 Yanyun Pan1
Yanyun Pan1 Xinbin Zhou
Xinbin Zhou Wei Mao
Wei Mao Xiaoming Xu
Xiaoming Xu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 03 April 2025
Sec. Cancer Cell Signaling
Volume 15 - 2025 | https://doi.org/10.3389/fonc.2025.1454546
Cancer-associated fibroblasts (CAFs), the most abundant stromal cells in the tumor microenvironment (TME), control tumor growth through production and organization of the extracellular matrix (ECM) for a long time. However, the results from different studies that have focused on targeting CAFs to disturb tumor progression are extremely controversial. Recent studies using advanced single-cell RNA sequencing technology (scRNAseq) combined with multiple genetically engineered mouse models have identified diverse CAF subpopulations in the premalignant liver microenvironment (PME) of hepatocellular carcinoma (HCC) and TME of intrahepatic cholangiocarcinoma (ICC), providing a deeper understanding of the exact roles of each CAF subpopulation in cancer development. This review focuses on the specific protein markers, signaling pathways, and functions of various emerging CAF subclusters that contribute to the development of ICC and HCC. Elucidating the role and regulation of CAF subpopulations under different pathophysiological conditions will facilitate the discovery of new therapeutics that modulate CAF activity.
Cancer treatments have shifted from a tumor-centric view to a microenvironment-centric view because of the vital role of the tumor microenvironment (TME) or premalignant microenvironment (PME) in regulating tumor progression (1–4). The TME or PME comprises all non-cancerous host cells, including fibroblasts, endothelial cells, neurons, adipocytes, and immune cells, as well as its non-cellular components, including the extracellular matrix (ECM) and soluble products, such as chemokines, cytokines, growth factors, and extracellular vesicles (5, 6). The dynamic architecture of the TME or PME has a profound effect on tumor growth, which makes the interaction between malignant cells and their microenvironment a promising target for anti-cancer therapies, particularly those associated with excessive fibrosis.
Primary liver cancer remains among the deadliest human neoplasms worldwide owing to its poor prognosis and lack of effective therapeutics, despite numerous improvements in drugs and other medical therapies during the past decades (7). Hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (ICC), which occur in the setting of chronic liver injury and inflammation, are the two main types of primary liver cancers, accounting for approximately 80% and 15% of cases, respectively (8). While most HCC at their early stage are characterized by fibrosis or cirrhosis formed by hepatic stellate cells (HSC) or cancer-associated fibroblasts (CAFs) in the PME, ICC is characterized by its desmoplastic TME in which CAFs are the main stromal components (9). CAFs are involved in tumor cell proliferation and metastasis, angiogenesis, immunomodulation, and chemoresistance via ECM deposition, interacting with other cells by secreting multiple cytokines and growth factors, or directly interacting with other cell types via ligand–receptor interactions in liver cancer (10), suggesting a key role of CAFs in tumor progression. Targeting CAFs may provide strong therapeutic effects for cancer treatment and biomarkers for diagnosis and prognosis. However, results obtained from distinct studies focused on disturbing ECM production or targeting CAF using genetically engineered mouse cancer models to treat cancer are contradictory, implying that the roles of CAFs in tumor progression are diverse and complex (11–14). For example, while CAF-produced type I collagen (COL-I) promotes the malignant phenotype of tumor cells, depletion of CAF leads to escape from immune surveillance and accelerates tumor progression in pancreatic cancer mouse models (15, 16). Recent breakthroughs in integrating cutting-edge methods involving high-resolution single-cell profiling coupled with sophisticated animal model manipulation strategies have shed light on various functional subsets belonging to the CAF population that contribute unique regulatory features towards shaping malignant cells or their microenvironment, offering new opportunities for targeting the trajectory of CAFs (17–21).
CAFs are thought to be a group of cells negative for epithelial, endothelial, and leukocyte markers, with an elongated spindle morphology and lack of mutations found within cancer cells (10). CAFs are activated fibroblasts characterized by higher expression of α-smooth muscle actin (αSMA) than quiescent fibroblasts or normal fibroblasts in the TME or PME (22, 23). Heterogeneity and plasticity, well-known hallmarks of CAFs, make only αSMA (+) available to define CAFs (24). However, the specific markers that can define all types of CAFs remain unknown because of the diverse cellular origins and functions of distinct CAF subclusters (24, 25). Most CAFs in the liver originate from HSCs which are quiescent fibroblasts constituting the native extracellular matrix (ECM) and maintaining liver homeostasis under basal circumstances (10, 24, 26). HSC can be activated and transformed into various subpopulations of CAFs in ICC and HCC after receiving stimuli from cancer cells or TME including cytokines and growth factors (17, 19, 20, 27). Another source of CAF in the liver are resident portal fibroblasts (PFs) with mesothelial characteristics that reside in portal spaces where they produce connective tissue containing the bile duct, portal vein, and artery (28). PF is transformed into a small subpopulation of CAFs, designated as mesCAFs with unknown functions in ICC (21). Interestingly, PF, rather than HSC, is the dominant origin of CAFs in colorectal cancer liver metastases, suggesting the complex origin and functions of CAFs in different TMEs (29). Apart from HSC and PF, other cells in the TME or PME can be alternative origins of CAF in the liver (25, 28, 29) (Figure 1). For example, a subpopulation of CAF is derived from endothelial cells, defined as CAFEndMT, which exhibits dual expression of canonical lineage markers of fibroblasts and endothelial cells, highlighting the possibility of its involvement in endothelial–mesenchymal transition (EMT) (30). In addition, CAF can also originate from adipose cells with unclear function (30) or be transformed from bone marrow (BM)-derived mesenchymal stromal cells (MSCs) that are recruited to primary breast tumors and lung metastases (31). However, whether BM-derived CAF exist in primary liver cancers remains unknown. Notably, although many new subpopulations of CAFs with specific markers have been identified using advanced scRNA-seq, the function of some CAFs remains unexplained. One possible reason is that these CAFs are in the intermediate state transited from the original CAFs and need to be differentiated into terminal effective fibroblasts to execute functions (18).
