 Oliver Tomkins
Oliver Tomkins- UCLH Centre for Waldenström’s Macroglobulinaemia and Related Conditions, Department of Haematology, University College London Hospitals NHS Foundation Trust, London, United Kingdom
Lymphoplasmacytic lymphoma (LPL) is a relatively rare form of indolent B-cell non-Hodgkin’s lymphoma, termed Waldenström’s macroglobulinaemia (WM) in the presence of an IgM paraprotein. Although traditionally treated with combination chemoimmunotherapy, the management is evolving in the era of targeted molecular therapies including Bruton’s tyrosine kinase inhibitors (BTKi). However, intolerance and refractoriness to BTKi mean newer agents are required, and the prognosis of so-called quadruple-refractory patients is poor. BCL2 is an anti-apoptotic, pro-survival protein that promotes lymphoma cell survival. Inhibition of BCL2 using first-in-class agent venetoclax has already altered the treatment paradigm in other conditions, including chronic lymphocytic leukaemia (CLL). In-vivo inhibition of BCL2 has been shown to lead to apoptosis of LPL/WM cells. Five studies have published results on the use of BCL2 inhibitors in WM to date, including oblimersen sodium, venetoclax, and sonrotoclax. Fixed-duration venetoclax resulted in high response rates, but many patients relapsed following the completion of therapy. The combination of venetoclax with ibrutinib resulted in higher and relatively deep response rates, but unexpected deaths due to ventricular events mean this combination cannot be explored. Two pivotal trials are currently evaluating the use of fixed-duration venetoclax, either in combination with rituximab or pirtobrutinib, whereas another multi-arm study is studying the use of continuous sonrotoclax monotherapy for R/R WM or in fixed-duration combination with Zanubrutinib for treatment-naïve patients. The potential role of BCL2 inhibitors in WM/LPL remains under study, with many hopeful that they may provide an additional chemotherapy-free oral alternative for patients requiring treatment. In an indolent condition with existing effective treatment regimens, including CIT and cBTKi, cost-effectiveness and toxicity profile will be key, although an additional treatment modality for quadruple-refractory patients with limited treatment options is urgently required.
1 Introduction
Lymphoplasmacytic lymphoma (LPL) is a relatively rare form of indolent B-cell non-Hodgkin’s lymphoma and a distinct pathophysiological disease entity (1). In the great majority of patients, the lymphoma secretes a monoclonal immunoglobulin M (IgM), and the disease is eponymously termed Waldenström’s macroglobulinaemia (WM). There is an additional recognised subtype, termed non-WM–type LPL that represents approximately 5% of cases and includes those with non-secretory disease, IgG or IgA paraproteins, and technically IgM LPL without bone marrow involvement (2). All are indolent but ultimately incurable conditions. The median age at diagnosis is approximately 70 years, and there is a 2:1 male preponderance. The incidence is estimated at 0.38 per 100,000 persons per year (3).
2 Pathophysiology of LPL/WM
WM/LPL arises through the neoplastic proliferation of terminal B cells, which retain the ability to undergo plasmacytic differentiation. This results in an infiltrate of both clonal B and plasma cells in varying proportions. There is a precursor phase, termed monoclonal gammopathy of undetermined significance (MGUS), which is followed by progression to asymptomatic and finally symptomatic LPL/WM. During this process, there is gradual accumulation of LPL with clonal and subclonal molecular evolution. Gradual infiltration in the bone marrow results in cytopaenias, most commonly anemia, but infiltration of other tissues can also occur, including lymph nodes, spleen, bones, and the central nervous system.
Serum IgM concentration appears to correlate with the degree of plasmacytic differentiation of the lymphoma, rather than simply overall disease burden (4). High levels of monoclonal IgM in the plasma result in symptoms of hyperviscosity and acquired von Willebrand syndrome, owing to the immunoglobulin’s large pentameric and hexameric configuration (5). Monoclonal IgM can also cause autoimmune phenomena according to its antigenic target, resulting in complement-mediated destruction, such cold agglutinin syndrome (CAS) and anti-myelin-associated-glycoprotein (MAG) neuropathy. Precipitation of IgM in the form of types I and II cryoglobulin is seen, resulting in cryoglobulinaemic vasculitis (6).
Transformation to a more aggressive, high-grade B-cell lymphoma occurs in up to 4% of patients and has a more adverse prognosis (7).
2.1 Disease genomics
Recurring somatic mutations have been identified in LPL cells (8). The activating single-point mutation L265P in the gene myeloid differentiation primary response 88 (MYD88) is present in >90% of patients with WM and non-IgM LPL. MYD88L265P complexes with downstream effectors of B cell receptor signalling, such as IRAK1 and 4, leading to increased activation of the NFκB pathway. In addition, mutated MYD88 causes hyperactivation of haematopoetic cell kinase (HCK), in turn inducing PI3K/AKT, MAPK/ERK, and BTK signalling (9). These pathways drive cell survival and proliferation. Other MYD88 point mutations have also been described, including S219C, M232T, and S243N (10). MYD88 mutations are absent in patients with IgM multiple myeloma, a wholly separate disease entity characterised by t(11;14) (11).
