
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 24 December 2024
Sec. Cancer Metabolism
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1519046
 Huan Peng1,2,3,4
Huan Peng1,2,3,4 Huihong Dou1,2,3
Huihong Dou1,2,3 Sheng He1,2,3,5Yu-an Xie1,2,3,4
Sheng He1,2,3,5Yu-an Xie1,2,3,4 Qinle Zhang1,2,3,4*Jianqiu Zheng1,2,3*
Qinle Zhang1,2,3,4*Jianqiu Zheng1,2,3*GOT1, a cytoplasmic glutamic oxaloacetic transaminase, plays a critical role in various metabolic pathways essential for cellular homeostasis and dysregulated metabolism. Recent studies have highlighted the significant plasticity and roles of GOT1 in metabolic reprogramming through participating in both classical and non-classical glutamine metabolism, glycolytic metabolism, and other metabolic pathways. This review summarizes emerging insights on the metabolic roles of GOT1 in cancer cells and emphasizes the response of cancer cells to altered metabolism when the expression of GOT1 is altered. We review how cancer cells repurpose cell intrinsic metabolism and their flexibility when GOT1 is inhibited and delineate the molecular mechanisms of GOT1’s interaction with specific oncogenes and regulators at multiple levels, including transcriptional and epigenetic regulation, which govern cellular growth and metabolism. These insights may provide new directions for cancer metabolism research and novel targets for cancer treatment.
Cancers reprogram cellular metabolism to meet the bioenergetic, biosynthetic, and redox demands of malignant cells, supporting their aberrant proliferation and survival (1, 2). These reprogrammed activities are recognized as hallmarks of cancers, as many cancer types exhibit general metabolic alterations (2, 3). Normal cell metabolism generates energy for maintaining physiological activities and cellular functions through complex reactions (4). However, cancer cells reprogram metabolism to acquire necessary nutrients from a frequently nutrient-poor environment and repurpose these nutrients to sustain viability and build new biomass (1, 2, 5, 6). Understanding how metabolism is rewired in cancer cells, and utilizing these metabolic changes for therapeutic benefits are key research areas. Accruing evidence suggests that metabolic regulation plays a predominant role in determining cell states, including proliferation and senescence. Therefore, targeting cell metabolism or the key enzymes involved in these processes may be a new strategy for treating refractory cancers.
GOT1 is a key enzyme in cell metabolism, distributed in the liver, muscle, heart, kidney, brain, and other tissues (7). The GOT1 gene, located on human chromosome 10q24.1–25, consists of 9 exons, and encodes a 46.2 kDa protein composed of 413 amino acids (8). GOT1 exhibits abnormal expression in numerous cancers. The expression of GOT1 was up-regulated in pancreatic ductal adenocarcinoma (PDAC) (9–11), colorectal cancer (12, 13), breast cancer (14–16), lung adenocarcinoma (17), glioblastoma (17), prostate cancer (17–19), acute myeloid leukemia (20), and multiple myeloma (21), while down-regulated in poorly-differentiated hepatocellular carcinoma cells (22). Aberrant expression of GOT1 serves as a candidate biomarker. In most cases, elevated expression of GOT1 in cancer is associated with poor prognosis (23, 24). For example, GOT1 expression could be served as an independent prognostic biomarker in PDAC (9). Additionally, serum GOT1 levels are indicators of liver dysfunction in both liver and non-liver tumors, cardiovascular diseases, type 2 diabetes, and all-cause mortality (25–31).
Cytoplasmic GOT1 and its mitochondrial isozyme GOT2 usually occur together and interact with each other in metabolic processes (32–34). GOT1 is mainly present in the cytoplasm and GOT2 exists in the mitochondrial matrix. GOT1 catalyzes aspartate (Asp) and α-ketoglutarate (α-KG) into oxaloacetate (OAA) and glutamate (Glu) in cytoplasm. Then, Glu can generate Asp or be metabolized into other products through tricarboxylic acid (TCA) cycle. Glu can be converted into α-KG via the deamination of glutamate dehydrogenase 1 (GLUD1) and catalyzed to generate Asp by GOT2 in mitochondrial matrix. And then Asp from mitochondrial matrix is converted to OAA via GOT1 in cytoplasm (35). OAA is catalyzed by malate dehydrogenase 1 (MDH1) to generate malate, which is catalyzed to pyruvate through malic enzyme (ME). In this process, NADPH is produced and NADPH/NADP+ ratio is increased to maintain reactive oxygen species (ROS) balance (36). GOT2 reversibly catalyzes OAA and Glu into Asp and α-KG, contributing to the TCA cycle and energy production (37–43) (Figure 1). Both GOT1 and GOT2 are crucial components of the malate-aspartate shuttle (MAS), transferring reducing equivalents from NADH into the mitochondria for oxidative phosphorylation (44). Cancer cells utilize the amino acid Glu to support the anabolic processes and promote cell growth. The expression of GOT1 could be regulated by oncogene KRAS (10, 45) or other regulators (43, 45–47) like non-coding RNAs (24, 48–52), affecting Asp synthesis, NADPH production, glucose metabolism, as well as other metabolic pathways, and then tumor cells rewired metabolism to support their proliferation. This process links the expression of GOT1 closely to cancer progression. This review will dissect GOT1-related metabolic mechanisms and the molecular pathways about how GOT1 influences cancer progression. We also discuss potential limitations, challenges, and practical implications in targeting GOT1 therapeutically.
