 Pei Wu
Pei Wu Zelin Wen
Zelin Wen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Oncol. , 12 March 2025
Sec. Gastrointestinal Cancers: Colorectal Cancer
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1470939
This article is part of the Research Topic Primary and Secondary Chemotherapy Resistance in Gastrointestinal Tumors: Key Mechanisms and Ways to Overcome Resistance View all 5 articles
Objective: Chemosensitivity and radiosensitivity are associated with the prognosis of colorectal cancer, and the expression of the ataxia-telangiectasia mutated (ATM) protein plays an essential role in these processes. The present study examined the relationship between ATM expression and the survival outcomes of colorectal cancer patients and explored the underlying mechanism and promising therapeutic strategies.
Method: A search including medical subject headings (MeSH), free terms, and combined words was conducted using Pubmed, EMBASE, and Cochrane. Studies had to meet the inclusion criteria as well as include processes such as data extraction and quality evaluation. The survival outcomes were assessed using hazard ratio (HR) and 95% confidence interval (CI). Heterogeneity, and publication bias were analyzed, and a P value <0.05 was considered statistically significant.
Results: Nine studies with 2883 patients were included in the meta-analysis. Low ATM expression level was related to poor overall survival (HR=0.542, 95% CI=0.447–0.637; P=0.000). Disease-free, progression-free, and recurrence-free survival rates were lower in patients with low ATM expression than in those with high ATM expression. There was no significant difference between Stage I–II and Stage III–IV colorectal cancer patients [risk ratio (RR)=1.173, 95% CI=0.970–1.417, P=0.690].
Conclusions: Low ATM expression level may be a marker of poor survival in colorectal cancer and contributes to resistance to therapy. Targeting related factors in these pathways to sensitize tumors to treatment is a potential therapeutic strategy, and monitoring ATM status could be a valuable guide independent of the immunotherapy or chemotherapy strategy used.
Colorectal cancer (CRC) is one of the most common cancers worldwide and a leading cause of cancer-related mortality among both men and women (1). In China, CRC ranks fifth as a cause of cancer-related death among all cancers (2). According to the latest epidemiology studies in China, CRC incidence and mortality are increasing compared with the latest Cancer Statistics report in 2015 (3). The treatment of cancer consists primarily of radical surgery combined with adjuvant therapy such as chemotherapy, radiotherapy, targeted therapy, and immunotherapy (4). The development of next generation sequencing led to the identification a variety of mutations, and precision medicine addressed the relationship between clinical treatment and gene alterations, resulting in decreased treatment toxicity, better survival, and improved quality of life of patients (5). Because of the increased incidence and mortality of CRC, identifying therapeutic targets is essential to open the era of precision medicine based on gene expression status.
The protein kinase of ataxia-telangiectasia mutated (ATM), a serine/threonine-protein kinase, is a critical repair factor that is recruited to and activated by double-stranded DNA breaks (DSBs). This is a vital process for maintaining normal cell division, and disruption of this system can promote carcinogenesis and is associated with poor prognosis in cancer (6, 7). Genetic variation in ATM is associated with poor survival in CRC; however, Sundar et al. (8) reported that patients with deficient ATM expression have better survival outcomes because they show increased sensitivity to DNA damage agents such as oxaliplatin (8). This underscores the need to explore the mechanisms underlying the role of ATM expression in CRC.
The most effective adjuvant treatments for patients with CRC are radiotherapy; chemotherapy regimens composed of 5-fluorouracil, oxaliplatin, and irinotecan; targeted agents such as anti-angiogenic compounds (bevacizumab or aflibercept) or anti-epidermal growth factor receptor (EGFR) drugs (cetuximab or panitumumab) according to the RAS/BRAF status of the tumor; and immunotherapy according to mismatch repair (MMR) alterations and microsatellite status (9). ATM expression status can influence the effect of therapy through multiple processes, although the association between ATM status and CRC-related processes has not been systematically reviewed to date. In order to verify the effect of ATM status on the prognosis of CRC patients, we performed a systematic review of the literature. Studies containing data on ATM expression status and survival outcomes were analyzed to explore the effect of ATM on the prognosis of CRC patients. Tumor stages were analyzed further to determine their importance for patient survival. Related studies were summarized to identify underlying mechanisms and pathways affecting survival outcomes.
The Pubmed, Cochrane, and Embase databases were searched for related studies. The search included MeSH terms, free terms, and combined words. After the literature search, all studies were screened in Endnote according to the keywords and objectives. The selected studies were subjected to quality evaluation according to the Newcastle–Ottawa Scale (NOS), which is one of the most useful methods for assessing the quality of nonrandomized studies, and only high or medium quality studies were included (Table 1). The acquired studies were used for data extraction, which was performed by two authors (ZW and PW) according to a predesigned data extraction form, including the name of the first author, sample size, tumor stage, and survival outcome (Table 1).
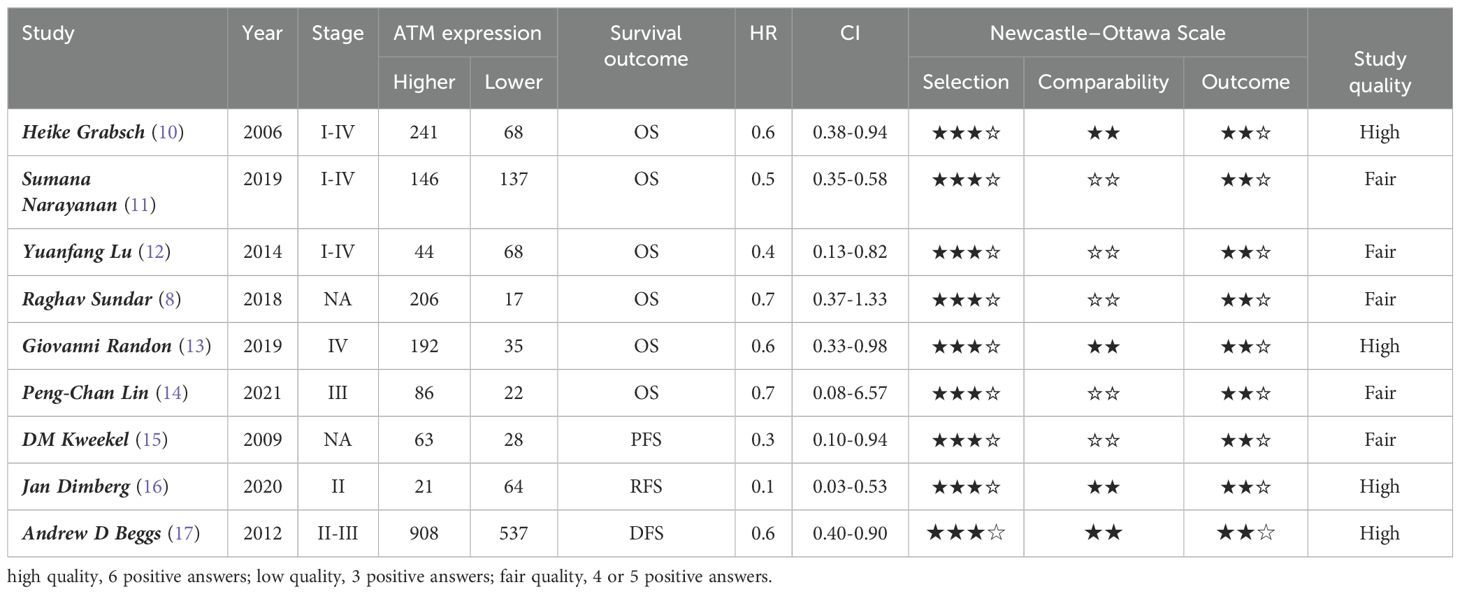
Table 1. Characteristics of all studies.
