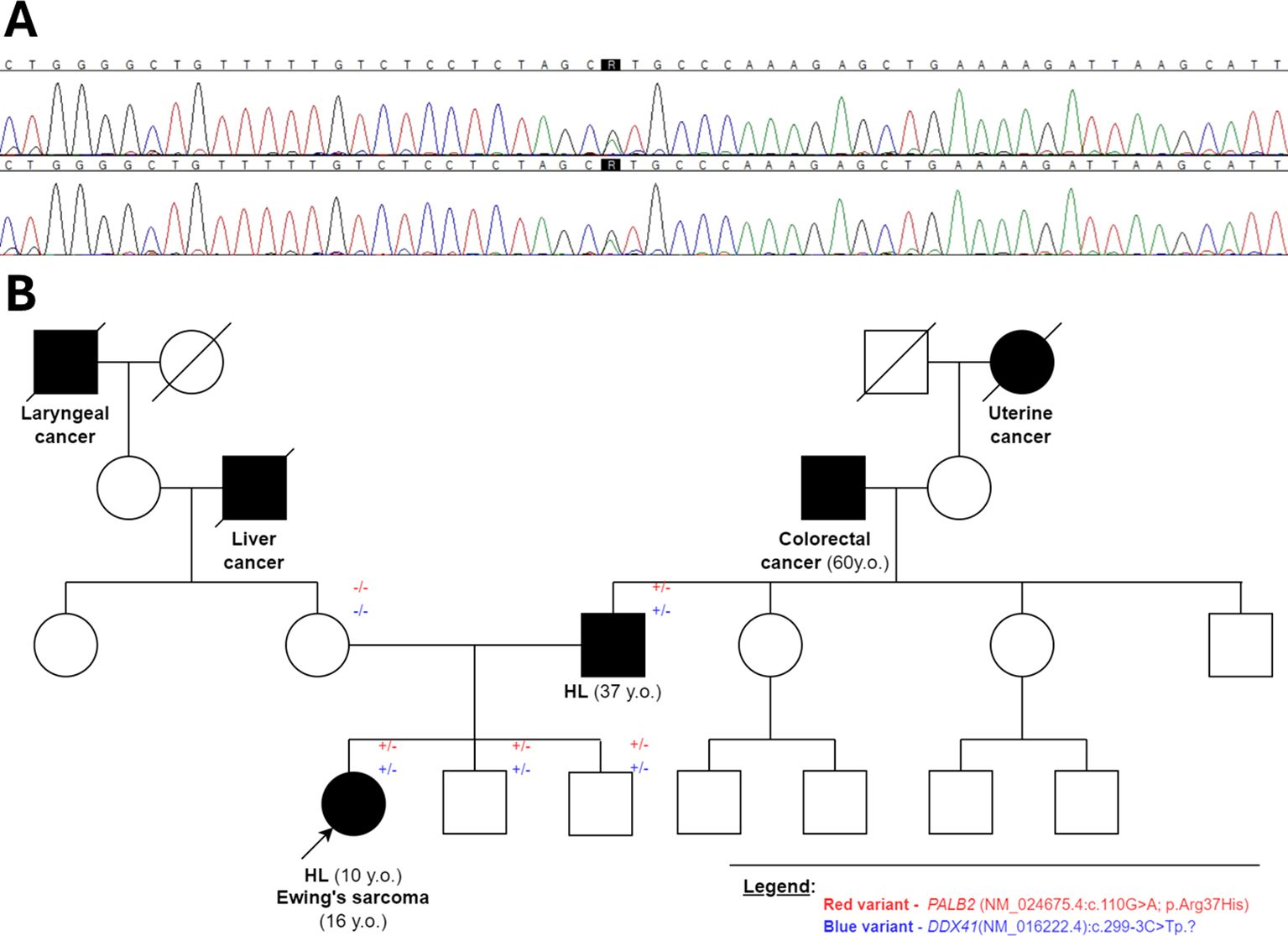
Figure 4. (A) Chromatogram confirming the presence of heterozygous missense variant (NM_024675.4):c.110G>A;p.Arg37His within the PALB2 gene sequence in the child and the father; (B) the pedigree showing the carriership of identified variants in proband’s family members.
