 Marju Puurand
Marju Puurand Alicia Llorente
Alicia Llorente Aija Linē
Aija Linē Tuuli Kaambre
Tuuli Kaambre
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 06 January 2025
Sec. Cancer Metabolism
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1480074
Cancer is caused by complex interactions between genetic, environmental, and lifestyle factors, making prevention strategies, including exercise, a promising avenue for intervention. Physical activity is associated with reduced cancer incidence and progression and systemic anti-cancer effects, including improved tumor suppression and prolonged survival in preclinical models. Exercise impacts the body’s nutrient balance and stimulates the release of several exercise-induced factors into circulation. The mechanisms of how exercise modulates cancer energy metabolism and the tumor microenvironment through systemic effects mediated, in part, by extracellular vesicles (EVs) are still unknown. By transferring bioactive cargo such as miRNAs, proteins and metabolites, exercise-induced EVs may influence cancer cells by altering glycolysis and oxidative phosphorylation, potentially shifting metabolic plasticity – a hallmark of cancer. This short review explores the roles of EVs in cancer as mediators to reprogram cellular energy metabolism through exchanging information inside the tumor microenvironment, influencing immune cells, fibroblast and distant cells. Considering this knowledge, further functional studies into exercise-induced EVs and cellular energy production pathways could inform more specific exercise interventions to enhance cancer therapy and improve patient outcomes.
Most cancers arise from a complex etiology involving genetic, environmental and lifestyle factors and their interactions (1, 2), and there is a great need and opportunity for cancer prevention through lifestyle change. Numerous epidemiological studies have provided evidence supporting the link between higher physical activity levels and a lower incidence of multiple cancer types (3, 4). Systemic reviews of the preclinical animal studies have demonstrated the anti-cancer effect (a decrease in cancer incidence and tumor size and a prolonged life span of animals) of physical exercise (5–7). The exact doses, types and routines/protocols of physical activity in decreasing cancer risk and mortality continue to be under active investigation. Elucidation of the mechanisms linking bodily activity interventions to cancer may have great potential in fighting this debilitating disease.
Profound changes in both cell genome and gene expression during malignant transformation are likely to be accompanied by changes in metabolism, including pathways for cellular energy production (8). The metabolomic analysis of human patients’ colorectal cancer xenografts in mice models revealed exercise-induced changes in intratumoral central carbon metabolism (especially mitochondrial metabolism), independently of whether exercise inhibited tumor growth (9). Investigating the mechanisms of exercise`s effect on cancer cells and tumors is complicated. This is because cancer cells inside the tumor are heterogeneous, and in addition they are surrounded and dependent on the functioning of cells forming the tumor microenvironment (10, 11), which can also be affected by exercise.
In general, the explanations of how exercise and muscle-derived bioactive molecules secreted into circulation can affect cancer cells (summarizing the review by Kang et al. (12)) are (1) enhancement of cancer immunosurveillance through increased innate immune function to boost the body’s ability to detect tumor cells and inhibit their growth; (2) regulation of inflammation and oxidative stress in conjunction with immune functions; (3) activation of physiological mechanisms such as changes in nutrient availability and metabolism. In parallel, physical exercise can stimulate tumor vascularization and improve the infiltration of systemic biomolecules and immune cells, thus affecting the entire tumor microenvironment.
One of the communication forms between cells across the body is via extracellular vesicles (EVs). EVs are lipid bilayer-enclosed nano to micro-sized particles that cannot replicate independently and are released by virtually all cell types in the body (13). Mounting evidence supports the hypothesis that EVs are not a random waste elimination mechanism but mediate intercellular communication, allowing the specific intercellular exchange of proteins, lipids, and genetic material (14, 15). Several recent studies have shown that the amount of circulating EVs rapidly increases due to physical exercise (16–18). According to the study by Frühlbeis et al., 2015, the increase in plasma EV peaks at 10 min after acute exercise, starts to decline within 30 min after training and returns to the baseline levels after 90 min (16). However, the increase in EV levels after exercise does not seem to be related to exercise intensity per se (16, 19). It has been suggested that EVs released from contracting skeletal muscles play a central role in mediating the systemic benefits of exercise by transporting bioactive molecules, which account for only about 5% of plasma EVs, although EVs released by immune cells contribute more (20). The results of several studies with human subjects suggest that exercise affects the content of circulating EVs in the blood (16–18, 21–23). A comprehensive review of the content of exercise-induced EVs (especially regarding nucleic acid content) can be found in Llorente et al., 2024 (23). Exercise-conditioned serum and myokines have been suggested to impact several hallmark features of cancer and act as tumor suppressors in breast, colon and lung cancer (24, 25). The role of EVs released into the systemic circulation during exercise in influencing the reprogramming of cancer energy metabolism is unknown. Considering the current understanding of EV circulation kinetics, the systemic effects of exercise-induced EVs seem to be relatively short-lived. However, localized increases in EVs within tissues and tumors may have more enduring effects and influence cellular energy metabolism.
The EVs secreted by tumors are abundant in plasma, and their use to find biomarkers for assessing disease progression or treatment efficacy is widely explored (26, 27). It also has to be considered that during exercise, the same stimuli that trigger the EV release from normal cells could also stimulate the release of EVs from cancer cells. However, this can have unintended consequences. EVs released by drug-resistant tumor cells or neighboring cells from the tumor microenvironment have an established role in the intercellular transfer of drug-resistance traits, with accompanying metabolic reprogramming, to drug-sensitive tumor cells (28) as reviewed by Polonia and Xavier (29, 30). In this context, a detailed knowledge of how different exercise types and doses affect the energy metabolism of cancer cells and other cells in the cancer microenvironment could help determine the type of exercise suitable for the patient during treatment, allowing better use or mimicking of the positive effects of exercise. Investigating the role of EV cargo in the modulation of tumor energy metabolism represents a new opportunity to target and stop disease development, particularly regarding antibodies’ against specific putative membrane receptors.
This mini-review analyzes examples of functional studies of how EV content can reprogram recipient cell glycolysis and OXPHOS in cancer. This knowledge also reflects on the potential role of exercise-induced EVs in reprogramming cellular energy metabolism in cancer.
Changes in cell metabolism contribute to transformation and tumor progression. There are only two central energy-producing metabolic pathways, glycolysis and oxidative phosphorylation (OXPHOS), which are, at the same time, sources for essential precursors for nucleotide, polysaccharides, amino saccharides, amino acids and lipid biosynthesis (31–33). The constant need of cancer cells for resources to ensure proliferation keeps both pathways working in cancer cells as well, but often, the proportion of usage and partly also the direction of the pathway is different in cancer cells than in normal tissue cells (34).
