 Edgar Yebran Villegas-Vazquez1†Francisco Pável Marín-Carrasco1†Octavio Daniel Reyes-Hernández1†
Edgar Yebran Villegas-Vazquez1†Francisco Pável Marín-Carrasco1†Octavio Daniel Reyes-Hernández1† Andrea S. Báez-González2Lilia Patricia Bustamante-Montes3
Andrea S. Báez-González2Lilia Patricia Bustamante-Montes3 Teresita Padilla-Benavides2
Teresita Padilla-Benavides2 Laura Itzel Quintas-Granados4*
Laura Itzel Quintas-Granados4* Gabriela Figueroa-González1*
Gabriela Figueroa-González1*- 1Laboratorio de Farmacogenética, UMIEZ, Facultad de Estudios Superiores Zaragoza, Universidad Nacional Autónoma de México, Ciudad de México, Mexico
- 2Department of Molecular Biology and Biochemistry, Wesleyan University, Middletown, CT, United States
- 3Coordinación de Investigación, Centro Universitario siglo XXI, Estado de México, Toluca, Mexico
- 4Colegio de Ciencias y Humanidades, Plantel Cuautepec, Universidad Autónoma de la Ciudad de México, Ciudad de México, Mexico
Drug repositioning, the practice of identifying novel applications for existing drugs beyond their originally intended medical indications, stands as a transformative strategy revolutionizing pharmaceutical productivity. In contrast to conventional drug development approaches, this innovative method has proven to be exceptionally effective. This is particularly relevant for cancer therapy, where the demand for groundbreaking treatments continues to grow. This review focuses on drug repositioning for ovarian cancer treatment, showcasing a comprehensive exploration grounded in thorough in vitro experiments across diverse cancer cell lines, which are validated through preclinical in vivo models. These insights not only shed light on the efficacy of these drugs but also expand in potential synergies with other pharmaceutical agents, favoring the development of cost-effective treatments for cancer patients.
Introduction
Drug repositioning, also known as drug repurposing, is a strategy that involves identifying new therapeutic uses for existing drugs beyond their original indications. This approach has gained the attention of the scientific community due to its potential to expedite the drug development process, reduce costs, and maximize the utility of existing pharmaceutical agents (Figure 1). Drug repositioning possesses multiple advantages, as it presents increased efficiency by shortening the drug development timeline, as existing drugs have already endured various stages of testing for safety and efficacy. It capitalizes on the existing safety, toxicity, and pharmacokinetic data of approved drugs, significantly reducing the time and financial investment compared to de novo drug discovery. This approach has been especially valuable in addressing unmet medical needs, such as rare diseases and conditions lacking effective treatments. Thus, repurposing of drugs often results in a more cost-effective process than developing entirely new compounds, as it bypasses the extensive research and development phases, as repositioned drugs usually have established safety profiles, minimizing the risks associated with introducing entirely new substances. In fact, in recent years, the introduction of new drugs to the market has seen a decline owing to the adverse outcomes witnessed in medical trials and challenges in pharmacokinetics (1). However, significant progress in computational sciences, including bioinformatics, machine learning and computational chemistry, coupled with advancements in -omics sciences and high-throughput screening technologies, has enabled the exploration of drugs with multiple target molecules. These have broadened their potential applications and pharmacological benefits (2).
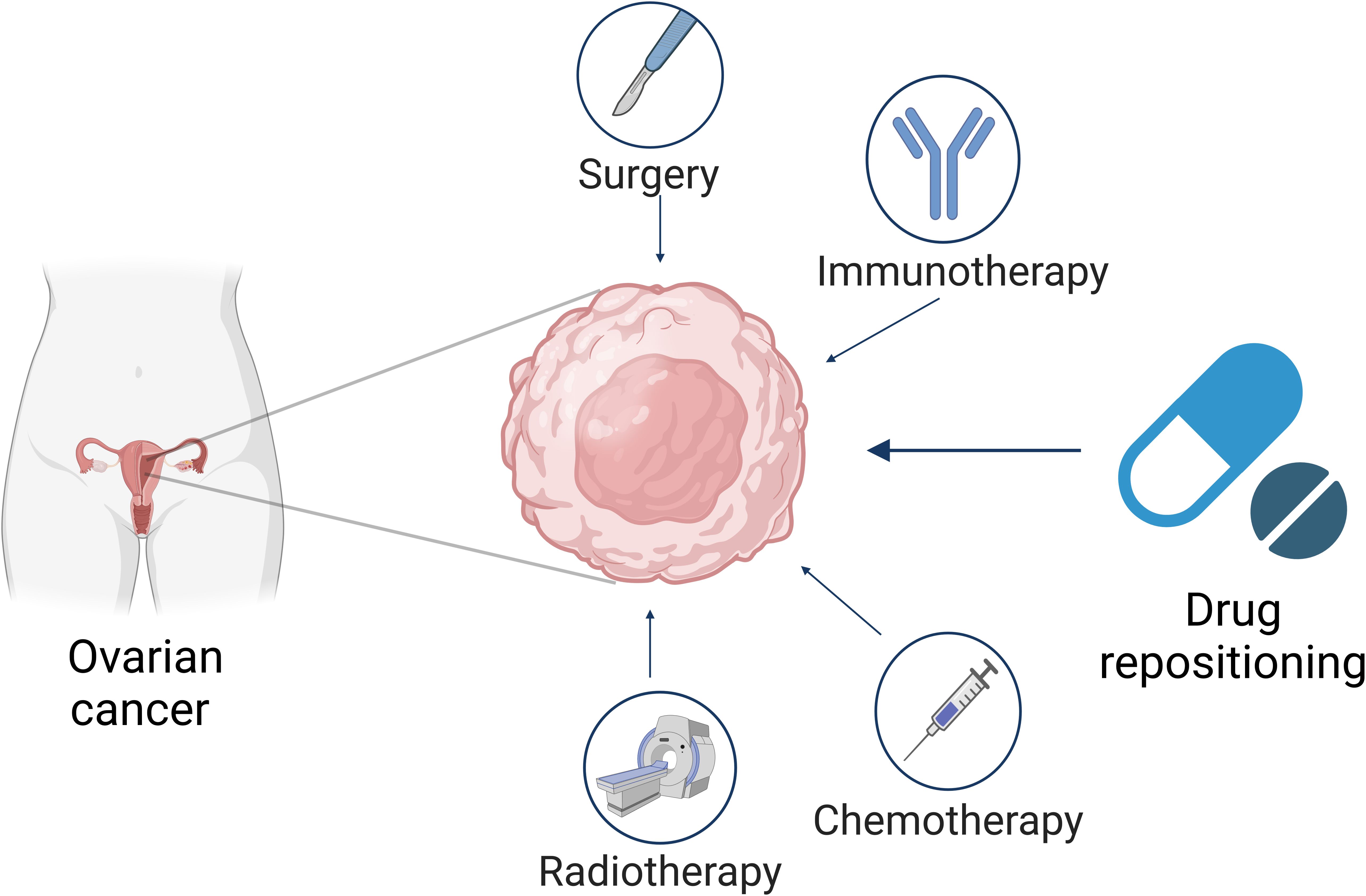
Figure 1. Schematic representation of ovarian cancer treatments. This diagram illustrates the general approach to ovarian cancer therapy. Outline the importance of drug repositioning besides surgery, immunotherapy, chemotherapy, and radiotherapy (Figure created with Biorender).
Drug repurposing has multiple applications in cancer treatment. For instance, repositioned drugs offer the opportunity to target specific pathways or mechanisms relevant to cancer, potentially introducing alternative treatment options. In addition, this strategy allows the identification of synergies between repositioned drugs and existing cancer treatments, which can lead to the development of more effective combination therapies. However, there are limitations associated with drug repurposing, such as patent protection for the original application of the drugs, which may pose challenges for repositioning efforts. Ultimately, administration of repurposed drugs in patients may also require adjustments and further investigation of the pharmacological conditions through in vitro, in vivo and clinical studies. Thus, designing appropriate clinical trials for repositioned drugs requires careful consideration of the new therapeutic context and side effects, as well as dosing regimens and possible coadjuvant treatments must also be considered when treating patients. Despite these limitations, since the safety and pharmacokinetic parameters of many drugs are well known, the increasing interest in drug repositioning is helping to determine new favorable outcomes of these drugs. Emerging approaches, such as molecular docking studies and other computer-aided methods, are helping to model novel ligand-targeting strategies and to help drug repositioning take a better landscape in cancer therapy and unlocking the potential of already existed drugs.
In addition, combination therapies that integrate repurposed drugs with existing pharmaceutical agents hold significant promise for enhancing treatment efficacy in cancer and other malignancies. These synergies can target multiple pathways simultaneously, overcoming limitations such as drug resistance and heterogeneity within tumors, as discussed in specific cases below. However, the complexity of drug-drug interactions in combination therapies poses challenges. In this regard, additional limitations of drug repurposing combined with existing therapies include variations in pharmacokinetics, off-target effects, and potential antagonistic interactions. Moreover, the tumor microenvironment, patient-specific genetic factors, and dosage optimization add further layers of complexity. Thus, in order to utilize this strategy in the clinic, it is essential to perform rigorous preclinical and clinical studies, to avoid adverse effects in patients.
This review offers a comprehensive analysis of drug repositioning in ovarian cancer (OC) therapy, synthesizing findings from in vitro and preclinical models while also acknowledging the limited data from clinical studies. Despite inherent limitations, the contributions of these models are invaluable for advancing our understanding of potential treatments. In vitro studies, particularly with cancer cell lines, continue to be essential tools in screening and identifying promising drug candidates. Although these models cannot fully mimic the complexities of human tumors, they provide controlled environments that allow for detailed investigation of drug effects on cancer cell biology, offering critical insights into drug responses, mechanisms of action, and preliminary efficacy (3–5). These findings can then be validated through more sophisticated in vivo models. Preclinical mouse models, for example, provide vital data on the pharmacokinetics, pharmacodynamics, and toxicity of repositioned drugs. These models allow for a comprehensive evaluation of drug responses within the context of tumor microenvironments, immune responses, and metabolism, even though they do not perfectly replicate human disease (6–8). Preclinical models thus serve as an important bridge between in vitro assays and clinical applications, often informing the design of clinical trials. Clinical translation remains a significant challenge, but the few studies conducted on repurposed drugs for OC in patients have demonstrated encouraging results. These trials, although limited in scale, provide critical insights into the pharmacokinetic properties, dosing regimens, and side effect profiles of repositioned drugs, underscoring the importance of further research to confirm their clinical efficacy. However, the current focus on short-term experimental models limits our understanding of the long-term therapeutic benefits and safety of these drugs. Longitudinal clinical studies are crucial to fully evaluate their sustained effectiveness and potential side effects, paving the way for the future of OC therapy.
Ovarian cancer
Epithelial ovarian cancer is the rapid growth of cells with abnormal function and structure with the potential to invade and destroy other healthy tissues (9). Among gynecological cancers, OC has a superior mortality rate because of its difficult early diagnosis resulting widely metastatic within the abdomen (10), placing OC as the 3rd most common gynecological cancer around the world in 2020 (11). Low- and middle-income countries presented the highest mortality rates of OC but its incidence was highest in high-income countries (12).
The nomenclature for OC subclassification includes five main histological types: high-grade serous (HGSOC), low-grade serous (LGSOC), endometrioid (ENOC), clear cell (CCOC), and mucinous (MOC) (13–15). HGSOC tumors are solid masses of cells characterized by slit-like fenestrations and structured with papillary, glandular or cribriform architecture (16). LGSOC tumors are distinguished by a monotonous proliferation of cuboidal, low columnar cells, mild to moderate atypia without nuclear pleomorphism, a mitotic index reaching up to 12 mitoses per 10 high-power fields (HPF) and invasion (17–19). Histopathological distinction of ENOC tumors from HGSOC is challenging, but the use of some discriminatory immunohistochemistry tools such as Wilms’ tumor 1 (WT1) lead to better tumor classification. ENOC tumors generally lack WT1 expression, whereas HGSOC tumors overwhelmingly exhibit WT1 positivity. In addition, ENOC is positive for the estrogen receptor (ER) in ≥75% of cases and for the progesterone receptor (PR) in over 60% of cases, with the majority (80%) of patients being also positive for wild-type Tumor Protein p53 (TP53) (20–23). Immunohistochemical markers, including WT1, Napsin A, hepatocyte nuclear factor-1-beta (HNF-1β), ER, and PR, are employed to distinguish CCOC from HGSOC and ENOC tumors. CCOC tumors are WT1 negative, Napsin A/HNF-1β positive, and EP/PR negative (22–24). MOC tumors are large, unilateral mucinous growths that negative for the WT1 and Napsin A markers, and approximately 60% of cases express a mutant version of p53 (23, 25).
Ovarian tumors are histopathologically heterogeneous, resulting genetic mutations specific for each epithelial OC subtype, which can be used as targets of treatment (10). For instance, near-ubiquitous mutations of p53, Breast Cancer Gene 1/2 (BRCA1/2), Neurofibromin 1 (NF1), and Cyclin-Dependent Kinase 12 (CDK12) are characteristic of HGSOC subtype (10, 26). LGSOC subtype is characterized by mutations in Kirsten Rat Sarcoma Viral Oncogene Homolog (KRAS), B-Raf Proto-Oncogene, Serine/Threonine Kinase (BRAF), Neuroblastoma RAS Viral Oncogene Homolog (NRAS), and Erb-B2 Receptor Tyrosine Kinase 2 (ERBB2, also known as HER2) mutations, while Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit α (PI3KCA), Phosphatase and Tensin Homolog (PTEN), AT-Rich Interaction Domain 1A (ARID1A), and Protein Phosphatase 2 Scaffold Subunit α (PPP2R1A) mutations are present in ENOC subtypes. Moreover, mutations in PI3KCA, ARID1A, Catenin β1 (CTNNB1), PTEN, and PP2R1A are also found in the CCOC subtype. Finally, the MOC subtype is characterized by KRAS and ERBB2 mutations (10). Changes in DNA methylation patterns also contribute to the development of OC, being the hypermethylation of the BRCA promoter a common example detected in 15-30% of patients (27–30). Figure 2 shows the recorded percentage of cases where mutations on the genes outlined here, as reported by the Integrated Genomic Analyses of Ovarian Carcinoma (Figure 2A) and the Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas (Figure 2B) from The Cancer Genome Atlas (TGCA) Human Cancer Models Initiative (HCMI) Cancer Model Development Center (Figure 2C). This information highlights the relevance of the presence of the mutations described above in the development of OC.
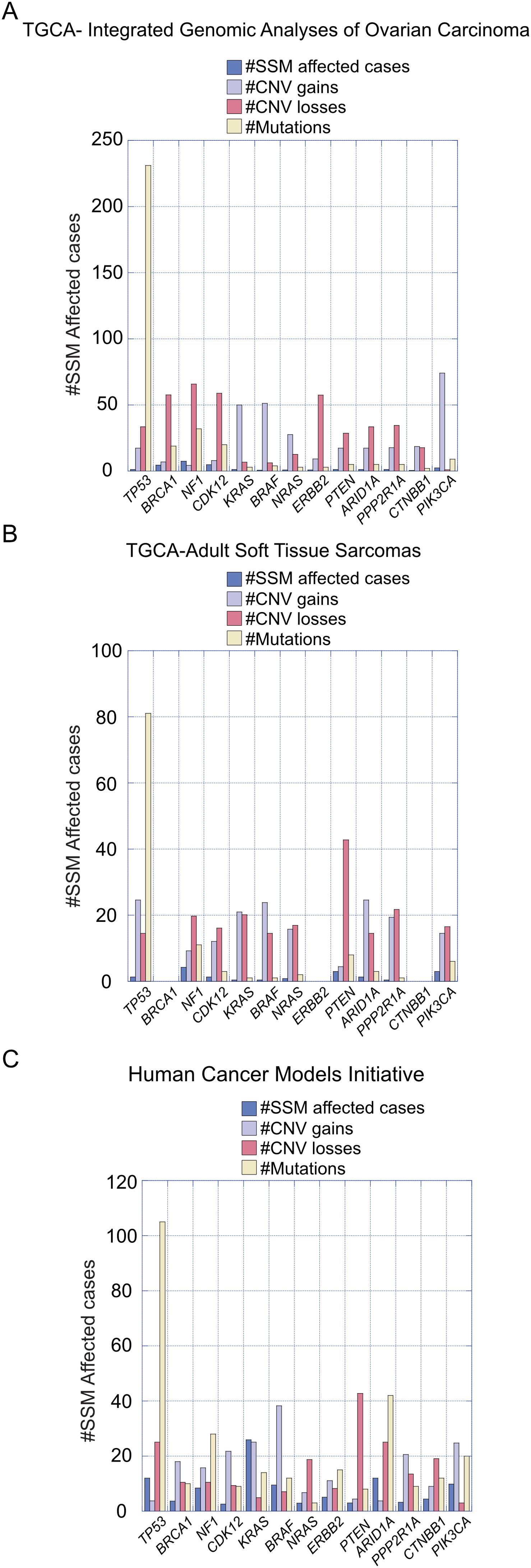
Figure 2. Recorded percentage of cases with mutations in specified genes relevant to ovarian carcinoma development. (A) Data presented are from the Integrated Genomic Analyses of Ovarian Carcinoma and (B) from the Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas, sourced from The Cancer Genome Atlas (TCGA). (C) Information on relevant genes was obtained from the Human Cancer Models Initiative (HCMI) Cancer Model Development Center.
Ovarian cancer staging is determined by the severity of the disease, considering factors such as tumor size, spread to nearby tissues, and the presence of distant metastasis (Figure 3). The International Federation of Gynecology and Obstetrics (FIGO) system is commonly used for ovarian cancer staging. Stage I is when cancer is confined to one or both ovaries, being sub-stage IA limited to one ovary, no tumor on the external surface; no ascites (fluid in the peritoneal cavity) present containing malignant cells, and substage IB when both ovaries are affected but no tumor is found on the external surfaces; no ascites containing malignant cells. Stage II is when cancerous tissue is found in one or both ovaries with pelvic extension. Substage IIA includes an extension to the uterus and/or fallopian tubes with no evident tumor on the external surfaces and neither ascites containing malignant cells. Substage IIB is when malignant tissue is extended to other pelvic tissues, but no tumor is found on the external surfaces and there are still no ascites containing malignant cells. Stage III OC involves one or both ovaries, the tumor may be spread to the peritoneum outside the pelvis and/or metastasis to the retroperitoneal lymph nodes and/or the omentum. Patients in stage III can be further classified in three substages according to the severity of metastasis. For instance, substage IIIA is determined by microscopic peritoneal metastasis beyond the pelvis; substage IIIB involves macroscopic peritoneal metastasis beyond the pelvis less than 2 cm in size; and substage IIIC includes abdominal metastasis greater than 2 cm in size and/or positive retroperitoneal lymph nodes. Finally, stage IV is diagnosed once malignant cancer cells have spread to distant organs or tissues, being predominant in the pleural fluid (substage IVA) and in the parenchyma of the liver and other distant organs (Substage IVB) (31–33).
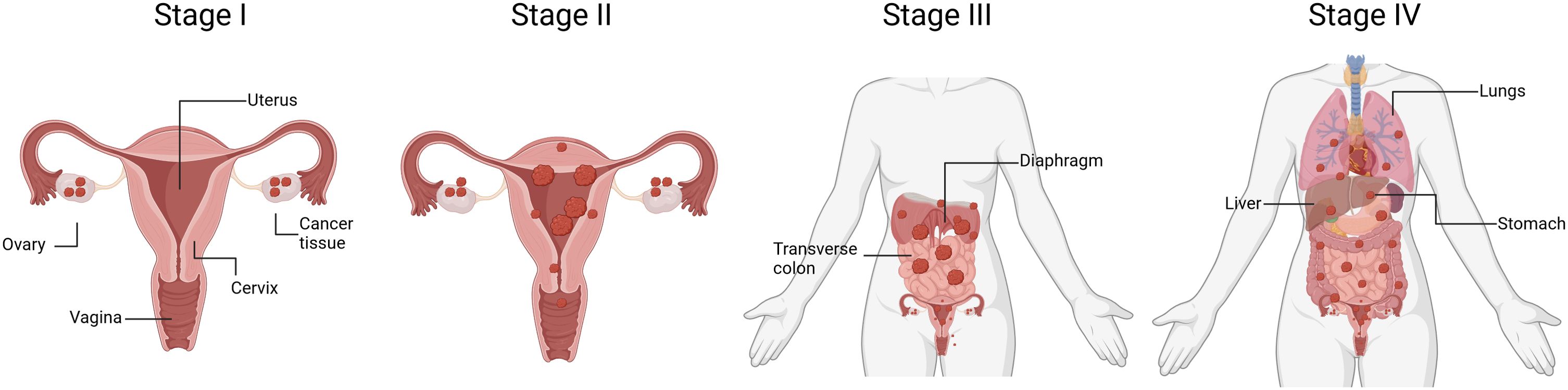
Figure 3. Schematic representation of ovarian cancer progression. The diagram outlines the four stages of OC, ranging from Stage I to Stage IV. Stage I signifies cancer that is confined to one or both ovaries. Stage II indicates cancer that has spread beyond the ovaries but is still within the pelvis. Stage III represents cancer that has advanced beyond the pelvis and has spread to the abdominal lining or nearby lymph nodes. Stage IV denotes cancer that has metastasized to distant organs beyond the abdominal cavity, such as the liver or lungs. Understanding the stage of OC is crucial for determining treatment options and predicting prognosis. (Figure created with Biorender).