Considering all of the above, according to the ACMG classification (8), we observed one strong pathogenic criterion and two supporting ones, along with one moderate benign criterion and two supporting benign criteria. This combination results in a total score of two points, classifying the variant as one of uncertain significance (see Table 1 for further details).

Table 1. The classification of variant significance according to the American College of Medical Genetics Criteria.
We describe the patient who suffered from Hodgkin lymphoma and met two criteria of inclusion to testing for inherited cancer predisposition: diagnosis of secondary Ewing sarcoma during childhood and positive family history of cancer (4, 5, 9, 10). The patient is a carrier of a germline variant of uncertain significance within the PALB2 gene. Although variants in this gene are not widely linked to increased lymphoma risk, its dysfunction, according to functional studies, may affect DNA repair and therefore promote tumorigenesis (11). In fact, genetic susceptibility to lymphomas is associated with various inborn errors, especially in primary immunodeficiencies and DNA repair disorders. Regarding genetic aberrations predisposing to sarcomas, the American College of Medical Genetics indicates the need of genetic testing among patients who developed non-Ewing sarcomas during childhood, in case of the incidence of sarcoma and other Li-Fraumeni syndrome-associated tumors in one family member or two close relatives at the age up to 45 years (8). However, some studies have reported that heterozygous pathogenic or potentially pathogenic germline mutations in DNA repair genes, for example, FANCC, FANCA, ERCC2, and BRCA1, may contribute to Ewing sarcomas because they enable the occurrence of DNA breaks leading to oncogenic gene fusions, such as EWSR1::ETS (12–15). Moreover, germline mutations in the CHEK2 gene were overrepresented in some cohorts of patients diagnosed with Ewing sarcoma (13, 16). In addition, Qin et al. revealed that mutations involving homologous recombination (HR) genes are associated with an increased risk of subsequent sarcoma after treatment with alkylating agents in the third tertile (17). Our patient was administered dacarbazine and cyclophosphamide due to Hodgkin lymphoma, which could have a significant impact on the development of secondary malignancy.
PALB2 (also known as partner and localizer of BRCA2 or FANCN) is crucial for DNA repair through HR, collaborating with key effectors like BRCA1, BRCA2, RAD51, and RAD51C at DNA damage sites. Disruption of PALB2’s structure leads to genomic instability by impairing HR repair, thereby elevating the tumor mutational burden (18–21). Thus, PALB2 functions as a tumor suppressor. Heterozygous germline variants in PALB2 significantly increase the risk for various cancers, including breast (22–32), ovarian (22, 24, 28, 31), pancreatic (22, 24, 30, 33), prostate (22, 34), colorectal (35) cancer, and so forth, with a significant decrease in these patients’ survival (34). Moreover, studies show a good response to the treatment of HR deficient (including PALB2-mutated tumors) with PARP inhibitors and platinum-based chemotherapy (21, 36–42). PALB2 gene variants with defective HR are associated with higher sensitivity to cisplatin and PARP inhibitors (40). The role of PALB2 variants in the pathogenesis of sarcomas has not been proved. However, PALB2 variant has been observed in the patient treated due to sarcoma who also carried the variants within PALB2 and MITF genes (43). This case highlights again that adult-onset cancer predisposition genes germline variants may possibly influence the presence of pediatric-onset neoplasms.
The variant detected in our patient is localized at the second exonic base from the 3’ site (44). It was previously reported in a Spanish family with non-BRCA1/BRCA2 early breast/ovarian cancer family history and the incidence of pancreatic cancer (44), as well as in a patient with unilateral breast cancer from the WECARE study (11). The functional analysis of this missense variant classified it as probably damaging, altering protein function. Two of the three prediction programs considered the variant as deleterious. The HR activity of PALB2 was proved to be impaired in this case but without the confirmed reduction of the PALB2-BRCA1 interaction. However, the decrease in the score predicted by the algorithms, the lack of variations near cryptic splice sites, and RNA analysis suggest a variant as a variant without a significant impact on the splicing process (43, 44). The variant may influence PALB2 function, therefore exacerbating DNA repair and changing the potential cancer risk after the exposure to cytostatic drugs (11, 42).