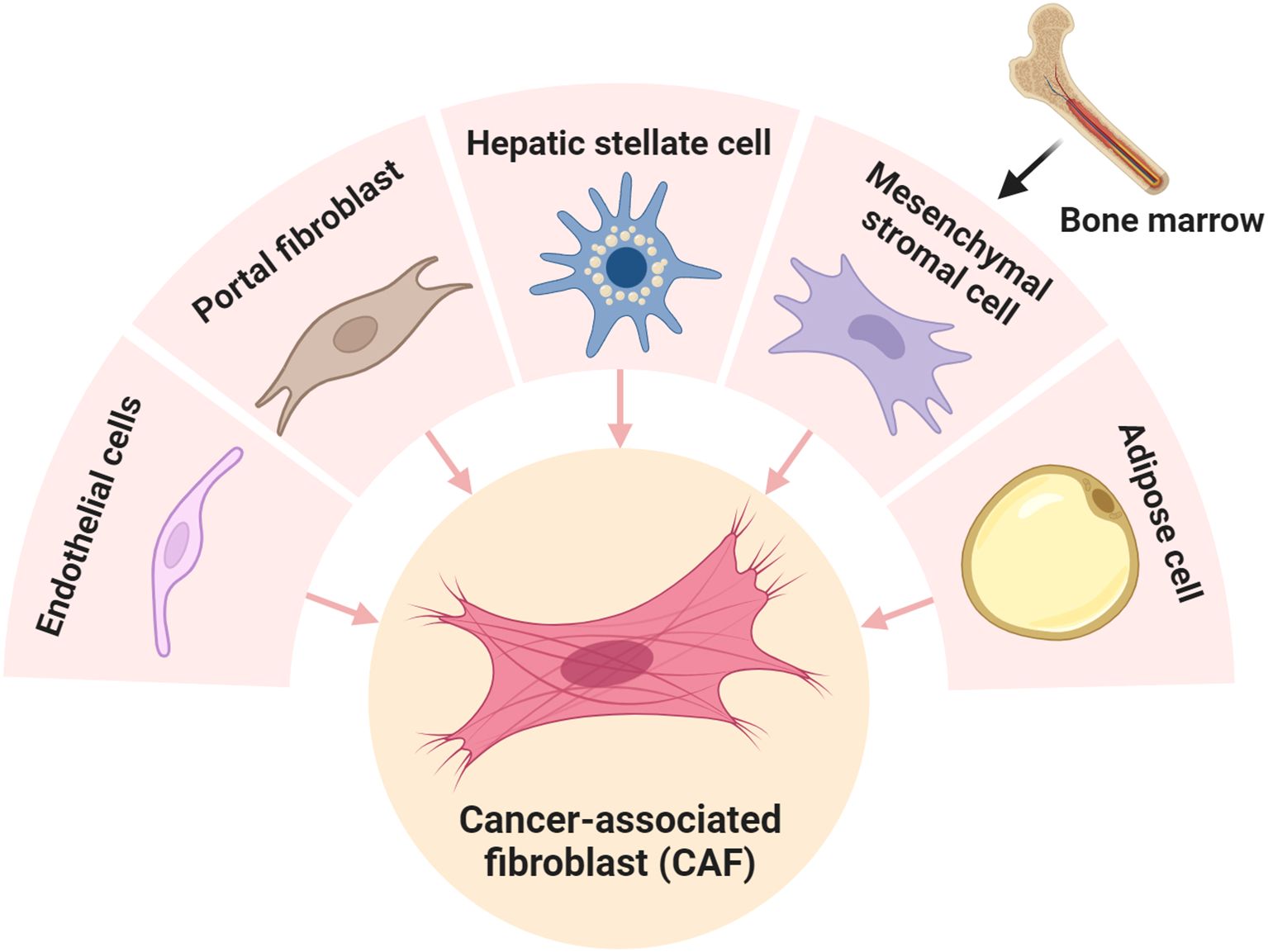
Figure 1. Cancer-associated fibroblasts (CAFs) originate from different cell types. CAFs can be driven from hepatic stellate cells (17, 19, 20, 27), portal fibroblast (21), endothelial cells (30), and bone marrow-derived mesenchymal stromal cells (31).
Lineage-tracking techniques combined with trajectory analyses and genetic ablation mouse models contribute to explaining how distinct subpopulations of CAFs are generated from their ancestors or transformed into each other, offering the possibility of targeting CAF transitions to anti-cancer (10, 17, 18, 20, 32–34). Recently, CAF transitions have been found to play an important role in liver fibrosis and HCC development. Through scRNA-seq in conjunction with pathway analysis and genetic ablation, two subpopulations of fibroblasts were identified in PME during HCC progression, termed myofibroblastic CAF (myCAF) and cytokine- and growth factor-expressing CAF (cyCAF) (20). The former was enriched in ECM-related molecules and pathways, whereas cytokines and growth factors were highly expressed in the latter. CyCAF is decreased and myCAF is increased in mice with liver fibrosis and cirrhosis, which promotes HCC development, whereas the shift in cyCAF-myCAF balance towards cyCAF inhibits HCC, suggesting that cyCAF–myCAF transitions play a fundamental role in orchestrating hepatocarcinogenesis. By investigating cellular heterogeneity and transcriptomic profiles during ICC initiation and progression (19), another study showed that while cytokine-producing inflammatory CAF (iCAF) are enriched in the early stage of ICC, myCAFs accumulate in the late stage of ICC, implying that the iCAF–myCAF transition promotes ICC progression. These findings suggest that CAF subpopulations and their functions vary among different malignant states, cancer types, and genetic contexts (17–20).
Different subpopulations of CAFs perform either tumor-supporting or -restraining roles by interacting with tumor cells, immune cells, and other components within the TME and producing multiple ECM proteins, cytokines, chemokines, growth factors, and exosomes (2, 17, 35–39). Given the sophisticated role of CAFs in regulating cancer progression, it is necessary to understand the features of various CAF clusters in the TME.
MyCAF and iCAF are two canonical CAF subpopulations in ICC: the former produces ECM and the latter secretes cytokines and growth factors (40–42). With improvements in scRNA-seq and a deeper understanding of CAF characteristics, CAFs can be classified into six subpopulations in ICC (Table 1): myCAF, iCAF, vascular CAF (vCAF), antigen-presenting CAF (apCAF), lipid metabolism and processing CAF (lpCAF), and epithelial CAF (eCAF) (27). All six subsets exhibited robust expression of canonical fibroblast markers like ACTA2 (α-SMA), COL1A2, and PDGFRβ, confirming their identity as fibroblasts; nevertheless, distinctive transcriptomic profiles were discernible among these groups. These subpopulations primarily derive from HSC, the quiescent or normal fibroblast in the liver parenchyma upon stimulation via various signals originating from neighboring cells (tumor cells or tumor-associated macrophage (TAM)) (17, 19, 39).