The primary bioenergetic profile of a cancer cell, which is different from its progenitor, may be as follows: (1) enhanced glycolysis under any conditions (Warburg effect); (2) unlike normal cells, cancer cells utilize both anaerobic and aerobic energy production with increased capacity (a hybrid phenotype); (3) be mainly OXPHOS users (34, 35). In addition, unlike non-neoplastic cells, a striking feature of cancer cells is their metabolic plasticity – the ability to readily switch their metabolic phenotypes to glycolysis or OXPHOS in response to changes in the microenvironment (i.e., variations in O2 concentration, lactate concentration, pH, carbon source and growth factors availability) or inhibition of one of these pathways, giving survival advantages during tumor progression (34, 36). This metabolic plasticity is promoted by the hybrid bioenergetic phenotype and is linked with metastasis and chemoresistance (35). However, it is still not fully understood how cancer cells regulate gene expression to maintain their hybrid energy metabolism state, although variations in the expression of key transcriptional factors and oncogenes like HIF-1α, p53, c-Myc, PGC-1α are clearly involved (37). Elucidation of intrinsic and extrinsic factors (e.g., EVs) in the metabolic reprogramming of cancer cells is beneficial for better treatment of disease.
There is a growing recognition that the metabolism of cells other than cancer cells within the tumor microenvironment, like endothelial cells, fibroblasts and immune cells, can modulate tumor progression, including supporting and shaping the metabolism of cancer cells (38, 39). In addition to the soluble factors found in the tumor microenvironment, cancer cells and their energy metabolism are also targets for active communication with EVs released into the tumor microenvironment by surrounding cells, mainly by cancer-associated fibroblasts (CAFs) and tumor-associated macrophages (TAMs) (40, 41). Indeed, in breast tumors, cancer cells and CAFs may establish the coupled metabolic pattern sometimes referred to as the „reverse Warburg effect”, by which glycolytic CAFs provide energy-rich metabolites like lactate and glutamine to fuel the mitochondrial respiration and anabolic metabolism of cancer cells (42). Mechanisms of this setup also involve EVs secreted into the tumor microenvironment by all cells involved.
There is evidence that patient-derived CAFs can extensively reprogram the energy metabolism of the recipient cancer cell in culture with EVs only (43). The relatively high abundance of miRNAs that target OXPHOS genes resulted in decreased mitochondrial respiration, upregulated glycolysis and enhanced reductive pathway of glutamine metabolism (reductive carboxylation) in prostate cancer cells cultivated with EVs released from prostate cancer patient-derived CAFs (43). Supplementation of cellular nutrition through the secretion of metabolites is another possible mechanism besides miRNAs in how the EVs from CAFs affect the metabolism of recipient tumor cells. Exosomes released by CAFs derived from prostate cancer patients are a vital source of metabolites that are essential contributors to central carbon metabolism under conditions of nutrient deprivation (43).
In turn, EV-packed miRNAs derived from cancer cells could be critical agents modulating the energy metabolism of target cells in the tumor microenvironment. For example, the breast cancer cell-secreted and EV-encapsulated miR-105 reprograms energy metabolism in CAFs (44). According to the model, a high MYC activity in cancer cells leads to a high secretion of miR-105 to activate MYC signaling in stromal cells, phenotypically extending the effects of MYC originating from cancer cells to non-cancer cells. The miR-105-EV-reprogrammed stromal cells increased the catabolism of glucose and glutamine to fuel adjacent cancer cells when nutrients were sufficient. When nutrients are in shortage and metabolic by-products accumulate, these reprogrammed CAFs detoxify metabolic wastes, including lactic acid and ammonium, by converting them into fuel metabolites, making them substrates for the surrounding cancer cells (44).
It has been suggested that one of the possible mechanisms of exercise for cancer prevention or in inhibiting cancer development might be the intervention of the regulation of TAM polarization (45). TAM polarization is a process actively driven by cancer cell-derived EVs. A previous investigation revealed that EVs derived from human hepatocellular carcinoma carrying glycolytic enzyme pyruvate kinase M2 isoform (PKM2) foster the progression of carcinoma by promoting PKM2 polarization of macrophages and reconfiguring the tumor immune microenvironment (46). A similar phenomenon is demonstrated also in gastric cancer (47). Wu et al. showed that gastric cancer cells can deliver PKM2 packed into EVs to macrophages, leading to the differentiation of macrophages into the pro-inflammatory M2 subtype, consequently promoting the progression of gastric cancer. In addition, in a study involving patients with neuroblastoma, glycolytic enzymes PKM2 and Hexokinase II were detected in circulating EVs isolated from blood using mass spectrometry (48). Overall, tumor cell-derived PKM2 could promote macrophage differentiation through glycolytic reprogramming. However, the non-metabolic functions of PKM2, such as its activity as a protein kinase or transcriptional coactivator (49, 50), could also participate in metabolic reprogramming. How PKM2 levels in EVs are affected by exercise needs further investigation.
An example is also found in EVs from TAMs’ participation in reprogramming breast cancer cell metabolism (51). Primary TAMs isolated from breast cancer patients were co-cultured with breast cancer cells without physical contact between them. TAM-derived EVs enhance breast cancer cells’ glycolysis and apoptotic resistance by transmitting long noncoding RNAs that stabilize HIF1-alpha and enhance glycolysis in the target cells (51). Unfortunately, the function of OXPHOS was not evaluated in this study. However, there is a lack of studies showing the direct functional effect of factors contained in EVs on the energy metabolism of recipient cancer cells. The profile of EVs probably also affects the metabolic plasticity of cancer cells, which changes more unfavorable for cancer. Functional investigation of mechanistic basis and intercellular codependencies in tumor microenvironments may allow the identification of specific vulnerabilities and targets for eradicating cancer cells. How physical exercise affects these relationships is a challenging research topic for the future.
Physical activity (particularly intense exercise) affects the whole body, is safe for patients, and improves their quality of life. Exercise-induced overall systemic effects on cellular bioenergetics can be realized through fluctuations in nutrient balance. At the molecular level, an exercise-induced deficit of nutrients such as glucose and glutamine downregulates intrinsic cellular cancer-promoting signaling pathways like PI3K/AKT/mTOR and RAS-ERK and thereby increases AMP-activated protein kinase (AMPK) activity (52, 53). As AMPK acts as a master regulator of OXPHOS and mitochondrial biogenesis (54), which generally inhibits anabolism under energetic stress, the bioenergetic consequences of such changes involve the downregulation of (aerobic) glycolysis and activation of mitochondrial biogenesis and OXPHOS (55).
Other mechanisms influencing metabolism are driven by agents secreted by cells during exercise. The results of several studies with human subjects suggest that exercise affects the content of circulating EVs in the blood (16–18, 21, 22). Further, exercise-induced EVs also carry active enzymes, critical regulatory factors and metabolites that affect OXPHOS and glycolysis fluxes (Figure 1), researching the effects of which would help implement exercise programs that support cancer treatment.