Notably, a heterozygous pathogenic PALB2 variant has been documented in cases of acute lymphoblastic leukemia in a 6-year-old patient diagnosed with acute lymphoblastic leukemia and secondary Ewing sarcoma at the age of 12 years, originating from a family with a notable history of cancer across both paternal and maternal lineages. Genetic analysis revealed a heterozygous constitutional deletion within the PALB2 gene (NM_024675.3:c.(1684 + ?1685-)?(2586 +?_2587-)?del). This deletion encompassed entire exons 5 and 6, leading to the premature termination of mRNA translation or the production of a truncated, dysfunctional PALB2 protein. Unfortunately, information on the familial segregation of this variant remains unavailable, precluding further insights into its hereditary transmission and phenotypic consequences of the variant (45).
Our findings reveal a variant situated at the second exonic base from the 3’ end. Initial functional analysis indicates that the R37H variant diminishes PALB2’s HR activity. This reduction suggests potential disruption to the coiled-coil motif’s integrity, implying that even minimal structural distortions could impair HR activity without necessarily affecting BRCA1 binding (11). Remarkably, this variant is the first to demonstrate a relative HR efficiency below 50%, significantly higher than the ~10% or less efficiency observed in truncating variants (46). The penetrance of this genetic variant and its role in phenotype severity remain poorly understood, necessitating further study. However, the observed decrease in HR efficiency is associated with an enhanced response to PARP inhibitors, suggesting potential meaning for therapeutic intervention (47).
Finally, it should be mentioned that the discussed variant was observed in two brothers without cancer. They are currently 6 and 9 years old, which means they are younger than their sister at the time of her cancer development and significantly younger than their father, who developed HL at the age of 37. In the further analysis, we have to consider not only age-related cancer risk but also hormonal factors (cancer risk can be influenced by hormonal changes or developmental processes that occur later in life; the brothers may not yet have reached this stage), possibly environmental factors, as well as the impact of modifier genes (the brothers may carry protective genetic variants in other genes that modulate the effects of the PALB2 variant, reducing their overall cancer susceptibility) juxtaposed with the impact of epigenetic regulation.
The reported patient exemplifies the possible interplay between germline genetic predisposition to cancer and exposure to chemical compounds in promoting secondary tumors during childhood. It suggests that the causative role of variants in adult cancer-predisposing genes in the development of pediatric malignancies needs to be functionally studied.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethical approval was not required for the studies involving humans because all data come from the medical diagnosis process carried out on the described patient and her family- they gave informed consent for each procedure. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from a by-product of routine care or industry. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
