 Lifei Qin
Lifei Qin Xinya Zeng
Xinya Zeng Xinze Qiu
Xinze Qiu Shiquan Liu
Shiquan Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 03 December 2024
Sec. Cancer Molecular Targets and Therapeutics
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1467850
This article is part of the Research TopicAbnormal Angiogenesis in Tumors and the Improvement of Antiangiogenic DrugsView all 5 articles
Tumor angiogenesis is a characteristics of malignant cancer progression that facilitates cancer cell growth, diffusion and metastasis, and has an indispensable role in cancer development. N6-methyladenosine (m6A) is among the most prevalent internal modifications in eukaryotic RNAs, and has considerable influence on RNA metabolism, including its transcription, splicing, localization, translation, recognition, and degradation. The m6A modification is generated by m6A methyltransferases (“writers”), removed by m6A demethylases (“erasers”), and recognized by m6A-binding proteins (“readers”). There is accumulating evidence that abnormal m6A modification is involved in the pathogenesis of multiple diseases, including cancers, and promotes cancer occurrence, development, and progression through its considerable impact on oncoprotein expression. Furthermore, increasing studies have demonstrated that m6A modification can influence angiogenesis in cancers through multiple pathways to regulate malignant processes. In this review, we elaborate the role of m6A modification in tumor angiogenesis-related molecules and pathways in detail, providing insights into the interactions between m6A and tumor angiogenesis. Moreover, we describe how targeting m6A modification in combination with anti-angiogenesis drugs is expected to be a promising anti-tumor treatment strategy, with potential value for addressing the challenge of drug resistance.
Tumor cell proliferation relies on provision of sufficient oxygen and nutrients to meet the metabolic needs of the tissue via blood vessels; however, when tumors grow beyond a certain extent, existing blood vessels are insufficient to satisfy the demands of the tumor, and new blood vessel formation is required (1). During tumor angiogenesis, the “angiogenesis switch” is activated due to the imbalance of pro- and anti-angiogenic factors in the tumor microenvironment, resulting in formation of an abnormal tumor vascular system (2). Cancer vascular networks are also integral to the metastasis and spread of cancer tissue to distant organs.
In recent years, there has been considerable research interest in N6-methyladenosine (m6A) RNA methylation, which is an internal RNA modification occurring extensively in mammalian eukaryotic cells as an epigenetic gene expression regulatory mechanism (3). The m6A modification is most frequent found in highly-conserved RRACH (R=G/A, H=A/C/U) consensus sequences, and is predominantly enriched in 3’-untranslated terminal regions (3’-UTRs), close to termination codons, and in internal exons (4, 5). Further, m6A modification is a dynamic and reversible process that methylated at the N-6 site of adenosine of RNA molecule, not only presenting in messenger RNAs (mRNAs), but also in non-coding RNAs (ncRNAs), such as long ncRNAs (lncRNAs), microRNAs (miRNAs), and circular RNAs (circRNAs) (3, 6). The basic processes involved in m6A methylation are regulated by interactions among three factors: “writers” (m6A methyltransferases), “erasers” (m6A demethylases), and “readers” (RNA-binding proteins) (7), where m6A methyltransferases catalyze methylation modifications, m6A demethylases are responsible for removal of methylation modifications, and RNA-binding proteins are mainly responsible for recognizing and binding to specific m6A binding sites (8). Through its roles in processes including RNA transcription, splicing, localization, translation, recognition, and degradation, m6A methylation modification participates widely in the regulation of target RNA expression, which contributes to tumorigenesis and tumor progression (9). Research into m6A modification in the context of tumor angiogenesis has gradually increased and numerous molecular mechanisms related to tumor angiogenesis regulation by m6A have been preliminarily validated.
In this review, we provide a brief introduction to the biological processes of m6A modification and tumor angiogenesis. Then, we summarize studies on the mechanisms by which m6A modification impacts the development of diverse tumors through regulation of angiogenesis. Finally, we discuss the potential clinical application value of targeting m6A modification combined with drugs inhibiting angiogenesis in the treatment of cancer.
As an epigenetic posttranscriptional regulatory mechanism and an emerging research frontier, m6A modification is the most prevalent and abundant internal chemical modification occurring in mammalian mRNAs and ncRNAs (10–13). m6A methylation was first discovered in 1974, and its study has increased considerably in recent years, owing to improvements in detection methods (3). m6A modification is involved in the regulation of almost all RNA metabolism processes, and influences various biological functions (8, 9, 14), including RNA transcription (15, 16), splicing (17–19), subcellular location (20, 21), translation (22, 23), stability (24–26) and binding capacity (27), thus affecting RNA expression and functions. In mammalian RNA, m6A accounts for approximately 0.1%–0.4% of adenylate residues, representing an average of 3-5 m6A-modified sites per transcript (5, 11, 28). m6A modification is defined as methylation of the sixth N atom of RNA adenylate (A) (3). With the development and application of high-throughput sequencing technologies, abundant studies have reported that m6A sites are preferentially enriched in 3’-UTRs, near termination codons, and in internal exons, at the highly-conserved consensus motif, RRACH (4, 5, 26, 29). In recent years, m6A modification sites have also been discovered 5’-UTRs, and play critical roles in commencement of cap-independent translation (30, 31).
Similar to DNA methylation and histone modifications, m6A RNA modification is dynamic and reversible, and is mediated by three categories of enzymes: methyltransferases, also referred to as “writers”, which catalyze methylation; demethylases, or “erasers”, which removed the modification; and RNA binding-proteins, known as “readers”, which recognize and bind to m6A sites (8, 32, 33). The functions of these proteins separately ensure the expression of RNAs (Figure 1; Table 1).
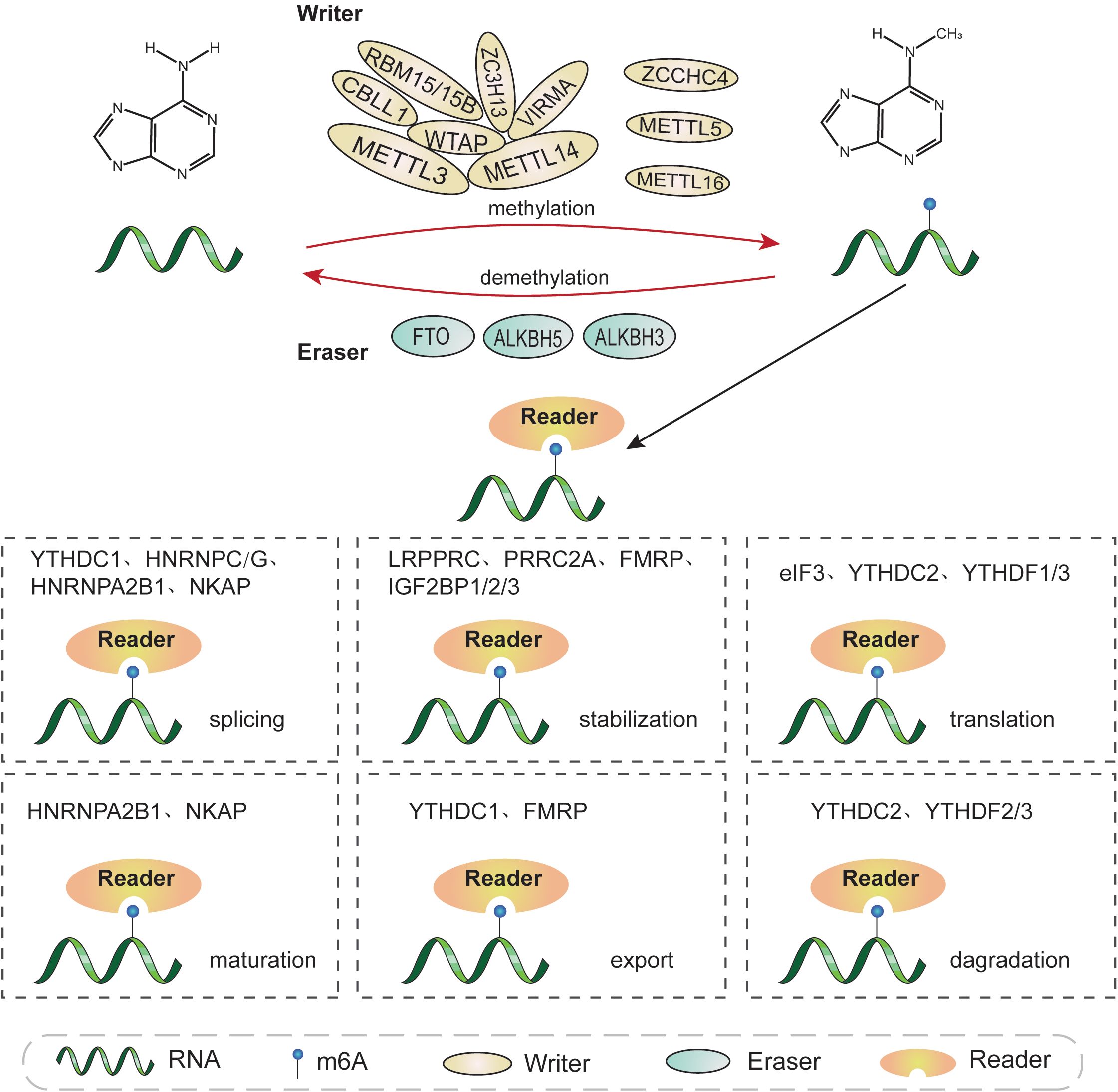
Figure 1. The molecular mechanisms involved in m6A methylation. m6A modification is a dynamic and reversible process conducted by m6A “writers”, “erasers”, and “readers”. m6A “writers” (METTL3, METTL14, WTAP, RBM15/15B, VIRMA, ZC3H13, CBLL1, ZCCHC4, METTL5, and METTL16) catalyze RNA methylation. m6A modification can be removed by m6A “erasers”, including FTO, ALKBH5, and ALKBH3. m6A “readers” recognize and combine with m6A sites on targeted RNA to affect its fate, and primarily include YTHDC1/2, YTHDF1/2/3, IGF2BP1/2/3, HNRNPA2B1, HNRNPC/G, eIF3, FMRP, PRRC2A, LRPPRC, and NKPA. m6A modifications are involved in almost all RNA metabolic processes, including transcription, splicing, translation, degradation, and nuclear export, among others.