The inclusion criteria were patients pathologically diagnosed primary CRC, data on ATM detection, data on survival outcome acquired directly from studies or extracted from the Kaplan–Meier curve generated with the Engauge Digitizer (http://digitizer.sourceforge.net/), which were used to calculate the hazard ratios (HRs) using the method described by Tierney et al. (18), and studies with a sample size of >30 patients. The Newcastle Ottawa Scale was used to assess the quality of included studies; if the study quality after evaluation of the “selection,” “comparability,” and “outcome” items in the scale was low (positive answers ≤3) or the survival outcome data could not be obtained, the study was excluded.
The endpoints included overall survival (OS), disease-free survival (DFS), progression-free survival (PFS), and recurrence-free survival (RFS). The hazard ratio (HR) and 95% confidence interval (CI) were used to express survival outcomes; the risk ratio (RR) and 95% CI were used to analyze differences in tumor stage between two groups. Heterogeneity was quantified using the I2 statistic. P <0.05 and I2 >50% were considered substantial heterogeneity, and a random effects model was then used. Publication bias was detected using Egger’s and Begg’s tests, where P <0.1 was regarded as confirmation of significant publication bias.
Images were processed with the Engauge Digitizer and Adobe Photoshop CC 2018 (Adobe Inc., San Jose, CA, USA). Survival analyses were performed using STATA 14.0 software (Stata LLC, College Station, TX, USA). A P value <0.05 was regarded as statistically significant.
After a literature review, nine studies with 2883 patients were included in the meta-analysis (Figure 1). Excluding Sundar et al. (8) and Lin et al. (14), studies supported the association between low ATM expression level and poor survival, including OS, PFS, DFS, and RFS. All cohort studies were evaluated based on the Newcastle–Ottawa Scale (NOS) to be assessed as high or median stages (Table 1).
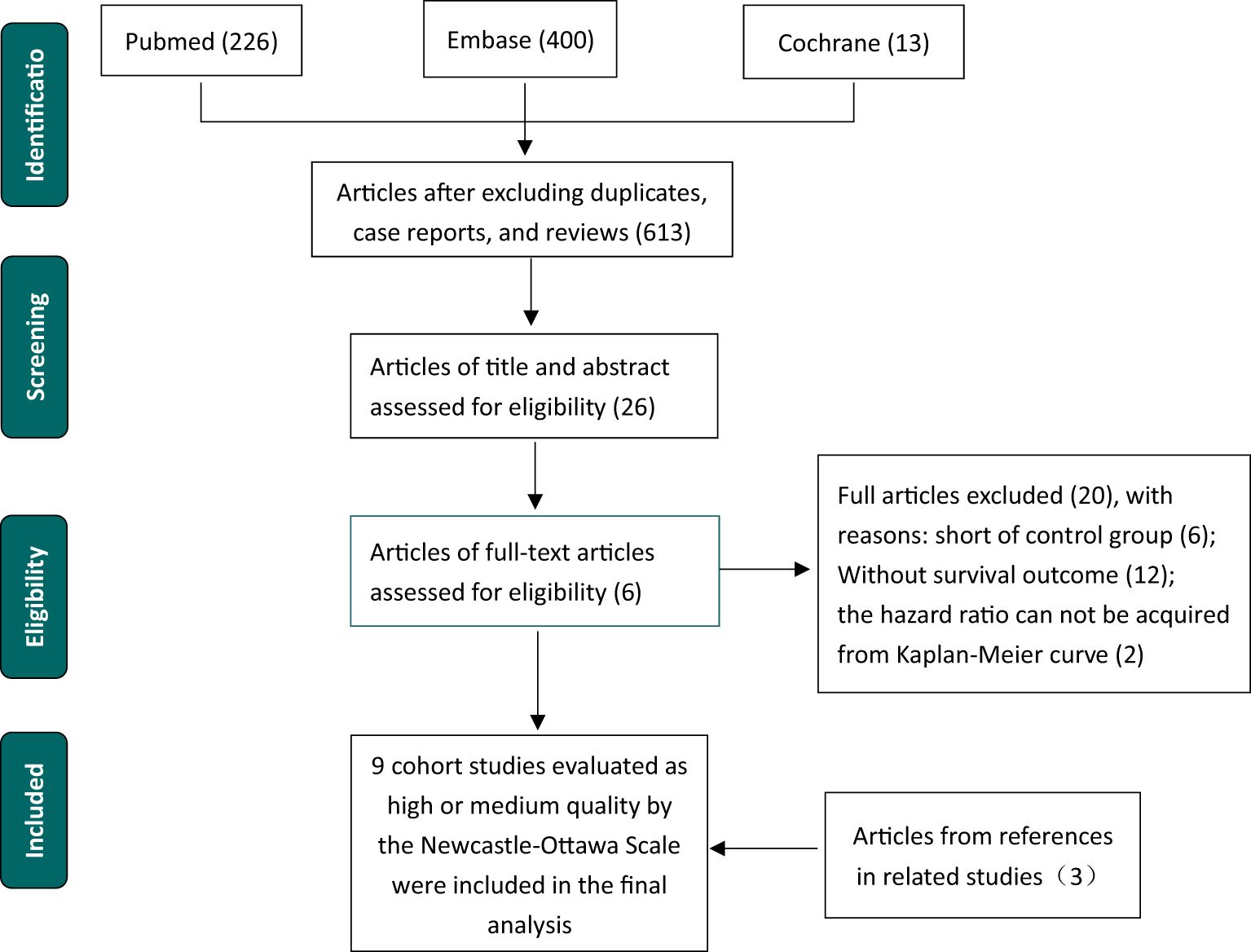
Figure 1. Flow chart of selection procedure literature.
We pooled the HR for OS according to the initial results, which suggested that low ATM expression is related to poor OS (HR = 0.542, 95% CI = 0.447–0.637; P = 0.000) (Figure 2A). DFS, PFS, and RFS were lower in patients with low ATM expression than in those with high expression. Finally, we explored the connection between ATM expression and tumor stage. The results showed no significant difference between Stage I–II and Stage III–IV CRC patients regarding ATM expression (RR = 1.173, 95% CI = 0.970–1.417, P = 0.690) (Figure 2B). No heterogeneity or publication bias was detected based on Egger’s test (P> |t| = 0.875) and Begg’s test (Pr > |z| = 0.851) (Figure 2C), and I2 (P = 0.892).