JC: Conceptualization, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. DG: Conceptualization, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. AW: Conceptualization, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. AP: Writing – original draft, Writing – review & editing. BS: Writing – original draft, Writing – review & editing. BP: Writing – review & editing. AR: Writing – review & editing. KJ-P: Writing – review & editing. KD: Conceptualization, Formal analysis, Methodology, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The genetic testing was funded by the Ministry of Education and Science under the “Diamond Grant” program, grant number 0136/DIA/2020/49.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1514697/full#supplementary-material
1. Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. (2015) 373:2336–46. doi: 10.1056/NEJMoa1508054
2. Rahman N. Realising the promise of cancer predisposition genes. Nature. (2014) 505:302–8. doi: 10.1038/nature12981
3. Jongmans MCJ, Loeffen JLCM, Waanders E, Hoogerbrugge PM, Ligtenberg MJL, Kuiper RP, et al. Recognition of genetic predisposition in pediatric cancer patients: An easy-to-use selection tool. Eur J Med Genet. (2016) 59:116–25. doi: 10.1016/j.ejmg.2016.01.008
4. Waespe N, Strebel S, Marino D, Mattiello V, Muet F, Nava T, et al. Predictors for participation in DNA self-sampling of childhood cancer survivors in Switzerland. BMC Med Res Methodol. (2021) 21:236. doi: 10.1186/s12874-021-01428-1
5. Wang Z, Wilson CL, Easton J, Thrasher A, Mulder H, Liu Q, et al. Genetic risk for subsequent neoplasms among long-term survivors of childhood cancer. J Clin Oncol. (2018) 36:2078–87. doi: 10.1200/JCO.2018.77.8589
6. Kim J, Gianferante M, Karyadi DM, Hartley SW, Frone MN, Luo W, et al. Frequency of pathogenic germline variants in cancer-susceptibility genes in the childhood cancer survivor study. JNCI Cancer Spectr. (2021) 5:pkab007. doi: 10.1093/jncics/pkab007
7. Makishima H, Saiki R, Nannya Y, Korotev S, Gurnari C, Takeda J, et al. Germ line DDX41 mutations define a unique subtype of myeloid neoplasms. Blood. (2023) 141:534–49. doi: 10.1182/blood.2022018221
8. Hampel H, Bennett RL, Buchanan A, Pearlman R, Wiesner GL, Guideline Development Group, American College of Medical Genetics and Genomics Professional Practice and Guidelines Committee and National Society of Genetic Counselors Practice Guidelines Committee. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med. (2015) 17:70–87. doi: 10.1038/gim.2014.147
9. Waespe N, Belle FN, Redmond S, Schindera C, Spycher BD, Rössler J, et al. Cancer predisposition syndromes as a risk factor for early second primary neoplasms after childhood cancer - A national cohort study. Eur J Cancer. (2021) 145:71–80. doi: 10.1016/j.ejca.2020.11.042
10. Byrjalsen A, Diets IJ, Bakhuizen JJ, Thomas, Schmiegelow K, Gerdes AM, et al. Selection criteria for assembling a pediatric cancer predisposition syndrome gene panel. Familial Cancer. (2021) 20:279–87. doi: 10.1007/s10689-021-00254-0
11. Foo TK, Tischkowitz M, Simhadri S, Boshari T, Zayed N, Burke KA, et al. Compromised BRCA1-PALB2 interaction is associated with breast cancer risk. Oncogene. (2017) 36:4161–70. doi: 10.1038/onc.2017.46
12. Kratz CP, Jongmans MC, Cavé H, Wimmer K, Behjati S, Guerrini-Rousseau L, et al. Predisposition to cancer in children and adolescents. Lancet Child Adolesc Health. (2021) 5:142–54. doi: 10.1016/S2352-4642(20)30275-3
13. Gillani R, Camp SY, Han S, Jones JK, Chu H, O’Brien S, et al. Germline predisposition to pediatric Ewing sarcoma is characterized by inherited pathogenic variants in DNA damage repair genes. Am J Hum Genet. (2022) 109:1026–37. doi: 10.1016/j.ajhg.2022.04.007
14. Brohl AS, Patidar R, Turner CE, Wen X, Song YK, Wei JS, et al. Frequent inactivating germline mutations in DNA repair genes in patients with Ewing sarcoma. Genet Med. (2017) 19:955–8. doi: 10.1038/gim.2016.206