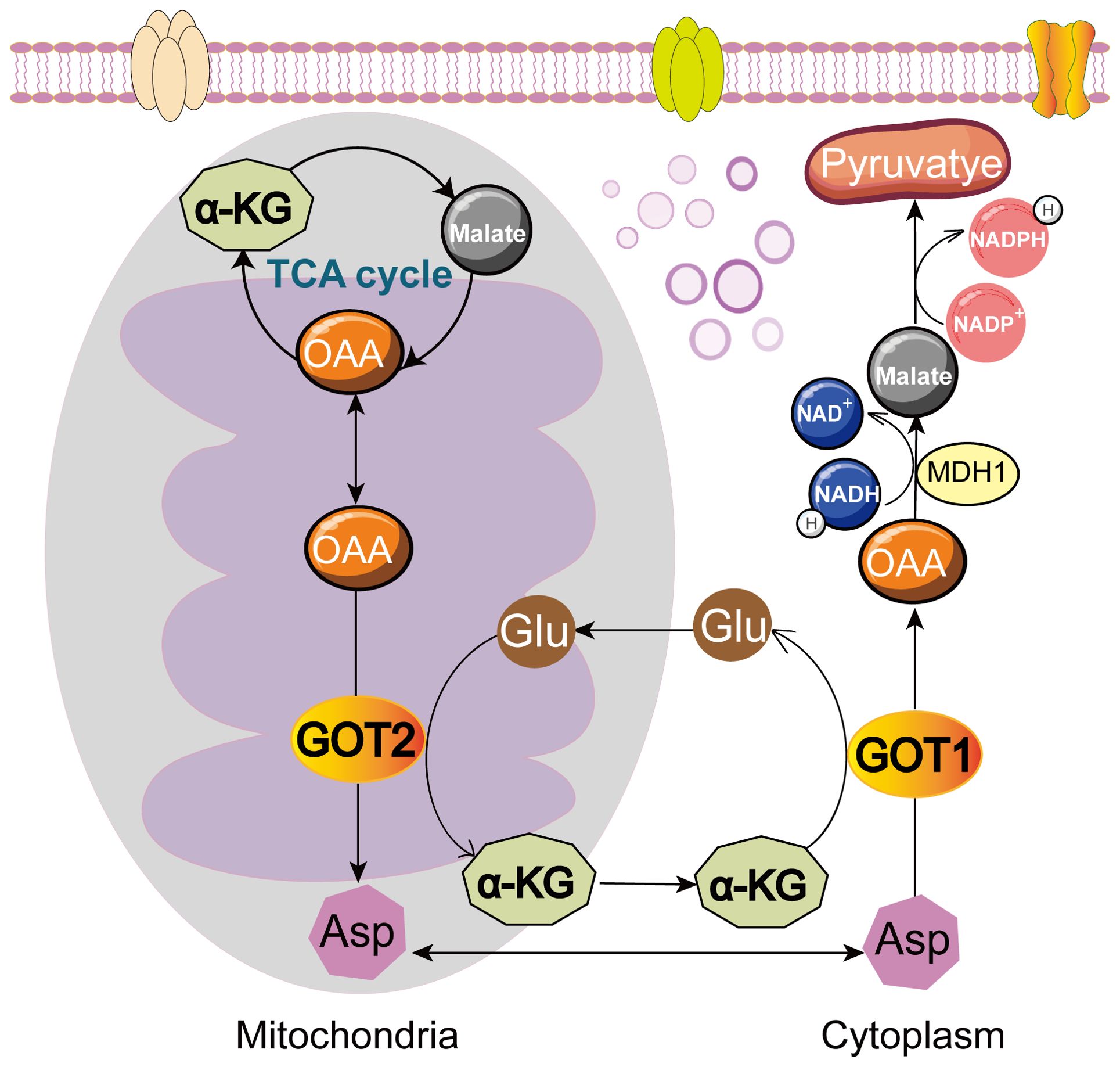
Figure 1. Roles of GOT1 and GOT2 in cell metabolism. GOT1 catalyzes Asp and α-KG into OAA and Glu in cytoplasm. OAA is further converted to malate by MDH1, and then to pyruvate, producing NADPH. GOT2 reversibly catalyzes OAA and Glu into Asp and α-KG, fueling TCA cycle. Glu, glutamate; Asp, aspartate; OAA, oxaloacetate; α-KG, α-ketoglutarate; MDH1, malate dehydrogenase 1.
Cancer cells utilize Asp and glutamine (Gln) to support their metabolic process, producing energy, and promote cell proliferation. GOT1, involved in this process, participates in several critical metabolic functions: Asp synthesis, Gln metabolism, glycolysis pathway regulation, immune cell function regulation, and epigenetic regulation (Figure 2). Therefore, GOT1 has become a key consideration when interrogating cancer metabolism.
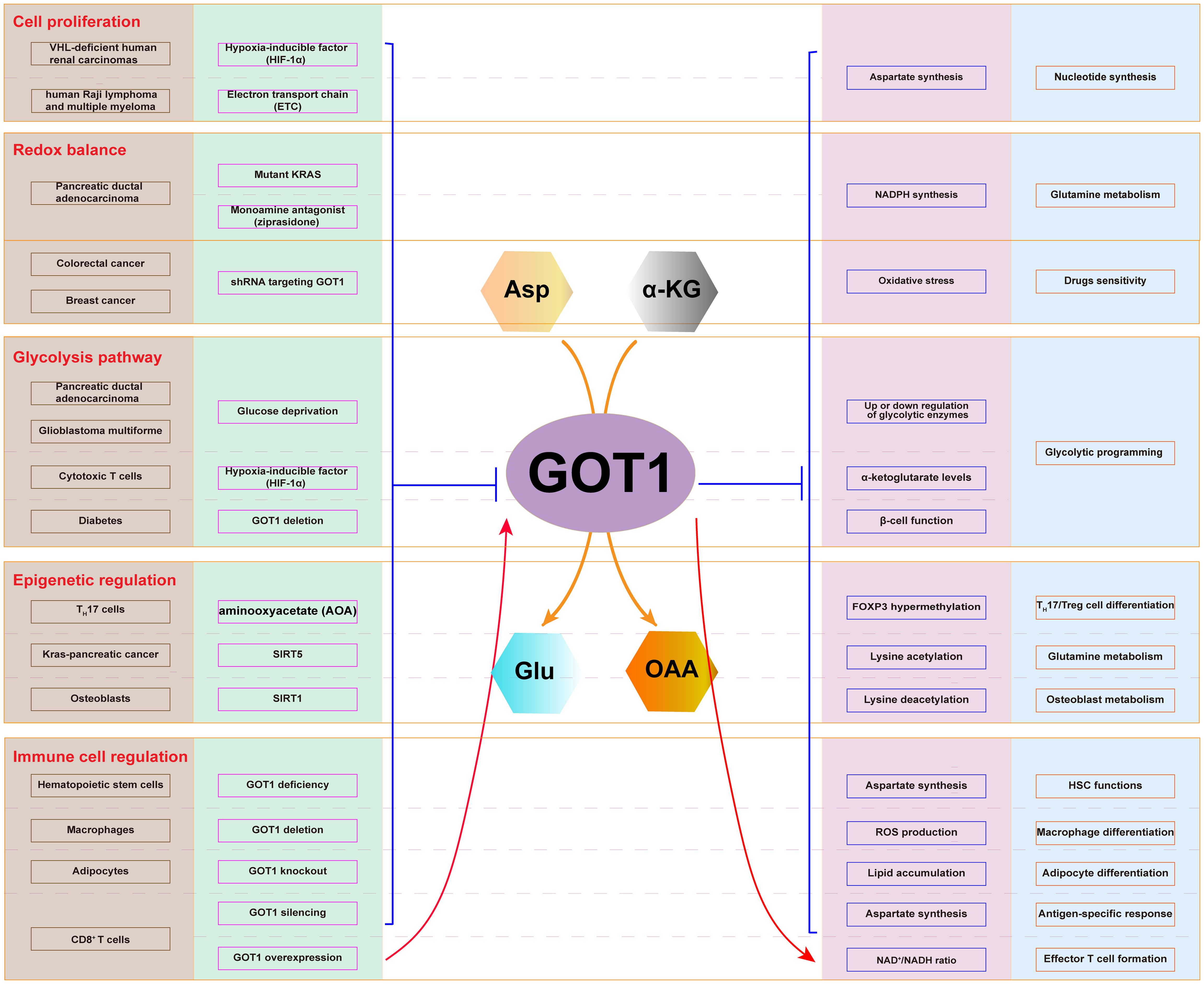
Figure 2. Cellular metabolic pathway involving GOT1. The expression of GOT1 is regulated by HIF-1α and ETC, which affects Asp synthesis and nucleotide production in cancer cells. Mutant KRAS and ziprasidone regulate the expression of GOT1, which influence NADPH production, oxidative stress, and Gln metabolism. GOT1 participates in glycolysis upon glucose deprivation. GOT1 participates in epigenetic regulation through lysine acetylation and deacetylation. GOT1 is involved in hematopoietic stem cell and T cell function, as well as T cell, macrophage, and adipocyte differentiation.