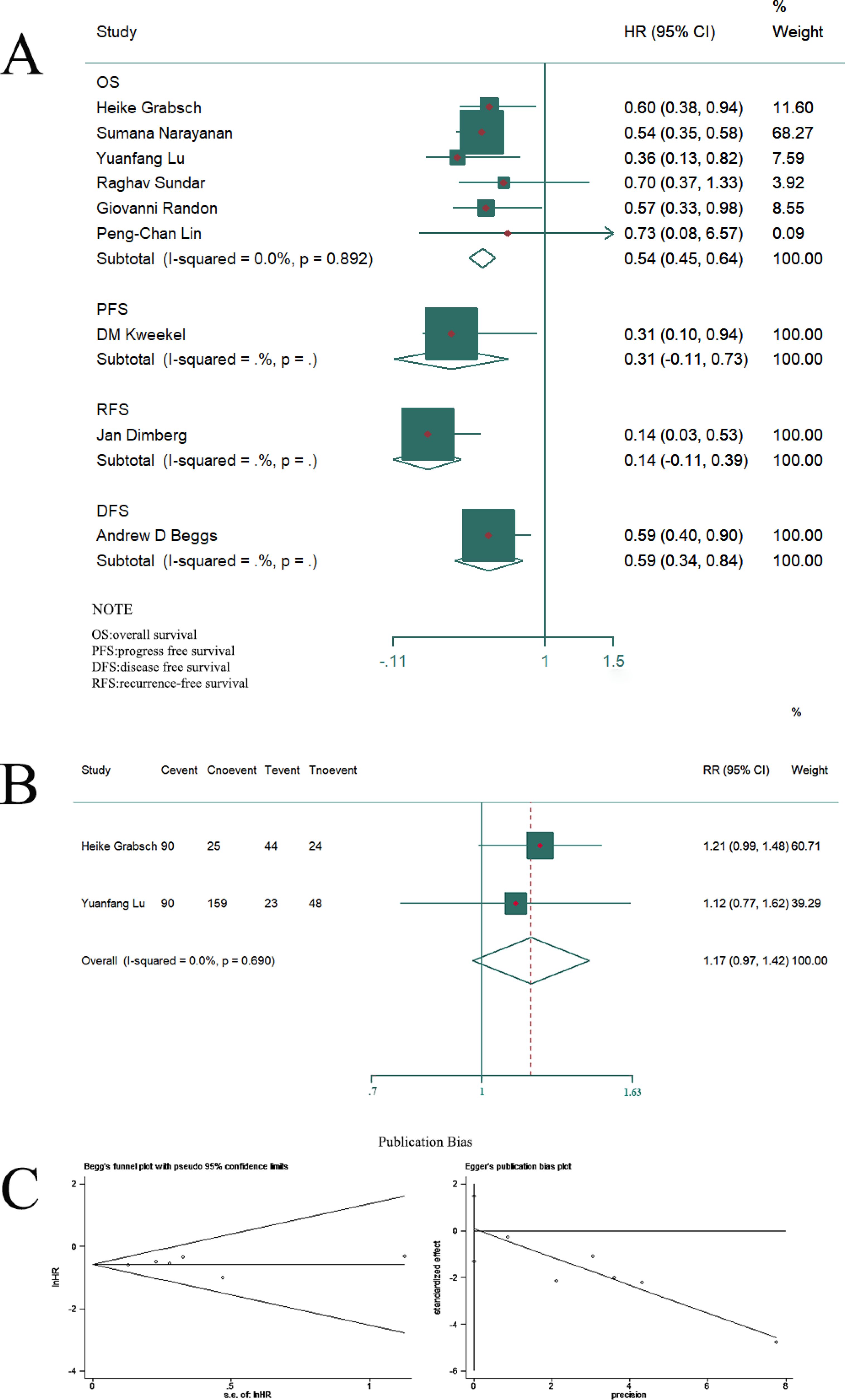
Figure 2. Compared with the low expression group, high expression of ATM was associated with superior survival outcomes, including OS, PFS, DFS, and RFS (A). There was no significant difference in tumor stage between ATM mutation or not (B). There was no publication bias according to Egger’s and Begg’s tests (C).
In the final result, although we concluded that Low ATM expression level was associated with poor survival outcomes, a systematic review is necessary to explore the relationship between ATM status and treatment strategies because of the shortage of related subgroup results in present studies and already existed promising study results about ATM status in colorectal cancer patients. Subsequently, aiming at the clinical treatment, the effects of ATM status on radiotherapy, Mismatch repair/microsatellite status, and chemotherapy were systematically summarized and provided novel insights for further studies and treatments.
CRC is a highly heterogeneous disease with respect to its clinical and biological features, resulting in striking differences in disease progression and treatment response among patients (19). CRC patients are categorized into four subtypes according to the consensus molecular subtype (CMS 1–4), a thoroughly studied and robust stratification strategy for CRC. ATM mutations are present in 7% of non-hypermutated tumors in patients with the CMS3 subtype and are associated with a poor prognosis and limited treatment options (7, 20). In the present study, we performed a systematic review to summarize the effect of ATM expression level on survival outcomes in CRC patients. Nine studies with 2883 patients were included, and patients were classified into high ATM expression (n = 1907) and low ATM expression (n = 976) groups. Most of the studies analyzed found an association between low ATM expression level and inferior survival in CRC patients except the studies by Sundar et al. (8) and Lin et al. (14) After combining survival outcomes, low ATM expression was associated with poor survival, whereas no significant difference in tumor stage was observed between the low and high ATM expression groups. This result is consistent with the function of ATM in the response to DSBs and poor molecular subtypes. Understanding the relationship between ATM and known prognostic factors may provide insight into the utility of ATM expression level as a potential therapeutic approach. For example, in the studies analyzed, high ATM expression levels in CRC were related to better survival outcomes than low ATM expression levels because of the role of ATM in DSB repair and MMR.
The mismatch repair (MMR) system is mainly composed of four proteins (MLH1, MSH2, MSH6, and PMS2) involved in the repair of base-base mismatch that occurs during DNA replication in proliferating cells, which can lead to the accumulation of mutations that fuel carcinogenesis. Microsatellite instability (MSI) is a molecular marker of MMR deficiency (dMMR) and occurs in approximately 15% of CRCs (21, 22). Microsatellite status is considered as a prognostic indicator and a predictor of the response to immunotherapy against targets such as programmed cell death-1 (PD-1) and programmed death-ligand 1 (PD-L1). Grabsch et al. reported that loss of the MMR proteins MLH1 and MSH2 is related to reduced expression of ATM because of effects on the respective gene loci and is associated with significantly longer overall survival (10). Narayanan et al. reported that low ATM expression is associated with high MSI (MSI-H)/dMMR, which is related to helper T-cells and M1 macrophages, finally leading to improved survival (11). Immune checkpoint blockade with PD1 inhibitors is the standard of care for the first-line treatment of MSI-H/dMMR metastatic CRC; however, MSI-H/dMMR represents a small proportion of CRCs, and immune checkpoint inhibitor therapy is largely ineffective for metastatic microsatellite-stable (MSS)/proficient MMR (pMMR) patients (23). Zhou et al. reported that ATM mutations significantly increase immune activity in MSS colon adenocarcinoma (COAD) patients, supporting the feasibility of using ATM mutation status as a predictor of the immunotherapy response in MSS COAD (24). Alterations in ATM expression often manifest as an ultra-high tumor mutational burden, which is associated with a better response to immunotherapies (25–27). The relationship between ATM mutation and MMR/microsatellite status may provide novel insight into the response to immune therapy in CRC, especially for MSS CRC (27). However, additional clinical studies are necessary to elucidate the underlying mechanisms. The connection between ATM and microsatellite status will be a promising research direction considering its immediate relevance to clinical treatments.
Radiation therapy (RT) is a mainstay of colorectal cancer treatment (CRC treatment), especially for rectal cancer; however, resistance to RT limits the efficacy of treatment, especially in patients with advanced disease. DSB repair plays a vital role in acquired resistance to radiotherapy and chemotherapy in many cancers (28, 29); therefore, it is essential to clarify the involvement of ATM in these processes. When DNA DSBs appear after exposure to factors such as chemotherapy or radiotherapy, ATM is recruited and binds to the location of DSBs to repair the damage; therefore the expression level of ATM thus plays a critical role in this process (30). However, this repair mechanism is also responsible for resistance, leading to poor survival outcomes (31). Given that the relationship between ATM and DBSs is associated with radioresistance, targeting this response represents a promising approach to improving the efficacy of RT (32, 33). This process includes various mechanisms influencing radiosensitivity according to previous studies (34, 35) (Figure 3), and cell cycle arrest is a vital alteration among these. Cell cycle checkpoints control various mechanisms in the eukaryotic cell cycle by examining internal and external cues at every stage and determining whether cells move forward with division. These include DNA structure checkpoints (DSCs or DDCs) and spindle assembly checkpoints (SACs) (36). Cells can undergo arrest at G1, S, and G2/M points after ionizing radiation (IR)-induced DNA damage (37). Radiosensitivity is closely associated with cell cycle arrest in these processes (38).