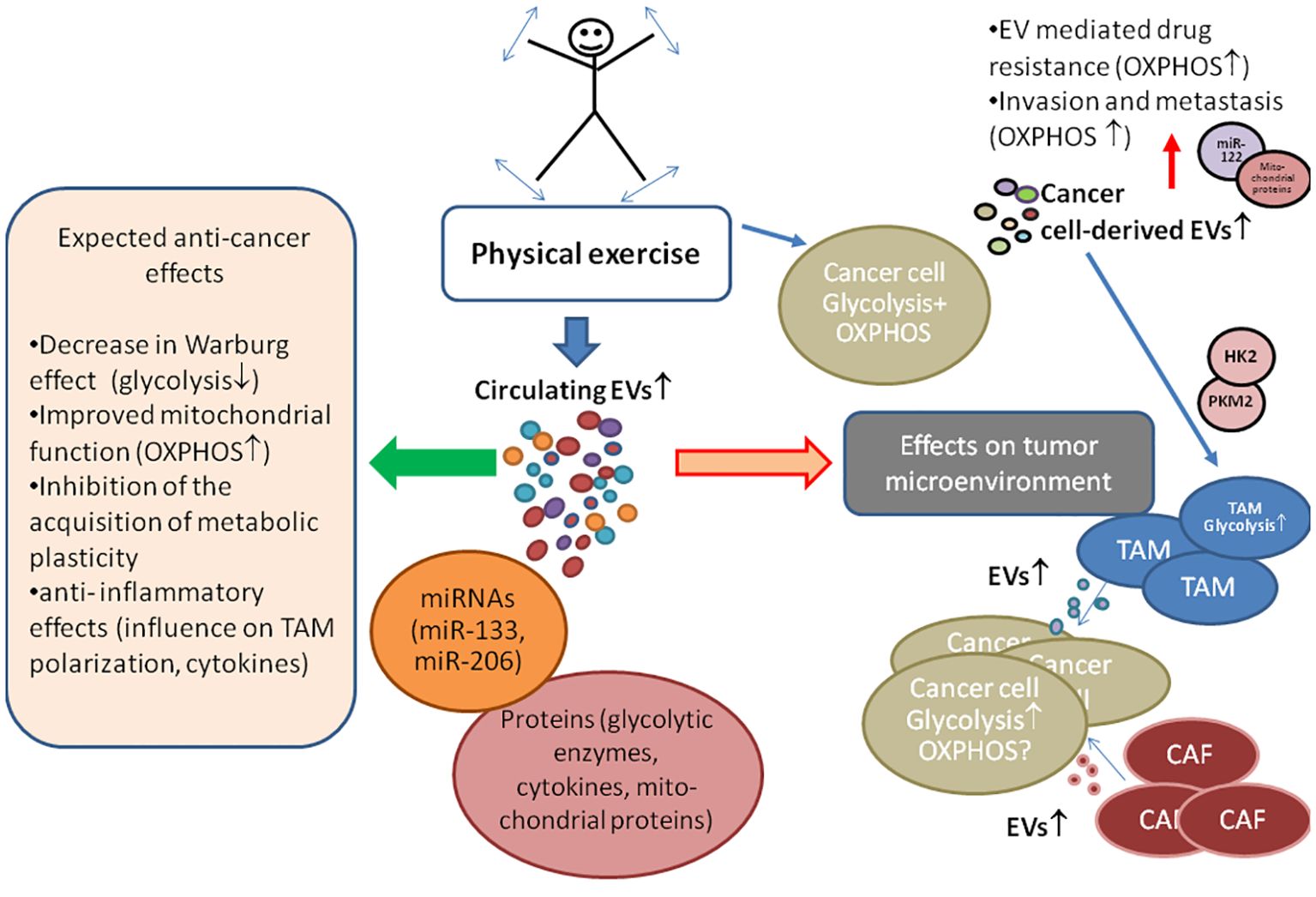
Figure 1. The role of exercise-induced extracellular vesicles (EVs) in cancer energy metabolism and tumor microenvironment. Experimental animal studies have consistently reported that physical exercise leads to a reduction in cancer incidence, a decrease in tumor size, and a prolonged life span of animals, suggesting similar benefits for humans. Exercise prompts various cells, including cancer cells, to release EVs containing bioactive molecules like glycolytic enzymes (pyruvate kinase II (PKM2), hexokinase II (HK2)), mitochondrial proteins, and miRNAs, which influence recipient cell metabolism. Cancer energy metabolism is often characterized by high glycolysis activity, with oxidative phosphorylation (OXPHOS) also not being defective. Unlike normal cells, cancer cells acquire the ability to switch their primary energy production pathway, i.e., they acquire metabolic plasticity supporting growth and drug resistance. EVs secreted by tumor microenvironment cells, such as cancer-associated fibroblasts (CAF) and tumor-associated fibroblasts (TAM), can enhance glycolysis in cancer cells. However, cancer cell-derived EVs may have a pro-OXPHOs effect (demonstrated through miR-122 and mitochondrial component exchange), contributing to drug resistance and supporting cellular metabolic plasticity, effects that can be minimized with the proper exercise regimens. The symbols ↓,↑ indicate up- and down-regulation, respectively.
A recent study demonstrated that exercise induces the secretion of over 300 proteins into EVs, including glycolytic enzymes and cytokines secreted by skeletal muscle (18). Proteome data show that small EV-delivered glycolytic enzymes can influence the glycolysis rate of recipient cells (56), but the exact extent of such changes is not well known.
Within the various exercise- and cancer-related EV cargos, miRNAs are some of the most extensively studied candidates for mediating specific muscle-to-cancer crosstalk (57–59). Among the exercise-induced miRNA families, miR-1, -133 and -206 are the most widely investigated (60, 61). In several studies, members of these families have been shown to be enriched in EVs released after exercise (21, 62–64). However, miR-1, miR-133a, miR-133b and miR-206 are primarily down-regulated in various cancers where they function as tumor suppressors (65, 66). A direct effect on cancer energy metabolism has only been shown for miR-133b. The regulatory network of miR-133b is involved in the altered energy metabolism of cancer cells. MiR-133b controls pyruvate kinase expression, thereby repressing glycolysis (67–69), a property that can confuse the metabolism of cancer cells. This process reduces the supply of pyruvate, which may decrease OXPHOS activity along with higher production of oxygen free radicals. Under these conditions, cancer cells could become vulnerable to treatments that induce oxidative stress, such as doxorubicin or radiation.
