
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 31 March 2025
Sec. Thoracic Oncology
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1461530
This article is part of the Research Topic Advancing Precision Medicine in Lung Cancer: Integrating Genomics, Liquid Biopsy and Novel Diagnostic Tools View all 3 articles
 Jiangang Liu1†
Jiangang Liu1† Hong Wang1Amit Aggarwal1Maria Jesus Ortiz-Ruiz1
Hong Wang1Amit Aggarwal1Maria Jesus Ortiz-Ruiz1 Elisabet Zapatero-Solana1
Elisabet Zapatero-Solana1 Maria Jose Lallena2
Maria Jose Lallena2 Sandra Peregrina3
Sandra Peregrina3 Gloria Martinez del Hoyo3Susana Velasco3Philip J. Ebert1Xueqian Gong1
Gloria Martinez del Hoyo3Susana Velasco3Philip J. Ebert1Xueqian Gong1 Anwar M. Hossain1
Anwar M. Hossain1 Shawn T. Estrem1*†
Shawn T. Estrem1*†Purpose: JUNIPER, a randomized, phase III trial of patients with stage IV non–small cell lung cancer and a detectable Kristen rat sarcoma (KRAS) mutation in codons 12 or 13 whose condition progressed after platinum-based chemotherapy and up to 1 additional therapy (could include immune checkpoint inhibitors), reported prolonged progression-free survival (PFS) but not overall survival (OS) among patients who were receiving abemaciclib versus those who were receiving erlotinib. To establish whether certain patient subgroups received an OS benefit from the addition of abemaciclib to best supportive care, JUNIPER patients were retrospectively evaluated for KRAS co-mutation gene expression subtype, and abemaciclib efficacy was assessed for each patient subgroup.
Materials and methods: Of the 453 patients enrolled in the JUNIPER trial, tumor specimens for biomarker analysis were available for 148 (abemaciclib arm, n=79; erlotinib arm, n=69). Samples were profiled for gene expression and classified into 3 previously identified expression subtypes (KL, KP, and K). Tumor response, OS, and PFS were assessed within each subtype.
Results: Retrospective analyses of expression subtypes revealed an OS advantage for patients with KL subtype tumors who were receiving abemaciclib versus those with the KL subtype who were receiving erlotinib (median, 13.05 vs 5.65 months; hazard ratio, 0.25; 95% confidence interval, 0.09–0.73; P=.011). KL and KP expression subtype groups derived a PFS benefit from abemaciclib versus erlotinib (KL median, 6.64 vs 2.1 months; hazard ratio, 0.12; 95% confidence interval, 0.03–0.41; P=.001, and KP median, 5.52 vs 2.24 months; hazard ratio, 0.44; 95% confidence interval, 0.23–0.84; P=.013). Patients with K subtype tumors received no OS or PFS benefit from abemaciclib treatment.
Conclusions: Patients with KL expression subtype tumors may derive better OS and PFS from abemaciclib versus erlotinib in KRAS-mutated non–small cell lung cancer. These results should be further validated in an independent dataset.
Non–small cell lung cancer (NSCLC) is a heterogeneous disease with numerous histologic and molecular subtypes (1). These molecular subtypes have distinct genomic alterations and clinical outcomes, which can be associated with differential response to treatment (2). Kirsten rat sarcoma (KRAS), the most commonly mutated oncogene in NSCLC, has a prevalence of 29% in lung adenocarcinoma (3). Recently, 3 different KRAS subtypes—KL, KP, and K—have been identified in lung adenocarcinoma based on gene expression and transcriptional profiling. Each is associated with co-mutations (4, 5); STK11/LKB1 and TP53 are co-mutations in KL and KP, respectively, whereas K includes CDKN2A/B inactivation and low expression of NKX2-1 (TTF1) transcription factor (4, 5). This classification distinguishes tumor subsets with distinct biology, immune profiles, and therapeutic vulnerabilities (6).
Recent reports detail co-mutation-dependent treatment variability in KRAS-mutant NSCLC. A retrospective analysis demonstrated that immune checkpoint inhibitors provide variable benefit for KRAS-mutant NSCLC, with the KRAS plus STK11/LKB1 co-mutation (KL subtype) being resistant to this treatment class (7). Additionally, the 2 recently approved therapies targeting KRAS G12C, the most common KRAS genetic variant detected in NSCLC, may provide variable benefit based on the specific co-mutations (6, 8).
The synthetic lethal interaction between KRAS mutations and cyclin-dependent kinase (CDK) 4 and 6 inhibition highlights a promising therapeutic strategy for using CDK4/6 inhibitors in NSCLC (9). The CDK4/6-retinoblastoma pathway is frequently dysregulated in NSCLC, further emphasizing its potential as an attractive therapeutic target (9, 10). Abemaciclib, a CDK4/6 inhibitor, has been approved by the United States Food and Drug Administration as monotherapy and in combination with endocrine therapy for women with hormone receptor–positive, human epidermal growth factor receptor-2–negative metastatic breast cancer across various treatment lines and settings and in patients with high-risk hormone receptor–positive, human epidermal growth factor receptor-2–negative early breast cancer (11–16).
In the phase III JUNIPER trial, abemaciclib did not demonstrate an overall survival (OS) benefit in patients with NSCLC who harbored a KRAS mutation (17). However, improved progression-free survival (PFS) and objective response rate (ORR) in the abemaciclib arm compared with that in the erlotinib arm suggested some activity. The objective of this retrospective analysis was to evaluate the clinical outcome of participants from the JUNIPER trial in the context of mRNA expression–based subtypes to identify potentially predictive associations.
The JUNIPER study design (NCT02152631) has been previously published (18). Briefly, JUNIPER was a randomized, open-label, parallel, comparator-controlled phase III trial that evaluated the efficacy and safety of abemaciclib versus erlotinib in patients with stage IV NSCLC with a detectable KRAS mutation (at the time of study design, erlotinib was used in second-line/subsequent-line treatment in NSCLC without any limitation regarding epidermal growth factor receptor (EGFR) mutation status) (17). Patients were stratified by number of prior chemotherapy regimens (1 vs 2), Eastern Cooperative Oncology Group performance status (ECOG PS [0 vs 1]), sex, and KRAS mutation (G12C vs all others) and were randomized 3:2 to receive best supportive care plus either abemaciclib (200 mg twice daily) or erlotinib (150 mg twice daily) for a 28-day cycle. Inclusion criteria were age ≥18 years; a confirmed diagnosis of stage IV NSCLC; a KRAS codon 12 or 13 mutation; and disease progression following platinum-based chemotherapy with the receipt of at least 1 other systemic therapy or deemed ineligible for further chemotherapy. The protocol was approved by each participating institutional ethical review board and was conducted in accordance with the ethical principles of the Declaration of Helsinki, Council for International Organizations of Medical Sciences International Ethical Guidelines, and good clinical practice. All patients provided written informed consent prior to treatment.
At screening, KRAS mutation status was determined by the central laboratory using the QIAGEN® therascreen® KRAS Rotor-Gene Q Polymerase Chain Reaction (Qiagen, Germany) kit for NSCLC with formalin-fixed paraffin-embedded (FFPE) tumor tissue collected by surgical biopsy, fine needle aspiration, or core needle biopsy. FFPE tumor tissue samples were obtained prior to enrollment from patients who consented to participate.
Archival FFPE tumor tissue samples from 167 patients were macro-dissected to minimize the amount of adjacent normal tissue (Almac Diagnostics Ltd, USA). RNA extraction was performed using the Qiagen RNeasy extraction kit (Qiagen, Germany) followed by quality assessments using NanoDrop (Thermo Fischer Scientific, USA), Bioanalyzer (Agilent, USA), and RNA sequencing (an Almac Diagnostics Ltd, USA proprietary quality control step). Illumina TruSeq RNA Exome sequencing was performed by Almac with the Illumina TruSeq RNA Sample Preparation Kit v2 as described previously (4). Paired-end sequencing with a read length of 100 base pairs and targeted read depth of 50 million reads/sample was performed. Data were filtered to remove genes with fewer than 5 counts across 80% of the samples from the analysis. The resulting data were quantile-normalized and summarized across samples. Analyses were performed using the programming language R (version 3.1; http://www.r-project.org). Quality control failures were flagged for 11 tumor samples due to low overall read depth, indicating insufficient input material or other sources of assay failures. Raw counts for 156 samples were log2 transformed and normalized. Eight outlier samples were identified using principal component analysis of normalized counts and were removed from further analysis, leaving 148 translational marker-evaluable samples.
We applied a single sample predictor (SSP) algorithm to obtain subtype labels for the 148 translational marker-evaluable samples according to the Skoulidis classification (5). The SSP utilizes correlations between each sample and the molecular subtype “centroids,” which are representative vectors of average gene expression values for subtype-specific genes (19). To classify a new sample, its distance to each of the centroids is calculated and it is assigned to the subtype corresponding to the nearest centroid (20). For the Skoulidis expression subtypes (KL, KP, and K), the centroids consisting of an average expression of 384 genes generated from RNA sequences were used (4). Pearson correlation coefficient was used for the samples of each subtype centroid to assign the sample to 1 of 3 tumor subtypes (KL, KP, and K). We also calculated the difference between the highest and second-highest correlations to the centroids for each sample. Additionally, the highest correlations to these centroids were computed for virtual tumor sample arrays obtained by random permutations of the data for each gene. Tumors with a correlation ≥0.15 (a default parameter from the “CMSclassifier” R package; https://github.com/Sage-Bionetworks/CMSclassifier/blob/master/R/cmsClassifier.R) with any of the 3 centroids were classified, and tumors with a correlation of <0.15 remained unclassified. Each sample was assigned to the subtype with which it had the highest Pearson correlation coefficient.