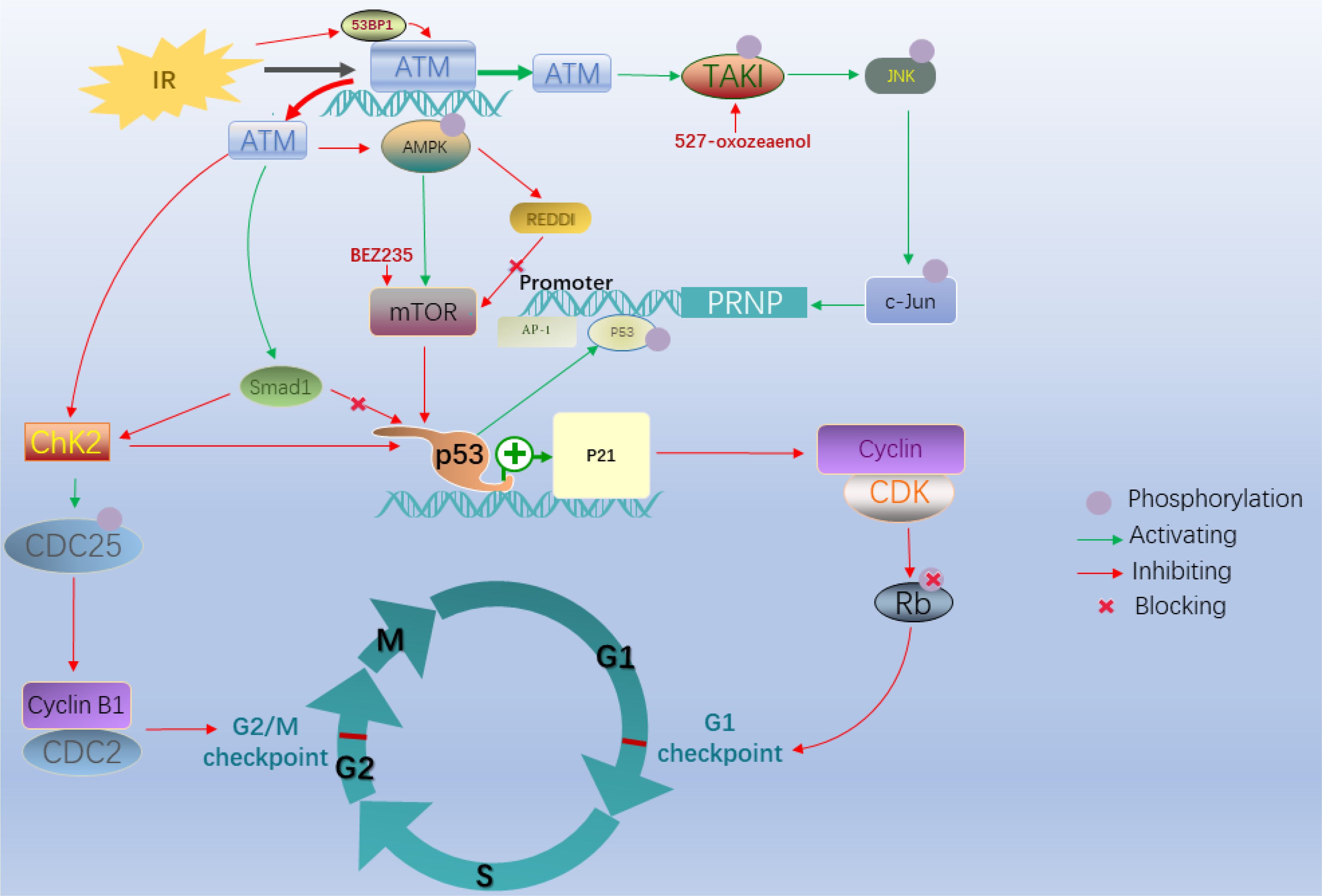
Figure 3. After ionizing radiation, G1/S and G2/M cell cycle arrest plays an essential role in radioresistance. In these processes, the P53-dependent ATM/p53/p21 pathway and ATM–Chk2 pathway with the absence of P53 are major signaling pathways leading to G1/S and G2/M cell cycle arrest, respectively. The ATM-CHK2-p53 pathway is activated when vital factors alternate including degraded CDC25C and deficiency of 53BP1. The ATM-TAK1-PrPC pathway is another essential pathway mediating radioresistance in colorectal cancer cells.
Regarding the mechanisms responsible for the decreased radiosensitizing effects of ATM alteration, cell cycle arrest plays a vital role. The tumor suppressor p53 is critical for these checkpoint pathways (39–41). The different status of p53 determines the distinct ATM pathway that decreases radiosensitivity because of IR-induced DNA damage in CRC cells (39, 42). In the presence of p53 deficiency, the checkpoint arrest primarily relies on the CHK (Checkpoint kinase)–dependent pathway (43). For example, in the ATM–checkpoint kinase 2 (CHK2) pathway, ATM mutation induced by DSBs initially phosphorylates the CHK2 protein kinase encoded by the tumor suppressor gene CHEK2, which is involved in cell cycle arrest, DNA repair, or apoptosis in response to DNA damage (44); subsequently, this active kinase phosphorylates cell division cycle 25C (CDC25C) at Ser216 and inactivates it, which in turn inhibits the activity of the CDC2-cyclin B1 complex and finally triggers G2/M phase arrest. However, if CDC25C is degraded in the IR response, the ATM-CHK2-p53 pathway would replace the above mechanism to monitor G1 checkpoint arrest when p53 is proficient, which is an essential regulatory mechanism used by cells to block mitotic entry in response to DNA damage (45). CHK2 is a traditional target of ATM in the DNA damage response, although the ATR-CHK1 pathway is more common (46). Nevertheless, ATM is also required for CHK1 activation under certain circumstances (47).
Compared with the absence of p53 expression, cells expressing p53 (+/+) are primarily dependent on the ATM/p53/p21 pathway (48, 49). Regarding cell cycle checkpoints, multiple downstream targets of ATM determine the different outcomes. For example, adenosine monophosphate-activated kinase (AMPK), a metabolic sensor, acts as an upstream inhibitor of mTOR activity, which regulates cellular responses to IR (50–52). In this pathway, IR contributes to inhibiting the phosphorylation of both AMPK and its substrate in an ATM-dependent manner; the absence of AMPK stimulates mTOR activity by inhibiting the expression of the mTOR inhibitor REDD1, which not only acts as a classical pathway inducing tolerance to radiotherapy, but is also associated with the canonical p53-dependent G1 phase arrest pathway (50). Overexpression of p53 may cause p21 to accumulate, leading to inhibition of cyclin-CDK complex activity, which prevents the phosphorylation of retinoblastoma protein, which is required for progression to the S phase (53). This process ultimately results in G1/S arrest. However, when ATM upregulates Smad1 in the DNA damage response, ATM–CHK2 is activated after stabilization of p53, resulting in G2/M phase arrest (54). Decreased expression of p53 binding protein 1 downregulates ATM and CHK2, which affects the cell cycle leading to radiotolerance of CRC cells through the ATM-CHK2-p53 pathway (55, 56).