So far, glycolysis in cancer has received more attention. However, OXPHOS is also active in cancer cells, which has yet to be directly measured and investigated. Different approaches involving high-resolution respirometry in cells and tissues allow the determination of OXPHOS activity and efficiency and cancer-specific alterations in OXPHOS donor pathways such as the Krebs cycle (70, 71). Such studies in the context of exercise-induced EVs would contribute to implementing the positive effects of exercise in clinical practice.
Mitochondria are dynamic organelles, and the constant remodeling of mitochondria is driven mainly by the outcome of continuous maintenance and quality control. The number of mitochondria and their activity can change in response to various physiological or pathological conditions. As a part of routine mitochondrial maintenance, damaged mitochondria undergo fission, and resulting vesicles are directed to mitophagy by lysosomes (72). In various cell types, the generation of mitochondria-derived vesicles is accelerated by oxidative stress and their cargo is enriched with oxidized proteins (73, 74). As the biogenesis of mitochondria-derived vesicles is faster than fission, resulting vesicles are likely to be the first round of defense for the mitochondria to eject damaged proteins and avoid the complete failure of the organelle (75, 76). Therefore, it is unsurprising that specific EV subpopulations isolated from blood plasma have been found to contain mitochondrial proteins, fragments, and, in some cases, full-length mitochondrial DNA (77).
Consequently, an increase in mitochondrial proteins in EVs can reflect the presence and degree of disease. Notably, some evidence suggests that EVs isolated from the blood plasma of cancer patients are enriched with mitochondrial membrane proteins compared to EVs derived from control patients (15). Plasma EVs from breast cancer patients contain higher levels of mitochondrially encoded mRNAs for subunits of NADH dehydrogenase, ATP synthase, and cytochrome C oxidase (78). Enrichment of plasma EVs with mitochondrial proteins and mRNAs in cancer suggests that mitochondria-derived vesicles may play an important role in modulating the cellular stress response.
However, which cells exactly produce these vesicles and whether these vesicles also have specific recipients with specific receptors is not known.
At the local level, within the tumor microenvironment, there is evidence of the intercellular transfer of mitochondria as an additional mechanism to replace damaged mitochondria. Mitochondrial transfer between astrocytes and neurons has been observed after acute stress like an ischemic insult (79), from mesenchymal stem cells to injured pulmonary epithelial cells (80–82), both in vivo and in vitro, and from bone marrow stromal cells to acute myeloid leukemia cells during chemotherapy (83). Mitochondrial transfer between mesenchymal stem cells, endothelial cells, and cancer cells by tunneling via nanotubes occurs probably more often than one might think and is an influencing factor in cancer treatment by promoting the development of chemoresistance (84–88).
The cancer-promoting effect of mitochondria transfer is suggested to occur mainly by potentiating OXPHOS through the influx of mutation-free mitochondrial DNA, which ensures efficient energy production and reduces oxygen free radical generation (82, 89). How exercise-induced EVs might interfere with intercellular mitochondrial information transfer remains unknown.
Rapid and progressive loss of muscle mass (sarcopenia) is a distinctive characteristic of a cancer-related multifactorial syndrome called cachexia (90). Decreased mitochondrial OXPHOS and mitochondrial damage are linked to diminished muscle function and muscle weakness in cancer patients (91–93). Altered mitochondrial morphology, decreased interaction of mitochondria with endoplasmic reticulum, decreased ATP synthesis, and augmented uncoupling seem to be the main trends associated with skeletal muscle mitochondria during cancer states (91, 93, 94). Poor muscle quality and low muscle mass have been related to worse treatment outcomes and higher mortality in patients with metastatic and nonmetastatic breast cancer (95–97). Sarcopenia is reported in one-third (34%) of nonmetastatic, newly diagnosed breast cancer patients before chemotherapy or radiation (95). Also, the extent of the association of low muscle mass with poor survival is similar in stage II and III breast cancer (95). In addition, there is no relationship between muscle gene expression and anti-cancer treatment (92).
Evidence shows that changes in metabolism and aspects of the inflammatory response in cancer-associated muscle wasting is modulated by miRNA-containing EVs (58, 98, 99). It has been shown that breast cancer cell-derived EVs containing miR-122 play an important role in reprogramming systemic energy metabolism, including mitochondrial metabolism in skeletal muscle (100–102). The levels of miR-122 in total circulating EVs were higher in the breast cancer group compared to the control group (102). EVs containing miR-122 released by breast cancer MDA-MB-231 cells suppressed p53 signaling in skeletal muscle cells. Consequently, suppression of p53 signaling by miR-122 decreased the expression of TP53 target genes related to mitochondrial homeostasis, like Tfam, Pgc-1a, Sco2, and 16S ribosomal RNA, resulting in impaired energy production in skeletal muscles, promoting muscle deconditioning and cancer-associated cachexia development (101).
The same breast cancer cell-derived miR-122 can modify potential pre-metastatic niches in the lung and brain by downregulating glucose uptake and glycolysis by decreasing pyruvate kinase and GLUT1 expression and increasing the local nutrient availability for metastatic cancer cells (100). However, another study shows that higher levels of circulating miR-122 were specifically associated with predicting metastatic recurrence in stage II-III breast cancer patients (103). The effects of miR-122 may explain the production of specific miRNA-loaded EVs, which is the reason for the heterogeneity of metastasis and cachexia. How and to what extent a patient’s physical activity or different nutritional and conditional settings mimicking exercise-induced changes affect miRNA production in cancer cells requires further investigation.
Since EV content and type of generation are intrinsically linked to cellular processes, they offer potential as biomarkers or therapeutics for a range of diseases (104).
Limitations and challenges in the study of EVs are primarily technical. The lack of methods to rapidly and reliably quantify and trace EVs from different origins makes studying the effects of exercise-induced EVs challenging. Due to the exosome heterogeneity, non-standardized isolation methods and proteomics/bioinformatics approaches (77), the information about the EV-s in intercellular communication is still unclear. The underlying molecular mechanisms of exercise training in cancer remain poorly understood. The incomplete nomenclature of EVs also limits the understanding of their functional role in cellular physiology (14).
The main challenges in determining the general effects of exercise on subjects include individual differences, comorbidities, and the wide variety of exercise types and loads. Continued technological advancements, alongside more standardized approaches, will be essential for addressing these limitations and furthering our understanding of the molecular mechanisms underlying the effects of exercise and disease.
Physical exercise stimulates cells to release EVs, and without a clear understanding of the specific EVs induced by exercise in cancer patients and their effects, it is difficult to recommend exercise with complete confidence. That is because EVs released from cancer cells have several effects, which could complicate the course of the disease. Regarding exercise in the context of cancer energy metabolism, it remains unclear which EVs are beneficial allies and which act as harmful foes.