To classify the tumors that remained unclassified by SSP, random forest, a well-established machine-learning method that operates by generating multiple bootstrapped versions of the training data to fit a decision tree to each of these bootstraps, was implemented (21). The random forest algorithm has been well studied in the context of gene expression classifiers, as it performs well with highly correlated, high-dimensional data and is less prone to overfitting because of the averaging effect across many models (22, 23). RNA sequence data of 67 TCGA samples with KL, KP, or K subtype labels were split at the ratio of 3:1 for training and validation, and a random forest classifier was generated from 500 balanced bootstraps of the training data (10). After model training, the classifier was applied to the validation samples and performance metrics were computed (sensitivity, specificity, and balanced accuracy). When applied to the validation data, the classifier demonstrated robust performance and evidenced subtype-specific characteristics.
The RNAseq data (reads per kilobase million) of 43 KRAS-mutated NSCLC cell lines were downloaded from Cancer Cell Line Encyclopedia (24). Gene expression profiles from the cell lines were correlated to the centroids reported by Skoulidis et al. for each of the KL, KP, and K subtypes using the same method used to classify the primary tumors. To assess the cell line subtype vulnerabilities, the antiproliferative sensitivity (half-maximal inhibitory concentration [IC50]) of abemaciclib for the 43 KRAS-mutated cell lines was retrieved from a novel cancer cell sensitivity profiling (CCSP) assay format developed internally (25).
Retroviruses were produced by transfection of HEK293T cells with pBABE-FLAG-LKB1 overexpression plasmid (plasmid 8592, Addgene, USA; gift from Lewis Cantley) (26) and packaging plasmid encoding VSV-G, gag-pol, using Lipofectamine 2000 (Invitrogen, USA). Viral particle–containing supernatants were collected 48 hours post-transfection, filtered through a 0.45 μm syringe filter, and used for infection immediately.
NCI-H1944 cells were plated the day before infection at approximately 70% confluence in complete growth medium (RPMI 1640 medium, ATCC modification 30-2001) and allowed to adhere overnight. The next day, cells were infected with viral supernatants in the presence of 6 μg/mL polybrene (Sigma-Aldrich, USA). After infection, successfully transduced cells were obtained by selection with 2 µg/mL puromycin (Millipore, USA).
Cells were plated at 2000 cells per well in 96-well plates in a total volume of 100 μL of growth media alone (RPMI 1640, 10% fetal bovine serum, 1% penicillin/streptomycin). Twenty-four hours post-seeding, cells were treated with either dimethyl sulfoxide or decreasing concentrations of test compounds in the range of 20 μM–1 nM. Test compounds were prepared at 2 times concentration in 100 µL of growth media using a dilution factor of 1:3 with a total of 0.4% dimethyl sulfoxide (0.2% final concentration in cells). After cells were incubated at 37°C for 2 doubling times (72 hours), cells were fixed with 70% ice-cold ethanol for 30 minutes and then treated with RNAse and nuclei stained with propidium iodide in phosphate-buffered saline for 1 hour. Cell nuclei per well were counted using Acumen Explorer™ (STP LabTech Ltd, UK) to determine the number of cells remaining after treatment. For data analyses, IC50 was determined by curve fitting to a 4-parameter logistic equation for each output normalized to dimethyl sulfoxide using GraphPad Prism®.
Cells were washed with phosphate-buffered saline and lysed in ice-cold lysis buffer (140 mM NaCl, 10 mM ethylenediaminetetraacetic acid, 10% glycerol, 1% Nonidet P40, 20 mM Tris, pH 8.0, cOmplete™ Mini Protease and PhosSTOP phosphatase Inhibitor Cocktail). Lysates were immediately frozen at -−-80°C and centrifuged at 12,500 rpm at 4°C for 10 minutes before protein quantification. A total of 40 µg of each sample quantified by Pierce Protein Assay Reagent (620 nm) (Thermo Fisher Scientific, USA) was loaded into Precast Midi Protein Gels (Bio-Rad, 5671095). Electrophoresis was run for 1.5 hours at 120 V in Running Buffer 1X (Sigma -Aldrich, USA), and protein transference was performed using Trans-Blot Turbo Transfer System and pre-assembled transfer packs (0.2 µm nitrocellulose membranes; Bio-Rad, USA). Immunoblotting was performed in a blocking buffer of 2% bovine serum albumin/phosphate-buffered saline-Tween 20 and detected by anti-LKB1 antibody (Cell Signaling Technology, USA) using enhanced chemiluminescence-horseradish peroxidase substrate (Thermo Fisher Scientific, USA) on Amersham Imager 600. Vinculin (Sigma-Aldrich, USA) was used as a loading control. Since cells were cultured under puromycin pressure and viability assays were performed in growth media alone, protein expression analyses were done in cells cultured with and without puromycin selection.
Efficacy outcomes, including ORR, disease control rate (DCR), and time-to-event variables (OS and PFS) were assessed in abemaciclib- versus erlotinib-treated patients, in the translational research (TR) population (patients with evaluable gene expression data available for association analysis with clinical efficacy), and for each of the subtypes (KL, KP, and K). A logistic regression model stratified by sex, ECOG PS, number of prior chemotherapies, and KRAS mutation was used to investigate the relationship between ORR, DCR, and treatment for each expression subtype; P-values were derived from the likelihood ratio test. ORR was defined as the combined rates of complete response and partial response (PR), and DCR was defined as the combined rates of complete response, PR, and stable disease (SD). The Kaplan-Meier product limit method was used to estimate the OS and PFS survival curves; 95% confidence intervals (CIs) for median OS and median PFS were computed by the Brookmeyer and Crowley method (27). A stratified Cox proportional hazards model was used to compare the treatment effect in each expression subtype, hazard ratio (HR) estimates, and their 95% CIs. Log-rank test P-values were reported on the Kaplan-Meier plot.
In addition, multivariable Cox proportional hazards models were performed including treatment, expression subgroups, treatment-by-subgroup interactions, key clinical covariates (age, prior immunotherapy, smoking status), and stratification factors as explanatory variables within each expression subtype. Univariate subgroup analyses for key clinical covariates and stratification factors were performed for each expression subtype and reported on forest plots. The effect size was calculated using a linear mixed-effects model implemented with the lme4 R package (version 1.1.26). All analyses in this study were exploratory in nature, and no multiplicity adjustments were performed.
RNA was obtained from 167 archival JUNIPER FFPE tumor samples, with high-quality data (data that passed quality control) from 148 samples (TR population; abemaciclib arm, n=79; erlotinib arm, n=69). Sample flow through the study is shown in Figure 1. Baseline demographic characteristics in the TR and intent-to-treat (ITT) populations were similar (Table 1), except that the TR population underrepresented Asian patients in the ITT population (2.5% and 7.2% for TR vs 20.0% and 22.4% for ITT in the abemaciclib and erlotinib arms, respectively). Within the TR population, notable differences in percentages were observed between the 2 arms for the categories of KRAS G12C mutation and ECOG PS of 0. In the abemaciclib arm, 63.3% had the KRAS G12C mutation (median OS: 7.61 months; median PFS: 3.91 months), compared to 49.3% in the erlotinib arm (median OS: 6.05 months; median PFS: 1.92 months; χ²test, P= .12). For ECOG PS of 0, 26.6% were in the abemaciclib arm (median OS: 10.2 months; median PFS: 6.61 months) while 18.8% were in the erlotinib arm (median OS: 11.9 months; median PFS: 2.20 months; χ² test, P=.357). However, these differences were not statistically significant. For participants receiving abemaciclib, numerically longer OS (Supplementary Figure 1A) and PFS (Supplementary Figure 1B) were observed in the TR population than in the ITT population, while erlotinib OS was numerically shorter in the TR verses the ITT population, and PFS was comparable between the TR and ITT populations.