The deregulated cell growth and division in cancers imposed more requirements for DNA. Asp, a vital rate-limiting product of GOT1-catalyzed reversible reaction, is crucial for cancer cells due to its roles in nucleotide synthesis. Cellular Asp drives cancer cell proliferation. Disruption of GOT1 significantly impacts Asp synthesis and consequently affects cancer cell growth. For example, the expression of GOT1 was suppressed by hypoxia-inducible factor-1α (HIF-1α) in VHL-deficient human renal carcinoma (43), and by electron transport chain (ETC) in human Raji lymphoma and KMS-26 multiple myeloma (38, 53), leading to impaired proliferation of cancer cells. Mechanistically, the abnormal proliferation of these tumor cells was due to the reduction of Asp synthesis through inhibiting Gln oxidation and attenuating reductive carboxylation following GOT1 deletion, which disrupted nucleotide generation within the cells. Additionally, GOT1 treatment reduced serum Glu concentration, and GOT1-induced protective effects in cerebral ischemia were mediated by Asp metabolism (54).
GOT1 is crucial for producing NADPH and maintaining redox homeostasis through Gln metabolism. Emerging studies showed that in PDAC, KRAS knockdown (10, 13, 55) or the use of selective monoamine antagonist ziprasidone (56, 57) directly targeted and inhibited the expression of GOT1, subsequently disrupting Gln metabolism, affecting NADPH production, elevating ROS levels, and disturbing the redox balance. Mechanistically, there are some differences between the two approaches. PDAC relied on a distinct KRAS-regulated metabolic pathway to fuel the TCA cycle. KRAS activated the expression of GOT1. At this moment, Gln-derived Glu was converted to Asp in the mitochondria by GOT2, and Asp was transported into the cytoplasm where it could be converted into OAA by GOT1. Furthermore, OAA is converted into malate, which is then metabolized by malic enzyme 1 (ME1) to produce pyruvate. These reactions produced NAPDH potentially to maintain the cellular redox state (11, 58). Ziprasidone induced cell cycle arrest at the G1 phase and inhibited the phosphorylation of p38 and Erk, two key steps of MAPKs pathway (57). These findings indicate that targeting GOT1 in Gln metabolism in PADC may be a potential strategy for treating cancers.
GOT1 also plays a role in balancing the oxidative stress response induced by therapeutic drugs. Inhibition of GOT1 could regulate NADPH synthesis and then regulated ROS levels to counteract oxidative stress, which making colorectal cancer cells sensitive to 5-fluorouracil (12) and breast cancer cells sensitive to doxorubicin (15), enhancing anti-cancer effects. Additionally, knockdown of GOT1 decreased NADPH production and suppressed prostate cancer cell growth (18). Gln deprivation activated the expression of c-Myc, which enhanced the transcriptional expression of GOT1 and Nrf2. These molecules increased the glutathione synthesis from Glu and inhibited ferroptosis in hepatocellular carcinoma (HCC) cells. Interestingly, the researchers found that a combination of Gln deprivation and GOT1 inhibition caused fatal damage of HCC in vitro and in vivo (47). These findings provide a novel strategy for improving the anti-cancer efficacy.
Emerging evidence has reported that GOT1 was also an important glycolytic enzyme that regulated the glycolysis pathway. In PDAC, shGOT1 cells displayed a consistent accumulation of glycolytic intermediates between the aldolase-catalyzed and pyruvate kinase-catalyzed steps of glycolysis, suggesting that GOT1 inhibition uniquely disrupted glycolysis pathways (13). Cancer cells obtained glucose through alternative metabolic pathways to meet their own requirements upon glucose deprivation. Inhibition of GOT1 sensitized the cancer cells to glucose deprivation, which was partially counteracted by metabolic intermediates downstream of GOT1 like OAA and phosphoenol pyruvate. GOT1 disruption in KRAS mutated cancer cells led to the up-regulated expression of the gluconeogenesis-pathway-gene glucose-6-phosphatase 3 (G6PC3) both during normal growth conditions and after glucose deprivation and the down-regulated expression of the glucose-controlling gene BIP after glucose deprivation, indicating GOT1’s roles in glucose metabolism (23). Non-coding RNA circGOT1 sponged miR-606 to promote GOT1, which induced glycolytic metabolism of esophageal squamous cell cancer cells, promoting cell proliferation (24). GOT1 inhibited glycolysis by interacting with pyruvate carboxylase and then inhibited malignant phenotypes of glioblastoma multiforme cells (59). GOT1 has also been reported to play roles in glycolysis in healthy cells. Sirt1 deletion in osteoblasts inhibited glycolysis by directly binding to and increasing the acetylation level of GOT1 to maintain bone homeostasis (60). Xu et al. found that GOT1 promoted the glycolytic programming and cytotoxic function of cytotoxic T lymphocytes via posttranslational regulation of HIF-1α protein, potentially by regulating the levels of α-KG (61). In early hypoxia, GOT1 participated in the regulation of glycolysis to maintain cells in a primed state that increased their chances of survival via sustaining cytoplasmic NAD+/NADH balance by sustaining flux through MDH1. In addition to its immediate contribution to glycolysis upon oxygen limitation, GOT1 activity contributed to α-KG turnover and thereby promoted HIF-1α stabilization (62, 63). Got1 also plays roles in diabetes. Got1 deletion in mouse β-cells impaired β-cell function by increased glycolysis and then led to phenocopying aging and diabetes (64).
Asp synthesis is required for cancer cell proliferation. However, it is unclear whether Asp is limiting in healthy immune cells. Previous studies have indicated that conditional deletion of GOT1 from hematopoietic cells would be expected to increase Asp levels (10, 38, 39). Qi et al. found that mouse hematopoietic stem cells (HSCs) depended entirely on cell-autonomous Asp synthesis, which increased upon HSC activation (65). They established Got1 deficient mice and Got2 deficient mice, and found that Got1 deficiency increased Asp levels and HSC function while Got2 deficiency reduced Asp levels and HSC function during hematopoietic regeneration. While Got1 deficiency and Got2 deficiency had opposite effects on Asp levels, both would be expected to disrupt the MAS. Deletion of both Got1 and Got2 was lethal for HSCs because that they were unable to synthesize nucleotides or other products producing energy (65). Ma et al. identified Got1 was a regulator of Gln-dependent Asp production in T cells and was required for CD8+ T cell responses in vivo (66). CD8+ T cells expressing Got1-targeting shRNA displayed reduced Gln-derived Asp and slight reduced cell expansion. In vivo experiments, they observed a marked reduction of percentage and number of antigen-specific CD8+ T cells responding to LmOVA infection. Moreover, Got1 silencing altered the CD8+ T cell effector response, as evidenced by an overall lower percentage and number of IFN-γ-producing CD8+ T cells in the spleens of LmOVA-infected animals, indicating a critical function for Got1 in mediating the expansion of CD8+ effector T cells in vivo (66). Recently published work by Xu et al. established similar requirements for GOT1 in mediating T cell-mediated anti-tumor responses. Xu et al. found that GOT1 was upregulated in effector CD8+ T cells, which promoted their differentiation and function by maintaining intracellular redox balance and serine-mediated purine nucleotide biosynthesis. Additionally, GOT1 promoted the glycolytic programming and cytotoxic function of cytotoxic T lymphocytes via posttranslationally regulating HIF-1α expression, potentially by regulating the levels of α-KG (61). The MAS including GOT1 detoxified ammonia in exhausted T cells by producing 2-ketoglutarate (2-KG) (67). CD8+ T cells expressed GOT1 during chronic infections to execute antiviral responses and to decrease the concentration of ammonia to promote the assimilation of free ammonia and cell survival by producing 2-KG. GOT1 promoted effector T cell formation in acute lymphocytic choriomeningitis virus infection dependent on the traditional function of GOT1 in maintaining the NAD+/NADH ratio. They also indicated that GOT1 deficiency influenced the transcriptional profiles and epigenetic landscapes of CD8+ T cells via decreasing the expression levels of genes involved in demethylation, such as Kdm6b and Tet1 (67). GOT1 could also regulate adipocyte differentiation by altering the NADPH content (68). Macrophages play a central role in host innate immune response defending against pathogens. A study has shown that Got1 was dispensable for M2 macrophage differentiation and did not influence the onset of lipopolysaccharides-induced immune tolerance in macrophages (69).