We are just beginning to explore direct associations between muscle health, exercise, and cancer cells and discovering complex cooperation mechanisms between different cell types in tumors. By studying the factors, including EVs, that affect the energy metabolism of cancer cells, we can target malignant cells with higher precision. The miRNAs, along with mitochondrial and glycolytic proteins present in EVs released into the tumor microenvironment and circulation, can influence the metabolic pathways utilized for energy production in recipient cells. Consequently, EVs released by cells after exercise certainly have the potential to affect the metabolism of the recipient cell. However, functional studies of cellular energy metabolism, which include measurements of OXPHOS and glycolysis fluxes of tumor tissues or cells, shall provide a better understanding of how to attack cancer cells and reverse the cancer-specific reprogramming of energy metabolism. This knowledge is essential for enhancing cancer treatment by amplifying or replicating the beneficial effects of exercise.
MP: Conceptualization, Investigation, Visualization, Writing – original draft, Writing – review & editing. ALl: Investigation, Writing – review & editing. ALi: Funding acquisition, Writing – review & editing, Conceptualization. TK: Conceptualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by EEA and Norway Grants under project No EEA-RESEARCH-164 (CancerBeat).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
CAF, cancer-associated fibroblast; EV, extracellular vesicle; OXPHOS, oxidative phosphorylation; TAM, tumor-associated macrophage.
1. Cogliano VJ, Baan R, Straif K, Grosse Y, Lauby-Secretan B, El Ghissassi F, et al. Preventable exposures associated with human cancers. J Natl Cancer Inst. (2011) 103:1827–39. doi: 10.1093/jnci/djr483
2. Jassim A, Rahrmann EP, Simons BD, Gilbertson RJ. Cancers make their own luck: theories of cancer origins. Nat Rev Cancer. (2023) 23:710–24. doi: 10.1038/s41568-023-00602-5
3. Moore SC, Lee IM, Weiderpass E, Campbell PT, Sampson JN, Kitahara CM, et al. Association of leisure-time physical activity with risk of 26 types of cancer in 1.44 million adults. JAMA Intern Med. (2016) 176:816–25. doi: 10.1001/jamainternmed.2016.1548
4. McTiernan A, Friedenreich CM, Katzmarzyk PT, Powell KE, Macko R, Buchner D, et al. Physical activity in cancer prevention and survival: A systematic review. Med Sci Sports Exerc. (2019) 51:1252–61. doi: 10.1249/MSS.0000000000001937
5. Eschke RK, Lampit A, Schenk A, Javelle F, Steindorf K, Diel P, et al. Impact of physical exercise on growth and progression of cancer in rodents-A systematic review and meta-analysis. Front Oncol. (2019) 9:35. doi: 10.3389/fonc.2019.00035
6. Li Y, Xiao X, Zhang Y, Tang W, Zhong D, Liu T, et al. Effect of exercise on breast cancer: A systematic review and meta-analysis of animal experiments. Front Mol Biosci. (2022) 9:843810. doi: 10.3389/fmolb.2022.843810
7. Ashcraft KA, Peace RM, Betof AS, Dewhirst MW, Jones LW. Efficacy and mechanisms of aerobic exercise on cancer initiation, progression, and metastasis: A critical systematic review of in vivo preclinical data. Cancer Res. (2016) 76:4032–50. doi: 10.1158/0008-5472.CAN-16-0887
8. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
9. Lu M, Sanderson SM, Zessin A, Ashcraft KA, Jones LW, Dewhirst MW, et al. Exercise inhibits tumor growth and central carbon metabolism in patient-derived xenograft models of colorectal cancer. Cancer Metab. (2018) 6:14. doi: 10.1186/s40170-018-0190-7
10. Zhao Y, Shen M, Wu L, Yang H, Yao Y, Yang Q, et al. Stromal cells in the tumor microenvironment: accomplices of tumor progression? Cell Death Dis. (2023) 14:587. doi: 10.1038/s41419-023-06110-6
11. Kroemer G, McQuade JL, Merad M, Andre F, Zitvogel L. Bodywide ecological interventions on cancer. Nat Med. (2023) 29:59–74. doi: 10.1038/s41591-022-02193-4
12. Kang DW, Barnes O, Vander Heiden MG, Dieli-Conwright CM. Effect of exercise on tumor markers - Is exercise anti-tumorigenic in humans?: A scoping review of preliminary clinical investigations. Crit Rev Oncol Hematol. (2022) 178:103779. doi: 10.1016/j.critrevonc.2022.103779
13. Welsh JA, Goberdhan DCI, O'Driscoll L, Buzas EI, Blenkiron C, Bussolati B, et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J extracellular vesicles. (2024) 13:e12404. doi: 10.1002/jev2.12404
14. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. (2014) 30:255–89. doi: 10.1146/annurev-cellbio-101512-122326
15. Jang SC, Crescitelli R, Cvjetkovic A, Belgrano V, Olofsson Bagge R, Sundfeldt K, et al. Mitochondrial protein enriched extracellular vesicles discovered in human melanoma tissues can be detected in patient plasma. J extracellular vesicles. (2019) 8:1635420. doi: 10.1080/20013078.2019.1635420
16. Frühbeis C, Helmig S, Tug S, Simon P, Kramer-Albers EM. Physical exercise induces rapid release of small extracellular vesicles into the circulation. J extracellular vesicles. (2015) 4:28239. doi: 10.3402/jev.v4.28239
17. Warnier G, De Groote E, Britto FA, Delcorte O, Nederveen JP, Nilsson MI, et al. Effects of an acute exercise bout in hypoxia on extracellular vesicle release in healthy and prediabetic subjects. Am J Physiol Regul Integr Comp Physiol. (2022) 322:R112–R22. doi: 10.1152/ajpregu.00220.2021
18. Whitham M, Parker BL, Friedrichsen M, Hingst JR, Hjorth M, Hughes WE, et al. Extracellular vesicles provide a means for tissue crosstalk during exercise. Cell Metab. (2018) 27:237–51 e4. doi: 10.1016/j.cmet.2017.12.001
19. Nederveen JP, Warnier G, Di Carlo A, Nilsson MI, Tarnopolsky MA. Extracellular vesicles and exosomes: insights from exercise science. Front Physiol. (2020) 11:604274. doi: 10.3389/fphys.2020.604274
20. Pinto AC, Tavares P, Neves B, Oliveira PF, Vitorino R, Moreira-Goncalves D, et al. Exploiting the therapeutic potential of contracting skeletal muscle-released extracellular vesicles in cancer: Current insights and future directions. J Mol Med (Berl). (2024) 102:617–28. doi: 10.1007/s00109-024-02427-7