Compared with decreased expression of ATM in many pathways, including the ATM/p53/p21, ATM–CHK2, and ATM-CHK2-p53 pathways, IR could activate ATM to respond to oxidative stress in tumors, resulting in resistance associated with the expression of proteins such as the cellular prion protein (PrPC), which is related to stemness, invasiveness, resistance to chemotherapy, and radiosensitivity (57–60). Increased levels of PrPC contribute to acquired resistance to radiotherapy in CRC. Jacqueline et al. found that the ATM-TAK1-PrPC pathway plays an essential role in mediating radioresistance in CRC cells (61). Exposure of CRC cells to irradiation activates c-Jun, followed by activation of ATM and induction of TAK1-dependent phosphorylation of JNK. An AP-1 binding site in the PRNP promoter results in increased levels of PrPC.
In theory, because the protein kinase activity of ATM mediates the cellular response to DNA damage induced by IR, ATM is involved in inducing radioresistance according to multiple pathways in CRC cells (62). Identifying novel targets and designing drugs according to these processes are promising strategies (63). For example, Sherri et al. demonstrated that 527-oxozeaenol, which acts as a pharmacological inhibitor of TAK1, enhances the radiotherapeutic effect in CRC cells according to the ATM-TAK1-PrPC pathway (61). BEZ235 acts as an effective radiosensitizer of CRC cells and prolongs the radiotherapeutic effects by suppressing the activation of ATM and DNA-PKcs-associated DNA repair (35, 64). Lin et al. reported that quercetin-induced radio-sensitization is mediated by the inhibition of ATM kinase (65). Although the efficacy of ATM inhibitors such as KU-55933, KU-60019, KU-59403, CP-466722, AZ31, AZ32, AZD0156, and AZD1390 has been demonstrated in different tumor types showing different responses to RT in preclinical studies (28, 66–68), systematic and comprehensive testing is lagging in CRC (7). A phase I clinical trial evaluating the safety and tolerability of AZD1390 in combination with RT (ClinicalTrials.gov Identifier: NCT03423628) is currently in the recruiting phase.
To the best of our knowledge, in addition to primary alterations and DNA damage induced by radiotherapy (69), chemotherapy-induced DNA alterations remain the underlying cause of ATM alteration. Fluoropyrimidine (5-fluorouracil or capecitabine) as a single agent or in combination with oxaliplatin is the standard first-line chemotherapy for CRC, which consolidates the curative effect and prolongs the survival time in CRC patients (70, 71). However, chemosensitivity and acquired drug resistance after repeated exposure to chemotherapy agents are the leading cause of cancer and even death (32, 72). A series of studies provide evidence that ATM regulates cellular defenses against certain cytotoxic agents (73–75). For example, although the DNA damage drug oxaliplatin acts as an effective chemotherapeutic drug (76, 77), Kweekel et al. reported that patients with mutated ATM have a 4.25-fold higher risk of progression on capecitabine plus oxaliplatin than patients with wild-type ATM (15), and concomitant chemoresistance after treatment limits the therapeutic efficacy in CRC patients (75). Previous studies found that the expression of Bmal1 (78), isocitrate dehydrogenase 2 (IDH2) (79), and miR-203 (80) is associated with resistance to oxaliplatin, and ATM is a crucial factor in these processes (80, 81). Mechanistically, overexpression of the circadian clock gene Bmal1 could increase sensitivity to oxaliplatin by regulating G2–M arrest through the activation of the ATM pathway (78); however, the abrogation of IDH2 could also increase the efficacy of oxaliplatin by promoting phosphorylation of the ATM protein; furthermore, the expression of miR-203 could contribute to oxaliplatin resistance by negatively regulating ATM kinase (80). 5-Fluorouracil (5-FU)-based chemotherapies are essential in the treatment of CRC (82). Morcillo et al. demonstrated that p38MAPK activation plays a role in the resistance to 5-FU, and ATM shows a redundant function in this mechanism (83). However, altered ATM expression is not the only decisive factor for the poor prognosis; epigenetic alteration of ATM, such as aberrant methylation of the ATM promoter, also plays an essential role in the resistance to treatment (34). Genetic variants of ATM also affect the survival outcome. For example, the G allele in ATM rs609429 is associated with longer OS than the C/C variant in refractory metastatic CRC patients receiving Tas-102 chemotherapy (84).
The clinical application of Poly (ADP-ribose) polymerase (PARP) inhibition in CRC has gained attention based on data showing that PARP inhibitors (PARPi) improve prognosis when combined with chemotherapeutic agents such as platinum agents and irinotecan, and ATM deficiency increases sensitivity to PARP inhibition (85). CRC patients with ATM mutation are highly sensitive to PARPi/chemotherapy combination at low doses, regardless of the CMS and microsatellite status. This is attributed to a delay in the resolution of DSBs through homologous recombination repair (HRR), which acts in coordination with the S and G2 checkpoint machinery to eliminate chromosomal breaks before cell division occurs (75). Therefore, ATM status may be a valuable strategy for determining the efficacy of anticancer agents.
Here, we performed a meta-analysis and showed that low ATM expression level is associated with poor survival and possibly decreased chemosensitivity and radiosensitivity. Although no significant heterogeneity or publication bias was detected in the present study, there were limitations, such as a lack of subgroup and data analysis of specific pathways. However, we systematically reviewed the related mechanisms presented intuitively, including a cell pathway map, explored the effects of ATM alteration on therapeutic strategies in CRC, and examined the promising application of ATM in clinical practice.
Although most of the studies included in the meta-analysis supported that patients with low ATM expression levels have shorter survival times than those with high ATM expression levels, multicenter randomized controlled trials are still needed to confirm these findings. In addition, further cell or animal experiments are necessary to explore the role of the relevant mechanisms in CRC, especially regarding MMR/microsatellite status, radiotherapy, and chemotherapeutic regimes. Finally, some studies suggest that ATM can be used as a marker to guide treatment or as a sensitizer. However, it is essential to conduct multicenter randomized trials to explore the relationship between ATM and chemotherapeutic regimes and screen the definite indications in clinical practice.
Low-level ATM expression may be a marker of poor survival in CRC and contributes to resistance to therapy, and targeting this mechanism could potentially increase sensitivity to treatments. ATM expression status was identified as a promising marker for guiding immunotherapies and chemotherapies, especially for MSS CRC patients, and for the design of chemotherapeutic regimes such as combination treatments with PARPi.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
PW: Conceptualization, Data curation, Formal analysis, Methodology, Project administration, Resources, Software, Writing – original draft, Writing – review & editing. ZW: Conceptualization, Data curation, Resources, Writing – original draft.
The author(s) declare that no financial support was received for the research and/or publication of this article.