Metabolism has been shown to integrate with epigenetics and transcription to modulate cell fate and function (70–72). Xu et al. identified a small molecule aminooxyacetate (AOA) reprogramed TH17 differentiation toward iTreg cells and Got1 was the main target of AOA during TH17 cell differentiation. Knock-down of Got1 in differentiation TH17 cells inhibited TH17 cell differentiation and reciprocally increased iTreg cell differentiation. TH17 cell differentiation was regulated mainly via Got1-dependent increased transamination leading to elevated 2-hydroxyglutarate (2-HG) level in differentiating TH17 cells. And accumulating 2-HG resulted in hypermethylation of FOXP3 gene locus and inhibited FOXP3 transcription, which was essential for fate determination towards TH17 cells (73). The study of Xu et al. suggested an important mechanistic link of Got1 activity in the fate determination of TH17 cell differentiation by an epigenetic mechanism. GOT1 activity could be enhanced by tumor suppressor SIRT5 deletion via facilitating GOT1’s lysine acetylation to regulate the non-canonical Gln and glutathione metabolism, and then promoted tumorigenesis of Kras-induced pancreatic cancer (45). SIRT1 inhibited GOT1 enzyme activity by catalyzing its lysine deacetylation in osteoblasts to regulate osteoblast metabolism (60). Future work into the regulatory underpinnings of GOT1 can provide a better understanding for differential metabolic activities and dependencies.
The expression of GOT1 can be regulated by non-coding RNAs including circular RNAs (circRNAs), microRNAs (miRNAs), and long non-coding RNAs (lncRNAs), and then affecting cell proliferation and metabolism (46, 48) (Table 1). In addition, lncRNAs and circRNAs usually affect cancer cell proliferation via sponging miRNAs targeted GOT1 directly. For example, circ-MBOAT2 silencing downregulated the expression of GOT1 via sponging miR-433-3p, modulating the tumor development of PDAC (51). In non-small cell lung cancer, hsa_circRNA_103809 sponged miR-337-3p to upregulate the expression of GOT1, affecting the cisplatin-resistance and cell growth (50). CircGOT1 was generated from exon 8 of its host gene GOT1 via back-splicing and then promoted GOT1 expression. CircGOT1 promoted the expression of GOT1 via sponging miR-606 to exert carcinogenesis including promoting tumor growth, migration, and glycolytic metabolism of esophageal squamous cell cancer cells (24). Furthermore, miR-9-5p could directly bind to the 3’-untranslated region (3’UTR) of GOT1 and then inhibited the expression of GOT1, which stunted the proliferation, invasion, Gln metabolism, and redox homeostasis of pancreatic cancer cells (48). Additionally, exosome-derived lncRNA NEAT1 functioned as a ceRNA of miR-9-5p to facilitate the expression of TFRC and GOT1, and then exacerbated ferroptosis of sepsis-associated encephalopathy (52). lncRNA TMPO-AS1 was highly expressed in HCC cells and expedited HCC progression via promoting cell proliferation, stemness as well as suppressing cell apoptosis. Molecular mechanism showed TMPO-AS1 functioned as a molecular sponge for miR-429 and GOT1 served as a downstream target gene of miR-429 in HCC. miR-429 could directly bind to GOT1 and negatively regulated the expression of GOT1 while TMPO-AS1 positively regulated the expression of GOT1. Additionally, GOT1 overexpression reversed the inhibition effects of TMPO-AS1 deficiency on HCC progression, indicating that the TMPO-AS1/miR-429/GOT1 axis may be an underlying treatment strategy for HCC (49).

Table 1. Non-coding RNAs regulate the expression of GOT1 in different tumors.
GOT1 is also a potential target of transcriptional regulators. Paralogous transcriptional regulators TAZ and YAP are considered as central factors in cancer biology and they play key roles in cell proliferation, survival, and cell fate determination (75). TAZ/YAP reprogram cellular energetics to promote the dependence of breast cancer cell growth on exogenous Gln, and TAZ/YAP induced GOT1 and phosphoserine aminotransferase (PSAT1) expression to promote the conversion of Gln to α-KG, and then to support cell growth (14). Additionally, targeting metabolic vulnerability to block of transamination of GOT1 using AOA suppressed the growth of breast cancer cells in a TAZ/YAP-dependent manner (14). These findings indicate more remains to be investigated regarding the role of GOT1 in transcriptional regulation in cancers.
Recent studies have indicated that GOT1 played critical roles in ferroptosis. For instance, in melanoma, miR-9 targeted GOT1 directly, inhibiting the expression of GOT1, which suppressed ferroptosis of melanoma cells (76). In multiple myeloma, GOT1 expression was inhibited by shikonin, enhancing ferroptosis in multiple myeloma cells by promoting the release of autophagic labile iron (21). Additionally, GOT1 inhibition increased labile iron availability through autophagy, enhancing the activity of ferroptosis and impaired proliferation and promoted cell death of pancreatic cancer cells (74). The expression of GOT1 was also up-regulated in pancreatic cancer cell-derived exosomes, promoting cancer cell proliferation, migration, and invasion (77–79). Experimental evidence found that pancreatic cancer cell-derived exosomes enriched with GOT1 inhibited cellular ferroptosis to promoted pancreatic cancer cell progression through mechanisms like upregulating CCR2 expression and activating the Nrf1/HO2559-1 pathway (80). These findings highlight the importance of GOT1 regulation through ferroptosis in cancer treatment strategies, and the necessity to study how genetic and environmental factors influence ferroptosis susceptibility in vivo.
Cellular metabolic reprogramming is a hallmark of cancer, serving as a therapeutic target to overcome limitations in current therapies of cancers. The body of this work involving GOT1 highlights several key points in cancer metabolism. First, cancer cells exhibit dynamic metabolism to support abnormal growth and functions. Second, the metabolic pathways utilized by cancer cells are influenced by various factors such as endogenous signals, tumor microenvironment, nutritional composition, external signals, and oncogenes. Third, using appropriate disease models in the suitable context and environment is crucial for identifying therapeutic targets in cancer metabolism.