Etiology of ovarian cancer
Ovarian cancer is a complex disease with multifactorial etiology, and its development involves a combination of genetic, hormonal, and environmental factors. Risk increases with age and genetic factors such as BRCA mutations (34, 35). Mutations in essential genes for DNA repair, such as MutL Homolog 1 (MLH1) and MutS Homolog 2 (MSH2), which are also predominant in Lynch syndrome patients, also increase the risk of ovarian cancer (36). Having descendants at an early or advanced age, nulliparity, and absence of full-term pregnancy are classic risk factors of OC (37). Long-term use of estrogen-only hormone replacement therapy, without progesterone, has also been linked to an increased risk of OC (38). Recently, an increase between 7 and 28% of OC incidence in women ranging from 15-40 years of age has been detected, likely due to the “normalization” of unhealthy lifestyles such as overweight and obesity (12). In addition, social determinants included the human development index and highest gross domestic product per capita and lifestyle characteristics such as physical inactivity, alcohol use, and prevalence of smoking (39). Some studies have suggested a potential link between the use of talcum powder in the genital area and an increased risk of OC as well (40). Pre-existing conditions such as lipid disorders, hypertension, diabetes, estrogen exposure, and metabolic syndrome are also associated with high OC incidence rates (12, 41–46). Evidence has also pointed to immune responses as potential contributors to OC, such as general dysregulation of the immune system and impaired immune surveillance (47), as well as chronic inflammation in the pelvic region (48).
OC incidence varies by race and ethnicity, with differences observed in the rates of diagnosis and outcomes, which in many instances correlate with the socioeconomic background. White females are the largest population affected, with 14.1 cases per 100,000 women. Then the next highest ethnicity incidence is in (Hispanic females, with a rate of 9.8 affected women per 100,000 individuals. This is followed by Asian/Pacific Islanders, African Americans, and American Indian/Alaska natives, whose incident rates are 9.0, 8.5, and 7.9 patients, respectively, per 100,000 women (49–51). Mortality rates are also dependent on different racial and ethnic groups. African American women often experience lower survival rates compared to non-Hispanic white women. This is largely due to limited access to healthcare and socioeconomic factors, which contribute to these important health disparities. The age at diagnosis and access to screening are also a relevant concern for appropriate treatment and survival, as the age at which OC is diagnosed may also vary by race and ethnicity. Some studies suggest that African American women may be diagnosed at a younger age compared to non-Hispanic white women (52–54).
Prognosis of ovarian cancer
Different histological subtypes of OC may have distinct prognoses. For example, HGSOC, the most common subtype, tends to be more aggressive. Overall, poor prognosis of OC is associated with the age and stage of disease at diagnosis (10, 55). Younger patients, particularly those diagnosed at premenopausal ages, may have a more favorable prognosis.; and the patient’s overall health and ability to tolerate treatment can also influence prognosis. In terms of disease stage, for stages I, II, III and IV, the 5-year relative survival rate after diagnosis is estimated at 89%, 70%, 36% and 17%, respectively; while the 10-year-relative survival rate was 84%, 59%, 23% and 8%, for those stages. In general, the overall relative survival rate at 2, 5 and 10 years after diagnosis was 65%, 44% and 36%, respectively (56). Thus, survival rate in women diagnosed at stage I in 5-year is 90% (55, 57). The five-year survival rate is about 80% in patients with disseminated disease to adjacent tissues. In metastatic patients the survival rate is 25% (55, 57). Unfortunately, the 5-year survival rate is less than 50% in patients diagnosed at an advanced OC stage (58–60). In 12-24 months, most patients relapse and die from progressive chemotherapy-resistant tumors (61).
Current therapies for OC treatment
The first-line treatments for OC are surgery, radiotherapy, and chemotherapy. Surgery is performed to remove the tumor tissue in its entirety. Surgery is also useful for histopathological diagnosis and staging of the tumor, according to the International FIGO (10). Surgical procedures include bilateral salpingo-oophorectomy, tumor debulking, total hysterectomy, and omentectomy (62). In addition, different trials used preoperative chemotherapy when interval debulking surgery is performed. Trials reported a reduction in postoperative morbidity (63, 64). Therefore, no difference in survival was observed when a second surgical procedure to complete tumor debulking (10).
For treatment of early-stage OC, cytotoxic chemotherapy improves survival (8%) (63, 64). Treatments with platinum (carboplatin or cisplatin) have been used as the first-line treatment (10). Cisplatin is a platinum-containing chemotherapy drug that is commonly used in the treatment of OC as it binds covalently to DNA in cancer cells, blocking replication and transcription (65, 66). Regimens with two cytotoxic agents improve survival, thus the standard treatment for OC is a combinatory therapy of paclitaxel or docetaxel with platinum-containing drugs (67–69). Rucaparib olaparib, niraparib, and talazoparib are poly(ADP-ribose) polymerase inhibitors (PARPi) that have been accepted by the FDA as chemotherapeutic drugs for OC treatment (70). However, platinum-containing drugs, paclitaxel, olaparib, niraparib, and bevacizumab, cause drug resistance in some OC patients (70).
Novel treatments for OC management employed several strategies (71) that include i) target morphomolecular OC types by using PARPi for HGSOC subtype, or the use of inhibitors of the Mitogen-activated protein kinase kinase (MEK) or aromatase inhibitors for LGSOC subtype (72, 73). ii) new clinical trial designs, such as umbrella and baskets studies (74, 75). iii) new inhibitors (ATRi) against the Ataxia telangiectasia and Rad-3 related kinase (ATR), such as prexasertib, adavosertib (76–82). iv) synergistic therapies combining drugs targeting both the tumor and its microenvironment, such as antiangiogenic compounds (e.g., bevacizumab, cediranib), immunotherapy, and chemotherapy (83–89). v) enhanced therapeutic delivery using antibody-directed conjugates or targeted radiotherapy (90–94). Among these, ATR serves as a promising target in cancer due to its role in signaling DNA lesions, replication stress, and regulating the S and G2/M checkpoints, offering potential for exploiting dysregulated DNA damage responses (95). Drugs that interfere with DNA repair, such as PARP inhibitors (Olaparib), are used particularly in patients with BRCA mutations (96).
Novel strategies to overcome drug resistance challenges against OC include the use of monoclonal antibodies. Specifically, the humanized IgG1 monoclonal antibody bevacizumab is directed against Vascular Endothelial Growth Factor (VEGF), and it is currently used for OC therapy (97). Immunotherapy is also an emerging strategy to overcome drug resistance. In this case, immune checkpoint inhibitors, such as pembrolizumab, are being investigated in clinical trials to boost the immune system to target cancer cells (98).
Treating OC presents significant challenges, including late diagnosis, chemoresistance, and the limited efficacy of available therapies. Thus, there is still a need to consider and develop alternative mechanisms to combat OC progression. In this regard, drug repositioning represents an excellent alternative therapy for patients who have developed drug resistance, which may result in failure to prevent recurrence, particularly in advanced stages. Moreover, the tumor microenvironment and genetic heterogeneity of OC further complicate treatment development. Drug repurposing offers a promising avenue to address these challenges by leveraging existing drugs with known safety profiles for new therapeutic uses. In consequence, these emerging approaches can expedite treatment availability and reduce development costs while exploring combinatorial strategies for enhanced efficacy. Drug repurposing studies hold promise as a bridge to personalized medicine, improving outcomes for OC patients.
Drug repositioning for OC treatment
Amid the evolving landscape of OC treatment, the exploration of innovative therapeutic strategies becomes imperative. Drug repositioning, a promising approach, involves repurposing existing drugs to uncover novel and effective treatments for this disease. As indicated above, this strategy harnesses the potential of compounds already approved for other indications, accelerating the development of cost-effective and targeted therapeutic options. In this review, we provide insights into drug repositioning specifically for OC, exploring its challenges, successes, and transformative therapeutic impact. Main highlights and structure of the compounds presented here are summarized in Table 1 and Figure 4.
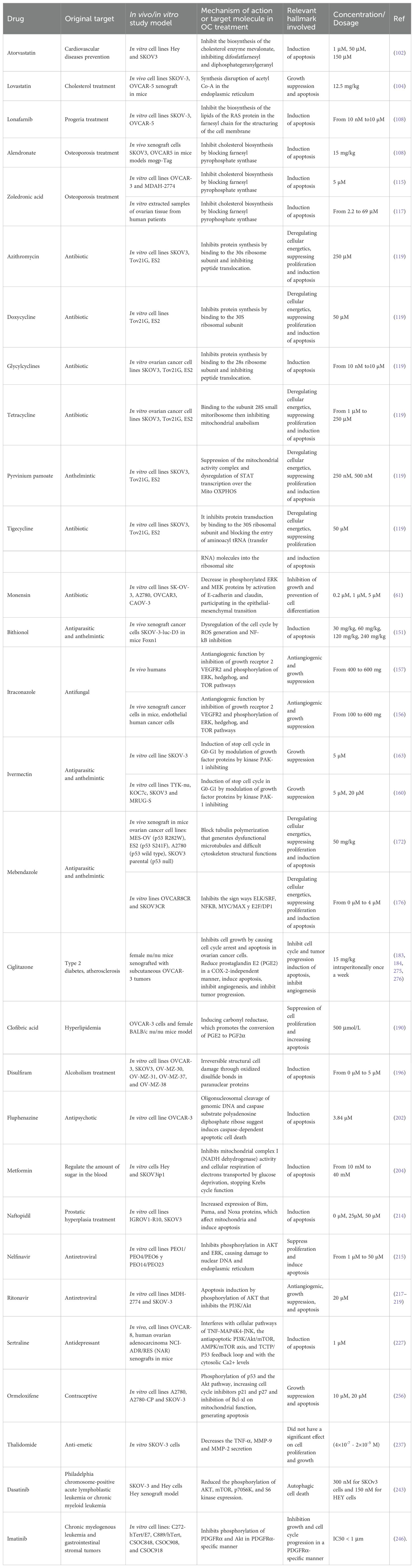
Table 1. Drug repositioned for ovarian cancer treatment.
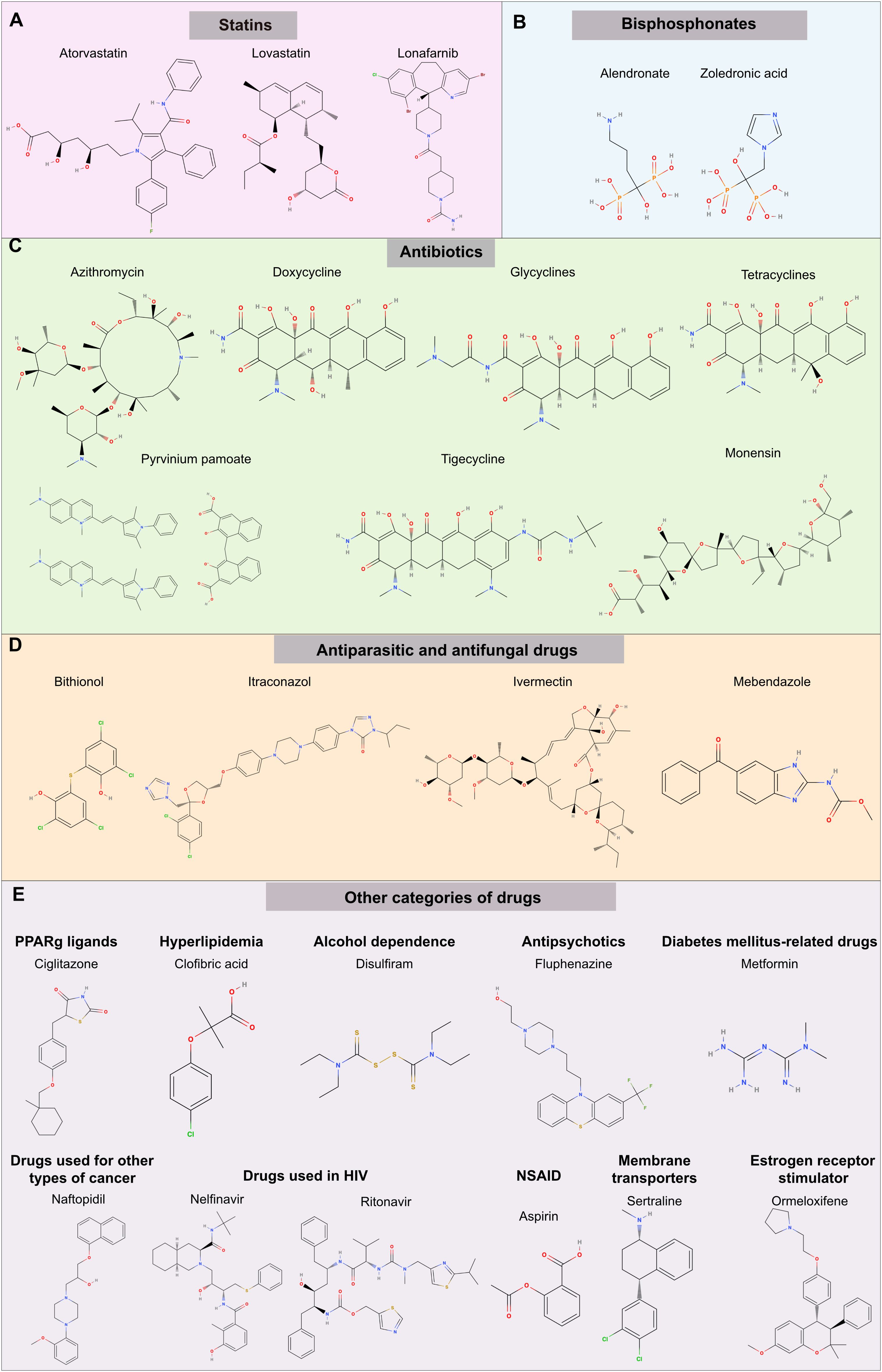
Figure 4. Chemical structure of potential drugs to be repurposed against ovarian cancer. The diagram illustrates potential drug candidates for repurposing in the treatment of OC. Various pharmaceutical agents target different pathways; see Table 1 for a summary of mechanisms. The groups represented are (A) statins, (B) bisphosphonates, (C) antibiotics (D) antiparasitic and antifungal drugs, (E) other categories of drugs including PPARγ inhibitors, compounds used to treat hyperlipidemia, alcoholism, and mental disorders, diabetes, other types of cancer (prostate), HIV infection, Non-steroidal anti-inflammatory drugs (NSAIDs), membrane transporters and estrogen receptors. Each drug offers a unique mode of action that could potentially enhance therapeutic outcomes in OC management.
Among relevant chemical compounds being repurposed towards treatment of OC, statins are medications commonly prescribed to lower cholesterol levels in the blood. Statins inhibit an enzyme involved in the production of cholesterol in the liver. By reducing cholesterol levels, statins help lower the risk of cardiovascular events such as heart attacks and strokes. Strategies to inhibit the mevalonate pathway have also been applied to dyslipidemic diseases. Statins reduce the hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase activity, which is enzymatically essential in the upstream part of the mevalonate pathway, resulting in a reduction in cholesterol levels in blood (99). Thus, considering the mechanism of action of statins related to mevalonate pathway inhibition, they are used to treat hypercholesterolemia (100). However, recent findings suggested that these molecules have antitumoral activities by causing apoptosis in tumor cells (101). For instance, atorvastatin (ATO) inhibits cell proliferation and invasion, while decreasing cell adhesion of cultured OC cells. Besides, ATO induces cellular stress, autophagy, apoptosis, and arrest cell cycle at G1 phase through Akt/mTOR pathway inhibition and MAPK pathway activation (102, 103). ATO also decreased the expression of VEGF, matrix-metalloproteinase-9 (MMP9), and the proto-oncogene cellular myelocytomatosis (c-Myc) in Hey and SKOV3 cultured OC cellular models (102). Experiments using the JQ1 selective inhibitor of bromodomain-containing proteins in Hey and SKOV3 OC cells also increased their sensitivity to the anti-proliferative activity of ATO (102). Another statin example that can be repurposed towards OC treatment is Lovastatin. This is another HMG-CoA reductase inhibitor that has been effective in reducing the proliferation of OC Hey and SKOV3 cells in vitro and in vivo murine models. Lovastatin delays tumorigenesis, proliferation, and cell cycle progression and suppresses tumor growth by influencing the cholesterol biosynthetic pathway (104, 105). Simvastatin, another HMG-CoA reductase inhibitor, reduces the production of cholesterol in the liver, thus lowering the levels of total cholesterol and low-density lipoprotein (LDL) cholesterol in the bloodstream. Simvastatin reduces the risk of cardiovascular events and has been shown to possess anti-metastatic and anti-tumorigenic effects in OC treatment. For instance, ID8, 28-2, and 30-2 cells treated with simvastatin had increased expression of apoptotic markers starting at 10 µM in 28-2 and 30-2 cells, and 50 µM for ID8 cell lines, suggesting that simvastatin induced cell death and decreased cell viability. Simvastatin was further shown to inhibit OC cell proliferation in a dose-dependent manner as measured by 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay in Hey and SKOV3 OC cells (106, 107). Other inhibitors of mevalonate pathway have been shown to promote autophagic responses. Examples of this are 6-fluoromevalonate, YM-53601, lonafarnib, and GGTI-298. These compounds can induce the expression of autophagy biomarkers such as LC3A (human microtubule-associated protein 1 light chain 3 gene LC3A and LC3B (human microtubule-associated protein 1 light chain 3 gene LC3B) and inhibit cell proliferation in a dose-dependent manner (108). Specifically, Lonafarnib inhibits the enzyme farnesyltransferase, which plays an important role in post-translational modification of proteins. Its primary function is to add a farnesyl group to specific proteins, in a process known as farnesylation.
Lonafarnib also inhibits protein prenylation in the mevalonate pathway, inhibiting cell proliferation with higher efficiency than 6-fluoromevalonate and YM-53601 (108). Besides, lonafarnib induces the expression of LC3A and LC3B genes, suggesting that the activation of autophagy impairs cell proliferation (108).
Bisphosphonates, such as alendronate, are anti-osteoporotic drugs that also inhibit the mevalonate pathway. Bisphosphonates delayed tumor formation and decreased tumor cell proliferation in a murine model of OC (108). Alendronate inhibits proliferation in OC SKOV3 and chemoresistant OVCAR5 cell lines in vitro. It induces the expression of LC3A and LC3B genes, indicating autophagy activation. In a transgenic OC mouse model (mogp-TAg), alendronate reduces total tumor mass, suggesting suppression of tumor initiation, and implies a potential chemo-preventive effect in OC development (108). In addition, alendronate reduces Rho activation by inhibiting the mevalonate pathway, resulting in the inhibition of cell migration in Caov-3, OC cells (109). Furthermore, the alendronate treatment (1mg/kg/d) reduced the tumor burden by ∼88% in female nude mice (BALB-c nu/nu) injected with Caov-3 (110). Together, in vitro and in vivo evidence strongly suggested findings that alendronate had potential as a drug for OC treatment (108–110).
Zoledronic acid is another example of a bisphosphonate drug, mainly prescribed for bone-related conditions like osteoporosis and cancer-induced bone complications. This drug inhibits bone resorption, promoting bone strengthening (111, 112). Specifically, zoledronic acid inhibits the activity of osteoclasts, the cells responsible for breaking down bone tissue, helping to maintain bone density and strength. It has also been used for the treatment of multiple myeloma and metastatic breast cancer and to treat hypercalcemia (high levels of calcium in the blood) associated with malignancy (113, 114). Zoledronic acid has been used in combination with gossypol, a natural polyphenolic compound used as a male contraceptive and with demonstrated anticancer properties in prostate cancer and leukemia. Combined, these two drugs render a synergistic cytotoxic and apoptotic effect in OC OVCAR-3 and MDAH-2774 cell lines (115). This combined treatment repressed the transcriptional expression of angiogenic molecules such as the inhibitor of differentiation or DNA binding (ID-1), EPH (Ephrin) receptors B2 and B4 (EPHB2/B4), laminin α-5 (LAMA-5), the fibroblastic growth factor (FGF2) and FGF receptor-3 (FGFR3), midkine (MDK), thymidine phosphorylase (TP), platelet-derived growth factor A (PDGF-A), and the cytokine CXCL-1, which plays a pivotal role in angiogenesis (115). Furthermore, experiments where NCI-H929, OPM-2, U266 and RPMI-8226 myeloma cell lines were pre-treated with simvastatin and then combined with antimyeloma drugs resulted in the apoptotic cascade (116). The combination of fluvastatin with zoledronic acid enhanced the chemosensitivity to the ATP-based tumor assay (ATP-TCA) in twenty-two pre-treated (mostly with platinum-based chemotherapy) ovarian carcinomas. Sequential drug experiments showed that pretreatment of tumor cells dissociated from solid carcinomas with fluvastatin resulted in decreased sensitivity to zoledronic acid (117). Mechanistically, zoledronic acid and fluvastatin treatment enhance the effects that involved Ras prenylation. Thus, implying that prior to bisphosphonate administration, statins would be expected to block the entry of mevalonate into the pathway, reducing the substrate concentration for the step that is blocked by zoledronic acid, potentially enhancing the effectiveness of the combination (117).