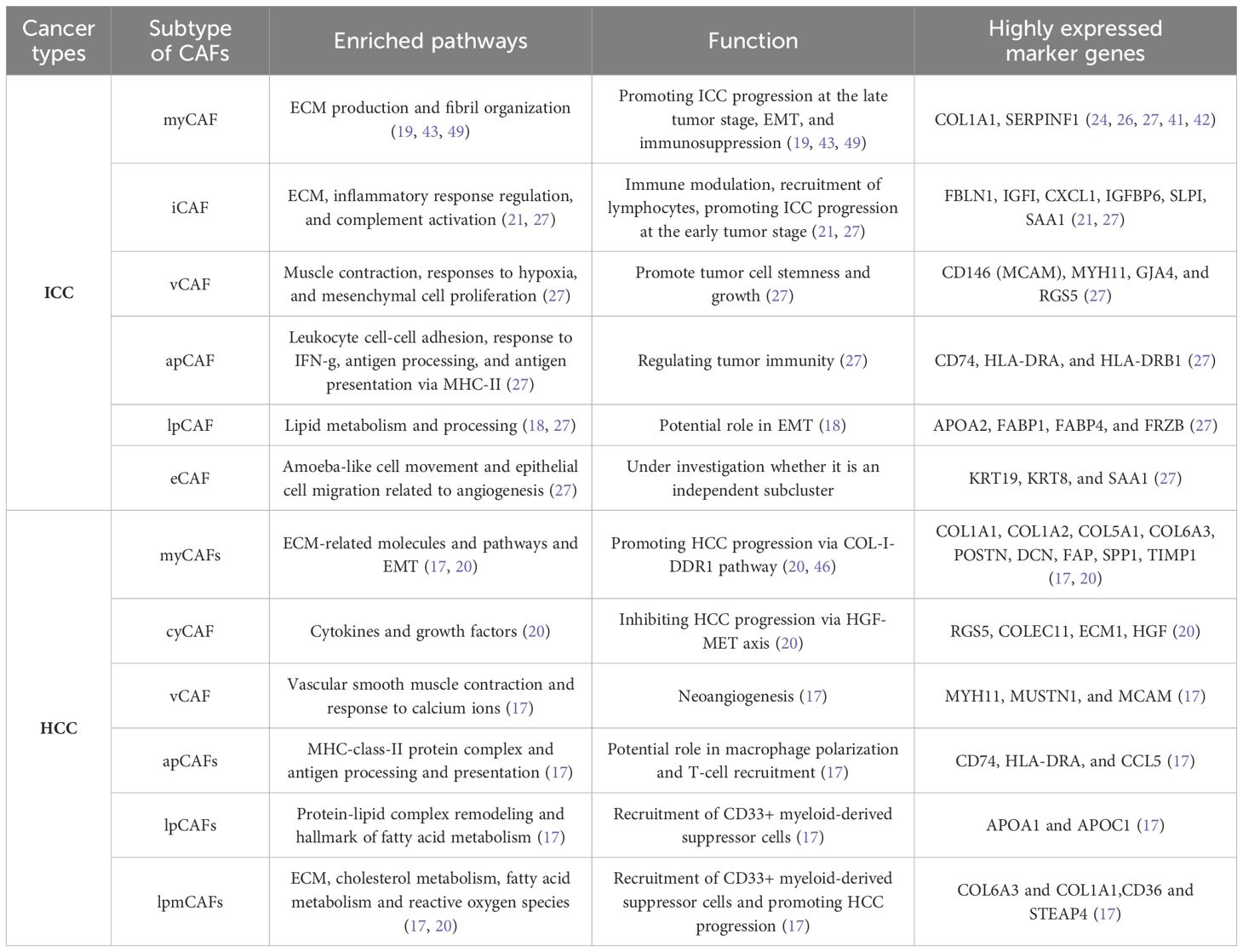
Table 1. CAF subpopulations of intrahepatic cholangiocarcinoma (ICC) and hepatocellular carcinoma (HCC).
MyCAF is a typical example of CAFs found in tumors, characterized by the involvement of specific genes linked to ECM production and fibril organization, such as COl1A1 and SERPINF1 (24, 26, 27, 43, 44). As a result, it receives an additional label for the matrix CAF (mCAF) (27, 45). MyCAFs promote ICC proliferation and are associated with intraneural invasion. High expression of myCAF signature genes was correlated with poor survival and displayed a trend toward higher recurrence in the Sia cohort (46). Type I collagen (COL-I) is enriched in myCAF while its cognate receptor DDR1, is expressed in tumor cells, suggesting a link between myCAF and tumor cells via COL-I-DDR1 interaction, which has been verified in PDAC where COL-I-DDR1 axis orchestrates tumor growth and metastasis via regulating macropinocytosis, a nutrient scavenging pathway, and mitochondria metabolism (1, 2, 21, 47, 48). However, despite the ablation of COL-I in either fibroblasts or all liver cells in mouse ICC models using hydrodynamic delivery of oncogenic drivers into hepatocytes reducing stiffness and mechanosensitive signals, loss of COL-I does not prevent tumor growth, implying that COL1-DDR1 signaling is not essential for ICC development (21). Deletion of the other most upregulated gene in myCAF, hyaluronan synthase 2 (Has2), significantly reduces ICC tumor growth with an unknown mechanism due to the multiple hyaluronan receptors and multiple cell types of Has2-expressing myCAF (21). However, it is difficult to rule out DDR1’s involvement in ICC development because hydrodynamic transfection cannot be applied to study tumors originating from hepatic stem cells or biliary epithelial cells and cannot create a fibrotic microenvironment, a typical feature of human ICC (49). Another possible explanation of why myCAF-produced COL-I might not contribute significantly to ICC growth involves the apparent abundance of myCAF observed recently in the late stage of ICC combined with driving ICC progression via Igf1/Igfr signaling (19). In addition, myCAF is found to potentially promote epithelial mesenchymal transition (EMT) and immunosuppression via TGFβ signaling (45, 50, 51). Together, these results suggest that myCAF is an oncogenic biomarker for advanced ICC stages and poor prognosis, and targeting Has2, TGFβ, or other key regulators that can disturb the interaction between myCAF and other cell types may offer strong therapeutic effects. PEGPH20, a PEGylated human hyaluronidase that can degrade excessive stromal hyaluronan, remodels the TME and consistently achieves objective tumor responses in PDAC (52). Galunisertib, a small-molecule inhibitor of TGFβ receptor I kinase that blocks TGFβ signaling, has been under Phase I/II trials for multiple cancer types (26, 53). Whether PEGPH20 and Galunisertib could inhibit myCAF in ICC should be further investigated.