A major challenge in targeting cancer metabolism lies in the rapid adaptability of cancer cells to adjust their metabolic pathways (81). Cancer cells can quickly modify their metabolic strategies to utilize available metabolic intermediates from nutrient-deficient environments, synthesizing the energy necessary for their growth. As a component of MAS, the inhibition of GOT1 leads to a decrease in NADPH production. NADPH is essential for multiple metabolic pathways, including glycolysis, and its reduction results in decreased flux into these metabolic routes. Under such circumstances, tumor cells activate a specific metabolic pathway known as non-canonical Gln metabolism replacing the canonical Gln metabolism (82) to meet their energetic demands. In PDAC, GOT1 inhibition resulted in Gln deprivation, dimethyl α-KG did not restore growth upon Gln deprivation, whereas the combination of α-KG and a non-essential amino acid mixture rescued proliferation, suggesting PDAC cells metabolize Gln in a model that is different from canonical models. Gln deprivation led to decreased Gln-derived malate, and Gln-derived OAA converted by GOT1 was metabolized into malate. Malate was utilized by ME1 to generate NADPH, maintaining cell growth (10). In PDAC cells, tumor Asp availability was maintained by both de novo synthesis and alternative extracellular sources in the absence of GOT1/GOT2 in vivo (83). Researchers found that PI3K/mTORC2 pathway promoted GOT1 expression via targeting hypoxia-inducible factor HIF-2α, and then activated the non-canonical Gln metabolism in vitro and in vivo, promoting progression of PDAC (46). GOT1 also plays an important role in an acidic tumor microenvironment. In pancreatic cancer, in response to chronic acidosis stress, the expression of GOT1 was increased in cancer cells to fuel oxidative metabolism by enhancing the non-canonical Gln metabolism (84). Cancer cells adapt to nutrient-deprived tumor microenvironment during progression via adjusting the level and function of metabolic enzymes. Inhibition of GOT1 significantly weakened HCC cell proliferation under high glucose conditions, while silencing of glutamate dehydrogenase 1(GDH1) did not take effect. Deletion of GDH1 impaired HCC cell proliferation under low glucose conditions, yet knock-down of GOT1 did not take effect. Moreover, GDH1 expression was elevated in glucose-poor HCC tissues while GOT1 expression was decreased. However, inhibiting Gln-dependent transaminases including GOT2, GPT-1, GPT-2, and PSAT-1 had essentially no impact on proliferation of cells no matter under high or low glucose conditions, suggesting that liver cancer cells maintained survival under different nutritional conditions through the metabolic flexibility of their Gln-related enzymes (63). Collectively, these studies indicate that in the absence of GOT1, cells possess both endogenous and exogenous compensatory mechanisms that allow cancer cells to survive. Thus, GOT1 may be served as a key source to fuel the rewired metabolic pathways.
Although metabolism-targeted therapy is not yet standard therapy for many cancers, several experimental and clinical trials targeting altered metabolism are currently underway. For example, WZB117, a specific GLUT1 inhibitor, could inhibit the tumor-initiating capacity of the cancer stem cells in vitro and inhibit tumor initiation in vivo (85). Adapalene, an approved drug clinically used in the therapy of acne vulgaris, could selectively inhibit the activity of GOT1 in a non-competitive manner and further suppress the proliferation of ovarian cancer ES-2 cells (86). AOA, an inhibitor of pyridoxal 5-phosphate-dependent enzymes, could also inhibit the activity of GOT1, causing amino acid deprivation (87, 88). Although numerous small-molecule drugs have been proven to inhibit the activity of GOT1 in a competitive or non-competitive manner (23, 57, 79, 86, 89–91), further efforts are still required for the development of specific inhibitors targeting GOT1. Moreover, none of these GOT1 inhibitors have progressed to clinical trials yet, necessitating further research to develop effective GOT1 inhibitors for cancer treatment. Considerable work remains to translate the basic research on GOT1-targeted drug therapy into clinical applications.
While attempts to treat cancers by targeting cellular metabolism have achieved some success, the development of drug resistance is still a significant challenge in the design of metabolic targeted therapies. It is worth noting that metabolic networks possess remarkable plasticity, allowing them to reconnect and circumvent targeted treatments. Furthermore, certain tumor cells exhibit heterogeneous metabolic subtypes, which exhibit varying prognoses and metabolic susceptibilities (92–94). Consequently, these factors should be thoroughly considered when designing effective metabolic targeted therapies. As research progresses, it is urgent to develop specific and effective drugs targeting metabolism. In the near future, more efficient metabolism-targeting drugs and treatment regimens are expected to develop to improve patient outcomes and extend survival, yielding clinical benefits for patients.
HP: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. HD: Conceptualization, Data curation, Formal analysis, Investigation, Writing – original draft, Methodology, Writing – review & editing. SH: Writing – original draft, Writing – review & editing, Supervision, Funding acquisition. YX: Supervision, Funding acquisition, Writing – original draft, Writing – review & editing. QZ: Conceptualization, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing. JZ: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Guangxi Science and Technology Project (Guangxi Science and Technology Base and Talent Special Project: Guike AD24010014), Guangxi Key Laboratory of reproductive health and birth defect prevention (21-220-22), Guangxi Clinical Research Center for Pediatric Diseases (Guike AD22035121), Self-Funded Program of Guangxi Health Commission (Z-A20240319), Guangxi Key Laboratory of Birth Defects and Stem Cell Biobank (ZTJ2020002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. (2016) 2:e1600200. doi: 10.1126/sciadv.1600200
2. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
4. Thakur C, Chen F. Connections between metabolism and epigenetics in cancers. Semin Cancer Biol. (2019) 57:52–8. doi: 10.1016/j.semcancer.2019.06.006
5. Kerk SA, Papagiannakopoulos T, Shah YM, Lyssiotis CA. Metabolic networks in mutant KRAS-driven tumours: tissue specificities and the microenvironment. Nat Rev Cancer. (2021) 21:510–25. doi: 10.1038/s41568-021-00375-9
6. Szkudliński M, Lewiński A, Sewerynek E, Wajs E. Factors stimulating and/or inhibiting growth processes of the adrenal cortex. I. The role of the anterior pituitary and hypothalamic hormones, insulin, sex steroids and certain neuropeptides. Postepy Hig Med Dosw. (1991) 45:335–47.
7. Ladue JS, Wroblewski F, Karmen A. Serum glutamic oxaloacetic transaminase activity in human acute transmural myocardial infarction. Science. (1954) 120:497–9. doi: 10.1126/science.120.3117.497
8. Choudhury BK, Setoyama C, Shimada K. Molecular cloning and sequence analysis of the human cytosolic aspartate aminotransferase gene. Biochem Int. (1990) 22:583–91.