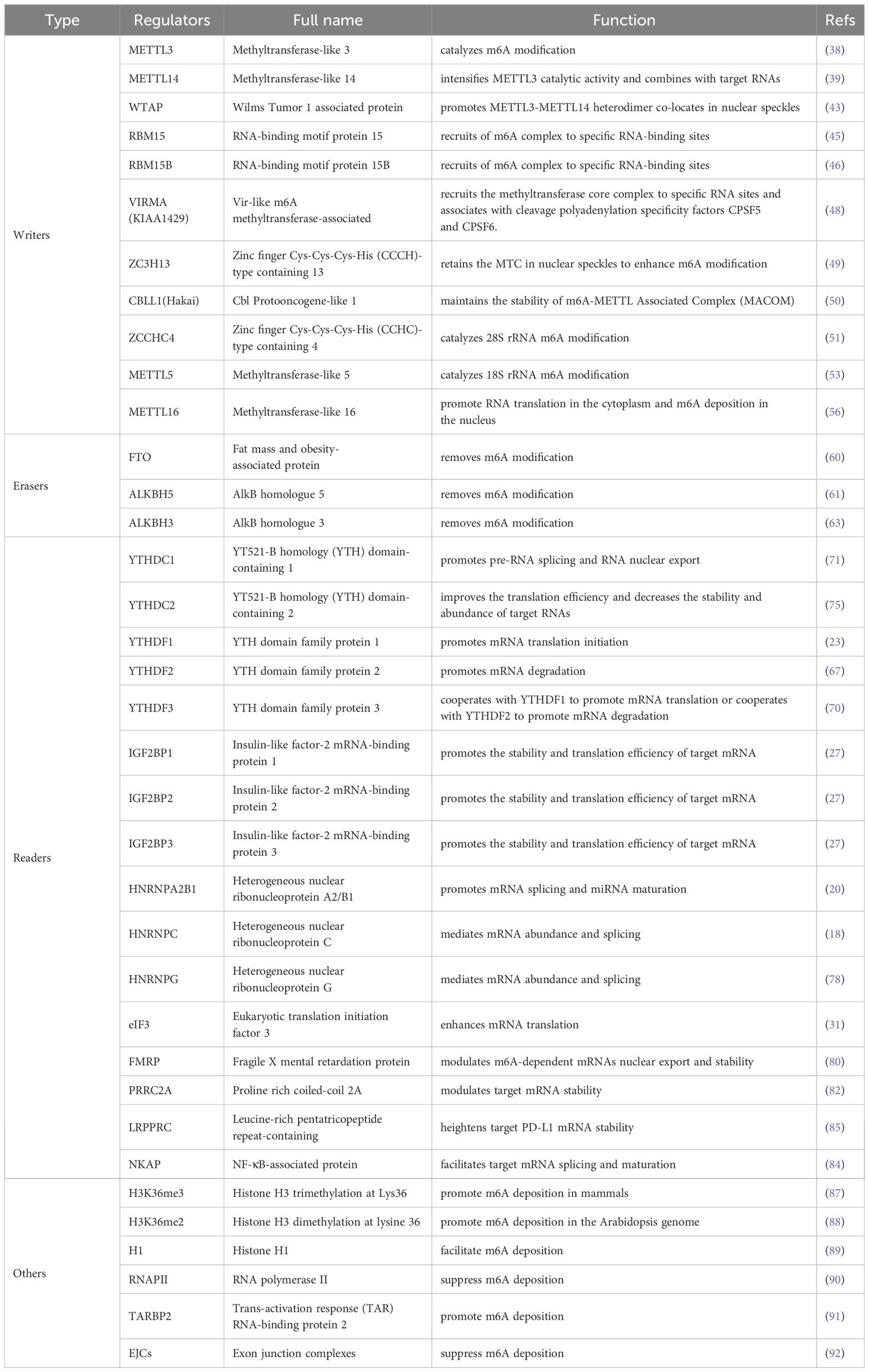
Table 1. The function of m6A methylation enzymes/regulators in RNA metabolism.
The core components of the methyltransferase complex (MTC) are methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14), and Wilms tumor 1 associated protein (WTAP) (34, 35). METTL3 is a highly conserved S-adenosyl methionine (SAM)-binding protein identified by Joseph et al. in 1997 (36), and the most significant component subunit of the MTC, able to catalyze the transfer of methyl groups in SAM to adenine bases in RNA (37). As a pseudo-methyltransferase, METTL14 exhibits no catalytic activity; however, it plays an essential role in allosteric activation of METTL3, to intensify its catalytic function and is also responsible for combining with target RNAs by recognizing the specific RRACH consensus sequence (38, 39). A 1:1 ratio of METTL3 and METTL14 come together to form a stable heterodimer and colocalize in nuclear speckles (40). A recent study found that METTL3 is vital for promoting METTL14 stabilization by protecting its ubiquitinated sites from STIP1 homology and U-box-containing protein 1 (STUB1)-induced ubiquitination degradation to maintain m6A homeostasis (41). WTAP, a partner of the Wilms tumor 1 (WT1) protein, can promote recruitment of the METTL3-METTL14 heterodimer and ensure colocalization of the complex in nuclear speckles to mediate m6A modification (42). WTAP is a bridging protein that occurs in two different complexes: the METTL3-METTL14-WTAP complex, also referred to as the m6A–METTL complex (MAC), which has a primarily catalytic role; and the RBM15/ZC3H13/WTAP/VIRMA/Hakai complex, known as the m6A-METTL-associated complex (MACOM), which mainly exerts a regulatory function (43).
RNA-binding motif protein 15 (RBM15) does not have a catalytic function and belongs to the split end (Spen) protein family. RBM15 can interact with METTL3 in a WTAP-dependent manner, and facilitates recruitment of the WTAP-METTL3 complex to specific RNA-binding sites, allowing m6A methylation of adjacent consensus motifs, as does its paralog, RBM15B (44, 45). Vir-like m6A methyltransferase associated (VIRMA, originally known as KIAA1429), localizes to nuclear speckles in humans, and modulates region-selective methylation modification by recruiting the catalytic core complex, METTL3-METTL14-WTAP, to specific RNA sites (46, 47). Alternatively, VIRMA associates with cleavage polyadenylation specificity factor subunit 5 (CPSF5) as well as cleavage polyadenylation specificity factor subunit 6 (CPSF6), in an m6A-dependent manner (47). In concert with WTAP, zinc finger Cys-Cys-Cys-His (CCCH)-type containing 13 (ZC3H13) retains the MTC in nuclear speckles, possibly due to its low-complexity domains, which target proteins to sub-cellular organelles enriched for RNA processing enzymes and pre-mRNA splicing factors, leading to enhanced m6A modification (48). Cbl protooncogene-like 1 (CBLL1, also called Hakai), a ring-finger type E3 ubiquitin ligase, has an essential role in maintaining the stability of MACOM (49).