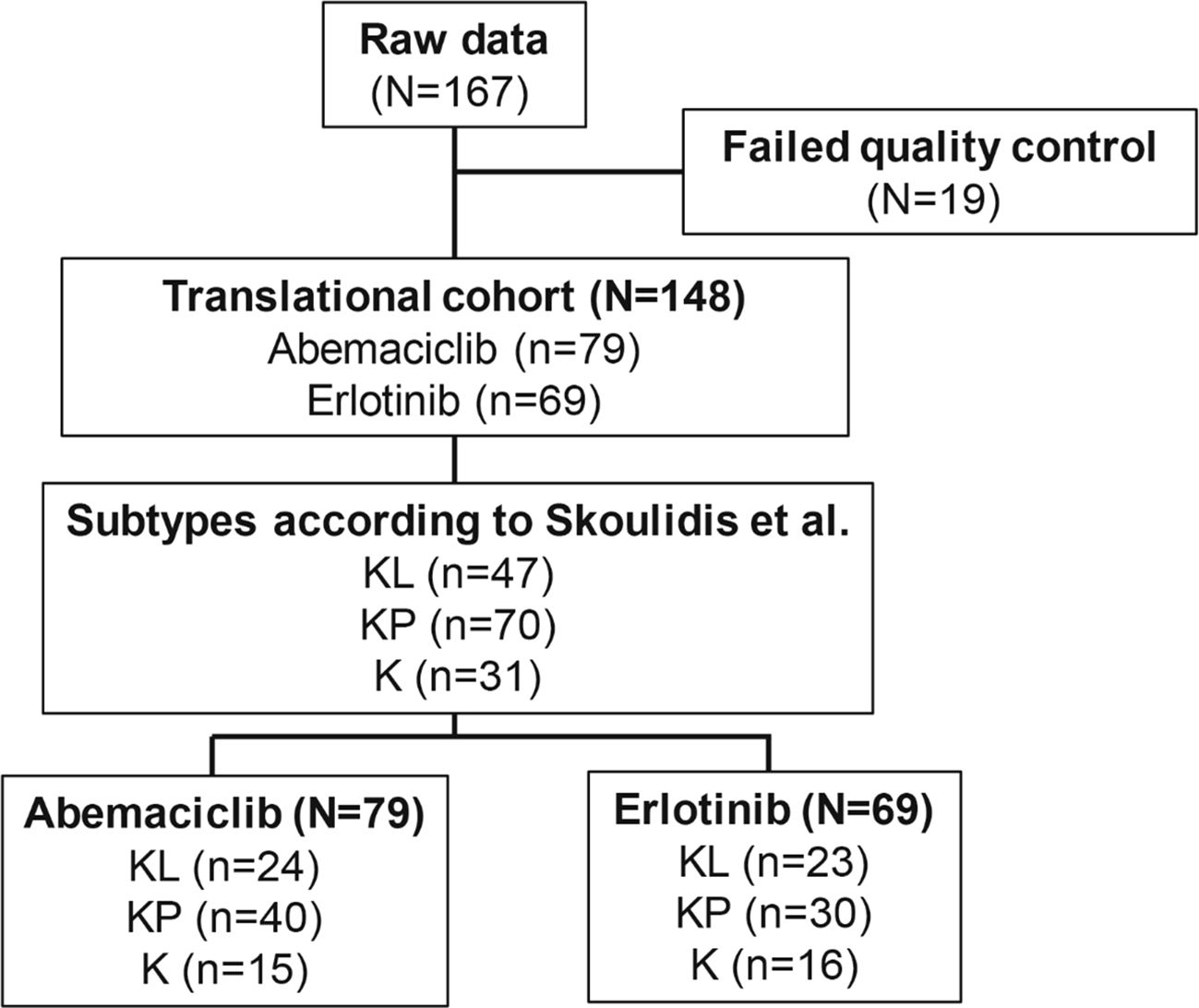
Figure 1. Flow diagram demonstrating sample derivation for biomarker analyses. N, number of patients in the population; n, number of patients in the specified category.
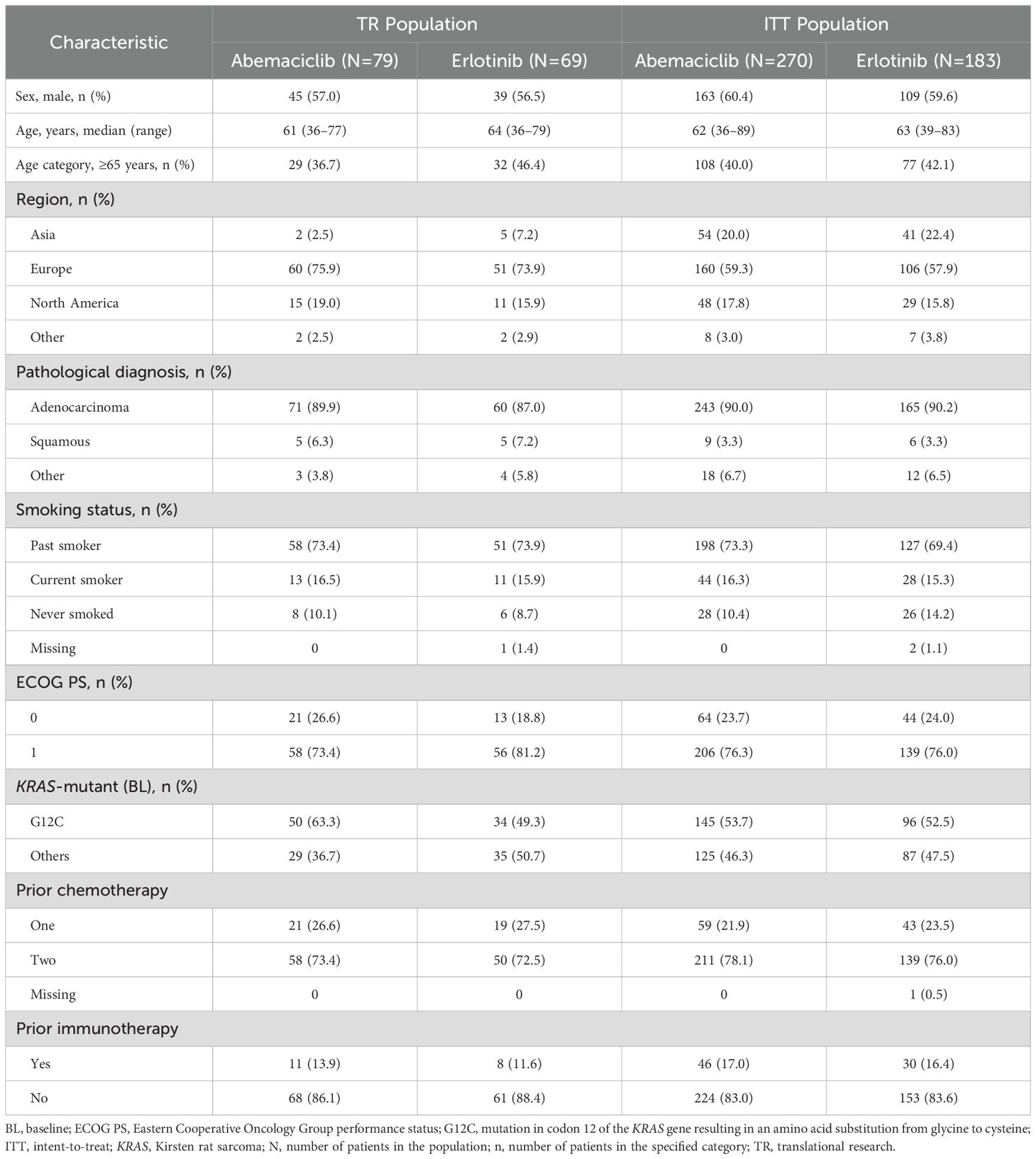
Table 1. Baseline demographic and disease characteristics of the TR and ITT populations.
Of the 148 samples, 113 (76.4%) were assigned to an expression subtype; 35 remained unclassified by SSP. Random forest was then applied to classify the remaining samples. The final classification assigned 70 KP (46.5%), 47 KL (32.7%), and 31 K (20.8%) subtypes. Hierarchical clustering with 18 core classifier genes, 3 additional characteristic genes (NKX2-1, STK11, CD274) identified by Skoulidis et al. (5), and 3 abemaciclib target-related genes (CDK4, CDK6, RB1) displayed as a heatmap (colored with a gradient from blue for low expression to red for high expression) highlights the selected subtype-specific gene enrichment in multiple subtypes (Figure 2A).
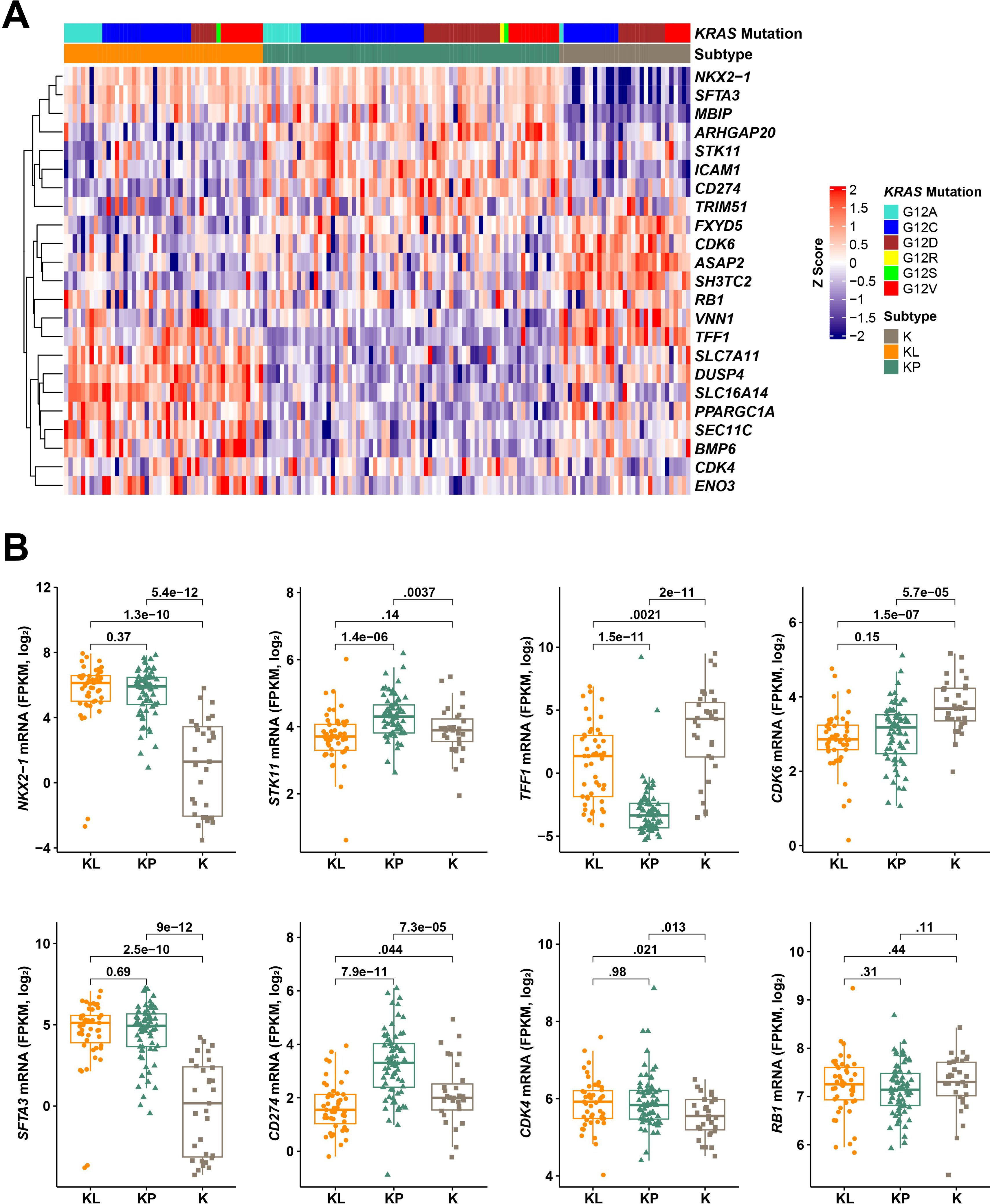
Figure 2. KRAS-mutant expression subtypes assigned based on Skoulidis et al.’s method. Heat map shows gene expression (z scores) of 18 core classifier genes and 3 additional characteristic genes (NKX2-1, STK11, CD274) identified by Skoulidis et al. and 3 abemaciclib target-related genes. Genes (rows) were clustered using Pearson correlation distance and hierarchical clustering. Samples (columns) were grouped by subtypes assigned based on Skoulidis et al.’s method (A). mRNA expression of 5 subtype specific genes (NKX2-1, SFTA3, TFF1, CD274 [PD-L1], STK11) and 3 abemaciclib target-related genes (CDK4, CDK6, RB1) in KRAS-mutant lung adenocarcinoma subsets. The Wilcoxon test was used for statistical comparison between the 3 subgroups (B). CDK, cyclin-dependent kinase; FPKM, fragments per kilobase of transcript per million mapped reads; G12C, mutation in codon 12 of the KRAS gene resulting in an amino acid substitution from glycine to cysteine; ITT, intent-to-treat; KRAS, Kirsten rat sarcoma; mRNA, messenger ribonucleic acid; PD-L1, programmed death-ligand 1; RB, retinoblastoma; RPM, revolutions per minute; SFTA3, surfactant associated 3; STK11, serine/threonine kinase 11; TFF1, trefoil factor 1.