Research has suggested cancer could be managed as an infectious disease, in other words, by taking advantage of antibiotics that inhibit mitochondrial biogenesis, which is essential for clonal expansion and survival of cancer stem cells (118). This idea arose from the anabolic nature of cancer stem cells, which require mitochondrial biogenesis for proliferation and survival (118). Thus, targeting mitochondrial biogenesis is an alternative avenue that might render anti-tumorigenic effects useful against cancer treatment. Examples of antibiotics that impair mitochondrial biogenesis as a side effect are pyrvinium pamoate, doxycycline, azithromycin, tigecycline, and chloramphenicol, which make these compounds potential candidates in the treatment of OC (119). Mechanistically, antibiotics such as erythromycin, chloramphenicol, tetracyclines, glycylcyclines and pyrvinium pamoate target three main mitochondrial molecules. These are the mitochondrial 39S/large and the 28S/small ribosome subunits and mitochondrial oxidative phosphorylation proteins (OXPHOS), such as complex I of the electron transport chain (119–121). Azithromycin was shown to inhibit the tumor-sphere formation of OC SKOV3, ES2 and Tov21G cells, demonstrating the potential for cancer management (119). Conventionally, doxycycline has been used in the treatment of prostatitis, urinary tract infections and acne due to its anti-inflammatory properties (122–124). Doxycycline also inhibits cell proliferation, migration and matrix metalloproteinase 2 (MMP-2) activity in vivo model (Male Sprague-Dawley rats) treated with 30-mg/kg/day doxycycline after arterial injury (125), suggesting that if this drug were to be used for cancer therapy, its toxic side effects might be negligible (119, 126, 127). Mechanistically, doxycycline inhibits matrix-metalloproteinases (MMPs) and the formation of the tumor-sphere in cellular models of OC like SKOV3, ES2 and Tov21G (119). Doxycycline from 50 μM to 500 μM did not affect the viability of normal fibroblasts (hTERT-BJ1) and MCF7 cells (119). Doxycycline treatment reduced tumor growth by 80% in pancreatic tumor xenografts of PANC-1 cells (128). The antibiotic also reduced bone and bone-associated soft-tissue tumor mass by >60% and ~80%, respectively, in a xenograft model of breast cancer bone metastasis that involved MDA-MB-231 cells (129). The anti-cancer activity of doxycycline was attributed to the inhibition of MMPs rather than the targeting of mitochondrial biogenesis (119). Doxycycline exhibited a marked suppression of both invasive and migratory behaviors in human oral squamous cell carcinoma (SCC-15 cells) in vitro, with inhibition levels exceeding 75% at a concentration of 10 μg/ml. Additionally, daily administration of doxycycline at a dosage of 3 mg/mice effectively impeded tumor progression in SCC-15 xenografted nude mice, resulting in an 85.6% inhibition rate. Following doxycycline treatment, MMP-9 mRNA levels in fresh tumor tissue notably decreased compared to the control group treated with normal saline (P < 0.01), while MMP-2 mRNA levels remained unchanged (130).
Glycylcyclines and tetracyclines impair protein synthesis in bacteria (131, 132). These molecules bind to the bacterial 30S ribosomal unit, inhibiting the binding of aminoacyl-tRNA to the ribosomal A-site. Thus, glycylcyclines could be used to inhibit mitochondrial biogenesis in a similar manner to the one discussed above (119). Pyrvinium pamoate is an anti-helminthic drug which inhibits OXPHOS under normoxic and hypoxic environments and also prevents the formation of the tumor-sphere (119). Finally, tigecycline was shown to also inhibit the formation of a mammo-sphere in SKOV3, ES2 and Tov21G OC cell lines (119).
Monensin is primarily used as a veterinary antibiotic and feed additive for livestock, especially in the prevention and control of coccidiosis. The use of Monensin in human medicine is not authorized, and it is not prescribed for human consumption. However, experiments in SKOV-3, A2780, OVCAR-3 and CAOV-3 cells yielded promising results as a potential repurposed drug against OC. Monensin was shown to regulate the expression molecules linked to the epithelial-mesenchymal transition (EMT) and the mitogen-activated protein kinase (MEK)-extracellular signal-regulated kinase (ERK) pathway (61). This drug inhibited the proliferation of A2780, OVCAR3 and CAOV-3 cell lines from 0.2 µM (low inhibitory effect) until 5 µM (complete inhibitory effect) and impaired the invasive properties of SKOV-3 cells. Furthermore, in vivo experiments where SKOV-3 cells were injected into nude mice, followed by monensin administration (0, 8, and 16 mg/kg), resulted in reduced tumor masses in monensein-treated animals, compared to control groups (61). Mechanistically, monensin stimulated the SUMOylation of MEK1, impaired the growth, migration, and invasive capabilities of the A2780, OVCAR3, CAOV-3 and SKOV-3 OC cell lines and in the in vivo murine ovarian cancer xenograft model. Thus, monensin holds promise for OC treatment by augmenting MEK1 SUMOylation by suppressing the MEK-ERK signaling pathway (61). To investigate the potential SUMOylation of MEK in OC cell lines, MEK1 and SUMO1 were co-expressed in the non-tumorous cell line HEK293. Immunoprecipitation and western blot analyses of MEK1 revealed that monensin augmented the SUMOylation of MEK1, in a dose- and time-dependent manner (61). However, this drug still needs to be evaluated for approval and usage in human patients.
Bithionol is an anthelmintic drug, historically used for the treatment of intestinal worm infections (133). Bithionol is believed to interfere with the energy metabolism of the parasites, leading to their death. Bithionol has also been used as an antibacterial and antifungal agent in some topical formulations (134, 135). However, due to potential side effects and the availability of other effective treatments, its use in medical practice has been limited. Bithonol causes cell death via caspases-3/7-mediated apoptosis, arrest cell cycle progression, promote the production of Reactive Oxygen Species (ROS) and inhibits Autotaxin (ATX) (136). Autotaxin is an enzyme involved in the production of the signaling molecule lysophosphatidic acid (LPA). ATX and LPA have been implicated in cancer progression, including tumor growth, invasion, and metastasis (137–146). ATX is often overexpressed in several types of cancer, and elevated levels of LPA have been associated with promoting cancer cell survival, migration, and angiogenesis (147–149). In human OC biopsies LPA2 and LPA3 are highly expressed in comparison with normal ovaries or low malignancy tumors. Furthermore, there is a significant correlation between the expression ratios of LPA2-3 and VEGF in patients with cancer (150). Thus, research has been focused to understand ways to inhibit ATX or block the LPA pathways to impede cancer progression. Since ATX is associated with an increase in invasiveness and aggressiveness of tumor cells, and with the grade of tumor development, the inhibition of ATX by bithionol might have an important repercussion in OC treatment (151). In addition, bithionol has been shown to also induce DNA fragmentation, loss of mitochondrial potential and overexpression and activation of apoptotic biomarkers, such as cleaved PARP and caspase-7 (136, 151).
Itraconazole, an anti-fungal drug, has an anti-angiogenic activity and inhibits the Hedgehog pathway inducing autophagic growth arrest (152–155). This drug has been proposed for the treatment of several cancers such as leukemia, ovarian, breast and pancreatic (156). Out of 55 patients with refractory OC, 19 individuals received a combination of itraconazole and chemotherapy. The median progression-free survival (PFS) was 103 days for those receiving chemotherapy (platinum and taxane administration) with itraconazole, compared to 53 days for those without itraconazole (p=0.014). Similarly, the median overall survival was 642 days and 139 days for patients with and without itraconazole, respectively (p=0.006). A proportional hazards regression model (Cox) was employed for multivariate analysis of progression-free survival (PFS) and overall survival (defined as the duration from the commencement of chemotherapy after becoming refractory to death by any cause) following itraconazole exposure alongside chemotherapy. The analysis was adjusted for factors including age, race, Eastern Cooperative Oncology Group (ECOG) performance status (PS), carcinoma histology, number of prior regimens, and platinum sensitivity status. The study demonstrated that the hazard ratio for PFS was 0.24 (p=0.002), and for overall survival, it was 0.27 (p=0.006) in the group receiving itraconazole therapy (157). This data strongly suggested that combining classic chemotherapy with itraconazole may improve the median overall survival rate due to a potential synergistic effect of itraconazole in the treatment of refractory OC (157).
The antiparasitic drug ivermectin, which binds to the glutamic acid operative chloride ion channel (GluCls) (158, 159) has been repurposed for OC treatment (160–162). Ivermectin arrests cell cycle at G0-G1 phase by increasing the synthesis of p21, reducing proliferating cell nuclear antigen (PCNA), cyclin E, and cyclin D protein levels in breast cancer cell lines (MCF-7, MDA-MB-231 and MDA-MB-468) (163). Ivermectin also reduces viability and colony-forming ability in cancer stem-like malignant populations in the SKOV-3 cellular model (163). Furthermore, ivermectin inactivates the P21 (RAC1) Activated Kinase 1 (PAK1), resulting in the inhibition of the phosphorylation of kinase Raf1 (RAF-1) in TYK-nu and RMUG-S OC cells (160). The proliferation rate of TYK-nu, KOC7c, SKOV3, and RMUG-S cell lines was also diminished (160). Ivermectin targets the yes-associated protein 1 (YAP1) (164), which promotes tumorigenesis in breast and liver cancers (165, 166), suggesting a potential application in the treatment of OC, where YAP1 is considered a prognostic marker of the disease (167, 168). Mechanistically, ivermectin blocks the activity of Karyopherin Subunit β1 (KPNB1) in the OC model SKOV3 and OVCAR3 cells, impairing proliferation by targeting several signal pathways, related to cell cycle progression and inducing apoptosis (169, 170). When combined with paclitaxel, these compounds present a synergistic anti-tumor action (171).
Mebendazole is another antiparasitic drug with anti-cancer activity. In several cultured cellular models of OC (MESOV, ES2, A2780, SKOV3 null p53, SKOV3 R248W p53, and SKOV3 R273H p53), mebendazole hindered cell proliferation and activated apoptosis via p53-independent induction of p21 and tubule depolymerization (172). The premise behind exploring these drugs as potential cancer treatments lies in their ability to destabilize microtubules (173–175). Importantly, mebendazole also inhibited cell proliferation and migration in the cisplatin-resistant human OC cell lines OVCAR8CR and OVCAR8 and further induced apoptosis in OVCAR8CR and SKOV3CR cells (176). Mechanistically, this drug modulates essential signaling pathways, such as MYC (Basic Helix-Loop-Helix protein transcription factor)/MAX (MYC Associated Factor X), ELK (ETS (E-twenty six) transcription factor)/SRF (Serum Response Factor), E2F (transcription factor)/DP1 (differentiation regulated transcription factor protein), and nuclear factor kappa B (NF-κB) (176). In addition, in a xenograft murine model of athymic nude mice injected with SKOV3CR cells, mebendazole acted cooperatively with cisplatin to inhibit proliferation, promote apoptosis, and decrease ovarian tumor growth (176). These findings support the possible application of mebendazole in the treatment and maintenance of OC (172), which in combination with cisplatin holds promise for treating chemoresistant OC cases (176).
Peroxisome proliferator-activated receptors (PPARs) have a crucial role in ovarian physiology by regulating the expression and activation of proteases (177–179). PPARγ is regulated by the luteinizing hormone and is highly expressed during ovulation (180). Furthermore, PPARγ(+/−) mice exhibited an approximately 3-fold rise in mammary adenocarcinomas (P<0.05), a more than 3-fold increase in ovarian granulosa cell carcinomas (P<0.05), a greater than 3-fold increase in malignant tumors (P<0.02), and a 4.6-fold elevation in metastatic incidence (181). In mice, PPARγ(+/−) has an increased susceptibility to ovarian carcinogenesis generated by dimethyl benzanthracene (DMBA, 7,12-dimethylbenz[α]anthracene) (181). In a murine model of PPARγ heterozygous female knockout (Pparγ+/−) and congenic wild-type littermate controls (Pparγ+/+), treated with the carcinogen DMBA, at the 25th week from the initiation of the study, KO mice exhibited significantly higher skin papilloma multiplicity (0.87 papillomas/mouse) compared to controls (0.52 papillomas/mouse; P<0.05) (182). By the end of the observation period, ∼41% (18 out of 44) of controls (Pparγ+/+) and ∼61% (24 out of 39) of knockout (Pparγ+/−) mice died or had to be killed due to morbidity resulting from tumor progression (181). Tumors in Pparγ+/− mice were found to be in a more advanced state compared to wild-type controls. Although the total ovarian tumor multiplicity did not differ between the two genotypes, Pparγ+/− mice displayed a significantly higher multiplicity of malignant tumors per mouse compared to wild-type controls when tumors were categorized as benign or malignant. In particular, among the total ovarian tumors, there were 3 carcinomas out of 12 in wild-type mice and 10 carcinomas out of 13 in Pparγ+/− mice, reaching a significance level of P < 0.02 (181). The increased susceptibility of Pparγ+/− mice to DMBA-induced carcinogenesis implies that PPARγ may function as a tumor modifier. Consequently, PPARγ-specific ligands could potentially play a beneficial role in chemo preventing ovarian carcinogenesis (181.
Activating ligands of PPARγ, such as ciglitazone, pioglitazone, and t-butyl [1,1-bis(3′-indolyl)-1-(p-t-butyl)methane (DIM-C-pPhtBu) have been proposed to inhibit OC by impairing proliferation and tumor development and also by triggering apoptosis (183–186). Specifically, ciglitazone inhibits cell proliferation by blocking cell cycle progression and promoting apoptosis (183, 184). In addition, ciglitazone enhanced PAR1-triggered prostaglandin E2 (PGE2) production and cyclooxygenase 2 (COX-2) expression in the normal rat gastric epithelial cell line (RGM1) (187).
Treatment with ciglitazone reduces Cox-2 mRNA expression and PGE2 production, while also decreasing COX-2 promoter activity. Additionally, it upregulates PPRE (putative PPAR response element) promoter activity in human non-small-cell lung cancer cells (A427 and A549) (188). Ciglitazone decreases expression levels of glucose transporter-1 (GLUT-1), inhibits glucose uptake, and increases tumor cell apoptosis in A2780 and OVCAR3 OC cells (189). Additionally, it reduces expressions of specific protein 1 (Sp-1) and β-catenin while increasing phosphorylation levels of adenosine monophosphate (AMP)-activated protein kinase and enhancing chromatin condensation and fragmentations (189). In an in vivo model utilizing eight-week-old female NOD-scid IL2R γ null (NSG) mice injected with A2780 OC cells, ciglitazone significantly decreases OC mass transplanted onto the back of the mice. GLUT-1 expression is increased in high-grade serous ovarian carcinoma, with expression levels proportional to cancer stage severity (189). Mechanistically, DIM-C-pPhtBu induces PPARγ-dependent p21 and reduces PPARγ-independent cyclin D1, resulting in cell cycle arrest, inhibition of cell proliferation, and apoptosis induction (185).
Clofibric acid, commonly used for the treatment of hyperlipidemia, was shown to reduce the growth of OVCAR-3 tumors transplanted subcutaneously and notably prolonged the survival of cancerous peritonitis mouse model with malignant ascites originating from DISS cells compared to the control group (190). Moreover, clofibric acid exhibited dose-dependent suppression of cell proliferation in OVCAR-3 cells. In both implanted OVCAR-3 tumors and cultured OVCAR-3 cells, clofibric acid treatment induced the expression of carbonyl reductase (CR), which promotes the conversion of PGE2 to PGF2α (prostaglandin F2α) (190). Clofibric acid treatment also reduced the levels of PGE2 and VEGF in OVCAR-3 tumors and DISS-derived ascites. Solid OVCAR-3 tumors treated with clofibric acid exhibited reduced microvessel density and increased apoptosis (190).
Disulfiram, a medication used to treat alcohol dependence, has been studied for its potential anticancer effects in OC. Research suggests that disulfiram may inhibit cancer cell growth and metastasis in copper (Cu)-dependent manner due to its ability to bind this ion (191, 192). Approximately 60% of breast cancer patients have elevated levels of Cu in serum (average 3.25 μg/mL) compared with healthy individuals (average 2 μg/mL) (193). The disulfiram-Cu complex was shown to be is a potent inhibitor of proteasomal activity and to trigger apoptosis in the cultured breast cancer cell lines MDA-MB-231 and MCF10DCIS.com, with no effect in non-tumorigenic immortalized MCF-10A cells (192). In mice with MDA-MB-231 tumor xenografts, disulfiram notably suppressed tumor growth by 74%. This effect was attributed to apoptosis induction and proteasome inhibition, which rendered accumulation of ubiquitinated proteins and the natural proteasome substrates p27 and Bax (BCL2 associated X, apoptosis regulator) (192). Disulfiram-Cu complex increases intracellular Cu concentration both in vitro and in vivo, bypassing the requirement for Cu-membrane transporters, such as Ctr1, suggesting that the classical transporter Ctr1 may not play a significant role in disulfiram-mediated Cu accumulation (194, 195). This complex antagonized NFκB signaling, suppressed aldehyde dehydrogenase activity and antioxidant levels, thereby inducing apoptosis mediated by oxidative stress in the inflammatory breast cancer model SUM149, rSUM149 cells (194, 195).
In a murine model, the disulfiram-Cu complex markedly suppressed tumor growth without notable toxicity, inducing apoptosis exclusively in tumor cells. This underscores that inflammatory breast cancer tumors are highly redox-adapted, potentially conferring resistance to ROS-inducing therapies (194, 195). Hence, the redox modulation capabilities of disulfiram represents a promising avenue for treating tumors enhancing the efficacy of traditional therapies (192). As such, in cultured OC models, disulfiram promoted oxidative stress through an Cu-dependent mechanism, resulting in death of OVCAR-3, SKOV-3, OVMZ-30, OVMZ-31, OVMZ-37, and OVMZ-38 cells (196). It has been reported that the ditiocarb-Cu complex, a metabolite of disulfiram, is responsible for the anti-cancer effects. Additionally, functional, and biophysical analyses identified NPL4 (nuclear protein localization protein 4 homolog) as the molecular target underlying the tumor-suppressing effects of disulfiram (191). NPL4 acts as an adaptor of p97, also known as ATPase valosin-containing protein (VCP) segregase, which is crucial for protein turnover in various regulatory and stress-response pathways within cells (191).
Disulfiram, combined with Cu, enhances cisplatin-induced apoptosis in IGROV1, SKOV3, and SKOV3IP1 cells, sensitizing cancer cells to cisplatin treatment and decreasing cell viability by 50-80%% (197). This combination targets acetaldehyde dehydrogenase (ALDH)+ cells, favoring cisplatin sensitivity in H460/CisR, H1299/CisR, and SKMES-1/CisR cells (198). Additionally, disulfiram and Cu supplementation reduces NF-κB activity and sensitizes H630WT and HCT116WT cell lines to gemcitabine (199). Disulfiram reverses doxorubicin (DOX) resistance by increasing c-Jun NH2-terminal kinase (JNK) expression and phosphorylation in HL60 cells (200). Effective cell death in OVCAR3 and SKOV3 cells is mediated by disulfiram, promoting an oxidative intracellular environment and causing irreversible cell damage associated with the expression of heat shock proteins HSP32, HSP40, and HSP70 (196). Furthermore, the combination of disulfiram and Cu, induces disulfide bond-mediated dimerization of HSP27, resulting in its inactivation and rapid detachment of OVCAR-3 cells, an effect not detected with disulfiram alone (196). Combinatory treatment of disulfiram with auranofin, an anti-rheumatic drug, enhances cytotoxic effects in OVCAR3 cells (188).
Fluphenazine is an antipsychotic drug that exerts its effects on the postsynaptic dopaminergic D1 and D2 receptors by inhibiting the release of dopamine. In OC OVCAR-3 cells, fluphenazine plays an essential role in the phosphorylation of AKT dependent on epidermal growth factor (EGF) (201, 202). Moreover, fluphenazine targets pyruvate dehydrogenase kinase 1 (PDK1), which is part of the PDK1/Akt pathway mediating cell survival, proliferation and tumorigenesis (201). Thus, the inhibition of PDK1/Akt kinase pathway suppressed the EFG-dependent proliferation phenotype and survival of cancer OVCAR-3 cells by inducing apoptosis (201). The proposed mechanism of action for fluphenazine is related to an enhancement of genomic DNA-oligonucleosomal cleavage, and to the activity of the caspase substrate polyadenosine diphosphatase ribose. These pathways trigger caspase-dependent apoptotic cell death (201).
Metformin, a frequently prescribed medication for type 2 diabetes mellitus, reduces proliferation of SKOV3ip1, OVCAR-5, HeyA8 and K-ras/PTEN cells. The K-ras/PTEN mouse OvCa cell line was established from ovarian tumors generated using a genetic mouse model (203, 204)). Mechanistically, metformin causes cell cycle arrest in G0/G1 phase by decreasing the expression of cyclin-dependent kinase 4 (CDK4) and Cyclin D1, with no evidence of triggering apoptosis (204). Metformin treatment results in reduced number and mass of ovarian tumors. For instance, female athymic nude mice pretreated with metformin (250 mg/kg/d) exhibited significantly fewer ovarian tumor implants compared to controls (mean number of tumors: placebo, 116; metformin, 47; P<.005) (204). In SKOV3ip1 xenograft mice, treatment with metformin in combination with paclitaxel resulted in a 60% reduction in tumor weight compared to controls (P=.02) (204). This combination demonstrated a stronger effect than each compound tested separately (204). Metabolically, metformin modifies adenosine monophosphate-activated protein kinase (AMPK) activity, lipid synthesis, and glycolysis. Notably, metformin induces apoptosis in OC cell lines in an AMPK-dependent manner (205–207).
Furthermore, a study of OC patients where a cohort of individuals was treated with metformin resulted in an increased survival rate (67%) of individuals treated with metformin compared to the non-treated group (47%) (208). Patients who consumed this drug exhibited a markedly enhanced 5-year survival rate (51%) compared to those who did not use metformin (8%) or those without diabetes (23%) (209). In combined metformin/cisplatin treatment, increasing metformin concentrations led to a notable reduction in the half-maximal inhibitory concentration of cisplatin (209). Consequently, research in ovarian cancer patients, alongside in vivo and in vitro models, highlights the inhibitory effect of metformin on tumor growth, its ability to enhance chemotherapy sensitivity, and its potential to prolong the life expectancy of affected individuals.