In addition, there are a number of newly-discovered methyltransferases that warrant research attention. For example, CCHC-type containing 4 (ZCCHC4), which has a conserved “DPPF” catalytic motif, was identified as a novel ribosomal RNA (rRNA)-adenosine-methyltransferase, with a critical role in catalyzing m6A modification of 28S rRNA and that mediates ribosome subunit distribution and global translation (50, 51). Methyltransferase-like 5 (METTL5) participates in 18S rRNA methylation and can form a heterodimeric complex together with TRMT112, which functions as an allosteric adaptor to enhance METTL5 stabilization (52, 53). Furthermore, methyltransferase-like 16 (METTL16) is a class-I methyltransferase considered to be a unique m6A regulatory factor, distinct from the METTL3/METTL14 complex (54). METTL16 interacts directly with eIF3a, eIF3b, and rRNAs in the cytosol, to promote translation of mRNAs and deposits m6A into mRNA targets in the nucleus (55, 56). The N-terminal methyltransferase domain and two C-terminus vertebrate-conserved regions of METTL16 are RNA-binding domains used to combine with its targets (57). It binds multiple RNAs, such as pre-mRNAs, ncRNAs and lncRNAs. METTL16 serves as a m6A methyltransferase that recognizes the conserved sequence UACAGAGAA, in MAT2A mRNA and U6 snRNA substrates (58); however, the interaction between METTL16 and its substrate lncRNA MALAT1, occurs through specific recognition and binding of triple helix RNA, and whether MALAT1 is methylated by METTL16 remains to be determined (59).
The above-mentioned “writers”, particularly those investigated in only a small number of studies, require further investigation.
m6A demethylases, also referred to as m6A “erasers”, can reverse m6A methylation and include: fat mass and obesity-associated protein (FTO), alkB homologue 5 (ALKBH5) and alkB homologue 3 (ALKBH3); these three proteins belong to the dioxygenase ALKB family, which have reserved α-ketoglutarate and iron(II)-dependent oxygenase domains, and can reduce m6A modification levels in RNA (60).
The first protein to be shown to harbor m6A demethylation capacity was FTO, which is located in the cytoplasm and has an important role in the regulation of adipogenesis (61). Under demethylase FTO catalysis, m6A can be sequentially oxidized to N6-hydroxymethyladenosine (hm6A) and then N6-formyladenosine (f6A), with subsequently conversion of f6A to adenosine (A), thereby regulating levels of m6A (62).
ALKBH5 was the second m6A demethylase to be discovered and is distributed in the nucleoplasm, where it is responsible for regulating mRNA export from the nucleus to the cytoplasm, as well as affecting mRNA metabolic processes, including splicing and stability (63, 64). ALKBH3, is located in both the cytoplasm and nucleus, and was recently identified as a novel m6A demethylase with a preference for demethylation of transfer RNA (tRNA), rather than mRNA or rRNA (65). Importantly, ALKBH3-demethylated tRNA prominently facilitated translation efficiency of protein (66).
In addition to “writers” and “erasers”, m6A modification requires another essential group of molecules, RNA binding proteins (RBPs), also termed “readers” (7). Although these proteins do not directly change the level of methylation like methyltransferases and demethylases, they can confer the destinies of RNAs by recognizing and preferentially combining the methylation sites of RNA (34).
The YT521-B homology (YTH) domain family members, YTHDF1/2/3 and YTHDC1/2, have conserved m6A-binding domains and selectively bind to RNA with m6A modification at RRACH consensus sequences, with numerous downstream effects (67, 68). YTHDF1, a translation promoter, can evoke m6A-modified mRNA translation through interaction with the translation complex, which comprises eukaryotic translation initiation factor 3 (eIF3), eIF4G, and poly (A) binding protein (PABP), among other molecules (22, 37). YTHDF2, as the first discovered and the most widely studied m6A “reader”, can recruit the deadenylase complex CCR4-NOT by direct interaction with the superfamily homology (SH) domain of CNOT1, thereby resulting in degradation of the transcripts (69, 70). with a synergistic role to those of YTHDF1 and YTHDF2, coordinating with YTHDF1 to trigger m6A-labelled mRNA translation, or cooperating with YTHDF2 to accelerate m6A-containing mRNA degradation (23, 71). Distinct from the dominant cytosol localization of other YTH domain family members, YTHDC1 is preferentially distributed in the nucleus, and contributes to pre-RNA splicing by recruiting serine- and arginine-rich splicing factor 3 (SRSF3) and antagonizing SRSF10 to promote exon inclusion (72). In addition, YTHDC1 facilitates RNA nuclear export via interacting with nuclear RNA export factor 1 (NXF1) and three prime repair exonuclease (TREX) (73, 74). YTHDC2 improves target RNA translation efficiency and elongation, as well as reducing their stability and abundance, by interacting with 5′-3′ exoribonuclease 1 (XRN1) (75, 76). The m6A “reader”, heterogeneous nuclear ribonucleoprotein (HNRNP) A2/B1 (HNRNPA2B1), localizes to the nucleus, and can not only recognize and combine with m6A-marked mRNA to regulate alternative splicing events, but also participates in facilitating miRNA maturation through binding to m6A-labelled primary miRNA transcripts and recruiting the microprocessor complex protein, DiGeorge syndrome critical region 8 (DGCR8) (19, 77, 78). Additionally, “m6A switch”, a special mechanism, has been proven to be related to both HNRNPC and HNRNPG, which can optionally bind to this structure switch, instead of directly binding to m6A sites, thereby influencing m6A-labelled transcript abundance and alternative splicing (17, 79). The three insulin-like factor-2 mRNA-binding protein (IGF2BP) paralogs, IGF2BP1/2/3, are a class of m6A “readers” located in the cytosol, and can stabilize m6A-modified target transcripts and enhance target mRNA translation efficiency by recognizing the consensus GG(m6A)C motif (26). A recent study showed that IGF2BP proteins may also be able to read the structural changes mediated by the “m6A-switch” (80).
In addition, eIF3 also serves as an m6A “reader”, located in both the nucleus and cytosol, that binds m6A sites in the 5′-UTR region of mRNAs and stimulates transcripts translation in a cap-independent manner (30). The cytosolic protein fragile X mental retardation protein (FMRP), can modulate m6A-dependent mRNA nuclear export and stability (81, 82). Further, proline rich coiled-coil 2A (PRRC2A) is a newly-discovered m6A modification “reader”, that localizes in both the nucleus and cytosol, whose GRE domain combines with a consensus GGACU motif in the Olig2 mRNA coding sequence, thereby post-transcriptionally modulating target mRNA stability in an m6A-dependent manner (83, 84). Most recently, the RBP NF-κB activating protein (NKAP) was shown to facilitate target mRNA splicing and maturation in an m6A-dependent manner (85). Further, leucine-rich pentatricopeptide repeat-containing (LRPPRC) was identified as a novel “reader” that can enhance target PD-L1 mRNA stabilization in an m6A-dependent manner (86). The specific mechanisms involved in m6A reader functions warrant further study and there remain many undiscovered “readers” to be explored.
In addition to the deposition of m6A methylation on RNA transcripts by m6A MTCs, recent studies have found that external factors, such as histone modifications, RBPs, transcription factors, and RNA polymerase II (RNAPII), are also involved in regulating m6A deposition. It was demonstrated that m6A deposition on RNAs in mammals was linked to the histone mark, H3K36me3, which interacts directly with METTL14 to recruit the m6A MTC to bind adjacent to RNAPII, thereby promoting m6A deposition on newly formed RNA (87). Another report has found that histone H3K36me2 distributed at the 3’-end of the genes is significantly correlated with m6A deposition in the Arabidopsis genome (88). Histone H1 modification is also proposed to mediate m6A modification of nascent RNAs. Genes transcribed slowly contain high levels of histone H1, which leads to reduced RNAPII recruitment at transcription start site-proximal regions, thus facilitating co-transcriptional m6A deposition (89). Further, RNAPII transcriptional dynamics are associated with m6A deposition; slow RNAPII transcription elongation and low rates of RNAPII pausing result in elevated m6A deposition on mRNAs and decreased translation efficiency (90). In addition, the RBP, TARBP2, promotes m6A deposition on transcripts by recruiting the MTC, leading to intron retention and nuclear decay (91). In addition to mechanisms involved in activation of m6A deposition, a recent study has found that exon junction complexes (EJCs) act as m6A “suppressors”, to prevent m6A deposition in average-length internal exons by packaging proximal RNA, thereby regulating global m6A specificity (92). Although we have gained new insights into the processes that guide and suppress m6A deposition, so far, the mechanisms underlying m6A specific enrichment in certain transcriptome regions remain unclear.