iCAF, another canonical subpopulation of CAFs in the TME of ICC, express high levels of FBLN1, IGFI, CXCL1, IGFBP6, SLPI, SAA1, and complement genes (C3 and C7). Gene ontology (GO) terms enriched for this subcluster are related to ECM, inflammatory response regulation, and complement activation, indicating that iCAF may engage in immune modulation (27). Based on bulk RNA sequencing data from TCGA gastric cohort, the expression levels of gene markers for iCAFs are associated with the abundance of lymphocytes, suggesting that iCAFs play an important role in the recruitment of lymphocytes (54). The main subpopulation of CAFs residing in lymphoid nodule-like structures are iCAFs, which are located around CD8+ and PD1+ T cells, indicating that iCAFs are involved in regulating T cells (54). Unlike myCAF, iCAF is enriched in early stage ICC and promotes ICC development through the hepatocyte growth factor (HGF)-MET (HGF receptor) axis as a key tumor-promoting ligand–receptor pair, directly linking iCAF to tumor cells via ERK-mediated tumor cell proliferation (21). This has been validated in HGF-ablated and MET-ablated mouse ICC models, as well as in CellPhoneDB, which is a publicly available repository of curated receptors, ligands, and their interactions (21, 55). However, the mechanism by which iCAF interacts with lymphocytes in ICC required further investigation. These results suggest that iCAF is an oncogenic biomarker of the early ICC stage and is correlated with a suppressed immune environment. Xentuzumab, a humanized monoclonal antibody targeting IGF1 and IGF2, has been in Phase I and II trials for multiple cancers. It showed preliminary antitumor activity in a heavily pretreated population (56). However, whether the therapeutic effects of Xentuzumab are derived from blocking iCAF remains unknown.
apCAFs express major histocompatibility complex II (MHC-II) genes such as CD74, HLA-DRA, and HLA-DRB1. The GO terms enriched in this subcluster are related to leukocyte cell–cell adhesion, response to IFN-γ, antigen processing, and antigen presentation via MHC-II, suggesting a function in regulating tumor immunity (27). apCAFs were first identified in pancreatic ductal adenocarcinoma (PDAC), which can present antigens to CD4+ T cells (57). By integrating multiple scRNA-seq studies and performing robust lineage-tracing assays, apCAFs were found to be derived from mesothelial cells during pancreatic cancer progression (58). This mesothelial cell-to-apCAF transition is induced by interleukin-1 and TGFβ by downregulating mesothelial features and upregulating fibroblastic features. However, a pan-cancer single-cell analysis revealed that apCAF, presenting TAM (such as MAFB and SPI1) and CAF (such as MYLK) regulons, is at an intermediate position during the TAM to myCAF transition. Further investigations are needed to determine whether apCAF is located in an intermediated state, where it originates, and how it contributes to tumor progression in ICC.
vCAF are the most abundant fibroblast population characterized by microvasculature signature genes such as CD146 (MCAM), MYH11, GJA4, and RGS5, as well as inflammatory chemokines such as IL-6 and CCL8 (27). vCAFs are significantly enriched in pathways related to muscle contraction, responses to hypoxia, and mesenchymal cell proliferation, all of which align with their microvasculature signatures. Of note, vCAFs are located in the tumor core and microvascular area and promote tumor cell stemness and growth through secretion of IL-6 which induces significant epigenetic alterations in tumor cells, particularly upregulating enhancer of zeste homolog 2 (EZH2) (27), suggesting that there is an intense interaction between vCAF and ICC tumor cells, and the anti-IL6 pathway is a potential treatment for ICC. However, IL-6 is also important for the differentiation of naïve CD4+ T cells into Th17 cells and for protecting T cells from apoptosis (59). Therefore, improving the therapeutic effects by targeting vCAF requires further investigation.
lpCAFs are mainly derived from adjacent ICC tumor tissues and express high levels of lipid metabolism- and processing-related genes, including APOA2, FABP1, FABP4, and FRZB (27). lpCAFs are found to express marker genes similar to adipogenic CAFs, which are abundant in genes associated with EMT across various cancers analyzed via single-cell sequencing analysis (18). Whether lpCAFs also contribute to EMT in ICC requires further investigation. eCAFs express both epithelium-specific marker genes, such as KRT19, KRT8, and SAA1, and activated fibroblast makers (27). The gene expression profile of eCAFs includes activities associated with amoeba-like cell movement and epithelial cell migration signals related to angiogenesis. However, it is still under investigation whether eCAFs should be considered an independent subgroup because they represent epithelial/CAF doublets (45).
Most HCCs arise in the context of fibrosis or cirrhosis, including those associated with non-alcoholic steatohepatitis (NASH), despite the absence of overt cirrhosis in 30%–50% of NASH-associated HCC (60). Emerging studies combining genetic CAF activation, inhibition, or depletion approaches in various mouse models of spontaneous, carcinogen-induced, oncogene-induced, and NASH-induced HCC have demonstrated that CAFs exert an overall HCC-promoting role (20). Co-implantation of CAFs with HCC cells also enhances HCC progression (17). However, deletion of CAF in mice with established tumors does not affect HCC development, suggesting that fibroblasts predominantly promote tumor growth by modulating PME and the early stages of hepatocarcinogenesis. A global view of fibroblasts in PME and TME during HCC progression is necessary and meaningful to deeply understand their precise role.