Naftopidil is an α1-adrenergic receptor antagonist that is primarily used for the treatment of benign prostatic hyperplasia (BPH), a condition characterized by an enlarged prostate gland in men (210–212). By blocking the α-1 adrenergic receptors in the prostate, naftopidil helps to relax the smooth muscles in the prostate and bladder neck, relieving symptoms of BPH and improving urine flow (213). In studies using cellular models of OC, naftopidil inhibited proliferation without eliciting apoptosis, leading to a cytostatic effect observed in SKOV-3 and IGROV1-R10 cell lines (214). Furthermore, this medication enhances the production of proapoptotic BH3-only proteins, namely Bim (BCL2-like 11, member of the Bcl-2 family that promotes apoptosis), Noxa (phorbol-12-myristate-13-acetate-induced protein 1), and Puma (BCL2 binding component 3). Two different mechanisms have been identified for naftopidil in OC-cultured models. For instance, in SKOV3 cells, an ER stress-induced response by the activating transcription factor 4 (ATF4), which is responsible for the phenotype, while in the IGROV1-R10 cell line, the JNK pathway is the leading pathway (214). Considering the mechanisms by which naftopidil induces the expression of Puma by the JNK/c-Jun pathway, resulting in a new alternative to OC management (214).
Nelfinavir is a protease inhibitor primarily used in the treatment of HIV (human immunodeficiency virus). Experiments in HGSOC cells showed that treatment with this drug reduces the cell number, clonogenic survival and viability (215). Additionally, nelfinavir favors a pro-apoptotic environment characterized by elevated levels of phospho-eIF2α (Eukaryotic Translation Initiation Factor 2A), DNA Damage Inducible Transcript 3 (DDIT3, also known as CHOP), and ATF4, as well as an increased ratio of Bax/Bcl-2 and cleaving of the executor caspase 7 (215). Nelfinavir triggered a dose-dependent reduction in the HGSOC cell number and viability and a parallel increase in hypo-diploid DNA content, independently of platinum sensitivity (215). DNA damage induced by nelfinavir was detected by the phosphorylation of the histone marker, H2AX (H2A.X variant histone) in PEO1 and PEO4 cell lines, in a process linked to reduced proliferation and survival mediated by the ERK and AKT pathways (215). In the PEO1 and PEO4 cellular models, a synergistic effect of nelfinavir with the protease inhibitor, bortezomib, enhanced the ability to induce short-term cell cycle arrest and long-term toxicity (215). So far, bortezomib has been used in the treatment of multiple myeloma and mantle cell lymphoma, but similarly to nelfinavir possess the potential as a treatment against OC.
Ritonavir is another protease inhibitor, largely used in the treatment of HIV, in combination with other antiretroviral medications to slow the progression of the disease. Ritonavir inhibits the activity of the HIV protease enzyme, which is necessary for the virus to replicate and produce new infectious viral particles (216). By inhibiting protease, Ritonavir helps reduce the viral load in the body. Additionally, ritonavir is often used as a “booster” medication, as it increases the levels of other protease inhibitors in the blood, enhancing their effectiveness. This boosting effect allows for lower doses of other protease inhibitors when used in combination with nucleoside analog reverse transcriptase inhibitors (NRTI), resulting in highly effective antiretroviral therapy. In the context of OC repurposing, ritonavir was shown to prevent cell cycle progression in MDH-2774 and SKOV-3 cultured models (217). Furthermore, in MDH-2774 and SKOV-3 cell lines, this drug promoted apoptosis and cell cycle arrest in G1 phase by depleting the phosphorylation of retinoblastoma (RB), and by reducing the expression of G1 cyclin and cyclin-dependent kinase (217). In MDAH 2774 and SKOV-3 cells, ritonavir also increased levels of phosphorylated AKT, thus inhibiting the PI3K-AKT pathway, which resulted in an antitumor effect that led to apoptosis (217–219). In xenograft models, nude mice injected with human ovarian adenocarcinoma A2780 cells and treated with ritonavir exhibited reduced tumor burden compared to untreated controls. Additionally, ritonavir-treated mice showed larger areas of necrosis and increased activated caspase-3 staining, indicating induction of apoptosis in the tumor cells (219).
Non-steroidal anti-inflammatory drugs (NSAIDs) are a class of medications commonly used to reduce inflammation, relieve pain, and lower fever. NSAIDs inhibit the production of prostaglandins by blocking cyclooxygenases (COX), which play a role in inflammation and pain (220). Thus, NSAIDs have anti-inflammatory and analgesic (pain-relieving) properties, often used to manage conditions characterized by inflammation, such as arthritis, osteoarthritis, rheumatoid arthritis, and to alleviate systemic pain in cases of menstrual cramps, headaches, muscle aches, and minor injuries (221–223).
Examples of NSAIDs include ibuprofen (e.g., Advil, Motrin), naproxen (e.g., Aleve), aspirin, diclofenac, and meloxicam. Among these, aspirin has been investigated for its potential to reduce the risk of ovarian cancer development and progression. Aspirin inhibits NF-κB, COX, and the PI3K/mTOR signaling pathway, concurrently activating AMPK (224). Some studies suggest that regular aspirin use may be associated with a reduced risk of OC incidence and mortality (225). Aspirin was shown to inhibit the proliferation of OCT2 and OVCAR-3 cells and to reduce PPARδ function by inhibiting ERK1/2 (226). Therefore, NSAIDs show promise as therapeutic treatments against OC; however, dosage seems to be a key feature requiring further investigation (158).
Cancer chemotherapeutic treatment often results in a significant upregulation of transmembrane efflux pumps, which contribute to the development of multiple drug resistance, a major impediment in effective cancer treatment. Highly resistant tumors might be eradicated using chemosensitizers that block the efflux of the drug and increase the entry of the drug into the cell (227). In this regard, sertraline is a selective serotonin reuptake inhibitor, commonly prescribed for the treatment of various mental health conditions, particularly depression, anxiety disorders, obsessive-compulsive disorder, and panic disorder (228). Sertraline increases the levels of serotonin, a neurotransmitter in the brain, which is believed to have a positive impact on mood and emotional well-being (229). For instance, P-glycoprotein (P-gp) is a transmembrane efflux pump that actively transports and eliminates drugs and other chemical compounds from cells. This protective function prevents the buildup of potentially harmful substances within cells, negatively impacting the therapeutic effectiveness of the drugs. Thus, P-gp is linked to multidrug resistance observed in cancer cells that develop resistance to multiple chemotherapeutic drugs (230). The significant implications for pharmacokinetics, where P-gp influences the absorption, distribution, and elimination of drugs, lead to altered bioavailability and distribution patterns for drugs that are substrates for P-gp (231). Certain drugs can either inhibit or induce P-gp activity, affecting the cellular concentrations of various substrate drugs. Ongoing research focuses on P-gp in drug development to enhance drug efficacy and address multidrug resistance, with efforts directed at designing drugs that can bypass or inhibit P-gp when necessary. This knowledge is essential for healthcare professionals and researchers navigating drug interactions and optimizing therapeutic outcomes. P-gp pumps are expressed and functional in the chemoresistant ovarian adenocarcinoma cell line OVCAR-8 and in the derived drug-resistant models (human ovarian adenocarcinoma cell line NCI/ADR-Res (NAR) cells) (227). Among these, sertraline has been shown to enhance the cytotoxicity of DOX and reduce DOX efflux in NAR cells (227). Studies conducted in human ovarian adenocarcinoma xenograft models demonstrated that combining sertraline with DOXIL® (pegylated liposomal DOX) effectively reverses multiple drug resistance (MDR). Sertraline acts as a chemosensitizer by blocking extrusion pumps, thereby allowing the drug delivered via the nanomedicine to accumulate inside the cell (227). Hence, the combined therapy of nanomedicine with chemosensitizers like sertraline is poised to amplify therapeutic responses in highly resistant tumors. This approach increases drug influx through nanomedicine while reducing drug efflux by employing a chemosensitizer (227). Moreover, findings from a xenograft murine model revealed that combining sertraline with DOX significantly enhances cytotoxicity, delaying tumor growth and improving survival rates by 1.5-fold (227). This combined treatment holds promise in mitigating multiple drug resistance phenotypes attributed to P-gp pumps, such as Multidrug Resistance 1 (MDR1, also known as ABCB1), which are ATP-dependent efflux pumps of the ABC protein superfamily (227, 232).
Thalidomide was initially marketed as a sedative-hypnotic drug with anti-emetic activity against morning sickness of early pregnancy, but was withdrawn from the market in the early sixties as it was found to cause severe fetal malformations (233–235). It is a medication with immunomodulatory and antiangiogenic properties that has been investigated for its potential to inhibit tumor growth and angiogenesis in ovarian cancer. Studies suggest that thalidomide may exert anticancer effects by modulating immune responses and disrupting tumor microenvironment interactions (10). Thalidomide inhibits TNF-α production in lipopolysaccharide-stimulated monocytes (236). Thalidomide decreased the capacity of SKOV-3 cells and primary epithelial ovarian carcinoma cells to secrete TNF-α, but this drug did not significantly affect the proliferation and growth of SKOV-3 cells (237). Thalidomide notably decreased the capacity of SKOV-3 cells to secrete MMP-9 and MMP-2, yet it did not have the same effect on primary epithelial ovarian carcinoma cells. However, thalidomide did not affect the secretion of IL-6 in either SKOV-3 cells or primary epithelial ovarian carcinoma cells (237). Thalidomide inhibits the processing of the TNF-α and the angiogenic factor VEGF transcripts (238). Sixty-six patients, comprising 37 women and 29 men, with advanced cancer (19 ovarian, 18 renal, 17 melanoma, 12 breast cancer) received daily treatments ofthalidomide at a dose of 100 mg. Out of the 18 patients with renal cancer, three showed partial responses, and an additional three patients experienced disease stabilization for up to 6 months. Although no conclusive responses were observed in patients, there was an improvement in the sleep quality (P<0.05) and preserved appetite (P<0.05) in these individuals (239). Women (138) diagnosed with biochemical-recurrent epithelial OC, primary peritoneal cancer, or fallopian tube carcinoma were eligible for a randomized phase III trial of tamoxifen versus thalidomide (240). Results suggested that thalidomide treatment was associated with a similar risk of progression (HR=1.31, 95% confidence interval [CI]=0.93–1.85), an increased risk of death (HR=1.76, 95% CI=1.16–2.68) and more grades 3 and 4 toxicities (55% versus 3%) in comparison with tamoxifen treatment (240). Therefore, thalidomide was not more effective than tamoxifen in delaying recurrence or death but was more toxic (240).
Repurposed kinase inhibitors
Several kinase inhibitors, originally developed for different pathologies, have been investigated for their potential to target specific signaling pathways implicated in OC. Examples include dasatinib, a Src kinase inhibitor, and imatinib, a BCR-ABL tyrosine kinase inhibitor, which have shown promise in preclinical studies of ovarian cancer (241).
Dasatinib is an inhibitor of Src/Abl family kinases used for the treatment of Philadelphia chromosome-positive acute lymphoblastic leukemia or chronic myeloid leukemia (242). Dasatinib inhibited cell growth by partially inducing apoptosis with a significant effect in autophagy activation in the SKOV3 and Hey cell lines (243). Dasatinib reduced the phosphorylation of AKT, mTOR, p70S6K, and S6 kinase expression and reduced Bcl-2 expression and activity. Dasatinib induces autophagy in Hey and SKOV3 cells that partially depends on beclin 1, AKT and Bcl-2. Overexpression of Bcl-2 partially prevented dasatinib-induced autophagy. In a Hey xenograft model, dasatinib inhibited tumor growth and induced both autophagy and apoptosis (243). Elevated levels of p-Src (phosphorylated Src family tyrosine kinases) protein expression were detected in A2780, HO8910, OVCAR3, CAOV3, and COC1 cell lines compared to healthy cells. This observation suggests activation of the Src signaling pathway (244). Combining dasatinib and paclitaxel significantly inhibited proliferation and boosted apoptosis in A2780 and HO8910 cells compared to controls. This combination showed tumor growth inhibitory rates of 76.7% and 58.5% in A2780 and HO8910 cell lines, respectively, outperforming paclitaxel treatment alone (244). In A2780 and HO8910 xenografts models, dasatinib treatment inhibited tumor growth by 43.2% and 34.0%, respectively (244). Paclitaxel treatment increased Src activation in Hey OC cells, inducing the expression of EpCAM (epithelial cell adhesion molecule) marker expression in Hey cells, while upregulated the expression of SSEA-4 (stage-specific embryonic antigen-4) and CD133 (prominin 1) markers (245). In this sense, dasatinib combined with paclitaxel significantly suppressed p-Src in Hey cells and xenografts but had no effect on the expression of these markers (245). However, this combination did not enhance the proliferative, tumorigenic, and vasculogenic of paclitaxel alone in HEY cell-induced ovarian tumors (245). Importantly, administration of dasatinib and paclitaxel in murine models reduced the invasion of cancer cells into the pancreas and liver, major organs affected by ovarian tumor metastasis. Thus, the evidence points to a significant potential of dasatinib in targeting intra-peritoneal dissemination of OC (245).
Imatinib inhibits the proliferation of several OC cell lines (C272-hTert/E7, C889/hTert, CSOC848, CSOC908, and CSOC918) that expressed elevated levels of PDGFRα (platelet-derived growth factor receptor α) (246). SKOV3 and CAOV3 cells do not express PDGFRα are insensitive to the effects of imatinib, suggesting that the inhibition of cell proliferation by imatinib is in a PDGFRα-specific manner. Imatinib induces antiproliferative effects by arresting cell progression at G0-G1 and impeding advancement through the S phase. Additionally, at a concentration of 1 μm, Imatinib inhibits both PDGFRα and Akt phosphorylation (246). However administration of imatinib to patients with epithelial OC, had minimal effect as a single treatment (247). A phase II trial of imatinib administered to patients with platinum-resistant OC, showed that imatinib mesylate, when used alone, lacks significant clinical efficacy in c-Kit and/or PDGFR positive, recurrent OC, particularly in heavily pretreated patients (248). Thus, imatinib may be considered as a supplementary drug to be used in combination with other treatments.
Hormonal therapy is an emerging treatment that utilizes hormones or hormone-blocking agents to interfere with the growth and progression of OC cells. While hormonal therapy is not a standard treatment for most OC, it may be considered in specific cases where the cancer cells express hormone receptors, such as ER and PR. This strategy can be particularly useful for endometrioid OC and some ovarian stromal tumors that may express these hormone receptors. Among the drugs utilized in hormonal therapy is tamoxifen, a Selective ER Modulator (SERM) commonly used in breast cancer treatment and has been investigated in some cases of OC with hormone receptor expression (249, 250). Aromatase inhibitors, such as letrozole and anastrozole, which prevent the synthesis of estrogen, are mainly used in the treatment of breast cancer and oftentimes as fertility treatments, are sometimes used in ER+ OC cases (251–255). In a phase II trial involving 50 patients with relapsed ovarian cancer, the antitumor activity of letrozole was assessed using Union International Contre Cancer (UICC) and CA125 (cancer antigen 125) marker criteria. Tumors categorized as stable disease by UICC criteria showed significantly higher ER (P=0.027) and PR (P=0.0066) values compared to those categorized as progressive disease (251). The combined presence of these receptors strongly correlated with stable disease (P<0.0001). Similarly, according to CA125 criteria, tumors with higher ER (P=0.013), lower erbB2 (P=0.026), and higher epidermal growth factor receptor (P=0.009) levels were associated with CA125 stable/responsive disease compared to progressive disease (251). In another phase II trial, letrozole was administered at a daily dosage of 2.5 mg until either clinical or marker evidence indicated disease progression. This trial focused on ER-positive OC patients with rising CA125 levels, indicative of progression according to Rustin’s criteria (252). Among the 42 patients assessed for CA125 response, 7 (17%) showed a response, defined as a decrease of more than 50%, while 11 (26%) patients did not experience progression, indicated by a doubling of CA125 levels, after 6 months of treatment (252). Of the 33 patients evaluable for radiological response, 3 (9%) had a partial remission, and 14 (42%) had stable disease at 12 weeks (252). Subgroup analysis based on ER status showed CA125 response rates of 0% (immunoscore of 150-199), 12% (immunoscore of 200-249), and 33% (immunoscore of 250-300), with a significant trend observed (P = 0.028, χ2 for trend). Additionally, expression levels of HER2, insulin-like growth factor binding protein 5 (IGFBP5), trefoil factor 1 (TFF1), and vimentin correlated with changes in CA125 levels during treatment (252). Finally, a 2.5 mg daily oral dose of letrozole was administrated to thirty-three women with recurrent ER+ epithelial ovary or peritoneum carcinoma enrolled in a phase II trial (253). Among these patients, 26% of the individuals diagnosed with ER-+, platinum- and taxane-resistant high-grade ovarian and primary peritoneal cancer who received letrozole treatment experienced a clinical benefit, defined as either stabilization of disease or partial response (3% of patients) (253).
Ormeloxifene is a SERM primarily used as an oral contraceptive and for the treatment of conditions related to the female reproductive system. It inhibits the action of estrogen on the uterus, leading to changes in the cervical mucus chemistry and endometrium. These physiological changes create a challenging environment for the sperm to reach the egg and for a fertilized egg to successfully implant in the uterus. In the context of OC repurposing, in vitro experiments showed that ormeloxifene hindered cell proliferation and triggered apoptosis in cisplatin-resistant in the A2780, A2780-CP and SKOV-3 cell lines (256). At the molecular level, ormeloxifene reduced AKT phosphorylation, enhanced p53 phosphorylation, and altered the synthesis and localization patterns of cyclin D1, cyclin E, p27, and CDK2 (256). In xenograft murine models, injecting 50 or 100 µg ormeloxifene once a week for 5 weeks reduced tumorigenesis and metastasis within the peritoneal cavity (256). Within 2 weeks of A2780-CP cell injection, all mice treated with vehicle displayed a swollen abdomen, indicative of ascites formation, along with significant peritoneal carcinomatosis and numerous solid tumors (256). Conversely, mice treated with 100 µg of ormeloxifene showed no detectable tumors (256). This suggests that ormeloxifene holds promise as a compound for OC treatment. Despite the potential benefits, hormonal therapy is not widely used in OC yet and is only considered when other standard treatments are not effective and in specific cases of OC patients expressing the hormone receptors. It is noteworthy that while hormonal therapy has potential in OC treatment, standard treatments such as surgery and chemotherapy remain the mainstay of ovarian cancer management. As clinical research and trials progress, this treatment option may become an efficient alternative to OC care.
Model-informed drug repurposing
Model-informed drug repurposing (MIDR) might be used to accelerate the repositioning of drugs (257). MIDR is a rapidly expanding in silico approach to drug discovery and development that involves mathematical models, computational tools, and data-driven techniques to identify new therapeutic uses for existing drugs (257). The development of powerful computational methods, such as bioinformatics, systems biology, and quantitative pharmacology modeling, and the combination of these techniques, allow the analysis of large datasets to identify potential connections between drugs and diseases. This is further achieved by consolidating diverse data sources, such as genomic, transcriptomic, proteomic, and clinical records. In addition, network pharmacology, as well as pharmacokinetic and pharmacodynamic modeling, further refine the understanding of complex interactions among drugs, targets, and diseases. Ultimately, these approaches are now being integrated into machine learning algorithms and artificial intelligence pipelines to combine complex datasets and efficiently predict drug-disease relationships. These in silico models can also help identify synergistic effects of drug combinations for improved therapeutic outcomes. This powerful experimental approach is now being utilized in the treatment of cancer.
As an example, a recent report that involved a literature search coupled to in silico analyses and screening process involving preclinical research, explored the testing of approved compounds for human use in treating OC (258). The combination of these approaches rendered four compounds used regularly in the clinic, metformin, celecoxib, lurbinectedin, and 5-azacytidine, as drugs with significant potential for repurposing in the context of myeloid-derived suppressor cells (MDSC) within the OC tumor microenvironment (258). MDSC suppresses the immune response in OC through several mechanisms; therefore, finding potential drug candidates for repositioning has been a challenging process (258). As example is the emerging evidence for lurbinectedin, a synthetic compound derived from the marine organism Ecteinascidia turbinate. lurbinectedin is an alkylating anticancer drug. It targets specific DNA repair mechanisms in cancer cells, leading to DNA damage and cell death (259). Lurbinectedin decreases myeloid-derived suppressor cell (MDSC) percentages in vitro in chronic lymphocytic leukemia (CLL) patient-derived studies (260). It induces cell death in a dose and time-dependent manner and reduces the expression of chemokine receptor CCR7 implicated in B-CLL cell migration (260). Notably, malignant B cells from patients with clinical lymph node involvement exhibit higher trans-endothelial cell migration (TEM) in response to CCL21 and CCL19 compared to those without such organomegaly (260). There is a correlation between CCR7 expression, receptor for both CCL21 and CCL19, and clinical lymphadenopathy, and blocking CCR7 suppresses TEM of CLL cells (260). Lurbinectedin has shown promise in treating various solid tumors, including small-cell lung cancer and relapsed OC (261). This drug received accelerated approval from the U.S. FDA for metastatic small-cell lung cancer that has progressed after platinum-based chemotherapy (262). In vitro studies revealed significant antitumor effects of lurbinectedin on both chemosensitive and chemoresistant clear cell carcinoma (CCC) of the ovary cells (RMG1, RMG2, KOC7C, and HAC2) (263). Evaluation of mouse CCC cell xenografts confirmed that lurbinectedin effectively suppressed tumor growth. Notably, combining lurbinectedin with SN-38 (7-ethyl-10-hydroxycamptothecin) demonstrated a significant synergistic effect, particularly evident in both cisplatin-resistant and paclitaxel-resistant CCC cell lines. These findings indicate potent antitumor activity of lurbinectedin in both cisplatin-sensitive and cisplatin-resistant OC (263). Furthermore, lurbinectedin is under investigation in clinical trials for potential efficacy in other cancer types, including advanced ovarian, endometrial, and breast cancers, relapsed hematological malignancies like acute myeloid leukemia (AML) and lymphomas, and soft tissue sarcomas including liposarcoma and leiomyosarcoma (261, 264, 265). Combination therapies involving lurbinectedin are being assessed, including with immune checkpoint inhibitors, PARP inhibitors, and other targeted therapies, to explore potential synergistic effects.