We thank International Science Editing (http://www.internationalscienceediting.com) for editing this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA: Cancer J Clin. (2021) 71:7–33. doi: 10.3322/caac.21654
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA: Cancer J Clin. (2016) 66:115–32. doi: 10.3322/caac.21338
3. Zhang S, Sun K, Zheng R, Zeng H, Wang S, Chen R, et al. Cancer incidence and mortality in China, 2015. J Natl Cancer Center. (2021) 1:2–11. doi: 10.1016/j.jncc.2020.12.001
4. Benson AB, Venook AP, Adam M, Chang G, Chen YJ, Ciombor KK, et al. Nccn guidelines® Insights: rectal cancer, version 3.2024. J Natl Compr Cancer Netw: JNCCN. (2024) 22:366–75. doi: 10.6004/jnccn.2024.0041
5. Hayashi H, Takiguchi Y, Minami H, Akiyoshi K, Segawa Y, Ueda H, et al. Site-specific and targeted therapy based on molecular profiling by next-generation sequencing for cancer of unknown primary site: A nonrandomized phase 2 clinical trial. JAMA Oncol. (2020) 6:1931–8. doi: 10.1001/jamaoncol.2020.4643
6. Hulshof EC, Lim L, de Hingh I, Gelderblom H, Guchelaar HJ, Deenen MJ. Genetic variants in DNA repair pathways as potential biomarkers in predicting treatment outcome of intraperitoneal chemotherapy in patients with colorectal peritoneal metastasis: A systematic review. Front Pharmacol. (2020) 11:577968. doi: 10.3389/fphar.2020.577968
7. Mauri G, Arena S, Siena S, Bardelli A, Sartore-Bianchi A. The DNA damage response pathway as a land of therapeutic opportunities for colorectal cancer. Ann Oncol: Off J Eur Soc Med Oncol. (2020) 31:1135–47. doi: 10.1016/j.annonc.2020.05.027
8. Sundar R, Miranda S, Rodrigues DN, Chénard-Poirier M, Dolling D, Clarke M, et al. Ataxia telangiectasia mutated protein loss and benefit from oxaliplatin-based chemotherapy in colorectal cancer. Clin Colorectal Cancer. (2018) 17:280–4. doi: 10.1016/j.clcc.2018.05.011
9. Ness RM, Llor X, Abbass MA, Bishu S, Chen CT, Cooper G, et al. Nccn guidelines® Insights: colorectal cancer screening, version 1.2024. J Natl Compr Cancer Netw: JNCCN. (2024) 22:438–46. doi: 10.6004/jnccn.2024.0047
10. Grabsch H, Dattani M, Barker L, Maughan N, Maude K, Hansen O, et al. Expression of DNA double-strand break repair proteins Atm and Brca1 predicts survival in colorectal cancer. Clin Cancer Res: an Off J Am Assoc Cancer Res. (2006) 12:1494–500. doi: 10.1158/1078-0432.Ccr-05-2105
11. Narayanan S, Kawaguchi T, Peng X, Qi Q, Liu S, Yan L, et al. Tumor infiltrating lymphocytes and macrophages improve survival in microsatellite unstable colorectal cancer. Sci Rep. (2019) 9:13455. doi: 10.1038/s41598-019-49878-4
12. Lu Y, Gao J, Lu Y. Downregulated Ku70 and ATM associated to poor prognosis in colorectal cancer among chinese patients. Onco Targets Ther (2014) 7:1955–61. doi: 10.2147/OTT.S67814
13. Randon G, Fucà G, Rossini D, Raimondi A, Pagani F, Perrone F, et al. Prognostic impact of ATM mutations in patients with metastatic colorectal cancer. Sci Rep (2019) 9(1):2858. doi: 10.1038/s41598-019-39525-3
14. Lin PC, Yeh YM, Chan RH, Lin BW, Chen PC, Pan CC, et al. Sequential and co-occurring DNA damage response genetic mutations impact survival in stage iii colorectal cancer patients receiving adjuvant oxaliplatin-based chemotherapy. BMC Cancer. (2021) 21:217. doi: 10.1186/s12885-021-07926-1
15. Kweekel DM, Antonini NF, Nortier JW, Punt CJ, Gelderblom H, Guchelaar HJ. Explorative study to identify novel candidate genes related to oxaliplatin efficacy and toxicity using a DNA repair array. Br J Cancer. (2009) 101:357–62. doi: 10.1038/sj.bjc.6605134
16. Dimberg J, Andersson RE, Haglund S. Genomic profiling of stage II colorectal cancer identifies candidate genes associated with recurrence-free survival, tumor location, and differentiation grade. Oncology. (2020) 98(8):575–82. doi: 10.1159/000507118
17. Beggs AD, Domingo E, McGregor M, Presz M, Johnstone E, Midgley R, et al. Loss of expression of the double strand break repair protein ATM is associated with worse prognosis in colorectal cancer and loss of Ku70 expression is associated with CIN. Oncotarget. (2012) 3(11):1348–55. doi: 10.18632/oncotarget.694
18. Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time-to-event data into meta-analysis. Trials. (2007) 8:16. doi: 10.1186/1745-6215-8-16
19. Punt CJ, Koopman M, Vermeulen L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat Rev Clin Oncol. (2017) 14:235–46. doi: 10.1038/nrclinonc.2016.171
20. Cancer Genome Atlas Network, Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. (2012) 487:330–7. doi: 10.1038/nature11252
21. Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol. (2010) 7:153–62. doi: 10.1038/nrclinonc.2009.237
22. Gelsomino F, Barbolini M, Spallanzani A, Pugliese G, Cascinu S. The evolving role of microsatellite instability in colorectal cancer: A review. Cancer Treat Rev. (2016) 51:19–26. doi: 10.1016/j.ctrv.2016.10.005
23. Ganesh K. Optimizing immunotherapy for colorectal cancer. Nat Rev Gastroenterol Hepatol. (2022) 19:93–4. doi: 10.1038/s41575-021-00569-4
24. Zhou P, Wu X, Chen H, Hu Y, Zhang H, Wu L, et al. The mutational pattern of homologous recombination-related (Hrr) genes in Chinese colon cancer and its relevance to immunotherapy responses. Aging. (2020) 13:2365–78. doi: 10.18632/aging.202267
25. Ying J, Yang L, Yin JC, Xia G, Xing M, Chen X, et al. Additive effects of variants of unknown significance in replication repair-associated DNA polymerase genes on mutational burden and prognosis across diverse cancers. J Immunother Cancer. (2021) 9(9):e002336. doi: 10.1136/jitc-2021-002336
26. Domingo E, Camps C, Kaisaki PJ, Parsons MJ, Mouradov D, Pentony MM, et al. Mutation burden and other molecular markers of prognosis in colorectal cancer treated with curative intent: results from the Quasar 2 clinical trial and an Australian community-based series. Lancet Gastroenterol Hepatol. (2018) 3:635–43. doi: 10.1016/s2468-1253(18)30117-1
27. Li J, Steffen P, Tse BCY, Ahadi MS, Gill AJ, Engel AF, et al. Deep sequencing of early T stage colorectal cancers reveals disruption of homologous recombination repair in microsatellite stable tumours with high mutational burdens. Cancers. (2022) 14(12):2933. doi: 10.3390/cancers14122933
28. Jin MH, Oh DY. Atm in DNA repair in cancer. Pharmacol Ther. (2019) 203:107391. doi: 10.1016/j.pharmthera.2019.07.002