15. Ballinger ML, Goode DL, Ray-Coquard I, James PA, Mitchell G, Niedermayr E, et al. Monogenic and polygenic determinants of sarcoma risk: an international genetic study. Lancet Oncol. (2016) 17:1261–71. doi: 10.1016/S1470-2045(16)30147-4
16. van Tilburg CM, Pfaff E, Pajtler KW, Langenberg KPS, Fiesel P, Jones BC, et al. The pediatric precision oncology INFORM registry: clinical outcome and benefit for patients with very high-evidence targets. Cancer Discovery. (2021) 11:2764–79. doi: 10.1158/2159-8290.CD-21-0094
17. Newman S, Nakitandwe J, Kesserwan CA, Azzato EM, Wheeler DA, Rusch M, et al. Genomes for kids: the scope of pathogenic mutations in pediatric cancer revealed by comprehensive DNA and RNA sequencing. Cancer Discovery. (2021) 11:3008–27. doi: 10.1158/2159-8290.CD-20-1631
18. Pauty J, Rodrigue A, Couturier A, Buisson R, Masson JY. Exploring the roles of PALB2 at the crossroads of DNA repair and cancer. Biochem J. (2014) 460:331–42. doi: 10.1042/BJ20140208
19. Belotserkovskaya R, Raga Gil E, Lawrence N, Butler R, Clifford G, Wilson MD, et al. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nat Commun. (2020) 11:819. doi: 10.1038/s41467-020-14563-y
20. Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol. (2009) 19:524–9. doi: 10.1016/j.cub.2009.02.018
21. Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. (2017) 18:495–506. doi: 10.1038/nrm.2017.48
22. Nepomuceno TC, De Gregoriis G, de Oliveira FMB, Suarez-Kurtz G, Monteiro AN, Carvalho MA. The role of PALB2 in the DNA damage response and cancer predisposition. Int J Mol Sci. (2017) 18:1886. doi: 10.3390/ijms18091886
23. Tischkowitz M, Capanu M, Sabbaghian N, Li L, Liang X, Vallée MP, et al. Rare germline mutations in PALB2 and breast cancer risk: a population-based study. Hum Mutat. (2012) 33:674–80. doi: 10.1002/humu.22022
24. Yang X, Leslie G, Doroszuk A, Schneider S, Allen J, Decker B, et al. Cancer risks associated with germline PALB2 pathogenic variants: an international study of 524 families. J Clin Oncol. (2020) 38:674–85. doi: 10.1200/JCO.19.01907
25. Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkäs K, Roberts J, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. (2014) 371:497–506. doi: 10.1056/NEJMoa1400382
26. Yadav S, Boddicker NJ, Na J, Polley EC, Hu C, Hart SN, et al. Contralateral breast cancer risk among carriers of germline pathogenic variants in ATM, BRCA1, BRCA2, CHEK2, and PALB2. J Clin Oncol. (2023) 41:1703–13. doi: 10.1200/JCO.22.01239
27. Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. (2017) 3:1190–6. doi: 10.1001/jamaoncol.2017.0424
28. Kwong A, Shin VY, Ho CYS, Khalid A, Au CH, Chan KKL, et al. Germline PALB2 mutation in high-risk chinese breast and/or ovarian cancer patients. Cancers (Basel). (2021) 13:4195. doi: 10.3390/cancers13164195
29. Wesoła M, Jeleń M. The risk of breast cancer due to PALB2 gene mutations. Adv Clin Exp Med. (2017) 26:339–42. doi: 10.17219/acem/59147
30. Hofstatter EW, Domchek SM, Miron A, Garber J, Wang M, Componeschi K, et al. PALB2 mutations in familial breast and pancreatic cancer. Fam Cancer. (2011) 10:225–31. doi: 10.1007/s10689-011-9426–1
31. Velázquez C, Esteban-Cardeñosa EM, Lastra E, Abella LE, de la Cruz V, Lobatón CD, et al. A PALB2 truncating mutation: Implication in cancer prevention and therapy of Hereditary Breast and Ovarian Cancer. Breast. (2019) 43:91–6. doi: 10.1016/j.breast.2018.11.010
32. Evans MK, Longo DL. PALB2 mutations and breast-cancer risk. N Engl J Med. (2014) 371:566–8. doi: 10.1056/NEJMe1405784
33. Slater E, Langer P, Niemczyk E, Strauch K, Butler J, Habbe N, et al. PALB2 mutations in European familial pancreatic cancer families. Clin Genet. (2010) 78:490–4. doi: 10.1111/j.1399-0004.2010.01425.x