Celecoxib, a selective COX-2 inhibitor used to treat pain and inflammation, has been investigated for its potential to inhibit OC growth and metastasis. Evidence suggested that celecoxib may exert anticancer effects in OC cells by inhibiting COX-2-mediated signaling pathways involved in tumor progression (266). In areas of active tumor growth in a murine model for mesothelioma, large numbers of MDSCs co-localize with COX-2 expression (267). Celecoxib effectively reduced PGE2 levels both in vitro (mesothelioma AB1 cell line) and in vivo (BALB/c mice) (267). Furthermore, celecoxib treatment decreased levels of ROS in immature myeloid subtypes (MO-MDSC, PMN-MDSC, and Gr-1lowSubset 2) from the spleen of tumor-bearing mice and improved cytotoxic T cell function (267). Ten days after injection with a lethal dose of 0.5×106 AB1 tumor cells, the absolute number of MDSCs was significantly lower in mice receiving the celecoxib diet compared with mice receiving the control diet. This difference was more pronounced at day 22 after tumor injection, and mice receiving the celecoxib diet did not exhibit any discernible side effects (267). These findings highlight the potential of celecoxib as an adjunctive therapy in cancer treatment strategies.
Finally, 5-azacytidine is a nucleoside analog that is incorporated into DNA and RNA and inhibits DNA methyltransferase enzymes leading to subsequent DNA hypomethylation. It is primarily used as a demethylating agent in the treatment of certain hematological malignancies, particularly myelodysplastic syndromes, where it can help to restore normal hematopoiesis by reversing aberrant DNA methylation patterns (268–270). The inhibition of methylation via 5-azacytidine increases the formation of invadopodia and enhances the extracellular matrix degradation in SKOV3 and A2780 cells and further promotes cell migration and invasion of SKOV3 cells (271). Moreover, in SKOV3 cells, 5-azacytidine induce the expression of genes and proteins involved in actin-regulating signaling pathways [PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha), SRC (SRC proto-oncogene, non-receptor tyrosine kinase), RhoC (ras homolog family member C), RhoA (ras homolog family member A), RAC1 (Rac family small GTPase 1), and AFAP (actin filament associated protein 1) (271). Furthermore, the 5-azacytidine increased the phosphorylation of AKT and p110 alpha (PI3-kinase isoform), suggesting that the PI3K-AKT pathway is activated in SKOV3 and A2780 cells (271). In mouse xenograft models, 5-azacytidine treatment suppressed tumor growth and increased the occurrence of metastatic nodules, indicating an enhanced metastatic potential due to DNA demethylation (271). Methylation inhibition led to increased transcription of PIK3CA and upregulation of genes associated with the PI3K-AKT signaling pathway (271). This induction likely occurs through epigenetic regulation of PIK3CA, as analysis of DNA methylation levels in the PIK3CA promoter region indicated decreased methylation of CpG islands in SKOV3 and A2780 cells following 5- azacytidine treatment (271). The impact of trichostatin A (TSA) and 5-azacytidine (5-aza-2′-deoxycytidine), either alone or in conjunction with low-dose cisplatin, was assessed on Hey, SKOV3 and A2780 lines in vitro (272). Combined treatment exhibited superior efficacy compared to individual drugs and notably suppressed cell viability, migration, and spheroid formation and growth in Hey, SKOV3 and A2780 cells (272). Sequential administration of cisplatin (1 mg kg−1) followed by TSA (0.3 mg kg−1) significantly suppressed the tumorigenicity of Hey xenografts by inhibiting the expression of epithelial to mesenchymal transition (EMT) markers (Twist, Snail, Slug, E-cadherin, and N-cadherin), and reducing the pluripotency of ovarian cancer cells (272). Finally, a clinical trial in patients with platinum-resistant OC showed the effect of oral 5-azacytidine in combination with pembrolizumab (NCT02900560). Pembrolizumab is a monoclonal antibody medication used as immune checkpoint inhibitor for immunotherapy of various types of cancer (273). This study helped to establish an optimal dosing schedule for oral azacitidine in combination with pembrolizumab for platinum-resistant/refractory OC patients (274). Additional preclinical studies using 5-azacytidine alone and in combination with a small molecule histone deacetylase (HDAC) inhibitor, entinostat, showed high potential in OC treatment as well (258).
Despite the enticing results from the in silico analyses, it is important to note that it is of the essence to perform subsequent in vitro and in vivo validations prior to clinical applications. However, computational approaches can provide information and further hypotheses for model-informed drug repurposing. As summarized in this review, Figure 5 provides a schematic representation of central metabolic pathways associated with the anti-tumor activities of repurposed drugs. This overview encompasses antiparasitic drugs, antiretrovirals, antibiotics, hypocholesterolemia treatments, and other drugs and metabolites.
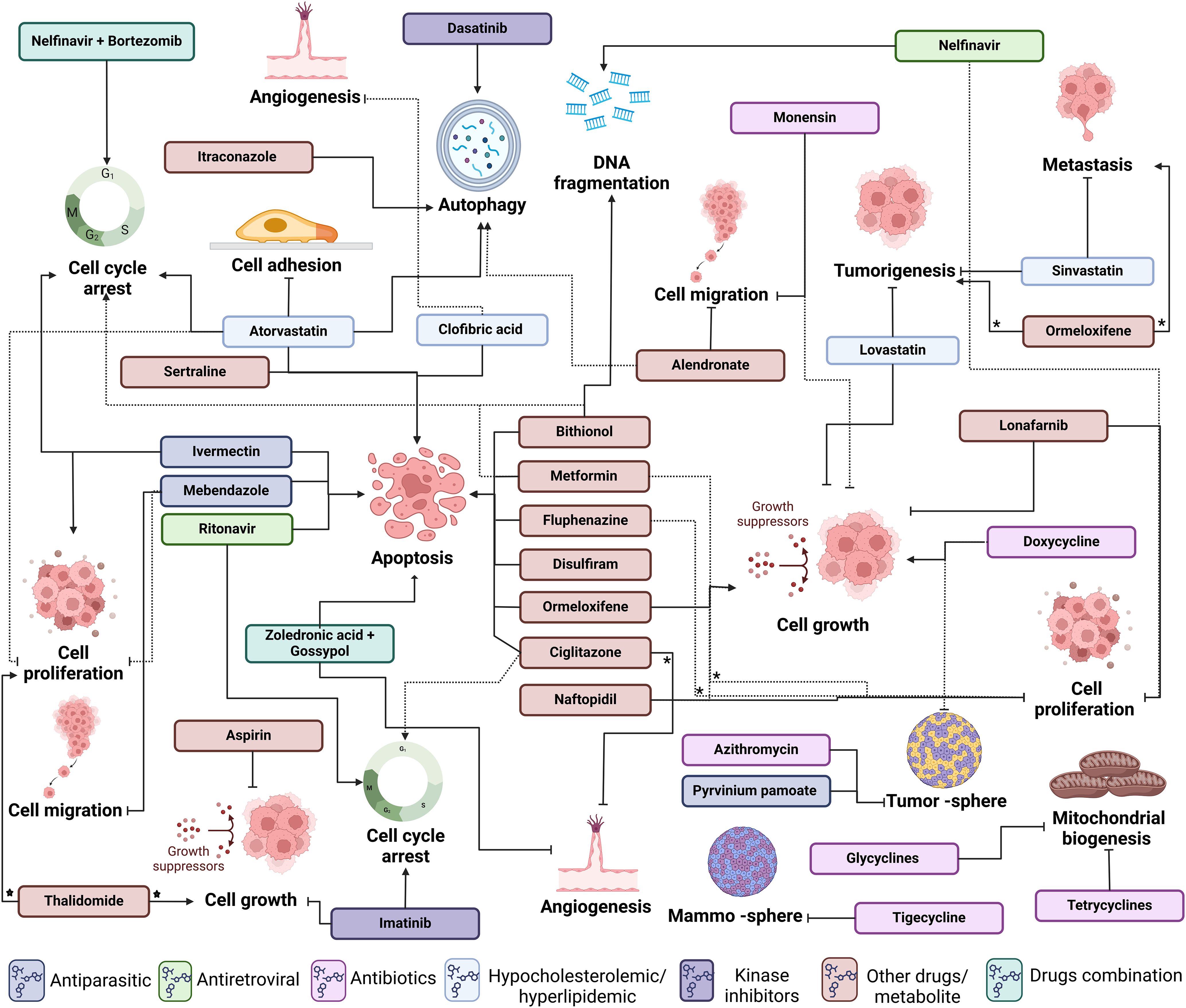
Figure 5. Cancer hallmarks involved in the drug reposition for ovarian cancer treatment. Schematic representation of the main metabolic pathways related to anti-tumor activities of repositioned drugs. The summary includes antiparasitic drugs (dark blue boxes), antiretroviral (light green boxes), antibiotics (grape boxes), compounds to treat hypocholesterolemia (light blue boxes), and other drugs and metabolites (red boxes). Compounds used in combinatorial treatments are also represented (dark green boxes). Induction (→), reduction (*), or inhibition (┤) of the cancer hallmarks is indicated within the figure (Figure created with Biorender).
Conclusion
This review provides valuable insights into the utility of drug repositioning for OC treatment, highlighting the importance of cell line and animal models in initial screening and drug testing. Although clinical translation remains in its early stages, these models form a crucial foundation for future studies. Overcoming the challenges of translating preclinical findings into successful human therapies will require careful consideration of drug interactions, personalized medicine approaches, and extensive clinical validation. We provided a critical discussion of the potential of drug repositioning for OC treatment, highlighting current advances in the area, which does not represent a clinical investigation. Our analysis focused on in vitro experiments across multiple cancer cell lines and short-term in vivo models, and the limited information available from clinical trials. The discussion revealed encouraging evidence of the efficacy of existing drugs in targeting OC. Despite these promising findings, we recognize the limitations inherent in in vitro and preclinical models. These systems, while informative, fail to capture the full complexity, heterogeneity, and microenvironment of OC in human patients, which limits the direct translation of findings to clinical practice. Key challenges include differences in pharmacokinetics and pharmacodynamics between preclinical models and human systems, which may influence therapeutic efficacy and safety profiles. Furthermore, in vivo and in vitro studies do not account for the complex pharmacological interactions that occur in real-world scenarios, such as those between drugs, dietary supplements, and food, which could either amplify or attenuate therapeutic effects. Careful optimization of dosing regimens is crucial to balance efficacy and minimize adverse outcomes. Looking forward, systematic approaches that integrate high-throughput screening, computational modeling, and patient-derived models offer a path to refining drug combinations and tailoring therapies to individual patients. These methodologies, combined with biomarker-driven approaches, enable the identification of molecular pathways most relevant to OC progression and therapeutic response. Leveraging genomics and proteomics can further clarify the pharmacokinetics and pharmacodynamics of repositioned drugs, supporting their clinical application.
Drug repositioning is particularly advantageous in oncology due to its ability to shorten the timeline for bringing effective therapies to patients, compared to the 10 - 15 years typically required for de novo drug development, as evidenced above. Despite the potential usage of a wide range of available drugs, the clinical validation of these therapies requires robust longitudinal studies and clinical trials to assess safety, efficacy, and optimal combination strategies in the context of OC. Ultimately, drug repositioning holds significant promise for overcoming challenges in OC treatment, particularly drug resistance. By addressing pharmacokinetic challenges, optimizing dosing strategies, and incorporating personalized medicine principles, repositioned drugs can offer cost-effective and innovative solutions. Nonetheless, clinical trials remain indispensable to substantiating preclinical findings and ensuring that repositioned therapies fulfill their potential in improving outcomes for OC patients. This review point to the critical role of multidisciplinary approaches in advancing the utility of drug repositioning for OC, with the ultimate goal of enhancing patient survival and quality of life.
Author contributions
EV-V: Data curation, Investigation, Writing – original draft. FM-C: Data curation, Investigation, Writing – original draft. OR-H: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing. AB-G: Data curation, Writing – original draft. LB-M: Data curation, Investigation, Software, Writing – original draft. TP-B: Data curation, Funding acquisition, Investigation, Writing – review & editing. LQ-G: Investigation, Writing – original draft, Writing – review & editing. GF-G: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by PAPIIT (projects IA208422 and IA206724 awarded to GF-G and projects IN222321 and IN221824 awarded to OR-H). EV-V is a recipient of a postdoctoral fellowship from Dirección General de Asuntos del Personal Académico (DGAPA) from Universidad Autónoma de Mexico (UNAM). TP-B and AB-G were supported by Wesleyan University institutional funds.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Banno K, Iida M, Yanokura M, Irie H, Masuda K, Kobayashi Y, et al. Drug repositioning for gynecologic tumors: a new therapeutic strategy for cancer. Sci World J. (2015) 2015:1–10. doi: 10.1155/tswj.v2015.1
2. Power A, Berger AC, Ginsburg GS. Genomics-enabled drug repositioning and repurposing: insights from an IOM Roundtable activity. Jama. (2014) 311:2063–4. doi: 10.1001/jama.2014.3002
3. Hernandez L, Kim MK, Lyle LT, Bunch KP, House CD, Ning F, et al. Characterization of ovarian cancer cell lines as in vivo models for preclinical studies. Gynecol Oncol. (2016) 142:332–40. doi: 10.1016/j.ygyno.2016.05.028
4. Qin T, Fan J, Lu F, Zhang L, Liu C, Xiong Q, et al. Harnessing preclinical models for the interrogation of ovarian cancer. J Exp Clin Cancer Res. (2022) 41:277. doi: 10.1186/s13046-022-02486-z
5. Mirabelli P, Coppola L, Salvatore M. Cancer cell lines are useful model systems for medical research. Cancers (Basel). (2019) 11:1–18. doi: 10.3390/cancers11081098
6. Li Z, Zheng W, Wang H, Cheng Y, Fang Y, Wu F, et al. Application of animal models in cancer research: recent progress and future prospects. Cancer Manag Res. (2021) 13:2455–75. doi: 10.2147/CMAR.S302565
7. Zhou Y, Xia J, Xu S, She T, Zhang Y, Sun Y, et al. Experimental mouse models for translational human cancer research. Front Immunol. (2023) 14:1095388. doi: 10.3389/fimmu.2023.1095388
8. Overgaard NH, Fan TM, Schachtschneider KM, Principe DR, Schook LB, Jungersen G. Of mice, dogs, pigs, and men: choosing the appropriate model for immuno-oncology research. ILAR J. (2018) 59:247–62. doi: 10.1093/ilar/ily014
9. Berek JS, Hacker NF. Berek and Hacker's gynecologic oncology. United States: Lippincott Williams & Wilkins (2010).
10. Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. (2014) 384:1376–88. doi: 10.1016/S0140-6736(13)62146-7
11. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
12. Huang J, Chan WC, Ngai CH, Lok V, Zhang L, Lucero-Prisno DE III, et al. Worldwide burden, risk factors, and temporal trends of ovarian cancer: A global study. Cancers. (2022) 14:2230. doi: 10.3390/cancers14092230
13. Young R. WHO classification of tumours of female reproductive organs. Monodermal teratomas and somatic-type tumours arising from a dermoid cyst. France: International Agency for Research on Cancer, Vol. 63-6. (2014).
14. Lheureux S, Gourley C, Vergote I, Oza AM. Epithelial ovarian cancer. Lancet. (2019) 393:1240–53. doi: 10.1016/S0140-6736(18)32552-2
15. Barnes BM, Nelson L, Tighe A, Burghel GJ, Lin I-H, Desai S, et al. Distinct transcriptional programs stratify ovarian cancer cell lines into the five major histological subtypes. Genome Med. (2021) 13:1–19. doi: 10.1186/s13073-021-00952-5
16. Kurman RJ, Carcangiu ML, Herrington CS. World Health Organisation classification of tumours of the female reproductive organs. Geneva, Switzerland: International agency for research on cancer (2014).
17. Malpica A, Deavers MT, Lu K, Bodurka DC, Atkinson EN, Gershenson DM, et al. Grading ovarian serous carcinoma using a two-tier system. Am J Surg Pathol. (2004) 28:496–504. doi: 10.1097/00000478-200404000-00009
18. Slomovitz B, Gourley C, Carey MS, Malpica A, Shih I-M, Huntsman D, et al. Low-grade serous ovarian cancer: state of the science. Gynecologic Oncol. (2020) 156:715–25. doi: 10.1016/j.ygyno.2019.12.033
19. Ahn G, Folkins AK, McKenney JK, Longacre TA. Low-grade serous carcinoma of the ovary. Am J Surg Pathol. (2016) 40:1165–76. doi: 10.1097/PAS.0000000000000693
20. Soovares P, Pasanen A, Similä-Maarala J, Bützow R, Lassus H. Clinical factors and biomarker profiles associated with patient outcome in endometrioid ovarian carcinoma-Emphasis on tumor grade. Gynecologic Oncol. (2022) 164:187–94. doi: 10.1016/j.ygyno.2021.10.078
21. Hollis RL, Thomson JP, Stanley B, Churchman M, Meynert AM, Rye T, et al. Molecular stratification of endometrioid ovarian carcinoma predicts clinical outcome. Nat Commun. (2020) 11:4995. doi: 10.1038/s41467-020-18819-5
22. Köbel M, Rahimi K, Rambau PF, Naugler C, Le Page C, Meunier L, et al. An immunohistochemical algorithm for ovarian carcinoma typing. Int J Gynecological Pathol. (2016) 35:430. doi: 10.1097/PGP.0000000000000274
23. McCluggage WG. Morphological subtypes of ovarian carcinoma: a review with emphasis on new developments and pathogenesis. Pathology. (2011) 43:420–32. doi: 10.1097/PAT.0b013e328348a6e7
24. Iida Y, Okamoto A, Hollis RL, Gourley C, Herrington CS. Clear cell carcinoma of the ovary: a clinical and molecular perspective. Int J Gynecologic Cancer. (2020) 34:ijgc–2020-001656. doi: 10.1136/ijgc-2020-001656
25. Kang EY, Cheasley D, LePage C, Wakefield MJ, da Cunha Torres M, Rowley S, et al. Refined cut-off for TP53 immunohistochemistry improves prediction of TP53 mutation status in ovarian mucinous tumors: implications for outcome analyses. Modern Pathol. (2021) 34:194–206. doi: 10.1038/s41379-020-0618-9
26. Network CGAR. Integrated genomic analyses of ovarian carcinoma. Nature. (2011) 474:609. doi: 10.1038/nature10166
27. Baldwin RL, Nemeth E, Tran H, Shvartsman H, Cass I, Narod S, et al. BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study. Cancer Res. (2000) 60:5329–33.
28. Hilton JL, Geisler JP, Rathe JA, Hattermann-Zogg MA, DeYoung B, Buller RE. Inactivation of BRCA1 and BRCA2 in ovarian cancer. J Natl Cancer Institute. (2002) 94:1396–406. doi: 10.1093/jnci/94.18.1396
29. Strathdee G, Appleton K, Illand M, Millan DW, Sargent J, Paul J, et al. Primary ovarian carcinomas display multiple methylator phenotypes involving known tumor suppressor genes. Am J Pathol. (2001) 158:1121–7. doi: 10.1016/S0002-9440(10)64059-X
30. Wang C, Horiuchi A, Imai T, Ohira S, Itoh K, Nikaido T, et al. Expression of BRCA1 protein in benign, borderline, and Malignant epithelial ovarian neoplasms and its relationship to methylation and allelic loss of the BRCA1 gene. J Pathology: A J Pathological Soc Great Britain Ireland. (2004) 202:215–23. doi: 10.1002/path.v202:2
31. Prat J, Oncology FCoG. Staging classification for cancer of the ovary, fallopian tube, and peritoneum. Int J Gynecology Obstetrics. (2014) 124:1–5. doi: 10.1016/j.ijgo.2013.10.001
32. Height T. Ovarian Epithelial, Fallopian Tube, and Primary Peritoneal Cancer Treatment (PDQ®) Health Professional Version. United States: National Cancer Institute (2024).