Integration of scRNA-seq of fibroblasts from various mouse models with fibrotic liver, normal and cirrhotic human livers, and pathway analyses, fibroblasts in PME can also be divided into two typical subpopulations, myCAF and cyCAF (20) (Table 1). Similar to myCAF in ICC, myCAF is characterized by enrichment of ECM-related molecules and pathways, whereas cyCAF, close to iCAF of ICC, is a cluster of quiescent to weakly activated CAFs enriched in genes and pathways related to cytokines and growth factors. These two clusters transition to each other to regulate hepatocarcinogenesis (20). The shift in cyCAF/myCAF balance towards myCAFs exhibits increased HCC numbers and towards cyCAFs exhibits decreased HCC numbers, implying that the ratio of cyCAF/myCAF is a strong prognostic indicator, which requires a wide examination of human HCC tumor specimens and survival for verification. Trajectory and pseudotime analyses of liver fibrosis models revealed that myCAFs might be derived from cyCAFs. However, given that activated CAFs may revert to quiescence, it is possible that myCAFs can transition to cyCAFs or shuttle back and forth, especially when tumor progression is dynamic. In addition, both clusters can strongly interact with hepatocytes and affect biology and tumorigenesis. MyCAF-secreted COL-I promotes proliferation and tumor development through increased stiffness and TAZ activation in pretumoral hepatocytes and through the activation of DDR1 in established tumors. Notably, consistent with the results obtained from PDAC (48), matrix metalloproteinase-cleaved COL-I rather than intact COL-I promoted DDR1 activity in HCC tumor cells, suggesting that targeting the components of cleaved COL-I-DDR1 signaling may provide an alternative therapeutic strategy for anti-HCC (20, 48). Indeed, DDR1 inhibitors have been widely used in preclinical tumor models. For example, 7rh a selective inhibitor of DDR1 has been shown strong inhibitory effects in tumor cell proliferation of breast cancer, pancreatic cancer, and gastric cancer (48, 61, 62). In contrast, cyCAF-produced HGF activates MET primarily expressed in hepatocytes to suppress hepatocarcinogenesis (20). However, future studies should focus on the mechanisms by which DDR1 activation accelerates and the HGF–MET axis decelerates HCC tumor growth.
Unlike ICC, in which CAF subpopulations from different studies are relatively consistent, CAF subpopulations vary in HCC studies by distinct research groups (17, 20). In addition to the classification mentioned above, the CAFs subpopulation in the TME of HCC can be grouped into five common subtypes: vCAF, myCAFs, lipid processing (lp)-myCAFs (lpmCAFs), lpCAFs, and apCAFs by scRNA-seq of human and mouse HCC tumors (Table 1). iCAF is not included. All five subpopulations expressed canonical fibroblast markers such as ACTA2, COL1A2, and COL1A1. Notably, apart from lpmCAFs, the characteristics of the other four CAF subclusters were similar to their ICC counterparts (Table 1). lpmCAFs expressed both ECM-related genes (such as COL6A3 and COL1A1) and lipid metabolism-related genes (such as CD36 and STEAP4) (Table 1). The GO and KEGG terms enriched for this cluster were related to ECM, cholesterol metabolism, fatty acid metabolism, and reactive oxygen species pathways, implying that this subpopulation is involved in both ECM and cholesterol metabolism. CD36 mediates oxidized LDL uptake-dependent expression of macrophage migration inhibitory factor (MIF) via the lipid peroxidation/p38/CEBPs axis in lpmCAFs, which may recruit CD33+ myeloid-derived suppressor cells (MDSCs) in an MIF- and CD74-dependent manner to promote HCC. lpmCAFs may be located in the intermediate state and transit to vCAFs, lpCAFs, and apCAFs, as determined by trajectory analysis. Infiltrating lpCAFs, especially in tumor tissues, can recruit CD33+ myeloid-derived suppressor cells to induce immunosuppression and correlate with poor prognosis in patients with HCC. Taken together, current research into subpopulations of HCC CAFs remains at the level of cell classification, and very little is known about the exact functions of each subset during tumor progression.
In addition, CAFs are key producers of vascular endothelial growth factor (VEGF), which stimulates the formation of new blood vessels to support tumor growth. CAFs can also produce angiopoietins and matrix metalloproteinases (MMPs), which further enhance angiogenesis by promoting vessel sprouting and degrading physical barriers. Moreover, CAFs secrete platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), and stromal-derived factor 1 (SDF-1/CXCL12), which recruit endothelial progenitor cells and promote vascularization.
Although CAF have been considered a potential therapeutic target for a long time, the results obtained from targeting CAF to treat cancer in preclinical models are extremely controversial. Advanced scRNA-seq combined with various genetically engineered mouse models and clinical species identify distinct CAF subpopulations displaying either tumor-promoting or -suppressing roles, which offers possibilities to target specific subclusters to cure cancer (17–21, 27, 43, 45). The tumor-promoting functions of CAF subpopulations make them promising targets for anticancer therapy, providing attractive potential therapeutic strategies that can be categorized as blockage of CAF subpopulation recruitment and activation, depletion of CAF populations, and blockage of interactions between CAF subpopulations and tumor cells or immune cells. To accomplish this, more efforts should be made to elucidate the molecular mechanisms by which CAF subpopulations influence tumor progression. Trajectory analysis revealed the origin of distinct subpopulations of CAF and subpopulations that are located in the intermediate state. However, further investigation is needed to confirm the origin of each subpopulation and the exact function of the intermediate clusters using new genetically engineered mouse cancer models. The mechanism by which the CAF transitions occur is still not fully understood. However, the dynamics of DNA methylation, histone acetylation, and histone methylation in several types of cancer have been found to play a potential role in CAF activation and transitions (63–67). For example, the loss of H3K27me3 activates CAFs to promote stem cell niche formation and tumor growth (67). In contrast to cancer cells carrying numerous mutations, CAFs possess greater genetic stability, another hallmark of CAFs (68). Based on this, it is necessary to investigate the epigenetic regulation of CAF transitions and CAF origin in different contexts, which will help develop novel strategies to interpose the shuttle between distinct CAF clusters and understand the origin and diversity of CAFs, as well as bring practical therapeutic efficacy and slight side effects. Notably, even though several CAF subpopulations are similar between ICC and HCC, the specific markers are not the same, which means targeting the “same” CAF subpopulation in the same organ may use distinct inhibitors in different cancers. Whether there is a long way to go to develop specific regulations to change the composition of CAF subpopulations in PME or TME for anticancer treatment.