21. Annibalini G, Contarelli S, Lucertini F, Guescini M, Maggio S, Ceccaroli P, et al. Muscle and systemic molecular responses to a single flywheel based iso-inertial training session in resistance-trained men. Front Physiol. (2019) 10:554. doi: 10.3389/fphys.2019.00554
22. Brahmer A, Neuberger E, Esch-Heisser L, Haller N, Jorgensen MM, Baek R, et al. Platelets, endothelial cells and leukocytes contribute to the exercise-triggered release of extracellular vesicles into the circulation. J extracellular vesicles. (2019) 8:1615820. doi: 10.1080/20013078.2019.1615820
23. Llorente A, Brokane A, Mlynska A, Puurand M, Sagini K, Folkmane S, et al. From sweat to hope: The role of exercise-induced extracellular vesicles in cancer prevention and treatment. J extracellular vesicles. (2024) 13:e12500. doi: 10.1002/jev2.12500
24. Hojman P, Dethlefsen C, Brandt C, Hansen J, Pedersen L, Pedersen BK. Exercise-induced muscle-derived cytokines inhibit mammary cancer cell growth. Am J Physiol Endocrinol Metab. (2011) 301:E504–10. doi: 10.1152/ajpendo.00520.2010
25. Ruiz-Casado A, Martin-Ruiz A, Perez LM, Provencio M, Fiuza-Luces C, Lucia A. Exercise and the hallmarks of cancer. Trends Cancer. (2017) 3:423–41. doi: 10.1016/j.trecan.2017.04.007
26. Westphal M, Lamszus K. Circulating biomarkers for gliomas. Nat Rev Neurol. (2015) 11:556–66. doi: 10.1038/nrneurol.2015.171
27. Moller A, Lobb RJ. The evolving translational potential of small extracellular vesicles in cancer. Nat Rev Cancer. (2020) 20:697–709. doi: 10.1038/s41568-020-00299-w
28. Lopes-Rodrigues V, Di Luca A, Mleczko J, Meleady P, Henry M, Pesic M, et al. Identification of the metabolic alterations associated with the multidrug resistant phenotype in cancer and their intercellular transfer mediated by extracellular vesicles. Sci Rep. (2017) 7:44541. doi: 10.1038/srep44541
29. Xavier CPR, Belisario DC, Rebelo R, Assaraf YG, Giovannetti E, Kopecka J, et al. The role of extracellular vesicles in the transfer of drug resistance competences to cancer cells. Drug Resist Update. (2022) 62:100833. doi: 10.1016/j.drup.2022.100833
30. Polonia B, Xavier CPR, Kopecka J, Riganti C, Vasconcelos MH. The role of Extracellular Vesicles in glycolytic and lipid metabolic reprogramming of cancer cells: Consequences for drug resistance. Cytokine Growth Factor Rev. (2023) 73:150–62. doi: 10.1016/j.cytogfr.2023.05.001
31. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. (2016) 2:e1600200. doi: 10.1126/sciadv.1600200
32. Moreno-Sanchez R, Marin-Hernandez A, Saavedra E, Pardo JP, Ralph SJ, Rodriguez-Enriquez S. Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int J Biochem Cell Biol. (2014) 50:10–23. doi: 10.1016/j.biocel.2014.01.025
33. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. (2011) 27:441–64. doi: 10.1146/annurev-cellbio-092910-154237
34. Paudel BB, Quaranta V. Metabolic plasticity meets gene regulation. Proc Natl Acad Sci United States America. (2019) 116:3370–2. doi: 10.1073/pnas.1900169116
35. Jia D, Park JH, Jung KH, Levine H, Kaipparettu BA. Elucidating the metabolic plasticity of cancer: mitochondrial reprogramming and hybrid metabolic states. Cells. (2018) 7:21. doi: 10.3390/cells7030021
36. Jia D, Lu M, Jung KH, Park JH, Yu L, Onuchic JN, et al. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc Natl Acad Sci United States America. (2019) 116:3909–18. doi: 10.1073/pnas.1816391116
37. Rodriguez-Enriquez S, Marin-Hernandez A, Gallardo-Perez JC, Pacheco-Velazquez SC, Belmont-Diaz JA, Robledo-Cadena DX, et al. Transcriptional regulation of energy metabolism in cancer cells. Cells. (2019) 8:1225. doi: 10.3390/cells8101225
38. Martinez-Reyes I, Chandel NS. Cancer metabolism: looking forward. Nat Rev Cancer. (2021) 21:669–80. doi: 10.1038/s41568-021-00378-6
39. Dey P, Kimmelman AC, DePinho RA. Metabolic codependencies in the tumor microenvironment. Cancer Discovery. (2021) 11:1067–81. doi: 10.1158/2159-8290.CD-20-1211
40. Li C, Teixeira AF, Zhu HJ, Ten Dijke P. Cancer associated-fibroblast-derived exosomes in cancer progression. Mol Cancer. (2021) 20:154. doi: 10.1186/s12943-021-01463-y
41. Peng L, Wang D, Han Y, Huang T, He X, Wang J, et al. Emerging role of cancer-associated fibroblasts-derived exosomes in tumorigenesis. Front Immunol. (2021) 12:795372. doi: 10.3389/fimmu.2021.795372
42. Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Salem AF, Tsirigos A, Lamb R, et al. Mitochondria "fuel" breast cancer metabolism: fifteen markers of mitochondrial biogenesis label epithelial cancer cells, but are excluded from adjacent stromal cells. Cell Cycle. (2012) 11:4390–401. doi: 10.4161/cc.22777
43. Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife. (2016) 5:e10250. doi: 10.7554/eLife.10250
44. Yan W, Wu X, Zhou W, Fong MY, Cao M, Liu J, et al. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat Cell Biol. (2018) 20:597–609. doi: 10.1038/s41556-018-0083-6
45. Kim MK, Kim Y, Park S, Kim E, Kim Y, Kim Y, et al. Effects of steady low-intensity exercise on high-fat diet stimulated breast cancer progression via the alteration of macrophage polarization. Integr Cancer Ther. (2020) 19:1534735420949678. doi: 10.1177/1534735420949678
46. Hou PP, Luo LJ, Chen HZ, Chen QT, Bian XL, Wu SF, et al. Ectosomal PKM2 promotes HCC by inducing macrophage differentiation and remodeling the tumor microenvironment. Mol Cell. (2020) 78:1192–206.e10. doi: 10.1016/j.molcel.2020.05.004
47. Wu J, Yuan M, Shen J, Chen Y, Zhang R, Chen X, et al. Effect of modified Jianpi Yangzheng on regulating content of PKM2 in gastric cancer cells-derived exosomes. Phytomedicine. (2022) 103:154229. doi: 10.1016/j.phymed.2022.154229
48. Tsakaneli A, Carregari VC, Morini M, Eva A, Cangemi G, Chayka O, et al. MYC regulates metabolism through vesicular transfer of glycolytic kinases. Open Biol. (2021) 11:210276. doi: 10.1098/rsob.210276
49. Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. (2012) 45:598–609. doi: 10.1016/j.molcel.2012.01.001
50. Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. (2011) 145:732–44. doi: 10.1016/j.cell.2011.03.054
51. Chen F, Chen J, Yang L, Liu J, Zhang X, Zhang Y, et al. Extracellular vesicle-packaged HIF-1alpha-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat Cell Biol. (2019) 21:498–510. doi: 10.1038/s41556-019-0299-0
52. Thompson HJ, Jiang W, Zhu Z. Candidate mechanisms accounting for effects of physical activity on breast carcinogenesis. IUBMB Life. (2009) 61:895–901. doi: 10.1002/iub.233
53. Zhu Z, Jiang W, Sells JL, Neil ES, McGinley JN, Thompson HJ. Effect of nonmotorized wheel running on mammary carcinogenesis: circulating biomarkers, cellular processes, and molecular mechanisms in rats. Cancer Epidemiol Biomarkers Prev. (2008) 17:1920–9. doi: 10.1158/1055-9965.EPI-08-0175
54. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. (2012) 13:251–62. doi: 10.1038/nrm3311
55. Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. (2017) 14:11–31. doi: 10.1038/nrclinonc.2016.60
56. Garcia NA, Moncayo-Arlandi J, Sepulveda P, Diez-Juan A. Cardiomyocyte exosomes regulate glycolytic flux in endothelium by direct transfer of GLUT transporters and glycolytic enzymes. Cardiovasc Res. (2016) 109:397–408. doi: 10.1093/cvr/cvv260
57. Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. (2014) 33:2142–56. doi: 10.15252/embj.201488104
58. Marinho R, Alcantara PSM, Ottoch JP, Seelaender M. Role of exosomal microRNAs and myomiRs in the development of cancer cachexia-associated muscle wasting. Front Nutr. (2017) 4:69. doi: 10.3389/fnut.2017.00069
59. Lai Z, Lin W, Yan X, Chen X, Xu G. Fatiguing freestyle swimming modifies miRNA profiles of circulating extracellular vesicles in athletes. Eur J Appl Physiol. (2023) 123:2041–51. doi: 10.1007/s00421-023-05167-7
60. Mitchelson KR, Qin WY. Roles of the canonical myomiRs miR-1, -133 and -206 in cell development and disease. World J Biol Chem. (2015) 6:162–208. doi: 10.4331/wjbc.v6.i3.162
61. Townley-Tilson WH, Callis TE, Wang D. MicroRNAs 1, 133, and 206: critical factors of skeletal and cardiac muscle development, function, and disease. Int J Biochem Cell Biol. (2010) 42:1252–5. doi: 10.1016/j.biocel.2009.03.002
62. Guescini M, Canonico B, Lucertini F, Maggio S, Annibalini G, Barbieri E, et al. Muscle releases alpha-sarcoglycan positive extracellular vesicles carrying miRNAs in the bloodstream. PLoS One. (2015) 10:e0125094. doi: 10.1371/journal.pone.0125094
63. D'Souza RF, Woodhead JST, Zeng N, Blenkiron C, Merry TL, Cameron-Smith D, et al. Circulatory exosomal miRNA following intense exercise is unrelated to muscle and plasma miRNA abundances. Am J Physiol Endocrinol Metab. (2018) 315:E723–E33. doi: 10.1152/ajpendo.00138.2018
64. Pietrangelo T, Santangelo C, Bondi D, Cocci P, Piccinelli R, Piacenza F, et al. Endurance-dependent urinary extracellular vesicle signature: shape, metabolic miRNAs, and purine content distinguish triathletes from inactive people. Pflugers Arch. (2023) 475:691–709. doi: 10.1007/s00424-023-02815-x
65. Nohata N, Hanazawa T, Enokida H, Seki N. microRNA-1/133a and microRNA-206/133b clusters: dysregulation and functional roles in human cancers. Oncotarget. (2012) 3:9–21. doi: 10.18632/oncotarget.424
66. Bahari Khasraghi L, Nouri M, Vazirzadeh M, Hashemipour N, Talebi M, Aghaei Zarch F, et al. MicroRNA-206 in human cancer: Mechanistic and clinical perspectives. Cell Signal. (2023) 101:110525. doi: 10.1016/j.cellsig.2022.110525
67. Sugiyama T, Taniguchi K, Matsuhashi N, Tajirika T, Futamura M, Takai T, et al. MiR-133b inhibits growth of human gastric cancer cells by silencing pyruvate kinase muscle-splicer polypyrimidine tract-binding protein 1. Cancer Sci. (2016) 107:1767–75. doi: 10.1111/cas.13091
68. Li D, Xia L, Chen M, Lin C, Wu H, Zhang Y, et al. miR-133b, a particular member of myomiRs, coming into playing its unique pathological role in human cancer. Oncotarget. (2017) 8:50193–208. doi: 10.18632/oncotarget.16745
69. Wu T, Han N, Zhao C, Huang X, Su P, Li X. The long non-sacoding RNA TMEM147-AS1/miR-133b/ZNF587 axis regulates the Warburg effect and promotes prostatic carcinoma invasion and proliferation. J Gene Med. (2022) 24:e3453. doi: 10.1002/jgm.3453
70. Koit A, Shevchuk I, Ounpuu L, Klepinin A, Chekulayev V, Timohhina N, et al. Mitochondrial respiration in human colorectal and breast cancer clinical material is regulated differently. Oxid Med Cell Longev. (2017) 2017:1372640. doi: 10.1155/2017/1372640
71. Koit A, Timohhina N, Truu L, Chekulayev V, Gudlawar S, Shevchuk I, et al. Metabolic and OXPHOS Activities Quantified by Temporal ex vivo Analysis Display Patient-Specific Metabolic Vulnerabilities in Human Breast Cancers. Front Oncol. (2020) 10:1053. doi: 10.3389/fonc.2020.01053
72. Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. (2012) 337:1062–5. doi: 10.1126/science.1219855
73. Cadete VJ, Deschenes S, Cuillerier A, Brisebois F, Sugiura A, Vincent A, et al. Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J Physiol. (2016) 594:5343–62. doi: 10.1113/JP272703
74. Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, McBride HM. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One. (2012) 7:e52830. doi: 10.1371/journal.pone.0052830
75. McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. (2014) 33:282–95. doi: 10.1002/embj.201385902
76. Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. (2012) 22:135–41. doi: 10.1016/j.cub.2011.11.057
77. Yang H, Zhang H, Gu H, Wang J, Zhang J, Zen K, et al. Comparative analyses of human exosome proteomes. Protein J. (2023) 365–373. doi: 10.1007/s10930-023-10100-0