Additionally, WHIM-like mutations in the gene C-X-C chemokine receptor type 4 (CXCR4) are seen in approximately 30%–40% of cases of LPL/WM. These are subclonal and acquired after MYD88-mutations in disease pathogenesis (10, 12). Half are CXCR4S338X, but >40 frameshift or nonsense mutations are described (13). These mutations result in a CXR4 protein resistant to intracellular inactivation, thereby driving AKT, ERK, and MAPK1/2 pathway signaling, again promoting cellular survival. These mutations are associated with a degree of resistance to BKTi (14), higher serum IgM, and increased incidence of hyperviscosity and acquired von Willebrand syndrome (15, 16).
Other, uncommonly identified somatic mutations include in AT-rich interactive domain 1A (ARID1A), cluster of differentiation (CD)79B, and lysine methyltransferase 2D (KMT2D) (8, 17, 18). TP53 aberrations are seen in <10% of patients, associated with resistance to chemotherapy and shorter overall survival (18–21). Most cases of WM also feature multiple chromosomal abnormalities, most frequently deletion 6q, which is associated with progression to symptomatic disease (22, 23).
3 Current therapeutic approach
Treatment for WM/LPL is instituted when there are complications or symptoms related to the disease. Many patients can initially be monitored following the diagnosis, in the absence of disease-related symptoms or organ compromise, but the majority come to require therapy (24). The disease is ultimately incurable, but remissions lasting several years following treatment are often seen (25). There is international heterogeneity in the choice of anti-cancer therapy regimen, according to patient age, fitness, comorbidities, disease burden, genomic findings, and local health system reimbursement arrangements.
3.1 Combination chemoimmunotherapy
Combination chemoimmunotherapy (CIT) with an anti-CD20 monoclonal antibody (typically rituximab) remains the cornerstone of frontline therapy. Cytotoxic agents, such as purine analogue bendamustine (BR), alkylator cyclophosphamide with dexamethasone (DRC), and the proteasome inhibitor (PI) bortezomib with dexamethasone and cyclophosphamide (B-DRC) are widely used with good efficacy (26, 27). BR results in 2-year overall survival (OS) of 97% and progression-free survival (PFS) 89%, DRC 91%, and 69%, and B-DRC 94 and 81%, respectively (28–30).
3.2 Bruton’s tyrosine kinase inhibitors
Targeted agents are dramatically changing the treatment landscape, particularly in the relapsed and refractory (R/R) setting. Following the discovery of MYD88L265P, the use of targeted covalent BTK inhibitors (cBTKi) was shown to lead to apoptosis of WM cells (31, 32) leading to successful clinical trials (33). cBTKis are now commonly employed at relapse and continued until disease progression. In some healthcare systems, it is also available for frontline use without prior exposure to CIT and may be preferable to CIT in those with significant comorbidities or frailty (27). First-generation cBTKi ibrutinib demonstrated a 90.5% ORR and 79.4% MRR in R/R WM, with 5-year OS 87% and PFS 54% (34).
Subsequent iterations of cBKTis include next-generation agents Zanubrutinib and Acalabrutinib. Ibrutinib and Zanubrutinib have received Food and Drug Administration and European Medicines Agency approvals for use in WM, whereas Acalabrutinib is not available in Europe for this indication. There was no statistically significant difference in response rates between Ibrutinib and Zanubrutinib in the randomised phase III clinical trial ASPEN (94% vs. 95%, respectively) for R/R WM, with a median duration of response not reached at 44 months follow-up. The observed side effect profile was generally more favourable with Zanubrutinib (35). Acalabrutinib demonstrated efficacy in WM in a single-arm phase II study, with a 93% ORR of 93% both frontline and in R/R WM. cBTKi orelabrutinib and tirabrutinib have also demonstrated efficacy in Chinese and Japanese multicentre studies (36, 37).
However, complete responses are not attained with cBTKi and drug discontinuation often leads to rapid disease flare, necessitating continuous therapy until progression (38). Disease progression and resistance to cBTKi occur through several mechanisms. Half of cases due to the subclonal mutation BTKC481S, affecting the drug binding site, as well mutations in the downstream protein PLCγ2 (39). Patients who are quadruple-agent refractory (i.e., to an alkylating agent, rituximab, cBKTi, and PI) have a poor prognosis, with a median OS of 13.2 months (38).
Novel targeted agents under investigation in clinical trials include BCL2 inhibitors (BCL2i), non-covalent BTKi (ncBTKi) [such as nemtabrutinib and pritobrutinib (40, 41)], BTK degraders, and cellular therapies with bispecific T-cell engagers (such as epcoritamab) and chimeric antigen receptor T-cell (CAR-T) therapy (42, 43).
This review will focus on the role of BCL2 inhibitors.
4 Rationale for BCL2 inhibitor use in LPL/WM
The B-cell lymphoma 2 (Bcl-2) family of proteins has long been known to be key cellular apoptotic regulators (44), and the relationship between its pro- and anti-apoptotic members largely determines whether a cell survives or dies (45). Bcl-2 is the name of one of the constituent anti-apoptotic, pro-survival proteins within the wider Bcl2-group, which is capable of sequestering the pro-apoptotic proteins BAK, BAX, and other Bcl-2 homology domain 3(BH3)–only proteins. The regulation of Bcl-2 family proteins is complex but ultimately results in a binary decision whether a cell lives or undergoes apoptosis.
The therapeutic use of BCL2i has achieved impressive results in other B-cell disorders, notably chronic lymphocytic leukaemia (CLL), and drastically shifted the treatment landscape away from CIT (46). Gene expression profiling in LPL/WM cells has identified upregulation of BCL2, similar to that seen in CLL (47). In-vitro exposure of MYD88MUT cells, both CXCRWT and CXCRWHIM, resulted in apoptosis and has paved the way for therapeutic studies (47).