Although scRNA-seq greatly advances our understanding of tumor cell heterogeneity, it has some limitations. Spatial and morphological information is lost after tissue dissociation into a single-cell suspension, making it difficult to investigate the tumor spatial architecture. Recently developed spatial transcriptomics technology (STT) may overcome these limitations because it provides information on spatial TME characteristics from non-tumor to leading-edge tumor regions of primary liver cancers (69). Additionally, as compared to studies of a single omics type, multi-omics that are capable of interrogating entire pools of transcripts, proteins, and metabolites, as well as the genome, offer the opportunity to understand the flow of information, from the functional consequences or relevant interactions. However, the importance of the tremendous amount of data produced by STT or multi-omics in practice requires the development of new preclinical models for verification. Another major challenge in current CAF research is nomenclature. Unified naming rules to define CAF subpopulations or other cell types with specific markers and developmental trajectories will benefit all researchers in understanding the unique aspects of each cancer type, avoid repetitive research, and allow researchers to easily follow the findings from different study groups. A simple and efficient solution is to reach expert consensus. In addition, accurate recording of CAF subpopulations in clinical specimens limits the application of targeting CAF subpopulations in clinical settings. This depends on high-quality antibodies against marker proteins of each CAF subcluster, which in many cases is lacking. Technology for multiplexed mRNA probes characterized by high sensitivity and specificity is developing rapidly and may offer a better and more flexible solution than antibody-based methods. Moreover, identifying specific biomarkers for each CAF subpopulation and combined inhibition of the CAF subpopulation with immunotherapy or chemotherapy will expedite the application of CAFs in liver cancer treatment.
YP: Writing – original draft, Writing – review & editing, Investigation. YQ: Writing – review & editing, Investigation. XZ: Investigation, Writing – review & editing. WM: Investigation, Writing – review & editing. XX: Writing – review & editing, Writing – original draft.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from the TCM Science and Technology Foundation Project of Zhejiang Province (grant number 2023ZL411), Zhejiang Provincial Science and Technology Project of Traditional Chinese Medicine (grant number 2024ZR012), Natural Science Foundation of Zhejiang Province (no. LY21H290007), and the Scientific Research Foundation of Zhejiang University of Traditional Chinese Medicine (no. 2020ZG45).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
CAFs, cancer-associated fibroblasts; TME, tumor microenvironment; ECM, extracellular matrix; scRNA-seq, single-cell RNA sequencing technology; PME, premalignant liver microenvironment;HCC, hepatocellular carcinoma; ICC, intrahepatic cholangiocarcinoma; HSC, hepatic stellate cell; αSMA, α-smooth muscle actin; PF, portal fibroblast; BM, bone marrow; MSCs, mesenchymal stromal cells; iCAF, inflammatory CAF; vCAF, vascular CAF; myCAF, myofibroblastic CAF; apCAF, antigen-presenting CAF; lpCAF, lipid metabolism and processing CAF; eCAF, epithelial CAF; STT, spatial transcriptomics technology; TAM, tumor-associated macrophage; mCAF, matrix CAF; COL-I, type I collagen; EMT, epithelial–mesenchymal-transition; MHC-II, major histocompatibility complex II; PDAC, pancreatic ductal adenocarcinoma; GO, gene ontology; NASH, non-alcoholic steatohepatitis; lpmCAFs, lipid processing (lp)-myCAFs.
1. Su H, Karin M. Multifaceted collagen-DDR1 signaling in cancer. Trends Cell Biol. (2023) 34:406–15. doi: 10.1016/j.tcb.2023.08.003
2. Su H, Karin M. Collagen architecture and signaling orchestrate cancer development. Trends Cancer. (2023) 9:764–73. doi: 10.1016/j.trecan.2023.06.002
3. Kraehenbuehl L, Weng C-H, Eghbali S, Wolchok JD, Merghoub T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat Rev Clin Oncol. (2022) 19:37–50. doi: 10.1038/s41571-021-00552-7
4. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. (2018) 359:1350–5. doi: 10.1126/science.aar4060
5. de Visser KE, Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. (2023) 41:374–403. doi: 10.1016/j.ccell.2023.02.016
6. Bader JE, Voss K, Rathmell JC. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol Cell. (2020) 78:1019–33. doi: 10.1016/j.molcel.2020.05.034
7. Villanueva A. Hepatocellular carcinoma. Longo DL editor. N Engl J Med. (2019) 380:1450–62. doi: 10.1056/NEJMra1713263
8. Li X, Ramadori P, Pfister D, Seehawer M, Zender L, Heikenwalder M. The immunological and metabolic landscape in primary and metastatic liver cancer. Nat Rev Cancer. (2021) 21:541–57. doi: 10.1038/s41568-021-00383-9
9. Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S, et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol. (2018) 69:385–95. doi: 10.1016/j.jhep.2018.03.011
10. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. (2020) 20:174–86. doi: 10.1038/s41568-019-0238-1
11. Miyai Y, Esaki N, Takahashi M, Enomoto A. Cancer-associated fibroblasts that restrain cancer progression: Hypotheses and perspectives. Cancer Sci. (2020) 111:1047–57. doi: 10.1111/cas.v111.4
12. Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. (2008) 68:918–26. doi: 10.1158/0008-5472.CAN-07-5714
13. Rice AJ, Cortes E, Lachowski D, Cheung BCH, Karim SA, Morton JP, et al. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis. (2017) 6:e352. doi: 10.1038/oncsis.2017.54
14. Chen Y, Kim J, Yang S, Wang H, Wu C-J, Sugimoto H, et al. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell. (2021) 39:548–565.e6. doi: 10.1016/j.ccell.2021.02.007