In the K subtype, the NKX2-1 (TTF1) transcription factor showed low or no expression in log2-transformed fragments per kilobase of transcript per million mapped reads [FPKM]: 1.08 vs 5.61 for KL, 5.54 for KP; P<1.3e–10), along with its downstream lung-specific gene SFTA3 (log2-transformed FPKM: −0.09 vs 4.52 for KL, 4.55 for KP; P<2.5e–10). The immune checkpoint ligand CD274 (programmed death-ligand 1) was expressed at a higher level in KP tumors (log2-transformed FPKM: 2.16 for K, 1.66 for KL, 3.27 for KP; P=7.9e–11 KP vs KL). STK11/LKB1 mRNA expression was lower in the KL subtype than in the KP subtype (log2-transformed FPKM: 3.89 for K, 3.69 for KL, 4.3 for KP; P=1.4e–06). CDK4 and CDK6 expression levels did not show statistically significant differences between the KL and KP tumors; however, patients with K tumors had lower CDK4 (log2-transformed FPKM: 5.55 for K, 5.88 for KL, 5.94 for KP) and higher CDK6 expression (log2-transformed FPKM: 3.75 for K, 2.86 for KL, 3.02 for KP) compared to those with other tumor subtypes (Figure 2B). Additionally, RB1 expression levels were relatively consistent across subtypes (log2-transformed FPKM: 7.27 for K, 7.23 for KL, 7.15 for KP), and TFF1 expression was significantly lower in the KP subtype (log2-transformed FPKM: 3.32 for K, 0.81 for KL, −2.99 for KP; P=1.5e–11 KP vs KL).
Among abemaciclib recipients with the KL subtype, 2 (8.3%) experienced a best overall response (BOR) of PR, 20 (83.3%) had a BOR of SD, and 1 (4.2%) experienced a BOR of progressive disease (PD; Table 2). Among erlotinib recipients with the KL subtype, none experienced a BOR of PR, 8 (34.8%) had a BOR of SD, and 10 (43.5%) patients experienced a BOR of PD (Table 2). A significant treatment-arm DCR difference was observed among patients with the KL subtype (abemaciclib arm, 91.7% vs erlotinib arm, 34.8%, P=.001). The treatment-arm ORR difference was not statistically significant (8.3% vs 0.0%, P=.62; Table 2).
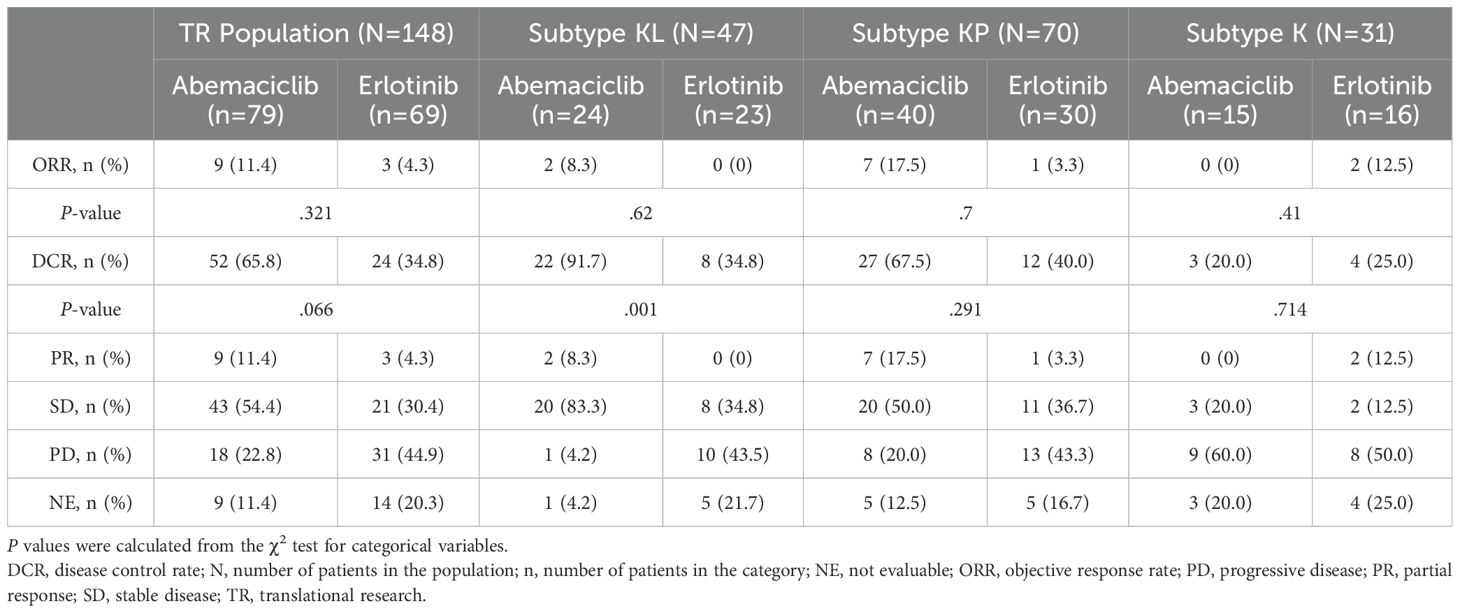
Table 2. Tumor subtype response to treatment.
Abemaciclib-treated patients with the KP subtype had numerically better DCR and ORR than erlotinib-treated patients, whereas the ORR and DCR were lower, though not statistically significant, for abemaciclib- compared to erlotinib-treated patients with the K subtype (Table 2). Furthermore, in the TR population, no statistically significant ORR or DCR differences between treatment arms were observed (Table 2).
Comparison of survival differences among the expression subtypes regardless of treatments suggests a subtype-specific prognosis. The KL, KP, and K subtypes had a median OS of 8.71, 11.93, and 4.57 months, respectively (Figure 3A), and a median PFS of 3.72, 3.91, and 1.89 months, respectively (Figure 3B). These data demonstrate that the K subtype may be a poor prognostic indicator compared to the KP subtype, which had the best OS and PFS, and the KL subtype.