34. Momenimovahed Z, Tiznobaik A, Taheri S, Salehiniya H. Ovarian cancer in the world: epidemiology and risk factors. Int J women's Health. (2019) 11:287–99. doi: 10.2147/IJWH.S197604
35. Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. (2003) 72:1117–30. doi: 10.1086/375033
36. Lu HK, Broaddus RR. Gynecologic cancers in Lynch syndrome/HNPCC. Familial Cancer. (2005) 4:249–54. doi: 10.1007/s10689-005-1838-3
37. Salehi F, Dunfield L, Phillips KP, Krewski D, Vanderhyden BC. Risk factors for ovarian cancer: an overview with emphasis on hormonal factors. J Toxicol Environ Health Part B. (2008) 11:301–21. doi: 10.1080/10937400701876095
38. Beral V, Bull D, Green J, Reeves G. Ovarian cancer and hormone replacement therapy–Authors' reply. Lancet. (2007) 370:932–3. doi: 10.1016/S0140-6736(07)61437-8
39. Cancer CGoESoO. Ovarian cancer and smoking: individual participant meta-analysis including 28 114 women with ovarian cancer from 51 epidemiological studies. Lancet Oncol. (2012) 13:946–56. doi: 10.1016/S1470-2045(12)70322-4
40. Terry KL, Karageorgi S, Shvetsov YB, Merritt MA, Lurie G, Thompson PJ, et al. Genital powder use and risk of ovarian cancer: a pooled analysis of 8,525 cases and 9,859 controls. Cancer Prev Res. (2013) 6:811–21. doi: 10.1158/1940-6207.CAPR-13-0037
41. Braicu EI, Darb-Esfahani S, Schmitt WD, Koistinen KM, Heiskanen L, Pöhö P, et al. High-grade ovarian serous carcinoma patients exhibit profound alterations in lipid metabolism. Oncotarget. (2017) 8:102912. doi: 10.18632/oncotarget.22076
42. Minlikeeva AN, Freudenheim JL, Cannioto RA, Szender JB, Eng KH, Modugno F, et al. History of hypertension, heart disease, and diabetes and ovarian cancer patient survival: evidence from the ovarian cancer association consortium. Cancer causes control. (2017) 28:469–86. doi: 10.1007/s10552-017-0867-1
43. Shah MM, Erickson BK, Matin T, McGwin G Jr, Martin JY, Daily LB, et al. Diabetes mellitus and ovarian cancer: more complex than just increasing risk. Gynecologic Oncol. (2014) 135:273–7. doi: 10.1016/j.ygyno.2014.09.004
44. Zhang D, Li N, Xi Y, Zhao Y, Wang T. Diabetes mellitus and risk of ovarian cancer. A systematic review and meta-analysis of 15 cohort studies. Diabetes Res Clin Pract. (2017) 130:43–52. doi: 10.1016/j.diabres.2017.04.005
45. O’Donnell AJ, Macleod KG, Burns DJ, Smyth JF, Langdon SP. Estrogen receptor-α mediates gene expression changes and growth response in ovarian cancer cells exposed to estrogen. Endocrine-related Cancer. (2005) 12:851–66. doi: 10.1677/erc.1.01039
46. Chen Y, Zhang L, Liu W, Wang K. Case-control study of metabolic syndrome and ovarian cancer in Chinese population. Nutr Metab. (2017) 14:1–9. doi: 10.1186/s12986-017-0176-4
47. Ostrand-Rosenberg S. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer immunology immunotherapy. (2010) 59:1593–600. doi: 10.1007/s00262-010-0855-8
48. Ness RB, Grisso JA, Cottreau C, Klapper J, Vergona R, Wheeler JE, et al. Factors related to inflammation of the ovarian epithelium and risk of ovarian cancer. Epidemiology. (2000) 11:111–7. doi: 10.1097/00001648-200003000-00006
49. Group UCSW. US cancer statistics data visualizations tool, based on November 2018 submission data (1999–2016): US Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute. United States: Centers for Disease Control and Prevention and National Cancer Institute (2019).
50. Grossman DC, Curry SJ, Owens DK, Barry MJ, Davidson KW, Doubeni CA, et al. Screening for ovarian cancer: US preventive services task force recommendation statement. Jama. (2018) 319:588–94. doi: 10.1001/jama.2017.21926
51. Moorman PG, Palmieri RT, Akushevich L, Berchuck A, Schildkraut JM. Ovarian cancer risk factors in African-American and white women. Am J Epidemiol. (2009) 170:598–606. doi: 10.1093/aje/kwp176
52. Zheng L, Cui C, Shi O, Lu X, Li Y-K, Wang W, et al. Incidence and mortality of ovarian cancer at the global, regional, and national levels, 1990–2017. Gynecologic Oncol. (2020) 159:239–47. doi: 10.1016/j.ygyno.2020.07.008
53. Cabasag CJ, Fagan PJ, Ferlay J, Vignat J, Laversanne M, Liu L, et al. Ovarian cancer today and tomorrow: A global assessment by world region and Human Development Index using GLOBOCAN 2020. Int J Cancer. (2022) 151:1535–41. doi: 10.1002/ijc.v151.9
54. Gaona-Luviano P, Medina-Gaona LA, Magaña-Pérez K. Epidemiology of ovarian cancer. Chin Clin Oncol. (2020) 9:47. doi: 10.21037/cco-20-34
55. Chien J, Poole EM. Ovarian cancer prevention, screening, and early detection: report from the 11th biennial ovarian cancer research symposium. Int J Gynecologic Cancer. (2017) 27:S20–2. doi: 10.1097/IGC.0000000000001118
56. Baldwin LA, Huang B, Miller RW, Tucker T, Goodrich ST, Podzielinski I, et al. Ten-year relative survival for epithelial ovarian cancer. Obstetrics Gynecology. (2012) 120:612–8. doi: 10.1097/AOG.0b013e318264f794
57. Vargas AN. Natural history of ovarian cancer. ecancermedicalscience. (2014) 8:780–96. doi: 10.3332/ecancer.2014.465
58. Oncology NCPGi. Ovarian Cancer. Including Fallopian Tube Cancer and Primary Peritoneal Cancer. Version 2.2020. Plymouth Meeting, United States: National Comprehensive Cancer Network (2021).
59. Colombo N, Sessa C, du Bois A, Ledermann J, McCluggage WG, McNeish I, et al. ESMO–ESGO consensus conference recommendations on ovarian cancer: pathology and molecular biology, early and advanced stages, borderline tumours and recurrent disease. Ann Oncol. (2019) 30:672–705. doi: 10.1093/annonc/mdz062
60. Arora N, Talhouk A, McAlpine JN, Law MR, Hanley GE. Long-term mortality among women with epithelial ovarian cancer: a population-based study in British Columbia, Canada. BMC cancer. (2018) 18:1–9. doi: 10.1186/s12885-018-4970-9
61. Yao S, Wang W, Zhou B, Cui X, Yang H, Zhang S. Monensin suppresses cell proliferation and invasion in ovarian cancer by enhancing MEK1 SUMOylation. Exp Ther Med. (2021) 22:1–10. doi: 10.3892/etm.2021.10826
62. King M-C, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. (2003) 302:643–6. doi: 10.1126/science.1088759
63. ICON, Research EOf, Neoplasm ToCCACIO. International Collaborative Ovarian Neoplasm trial 1 and Adjuvant ChemoTherapy In Ovarian Neoplasm trial: two parallel randomized phase III trials of adjuvant chemotherapy in patients with early-stage ovarian carcinoma. J Natl Cancer Institute. (2003) 95:105–12. doi: 10.1093/jnci/95.2.105
64. Trimbos JB, Vergote I, Bolis G, Vermorken JB, Mangioni C, Madronal C, et al. Impact of adjuvant chemotherapy and surgical staging in early-stage ovarian carcinoma: European Organisation for Research and Treatment of Cancer–Adjuvant ChemoTherapy In Ovarian Neoplasm trial. J Natl Cancer Institute. (2003) 95:113–25. doi: 10.1093/jnci/95.2.113
65. Ledermann J, Raja F, Fotopoulou C, Gonzalez-Martin A, Colombo N, Sessa C. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2013) 24:vi24–32. doi: 10.1093/annonc/mdt333
66. Ashing-Giwa K, Ganz PA, Petersen L. Quality of life of African-American and white long term breast carcinoma survivors. Cancer: Interdiscip Int J Am Cancer Soc. (1999) 85:418–26. doi: 10.1002/(SICI)1097-0142(19990115)85:2<418::AID-CNCR20>3.0.CO;2-9
67. McGuire WP, Hoskins WJ, Brady MF, Kucera PR, Partridge EE, Look KY, et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. New Engl J Med. (1996) 334:1–6. doi: 10.1056/NEJM199601043340101
68. Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke-Pearson D, Burger RA, et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol. (2003) 21:3194–200. doi: 10.1200/JCO.2003.02.153
69. Vasey PA, Jayson GC, Gordon A, Gabra H, Coleman R, Atkinson R, et al. Phase III randomized trial of docetaxel–carboplatin versus paclitaxel–carboplatin as first-line chemotherapy for ovarian carcinoma. J Natl Cancer Institute. (2004) 96:1682–91. doi: 10.1093/jnci/djh323
70. Ortiz M, Wabel E, Mitchell K, Horibata S. Mechanisms of chemotherapy resistance in ovarian cancer. Cancer Drug Resistance. (2022) 5:304. doi: 10.20517/cdr.2021.147
71. Lee JM, Minasian L, Kohn EC. New strategies in ovarian cancer treatment. Cancer. (2019) 125:4623–9. doi: 10.1002/cncr.v125.s24
72. Conic I, Stanojevic Z, Jankovic Velickovic L, Stojnev S, Ristic Petrovic A, Krstic M, et al. Epithelial ovarian cancer with CD117 phenotype is highly aggressive and resistant to chemotherapy. J Obstetrics Gynaecology Res. (2015) 41:1630–7. doi: 10.1111/jog.2015.41.issue-10
73. Farley J, Brady WE, Vathipadiekal V, Lankes HA, Coleman R, Morgan MA, et al. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: an open-label, single-arm, phase 2 study. Lancet Oncol. (2013) 14:134–40. doi: 10.1016/S1470-2045(12)70572-7
74. Parmar MK, Sydes MR, Cafferty FH, Choodari-Oskooei B, Langley RE, Brown L, et al. Testing many treatments within a single protocol over 10 years at MRC Clinical Trials Unit at UCL: Multi-arm, multi-stage platform, umbrella and basket protocols. Clin trials. (2017) 14:451–61. doi: 10.1177/1740774517725697
75. Lheureux S, McCourt C, Rimel B, Duska L, Fleming G, Mackay H, et al. Moving forward with actionable therapeutic targets and opportunities in endometrial cancer: a NCI clinical trials planning meeting report. Gynecologic Oncol. (2018) 149:442–6. doi: 10.1016/j.ygyno.2018.02.005
76. Rundle S, Bradbury A, Drew Y, Curtin NJ. Targeting the ATR-CHK1 axis in cancer therapy. Cancers. (2017) 9:41. doi: 10.3390/cancers9050041
77. Haynes B, Murai J, Lee J-M. Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition. Cancer Treat Rev. (2018) 71:1–7. doi: 10.1016/j.ctrv.2018.09.003
78. Lin AB, McNeely SC, Beckmann RP. Achieving precision death with cell-cycle inhibitors that target DNA replication and repair. Clin Cancer Res. (2017) 23:3232–40. doi: 10.1158/1078-0432.CCR-16-0083
79. Lee J-M, Nair J, Zimmer A, Lipkowitz S, Annunziata CM, Merino MJ, et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. Lancet Oncol. (2018) 19:207–15. doi: 10.1016/S1470-2045(18)30009-3
80. Carrassa L, Damia G. DNA damage response inhibitors: Mechanisms and potential applications in cancer therapy. Cancer Treat Rev. (2017) 60:139–51. doi: 10.1016/j.ctrv.2017.08.013
81. Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, et al. Phase I study of single-agent AZD1775 (MK-1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol. (2015) 33:3409. doi: 10.1200/JCO.2014.60.4009
82. Parsels LA, Karnak D, Parsels JD, Zhang Q, Vélez-Padilla J, Reichert ZR, et al. PARP1 trapping and DNA replication stress enhance radiosensitization with combined WEE1 and PARP inhibitors. Mol Cancer Res. (2018) 16:222–32. doi: 10.1158/1541-7786.MCR-17-0455
83. Burger RA, Brady MF, Bookman MA, Fleming GF, Monk BJ, Huang H, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. New Engl J Med. (2011) 365:2473–83. doi: 10.1056/NEJMoa1104390
84. Oza AM, Cook AD, Pfisterer J, Embleton A, Ledermann JA, Pujade-Lauraine E, et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): overall survival results of a phase 3 randomised trial. Lancet Oncol. (2015) 16:928–36. doi: 10.1016/S1470-2045(15)00086-8
85. Aghajanian C, Blank SV, Goff BA, Judson PL, Teneriello MG, Husain A, et al. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. (2012) 30:2039. doi: 10.1200/JCO.2012.42.0505
86. Coleman RL, Brady MF, Herzog TJ, Sabbatini P, Armstrong DK, Walker JL, et al. Bevacizumab and paclitaxel–carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. (2017) 18:779–91. doi: 10.1016/S1470-2045(17)30279-6
87. Pujade-Lauraine E, Hilpert F, Weber B, Reuss A, Poveda A, Kristensen G, et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: the AURELIA open-label randomized phase III trial. Obstetrical Gynecological Survey. (2014) 69:402–4. doi: 10.1097/01.ogx.0000452705.82050.e4
88. Liu JF, Barry WT, Birrer M, Lee J-M, Buckanovich RJ, Fleming GF, et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol. (2014) 15:1207–14. doi: 10.1016/S1470-2045(14)70391-2
89. Liu J, Barry W, Birrer M, Lee J-M, Buckanovich R, Fleming G, et al. Overall survival and updated progression-free survival outcomes in a randomized phase II study of combination cediranib and olaparib versus olaparib in relapsed platinum-sensitive ovarian cancer. Ann Oncol. (2019) 30:551–7. doi: 10.1093/annonc/mdz018
90. Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. New Engl J Med. (2012) 367:1783–91. doi: 10.1056/NEJMoa1209124
91. Vergote I, Armstrong D, Scambia G, Teneriello M, Sehouli J, Schweizer C, et al. A randomized, double-blind, placebo-controlled, phase III study to assess efficacy and safety of weekly farletuzumab in combination with carboplatin and taxane in patients with ovarian cancer in first platinum-sensitive relapse. J Clin Oncol. (2016) 34:2271–8. doi: 10.1200/JCO.2015.63.2596
92. Ab O, Whiteman KR, Bartle LM, Sun X, Singh R, Tavares D, et al. IMGN853, a folate receptor-α (FRα)–targeting antibody–drug conjugate, exhibits potent targeted antitumor activity against FRα-expressing tumors. Mol Cancer Ther. (2015) 14:1605–13. doi: 10.1158/1535-7163.MCT-14-1095
93. Martin LP, Konner JA, Moore KN, Seward SM, Matulonis UA, Perez RP, et al. Characterization of folate receptor alpha (FRα) expression in archival tumor and biopsy samples from relapsed epithelial ovarian cancer patients: A phase I expansion study of the FRα-targeting antibody-drug conjugate mirvetuximab soravtansine. Gynecologic Oncol. (2017) 147:402–7. doi: 10.1016/j.ygyno.2017.08.015
94. Moore KN, Vergote I, Oaknin A, Colombo N, Banerjee S, Oza A, et al. FORWARD I: a Phase III study of mirvetuximab soravtansine versus chemotherapy in platinum-resistant ovarian cancer. Future Oncol. (2018) 14:1669–78. doi: 10.2217/fon-2017-0646
95. Bradbury A, Zenke FT, Curtin NJ, Drew Y. The role of ATR inhibitors in ovarian cancer: investigating predictive biomarkers of response. Cells. (2022) 11:2361. doi: 10.3390/cells11152361
96. Montemorano L, Lightfoot MD, Bixel K. Role of olaparib as maintenance treatment for ovarian cancer: the evidence to date. OncoTargets Ther. (2019) 12:11497. doi: 10.2147/OTT.S195552
97. Kazazi-Hyseni F, Beijnen JH, Schellens JH. Bevacizumab. oncologist. (2010) 15:819. doi: 10.1634/theoncologist.2009-0317
98. Brahmer JR, Tykodi SS, Chow LQ, Hwu W-J, Topalian SL, Hwu P, et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. New Engl J Med. (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
99. Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, et al. Endotext. South Dartmouth (MA: MDText.com, Inc (2000). Available at: https://www.ncbi.nlm.nih.gov/sites/books/NBK278943/.
100. Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. (1990) 343:425–30. doi: 10.1038/343425a0
101. Wong WW, Dimitroulakos J, Minden M, Penn L. HMG-CoA reductase inhibitors and the Malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis. Leukemia. (2002) 16:508–19. doi: 10.1038/sj.leu.2402476
102. Jones HM, Fang Z, Sun W, Clark LH, Stine JE, Tran A-Q, et al. Atorvastatin exhibits anti-tumorigenic and anti-metastatic effects in ovarian cancer in vitro. Am J Cancer Res. (2017) 7:2478.
103. Toepfer N, Childress C, Parikh A, Rukstalis D, Yang W. Atorvastatin induces autophagy in prostate cancer PC3 cells through activation of LC3 transcription. Cancer Biol Ther. (2011) 12:691–9. doi: 10.4161/cbt.12.8.15978
104. Kobayashi Y, Kashima H, Wu R-C, Jung J-G, Kuan J-C, Gu J, et al. Mevalonate pathway antagonist suppresses formation of serous tubal intraepithelial carcinoma and ovarian carcinoma in mouse models. Clin Cancer Res. (2015) 21:4652–62. doi: 10.1158/1078-0432.CCR-14-3368
105. Martirosyan A, Clendening JW, Goard CA, Penn LZ. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: potential therapeutic relevance. BMC Cancer. (2010) 10:1–13. doi: 10.1186/1471-2407-10-103
106. Greenaway JB, Virtanen C, Revay T, Hardy D, Shepherd T, DiMattia G, et al. Ovarian tumour growth is characterized by mevalonate pathway gene signature in an orthotopic, syngeneic model of epithelial ovarian cancer. Oncotarget. (2016) 7:47343. doi: 10.18632/oncotarget.10121
107. Stine JE, Guo H, Sheng X, Han X, Schointuch MN, Gilliam TP, et al. The HMG-CoA reductase inhibitor, simvastatin, exhibits anti-metastatic and anti-tumorigenic effects in ovarian cancer. Oncotarget. (2016) 7:946. doi: 10.18632/oncotarget.5834
108. Kobayashi Y, Kashima H, Rahmanto YS, Banno K, Yu Y, Matoba Y, et al. Drug repositioning of mevalonate pathway inhibitors as antitumor agents for ovarian cancer. Oncotarget. (2017) 8:72147. doi: 10.18632/oncotarget.20046
109. Sawada K, Morishige K-i, Tahara M, Kawagishi R, Ikebuchi Y, Tasaka K, et al. Alendronate inhibits lysophosphatidic acid-induced migration of human ovarian cancer cells by attenuating the activation of rho. Cancer Res. (2002) 62:6015–20.
110. Hashimoto K, Morishige K-I, Sawada K, Tahara M, Kawagishi R, Ikebuchi Y, et al. Alendronate inhibits intraperitoneal dissemination in in vivo ovarian cancer model. Cancer Res. (2005) 65:540–5. doi: 10.1158/0008-5472.540.65.2
111. Boonen S, Reginster J-Y, Kaufman J-M, Lippuner K, Zanchetta J, Langdahl B, et al. Fracture risk and zoledronic acid therapy in men with osteoporosis. New Engl J Med. (2012) 367:1714–23. doi: 10.1056/NEJMoa1204061
112. Crawford BA, Kam C, Pavlovic J, Byth K, Handelsman DJ, Angus PW, et al. Zoledronic acid prevents bone loss after liver transplantation: a randomized, double-blind, placebo-controlled trial. Ann Internal Med. (2006) 144:239–48. doi: 10.7326/0003-4819-144-4-200602210-00005
113. Major PP, Coleman RE. Zoledronic acid in the treatment of hypercalcemia of Malignancy: results of the international clinical development program. Semin Oncol. (2001) 28:17–24. doi: 10.1016/S0093-7754(01)90261-1
114. Rosen LS, Gordon D, Kaminski M, Howell A, Belch A, Mackey J, et al. Zoledronic acid versus pamidronate in the treatment of skeletal metastases in patients with breast cancer or osteolytic lesions of multiple myeloma: a phase III, double-blind, comparative trial. Cancer J (Sudbury Mass). (2001) 7:377–87.