15. Armstrong T, Packham G, Murphy LB, Bateman AC, Conti JA, Fine DR, et al. Type I collagen promotes the Malignant phenotype of pancreatic ductal adenocarcinoma. Clin Cancer Res. (2004) 10:7427–37. doi: 10.1158/1078-0432.CCR-03-0825
16. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. (2014) 25:719–34. doi: 10.1016/j.ccr.2014.04.005
17. Zhu G-Q, Tang Z, Huang R, Qu W-F, Fang Y, Yang R, et al. CD36+ cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor. Cell Discovery. (2023) 9:25. doi: 10.1038/s41421-023-00529-z
18. Luo H, Xia X, Huang L-B, An H, Cao M, Kim GD, et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat Commun. (2022) 13:6619. doi: 10.1038/s41467-022-34395-2
19. Wang T, Xu C, Zhang Z, Wu H, Li X, Zhang Y, et al. Cellular heterogeneity and transcriptomic profiles during intrahepatic cholangiocarcinoma initiation and progression. Hepatology. (2022) 76:1302–17. doi: 10.1002/hep.32483
20. Filliol A, Saito Y, Nair A, Dapito DH, Yu L-X, Ravichandra A, et al. Opposing roles of hepatic stellate cell subpopulations in hepatocarcinogenesis. Nature. (2022) 610:356–65. doi: 10.1038/s41586-022-05289-6
21. Affo S, Nair A, Brundu F, Ravichandra A, Bhattacharjee S, Matsuda M, et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell. (2021) 39:866–882.e11. doi: 10.1016/j.ccell.2021.05.010
22. Glaire MA, El-Omar EM, Wang TC, Worthley DL. The mesenchyme in Malignancy: a partner in the initiation, progression and dissemination of cancer. Pharmacol Ther. (2012) 136:131–41. doi: 10.1016/j.pharmthera.2012.08.007
23. Nurmik M, Ullmann P, Rodriguez F, Haan S, Letellier E. In search of definitions: Cancer-associated fibroblasts and their markers. Int J Cancer. (2020) 146:895–905. doi: 10.1002/ijc.v146.4
24. Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discovery. (2019) 18:99–115. doi: 10.1038/s41573-018-0004-1
25. Biffi G, Tuveson DA. Diversity and biology of cancer-associated fibroblasts. Physiol Rev. (2021) 101:147–76. doi: 10.1152/physrev.00048.2019
26. Chen Y, McAndrews KM, Kalluri R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol. (2021) 18:792–804. doi: 10.1038/s41571-021-00546-5
27. Zhang M, Yang H, Wan L, Wang Z, Wang H, Ge C, et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J Hepatol. (2020) 73:1118–30. doi: 10.1016/j.jhep.2020.05.039
28. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. (2019) 575:512–8. doi: 10.1038/s41586-019-1631-3
29. Giguelay A, Turtoi E, Khelaf L, Tosato G, Dadi I, Chastel T, et al. The landscape of cancer-associated fibroblasts in colorectal cancer liver metastases. Theranostics. (2022) 12:7624–39. doi: 10.7150/thno.72853
30. McAndrews KM, Chen Y, Darpolor JK, Zheng X, Yang S, Carstens JL, et al. Identification of functional heterogeneity of carcinoma-associated fibroblasts with distinct IL6-mediated therapy resistance in pancreatic cancer. Cancer Discovery. (2022) 12:1580–97. doi: 10.1158/2159-8290.CD-20-1484
31. Raz Y, Cohen N, Shani O, Bell RE, Novitskiy SV, Abramovitz L, et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J Exp Med. (2018) 215:3075–93. doi: 10.1084/jem.20180818
32. Schönhuber N, Seidler B, Schuck K, Veltkamp C, Schachtler C, Zukowska M, et al. A next-generation dual-recombinase system for time- and host-specific targeting of pancreatic cancer. Nat Med. (2014) 20:1340–7. doi: 10.1038/nm.3646
33. Chen Y, LeBleu VS, Carstens JL, Sugimoto H, Zheng X, Malasi S, et al. Dual reporter genetic mouse models of pancreatic cancer identify an epithelial-to-mesenchymal transition-independent metastasis program. EMBO Mol Med. (2018) 10:e9085. doi: 10.15252/emmm.201809085
34. Steele NG, Biffi G, Kemp SB, Zhang Y, Drouillard D, Syu L, et al. Inhibition of hedgehog signaling alters fibroblast composition in pancreatic cancer. Clin Cancer Res. (2021) 27:2023–37. doi: 10.1158/1078-0432.CCR-20-3715
35. Ma Z, Li X, Mao Y, Wei C, Huang Z, Li G, et al. Interferon-dependent SLC14A1+ cancer-associated fibroblasts promote cancer stemness via WNT5A in bladder cancer. Cancer Cell. (2022) 40:1550–1565.e7. doi: 10.1016/j.ccell.2022.11.005
36. Bianchi A, De Castro Silva I, Deshpande NU, Singh S, Mehra S, Garrido VT, et al. Cell-autonomous cxcl1 sustains tolerogenic circuitries and stromal inflammation via neutrophil-derived TNF in pancreatic cancer. Cancer Discovery. (2023) 13:1428–53. doi: 10.1158/2159-8290.CD-22-1046
37. Zarin B, Rafiee L, Daneshpajouhnejad P, Haghjooy Javanmard S. A review on the role of CAFs and CAF-derived exosomes in progression and metastasis of digestive system cancers. Tumour Biol. (2021) 43:141–57. doi: 10.3233/TUB-200075
38. Jia W, Liang S, Lin W, Li S, Yuan J, Jin M, et al. Hypoxia-induced exosomes facilitate lung pre-metastatic niche formation in hepatocellular carcinoma through the miR-4508-RFX1-IL17A-p38 MAPK-NF-κB pathway. Int J Biol Sci. (2023) 19:4744–62. doi: 10.7150/ijbs.86767
39. Fang T, Lv H, Lv G, Li T, Wang C, Han Q, et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat Commun. (2018) 9:191. doi: 10.1038/s41467-017-02583-0
40. Tsoumakidou M. The advent of immune stimulating CAFs in cancer. Nat Rev Cancer. (2023) 23:258–69. doi: 10.1038/s41568-023-00549-7
41. Affo S, Filliol A, Gores GJ, Schwabe RF. Fibroblasts in liver cancer: functions and therapeutic translation. Lancet Gastroenterol Hepatol. (2023) 8:748–59. doi: 10.1016/S2468-1253(23)00111-5
42. Pereira BA, Vennin C, Papanicolaou M, Chambers CR, Herrmann D, Morton JP, et al. CAF subpopulations: A new reservoir of stromal targets in pancreatic cancer. Trends Cancer. (2019) 5:724–41. doi: 10.1016/j.trecan.2019.09.010
43. Lavie D, Ben-Shmuel A, Erez N, Scherz-Shouval R. Cancer-associated fibroblasts in the single-cell era. Nat Cancer. (2022) 3:793–807. doi: 10.1038/s43018-022-00411-z