29. Weber AM, Ryan AJ. Atm and Atr as therapeutic targets in cancer. Pharmacol Ther. (2015) 149:124–38. doi: 10.1016/j.pharmthera.2014.12.001
30. Hu W, Lei L, Xie X, Huang L, Cui Q, Dang T, et al. Heterogeneous nuclear ribonucleoprotein L facilitates recruitment of 53bp1 and Brca1 at the DNA break sites induced by oxaliplatin in colorectal cancer. Cell Death Dis. (2019) 10:550. doi: 10.1038/s41419-019-1784-x
31. Stover EH, Konstantinopoulos PA, Matulonis UA, Swisher EM. Biomarkers of response and resistance to DNA repair targeted therapies. Clin Cancer Res: an Off J Am Assoc Cancer Res. (2016) 22:5651–60. doi: 10.1158/1078-0432.Ccr-16-0247
32. Panczyk M. Pharmacogenetics research on chemotherapy resistance in colorectal cancer over the last 20 years. World J Gastroenterol. (2014) 20:9775–827. doi: 10.3748/wjg.v20.i29.9775
33. García MEG, Kirsch DG, Reitman ZJ. Targeting the ATM kinase to enhance the efficacy of radiotherapy and outcomes for cancer patients. Semin Radiat Oncol. (2022) 32(1):3–14. doi: 10.1016/j.semradonc.2021.09.008
34. Kim WJ, Vo QN, Shrivastav M, Lataxes TA, Brown KD. Aberrant methylation of the ATM promoter correlates with increased radiosensitivity in a human colorectal tumor cell line. Oncogene. (2002) 21(24):3864–71. doi: 10.1038/sj.onc.1205485. PMID: 12032824
35. Chen YH, Wei MF, Wang CW, Lee HW, Pan SL, Gao M, et al. Dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitor is an effective radiosensitizer for colorectal cancer. Cancer Lett. (2015) 357:582–90. doi: 10.1016/j.canlet.2014.12.015
36. Matthews HK, Bertoli C, de Bruin RAM. Cell cycle control in cancer. Nat Rev Mol Cell Biol. (2022) 23:74–88. doi: 10.1038/s41580-021-00404-3
37. Guo XX, Guo ZH, Lu JS, Xie WS, Zhong QZ, Sun XD, et al. All-purpose nanostrategy based on dose deposition enhancement, cell cycle arrest, DNA damage, and Ros production as prostate cancer radiosensitizer for potential clinical translation. Nanoscale. (2021) 13:14525–37. doi: 10.1039/d1nr03869a
38. Waterman DP, Haber JE, Smolka MB. Checkpoint responses to DNA double-strand breaks. Annu Rev Biochem. (2020) 89:103–33. doi: 10.1146/annurev-biochem-011520-104722
39. Fernández-Aroca DM, Roche O, Sabater S, Pascual-Serra R, Ortega-Muelas M, Sánchez Pérez I, et al. P53 pathway is a major determinant in the radiosensitizing effect of palbociclib: implication in cancer therapy. Cancer Lett. (2019) 451:23–33. doi: 10.1016/j.canlet.2019.02.049
40. Boutelle AM, Attardi LD. P53 and tumor suppression: it takes a network. Trends Cell Biol. (2021) 31:298–310. doi: 10.1016/j.tcb.2020.12.011
41. Hu J, Cao J, Topatana W, Juengpanich S, Li S, Zhang B, et al. Targeting mutant P53 for cancer therapy: direct and indirect strategies. J Hematol Oncol. (2021) 14:157. doi: 10.1186/s13045-021-01169-0
42. Shiloh Y, Ziv Y. The Atm protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. (2013) 14:197–210. doi: 10.1038/nrm3546
43. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. (2009) 461(7267):1071–8. doi: 10.1038/nature08467
44. Bartek J, Falck J, Lukas J. Chk2 kinase–a busy messenger. Nat Rev Mol Cell Biol. (2001) 2:877–86. doi: 10.1038/35103059
45. Wang Y, Yang L, Zhang J, Zhou M, Shen L, Deng W, et al. Radiosensitization by irinotecan is attributed to G2/M phase arrest, followed by enhanced apoptosis, probably through the Atm/Chk/Cdc25c/Cdc2 pathway in P53-mutant colorectal cancer cells. Int J Oncol. (2018) 53:1667–80. doi: 10.3892/ijo.2018.4514
46. Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and Cancer. Cancer Cell. (2003) 3:421–9. doi: 10.1016/s1535-6108(03)00110-7
47. Akasaka T, Tsujii M, Kondo J, Hayashi Y, Ying J, Lu Y, et al. 5−Fu resistance abrogates the amplified cytotoxic effects induced by inhibiting checkpoint kinase 1 in P53−Mutated colon cancer cells. Int J Oncol. (2015) 46:63–70. doi: 10.3892/ijo.2014.2693
48. Lin K, Adamson J, Johnson GG, Carter A, Oates M, Wade R, et al. Functional analysis of the Atm-P53-P21 pathway in the Lrf Cll4 trial: blockade at the level of P21 is associated with short response duration. Clin Cancer Res: an Off J Am Assoc Cancer Res. (2012) 18:4191–200. doi: 10.1158/1078-0432.Ccr-11-2936
49. Xia F, Powell SN. The molecular basis of radiosensitivity and chemosensitivity in the treatment of breast cancer. Semin Radiat Oncol. (2002) 12:296–304. doi: 10.1053/srao.2002.35250
50. Zannella VE, Cojocari D, Hilgendorf S, Vellanki RN, Chung S, Wouters BG, et al. Ampk regulates metabolism and survival in response to ionizing radiation. Radiother Oncol: J Eur Soc Ther Radiol Oncol. (2011) 99:293–9. doi: 10.1016/j.radonc.2011.05.049
51. Sanli T, Rashid A, Liu C, Harding S, Bristow RG, Cutz JC, et al. Ionizing radiation activates Amp-activated kinase (Ampk): A target for radiosensitization of human cancer cells. Int J Radiat Oncol Biol Phys. (2010) 78:221–9. doi: 10.1016/j.ijrobp.2010.03.005
52. Fu X, Wan S, Lyu YL, Liu LF, Qi H. Etoposide induces Atm-dependent mitochondrial biogenesis through Ampk activation. PloS One. (2008) 3:e2009. doi: 10.1371/journal.pone.0002009
53. Rubin SM, Sage J, Skotheim JM. Integrating old and new paradigms of G1/S control. Mol Cell. (2020) 80:183–92. doi: 10.1016/j.molcel.2020.08.020
54. Ruan X, Zuo Q, Jia H, Chau J, Lin J, Ao J, et al. P53 deficiency-induced smad1 upregulation suppresses tumorigenesis and causes chemoresistance in colorectal cancers. J Mol Cell Biol. (2015) 7:105–18. doi: 10.1093/jmcb/mjv015
55. Xiao Y, Zheng X, Huang A, Liu T, Zhang T, Ma H. Deficiency of 53bp1 inhibits the radiosensitivity of colorectal cancer. Int J Oncol. (2016) 49:1600–8. doi: 10.3892/ijo.2016.3629
56. Squatrito M, Brennan CW, Helmy K, Huse JT, Petrini JH, Holland EC. Loss of Atm/Chk2/P53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell. (2010) 18:619–29. doi: 10.1016/j.ccr.2010.10.034
57. Qin K, Zhao L, Ash RD, McDonough WF, Zhao RY. Atm-mediated transcriptional elevation of prion in response to copper-induced oxidative stress. J Biol Chem. (2009) 284:4582–93. doi: 10.1074/jbc.M808410200
58. Martin-Lannerée S, Hirsch TZ, Hernandez-Rapp J, Halliez S, Vilotte JL, Launay JM, et al. PrP(C) from stem cells to cancer. Front Cell Dev Biol. (2014) 2:55. doi: 10.3389/fcell.2014.00055
59. Ryskalin L, Busceti CL, Biagioni F, Limanaqi F, Familiari P, Frati A, et al. Prion protein in glioblastoma multiforme. Int J Mol Sci. (2019) 20(20):5107. doi: 10.3390/ijms20205107