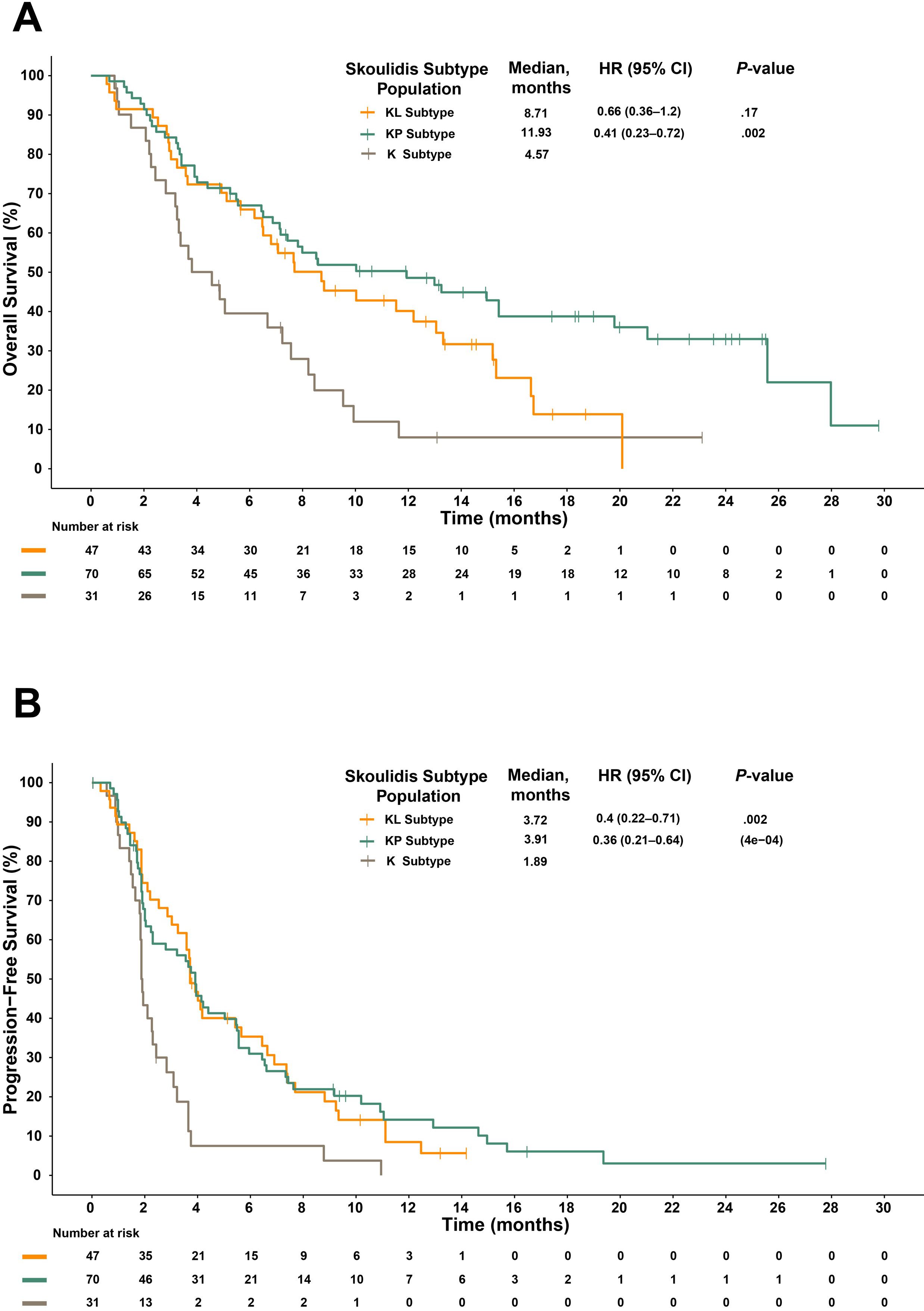
Figure 3. Kaplan-Meier plots of OS (A) and PFS (B) stratified by subtypes identified by Skoulidis et al. regardless of treatment arms. CI, confidence interval; HR, hazard ratio; OS, overall survival; PFS, progression-free survival.
Patients with the KL subtype benefitted most from abemaciclib treatment (OS, 13.05 vs 5.65 months for erlotinib; HR, 0.25; 95% CI, 0.09–0.73; P=.011; Figure 4A); this differed from the KP or K subtypes, where no OS benefit was observed (Figures 4C, E).
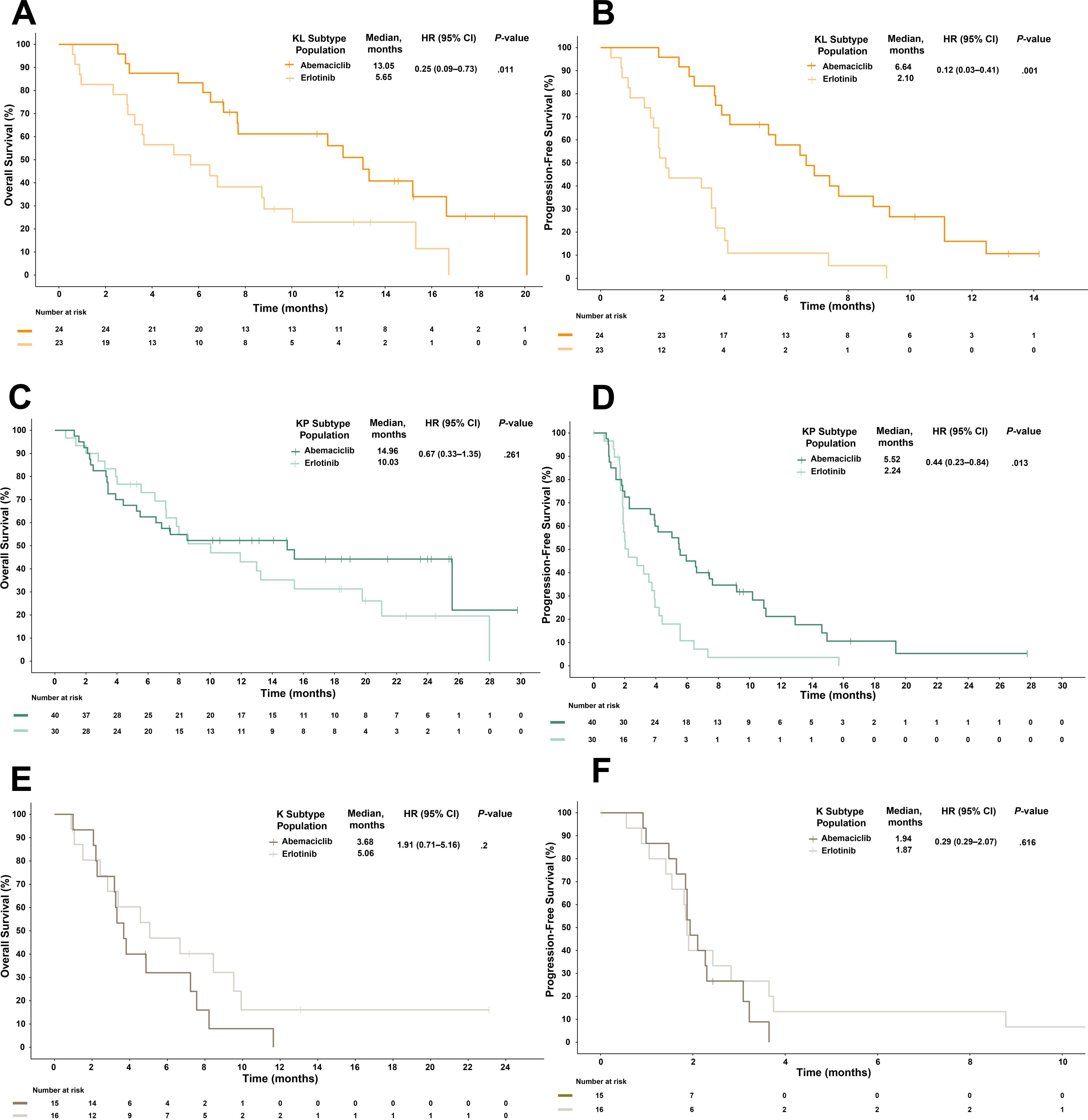
Figure 4. Overall survival (A, C, E) and progression-free survival (B, D, F) stratified by subtypes identified by Skoulidis et al. and treatment arm. The KL subtype is associated with better overall survival (A) and progression-free survival (B) after abemaciclib treatment than after erlotinib treatment among patients with KRAS-mutant lung adenocarcinoma in JUNIPER. Multivariate analysis adjusted for stratification factors was performed to compute the HRs. CI, confidence interval; HR, hazard ratio; KRAS, Kirsten rat sarcoma.
Patients with the KL and KP subtypes in the abemaciclib arm had increased PFS versus those in the erlotinib arm (Figures 4B, D; KL: 6.64 vs 2.1 months; HR, 0.12; 95% CI, 0.03–0.49; P=.001; KP: 5.52 vs 2.24 months; HR, 0.44; 95% CI, 0.23–0.84; P=.013), while those with the K subtype had little PFS benefit (1.94 vs 1.87 months; HR, 0.78; 95% CI, 0.29–2.07; P=.616; Figure 4F).
Both univariate and multivariate analyses demonstrated consistent survival benefits with abemaciclib treatment specifically in the KL molecular subtype. The multivariate Cox regression analysis confirmed the OS benefit in KL subtype patients (HR, 0.35; 95% CI, 0.15–0.79; P=.02) but not in KP (HR, 0.76; 95% CI, 0.38–1.51; P=.84) or K subtypes (HR, 1.27; 95% CI, 0.52–3.18; P=.31; Table 3). This pattern was also observed for PFS in the KL subtype (HR, 0.39; 95% CI, 0.20–0.72; P=.004; Supplementary Table 1).

Table 3. Multivariable analysis for OS by expression subtype.
The univariate subgroup analysis for clinical covariates and stratification factors in the KL subtype population showed consistency across subgroups, with expected variations. Notably, KL subtype patients with KRAS G12C mutation demonstrated superior survival with abemaciclib compared to erlotinib (median OS: 15.19 vs 4.62 months; HR, 0.183; 95% CI, 0.059–0.572; P=.001; Figure 5), despite the limited sample size (abemaciclib n=13 vs erlotinib n=8). This KRAS G12C-associated survival benefit was not observed in either the KP or K subtypes (Supplementary Figures 2A, B).