The critically traditional tumor angiogenesis, also termed as sprouting angiogenesis, was first proposed by Professor Judah Folkman in 1971 suggesting that tumor growth was closely associated with angiogenesis (93). Since then, there have been numerous studies worldwide focused on confirming the theory of Folkman, which have led to considerable achievements based on targeting tumor angiogenesis (94, 95). Sprouting angiogenesis, existing in physiological and pathological processes, is the development of new blood vessels from pre-existing vascular networks involved in the survival and development of tumor through the supply of oxygen, nutrients as well as removal of metabolic waste, and plays a pivotal role in tumor invasion and metastasis (1, 96). Solid tumor survival and growth rely on sufficient blood supply, and when tumors reach > 2 mm in diameter, diffusion alone cannot meet their need to obtain oxygen and nutrients, so that neovascularization becomes necessary to meet the requirements of tumor tissues, which will otherwise undergo necrosis as a result of ischemia and hypoxia (1, 97). Hypoxia in cells leads to increased expression of hypoxia-inducible factor (HIF) (98), which induces upregulation of vascular endothelial (VE) growth factor (VEGF), angiopoietin (Ang), and other angiogenic molecules (99, 100). Other drivers of tumor angiogenesis include genetic mutations, inflammatory responses, and mechanical stress (101). The normal process of angiogenesis is strictly controlled by pro- and anti-angiogenic regulatory factors, to maintain a relatively dynamic homeostasis (102), disruption of which within the tumor microenvironment activates the so-called “angiogenic switch”, thus promoting tumor angiogenesis (103). Among them, pro-angiogenic factors include VEGF, platelet-derived growth factor (PDGF), epidermal growth factor (EGF), tumor necrosis factor-α (TNF-α), interleukin-8 (IL-8) etc., while anti-angiogenic factors comprise angiostatin, endostatin, thrombospondin-1 (TSP-1), tissue inhibitors of metalloproteinases (TIMPs) and so on (101, 104). Unlike normal blood vessels, tumor vasculature is abnormally variable in shape and structure, resulting in dense and disordered vascular networks (105). This is because tumor endothelial cells are not organized according to traditional grading arrangements. The characteristics of tumor blood vessels are also reflected in the permeability of curled and dilated abnormal blood vessels and the efficiency of tissue perfusion, resulting in irregular blood flow (106). Therefore, abnormalities in tumor blood vessels affect both the delivery of oxygen and nutrients to the tumor, and create a hypoxic, acidic, and inflammatory tumor microenvironment, which activates tumor angiogenesis by promoting secretion of proangiogenic factors and further inducing malignant tumor development (107, 108).
More than 20 years after the concept of classical tumor angiogenesis was proposed, the distinct process of vasculogenic mimicry (VM) was first defined by Maniotis et al. in human melanoma (109). VM relies on pluripotent embryonic stem cells, highly invasive tumor cells and the extra-cellular matrix in aggressive primary and metastatic tumors rather than depending on vascular ECs; however, it can also play an important role in supplying malignant tumors with sufficient blood, thus promoting tumor survival, invasion and metastasis (110). Furthermore, VM is significantly linked with tumor grade and poor prognosis in patients with aggressively malignant cancers (111, 112), consisting of gastric cancer (113), colorectal cancer (114), hepatocellular carcinoma (115), glioblastoma (116), lung cancer (117), breast cancer (118), ovarian cancer (119), among others. This explains one of the reasons why some current clinical treatments against tumor angiogenesis have not achieved satisfactory efficacy (120, 121). Epithelial-mesenchymal transition (EMT) and cancer stem cells (CSCs) are consider significant factors contributing to the relatively complex process of VM in tumors (122). Molecules essential for development of VM include: VE-cadherin, phosphatidyl inositol 3-kinase (PI3K), erythropoietin-producing hepatocellular receptor A2 (EphA2), matrix metalloproteinases (MMPs), and VE growth factor receptor (VEGFR1) (122–124). Mounting evidence has pointed out that hypoxia is also inseparable from VM formation in multiple types of solid tumors (125, 126). In addition, vascular co-option and glomeruloid angiogenesis function in tumor angiogenesis initiation. Vascular co-option refers to the process in which tumor cells attach themselves to host capillaries to obtain a blood supply providing oxygen and nutrients for tumor growth and development, without the formation of new blood vessels, and mainly occurs in organs with an extremely high degree of vascularization, such as the brain, liver, and lungs (127). Glomeruloid angiogenesis has also been reported in melanoma, breast cancer, meningioma, and GBM (128–131). However, there is relatively little research into tumor angiogenesis and malignant processes in this context, and further in-depth analysis of the mechanisms involved is needed.
There is accumulating evidence that m6A modification regulates tumor angiogenesis in various ways, and the relationship between them is of great significance for tumor proliferation, invasion and metastasis (Figure 2; Table 2)
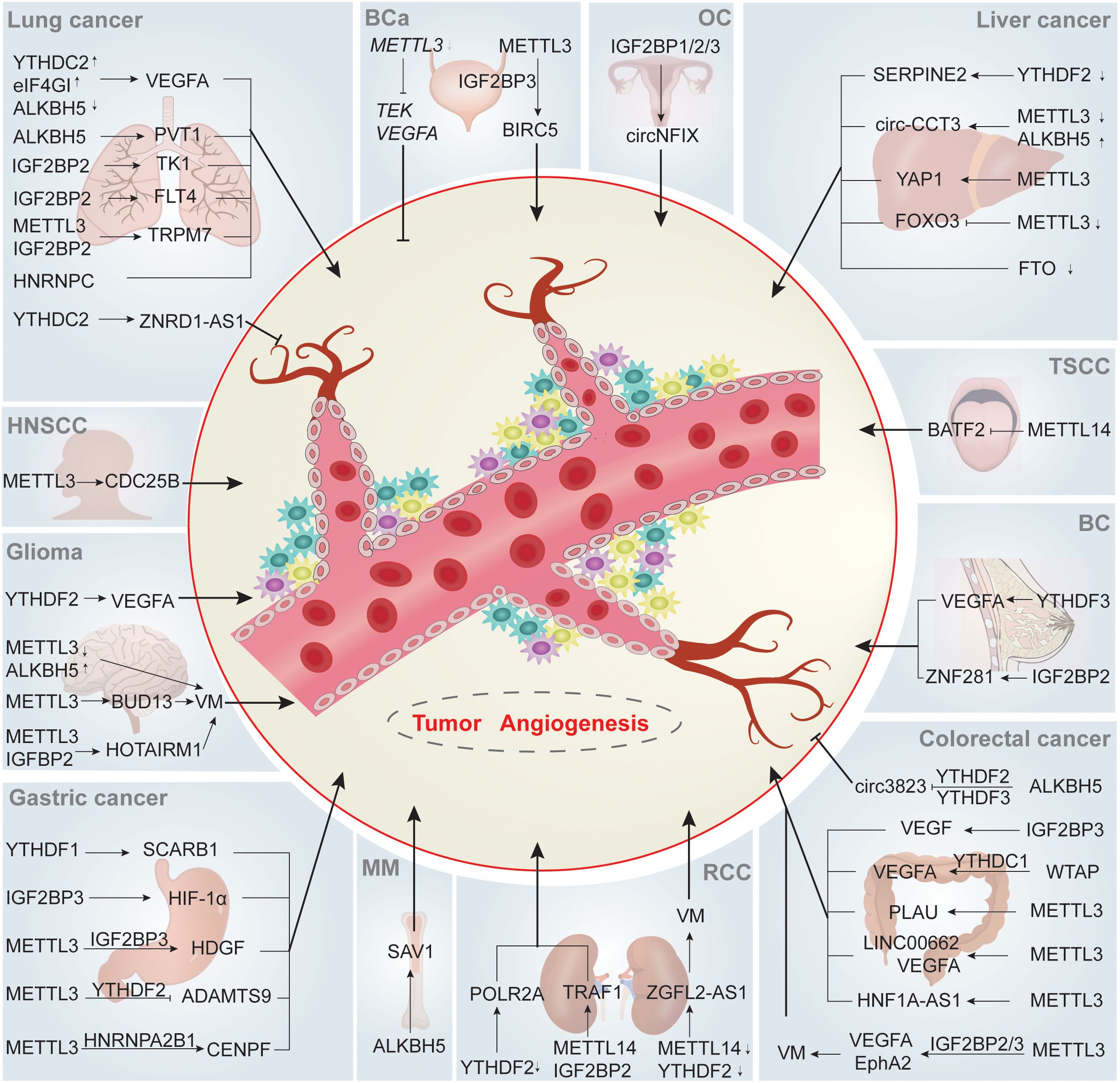
Figure 2. m6A regulate tumor angiogenesis. m6A writers, erasers and readers participate in tumor angiogenesis through regulating angiogenesis-related targeted proteins or pathways in an array of direct or indirect ways, ultimately influencing the tumorigenesis and development of various human tumors. ↑ and ↓ indicate upregulation and downregulation of m6A regulators, respectively. Abbreviations: BC, breast cancer; OC, ovarian cancer; HNSCC, head and neck squamous cell carcinoma; TSCC, tongue squamous cell carcinoma; BCa, bladder cancer; MM, multiple myeloma; RCC, renal cell carcinoma.