5 Existing evidence for BCL2 inhibitors in LPL/WM
No BCL2i is currently licenced for the treatment of WM/LPL. The only licensed BCL2i in haematological disorders is venetoclax, a small molecule that binds to BH3 and prevents the sequestering of anti-apoptotic proteins, thereby promoting apoptosis (48). It is licenced for and widely used in, CLL and acute myeloid leukaemia (AML). Several other BCL2 inhibitors are in development. Sonrotoclax is a second-generation, highly potent, and selective inhibitor of BCL2. It has shown greater in vitro inhibition of BCL2 than venetoclax, and currently undergoing testing in clinical trials. Interestingly, it has shown the ability to overcome several common venetoclax-resistance mutations, such as G101V in pre-clinical mouse models (49, 50).
To date, five studies have evaluated the use of BCL2i in LPL/WM (Table 1):
● The first was a phase 1/2 multicentre dose-escalation trial (NCT00062244), by Gertz and colleagues at the Mayo clinic, in patients with WM who received oblimersen sodium. This was an antisense oligonucleotide for the first six codons of the BCL2 open reading frame, which prevents the expression of the gene product. The results of the phase 1 portion were published in 2005, with a partial response in one of nine enrolled patients, but with grade 3 or higher haematological toxicities in five patients (51). A total of 58 patients were enrolled between 2003 and 2007, but no formal results of the phase 2 portion have been published to date (52).
● A phase I dose-escalation study (NCT01328626) by Davids et al. sought to evaluate the safety, pharmacokinetics, and preliminary efficacy of venetoclax in patients with R/R non-Hodgkin lymphomas. Venetoclax was given once daily until progressive disease or unacceptable toxicity, with stepwise dose titration to a maximal dose of 200–1200 mg. Treatment was given on a 3-week dose escalation protocol in most patients. The study began recruiting in 2011 and primary completion occurred in 2020. A total of 106 patients were enrolled, but only four patients with WM. The median age of WM patients was 67 years (range: 58–73 years), with a median of four prior lines of therapy. ORR and MRR were 100%, with all four patients attaining a partial response (PR). Median time to first response was 2.6 months and median duration of response 25.3 months. No patient with WM experienced tumour lysis syndrome (TLS). Study-wide rates of haematological toxicity were <20%, with 49% patients experiencing nausea, 46% diarrhoea, and 44% fatigue (53).
● A multicentre, prospective phase II study of fixed-duration venetoclax monotherapy in patients with previously treated WM (NCT02677324), by Castillo et al., recruited between 2016 and 2018. A total of 32 patients with WM were recruited, with a median of one prior treatment line. Sixteen patients had received previous cBTKi and seven were cBTKi-refractory. All patients harboured MYD88L265P and 17/32 CXCR4WHIM mutations. Venetoclax was administered orally once daily at 200 mg for 1 week, followed by 400 mg for 1 week, and then 800 mg for a total of 24 months. A study amendment allowed venetoclax to be administered orally once daily at 400 mg for one week followed by 800 mg for 24 months. ORR was 84% and MRR 81%. Categorical responses included 19% very good partial response (VGPR), 61% PR, and 3% minor response (MR). No patient attained a CR. Median PFS was 30 months, with 12- and 24-month PFS rates were 83% and 80%, respectively. Six patients progressed within the first 24 months, and 13 after completion of 24 months of venetoclax therapy. Prior cBTKi exposure was associated with longer duration to response but did not affect the maximum depth of response or PFS. All 32 patients were alive at time of data cutoff, with a 30-month OS rate of 100%. One patient experienced biochemical TLS, in the context of moderate nodal disease, splenomegaly, and peripheral lymphocytosis. Neutropaenia was common, including 14/32 grade ≥3 neutropaenias (54).
● A multicentre, single-arm prospective phase II study evaluated ibrutinib and venetoclax combination therapy in patients with previously untreated WM for a fixed duration of 24 months, by Castillo et al. (NCT04273139) (16). A total of 45 patients were recruited between 2020 and 2022. MYD88WT patients were excluded. Treatment cycles were administered every 28 days. Cycle one consisted of ibrutinib 420 mg, with the addition of venetoclax from cycle 2. Venetoclax was administered once daily at 100 mg for 1 week, 200 mg for another week, and 400 mg for 2 weeks. From cycles 3 to 24, participants received ibrutinib 420 mg and venetoclax 400 mg once daily, unless there was disease progression or unacceptable toxicity. TLS prophylaxis was provided in the form of outpatient oral hydration and allopurinol. The ORR was 100% and MRR 96%, with 42% attaining a VGPR, 53% PR, and 4% MR. The 24-month OS rate was 96% and PFS 76%. Crucially, ventricular arrhythmias occurred in three patients, with two deaths and one grade 4 event. The affected patients were all male, >65 years, and had cardiac comorbidities including a history of arrhythmia, coronary artery disease, hypertension, hypercholesterolemia, diabetes, or obesity. The study therapy was subsequently terminated after a grade 2 ventricular arrhythmia occurred in a participant undergoing a precautionary cardiac stress test, resulting in a concerning overall rate of ventricular arrhythmias of 9% in this study. Neutropaenia was common, including 17/45 grade ≥3 neutropaenias. The study authors concluded that there is a likely additive effect of combining BTK and BCL2 inhibitors in WM, with high rates of VGPR, rapid responses, and significant reduction in bone marrow burden (decreasing from 60% to 5% at best response), but that there was an unacceptably high incidence of ventricular events, including two deaths. Intriguingly, such events were not seen in other trials using ibrutinib and venetoclax combinations for other B-cell neoplasms (55, 56). Possible explanations include undetected cardiac involvement by AL amyloid, other cardiac paraprotein deposition, or an inherently higher risk of cardiac events in the patient cohort due to co-morbidities. The combination of ibrutinib and venetoclax for WM/LPL is therefore not recommended or currently planned for further study (16).