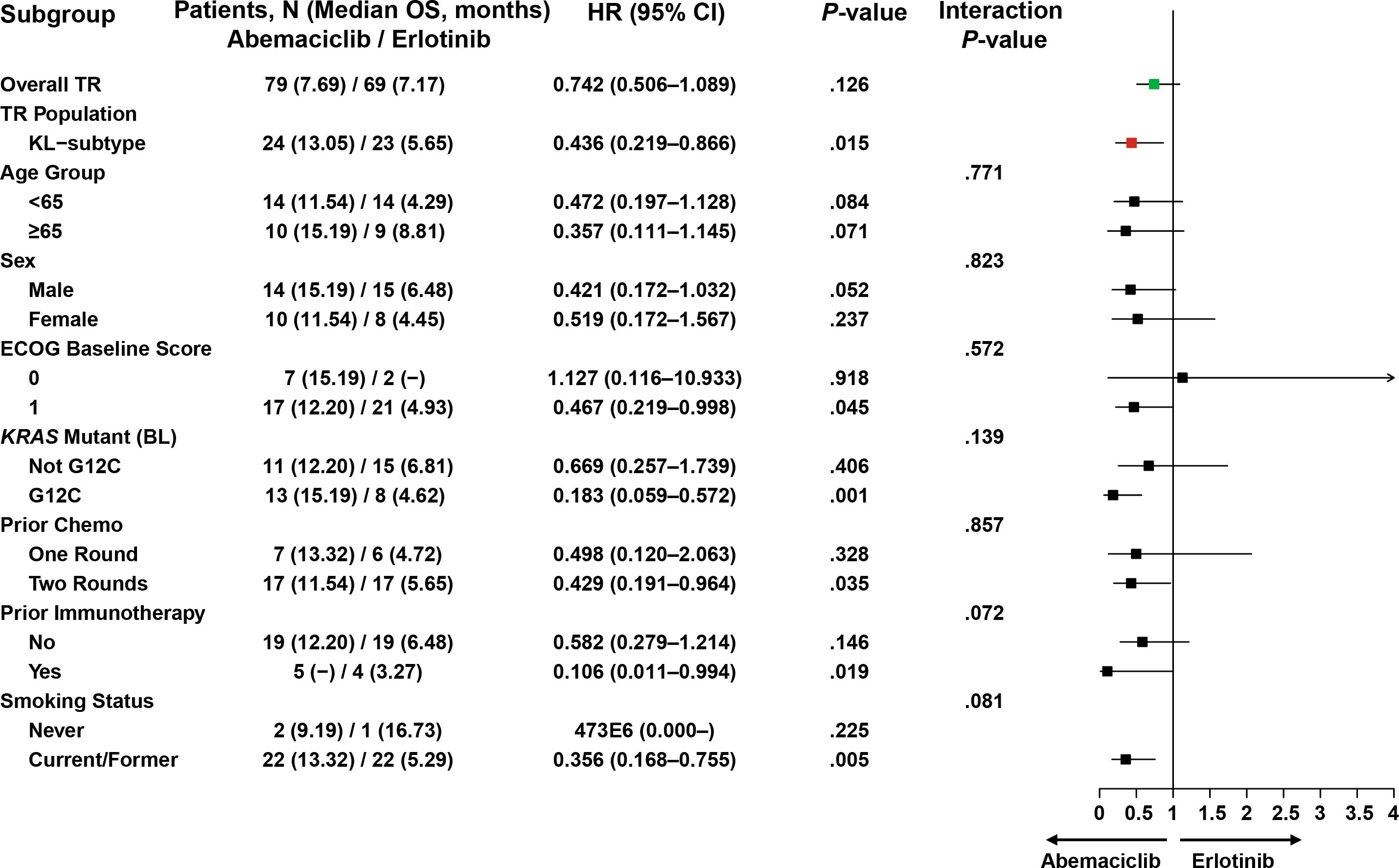
Figure 5. Forest plot for OS treatment effect and univariate subgroup analysis for clinical covariates and stratification factors within the KL subtype. –, Not evaluable due to the small sample size; Green: HR for the ITT population; Red: HR for the TR population; Black; subgroups; arrow line, CI out of the x-axis. Univariate analysis was performed to compute the HRs. BL, baseline; CI, confidence interval; ECOG, Eastern Cooperative Oncology Group; G12C, mutation in codon 12 of the KRAS gene resulting in an amino acid substitution from glycine to cysteine; HR, hazard ratio; ITT, intent-to-treat; KRAS, Kirsten rat sarcoma; OS, overall survival; TR, translational research.
Effect Size Analysis: For OS, the abemaciclib arm showed a consistent positive effect (1.27–1.30) across various models (Supplementary Figures 3A, C), with clinical parameters like baseline ECOG PS of 1 (−3.77, P<.05) and stage IV disease (−6.28) demonstrating substantial negative effects, while the age category of ≥65 years showing a measurable positive effect (1.85). For PFS, the abemaciclib arm exhibited a stronger positive effect (2.95, P< 0.001) compared to OS (Supplementary Figures 3B, D), with sex (male) demonstrating a significant positive effect (1.61–1.63, P<.05) and Stage IV disease maintaining a consistent negative effect (−2.65 to −4.36). Notably, both KP and KL subtypes demonstrated significant effects for OS (KP: 5.81, P<.001; KL: 3.57, P<.05) and PFS (KP: 2.76, P<.01; KL: 2.38, P<.05).
The abemaciclib activity data from CCSP was used to evaluate differential sensitivity of expression subtypes within the KRAS-mutated NSCLC cell lines. Gene expression profiles from cell lines were correlated to centroids identified by Skoulidis et al. (4) for the KL, KP, and K subtypes using the same method used to classify primary tumors. In CCSP, 24 of 43 KRAS-mutated cell lines had abemaciclib activity data and were classified: 9 KL, 12 KP, and 3 K subtypes. The 3 K and 12 KP subtype cell lines were combined into the non-KL group to increase statistical power. KL subtype cell lines were more sensitive to abemaciclib treatment than the non-KL subtype cell lines (Figure 6A).
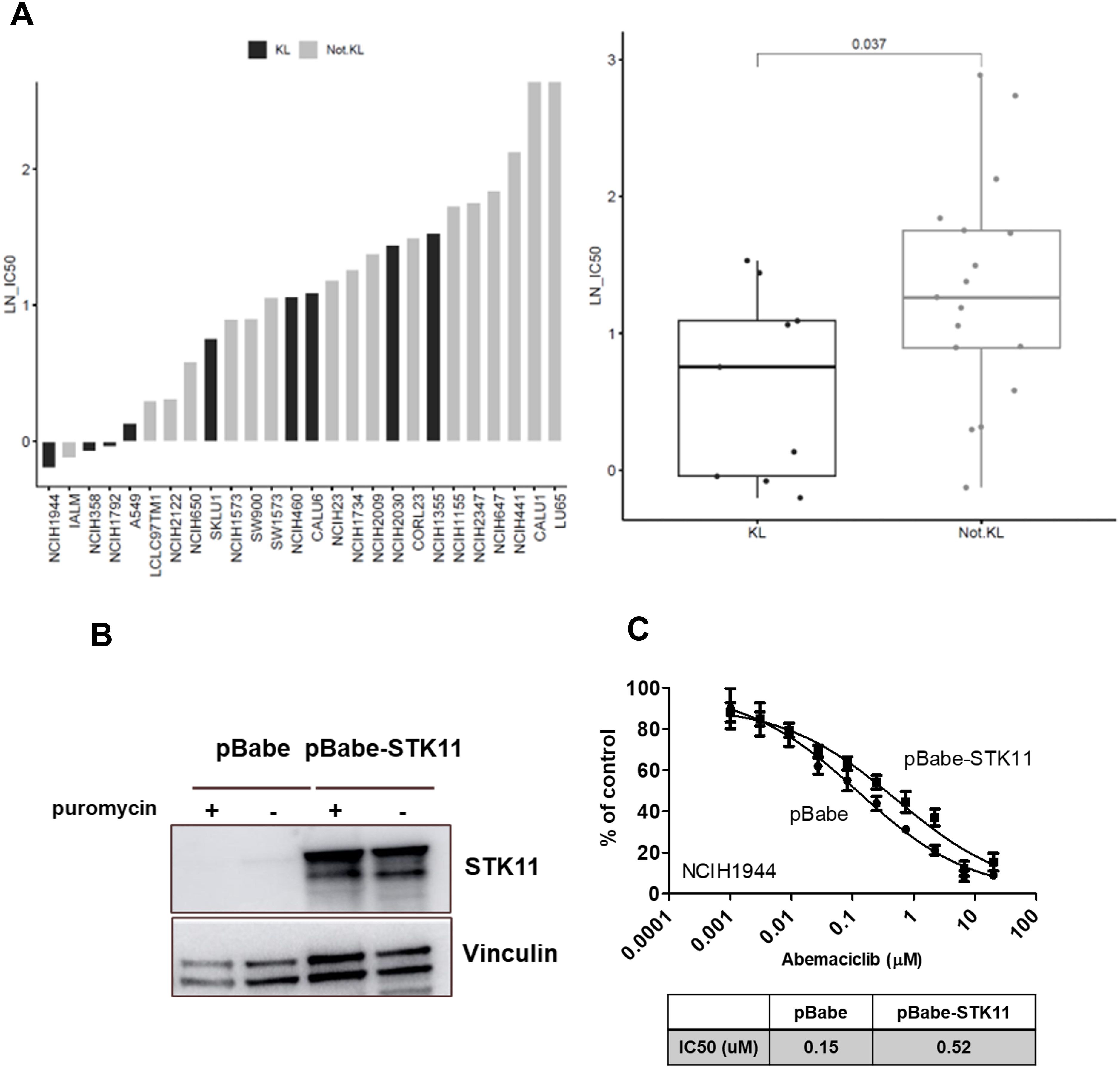
Figure 6. KRAS-mutated NSCLC cell line subtypes have differential sensitivity to abemaciclib. IC50 values for the cell lines treated with the CDK4/6 inhibitor abemaciclib. (A) Antiproliferative activity of abemaciclib against 24 KRAS-mutated NSCLC cell lines. Dot plot shows the log distribution of drug sensitivity to abemaciclib in the KL subtypes and the non-KL subtype (Not-KL = K+KP). Statistically significant differences in the IC50 values of KL compared with non-KL (P=.037) as determined by Mann-Whitney U test. Overexpression of STK11/LKB1 in KL-classified cells (NCI-H1941). Western blots are representative of 2 independent experiments. (B) Reduced sensitivity of pBABE-STK11 NCI-H1941 cells to abemaciclib. (C) Cell viability is expressed as % of control (mean ± standard deviation; n=2). CDK, cyclin-dependent kinase; IC50, half-maximal inhibitory concentration; KRAS, Kirsten rat sarcoma; NSCLC, non–small cell lung cancer; STK11, serine/threonine kinase 11.