9. Feld FM, Nagel PD, Weissinger SE, Welke C, Stenzinger A, M ller P, et al. GOT1/AST1 expression status as a prognostic biomarker in pancreatic ductal adenocarcinoma. Oncotarget. (2015) 6:4516–26. doi: 10.18632/oncotarget.2799
10. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. (2013) 496:101–5. doi: 10.1038/nature12040
11. Chakrabarti G, Moore ZR, Luo X, Ilcheva M, Ali A, Padanad M, et al. Targeting glutamine metabolism sensitizes pancreatic cancer to PARP-driven metabolic catastrophe induced by ß-lapachone. Cancer Metab. (2015) 3:12. doi: 10.1186/s40170-015-0137-1
12. Hong C, Zheng J, Li X. Inhibition of GOT1 sensitizes colorectal cancer cells to 5-fluorouracil. Cancer Chemother Pharmacol. (2017) 79:835–40. doi: 10.1007/s00280-017-3282-0
13. Nelson BS, Lin L, Kremer DM, Sousa CM, Cotta-Ramusino C, Myers A, et al. Tissue of origin dictates GOT1 dependence and confers synthetic lethality to radiotherapy. Cancer Metab. (2020) 8:1. doi: 10.1186/s40170-019-0202-2
14. Yang CS, Stampouloglou E, Kingston NM, Zhang L, Monti S, Varelas X, et al. Glutamine-utilizing transaminases are a metabolic vulnerability of TAZ/YAP-activated cancer cells. EMBO Rep. (2018) 19:e43577. doi: 10.15252/embr.201643577
15. Yang Y. Enhancing doxorubicin efficacy through inhibition of aspartate transaminase in triple-negative breast cancer cells. Biochem Biophys Res Commun. (2016) 473:1295–300. doi: 10.1016/j.bbrc.2016.04.061
16. He Y, Deng F, Zhao S, Zhong S, Zhao J, Wang D, et al. Analysis of miRNA-mRNA network reveals miR-140-5p as a suppressor of breast cancer glycolysis via targeting GLUT1. Epigenomics. (2019) 11:1021–36. doi: 10.2217/epi-2019-0072
17. Matschke J, Riffkin H, Klein D, Handrick R, L demann L, Metzen E, et al. Targeted inhibition of glutamine-dependent glutathione metabolism overcomes death resistance induced by chronic cycling hypoxia. Antioxid Redox Signal. (2016) 25:89–107. doi: 10.1089/ars.2015.6589
18. Lee B, Mahmud I, Marchica J, Derezinski P, Qi F, Wang F, et al. Integrated RNA and metabolite profiling of urine liquid biopsies for prostate cancer biomarker discovery. Sci Rep. (2020) 10:3716. doi: 10.1038/s41598-020-60616-z
19. Singh AN, Sharma N. Quantitative SWATH-based proteomic profiling for identification of mechanism-driven diagnostic biomarkers conferring in the progression of metastatic prostate cancer. Front Oncol. (2020) 10:493. doi: 10.3389/fonc.2020.00493
20. Cheng Z, Dai Y, Zeng T, Liu Y, Cui L, Qian T, et al. Upregulation of glutamic-oxaloacetic transaminase 1 predicts poor prognosis in acute myeloid leukemia. Front Oncol. (2020) 10:379. doi: 10.3389/fonc.2020.00379
21. Li W, Fu H, Fang L, Chai H, Gao T, Chen Z, et al. Shikonin induces ferroptosis in multiple myeloma via GOT1-mediated ferritinophagy. Front Oncol. (2022) 12:1025067. doi: 10.3389/fonc.2022.1025067
22. Nwosu ZC, Battello N, Rothley M, Pioronska W, Sitek B, Ebert MP, et al. Liver cancer cell lines distinctly mimic the metabolic gene expression pattern of the corresponding human tumours. J Exp Clin Cancer Res. (2018) 37:211. doi: 10.1186/s13046-018-0872-6
23. Zhou X, Curbo S, Li F, Krishnan S, Karlsson A. Inhibition of glutamate oxaloacetate transaminase 1 in cancer cell lines results in altered metabolism with increased dependency of glucose. BMC Cancer. (2018) 18:559. doi: 10.1186/s12885-018-4443-1
24. Zhou S, Guo Z, Lv X, Zhang X. CircGOT1 promotes cell proliferation, mobility, and glycolysis-mediated cisplatin resistance via inhibiting its host gene GOT1 in esophageal squamous cell cancer. Cell Cycle. (2022) 21:247–60. doi: 10.1080/15384101.2021.2015671
25. Luo P, Yan H, Du J, Chen X, Shao J, Zhang Y, et al. PLK1 (polo like kinase 1)-dependent autophagy facilitates gefitinib-induced hepatotoxicity by degrading COX6A1 (cytochrome c oxidase subunit 6A1). Autophagy. (2021) 17:3221–37. doi: 10.1080/15548627.2020.1851492
26. Song YM, Lee YH, Kim JW, Ham DS, Kang ES, Cha BS, et al. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy. (2015) 11:46–59. doi: 10.4161/15548627.2014.984271
27. Zheng ZG, Zhu ST, Cheng HM, Zhang X, Cheng G, Thu PM, et al. Discovery of a potent SCAP degrader that ameliorates HFD-induced obesity, hyperlipidemia and insulin resistance via an autophagy-independent lysosomal pathway. Autophagy. (2021) 17:1592–613. doi: 10.1080/15548627.2020.1757955
28. Lee TH, Kim WR, Benson JT, Therneau TM, Melton LJ 3rd. Serum aminotransferase activity and mortality risk in a United States community. Hepatology. (2008) 47:880–7. doi: 10.1002/hep.22090
29. Kim HC, Nam CM, Jee SH, Han KH, Oh DK, Suh I. Normal serum aminotransferase concentration and risk of mortality from liver diseases: prospective cohort study. BMJ. (2004) 328:983. doi: 10.1136/bmj.38050.593634.63
30. Monami M, Bardini G, Lamanna C, Pala L, Cresci B, Francesconi P, et al. Liver enzymes and risk of diabetes and cardiovascular disease: results of the Firenze Bagno a Ripoli (FIBAR) study. Metabolism. (2008) 57:387–92. doi: 10.1016/j.metabol.2007.10.015
31. Siwo GH, Singal AG, Waljee AK. Pan-cancer molecular signatures connecting aspartate transaminase (AST) to cancer prognosis, metabolic and immune signatures. arXiv [preprint]. (2024). doi: 10.1101/2024.03.01.582939
32. Otto-Ślusarczyk D, Graboń W, Mielczarek-Puta M. Aspartate aminotransferase–key enzyme in the human systemic metabolism. Postepy Hig Med Dosw. (2016) 70:219–30. doi: 10.5604/17322693.1197373
33. Song Z, Yang Y, Wu Y, Zheng M, Sun D, Li H, et al. Glutamic oxaloacetic transaminase 1 as a potential target in human cancer. Eur J Pharmacol. (2022) 917:174754. doi: 10.1016/j.ejphar.2022.174754
34. Chapman VM, Ruddle FH. Glutamate oxaloacetate transaminase (got) genetics in the mouse: polymorphism of got-1. Genetics. (1972) 70:299–305. doi: 10.1093/genetics/70.2.299
35. Li T, Copeland C, Le A. Glutamine metabolism in cancer. Adv Exp Med Biol. (2021) 1311:17–38. doi: 10.1007/978-3-030-65768-0_2
36. Chae YC, Kim JH. Cancer stem cell metabolism: target for cancer therapy. BMB Rep. (2018) 51:319–26. doi: 10.5483/BMBRep.2018.51.7.112
37. Toney MD. Aspartate aminotransferase: an old dog teaches new tricks. Arch Biochem Biophys. (2014) 544:119–27. doi: 10.1016/j.abb.2013.10.002
38. Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. (2015) 162:540–51. doi: 10.1016/j.cell.2015.07.016
39. Safer B. The metabolic significance of the malate-aspartate cycle in heart. Circ Res. (1975) 37:527–33. doi: 10.1161/01.RES.37.5.527
40. Hayashi H, Mizuguchi H, Miyahara I, Nakajima Y, Hirotsu K, Kagamiyama H. Conformational change in aspartate aminotransferase on substrate binding induces strain in the catalytic group and enhances catalysis. J Biol Chem. (2003) 278:9481–8. doi: 10.1074/jbc.M209235200
41. Li T, Le A. Glutamine metabolism in cancer. Adv Exp Med Biol. (2018) 1063:13–32. doi: 10.1007/978-3-319-77736-8_2
42. Jiang X, Chang H, Zhou Y. Expression, purification and preliminary crystallographic studies of human glutamate oxaloacetate transaminase 1 (GOT1). Protein Expr Purif. (2015) 113:102–6. doi: 10.1016/j.pep.2015.05.010
43. Meléndez-Rodríguez F, Urrutia AA, Lorendeau D, Rinaldi G, Roche O, B g rc -Seidel N, et al. HIF1α Suppresses tumor cell proliferation through inhibition of aspartate biosynthesis. Cell Rep. (2019) 26:2257–2265.e2254. doi: 10.1016/j.celrep.2019.01.106
44. Borst P. The malate-aspartate shuttle (Borst cycle): How it started and developed into a major metabolic pathway. IUBMB Life. (2020) 72:2241–59. doi: 10.1002/iub.v72.11
45. Hu T, Shukla SK, Vernucci E, He C, Wang D, King RJ, et al. Metabolic rewiring by loss of sirt5 promotes kras-induced pancreatic cancer progression. Gastroenterology. (2021) 161:1584–600. doi: 10.1053/j.gastro.2021.06.045
46. Li W, Chen C, Zhao X, Ye H, Zhao Y, Fu Z, et al. HIF-2α regulates non-canonical glutamine metabolism via activation of PI3K/mTORC2 pathway in human pancreatic ductal adenocarcinoma. J Cell Mol Med. (2017) 21:2896–908. doi: 10.1111/jcmm.2017.21.issue-11
47. Zhao Y, Wang Y, Miao Z, Liu Y, Yang Q. c-Myc protects hepatocellular carcinoma cell from ferroptosis induced by glutamine deprivation via upregulating GOT1 and Nrf2. Mol Biol Rep. (2023) 50:6627–41. doi: 10.1007/s11033-023-08495-1
48. Wang J, Wang B, Ren H, Chen W. miR-9-5p inhibits pancreatic cancer cell proliferation, invasion and glutamine metabolism by targeting GOT1. Biochem Biophys Res Commun. (2019) 509:241–8. doi: 10.1016/j.bbrc.2018.12.114
49. Liu X, Shen Z. LncRNA TMPO-AS1 Aggravates the Development of Hepatocellular Carcinoma via miR-429/GOT1 Axis. Am J Med Sci. (2020) 360:711–20. doi: 10.1016/j.amjms.2020.08.010
50. Zhu X, Han J, Lan H, Lin Q, Wang Y, Sun X. A novel circular RNA hsa_circRNA_103809/miR-377-3p/GOT1 pathway regulates cisplatin-resistance in non-small cell lung cancer (NSCLC). BMC Cancer. (2020) 20:1190. doi: 10.1186/s12885-020-07680-w
51. Zhou X, Liu K, Cui J, Xiong J, Wu H, Peng T, et al. Circ-MBOAT2 knockdown represses tumor progression and glutamine catabolism by miR-433-3p/GOT1 axis in pancreatic cancer. J Exp Clin Cancer Res. (2021) 40:124. doi: 10.1186/s13046-021-01894-x
52. Wei XB, Jiang WQ, Zeng JH, Huang LQ, Ding HG, Jing YW, et al. Exosome-Derived lncRNA NEAT1 Exacerbates Sepsis-Associated Encephalopathy by Promoting Ferroptosis Through Regulating miR-9-5p/TFRC and GOT1 Axis. Mol Neurobiol. (2022) 59:1954–69. doi: 10.1007/s12035-022-02738-1
53. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell. (2015) 162:552–63. doi: 10.1016/j.cell.2015.07.017
54. Pérez-Mato M, Ramos-Cabrer P, Sobrino T, Blanco M, Ruban A, Mirelman D, et al. Human recombinant glutamate oxaloacetate transaminase 1 (GOT1) supplemented with oxaloacetate induces a protective effect after cerebral ischemia. Cell Death Dis. (2014) 5:e992. doi: 10.1038/cddis.2013.507
55. Kerk SA, Lin L, Myers AL, Sutton DJ, Andren A, Sajjakulnukit P, et al. Metabolic requirement for GOT2 in pancreatic cancer depends on environmental context. Elife. (2022) 11:e73245. doi: 10.7554/eLife.73245
56. Duarte T, Barbisan F, do Prado-Lima PAS, Azzolin VF, da Cruz Jung IE, Duarte MMMF, et al. Ziprasidone, a second-generation antipsychotic drug, triggers a macrophage inflammatory response in vitro. Cytokine. (2018) 106:101–7. doi: 10.1016/j.cyto.2017.10.017
57. Yang Y, Zheng M, Han F, Shang L, Li M, Gu X, et al. Ziprasidone suppresses pancreatic adenocarcinoma cell proliferation by targeting GOT1 to trigger glutamine metabolism reprogramming. J Mol Med (Berl). (2022) 100:599–612. doi: 10.1007/s00109-022-02181-8
58. Suzuki T, Otsuka M, Seimiya T, Iwata T, Kishikawa T, Koike K, et al. The biological role of metabolic reprogramming in pancreatic cancer. MedComm. (2020) 1:302–10. doi: 10.1002/mco2.v1.3
59. Tang T, Liu Y, Yang M, Tu M, Zhu W, Chen M. Glutamate-oxaloacetate transaminase 1 impairs glycolysis by interacting with pyruvate carboxylase and further inhibits the Malignant phenotypes of glioblastoma cells. World Neurosurg. (2021) 154:e616–26. doi: 10.1016/j.wneu.2021.07.097
60. Jin X, Sun X, Ma X, Qin Z, Gao X, Kang X, Li H, et al. SIRT1 maintains bone homeostasis by regulating osteoblast glycolysis through GOT1. Cell Mol Life Sci. (2024) 81:204. doi: 10.1007/s00018-023-05043-9
61. Xu W, Patel CH, Zhao L, Sun IH, Oh MH, Sun IM, et al. GOT1 regulates CD8+ effector and memory T cell generation. Cell Rep. (2023) 42:111987. doi: 10.1016/j.celrep.2022.111987
62. Grimm F, Asuaje A, Jain A, Silva Dos Santos M, Kleinjung J, Nunes PM, et al. Metabolic priming by multiple enzyme systems supports glycolysis, HIF1α stabilisation, and human cancer cell survival in early hypoxia. EMBO J. (2024) 43:1545–69. doi: 10.1038/s44318-024-00065-w
63. Zhou Y, Yu H, Cheng S, Chen Y, He L, Ren J, et al. Glutamate dehydrogenase 1 mediated glutaminolysis sustains HCC cells survival under glucose deprivation. J Cancer. (2022) 13:1061–72. doi: 10.7150/jca.64195
64. Murao N, Yokoi N, Takahashi H, Hayami T, Minami Y, Seino S. Increased glycolysis affects β-cell function and identity in aging and diabetes. Mol Metab. (2022) 55:101414. doi: 10.1016/j.molmet.2021.101414
65. Qi L, Martin-Sandoval MS, Merchant S, Gu W, Eckhardt M, Mathews TP, et al. Aspartate availability limits hematopoietic stem cell function during hematopoietic regeneration. Cell Stem Cell. (2021) 28:1982-1999.e1988. doi: 10.1016/j.stem.2021.07.011