● A phase 1a/1b open-label dose escalation and expansion study is examining the use of second-generation sonrotoclax in patients with mature B-cell malignancies (NCT04277637) (57). Interim results were reported on a dedicated cohort of 17 patients with R/R WM, enrolled into three dose escalation cohorts. Previous cBTKi-exposure was noted in 10 patients. At a median follow-up of 10.6 months, four (24%) patients had progressed and two had discontinued due to adverse events. ORR was 76%, MRR 41%, and VGPR 12%. The study aims to complete by 2027.
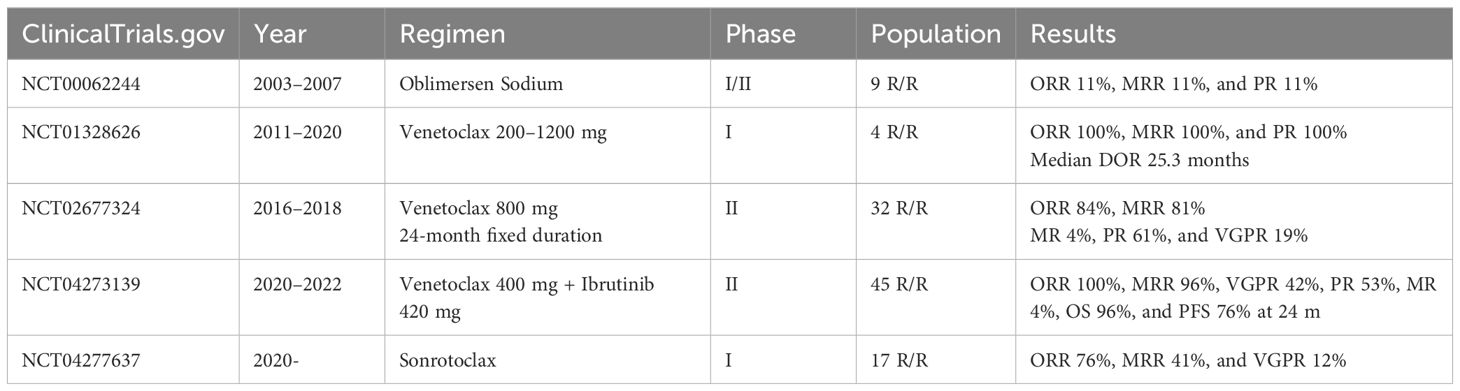
Table 1. Existing trials of BCL2 inhibition in WL/LPL.
6 Trials in progress for BCL2 inhibitors in LPL/WM
There are currently three trials in progress, evaluating the use of BCLi in LPL/WM (Table 2):
● A multicentre randomised phase II study is testing the combination of fixed-duration venetoclax and rituximab versus ibrutinib and rituximab for treatment naïve WM/LPL, given for a fixed-duration of 24 months (NCT04840602). The study is led by Dr. Sikander Ailawadhi at the SWOG Cancer Research Network. Enrolment commenced in 2022, with the aim to recruit 92 patients in total. Study completion is estimated by 2028. The primary objective is to compare the rates of ≥VGPR, with secondary objectives including the ORR, PFS, and OS rates (58).
● A single-arm open-label phase II study is evaluating the safety and efficacy of venetoclax in combination with the ncBTKi pirtobrutinib in R/R WM, for a total duration of 24 months (NCT05734495). The study is led by Dr. Jorge Castillo at the Dana Farber and began recruiting in 2023, aiming to enrol 42 patients. Study completion is estimated by 2033. The primary outcome measure is the incidence of ≥VGPR, with secondary objectives including PFS and OS rates (59).
● A multi-arm open label phase II study is evaluating the efficacy and safety of sonrotoclax in patients with WM who are refractory or intolerant to cBKTi (NCT05952037). It seeks to enrol a total of 105, with study completion by 2028. There are three cohorts: cohort 1 is for participants with R/R disease to both cBTKi and anti-CD20–containing CIT, cohort 2 R/R disease to anti-CD20–containing CIT with intolerance to BTKi, and cohort 3 R/R disease to BTKi and unsuitable for CIT. In addition, a fourth subcohort will evaluate the use of sonrotoclax in combination with cBTKi zanubrutinib in treatment-naïve patients for a fixed duration. The primary outcome measure is MRR, with secondary outcomes including ORR, rates of ≥VGPR, duration of response, PFS, and OS (60).