78. Zayakin P, Sadovska L, Eglitis K, Romanchikova N, Radovica-Spalvina I, Endzelins E, et al. Extracellular vesicles-A source of RNA biomarkers for the detection of breast cancer in liquid biopsies. Cancers (Basel). (2023) 15:4329. doi: 10.3390/cancers15174329
79. Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. (2016) 535:551–5. doi: 10.1038/nature18928
80. Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, et al. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med. (2012) 18:759–65. doi: 10.1038/nm.2736
81. Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Kumar M, et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. (2014) 33:994–1010. doi: 10.1002/embj.201386030
82. Spees JL, Olson SD, Whitney MJ, Prockop DJ. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci United States America. (2006) 103:1283–8. doi: 10.1073/pnas.0510511103
83. Moschoi R, Imbert V, Nebout M, Chiche J, Mary D, Prebet T, et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood. (2016) 128:253–64. doi: 10.1182/blood-2015-07-655860
84. Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med. (2013) 11:94. doi: 10.1186/1479-5876-11-94
85. Dong LF, Rohlena J, Zobalova R, Nahacka Z, Rodriguez AM, Berridge MV, et al. Mitochondria on the move: Horizontal mitochondrial transfer in disease and health. J Cell Biol. (2023) 222:e202211044. doi: 10.1083/jcb.202211044
86. Nakhle J, Khattar K, Ozkan T, Boughlita A, Abba Moussa D, Darlix A, et al. Mitochondria transfer from mesenchymal stem cells confers chemoresistance to glioblastoma stem cells through metabolic rewiring. Cancer Res Commun. (2023) 3:1041–56. doi: 10.1158/2767-9764.CRC-23-0144
87. Abad E, Lyakhovich A. Movement of mitochondria with mutant DNA through extracellular vesicles helps cancer cells acquire chemoresistance. ChemMedChem. (2022) 17:e202100642. doi: 10.1002/cmdc.202100642
88. Zampieri LX, Silva-Almeida C, Rondeau JD, Sonveaux P. Mitochondrial transfer in cancer: A comprehensive review. Int J Mol Sci. (2021) 22:23245. doi: 10.3390/ijms22063245
89. Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. (2015) 21:81–94. doi: 10.1016/j.cmet.2014.12.003
90. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. (2011) 12:489–95. doi: 10.1016/S1470-2045(10)70218-7
91. Migliavacca E, Tay SKH, Patel HP, Sonntag T, Civiletto G, McFarlane C, et al. Mitochondrial oxidative capacity and NAD(+) biosynthesis are reduced in human sarcopenia across ethnicities. Nat Commun. (2019) 10:5808. doi: 10.1038/s41467-019-13694-1
92. Wilson HE, Stanton DA, Montgomery C, Infante AM, Taylor M, Hazard-Jenkins H, et al. Skeletal muscle reprogramming by breast cancer regardless of treatment history or tumor molecular subtype. NPJ Breast Cancer. (2020) 6:18. doi: 10.1038/s41523-020-0162-2
93. Argiles JM, Lopez-Soriano FJ, Busquets S. Muscle wasting in cancer: the role of mitochondria. Curr Opin Clin Nutr Metab Care. (2015) 18:221–5. doi: 10.1097/MCO.0000000000000164
94. Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab. (2012) 16:153–66. doi: 10.1016/j.cmet.2012.06.011
95. Caan BJ, Cespedes Feliciano EM, Prado CM, Alexeeff S, Kroenke CH, Bradshaw P, et al. Association of muscle and adiposity measured by computed tomography with survival in patients with nonmetastatic breast cancer. JAMA Oncol. (2018) 4:798–804. doi: 10.1001/jamaoncol.2018.0137
96. Prado CM, Baracos VE, McCargar LJ, Reiman T, Mourtzakis M, Tonkin K, et al. Sarcopenia as a determinant of chemotherapy toxicity and time to tumor progression in metastatic breast cancer patients receiving capecitabine treatment. Clin Cancer Res. (2009) 15:2920–6. doi: 10.1158/1078-0432.CCR-08-2242
97. Rossi F, Valdora F, Bignotti B, Torri L, Succio G, Tagliafico AS. Evaluation of body Computed Tomography-determined sarcopenia in breast cancer patients and clinical outcomes: A systematic review. Cancer Treat Res Commun. (2019) 21:100154. doi: 10.1016/j.ctarc.2019.100154
98. Camargo RG, Quintas Teixeira Ribeiro H, Geraldo MV, Matos-Neto E, Neves RX, Carnevali LC Jr., et al. Cancer cachexia and micrornas. Mediators Inflammation. (2015) 2015:367561. doi: 10.1155/2015/367561
99. Li C, Wu Q, Li Z, Wang Z, Tu Y, Chen C, et al. Exosomal microRNAs in cancer-related sarcopenia: Tumor-derived exosomal microRNAs in muscle atrophy. Exp Biol Med (Maywood). (2021) 246:1156–66. doi: 10.1177/1535370221990322
100. Fong MY, Zhou W, Liu L, Alontaga AY, Chandra M, Ashby J, et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol. (2015) 17:183–94. doi: 10.1038/ncb3094
101. Ruan X, Cao M, Yan W, Jones YZ, Gustafsson AB, Patel HH, et al. Cancer-cell-secreted extracellular vesicles target p53 to impair mitochondrial function in muscle. EMBO Rep. (2023) 24:e56464. doi: 10.15252/embr.202256464
102. Cao M, Isaac R, Yan W, Ruan X, Jiang L, Wan Y, et al. Cancer-cell-secreted extracellular vesicles suppress insulin secretion through miR-122 to impair systemic glucose homeostasis and contribute to tumour growth. Nat Cell Biol. (2022) 24:954–67. doi: 10.1038/s41556-022-00919-7
103. Wu X, Somlo G, Yu Y, Palomares MR, Li AX, Zhou W, et al. De novo sequencing of circulating miRNAs identifies novel markers predicting clinical outcome of locally advanced breast cancer. J Transl Med. (2012) 10:42. doi: 10.1186/1479-5876-10-42
Keywords: extracellular vesicles, cancer, energy metabolism, cancer microenvironment, physical exercise
Citation: Puurand M, Llorente A, Linē A and Kaambre T (2025) Exercise-induced extracellular vesicles in reprogramming energy metabolism in cancer. Front. Oncol. 14:1480074. doi: 10.3389/fonc.2024.1480074
Received: 13 August 2024; Accepted: 06 December 2024;
Published: 06 January 2025.
Edited by:
Juan Carlos Gallardo-Pérez, National Institute of Cardiology Ignacio Chavez, MexicoReviewed by:
Erika Monserrat Navarro Araujo, National Autonomous University of Mexico, MexicoCopyright © 2025 Puurand, Llorente, Linē and Kaambre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marju Puurand, bWFyanUucHV1cmFuZEBrYmZpLmVl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.