The NCI-H1944 cell line with the inactivating STK11/LKB1 mutation was most sensitive to abemaciclib. To evaluate the relationship between STK11/LKB1 activity and abemaciclib sensitivity, STK11/LKB1 was overexpressed in the NCI-H1944 KL cell line, which lacks LKB1 expression (Figure 6B). A cell viability assay of the NCI-H1944 KL cell line with STK11/LKB1 overexpression showed reduced abemaciclib sensitivity, with an approximate 3.5-fold IC50 relative to the parental cell line (Figure 6C).
Despite recent advances in KRAS-targeted NSCLC therapy, many tumors do not respond to current treatments, including immune checkpoint inhibitors. There are no targeted therapies for KRAS-mutant tumors lacking the G12C variant. In particular, KRAS plus STK11/LKB1 co-mutated (KL subgroup) tumors represent an especially poor prognostic subgroup, with lower responses to immune checkpoint and KRAS G12C inhibitors (8, 17). Our study results indicate that patients with the expression-derived KL subtype of KRAS-mutant NSCLC treated with abemaciclib have significantly improved OS and PFS compared with those treated with erlotinib. This association was further supported by cell line analyses and an STK11/LKB1 knock-in model. The effect size analyses highlighted meaningful differences across both survival endpoints, with consistent effects and stable variables when subtypes were excluded, supporting the robustness of these findings. These results should be considered preliminary, as they are based on a retrospective analysis of a subset of patients in the JUNIPER trial, where the comparator arm received erlotinib treatment.
We previously reported no association between mutation subgroups and response to abemaciclib in a subset of patients from the JUNIPER trial (17). In contrast, this study identified clinically relevant subgroups and demonstrates a significant association between abemaciclib and clinical outcome in the KL subtype. A possible explanation for this difference is an imperfect association of KL mutation–defined tumors and the associated RNA-defined cluster in earlier reports. The previous analysis examined cancer gene DNA sequencing in a subset of JUNIPER samples after abemaciclib treatment (17). Associated RNA expression data were available for 16 of these genetically KRAS plus STK11/LKB1–mutated samples. In this sample set, 10 were KL, 1 was KP, and 3 were K subtype tumor samples. Similar to the originally reported data, these results suggest an imperfect association between the genetic (mutation)- and expression (RNA)-derived subtypes. Furthermore, there were differences in clinical parameters associated with abemaciclib treatment between the mutation-defined and expression-defined KL subtypes. The abemaciclib-treated patients with samples characterized as KL by both mutation- and expression-based subtyping (n=10) had an average PFS of 8.42 months, and only 2 patients had an increase in tumor size. In addition, 2 patients experienced PR, and the remaining patients had a best response of SD. In contrast, patients who were classified as having KL subtype tumors by mutation only (n=6) had an average PFS of 1.99 months, an increase in tumor size of ≥14%, and all but one had PD. Another potential explanation for the discordance between the predictive utility of mutation and RNA clustering is that RNA-based clustering performed here was not identical to the original methodology. However, we observed a similar prevalence of each expression-defined cluster with the same relative prognosis as previously reported (4).
Although patients with KL and KP subtype tumors experienced longer PFS with abemaciclib, only those with the KL pattern experienced longer OS after treatment with abemaciclib. The reason for the longer OS observed in patients with the KL subtype is unclear; however, STK11/LKB1 and CDK4 may play a role in this. The CDK4/6 inhibitor palbociclib was shown to be more potent in cell lines after knockdown of STK11/LKB1 expression or had decreased sensitivity when STK11/LKB1 was expressed in STK11/LKB1-mutant cell lines (28). Aligned with this observation, we showed that abemaciclib sensitivity was reduced on overexpression of STK11/LKB1 in a sensitive KL cell line. Furthermore, downregulation of STK11/LKB1 facilitates the G1/S transition, which is regulated by CDK4 (29). These observations are consistent with the increased sensitivity to the CDK4/6 inhibitor abemaciclib seen in tumors with disrupted STK11/LKB1 function associated with KL expression subtype tumors.
In patients with the K subtype, no clinical benefit was observed with abemaciclib compared with that after erlotinib treatment. The K subtype is coupled with low level or no expression of the NKX2-1(TTF1) transcription factor and higher CDK6 expression. CDK6 amplification or overexpression is one possible resistance mechanism to CDK4/6 inhibitors. Thus, this is consistent with the lack of clinical benefit observed with abemaciclib treatment in this study (30).
Our study had certain limitations that should be acknowledged. First, only a subset of the trial population could be included in the retrospective analysis; this introduced bias. The abemaciclib arm of the TR population had longer OS and PFS than that of the ITT population, while the erlotinib arms were balanced. This bias of the TR population needs to be considered when interpreting the KL expression subtype predictive analysis reported. However, the significance associated with the predictive value relative to the magnitude of the bias suggests the predictive value is relevant. Second, the available clinical data on erlotinib as a comparator in this population are limited and inconsistent. Erlotinib was acceptable in the JUNIPER trial given that it was the only Food and Drug Administration-approved third-line treatment at the time of study initiation and its use was not dependent on the presence of an epidermal growth factor receptor mutation. However, during accrual, in 2016, the Food and Drug Administration label for erlotinib was modified to restrict use to patients with tumors with epidermal growth factor receptor exon 19 deletions or exon 21 (L858R) substitution mutations (31). Furthermore, the subsequent availability of immune checkpoint inhibitors changed the second and later line standard of care. Despite the limitations associated with the use of erlotinib as a comparator, docetaxel, a common second-line treatment for KRAS-mutant NSCLC, showed similar median OS (7.9 months) and PFS (2.8 months) (32) to what was observed with second- and third-line erlotinib treatment in the JUNIPER trial (OS, 7.8 months; PFS, 1.9 months) (17). Moreover, our investigation focused solely on drug sensitivity within the 2 cell lines. It would be advantageous to incorporate additional cell lines to elucidate the correlation between STK11/LKB1 activity and sensitivity to abemaciclib.
In conclusion, this retrospective analysis of the phase III JUNIPER trial identified the KL subtype as a candidate predictive biomarker for abemaciclib efficacy in platinum-refractory KRAS-mutant lung adenocarcinoma. Although KRAS G12C mutations can be targeted, the other KRAS mutations, which represent approximately half of KRAS-mutated NSCLC, have no approved target-specific therapy. So-called “pan-RAS” and other mutation-specific agents are in development, and if approved, these will be treatment options for subsets of patients with KRAS-mutant NSCLC. Our findings can potentially identify a patient subpopulation that may derive a OS benefit from the addition of abemaciclib to best supportive therapy. These findings require independent validation and the development of a robust KL-subtyping companion diagnostic assay before they can be implemented in practice.
Eli Lilly and Company provides access to all individual data collected during the trial, after anonymization, with the exception of pharmacokinetic, genomic, or genetic data. Access to the data is provided after a proposal has been approved by an independent review committee and after receipt of a signed data sharing agreement. Data and documents (such as study protocol, statistical analysis plan, clinical study report, and blank or annotated case report forms) will be provided in a secure data sharing environment.
The protocol was approved by each participating institutional ethical review board and was conducted in accordance with the ethical principles of the Declaration of Helsinki, Council for International Organizations of Medical Sciences International Ethical Guidelines, and good clinical practice. All patients provided written informed consent prior to treatment. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
JL: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Methodology. HW: Writing – original draft, Writing – review & editing, Formal analysis, Methodology, Visualization. AA: Writing – original draft, Writing – review & editing. MO: Writing – original draft, Writing – review & editing, Formal analysis, Methodology, Validation, Visualization. EZ: Writing – original draft, Writing – review & editing, Formal analysis, Methodology, Visualization. ML: Writing – original draft, Writing – review & editing, Formal analysis, Methodology, Visualization. SP: Writing – original draft, Writing – review & editing, Formal analysis, Software, Validation, Visualization. GD: Writing – original draft, Writing – review & editing, Formal analysis, Software, Validation, Visualization. SV: Writing – original draft, Writing – review & editing, Formal analysis, Software, Validation, Visualization. PE: Writing – original draft, Writing – review & editing. XG: Writing – original draft, Writing – review & editing. AH: Writing – original draft, Writing – review & editing, Formal Analysis, Methodology, Validation. SE: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Supervision, Validation.