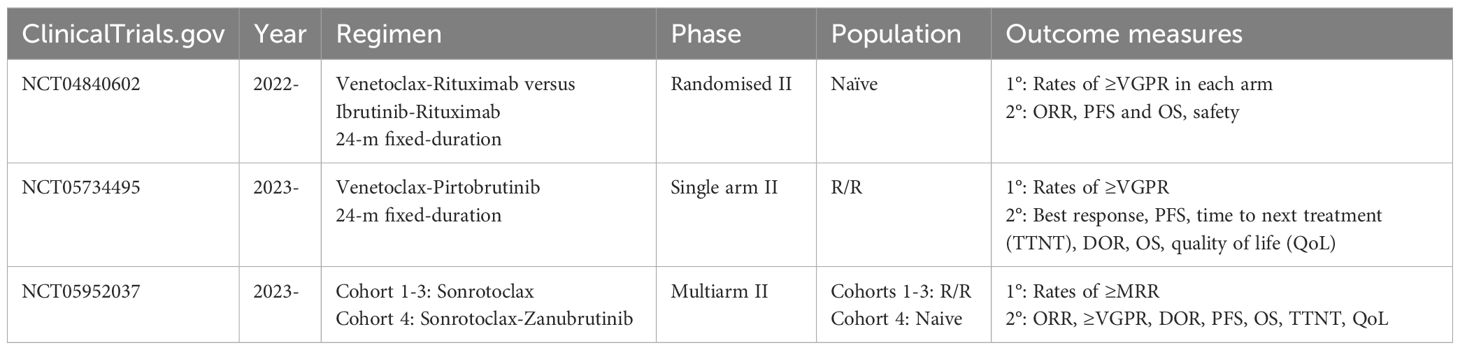
Table 2. Trials in progress of BCL2 inhibition in WL/LPL.
7 Conclusion
WM/LPL is an indolent but incurable condition, typically requiring several lines of therapy punctuated by periods of relative remission. Although CIT continues to be used, particularly for treatment-naïve patients where it has demonstrated good response rates and PFS, cBTKis are the mainstay of R/R WM. With increasing duration of use, resistance to cBTKis is an emerging problem and other oral agents are needed. A proportion of patients are also unable to tolerate BTKis due to class-specific side effects, such as bleeding. In addition, the prospect of a “chemotherapy-free,” fixed-duration frontline therapy would no doubt be appealing to patients and clinicians.
A total of five studies have examined the use of BCL2 inhibitors in WM/LPL: one oblimersen sodium, three venetoclax, and one sonrotoclax. Three pivotal trials are currently in progress, with the potential to alter practice in the treatment of LPL/WM. Fixed-duration venetoclax monotherapy for 24 months resulted in high-response rates but significant progression within 12 months of completion. This suggests that either continuous or combination therapy is required for the use of BCL2i in this condition. The combination of a BTKi ibrutinib and BCL2i venetoclax in LPL/WM, unfortunately, resulted in unacceptably high rates of ventricular events and death (16), essentially excluding this drug combination from further evaluation in WM/LPL. A cautious interpretation of the rapid responses and high rates of VGPR with this combination suggests that BCL2i in combination with other covalent- and non-covalent BTKis, with less cardiotoxic side effect profiles and in selected patients, may be of interest. The combination may particularly have a role in the context of heavy bone marrow infiltration, given the high rates of bone marrow clearance seen. This combination is currently being evaluated in two studies, one using sonrotoclax-zanubrutinib frontline and another using venetoclax-pirtobrutinib for R/R WM. The combination of a BCL2i with an anti-CD20 antibody, such as rituximab, may also prove efficacious.
The role of BCL2 inhibitors in WM/LPL remains under study, with many hopeful that they may provide an additional chemotherapy-free oral alternative for patients requiring treatment, both frontline and in the R/R setting. In an indolent condition with existing effective options, including CIT and cBTKi, cost-effectiveness and toxicity profile will be key. However, they are likely to find an important role if capable of offering an effective therapeutic mechanism for patients’ refractory to existing therapeutic options with limited options, particularly quadruple refractory patients.
Author contributions
OT: Writing – original draft, Writing – review & editing. SD’S: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
SD’S has received research support from Janssen and BeiGene, and advisory board honoraria from Janssen, BeiGene, HillStarBio, Kite and Sanofi.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, IBdO A, Berti E, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. (2022) 36:1720–48. doi: 10.1038/s41375-022-01620-2
2. Owen RG, Treon SP, Al-Katib A, Fonseca R, Greipp PR, McMaster ML, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol. (2003) 30:110–5. doi: 10.1053/sonc.2003.50082
3. Wang H, Chen Y, Li F, Delasalle K, Wang J, Alexanian R, et al. Temporal and geographic variations of Waldenstrom macroglobulinemia incidence: A large population-based study. Cancer. (2012) 118:3793–800. doi: 10.1002/cncr.v118.15
4. de Tute RM, Rawstron AC, Owen RG. Immunoglobulin M concentration in Waldenström macroglobulinemia: correlation with bone marrow B cells and plasma cells. Clin Lymphoma Myeloma Leuk. (2013) 13:211–3. doi: 10.1016/j.clml.2013.02.018
5. Eskeland T, Christensen TB. IgM molecules with and without J chain in serum and after purification, studied by ultracentrifugation, electrophoresis, and electron microscopy. Scand J Immunol. (1975) 4:217–28. doi: 10.1111/j.1365-3083.1975.tb02620.x
6. Khwaja J, D’Sa S, Minnema MC, Kersten MJ, Wechalekar A, Vos JM. IgM monoclonal gammopathies of clinical significance: diagnosis and management. Haematologica. (2022) 107:2037–50. doi: 10.3324/haematol.2022.280953
7. Zanwar S, Abeykoon JP, Durot E, King R, Perez Burbano GE, Kumar S, et al. Impact of MYD88L265P mutation status on histological transformation of Waldenström Macroglobulinemia. Am J Hematology. (2020) 95:274–81. doi: 10.1002/ajh.25697