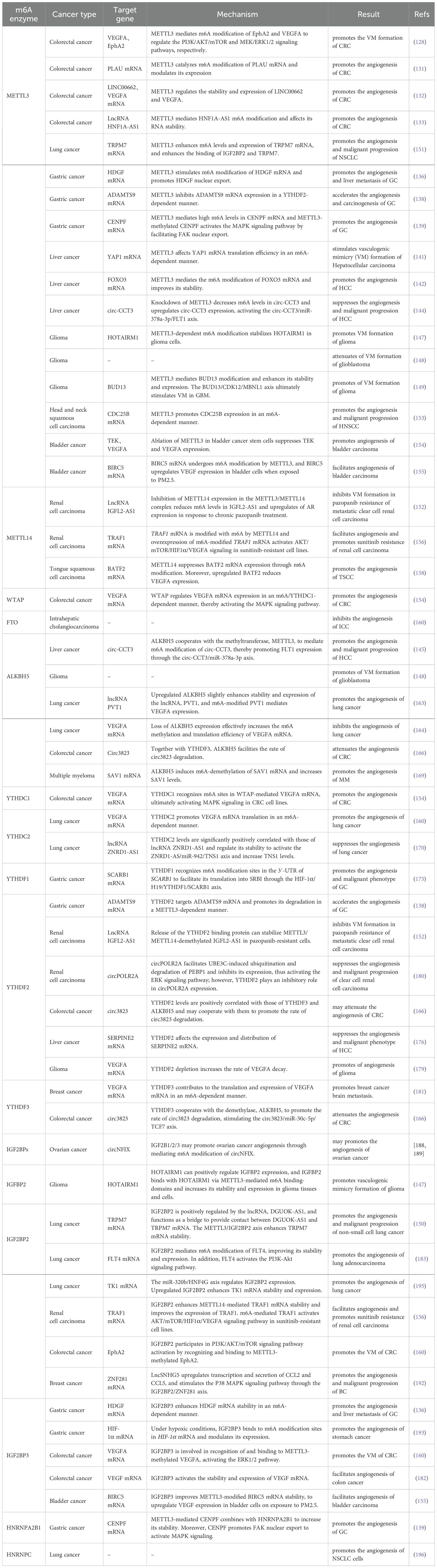
Table 2. m6A methylation in tumor angiogenesis.
Previous publications have well documented that vascular endothelial growth factor A (VEGFA) plays a pivotal role in the formation of new blood vessels (132). Colorectal cancer (CRC) is among the most common types of cancer and presents significant challenges in terms of morbidity and mortality (133). In CRC, EphA2 and VEGFA mRNA are transcripts downstream of the “writer”, METTL3, and undergo METTL3-mediated m6A modification via different IGF2BP-dependent mechanisms, resulting in VM formation (134). Urokinase plasminogen activator (uPA, PLAU) mRNA, a classical molecule of the plasminogen activation system, contributes to various cancer processes, including tumor proliferation, invasion, metastasis, and angiogenesis (135, 136). Yu et al. demonstrated that METTL3 could facilitate angiogenesis through directly targeting and specifically catalyzing the m6A binding sites in the 3′-UTR coding region of PLAU mRNA and modulating its expression (137). m6A modification not only also functions on mRNAs, but also acts on some lncRNAs to regulate tumor angiogenesis. For example, m6A enzyme METTL3 was confirmed to modify targets―LINC00662 and VEGFA to stabilize them and positively regulate their expression levels, thus promoting angiogenesis in CRC, which was demonstrated by the levels of CD31, CD34 and VEGF (138). In addition, METTL3 mediated upregulation of lncRNA HNF1A-AS1 through influencing its stability in CRC cells, and HNF1A-AS1 overexpression can clearly promoted angiogenesis, while its deficiency has the opposite effect (139). however, the cited research was focused on exploring the mechanism underlying HNF1A-AS1 involvement in CRC cell cycle progression, while the role of HNF1A-AS1 in angiogenesis in the context of CRC has not been thoroughly elucidated.
Gastric cancer (GC) is one of the most prevalent malignant cancers worldwide associated with high morbidity and mortality (140). Hepatoma-derived growth factor (HDGF) expression is associated with aggressive biological characteristics, including cancer cell proliferation, apoptosis, angiogenesis, and metastasis (141). In GC, METTL3 stimulates m6A modification of HDGF mRNA, while the m6A “reader”, IGF2BP3 (also known as IMP3 or KOC), can stabilize HDGF mRNA through direct recognition and binding with its m6A sites. HDGF can be translocated from the nucleus to the cytoplasm and contributes to facilitating angiogenesis, ultimately promoting tumor cell growth and liver metastasis (142). ADAMTS9 mRNA, which is an independent prognostic factor in patients with GC (143), is downstream of METTL3, and ADAMTS9 mRNA m6A levels were clearly decreased after METTL3 knockdown, while the opposite effect occurred on METTL3 overexpression. Further, ADAMTS9 mRNA is inhibited by METTL3 via a YTHDF2-dependent pathway, thus accelerating GC angiogenesis (144). In addition, Xu et al. revealed that METTL3-mediated m6A modification of centromere protein F (CENPF) mRNA can enhance GC angiogenesis both in vitro and in vivo; METTL3-methylated CENPF mRNA had increased stability after combining with the m6A “reader”, HNRNPA2B1. Moreover, CENPF mRNA can promote focal adhesion kinase (FAK) nuclear export, leading to MAP kinase (MAPK) signaling pathway activation, and thereby inducing angiogenesis in GC (145).
Liver cancer is a highly aggressive tumor in humans that contributes to significant cancer-related mortality and morbidity in the world (133). Activation of yes-associated protein (YAP) mRNA has been demonstrated to contribute to tumor cell proliferation, angiogenesis and invasion in various types of tumors (146). In HCC, Qiao et al. found that YAP1 mRNA stimulates VM in an m6A-dependent manner, both in vitro and in vivo, where METTL3 mediates YAP1 m6A modification, affecting its translation efficiency (147). Lin et al. found that, under hypoxia, decreased METTL3 leads to increased expression of angiogenic markers, including FGF, PDGF-B, STAT3, and VEGFA. The mechanism underlying angiogenesis in liver cancer involves METTL3-mediated m6A modification of FOXO3 mRNA to increase its stability through a YTHDF1-dependent mechanism, which ultimately enhances sorafenib resistance of HCC (148). Further, an oncogenic role of circ−CCT3 was validated in HCC cells (149). Qian et al. conducted tube formation assays, demonstrating human umbilical vein EC (HUVEC) angiogenesis inhibition after circ-CCT3 knockdown. Mechanistically, circ-CCT3 functions as a sponge for miR-378a-3p, thereby regulating the expression of FLT1, which acts as a cell-surface receptor for VEGFA, participating in angiogenesis (150), and has a critical role in promoting HCC progression. Knockdown of the m6A methyltransferase, METTL3, caused elevated circ-CCT3 expression and a decrease of its m6A levels (151).