34. Wokołorczyk D, Kluźniak W, Stempa K, Rusak B, Huzarski T, Gronwald J, et al. PALB2 mutations and prostate cancer risk and survival. Br J Cancer. (2021) 125:569–75. doi: 10.1038/s41416-021-01410-0
35. Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. (2017) 3:464–71. doi: 10.1001/jamaoncol.2016.5194
36. Dillon KM, Bekele RT, Sztupinszki Z, Hanlon T, Rafiei S, Szallasi Z, et al. PALB2 or BARD1 loss confers homologous recombination deficiency and PARP inhibitor sensitivity in prostate cancer. NPJ Precis Oncol. (2022) 6:49. doi: 10.1038/s41698-022-00291-7
37. Nepomuceno TC, Carvalho MA, Rodrigue A, Simard J, Masson JY, Monteiro ANA. PALB2 variants: protein domains and cancer susceptibility. Trends Cancer. (2021) 7:188–97. doi: 10.1016/j.trecan.2020.10.002
38. Li A, Geyer FC, Blecua P, Lee JY, Selenica P, Brown DN, et al. Homologous recombination DNA repair defects in PALB2-associated breast cancers. NPJ Breast Cancer. (2019) 5:23. doi: 10.1038/s41523-019-0115-9
39. Staaf J, Glodzik D, Bosch A, Vallon-Christersson J, Reuterswärd C, Häkkinen J, et al. Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study. Nat Med. (2019) 25:1526–33. doi: 10.1038/s41591-019-0582-4
40. Principe DR. Precision medicine for BRCA/PALB2-mutated pancreatic cancer and emerging strategies to improve therapeutic responses to PARP inhibition. Cancers (Basel). (2022) 14:897. doi: 10.3390/cancers14040897
41. Cilento MA, Poplawski NK, Paramasivam S, Thomas DM, Kichenadasse G. Germline PALB2 variants and PARP inhibitors in endometrial cancer. J Natl Compr Cancer Netw. (2021) 19:1212–7. doi: 10.6004/jnccn.2021.7067
42. Wiltshire T, Ducy M, Foo TK, Hu C, Lee KY, Belur Nagaraj A, et al. Functional characterization of 84 PALB2 variants of uncertain significance. Genet Med. (2020) 22:622–32. doi: 10.1038/s41436-019-0682-z
43. de Carvalho N de A, Santiago KM, Maia JML, Costa FD, Formiga MN, Soares Dc de Q, et al. Prevalence and clinical implications of germline pathogenic variants in cancer predisposing genes in young patients across sarcoma subtypes. J Med Genet. (2024) 61:61–8. doi: 10.1136/jmg-2023-109269
44. Blanco A, de la Hoya M, Osorio A, Diez O, Miramar MD, Infante M, et al. Analysis of PALB2 gene in BRCA1/BRCA2 negative Spanish hereditary breast/ovarian cancer families with pancreatic cancer cases. PloS One. (2013) 8:e67538. doi: 10.1371/journal.pone.0067538
45. Mehaffey C, Wahl D, Schaller T, Blattner-Johnson M, Claus R, Frühwald M, et al. Heterozygous PALB2 mutation in a boy with acute lymphoblastic leukemia and subsequent metastatic ewing sarcoma. Klin Padiatr. (2021) 233:141–4. doi: 10.1055/a-1404-3243
46. Boonen RACM, Rodrigue A, Stoepker C, Wiegant WW, Vroling B, Sharma M, et al. Functional analysis of genetic variants in the high-risk breast cancer susceptibility gene PALB2. Nat Commun. (2019) 10:5296. doi: 10.1038/s41467-019-13194-2
Keywords: PALB2, Hodgkin lymphoma, Ewing’s sarcoma, genetic predisposition, tumorigenesis
Citation: Czarny J, Galli D, Wziątek A, Pastorczak A, Szmyd B, Przybyszewski B, Raciborska A, Jończyk-Potoczna K and Derwich K (2025) Hodgkin lymphoma and Ewing sarcoma in pediatric patient carrying germline PALB2 variant: a case report and literature review. Front. Oncol. 15:1514697. doi: 10.3389/fonc.2025.1514697
Received: 21 October 2024; Accepted: 27 January 2025;
Published: 18 February 2025.
Edited by:
Oleg Tsodikov, University of Kentucky, United StatesReviewed by:
Luigi Boccuto, Clemson University, United StatesCopyright © 2025 Czarny, Galli, Wziątek, Pastorczak, Szmyd, Przybyszewski, Raciborska, Jończyk-Potoczna and Derwich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katarzyna Derwich, a2RlcndpY2hAdW1wLmVkdS5wbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.