The author(s) declare that financial support was received for the research and/or publication of this article.
We thank the patients, caregivers, and investigators who participated in this study. All authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work, and have given their approval for this manuscript to be published. Medical writing support was provided by Anchal Sood of and editorial support by Cynthia Abbott of Syneos Health, funded by Eli Lilly and Company.
JL, AH, SE, PE, XG, MO, EZ, and ML are full-time employees of Eli Lilly and Company and may own company stock. HW and AA are previous employees of Eli Lilly and Company.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
This work was supported by Eli Lilly and Company. The study sponsor provided the study drug and collaborated with the investigators to design the study; collect, analyze, and interpret the data; and write this report.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1461530/full#supplementary-material
BOR, best overall response; CCSP, cancer cell sensitivity profiling; CI, confidence interval; CDK, cyclin-dependent kinase; DCR, disease control rate; ECOG PS, Eastern Cooperative Oncology Group performance status; FFPE, formalin-fixed paraffin-embedded; FPKM, fragments per kilobase of transcript per million mapped reads; HR, hazard ratio; IC50, half-maximal inhibitory concentration; ITT, intent-to-treat; KRAS, Kirsten rat sarcoma; NSCLC, non–small cell lung cancer; ORR, objective response rate; OS, overall survival; PD, progressive disease; PFS, progression-free survival; PR, partial response; SD, stable disease; SSP, single sample predictor; TR, translational research.
1. Hayes DN, Monti S, Parmigiani G, Gilks B, Naoki K, Bhattacharjee A, et al. Gene expression profiling reveals reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. J Clin Oncol. (2006) 24:5079–90. doi: 10.1200/JCO.2005.05.1748
2. Wilkerson MD, Yin X, Walter V, Zhao N, Cabanski CR, Hayward MC, et al. Differential pathogenesis of lung adenocarcinoma subtypes involving sequence mutations, copy number, chromosomal instability, and methylation. PloS One. (2012) 7:e36530. doi: 10.1371/journal.pone.0036530
3. Barlesi F, Mazieres J, Merlio JP, Debieuvre D, Mosser J, Lena H, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet. (2016) 387:1415–26. doi: 10.1016/S0140-6736(16)00004-0
4. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discovery. (2015) 5:860–77. doi: 10.1158/2159-8290.CD-14-1236
5. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discovery. (2018) 8:822–35. doi: 10.1158/2159-8290.CD-18-0099
6. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. (2021) 384:2371–81. doi: 10.1056/NEJMoa2103695
7. Amanam I, Mambetsariev I, Gupta R, Achuthan S, Wang Y, Pharaon R, et al. Role of immunotherapy and co-mutations on KRAS-mutant non-small cell lung cancer survival. J Thorac Dis. (2020) 12:5086–95. doi: 10.21037/jtd.2020.04.18
8. Jänne PA, Riely GJ, Gadgeel SM, Heist RS, Ou SHI, Pacheco JM, et al. Adagrasib in non-small-cell lung cancer harboring a KRASG12C mutation. N Engl J Med. (2022) 387:120–31. doi: 10.1056/NEJMoa2204619
9. Puyol M, Martín A, Dubus P, Mulero F, Pizcueta P, Khan G, et al. A synthetic lethal interaction between K-Ras oncogenes and CDK4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. (2010) 18:63–73. doi: 10.1016/j.ccr.2010.05.025
10. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature (2014) 511(7511):543-50. doi: 10.1038/nature13385. Rogers, K [corrected to Rodgers, K. Nature. (2018) 559:E12. doi: 10.1038/s41586-018-0228-6
11. Dickler MN, Tolaney SM, Rugo HS, Cortés J, Diéras V, Patt D, et al. MONARCH 1, a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, in patients with refractory HR+/HER2- metastatic breast cancer. Clin Cancer Res. (2017) 23:5218–24. doi: 10.1158/1078-0432.CCR-17-0754
12. Sledge GW Jr, Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2- advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. (2017) 35:2875–84. doi: 10.1200/JCO.2017.73.7585
13. Johnston S, Martin M, Di Leo A, Im SA, Awada A, Forrester T, et al. MONARCH 3 final PFS: a randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer. (2019) 5:5. doi: 10.1038/s41523-018-0097-z
14. Neven P, Johnston SRD, Toi M, Sohn J, Inoue K, Pivot X, et al. MONARCH 2: subgroup analysis of patients receiving abemaciclib plus fulvestrant as first-line and second-line therapy for HR+, HER2–advanced breast cancer. Clin Cancer Res. (2021) 27:5801–9. doi: 10.1158/1078-0432.CCR-20-4685
15. Sledge GW Jr., Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. The effect of abemaciclib plus fulvestrant on overall survival in hormone receptor-positive, ERBB2-negative breast cancer that progressed on endocrine therapy-MONARCH 2: a randomized clinical trial. JAMA Oncol. (2020) 6:116–24. doi: 10.1001/jamaoncol.2019.4782
16. Johnston SRD, Toi M, O’Shaughnessy J, Rastogi P, Campone M, Neven P, et al. Abemaciclib plus endocrine therapy for hormone receptor-positive, HER2-negative, node-positive, high-risk early breast cancer (monarchE): results from a preplanned interim analysis of a randomised, open-label, phase 3 trial. Lancet Oncol. (2023) 24:77–90. doi: 10.1016/S1470-2045(22)00694-5
17. Goldman JW, Mazieres J, Barlesi F, Dragnev KH, Koczywas M, Göskel T, et al. A randomized phase III study of abemaciclib versus erlotinib in patients with stage IV non-small cell lung cancer with a detectable KRAS mutation who failed prior platinum-based therapy: JUNIPER. Front Oncol. (2020) 10:578756. doi: 10.3389/fonc.2020.578756
18. Goldman JW, Shi P, Reck M, Paz-Ares L, Koustenis A, Hurt KC. Treatment rationale and study design for the JUNIPER study: a randomized phase III study of abemaciclib with best supportive care versus erlotinib with best supportive care in patients with stage IV non-small-cell lung cancer with a detectable KRAS mutation whose disease has progressed after platinum-based chemotherapy. Clin Lung Cancer. (2016) 17:80–4. doi: 10.1016/j.cllc.2015.08.003
19. Hu Z, Fan C, Oh DS, Marron JS, He X, Qaqish BF, et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics. (2006) 7:96. doi: 10.1186/1471-2164-7-96
20. Sontrop HMJ, Reinders MJT, Moerland PD. Breast cancer subtype predictors revisited: from consensus to concordance? BMC Med Genomics. (2016) 9:26. doi: 10.1186/s12920-016-0185-6
22. Chen X, Ishwaran H. Random forests for genomic data analysis. Genomics. (2012) 99:323–9. doi: 10.1016/j.ygeno.2012.04.003
23. Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. (2015) 21:1350–6. doi: 10.1038/nm.3967
24. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. (2012) 483:603–7. doi: 10.1038/nature11003
25. Zhao G, Green CF, Hui YH, Prieto L, Shepard R, Dong S, et al. Discovery of a highly selective NAMPT inhibitor that demonstrates robust efficacy and improved retinal toxicity with nicotinic acid coadministration. Mol Cancer Ther. (2017) 16:2677–88. doi: 10.1158/1535-7163.MCT-16-0674
26. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U.S.A. (2004) 101:3329–35. doi: 10.1073/pnas.0308061100
27. Brookmeyer R, Crowley J. A confidence interval for the median survival time. Biometrics. (1982) 38:29–41. doi: 10.2307/2530286
28. Li Z, Ivanov AA, Su R, Gonzalez-Pecchi V, Qi Q, Liu S, et al. The OncoPPi network of cancer-focused protein-protein interactions to inform biological insights and therapeutic strategies. Nat Commun. (2017) 8:14356. doi: 10.1038/ncomms14356
29. Liang X, Wang P, Gao Q, Xiang T, Tao X. Endogenous LKB1 knockdown accelerates G(1)/S transition through p53 and p16 pathways. Cancer Biol Ther. (2010) 9:156–60. doi: 10.4161/cbt.9.2.10452
30. Yang C, Li Z, Bhatt T, Dickler M, Giri D, Scaltriti M, et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene. (2017) 36:2255–64. doi: 10.1038/onc.2016.379
32. Jänne PA, van den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crinò L, et al. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non-small cell lung cancer: the SELECT-1 randomized clinical trial. JAMA. (2017) 317:1844–53. doi: 10.1001/jama.2017.3438
Keywords: biomarkers, CDK4, CDK6, KRAS, non-small cell lung cancer clinical trial registration: NCT02152631
Citation: Liu J, Wang H, Aggarwal A, Ortiz-Ruiz MJ, Zapatero-Solana E, Lallena MJ, Peregrina S, del Hoyo GM, Velasco S, Ebert PJ, Gong X, Hossain AM and Estrem ST (2025) Survival benefit from abemaciclib in non–small cell lung cancer by Kirsten rat sarcoma–mutation gene expression subtype: retrospective analysis from the JUNIPER Trial. Front. Oncol. 15:1461530. doi: 10.3389/fonc.2025.1461530
Received: 08 July 2024; Accepted: 03 March 2025;
Published: 31 March 2025.
Edited by:
Beatrice Aramini, University of Bologna, ItalyCopyright © 2025 Liu, Wang, Aggarwal, Ortiz-Ruiz, Zapatero-Solana, Lallena, Peregrina, del Hoyo, Velasco, Ebert, Gong, Hossain and Estrem. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shawn T. Estrem, ZXN0cmVtX3NoYXduX3RAbGlsbHkuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.