8. Treon SP, Xu L, Guerrera ML, Jimenez C, Hunter ZR, Liu X, et al. Genomic landscape of waldenström macroglobulinemia and its impact on treatment strategies. J Clin Oncol. (2020) 38:1198–208. doi: 10.1200/JCO.19.02314
9. Yang G, Buhrlage SJ, Tan L, Liu X, Chen J, Xu L, et al. HCK is a survival determinant transactivated by mutated MYD88, and a direct target of ibrutinib. Blood. (2016) 127:3237–52. doi: 10.1182/blood-2016-01-695098
10. Hunter ZR, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. The genomic landscape of Waldenström macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. (2014) 123:1637–46. doi: 10.1182/blood-2013-09-525808
11. Willenbacher W, Willenbacher E, Brunner A, Manzl C. Improved accuracy of discrimination between IgM Multiple Myeloma and Waldenström Macroglobulinaemia by testing for MYD88 L265P mutations. Br J Haematology. (2013) 161:902–4. doi: 10.1111/bjh.2013.161.issue-6
12. Xu L, Hunter ZR, Tsakmaklis N, Cao Y, Yang G, Chen J, et al. Clonal architecture of CXCR4 WHIM-like mutations in Waldenström Macroglobulinaemia. Br J Haematol. (2016) 172:735–44. doi: 10.1111/bjh.2016.172.issue-5
13. Dogliotti I, Jiménez C, Varettoni M, Talaulikar D, Bagratuni T, Ferrante M, et al. Diagnostics in Waldenström’s macroglobulinemia: a consensus statement of the European Consortium for Waldenström’s Macroglobulinemia. Leukemia. (2023) 37:388–95. doi: 10.1038/s41375-022-01762-3
14. Cao Y, Hunter ZR, Liu X, Xu L, Yang G, Chen J, et al. The WHIM-like CXCR4(S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom’s Macroglobulinemia. Leukemia. (2015) 29:169–76. doi: 10.1038/leu.2014.187
15. Castillo JJ, Xu L, Gustine JN, Keezer A, Meid K, Dubeau TE, et al. CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenström macroglobulinaemia treated with ibrutinib. Br J Haematol. (2019) 187:356–63. doi: 10.1111/bjh.v187.3
16. Castillo JJ, Branagan AR, Sermer D, Flynn CA, Meid K, Little M, et al. Ibrutinib and venetoclax as primary therapy in symptomatic, treatment-naïve Waldenström macroglobulinemia. Blood. (2024) 143:582–91. doi: 10.1182/blood.2023022420
17. Varettoni M, Zibellini S, DeFrancesco I, Ferretti VV, Rizzo E, Malcovati L, et al. Pattern of somatic mutations in patients with Waldenström macroglobulinemia or IgM monoclonal gammopathy of undetermined significance. Haematologica. (2017) 102:2077–85. doi: 10.3324/haematol.2017.172718
18. Brunner A, Thalhammer-Thurner GC, Willenbacher W, Haun M, Zelger BG, Willenbacher E. In-depth molecular analysis of lymphomas with lymphoplasmacytic differentiation may provide more precise diagnosis and rational treatment allocation. Ann Hematol. (2024) 103:553–63. doi: 10.1007/s00277-023-05531-9
19. Gustine JN, Tsakmaklis N, Demos MG, Kofides A, Chen JG, Liu X, et al. TP53 mutations are associated with mutated MYD88 and CXCR4, and confer an adverse outcome in Waldenström macroglobulinaemia. Br J Haematology. (2019) 184:242–5. doi: 10.1111/bjh.15560
20. Poulain S, Roumier C, Bertrand E, Renneville A, Caillault-Venet A, Doye E, et al. TP53 mutation and its prognostic significance in waldenstrom’s macroglobulinemia. Clin Cancer Res. (2017) 23:6325–35. doi: 10.1158/1078-0432.CCR-17-0007
21. Krzisch D, Guedes N, Boccon-Gibod C, Baron M, Bravetti C, Davi F, et al. Cytogenetic and molecular abnormalities in Waldenström’s macroglobulinemia patients: Correlations and prognostic impact. Am J Hematol. (2021) 96:1569–79. doi: 10.1002/ajh.v96.12
22. Braggio E, Keats JJ, Leleu X, Van Wier S, Jimenez-Zepeda VH, Valdez R, et al. Identification of copy number abnormalities and inactivating mutations in two negative regulators of nuclear factor-κB signaling pathways in waldenstroüm’s macroglobulinemia. Cancer Res. (2009) 69:3579–88. doi: 10.1158/0008-5472.CAN-08-3701
23. García-Sanz R, Dogliotti I, Zaccaria GM, Ocio EM, Rubio A, Murillo I, et al. 6q deletion in Waldenström macroglobulinaemia negatively affects time to transformation and survival. Br J Haematology. (2021) 192:843–52. doi: 10.1111/bjh.17028
24. Bustoros M, Sklavenitis-Pistofidis R, Kapoor P, Liu CJ, Kastritis E, Zanwar S, et al. Progression risk stratification of asymptomatic Waldenström macroglobulinemia. J Clin Oncol. (2019) 37:1403–11. doi: 10.1200/JCO.19.00394
25. Buske C, Sadullah S, Kastritis E, Tedeschi A, García-Sanz R, Bolkun L, et al. Treatment and outcome patterns in European patients with Waldenström’s macroglobulinaemia: a large, observational, retrospective chart review. Lancet Haematol. (2018) 5:e299–309. doi: 10.1016/S2352-3026(18)30087-5