60. Manni G, Lewis V, Senesi M, Spagnolli G, Fallarino F, Collins SJ, et al. The cellular prion protein beyond prion diseases. Swiss Med Wkly. (2020) 150:w20222. doi: 10.4414/smw.2020.20222
61. Bernardino-Sgherri J, Siberchicot C, Auvré F, Busso D, Brocas C, El Masri G, et al. Tumor resistance to radiotherapy is triggered by an Atm/Tak1-dependent-increased expression of the cellular prion protein. Oncogene. (2021) 40:3460–9. doi: 10.1038/s41388-021-01746-0
62. Choi M, Kipps T, Kurzrock R. Atm mutations in cancer: therapeutic implications. Mol Cancer Ther. (2016) 15:1781–91. doi: 10.1158/1535-7163.Mct-15-0945
63. Ch’ang HJ, Maj JG, Paris F, Xing HR, Zhang J, Truman JP, et al. Atm regulates target switching to escalating doses of radiation in the intestines. Nat Med. (2005) 11:484–90. doi: 10.1038/nm1237
64. Chen YH, Wang CW, Wei MF, Tzeng YS, Lan KH, Cheng AL, et al. Maintenance Bez235 treatment prolongs the therapeutic effect of the combination of Bez235 and radiotherapy for colorectal cancer. Cancers. (2019) 11(8):1204. doi: 10.3390/cancers11081204
65. Lin C, Yu Y, Zhao HG, Yang A, Yan H, Cui Y. Combination of quercetin with radiotherapy enhances tumor radiosensitivity in vitro and in vivo. Radiother Oncol: J Eur Soc Ther Radiol Oncol. (2012) 104:395–400. doi: 10.1016/j.radonc.2011.10.023
66. Jucaite A, Stenkrona P, Cselényi Z, De Vita S, Buil-Bruna N, Varnäs K, et al. Brain exposure of the Atm inhibitor Azd1390 in humans-a positron emission tomography study. Neuro-oncology. (2021) 23:687–96. doi: 10.1093/neuonc/noaa238
67. Waqar SN, Robinson C, Olszanski AJ, Spira A, Hackmaster M, Lucas L, et al. Phase I trial of Atm inhibitor M3541 in combination with palliative radiotherapy in patients with solid tumors. Investigat New Drugs. (2022) 40:596–605. doi: 10.1007/s10637-022-01216-8
68. Durant ST, Zheng L, Wang Y, Chen K, Zhang L, Zhang T, et al. The brain-penetrant clinical Atm inhibitor Azd1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci Adv. (2018) 4:eaat1719. doi: 10.1126/sciadv.aat1719
69. Huang RX, Zhou PK. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct Target Ther. (2020) 5:60. doi: 10.1038/s41392-020-0150-x
70. André T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. New Engl J Med. (2004) 350:2343–51. doi: 10.1056/NEJMoa032709
71. Haller DG, Tabernero J, Maroun J, de Braud F, Price T, Van Cutsem E, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage iii colon cancer. J Clin Oncol: Off J Am Soc Clin Oncol. (2011) 29:1465–71. doi: 10.1200/jco.2010.33.6297
72. Zheng H, Liu J, Cheng Q, Zhang Q, Zhang Y, Jiang L, et al. Targeted activation of ferroptosis in colorectal cancer via Lgr4 targeting overcomes acquired drug resistance. Nat Cancer. (2024) 5:572–89. doi: 10.1038/s43018-023-00715-8
73. Fedier A, Schlamminger M, Schwarz VA, Haller U, Howell SB, Fink D. Loss of Atm sensitises P53-deficient cells to topoisomerase poisons and antimetabolites. Ann Oncol: Off J Eur Soc Med Oncol. (2003) 14:938–45. doi: 10.1093/annonc/mdg240
74. Knappskog S, Chrisanthar R, Løkkevik E, Anker G, Østenstad B, Lundgren S, et al. Low expression levels of Atm may substitute for Chek2/Tp53 mutations predicting resistance towards anthracycline and mitomycin chemotherapy in breast cancer. Breast Cancer Res: BCR. (2012) 14:R47. doi: 10.1186/bcr3147
75. Vitiello PP, Martini G, Mele L, Giunta EF, De Falco V, Ciardiello D, et al. Vulnerability to low-dose combination of irinotecan and niraparib in Atm-mutated colorectal cancer. J Exp Clin Cancer Res: CR. (2021) 40:15. doi: 10.1186/s13046-020-01811-8
76. O’Dwyer PJ, Stevenson JP, Johnson SW. Clinical pharmacokinetics and administration of established platinum drugs. Drugs. (2000) 59 Suppl 4:19–27. doi: 10.2165/00003495-200059004-00003
77. Farrell N. Progress in platinum-derived drug development. Drugs Future. (2012) 37:795. doi: 10.1358/dof.2012.37.11.1830167
78. Zeng ZL, Luo HY, Yang J, Wu WJ, Chen DL, Huang P, et al. Overexpression of the circadian clock gene Bmal1 increases sensitivity to oxaliplatin in colorectal cancer. Clin Cancer Res: an Off J Am Assoc Cancer Res. (2014) 20:1042–52. doi: 10.1158/1078-0432.Ccr-13-0171
79. Qiao S, Lu W, Glorieux C, Li J, Zeng P, Meng N, et al. Wild-type Idh2 protects nuclear DNA from oxidative damage and is a potential therapeutic target in colorectal cancer. Oncogene. (2021) 40:5880–92. doi: 10.1038/s41388-021-01968-2
80. Zhou Y, Wan G, Spizzo R, Ivan C, Mathur R, Hu X, et al. Mir-203 induces oxaliplatin resistance in colorectal cancer cells by negatively regulating Atm kinase. Mol Oncol. (2014) 8:83–92. doi: 10.1016/j.molonc.2013.09.004
81. Bakkenist CJ, Lee JJ, Schmitz JC. Atm is required for the repair of oxaliplatin-induced DNA damage in colorectal cancer. Clin Colorectal Cancer. (2018) 17:255–7. doi: 10.1016/j.clcc.2018.09.001
82. Vodenkova S, Buchler T, Cervena K, Veskrnova V, Vodicka P, Vymetalkova V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: past, present and future. Pharmacol Ther. (2020) 206:107447. doi: 10.1016/j.pharmthera.2019.107447
83. de-la-Cruz-Morcillo MA, Valero ML, Callejas-Valera JL, Arias-González L, Melgar-Rojas P, Galán-Moya EM, et al. P38mapk is a major determinant of the balance between apoptosis and autophagy triggered by 5-fluorouracil: implication in resistance. Oncogene. (2012) 31:1073–85. doi: 10.1038/onc.2011.321
84. Suenaga M, Schirripa M, Cao S, Zhang W, Yang D, Murgioni S, et al. Genetic variants of DNA repair-related genes predict efficacy of Tas-102 in patients with refractory metastatic colorectal cancer. Ann Oncol. (2017) 28(5):1015–22. doi: 10.1093/annonc/mdx035
Keywords: colorectal cancer, ataxia-telangiectasia mutated (ATM), prognosis, system review, meta-analysis, radiotherapy, chemotherapy, immunotherapy
Citation: Wu P and Wen Z (2025) ATM is associated with the prognosis of colorectal cancer: a systematic review. Front. Oncol. 15:1470939. doi: 10.3389/fonc.2025.1470939
Received: 26 July 2024; Accepted: 12 February 2025;
Published: 12 March 2025.
Edited by:
Kunpeng Hu, Third Affiliated Hospital of Sun Yat-sen University, ChinaReviewed by:
Jelena Pajić, Serbian Institute of Occupational Health, SerbiaCopyright © 2025 Wu and Wen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zelin Wen, dm90YXJ5X3dlbkAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.