44. Sun L, Wang Y, Wang X, Navarro-Corcuera A, Ilyas S, Jalan-Sakrikar N, et al. PD-L1 promotes myofibroblastic activation of hepatic stellate cells by distinct mechanisms selective for TGF-β receptor I versus II. Cell Rep. (2022) 38:110349. doi: 10.1016/j.celrep.2022.110349
45. Martin-Serrano MA, Kepecs B, Torres-Martin M, Bramel ER, Haber PK, Merritt E, et al. Novel microenvironment-based classification of intrahepatic cholangiocarcinoma with therapeutic implications. Gut. (2023) 72:736–48. doi: 10.1136/gutjnl-2021-326514
46. Sia D, Hoshida Y, Villanueva A, Roayaie S, Ferrer J, Tabak B, et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology. (2013) 144:829–40. doi: 10.1053/j.gastro.2013.01.001
47. Su H, Yang F, Fu R, Li X, French R, Mose E, et al. Cancer cells escape autophagy inhibition via NRF2-induced macropinocytosis. Cancer Cell. (2021) 39:678–693.e11. doi: 10.1016/j.ccell.2021.02.016
48. Su H, Yang F, Fu R, Trinh B, Sun N, Liu J, et al. Collagenolysis-dependent DDR1 signalling dictates pancreatic cancer outcome. Nature. (2022) 610:366–72. doi: 10.1038/s41586-022-05169-z
49. Zen Y. Intrahepatic cholangiocarcinoma: typical features, uncommon variants, and controversial related entities. Hum Pathol. (2023) 132:197–207. doi: 10.1016/j.humpath.2022.06.001
50. Deshmukh AP, Vasaikar SV, Tomczak K, Tripathi S, den Hollander P, Arslan E, et al. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc Natl Acad Sci U S A. (2021) 118:e2102050118. doi: 10.1073/pnas.2102050118
51. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. (2019) 20:69–84. doi: 10.1038/s41580-018-0080-4
52. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. (2012) 21:418–29. doi: 10.1016/j.ccr.2012.01.007
53. Melisi D, Garcia-Carbonero R, Macarulla T, Pezet D, Deplanque G, Fuchs M, et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer. (2018) 119:1208–14. doi: 10.1038/s41416-018-0246-z
54. Li X, Sun Z, Peng G, Xiao Y, Guo J, Wu B, et al. Single-cell RNA sequencing reveals a pro-invasive cancer-associated fibroblast subgroup associated with poor clinical outcomes in patients with gastric cancer. Theranostics. (2022) 12:620–38. doi: 10.7150/thno.60540
55. Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc. (2020) 15:1484–506. doi: 10.1038/s41596-020-0292-x
56. Haluska P, Menefee M, Plimack ER, Rosenberg J, Northfelt D, LaVallee T, et al. Phase I dose-escalation study of MEDI-573, a bispecific, antiligand monoclonal antibody against IGFI and IGFII, in patients with advanced solid tumors. Clin Cancer Res. (2014) 20:4747–57. doi: 10.1158/1078-0432.CCR-14-0114
57. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discovery. (2019) 9:1102–23. doi: 10.1158/2159-8290.CD-19-0094
58. Huang H, Wang Z, Zhang Y, Pradhan RN, Ganguly D, Chandra R, et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell. (2022) 40:656–673.e7. doi: 10.1016/j.ccell.2022.04.011
59. Li B, Jones LL, Geiger TL. IL-6 promotes T cell proliferation and expansion under inflammatory conditions in association with low-level RORγt expression. J Immunol. (2018) 201:2934–46. doi: 10.4049/jimmunol.1800016
60. Paradis V, Zalinski S, Chelbi E, Guedj N, Degos F, Vilgrain V, et al. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: a pathological analysis. Hepatology. (2009) 49:851–9. doi: 10.1002/hep.22734
61. Baltes F, Caspers J, Henze S, Schlesinger M, Bendas G. Targeting discoidin domain receptor 1 (DDR1) signaling and its crosstalk with β1-integrin emerges as a key factor for breast cancer chemosensitization upon collagen type 1 binding. Int J Mol Sci. (2020) 21:4956. doi: 10.3390/ijms21144956
62. Hur H, Ham I-H, Lee D, Jin H, Aguilera KY, Oh HJ, et al. Discoidin domain receptor 1 activity drives an aggressive phenotype in gastric carcinoma. BMC Cancer. (2017) 17:87. doi: 10.1186/s12885-017-3051-9
63. Su S-F, Ho H, Li J-H, Wu M-F, Wang H-C, Yeh H-Y, et al. DNA methylome and transcriptome landscapes of cancer-associated fibroblasts reveal a smoking-associated Malignancy index. J Clin Invest. (2021) 131:e139552. doi: 10.1172/JCI139552
64. Katarkar A, Bottoni G, Clocchiatti A, Goruppi S, Bordignon P, Lazzaroni F, et al. NOTCH1 gene amplification promotes expansion of Cancer Associated Fibroblast populations in human skin. Nat Commun. (2020) 11:5126. doi: 10.1038/s41467-020-18919-2
65. Dou C, Liu Z, Tu K, Zhang H, Chen C, Yaqoob U, et al. P300 acetyltransferase mediates stiffness-induced activation of hepatic stellate cells into tumor-promoting myofibroblasts. Gastroenterology. (2018) 154:2209–2221.e14. doi: 10.1053/j.gastro.2018.02.015
66. Mishra R, Haldar S, Placencio V, Madhav A, Rohena-Rivera K, Agarwal P, et al. Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. J Clin Invest. (2018) 128:4472–84. doi: 10.1172/JCI99397
67. Maeda M, Takeshima H, Iida N, Hattori N, Yamashita S, Moro H, et al. Cancer cell niche factors secreted from cancer-associated fibroblast by loss of H3K27me3. Gut. (2020) 69:243–51. doi: 10.1136/gutjnl-2018-317645
68. Kwa MQ, Herum KM, Brakebusch C. Cancer-associated fibroblasts: how do they contribute to metastasis? Clin Exp Metastasis. (2019) 36:71–86. doi: 10.1007/s10585-019-09959-0
Keywords: cancer-associated fibroblast, iCAF, myCAF, hepatocellular carcinoma, intrahepatic cholangiocarcinoma, tumor microenvironment
Citation: Pan Y, Qiu Y, Zhou X, Mao W and Xu X (2025) Cancer-associated fibroblasts: multidimensional players in liver cancer. Front. Oncol. 15:1454546. doi: 10.3389/fonc.2025.1454546
Received: 25 June 2024; Accepted: 19 February 2025;
Published: 03 April 2025.
Edited by:
Xuanmao Jiao, Baruch S. Blumberg Institute, United StatesReviewed by:
Yingpu Li, Harbin Medical University Cancer Hospital, ChinaCopyright © 2025 Pan, Qiu, Zhou, Mao and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoming Xu, eHV4aWFvMjQ1N0AxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.