115. Atmaca H, Gorumlu G, Karaca B, Degirmenci M, Tunali D, Cirak Y, et al. Combined gossypol and zoledronic acid treatment results in synergistic induction of cell death and regulates angiogenic molecules in ovarian cancer cells. Eur Cytokine Network. (2009) 20:121–30. doi: 10.1684/ecn.2009.0159
116. Schmidmaier R, Simsek M, Baumann P, Emmerich B, Meinhardt G. Synergistic antimyeloma effects of zoledronate and simvastatin. Anti-cancer Drugs. (2006) 17:621–9. doi: 10.1097/01.cad.0000215058.85813.02
117. Knight LA, Kurbacher CM, Glaysher S, Fernando A, Reichelt R, Dexel S, et al. Activity of mevalonate pathway inhibitors against breast and ovarian cancers in the ATP-based tumour chemosensitivity assay. BMC Cancer. (2009) 9:1–10. doi: 10.1186/1471-2407-9-38
118. Lamb R, Harrison H, Hulit J, Smith DL, Lisanti MP, Sotgia F. Mitochondria as new therapeutic targets for eradicating cancer stem cells: Quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget. (2014) 5:11029. doi: 10.18632/oncotarget.v5i22
119. Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, et al. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget. (2015) 6:4569. doi: 10.18632/oncotarget.3174
120. Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. (2009) 69:7507–11. doi: 10.1158/0008-5472.CAN-09-2994
121. Rattan R, Ali Fehmi R, Munkarah A. Metformin: an emerging new therapeutic option for targeting cancer stem cells and metastasis. J Oncol. (2012) 2012:1–12. doi: 10.1155/2012/928127
122. Sun X, Lin T, Fang J, Liu J, Yao W, Geng L, et al. Clinical efficacy analysis of biofeedback electrical stimulation combined with doxycycline in the treatment of type IIIA chronic prostatitis. Evidence-Based Complementary Altern Med. (2022) 2022:1–7. doi: 10.1155/2022/7150204
123. White CR, Jodlowski TZ, Atkins DT, Holland NG. Successful doxycycline therapy in a patient with Escherichia coli and multidrug-resistant Klebsiella pneumoniae urinary tract infection. J Pharm Pract. (2017) 30:464–7. doi: 10.1177/0897190016642362
124. Joseph BB. Subantimicrobial dose doxycycline for acne and rosacea. SKINmed: Dermatol Clinician. (2003) 2:234–46. doi: 10.1111/j.1540-9740.2003.03014.x
125. Bendeck MP, Conte M, Zhang M, Nili N, Strauss BH, Farwell SM. Doxycycline modulates smooth muscle cell growth, migration, and matrix remodeling after arterial injury. Am J Pathol. (2002) 160:1089–95. doi: 10.1016/S0002-9440(10)64929-2
126. Chang M-Y, Rhee Y-H, Yi S-H, Lee S-J, Kim R-K, Kim H, et al. Doxycycline enhances survival and self-renewal of human pluripotent stem cells. Stem Cell Rep. (2014) 3:353–64. doi: 10.1016/j.stemcr.2014.06.013
127. Krakauer T, Buckley M. Doxycycline is anti-inflammatory and inhibits staphylococcal exotoxin-induced cytokines and chemokines. Antimicrob Agents Chemother. (2003) 11:3630–3. doi: 10.1128/aac.47.11.3630-3633.2003
128. Son K, Fujioka S, Iida T, Furukawa K, Fujita T, Yamada H, et al. Doxycycline induces apoptosis in PANC-1 pancreatic cancer cells. Anticancer Res. (2009) 29:3995–4003.
129. Duivenvoorden WC, Popovic SV, Lhoták S, Seidlitz E, Hirte HW, Tozer RG, et al. Doxycycline decreases tumor burden in a bone metastasis model of human breast cancer. Cancer Res. (2002) 62:1588–91.
130. Shen L-C, Chen Y-K, Lin L-M, Shaw S-Y. Anti-invasion and anti-tumor growth effect of doxycycline treatment for human oral squamous-cell carcinoma–in vitro and in vivo studies. Oral Oncol. (2010) 46:178–84. doi: 10.1016/j.oraloncology.2009.11.013
131. Chopra I, Roberts M. Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol Mol Biol Rev. (2001) 65:232–60. doi: 10.1128/MMBR.65.2.232-260.2001
132. MacDougall C, Chambers H. Protein synthesis inhibitors and miscellaneous antibacterial agents. In: Goodman and Gilman's the pharmacological basis of therapeutics. McGraw-Hill, New York (2011). p. 1521–47.
133. Yang S-p, Lin C-c. Treatment of paragonimiasis with bithionol and bithionol sulfoxide. Dis Chest. (1967) 52:220–32. doi: 10.1378/chest.52.2.220
134. Guo W, Liu Y, Yao Z, Zhou H, Wang X, Huang Z, et al. Bithionol restores sensitivity of multidrug-resistant gram-negative bacteria to colistin with antimicrobial and anti-biofilm effects. ACS Infect Dis. (2023) 9:1634–46. doi: 10.1021/acsinfecdis.3c00257
135. Kim JH, Chan KL, Cheng LW, Tell LA, Byrne BA, Clothier K, et al. High efficiency drug repurposing design for new antifungal agents. Methods Protoc. (2019) 2:31. doi: 10.3390/mps2020031
136. Ayyagari VN, Brard L. Bithionol inhibits ovarian cancer cell growth in vitro-studies on mechanism (s) of action. BMC Cancer. (2014) 14:1–17. doi: 10.1186/1471-2407-14-61
137. Kawagoe H, Stracke ML, Nakamura H, Sano K. Expression and transcriptional regulation of the PD-Iα/autotaxin gene in neuroblastoma. Cancer Res. (1997) 57:2516–21.
138. Hoelzinger DB, Mariani L, Weis J, Woyke T, Berens TJ, McDonough W, et al. Gene expression profile of glioblastoma multiforme invasive phenotype points to new therapeutic targets. Neoplasia. (2005) 7:7–16. doi: 10.1593/neo.04535
139. Stassar M, Devitt G, Brosius M, Rinnab L, Prang J, Schradin T, et al. Identification of human renal cell carcinoma associated genes by suppression subtractive hybridization. Br J Cancer. (2001) 85:1372–82. doi: 10.1054/bjoc.2001.2074
140. Kehlen A, Englert N, Seifert A, Klonisch T, Dralle H, Langner J, et al. Expression, regulation and function of autotaxin in thyroid carcinomas. Int J Cancer. (2004) 109:833–8. doi: 10.1002/ijc.v109:6
141. Yang SY, Lee J, Park CG, Kim S, Hong S, Chung HC, et al. Expression of autotaxin (NPP-2) is closely linked to invasiveness of breast cancer cells. Clin Exp metastasis. (2002) 19:603–8. doi: 10.1023/A:1020950420196
142. Wu J-M, Xu Y, Skill NJ, Sheng H, Zhao Z, Yu M, et al. Autotaxin expression and its connection with the TNF-alpha-NF-κB axis in human hepatocellular carcinoma. Mol Cancer. (2010) 9:1–14. doi: 10.1186/1476-4598-9-71
143. Liu S, Umezu-Goto M, Murph M, Lu Y, Liu W, Zhang F, et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell. (2009) 15:539–50. doi: 10.1016/j.ccr.2009.03.027
144. Kato K, Yoshikawa K, Tanabe E, Kitayoshi M, Fukui R, Fukushima N, et al. Opposite roles of LPA 1 and LPA 3 on cell motile and invasive activities of pancreatic cancer cells. Tumor Biol. (2012) 33:1739–44. doi: 10.1007/s13277-012-0433-0
145. Nam SW, Clair T, Campo CK, Lee HY, Liotta LA, Stracke ML. Autotaxin (ATX), a potent tumor motogen, augments invasive and metastatic potential of ras-transformed cells. Oncogene. (2000) 19:241–7. doi: 10.1038/sj.onc.1203263
146. Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, et al. A stromal lysolipid–autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discovery. (2019) 9:617–27. doi: 10.1158/2159-8290.CD-18-1212
147. Yu S, Murph MM, Lu Y, Liu S, Hall HS, Liu J, et al. Lysophosphatidic acid receptors determine tumorigenicity and aggressiveness of ovarian cancer cells. JNCI: J Natl Cancer Institute. (2008) 100:1630–42. doi: 10.1093/jnci/djn378
148. Nam SW, Clair T, Kim Y-S, McMarlin A, Schiffmann E, Liotta LA, et al. Autotaxin (NPP-2), a metastasis-enhancing motogen, is an angiogenic factor. Cancer Res. (2001) 61:6938–44.
149. Hu Y-L, Tee M-K, Goetzl EJ, Auersperg N, Mills GB, Ferrara N, et al. Lysophosphatidic acid induction of vascular endothelial growth factor expression in human ovarian cancer cells. J Natl Cancer Institute. (2001) 93:762–7. doi: 10.1093/jnci/93.10.762
150. Fujita T, Miyamoto S, Onoyama I, Sonoda K, Mekada E, Nakano H. Expression of lysophosphatidic acid receptors and vascular endothelial growth factor mediating lysophosphatidic acid in the development of human ovarian cancer. Cancer Lett. (2003) 192:161–9. doi: 10.1016/S0304-3835(02)00713-9
151. Ayyagari VN, Johnston NA, Brard L. Assessment of the anti-tumor potential of Bithionol in vivo using a xenograft model of ovarian cancer. Anti-cancer Drugs. (2016) 27:547. doi: 10.1097/CAD.0000000000000364
152. Odds F, Bossche HV. Antifungal activity of itraconazole compared with hydroxy-itraconazole in vitro. J Antimicrobial Chemotherapy. (2000) 45:371–3. doi: 10.1093/jac/45.3.371
153. Aftab BT, Dobromilskaya I, Liu JO, Rudin CM. Itraconazole inhibits angiogenesis and tumor growth in non–small cell lung cancer. Cancer Res. (2011) 71:6764–72. doi: 10.1158/0008-5472.CAN-11-0691
154. Wei X, Liu W, Wang JQ, Tang Z. Hedgehog pathway”: a potential target of itraconazole in the treatment of cancer. J Cancer Res Clin Oncol. (2020) 146:297–304. doi: 10.1007/s00432-019-03117-5
155. Deng H, Huang L, Liao Z, Liu M, Li Q, Xu R. Itraconazole inhibits the Hedgehog signaling pathway thereby inducing autophagy-mediated apoptosis of colon cancer cells. Cell Death Dis. (2020) 11:539. doi: 10.1038/s41419-020-02742-0
156. Pantziarka P, Sukhatme V, Bouche G, Meheus L, Sukhatme VP. Repurposing Drugs in Oncology (ReDO)—itraconazole as an anti-cancer agent. Ecancermedicalscience. (2015) 9:1–16. doi: 10.3332/ecancer.2015.521
157. Tsubamoto H, Sonoda T, Yamasaki M, Inoue K. Impact of combination chemotherapy with itraconazole on survival of patients with refractory ovarian cancer. Anticancer Res. (2014) 34:2481–7.
158. McCavera S, Rogers AT, Yates DM, Woods DJ, Wolstenholme AJ. An ivermectin-sensitive glutamate-gated chloride channel from the parasitic nematode Haemonchus contortus. Mol Pharmacol. (2009) 75:1347–55. doi: 10.1124/mol.108.053363
159. Moreno Y, Nabhan JF, Solomon J, Mackenzie CD, Geary TG. Ivermectin disrupts the function of the excretory-secretory apparatus in microfilariae of Brugia malayi. Proc Natl Acad Sci. (2010) 107:20120–5. doi: 10.1073/pnas.1011983107
160. Hashimoto H, Messerli SM, Sudo T, Maruta H. Ivermectin inactivates the kinase PAK1 and blocks the PAK1-dependent growth of human ovarian cancer and NF2 tumor cell lines. Drug Discovery Ther. (2009) 3:243–6.
161. Li N, Zhan X. Anti-parasite drug ivermectin can suppress ovarian cancer by regulating lncRNA-EIF4A3-mRNA axes. EPMA J. (2020) 11:289–309. doi: 10.1007/s13167-020-00209-y
162. Zhang X, Qin T, Zhu Z, Hong F, Xu Y, Zhang X, et al. Ivermectin augments the in vitro and in vivo efficacy of cisplatin in epithelial ovarian cancer by suppressing Akt/mTOR signaling. Am J Med Sci. (2020) 359:123–9. doi: 10.1016/j.amjms.2019.11.001
163. Juarez M, Schcolnik-Cabrera A, Dominguez-Gomez G, Chavez-Blanco A, Diaz-Chavez J, Duenas-Gonzalez A. Antitumor effects of ivermectin at clinically feasible concentrations support its clinical development as a repositioned cancer drug. Cancer Chemotherapy Pharmacol. (2020) 85:1153–63. doi: 10.1007/s00280-020-04041-z
164. Nishio M, Sugimachi K, Goto H, Wang J, Morikawa T, Miyachi Y, et al. Dysregulated YAP1/TAZ and TGF-β signaling mediate hepatocarcinogenesis in Mob1a/1b-deficient mice. Proc Natl Acad Sci. (2016) 113:E71–80. doi: 10.1073/pnas.1517188113
165. Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci. (2006) 103:12405–10. doi: 10.1073/pnas.0605579103
166. Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. (2006) 125:1253–67. doi: 10.1016/j.cell.2006.05.030
167. Kang W, Tong JH, Chan AW, Lee T-L, Lung RW, Leung PP, et al. Yes-associated protein 1 exhibits oncogenic property in gastric cancer and its nuclear accumulation associates with poor prognosis. Clin Cancer Res. (2011) 17:2130–9. doi: 10.1158/1078-0432.CCR-10-2467
168. Xia Y, Chang T, Wang Y, Liu Y, Li W, Li M, et al. Correction: YAP promotes ovarian cancer cell tumorigenesis and is indicative of a poor prognosis for ovarian cancer patients. PloS One. (2016) 11:e0152712. doi: 10.1371/journal.pone.0152712
169. Kodama M, Kodama T, Newberg JY, Katayama H, Kobayashi M, Hanash SM, et al. In vivo loss-of-function screens identify KPNB1 as a new druggable oncogene in epithelial ovarian cancer. Proc Natl Acad Sci. (2017) 114:E7301–E10. doi: 10.1073/pnas.1705441114
170. Tang M, Hu X, Wang Y, Yao X, Zhang W, Yu C, et al. Ivermectin, a potential anticancer drug derived from an antiparasitic drug. Pharmacol Res. (2021) 163:105207. doi: 10.1016/j.phrs.2020.105207
171. Kobayashi Y, Banno K, Kunitomi H, Tominaga E, Aoki D. Current state and outlook for drug repositioning anticipated in the field of ovarian cancer. J gynecologic Oncol. (2019) 30:1–13. doi: 10.3802/jgo.2019.30.e10
172. Elayapillai S, Ramraj S, Benbrook DM, Bieniasz M, Wang L, Pathuri G, et al. Potential and mechanism of mebendazole for treatment and maintenance of ovarian cancer. Gynecologic Oncol. (2021) 160:302–11. doi: 10.1016/j.ygyno.2020.10.010
173. Spagnuolo PA, Hu J, Hurren R, Wang X, Gronda M, Sukhai MA, et al. The antihelmintic flubendazole inhibits microtubule function through a mechanism distinct from Vinca alkaloids and displays preclinical activity in leukemia and myeloma. Blood J Am Soc Hematol. (2010) 115:4824–33. doi: 10.1182/blood-2009-09-243055
174. Dogra N, Kumar A, Mukhopadhyay T. Fenbendazole acts as a moderate microtubule destabilizing agent and causes cancer cell death by modulating multiple cellular pathways. Sci Rep. (2018) 8:1–15. doi: 10.1038/s41598-018-30158-6
175. Doudican N, Rodriguez A, Osman I, Orlow SJ. Mebendazole induces apoptosis via Bcl-2 inactivation in chemoresistant melanoma cells. Mol Cancer Res. (2008) 6:1308–15. doi: 10.1158/1541-7786.MCR-07-2159
176. Huang L, Zhao L, Zhang J, He F, Wang H, Liu Q, et al. Antiparasitic mebendazole (MBZ) effectively overcomes cisplatin resistance in human ovarian cancer cells by inhibiting multiple cancer-associated signaling pathways. Aging (Albany NY). (2021) 13:17407. doi: 10.18632/aging.203232
177. Marx N, Schoünbeck U, Lazar MA, Libby P, Plutzky J. Peroxisome proliferator-activated receptor gamma activators inhibit gene expression and migration in human vascular smooth muscle cells. Circ Res. (1998) 83:1097–103. doi: 10.1161/01.RES.83.11.1097
178. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature. (1998) 391:79–82. doi: 10.1038/34178
179. Xin X, Yang S, Kowalski J, Gerritsen ME. Peroxisome proliferator-activated receptor γ ligands are potent inhibitors of angiogenesis in vitro and in vivo. J Biol Chem. (1999) 274:9116–21. doi: 10.1074/jbc.274.13.9116
180. Banerjee J, Komar CM. Effects of luteinizing hormone on peroxisome proliferator-activated receptor γ in the rat ovary before and after the gonadotropin surge. Reproduction. (2006) 131:93–101. doi: 10.1530/rep.1.00730
181. Nicol CJ, Yoon M, Ward JM, Yamashita M, Fukamachi K, Peters JM, et al. PPARγ influences susceptibility to DMBA-induced mammary, ovarian and skin carcinogenesis. Carcinogenesis. (2004) 25:1747–55. doi: 10.1093/carcin/bgh160
182. Fischer SM, Conti CJ, Locniskar M, Belury MA, Maldve RE, Lee ML, et al. The effect of dietary fat on the rapid development of mammary tumors induced by 7, 12-dimethylbenz (a) anthracene in SENCAR mice. Cancer Res. (1992) 52:662–6.
183. Vignati S, Albertini V, Rinaldi A, Kwee I, Riva C, Oldrini R, et al. Cellular, molecular consequences of peroxisome proliferator-activated receptor-δ activation in ovarian cancer cells. Neoplasia. (2006) 8:851–IN12. doi: 10.1593/neo.06433
184. Zhang X, Young HA. PPAR and immune system—what do we know? Int Immunopharmacol. (2002) 2:1029–44. doi: 10.1016/S1567-5769(02)00057-7
185. Lei P, Abdelrahim M, Safe S. 1, 1-Bis (3′-indolyl)-1-(p-substituted phenyl) methanes inhibit ovarian cancer cell growth through peroxisome proliferator–activated receptor–dependent and independent pathways. Mol Cancer Ther. (2006) 5:2324–36. doi: 10.1158/1535-7163.MCT-06-0184
186. Shigeto T, Yokoyama Y, Xin B, Mizunuma H. Peroxisome proliferator-activated receptor α and γ ligands inhibit the growth of human ovarian cancer. Oncol Rep. (2007) 18:833–40. doi: 10.3892/or.18.4.833
187. Takaoka K, Sekiguchi F, Shigi H, Maeda Y, Nishikawa H, Kawabata A. Opposite effects of two thiazolidinediones, ciglitazone and troglitazone, on proteinase-activated receptor-1-triggered prostaglandin E2 release. Toxicology. (2010) 268:40–5. doi: 10.1016/j.tox.2009.11.020
188. Hazra S, Dubinett SM. Ciglitazone mediates COX-2 dependent suppression of PGE2 in human non-small cell lung cancer cells. Prostaglandins leukotrienes essential Fatty Acids. (2007) 77:51–8. doi: 10.1016/j.plefa.2007.05.006
189. Shin SJ, Kim JY, Kwon SY, Mun K-C, Cho CH, Ha E. Ciglitazone enhances ovarian cancer cell death via inhibition of glucose transporter-1. Eur J Pharmacol. (2014) 743:17–23. doi: 10.1016/j.ejphar.2014.09.013
190. Yokoyama Y, Xin B, Shigeto T, Umemoto M, Kasai-Sakamoto A, Futagami M, et al. Clofibric acid, a peroxisome proliferator–activated receptor α ligand, inhibits growth of human ovarian cancer. Mol Cancer Ther. (2007) 6:1379–86. doi: 10.1158/1535-7163.MCT-06-0722
191. Skrott Z, Mistrik M, Andersen KK, Friis S, Majera D, Gursky J, et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature. (2017) 552:194–9. doi: 10.1038/nature25016
192. Chen D, Cui QC, Yang H, Dou QP. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. (2006) 66:10425–33. doi: 10.1158/0008-5472.CAN-06-2126
193. Majumder S, Chatterjee S, Pal S, Biswas J, Efferth T, Choudhuri SK. The role of copper in drug-resistant murine and human tumors. Biometals. (2009) 22:377–84. doi: 10.1007/s10534-008-9174-3
194. Guo W, Zhang X, Lin L, Wang H, He E, Wang G, et al. The disulfiram/copper complex induces apoptosis and inhibits tumour growth in human osteosarcoma by activating the ROS/JNK signalling pathway. J Biochem. (2021) 170:275–87. doi: 10.1093/jb/mvab045
195. Xu Y, Zhou Q, Feng X, Dai Y, Jiang Y, Jiang W, et al. Disulfiram/copper markedly induced myeloma cell apoptosis through activation of JNK and intrinsic and extrinsic apoptosis pathways. Biomedicine Pharmacotherapy. (2020) 126:110048. doi: 10.1016/j.biopha.2020.110048
196. Papaioannou M, Mylonas I, Kast RE, Brüning A. Disulfiram/copper causes redox-related proteotoxicity and concomitant heat shock response in ovarian cancer cells that is augmented by auranofin-mediated thioredoxin inhibition. Oncoscience. (2014) 1:21. doi: 10.18632/oncoscience.v1i1
197. Guo F, Yang Z, Kulbe H, Albers AE, Sehouli J, Kaufmann AM. Inhibitory effect on ovarian cancer ALDH+ stem-like cells by Disulfiram and Copper treatment through ALDH and ROS modulation. Biomedicine Pharmacotherapy. (2019) 118:109371. doi: 10.1016/j.biopha.2019.109371
198. MacDonagh L, Gallagher M, Ffrench B, Gasch C, Breen E, Gray S, et al. Targeting the cancer stem cell marker, aldehyde dehydrogenase 1, to circumvent cisplatin resistance in NSCLC. Oncotarget. (2017) 8:72544–63. doi: 10.18632/oncotarget.19881
199. Guo X, Xu B, Pandey S, Goessl E, Brown J, Armesilla AL, et al. Disulfiram/copper complex inhibiting NFκB activity and potentiating cytotoxic effect of gemcitabine on colon and breast cancer cell lines. Cancer Lett. (2010) 290:104–13. doi: 10.1016/j.canlet.2009.09.002
200. Xu B, Shi P, Fombon IS, Zhang Y, Huang F, Wang W, et al. Disulfiram/copper complex activated JNK/c-jun pathway and sensitized cytotoxicity of doxorubicin in doxorubicin resistant leukemia HL60 cells. Blood Cells Molecules Dis. (2011) 47:264–9. doi: 10.1016/j.bcmd.2011.08.004
201. Choi JH, Yang YR, Lee SK, Kim SH, Kim YH, Cha JY, et al. Potential inhibition of PDK1/Akt signaling by phenothiazines suppresses cancer cell proliferation and survival. Ann New York Acad Sci. (2008) 1138:393–403. doi: 10.1196/nyas.2008.1138.issue-1
202. Duarte D, Vale N. Antipsychotic drug fluphenazine against human cancer cells. Biomolecules. (2022) 12:1360. doi: 10.3390/biom12101360
203. Romero IL, Gordon IO, Jagadeeswaran S, Mui KL, Lee WS, Dinulescu DM, et al. Effects of oral contraceptives or a gonadotropin-releasing hormone agonist on ovarian carcinogenesis in genetically engineered mice. Cancer Prev Res. (2009) 2:792–9. doi: 10.1158/1940-6207.CAPR-08-0236
204. Lengyel E, Litchfield LM, Mitra AK, Nieman KM, Mukherjee A, Zhang Y, et al. Metformin inhibits ovarian cancer growth and increases sensitivity to paclitaxel in mouse models. Am J obstetrics gynecology. (2015) 212:479. e1–. e10. doi: 10.1016/j.ajog.2014.10.026
205. Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. (2006) 29:254–8. doi: 10.2337/diacare.29.02.06.dc05-1558
206. Landman GW, Kleefstra N, van Hateren KJ, Groenier KH, Gans RO, Bilo HJ. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care. (2010) 33:322–6. doi: 10.2337/dc09-1380
207. Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. (2009) 32:1620–5. doi: 10.2337/dc08-2175
208. Kumar S, Meuter A, Thapa P, Langstraat C, Giri S, Chien J, et al. Metformin intake is associated with better survival in ovarian cancer: A case-control study. Cancer. (2013) 119:555–62. doi: 10.1002/cncr.v119.3
209. Romero IL, McCormick A, McEwen KA, Park S, Karrison T, Yamada SD, et al. Relationship of type II diabetes and metformin use to ovarian cancer progression, survival, and chemosensitivity. Obstetrics gynecology. (2012) 119:61. doi: 10.1097/AOG.0b013e3182393ab3
210. Yokoyama T, Kumon H, Nasu Y, Takamoto H, Watanabe T. Comparison of 25 and 75 mg/day naftopidil for lower urinary tract symptoms associated with benign prostatic hyperplasia: a prospective, randomized controlled study. Int J Urol. (2006) 13:932–8. doi: 10.1111/j.1442-2042.2006.01443.x
211. Oh-oka H. Usefulness of naftopidil for dysuria in benign prostatic hyperplasia and its optimal dose–comparison between 75 and 50 mg. Urologia Internationalis. (2009) 82:136–42. doi: 10.1159/000200787
212. Funahashi Y, Hattori R, Matsukawa Y, Komatsu T, Sassa N, Gotoh M. Clinical efficacy of a loading dose of naftopidil for patients with benign prostate hyperplasia. World J Urol. (2011) 29:225–31. doi: 10.1007/s00345-010-0528-4
213. Thomas TS. Implementation of Lifestyle Modifications as First Line Therapies for the Management of Benign Prostatic Hyperplasia. Kent State University, United States: Kent State University (2023).