Glioma is the most common malignant primary brain tumor (152). Wu et al. found that HOXA transcript antisense RNA myeloid-specific 1 (HOTAIRM1), as an oncogene, is highly expressed in both glioma tissues and cell lines (the higher grade, the more expression). METTL3-dependent m6A modification imparts HOTAIRM1 stability in glioma cells, and m6A-modified HOTAIRM1 transcript plays a vital role in the promotion of VM formation (153). Tao et al. analyzed data from The Cancer Genome Atlas (TCGA) database and found that patients with relatively high METTL3 expression had prolonged overall survival (OS). Further, METTL3 downregulation, resulting in decreased RNA m6A methylation, strengthened VM formation in GBM (154). In another study, METTL3 was found to be important in enhancing the stability and expression of target BUD13 mRNA. Methylated BUD13 was bound by the downstream target, CDK12, to regulate its stability and expression, thereby promoting MBNL1 phosphorylation by CDK12, and ultimately stimulating VM in GBM (155).
Lung cancer is a significant cause of cancer-associated deaths, imposing a substantial health burden (133). Feng et al. showed that METTL3 increases m6A levels and expression of transient receptor potential melastatin 7 (TRPM7) mRNA, and enhances the binding of IGF2BP2 and TRPM7, ultimately facilitating angiogenesis in non-small cell lung cancer (NSCLC) (156). Renal cell carcinoma (RCC) is a common urological malignant cancer and represents a leading threat to healthcare (157). Cheng et al. demonstrated that METTL3/METTL14 complex-mediated lncRNA IGFL2-AS1 m6A methylation results in VM formation in pazopanib resistant metastatic clear cell RCC (ccRCC) (158). METTL3-mediated m6A modification of CDC25B is upregulated in head and neck squamous cell carcinoma (HNSCC), and promotes HNSCC malignant characteristics, including cell proliferation, migration, and invasion, as well as angiogenesis (159). In bladder cancer (BCa), Wang and coworkers found that METTL3 ablation in BCa CSCs suppressed tumor angiogenesis by regulating TEK and VEGFA (160). Liu et al. proposed that PM2.5 exposure may induce m6A methylation levels in BCa. Following exposure to PM2.5, aberrantly upregulated METTL3 modifies the 3′-UTR of BIRC5 with m6A, and IGF2BP3 combines with BIRC5 to improve its stability, ultimately accelerating angiogenesis in BCa in VEGF-dependent manner (161).
Overall, these studies illustrate the close connection between METTL3 and tumor angiogenesis, and demonstrate that METTL3 has potential as a target for cancer diagnosis and treatment. However, the differential expression of METTL3 in various cancers and its dual regulatory effect on tumor angiogenesis suggest that we should pay attention to the development and application of METTL3 activators and inhibitors.
In RCC, tumor necrosis factor receptor-associated factor 1 (TRAF1) mRNA is modified with m6A by METTL14, and its stability is enhanced after binding with IGF2BP2. m6A-modified TRAF1 mRNA overexpression obviously activated AKT/mTOR/HIF-1α/VEGFA signaling pathway to facilitate angiogenesis, thus promoting sunitinib resistance in RCC (162). Cheng et al. reported that IGFL2-AS1 is an m6A-modified lncRNA in pazopanib sensitive ccRCC cells. Chronic pazopanib treatment reduced the m6A level of IGFL2-AS1 and increased its expression through inhibiting METTL14 expression in METTL3/METTL14 complex. IGFL2-AS1 binds the 5′-UTR of androgen receptor (AR) mRNA and promoted AR expression, ultimately leading to VM formation and pazopanib resistance in ccRCC (158). In tongue squamous cell carcinoma (TSCC), Wen et al. found that low protein expression of basic leucine zipper ATF-like transcription factor 2 (BATF2), also termed as a suppressor of AP-1 regulated by interferon (SARI) (163), was correlated with poor patient OS duration. The m6A methylase METTL14 mediates m6A modification of BATF2 mRNA to suppress its expression. BATF2 mRNA can constrain TSCC cell angiogenesis through downregulating VEGFA (164). Further, METTL14 and the demethylase, ALKBH5, can control the expression of one another, block the demethylase activity of the m6A reader, YTHDF3, and regulate m6A modification levels of angiogenesis-associated transcripts, resulting in tumor angiogenesis and malignant processes (165). As an important component of an m6A MTC, the mechanisms underlying the effects of METTL14 on angiogenesis in other cancers warrant further investigation.
The methyltransferase WTAP acts as an oncogene and tumor promoter, and is significantly elevated in patients with CRC. WTAP has a pivotal role in regulation of VEGFA mRNA expression in an m6A/YTHDC1-dependent manner, which subsequently activates the MAPK signaling pathway to influence angiogenesis in CRC cell lines (166).
Whether other m6A methyltransferases are also involved in tumor angiogenesis, or affect tumor angiogenesis through other regulatory mechanisms, remains to be explored.
Through Kaplan-Meier analysis, Rong et al. found that low levels of FTO expression were associated with inferior OS of patients with intrahepatic cholangiocarcinoma (ICC). Unlike previous studies, which reported that FTO is highly expressed in various cancers (167), levels of FTO were reported to be downregulated in clinical ICC samples and cell lines. It was detected that CD34, an indicator representing angiogenesis ability and micro-vessel density (MVD), was highly expressed in low FTO expression samples (168). However, the specific functional mechanism of m6A modification involved requires further exploration. Although the roles of FTO in tumor occurrence and development, self-renewal of CSCs, immunity, and metabolism have been extensively explored, its function in tumor angiogenesis is still poorly understood.
In lung cancer, Shen et al. demonstrated that upregulated ALKBH5 slightly enhances the stability and expression of lncRNA plasmacytoma variant translocation 1 (lncRNA PVT1). Overexpression of PVT1 partially recuperates the lung cancer angiogenesis constrained by ALKBH5 knockdown through mediating VEGFA expression (169). In contrast, Zhang et al. found that loss of ALKBH5 promotes lung cancer angiogenesis in an m6A-dependent manner. Levels of ALKBH5 are negatively correlated with those of VEGFA in patients with lung cancer, and effectively decrease the m6A methylation and translation efficiency of VEGFA mRNA, but had no effect on its mRNA levels (170). Additionally, Jin et al. demonstrated that expression of the angiogenesis-related protein, YAP, is negatively associated with that of ALKBH5, and that ALKBH5 inhibits the malignant progression of NSCLC cells by reducing YTHDFs-mediated YAP expression and suppressing miR-107/LATS2-mediated YAP activity in a HuR-dependent manner (171). In colorectal cancer, Guo and his colleagues found that ALKBH5 was significantly decreased and promoted the degradation rate of circ3823 along with YTHDF2 and YTHDF3 (172). Mechanistically, circ3823, which is closely associated with inferior patient prognosis, inhibits expression of miR-30c-5p through functioning as a competing endogenous RNA, as widely reported in various cancers (173, 174), subsequently promoting the expression of TCF7 and regulating the downstream targets, MYC and CCND1. Tube junction formation of HUVEC observation indicated that the circ3823/miR-30c-5p/TCF7 axis might facilitate the ability of angiogenesis in CRC (172). In HCC, the ALKBH5 demethylase cooperates with the methyltransferase, METTL3, in regulating circ-CCT3 m6A levels and expression, contributing to HCC growth, migration, and angiogenesis (151). In GBM, Tao et al. conducted Kaplan-Meier survival analysis demonstrating that ALKBH5 overexpression was associated with reduced patient OS duration. Further, ALKBH5 upregulation enhances VM by reducing target RNA m6A methylation (154). Yu et al. found that angiogenesis was restrained in multiple myeloma (MM) after knockdown of the demethylase, ALKBH5, both in vivo in and vitro, as demonstrated by VEGF secretion ability. Mechanistically, ALKBH5 promotes MM angiogenesis by inducing m6A-demethylation of SAV1 mRNA (175). In summary, ALKBH5 has a dual regulatory effect on tumors angiogenesis.
In CRC, m6A “reader” YTHDC1 recognized specific m6A sites on VEGFA mRNA to activate WTAP/YTHDC1/VEGFA/MAPK axis, thus promoting the CRC angiogenesis (166). In lung cancer, Zhang et al. demonstrated that the m6A regulators, YTHDC2 and eIF4G, trigger angiogenesis by promoting VEGFA mRNA translation (170). Moreover, m6A “reader” YTHDC2, as a tumor suppressor that is significantly positively correlated with lncRNA zinc ribbon domain-containing 1-antisense 1 (lncRNA ZNRD1-AS1) and regulates its stability. The ZNRD1-AS1/miR-942/TNS1 axis participates in lung cancer angiogenesis regulation via YTHDC2 (176). In the early stage, the team has demonstrated that YTHDC2 was downregulated in lung cancer cells and contributed to cell proliferation, migration and the EMT process (76). In GC, YTHDC2 recognized at 5′-UTR of m6A-modified YAP mRNA, resulting in the enhancement of YAP translation efficiency, thus promoting the malignant progression of GC (177).