66. Ma EH, Dahabieh MS, DeCamp LM, Kaymak I, Kitchen-Goosen Susan M, Oswald BM, et al. 13C metabolite tracing reveals glutamine and acetate as critical in vivo fuels for CD8 T cells. Sci Adv. (2024) 10:eadj1431. doi: 10.1126/sciadv.adj1431
67. Weisshaar N, Ma S, Ming Y, Madi A, Mieg A, Hering M, et al. The malate shuttle detoxifies ammonia in exhausted T cells by producing 2-ketoglutarate. Nat Immunol. (2023) 24:1921–32. doi: 10.1038/s41590-023-01636-5
68. Yang Y, Cheng Z, Zhang W, Hei W, Lu C, Cai C, et al. Glutamic-oxaloacetic transaminase 1 regulates adipocyte differentiation by altering nicotinamide adenine dinucleotide phosphate content. Anim Biosci. (2022) 35:155–65. doi: 10.5713/ab.21.0174
69. Zhang L, Wu Z, Qiu X, Zhang J, Cheng S-C. Glutamate oxaloacetate transaminase 1 is dispensable in macrophage differentiation and anti-pathogen response. Commun Biol. (2024) 7:817. doi: 10.1038/s42003-024-06479-w
70. Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol. (2012) 13:270–6. doi: 10.1038/nrm3305
71. Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab. (2012) 16:9–17. doi: 10.1016/j.cmet.2012.06.001
72. Kaelin WG Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. (2013) 153:56–69. doi: 10.1016/j.cell.2013.03.004
73. Xu T, Stewart KM, Wang X, Liu K, Xie M, Ryu JK, et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature. (2017) 548:228–33. doi: 10.1038/nature23475
74. Kremer DM, Nelson BS, Lin L, Yarosz EL, Halbrook CJ, Kerk SA, et al. GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat Commun. (2021) 12:4860. doi: 10.1038/s41467-021-24859-2
75. Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. (2015) 15:73–9. doi: 10.1038/nrc3876
76. Zhang K, Wu L, Zhang P, Luo M, Du J, Gao T, et al. miR-9 regulates ferroptosis by targeting glutamic-oxaloacetic transaminase GOT1 in melanoma. Mol Carcinog. (2018) 57:1566–76. doi: 10.1002/mc.v57.11
77. Yan S, Qi C, Song W, Xu Q, Gu L, Sun W, et al. Discovery of GOT1 Inhibitors from a Marine-Derived Aspergillus terreus That Act against Pancreatic Ductal Adenocarcinoma. Mar Drugs. (2021) 19:588. doi: 10.3390/md19110588
78. Yoshida T, Yamasaki S, Kaneko O, Taoka N, Tomimoto Y, Namatame I, et al. A covalent small molecule inhibitor of glutamate-oxaloacetate transaminase 1 impairs pancreatic cancer growth. Biochem Biophys Res Commun. (2020) 522:633–8. doi: 10.1016/j.bbrc.2019.11.130
79. Sun W, Luan S, Qi C, Tong Q, Yan S, Li H, et al. Aspulvinone O, a natural inhibitor of GOT1 suppresses pancreatic ductal adenocarcinoma cells growth by interfering glutamine metabolism. Cell Commun Signal. (2019) 17:111. doi: 10.1186/s12964-019-0425-4
80. Guo Y, Chen T, Liang X, Gou S, Xiong J, Cui J, et al. Tumor cell derived exosomal GOT1 suppresses tumor cell ferroptosis to accelerate pancreatic cancer progression by activating nrf2/HO-1 axis via upregulating CCR2 expression. Cells. (2022) 11:3893. doi: 10.3390/cells11233893
81. Biancur DE, Paulo JA, Małachowska B, Del Rey MQ, Sousa CM, Wang X, et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun. (2017) 8:15965. doi: 10.1038/ncomms15965
82. Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem. (2002) 277:30409–12. doi: 10.1074/jbc.R200006200
83. Garcia-Bermudez J, Badgley MA, Prasad S, Baudrier L, Liu Y, La K, et al. Adaptive stimulation of macropinocytosis overcomes aspartate limitation in cancer cells under hypoxia. Nat Metab. (2022) 4:724–38. doi: 10.1038/s42255-022-00583-z
84. Abrego J, Gunda V, Vernucci E, Shukla SK, King RJ, Dasgupta A, et al. GOT1-mediated anaplerotic glutamine metabolism regulates chronic acidosis stress in pancreatic cancer cells. Cancer Lett. (2017) 400:37–46. doi: 10.1016/j.canlet.2017.04.029
85. Shibuya K, Okada M, Suzuki S, Seino M, Seino S, Takeda H, et al. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget. (2015) 6:651–61. doi: 10.18632/oncotarget.2892
86. Wang Q, Zhang Q, Luan S, Yang K, Zheng M, Li K, et al. Adapalene inhibits ovarian cancer ES-2 cells growth by targeting glutamic-oxaloacetic transaminase 1. Bioorg Chem. (2019) 93:103315. doi: 10.1016/j.bioorg.2019.103315
87. Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. (2006) 7:880–5. doi: 10.1038/sj.embor.7400779
88. Korangath P, Teo WW, Sadik H, Han L, Mori N, Huijts CM, et al. Targeting glutamine metabolism in breast cancer with aminooxyacetate. Clin Cancer Res. (2015) 21:3263–73. doi: 10.1158/1078-0432.CCR-14-1200
89. Anglin J, Zavareh RB, Sander PN, Haldar D, Mullarky E, Cantley LC, et al. Discovery and optimization of aspartate aminotransferase 1 inhibitors to target redox balance in pancreatic ductal adenocarcinoma. Bioorg Med Chem Lett. (2018) 28:2675–8. doi: 10.1016/j.bmcl.2018.04.061
90. Zhang BS, Li Y, Zhang Z, An Y, Wen YH, Gou XY, et al. Synthesis of C4-aminated indoles via a catellani and retro-diels-alder strategy. J Am Chem Soc. (2019) 141:9731–8. doi: 10.1021/jacs.9b05009
91. Holt MC, Assar Z, Beheshti Zavareh R, Lin L, Anglin J, Mashadova O, et al. Biochemical characterization and structure-based mutational analysis provide insight into the binding and mechanism of action of novel aspartate aminotransferase inhibitors. Biochemistry. (2018) 57:6604–14. doi: 10.1021/acs.biochem.8b00914
92. Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, et al. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. (2014) 508:108–12. doi: 10.1038/nature13110
93. Daemen A, Peterson D, Sahu N, McCord R, Du X, Liu B, et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci U S A. (2015) 112:E4410–4417. doi: 10.1073/pnas.1501605112
Keywords: cancer, cell metabolism, GOT1, therapeutic target, metabolic reprogramming
Citation: Peng H, Dou H, He S, Xie Y-a, Zhang Q and Zheng J (2024) The role of GOT1 in cancer metabolism. Front. Oncol. 14:1519046. doi: 10.3389/fonc.2024.1519046
Received: 29 October 2024; Accepted: 06 December 2024;
Published: 24 December 2024.
Edited by:
Alvaro Marín Hernández, Instituto Nacional de Cardiología, MexicoReviewed by:
Hanumantha Rao Madala, Harvard Medical School, United StatesCopyright © 2024 Peng, Dou, He, Xie, Zhang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qinle Zhang, cWlubGV6aGFuZ0Bob3RtYWlsLmNvbQ==; Jianqiu Zheng, empxLTEwMDhAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.