214. Florent R, Weiswald L-B, Lambert B, Brotin E, Abeilard E, Louis M-H, et al. Bim, Puma and Noxa upregulation by Naftopidil sensitizes ovarian cancer to the BH3-mimetic ABT-737 and the MEK inhibitor Trametinib. Cell Death Dis. (2020) 11:380. doi: 10.1038/s41419-020-2588-8
215. Subeha MR, Goyeneche AA, Bustamante P, Lisio MA, Burnier JV, Telleria CM. Nelfinavir induces cytotoxicity towards high-grade serous ovarian cancer cells, involving induction of the unfolded protein response, modulation of protein synthesis, DNA damage, lysosomal impairment, and potentiation of toxicity caused by proteasome inhibition. Cancers. (2021) 14:99. doi: 10.3390/cancers14010099
216. Markowitz M, Saag M, Powderly WG, Hurley AM, Hsu A, Valdes JM, et al. A preliminary study of ritonavir, an inhibitor of HIV-1 protease, to treat HIV-1 infection. New Engl J Med. (1995) 333:1534–40. doi: 10.1056/NEJM199512073332204
217. Kumar S, Bryant CS, Chamala S, Qazi A, Seward S, Pal J, et al. Ritonavir blocks AKT signaling, activates apoptosis and inhibits migration and invasion in ovarian cancer cells. Mol Cancer. (2009) 8:1–12. doi: 10.1186/1476-4598-8-26
218. Kumar S, Bryant C, Qazi A, Morris R, Imudia A, Steffes C, et al. Ritonavir blocks Akt signaling and exhibits antineoplastic activity in ovarian cancer cell lines. Cancer Res. (2008) 68:3381–.
219. Winterhoff B, Teoman A, Freyer L, Von Bismarck A, Dowdy S, Schmalfeldt B, et al. The HIV protease inhibitor ritonavir induces cell cycle arrest and apoptosis in the A2780 ovarian cancer cell line in vitro and in vivo. Gynecologic Oncol. (2013) 130:e138. doi: 10.1016/j.ygyno.2013.04.393
220. Wallace JL. Distribution and expression of cyclooxygenase (COX) isoenzymes, their physiological roles, and the categorization of nonsteroidal anti-inflammatory drugs (NSAIDs). Am J Med. (1999) 107:11–6. doi: 10.1016/S0002-9343(99)00363-0
221. Marjoribanks J, Ayeleke RO, Farquhar C, Proctor M, Gynaecology C, Group F. Nonsteroidal anti-inflammatory drugs for dysmenorrhoea. Cochrane Database systematic Rev. (1996) 2015:1–139. doi: 10.1002/14651858.CD001751
222. Affaitati G, Martelletti P, Lopopolo M, Tana C, Massimini F, Cipollone F, et al. Use of nonsteroidal anti-inflammatory drugs for symptomatic treatment of episodic headache. Pain Pract. (2017) 17:392–401. doi: 10.1111/papr.2017.17.issue-3
223. Malanga G, Wolff E. Evidence-informed management of chronic low back pain with nonsteroidal anti-inflammatory drugs, muscle relaxants, and simple analgesics. Spine J. (2008) 8:173–84. doi: 10.1016/j.spinee.2007.10.013
224. Thun MJ, Jacobs EJ, Patrono C. The role of aspirin in cancer prevention. Nat Rev Clin Oncol. (2012) 9:259–67. doi: 10.1038/nrclinonc.2011.199
225. Trabert B, Ness RB, Lo-Ciganic W-H, Murphy MA, Goode EL, Poole EM, et al. Aspirin, nonaspirin nonsteroidal anti-inflammatory drug, and acetaminophen use and risk of invasive epithelial ovarian cancer: a pooled analysis in the Ovarian Cancer Association Consortium. J Natl Cancer Institute. (2014) 106:djt431. doi: 10.1093/jnci/djt431
226. Daikoku T, Tranguch S, Chakrabarty A, Wang D, Khabele D, Orsulic S, et al. Extracellular signal-regulated kinase is a target of cyclooxygenase-1-peroxisome proliferator-activated receptor-δ signaling in epithelial ovarian cancer. Cancer Res. (2007) 67:5285–92. doi: 10.1158/0008-5472.CAN-07-0828
227. Drinberg V, Bitcover R, Rajchenbach W, Peer D. Modulating cancer multidrug resistance by sertraline in combination with a nanomedicine. Cancer Lett. (2014) 354:290–8. doi: 10.1016/j.canlet.2014.08.026
228. MacQueen G, Born L, Steiner M. The selective serotonin reuptake inhibitor sertraline: its profile and use in psychiatric disorders. CNS Drug Rev. (2001) 7:1–24. doi: 10.1111/j.1527-3458.2001.tb00188.x
229. Kitaichi Y, Inoue T, Nakagawa S, Boku S, Kakuta A, Izumi T, et al. Sertraline increases extracellular levels not only of serotonin, but also of dopamine in the nucleus accumbens and striatum of rats. Eur J Pharmacol. (2010) 647:90–6. doi: 10.1016/j.ejphar.2010.08.026
230. Gottesman MM, Pastan I, Ambudkar SV. P-glycoprotein and multidrug resistance. Curr Opin Genet Dev. (1996) 6:610–7. doi: 10.1016/S0959-437X(96)80091-8
231. Lin JH, Yamazaki M. Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet. (2003) 42:59–98. doi: 10.2165/00003088-200342010-00003
232. Fletcher JI, Haber M, Henderson MJ, Norris MD. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer. (2010) 10:147–56. doi: 10.1038/nrc2789
233. Franks ME, Macpherson GR, Figg WD. Thalidomide. Lancet. (2004) 363:1802–11. doi: 10.1016/S0140-6736(04)16308-3
234. Kumar S, Rajkumar SV. Thalidomide and lenalidomide in the treatment of multiple myeloma. Eur J Cancer. (2006) 42:1612–22. doi: 10.1016/j.ejca.2006.04.004
235. Braña MF, Acero N, Anorbe L, Mingarro DM, Llinares F, Domínguez G. Discovering a new analogue of thalidomide which may be used as a potent modulator of TNF-α production. Eur J medicinal Chem. (2009) 44:3533–42. doi: 10.1016/j.ejmech.2009.03.018
236. Sampaio EP, Sarno EN, Galilly R, Cohn ZA, Kaplan G. Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J Exp Med. (1991) 173:699–703. doi: 10.1084/jem.173.3.699
237. Piura B, Medina L, Rabinovich A, Dyomin V, Huleihel M. Thalidomide distinctly affected TNF-α, IL-6 and MMP secretion by an ovarian cancer cell line (SKOV-3) and primary ovarian cancer cells. Eur Cytokine Netw. (2013) 24:122–9. doi: 10.1684/ecn.2013.0342
238. Ching L, Xu Z, Gummer B, Palmer B, Joseph W, Baguley B. Effect of thalidomide on tumour necrosis factor production and anti-tumour activity induced by 5, 6-dimethylxanthenone-4-acetic acid. Br J Cancer. (1995) 72:339–43. doi: 10.1038/bjc.1995.335
239. Eisen T, Boshoff C, Mak I, Sapunar F, Vaughan M, Pyle L, et al. Continuous low dose thalidomide: a phase II study in advanced melanoma, renal cell, ovarian and breast cancer. Br J Cancer. (2000) 82:812–7. doi: 10.1054/bjoc.1999.1004
240. Hurteau JA, Brady MF, Darcy KM, McGuire WP, Edmonds P, Pearl ML, et al. Randomized phase III trial of tamoxifen versus thalidomide in women with biochemical-recurrent-only epithelial ovarian, fallopian tube or primary peritoneal carcinoma after a complete response to first-line platinum/taxane chemotherapy with an evaluation of serum vascular endothelial growth factor (VEGF): A Gynecologic Oncology Group Study. Gynecologic Oncol. (2010) 119:444–50. doi: 10.1016/j.ygyno.2010.08.002
241. Matei D, Emerson R, Lai Y, Baldridge L, Rao J, Yiannoutsos C, et al. Autocrine activation of PDGFRα promotes the progression of ovarian cancer. Oncogene. (2006) 25:2060–9. doi: 10.1038/sj.onc.1209232
242. Chen R, Chen B. The role of dasatinib in the management of chronic myeloid leukemia. Drug design Dev Ther. (2015) 9:773–9. doi: 10.2147/DDDT.S80207
243. Le XF, Mao W, Lu Z, Carter BZ, Bast RC Jr. Dasatinib induces autophagic cell death in human ovarian cancer. Cancer. (2010) 116:4980–90. doi: 10.1002/cncr.v116:21
244. Xiao J, Xu M, Hou T, Huang Y, Yang C, Li J. Dasatinib enhances antitumor activity of paclitaxel in ovarian cancer through Src signaling. Mol Med Rep. (2015) 12:3249–56. doi: 10.3892/mmr.2015.3784
245. Kadife E, Chan E, Luwor R, Kannourakis G, Findlay J, Ahmed N. Paclitaxel-induced Src activation is inhibited by dasatinib treatment, independently of cancer stem cell properties, in a mouse model of ovarian cancer. Cancers. (2019) 11:243. doi: 10.3390/cancers11020243
246. Matei D, Chang DD, Jeng M-H. Imatinib mesylate (Gleevec) inhibits ovarian cancer cell growth through a mechanism dependent on platelet-derived growth factor receptor α and Akt inactivation. Clin Cancer Res. (2004) 10:681–90. doi: 10.1158/1078-0432.CCR-0754-03
247. Posadas EM, Kwitkowski V, Kotz HL, Espina V, Minasian L, Tchabo N, et al. A prospective analysis of imatinib-induced c-KIT modulation in ovarian cancer: a phase II clinical study with proteomic profiling. Cancer. (2007) 110:309–17. doi: 10.1002/cncr.v110:2
248. Alberts DS, Liu PY, Wilczynski SP, Jang A, Moon J, Ward JH, et al. Phase II trial of imatinib mesylate in recurrent, biomarker positive, ovarian cancer (Southwest Oncology Group Protocol S0211). Int J Gynecologic Cancer. (2007) 17:784. doi: 10.1136/ijgc-00009577-200707000-00005
249. Trope C, Marth C, Kaern J. Tamoxifen in the treatment of recurrent ovarian carcinoma. Eur J Cancer. (2000) 36:59–61. doi: 10.1016/S0959-8049(00)00228-8
250. Trédan O, Provansal M, Abdeddaim C, Lardy-Cleaud A, Hardy-Bessard A-C, Kalbacher E, et al. Regorafenib or tamoxifen for platinum-sensitive recurrent ovarian cancer with rising CA125 and no evidence of clinical or RECIST progression: a GINECO randomized phase II trial (REGOVAR). Gynecologic Oncol. (2022) 164:18–26. doi: 10.1016/j.ygyno.2021.09.024
251. Bowman A, Gabra H, Langdon SP, Lessells A, Stewart M, Young A, et al. CA125 response is associated with estrogen receptor expression in a phase II trial of letrozole in ovarian cancer: identification of an endocrine-sensitive subgroup. Clin Cancer Res. (2002) 8:2233–9.
252. Smyth JF, Gourley C, Walker G, MacKean MJ, Stevenson A, Williams AR, et al. Antiestrogen therapy is active in selected ovarian cancer cases: the use of letrozole in estrogen receptor–positive patients. Clin Cancer Res. (2007) 13:3617–22. doi: 10.1158/1078-0432.CCR-06-2878
253. Ramirez PT, Schmeler KM, Milam MR, Slomovitz BM, Smith JA, Kavanagh JJ, et al. Efficacy of letrozole in the treatment of recurrent platinum-and taxane-resistant high-grade cancer of the ovary or peritoneum. Gynecologic Oncol. (2008) 110:56–9. doi: 10.1016/j.ygyno.2008.03.014
254. Papadimitriou CA, Markaki S, Siapkaras J, Vlachos G, Efstathiou E, Grimani I, et al. Hormonal therapy with letrozole for relapsed epithelial ovarian cancerLong-term results of a phase II study. Oncology. (2004) 66:112–7. doi: 10.1159/000077436
255. del Carmen MG, Fuller AF, Matulonis U, Horick NK, Goodman A, Duska LR, et al. Phase II trial of anastrozole in women with asymptomatic müllerian cancer. Gynecologic Oncol. (2003) 91:596–602. doi: 10.1016/j.ygyno.2003.08.021
256. Maher DM, Khan S, Nordquist JL, Ebeling MC, Bauer NA, Kopel L, et al. Ormeloxifene efficiently inhibits ovarian cancer growth. Cancer Lett. (2015) 356:606–12. doi: 10.1016/j.canlet.2014.10.009
257. Dodds M, Xiong Y, Mouksassi S, Kirkpatrick CM, Hui K, Doyle E, et al. Model-informed drug repurposing: a pharmacometric approach to novel pathogen preparedness, response and retrospection. Br J Clin Pharmacol. (2021) 87:3388–97. doi: 10.1111/bcp.14760
258. Berckmans Y, Hoffert Y, Vankerckhoven A, Dreesen E, Coosemans A. Drug repurposing for targeting myeloid-derived suppressor-cell-generated immunosuppression in ovarian cancer: A literature review of potential candidates. Pharmaceutics. (2023) 15:1792. doi: 10.3390/pharmaceutics15071792
259. Markham A. Lurbinectedin: first approval. Drugs. (2020) 80:1345–53. doi: 10.1007/s40265-020-01374-0
260. Till KJ, Lin K, Zuzel M, Cawley JC. The chemokine receptor CCR7 and α4 integrin are important for migration of chronic lymphocytic leukemia cells into lymph nodes. Blood J Am Soc Hematol. (2002) 99:2977–84. doi: 10.1182/blood.V99.8.2977
261. Trigo J, Subbiah V, Besse B, Moreno V, López R, Sala MA, et al. Lurbinectedin as second-line treatment for patients with small-cell lung cancer: a single-arm, open-label, phase 2 basket trial. Lancet Oncol. (2020) 21:645–54. doi: 10.1016/S1470-2045(20)30068-1
262. Singh S, Jaigirdar AA, Mulkey F, Cheng J, Hamed SS, Li Y, et al. FDA approval summary: lurbinectedin for the treatment of metastatic small cell lung cancer. Clin Cancer Res. (2021) 27:2378–82. doi: 10.1158/1078-0432.CCR-20-3901
263. Takahashi R, Mabuchi S, Kawano M, Sasano T, Matsumoto Y, Kuroda H, et al. Preclinical investigations of PM01183 (lurbinectedin) as a single agent or in combination with other anticancer agents for clear cell carcinoma of the ovary. PloS One. (2016) 11:e0151050. doi: 10.1371/journal.pone.0151050
264. Cruz C, Llop-Guevara A, Garber JE, Arun BK, Fidalgo JAP, Lluch A, et al. Multicenter phase II study of lurbinectedin in BRCA-mutated and unselected metastatic advanced breast cancer and biomarker assessment substudy. J Clin Oncol. (2018) 36:3134. doi: 10.1200/JCO.2018.78.6558
265. Elez ME, Tabernero J, Geary D, Macarulla T, Kang SP, Kahatt C, et al. First-in-human phase I study of Lurbinectedin (PM01183) in patients with advanced solid tumors. Clin Cancer Res. (2014) 20:2205–14. doi: 10.1158/1078-0432.CCR-13-1880
266. Dannenberg AJ, Altorki NK, Boyle JO, Dang C, Howe LR, Weksler BB, et al. Cyclo-oxygenase 2: a pharmacological target for the prevention of cancer. Lancet Oncol. (2001) 2:544–51. doi: 10.1016/S1470-2045(01)00488-0
267. Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC Funct BMC Cancer. (2010) 10:1–13. doi: 10.1186/1471-2407-10-464
268. Kantarjian H, Issa JPJ, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer: Interdiscip Int J Am Cancer Soc. (2006) 106:1794–803. doi: 10.1002/cncr.v106:8
269. Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. (2002) 20:2429–40. doi: 10.1200/JCO.2002.04.117
270. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. (2009) 10:223–32. doi: 10.1016/S1470-2045(09)70003-8
271. Cao D, Li D, Huang Y, Ma Y, Zhang B, Zhao C, et al. 5-Azacytidine promotes invadopodia formation and tumor metastasis through the upregulation of PI3K in ovarian cancer cells. Oncotarget. (2017) 8:60173. doi: 10.18632/oncotarget.18580
272. Meng F, Sun G, Zhong M, Yu Y, Brewer M. Anticancer efficacy of cisplatin and trichostatin A or 5-aza-2′-deoxycytidine on ovarian cancer. Br J Cancer. (2013) 108:579–86. doi: 10.1038/bjc.2013.10
273. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. (2014) 384:1109–17. doi: 10.1016/S0140-6736(14)60958-2
274. Zhao L, Chen X, Wu H, He Q, Ding L, Yang B. Strategies to synergize PD-1/PD-L1 targeted cancer immunotherapies to enhance antitumor responses in ovarian cancer. Biochem Pharmacol. (2023) 215:115724. doi: 10.1016/j.bcp.2023.115724
275. Xin B, Yokoyama Y, Shigeto T, Futagami M, Mizunuma H. Inhibitory effect of meloxicam, a selective cyclooxygenase-2 inhibitor, and ciglitazone, a peroxisome proliferator-activated receptor gamma ligand, on the growth of human ovarian cancers. Cancer: Interdiscip Int J Am Cancer Soc. (2007) 110:791–800. doi: 10.1002/cncr.v110:4
Keywords: cancer, ovarian cancer, conventional treatment, drug repositioning, cancer hallmarks
Citation: Villegas-Vazquez EY, Marín-Carrasco FP, Reyes-Hernández OD, Báez-González AS, Bustamante-Montes LP, Padilla-Benavides T, Quintas-Granados LI and Figueroa-González G (2025) Revolutionizing ovarian cancer therapy by drug repositioning for accelerated and cost-effective treatments. Front. Oncol. 14:1514120. doi: 10.3389/fonc.2024.1514120
Received: 20 October 2024; Accepted: 23 December 2024;
Published: 14 January 2025.
Edited by:
Tinghe Yu, Chongqing Medical University, ChinaReviewed by:
Eswari Dodagatta-Marri, University of California, San Francisco, United StatesPaula Rezende-Teixeira, University of São Paulo, Brazil
Copyright © 2025 Villegas-Vazquez, Marín-Carrasco, Reyes-Hernández, Báez-González, Bustamante-Montes, Padilla-Benavides, Quintas-Granados and Figueroa-González. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Itzel Quintas-Granados, aXR6ZWwucXVpbnRhc0B1YWNtLmVkdS5teA==; Gabriela Figueroa-González, Z2FicmllbGEuZmlndWVyb2FAemFyYWdvemEudW5hbS5teA==
†These authors have contributed equally to this work and share first authorship