It is established that HIF-1α mRNA is highly expressed in cancers (178) and that HIF-1α mRNA overexpression supports cancer progression through various mechanisms, including tumor cells proliferation, invasive, metastasis, as well as angiogenesis (179, 180). In GC, Bai et al. found that the HIF-1α/H19/YTHDF1/scavenger receptor class B member 1 (SCARB1) axis is involved in angiogenesis and malignant phenotype. Mechanistically, m6A “reader” YTHDF1, functions as a bridge between lncRNA H19 and SCARB1 mRNA, and can recognize m6A modification sites on 3′-UTR of SCARB1 to facilitate its translation into scavenger receptor class B type I (SR-BI), ultimately promoting GC cells angiogenesis (181). Further, the m6A “reader”, YTHDF2, targets ADAMTS9 mRNA by recognizing its m6A motifs and promotes its degradation in a METTL3-dependent manner. Suppression of ADAMTS9 expression facilitates angiogenesis and carcinogenesis in GC (144). In CRC, YTHDF3 cooperates with the demethylase, ALKBH5, to promote the rate of circ3823 degradation, contributing to CRC growth, metastasis and angiogenesis through the circ3823/miR-30c-5p/TCF7 axis. Although it has been confirmed that interaction between YTHDF2 and YTHDF3 can promote target mRNA degradation, whether YTHDF2 participates in circ3823 degradation along with YTHDF3 and ALKBH5 requires verification (172). Additionally, YTHDF2 in HCC specimens was downregulated in contrast to normal liver histiocytes. YTHDF2 deficiency resulted in the escalation of vessel abnormity and angiogenesis of HUVECs, while overexpression of YTHDF2 reduced vessel density and permeability (182). Moreover, serpin family E member 2 (SERPINE2) mRNA, which induces angiogenesis in breast cancer and oral squamous cell carcinoma (183, 184), is upregulated after YTHDF2 knockdown and responsible for the disruption of normal vascularization (182). In glioma, Dixit et al. reported that YTHDF2 was upregulated in mesenchymal GBM and its depletion increased the VEGFA transcript decay rate in GBM stem cells in an m6A-dependent manner, thus affecting tumor angiogenesis (185). In ccRCC, circPOLR2A acts as an oncogene closely correlated with malignancy, and YTHDF2 suppresses circPOLR2A expression in an m6A-dependent manner, where the circPOLR2A/PEBP1 axis positively affects angiogenesis in ccRCC (186). Moreover, Cheng et al. demonstrated that IGFL2-AS1 was demethylated by the METTL3/METTL14 complex and stabilized by release of the binding-protein YTHDF2 in pazopanib-resistant cells. Stabilized and highly expressed IGFL2-AS1 favored AR mRNA translation and expression, leading to VM formation and development in ccRCC (158). Chang and colleagues found that the YTHDF3 upregulates its own protein expression through automatic regulation and then binds to m6A-enriched VEGFA mRNA to increase VEGFA expression and angiogenesis in brain metastases of breast cancer in humans (187). Furthermore, In NSCLC cells, YTHDFs have a crucial role in regulation of YAP expression; YTHDF3 can bind YAP pre-mRNA, while YTHDF1 and YTHDF2 regulate YAP mRNA expression through competitively interacting with YTHDF3. Further, YTHDF2 promotes YAP mRNA decay by recruiting the AGO2 degradation system, whereas YTHDF1 interacts with eIF3a to facilitate YAP translation (171). Taken together, YTHDFs represent promising potential diagnostic biomarkers and therapeutic targets.
In CRC, both LINC00662 and VEGFA contribute to angiogenesis mediated through m6A by the enzyme METTL3. Interestingly, although high expression of IGF2BP1 in CRC was confirmed by analyzing data from TCGA CRC and the CPTAC protein prediction database, it has no effect on stabilization of RNAs (138). In patients with CRC, the m6A “reader” IGF2BP3 activates VEGF mRNA stability and expression by recognizing and combining with its m6A modification sites in an m6A-dependent manner. Moreover, interfering with IGF2BP3 represses angiogenesis in colon cancer, which was confirmed by HUVECs assay (188). Additionally, IGF2BP3 and IGF2BP2 are involved in recognition and binding to METTL3-methylated EphA2 and VEGFA, respectively, thereby enhancing the stability of these target genes and preventing their degradation. Subsequently, EphA2 and VEGFA activate the PI3K/AKT/mTOR and ERK1/2 signaling pathways, respectively, stimulating VM formation (166). AKT signaling pathway can be provoked to participate cancer angiogenesis. In RCC, In RCC, METTL14 activity modifies TRAF1 mRNA by m6A modification, and its stability was enhanced after binding with IGF2BP2. Further, m6A-modified TRAF1 mRNA overexpression obviously activated AKT/mTOR/HIF-1α/VEGFA signaling pathway to facilitate angiogenesis, thus promoting sunitinib resistance in RCC (162). In lung cancer, Shen and colleagues demonstrated that IGF2BP2 permeates ECs in the microenvironmental via lung adenocarcinoma (LUAD) cell-derived exosomes, subsequently mediating the m6A modification of FLT4 to improve its stability and expression. Then, FLT4 activates the PI3K-Akt signaling pathway, eventually promoting angiogenesis in LUAD cells (189).
Besides AKT effector, several factors are also involved in the regulation of angiogenic process, including p38 MAPK (190), FAK (191), signal transducer and activator of transcription 1 (STAT1) (192)and Rho GTPases (193). In ovarian cancer (OC), Ye et al. found that OC-derived exosomal circRNA nuclear factor IX (circNFIX) regulates the Janus-activated kinase (JAK)/STAT1 pathway via the miR-518a-3p/TRIM44 axis, thereby promoting tumor angiogenesis (194). The team further validated that IGF2BP1/2/3 recognizes m6A modification sites in circNFIX, leading to increased circNFIX expression in OC cells (195). Therefore, IGF2BP1/2/3 may promote OC angiogenesis by regulating circNFIX; however, the specific mechanism involved requires further in-depth investigation. Moreover, Zeng et al. showed that the LncSNHG5/ZNF281 axis upregulates transcription and secretion of CCL2 and CCL5, thereby activating P38 MAPK signaling in HUVECs, ultimately stimulating angiogenesis and vascular permeability in a VEGF-independent manner (196). Mechanistically, LncSNHG5 is highly increased in breast cancer-associated fibroblasts, which are an important subset of stromal fibroblasts in the tumor microenvironment (197). Enhanced LncSNHG5 has a key role in regulating angiogenesis and vascular leakiness by mediating recruitment of IGF2BP2 to augment ZNF281 mRNA stabilization, thus inducing lung premetastatic niche formation, which is reported to be essential for malignant tumor progression (198).
VEGF is key for tumor angiogenesis and is upregulated by HIF and other oncogenic factors. In GC, Jiang et al. demonstrated that IGF2BP3 binds to m6A sites on HIF-1α mRNA in stomach cancer (SC) cells to positively modulate HIF-1α mRNA expression in an m6A-dependent manner and promote angiogenesis in SC through the IGF2BP3/HIF-1α pathway under hypoxic conditions (199). Further, the HIF-1α/VEGF axis can contribute to mediation of angiogenesis and GC cell malignant growth in a hypoxic microenvironment (200). Inhibition of VEGF secretion by IGF2BP3 deficiency can be alleviated by HIF-1α mRNA overexpression (199). However, further exploration is needed to determine whether a methylase or demethylase regulates the IGF2BP3/HIF-1α/VEGF axis. In BCa, Liu et al. found that IGF2BP3 combines with METTL3-modified BIRC5 mRNA to improve its stability, ultimately accelerating angiogenesis in BCa exposed to PM2.5 in a VEGF-dependent manner (161). Wang and his colleagues verified that m6A “reader” IGF2BP3 recognized and bound m6A sites on METTL3-mediated HDGF mRNA to stabilize it and the subsequent GC angiogenesis and malignant progress (142).