26. Kastritis E, Leblond V, Dimopoulos MA, Kimby E, Staber P, Kersten MJ, et al. Waldenström’s macroglobulinaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann oncology: Off J Eur Soc Med Oncol. (2019) 29:iv41–50. doi: 10.1093/annonc/mdy466
27. Buske C, Castillo JJ, Abeykoon JP, Advani R, Arulogun SO, Branagan AR, et al. Report of consensus panel 1 from the 11th International Workshop on Waldenstrom’s Macroglobulinemia on management of symptomatic, treatment-naïve patients. Semin Hematology. (2023) 60:73–9. doi: 10.1053/j.seminhematol.2023.03.005
28. Chan W-L, Chong VCL, Wee IJY, Poon LM, Chan EHL, Lee J, et al. Efficacy and safety of front-line treatment regimens for Waldenstrom macroglobulinaemia: a systematic review and meta-analysis. Blood Cancer J. (2023) 13:140. doi: 10.1038/s41408-023-00916-5
29. Laribi K, Poulain S, Willems L, Merabet F, Le Calloch R, Eveillard JR, et al. Bendamustine plus rituximab in newly-diagnosed Waldenström macroglobulinaemia patients. A study on behalf of the French Innovative Leukaemia Organization (FILO). Br J Haematol. (2019) 186:146–9. doi: 10.1111/bjh.2019.186.issue-1
30. Paludo J, Ansell SM. Advances in the understanding of IgM monoclonal gammopathy of undetermined significance. F1000Res. (2017) 6:2142. doi: 10.12688/f1000research
31. Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. New Engl J Med. (2012) 367:826–33. doi: 10.1056/NEJMoa1200710
32. Yang G, Zhou Y, Liu X, Xu L, Cao Y, Manning RJ, et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia. Blood. (2013) 122:1222–32. doi: 10.1182/blood-2012-12-475111
33. Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, et al. Ibrutinib in previously treated waldenström’s macroglobulinemia. New Engl J Med. (2015) 372:1430–40. doi: 10.1056/NEJMoa1501548
34. Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, et al. Ibrutinib in previously treated Waldenström’s macroglobulinemia. New Engl J Med. (2015) 372:1430–40. doi: 10.1056/NEJMoa1501548
35. Dimopoulos MA, Opat S, D’Sa S, Jurczak W, Lee H-P, Cull G, et al. Zanubrutinib versus ibrutinib in symptomatic waldenström macroglobulinemia: final analysis from the randomized phase III ASPEN study. J Clin Oncol. (2023) 41:5099–106. doi: 10.1200/JCO.22.02830
36. Cao XX, Jin J, Fu CC, Yi SH, Zhao WL, Sun ZM, et al. Evaluation of orelabrutinib monotherapy in patients with relapsed or refractory Waldenström’s macroglobulinemia in a single-arm, multicenter, open-label, phase 2 study. EClinicalMedicine. (2022) 52:101682. doi: 10.1016/j.eclinm.2022.101682
37. Sekiguchi N, Rai S, Munakata W, Suzuki K, Handa H, Shibayama H, et al. A multicenter, open-label, phase II study of tirabrutinib (ONO/GS-4059) in patients with Waldenström’s macroglobulinemia. Cancer Sci. (2020) 111:3327–37. doi: 10.1111/cas.v111.9
38. Gustine JN, Sarosiek S, Flynn CA, Meid K, Leventoff C, White T, et al. Natural history of Waldenström macroglobulinemia following acquired resistance to ibrutinib monotherapy. Haematologica. (2022) 107:1163–71. doi: 10.3324/haematol.2021.279112
39. Xu L, Tsakmaklis N, Yang G, Chen JG, Liu X, Demos M, et al. Acquired mutations associated with ibrutinib resistance in Waldenström macroglobulinemia. Blood. (2017) 129:2519–25. doi: 10.1182/blood-2017-01-761726
40. Scarfò L, Patel MR, Eyre TA, Jurczak W, Lewis D, Gastinne T, et al. P1108: efficacy of pirtobrutinib, a highly selective, non-covalent (Reversible) btk inhibitor in relapsed/refractory waldenstrÖm macroglobulinemia: results from the phase 1/2 bruin study. Hemasphere. (2023) 7:e852670f. doi: 10.1097/01.HS9.0000971328.85267.0f
41. Woyach JA, Stephens DM, Flinn IW, Bhat SA, Savage RE, Chai F, et al. First-in-human study of the reversible BTK inhibitor nemtabrutinib in patients with relapsed/refractory chronic lymphocytic leukemia and B-cell non-hodgkin lymphoma. Cancer Discovery. (2024) 14:66–75. doi: 10.1158/2159-8290.CD-23-0670
42. A phase 2 trial investigating epcoritamab in patients with previously treated waldenstrom macroglobulinemia (WM)(2024). Available online at: https://clinicaltrials.gov/study/NCT06510491 (Accessed October 2, 2024).
43. A phase 2, open-label, multicenter, basket study evaluating the efficacy of brexucabtagene autoleucel in adults with rare B-cell Malignancies (ZUMA-25)(2022). Available online at: https://clinicaltrials.gov/study/NCT05537766 (Accessed October 2, 2024).
44. Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. (1988) 335:440–2. doi: 10.1038/335440a0
45. Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. (2019) 20:175–93. doi: 10.1038/s41580-018-0089-8
46. Wierda WG, Tambaro FP. How I manage CLL with venetoclax-based treatments. Blood. (2020) 135:1421–7. doi: 10.1182/blood.2019002841
47. Chng WJ, Schop RF, Price-Troska T, Ghobrial I, Kay N, Jelinek DF, et al. Gene-expression profiling of Waldenstrom macroglobulinemia reveals a phenotype more similar to chronic lymphocytic leukemia than multiple myeloma. Blood. (2006) 108:2755–63. doi: 10.1182/blood-2006-02-005488
48. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. (2013) 19:202–8. doi: 10.1038/nm.3048
49. Liu J, Li S, Wang Q, Feng Y, Xing H, Yang X, et al. Sonrotoclax overcomes BCL2 G101V mutation–induced venetoclax resistance in preclinical models of hematologic Malignancy. Blood. (2024) 143:1825–36. doi: 10.1182/blood.2023019706
50. Guo Y, Xue H, Hu N, Liu Y, Sun H, Yu D, et al. Discovery of the clinical candidate sonrotoclax (BGB-11417), a highly potent and selective inhibitor for both WT and G101V mutant bcl-2. J Med Chem. (2024) 67:7836–58. doi: 10.1021/acs.jmedchem.4c00027
51. Gertz MA, Geyer SM, Badros A, Kahl BS, Erlichman C. Early results of a phase I trial of oblimersen sodium for relapsed or refractory waldenström’s macroglobulinemia. Clin Lymphoma. (2005) 5:282–4. doi: 10.3816/CLM.2005.n.017
52. Phase I/II study of G3139 (Genasense) in patients with waldenstrom’s macroglobulinemia(2003). Available online at: https://clinicaltrials.gov/study/NCT00062244 (Accessed September 1, 2024).
53. Davids MS, Roberts AW, Kenkre VP, Wierda WG, Kumar A, Kipps TJ, et al. Long-term follow-up of patients with relapsed or refractory non-hodgkin lymphoma treated with venetoclax in a phase I, first-in-human study. Clin Cancer Res. (2021) 27:4690–5. doi: 10.1158/1078-0432.CCR-20-4842
54. Castillo JJ, Allan JN, Siddiqi T, Advani RH, Meid K, Leventoff C, et al. Venetoclax in previously treated waldenström macroglobulinemia. J Clin Oncol. (2022) 40:63–71. doi: 10.1200/JCO.21.01194
55. Eichhorst B, Niemann CU, Kater AP, Fürstenau M, von Tresckow J, Zhang C, et al. First-line venetoclax combinations in chronic lymphocytic leukemia. N Engl J Med. (2023) 388:1739–54. doi: 10.1056/NEJMoa2213093
56. Wang M, Ramchandren R, Chen R, Karlin L, Chong G, Jurczak W, et al. Concurrent ibrutinib plus venetoclax in relapsed/refractory mantle cell lymphoma: the safety run-in of the phase 3 SYMPATICO study. J Hematol Oncol. (2021) 14:179. doi: 10.1186/s13045-021-01188-x
57. A phase 1a/1b open-label dose escalation and expansion study of bcl-2 inhibitor BGB-11417 in patients with mature B-cell Malignancies(2020). Available online at: https://clinicaltrials.gov/study/NCT04277637 (Accessed September 1, 2024).
58. A Phase II Randomized Study Comparing Ibrutinib and Rituximab vs. Venetoclax and Rituximab in Previously Untreated Waldenström’s Macroglobulinemia (WM)/Lymphoplasmacytic Lymphoma (LPL)(2021). Available online at: https://clinicaltrials.gov/study/NCT04840602 (Accessed September 1, 2024).
59. A phase II study evaluating venetoclax and pirtobrutinib in previously treated waldenström macroglobulinemia(2023). Available online at: https://clinicaltrials.gov/study/NCT05734495 (Accessed September 1, 2024).
60. An open-label, multicenter phase 2 study to evaluate the efficacy and safety of the BCL2 inhibitor sonrotoclax (BGB-11417) as monotherapy and in combination with zanubrutinib (BGB-3111) in patients with waldenström macroglobulinemia(2023). Available online at: https://clinicaltrials.gov/study/NCT05952037 (Accessed September 1, 2024).
Keywords: BCL2, Waldenström’s macroglobulinaemia, lymphoplasmacytic lymphoma, venetoclax, sonrotoclax
Citation: Tomkins O and D’Sa S (2024) Review of BCL2 inhibitors for the treatment of Waldenström’s macroglobulinaemia and non-IgM lymphoplasmacytic lymphoma. Front. Oncol. 14:1490202. doi: 10.3389/fonc.2024.1490202
Received: 02 September 2024; Accepted: 14 October 2024;
Published: 04 November 2024.
Edited by:
Konstantinos Liapis, Democritus University of Thrace, GreeceReviewed by:
Stavros Papadakis, University Hospital of Heraklion, GreeceGeorge Vrachiolias, Democritus University of Thrace, Greece
Ioannis Liapis, Chania General Hospital St. George, Greece
Copyright © 2024 Tomkins and D’Sa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shirley D’Sa, s.d’sa@nhs.net