Ma et al. found that hepatocyte nuclear factor 4 gamma (HNF4G) is a target of miR-320b, where HNF4G expression is reduced on miR-320b overexpression. HNF4G upregulates the m6A reader, IGF2BP2, by combining with its promoter region, and upregulation of IGF2BP2 enhances the thymidine kinase 1 (TK1) stability and expression, thus facilitating angiogenesis in lung cancer (201). IIGF2BP2 functions as a bridge between the lncRNA, deoxyguanosine kinase antisense RNA 1 (DGUOK-AS1) and TRPM7 mRNA, where TRPM7 is positively regulated by DGUOK-AS1 in NSCLC; hence, the DGUOK-AS1/IGF2BP2/TRPM7 axis promotes angiogenesis in NSCLC (156). In glioma, IGFBP2 is closely associated with METTL3-mediated HOTAIRM1 via its m6A binding-domains and positively regulated by HOTAIRM1, thus promoting VM (153). Collectively, IGF2BPs appear to promote tumor angiogenesis, and targeted inhibition of IGF2BP expression may suppress tumor angiogenesis to some extent.
In gastric cancer, m6A reader HNRNPA2B1 directly bound m6A sites of METTL3-methylated CENPF mRNA and promoted the mRNA stability, ultimately GC angiogenesis and metastasis through activating the FAK/MAPK axis (145). Additionally, Gu et al. reported that HNRNPC predicted poor prognosis in patients with NSCLC, and was associated with NSCLC angiogenesis by Gene Ontology and Gene Set Enrichment Analysis, although the exact mechanism involved remains unclear (202).
He et al. analyzed 24 main m6A RNA methylation regulators from the TCGA breast cancer dataset, divided them into two subsets according to the height of RNA methylation modification-RNA methylation 1 (RM1) and RM2, and identified that angiogenesis was significantly comfortable in RM1 (203). Further, Li et al. showed that m6A modification in glioma stem cells is mainly distributed around sites of neovascularization in hypoxic environments. Further, the overall interaction between angiogenesis-related genes(ARGs) and m6A regulators(MAGs) is significantly correlated in low-grade gliomas (LGGs) (204). These findings provide potential new research directions for investigation of the relationship between m6A and angiogenesis, including deeper studies to generate more valuable and practical guidance that can inform cancer treatment. In summary, VEGF is the main factor involved in promotion of tumor angiogenesis, while other factors, such as Ang, can coordinate with VEGF in this context. Enzymes involved in m6A methylation modification and regulation play crucial roles in tumor angiogenesis, not only by direct regulation of VEGF expression, but also through mediating angiogenesis-related signaling pathways, such as PI3K/AKT, MAPK, and JAK/STATA, to affect VEGF expression (Figure 3). Nevertheless, numerous m6A regulators have not been investigated in the context of tumor angiogenesis and study of mechanisms involving such m6A regulators may be a fruitful research direction. Further, exploration of the interactions between the three types of m6A enzymes during tumor angiogenesis is of interest.
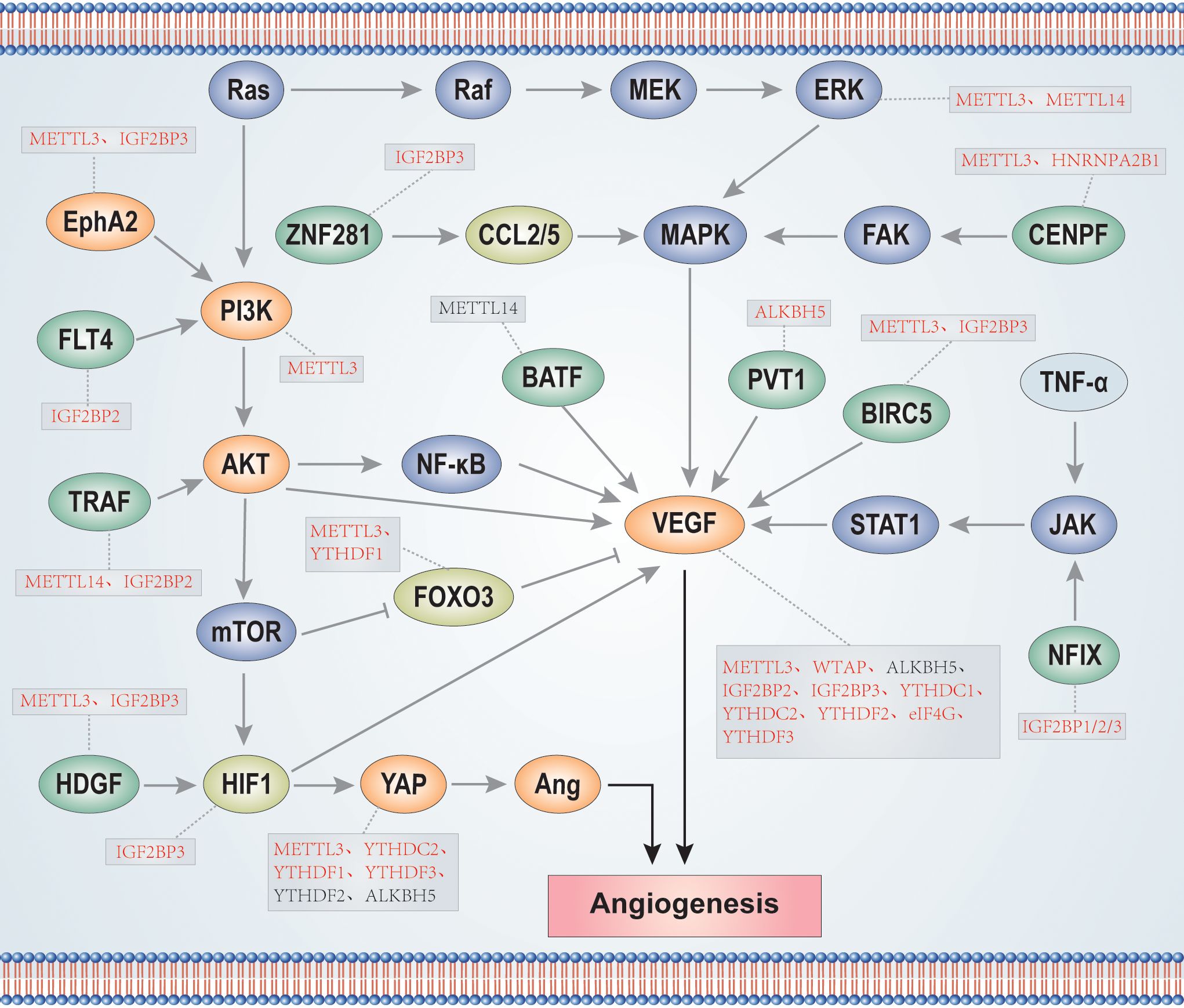
Figure 3. m6A enzymes participate in the regulation of signaling pathways and proteins related to tumor angiogenesis. The red font indicates that m6A enzymes positively regulate proteins, while the black font indicates that m6A enzymes negatively regulate proteins.
m6A modification has become a hot topic of current research, attracting various researchers who have focused on in-depth investigation into its roles in pathogenesis and malignant phenotypes. As described above, numerous studies have shown that m6A-regulated tumor angiogenesis and VM have crucial roles in tumor occurrence and development. In this section, we discuss the potential clinical significance of targeting m6A-regulated tumor angiogenesis.
Currently, the clinical therapeutic strategy of anti-angiogenesis is among the most commonly used treatment schemes to control tumor proliferation and distant metastasis. Since the concept of targeted angiogenesis was proposed, the US Food and Drug Administration has approved numerous anti-angiogenic drugs (93), which can be divided into three main categories: first, monoclonal antibodies, including bevacizumab and ramucirumab; second, fusion proteins, of which Ziv-aflibercept is an example; and third, VEGFR-targeting small molecules, including sorafenib, sunitinib, pazopanib, lenvatinib, and tivozanib, among others (205). For example, sunitinib is a tyrosine kinase inhibitor that shows potent anti-angiogenic activity, and was recommended as a first-line targeted drug for patients with recurrent and unresectable RCC (206–208). However, the majority of patients with RCC eventually develop drug resistance and malignant tumor progression, resulting in inability of sunitinib to effectively prolong their survival (209, 210). Angiogenesis switch is among the mechanisms elucidated as involved in the development of resistance to sunitinib (211), and is also associated with high expression of TRAF1 mRNA; increased TRAF1 expression contributes to activation of downstream anti-apoptotic and anti-angiogenic pathways in sunitinib-resistant cells, leading to drug resistance in patients with RCC (162). In combination with standard first-line chemotherapy drugs (including cisplatin and carboplatin), the monoclonal antibody, bevacizumab, can significantly prolong progression-free survival (PFS) and OS in patients with advanced NSCLC, as demonstrated by a meta-analysis of randomized studies (212). other drugs are currently under investigation and are expected to be used in clinical anti-angiogenesis treatment of tumors in the future. For example, using xenograft and chorioallantoic membrane angiogenesis models, as well as detection of CD31 via IHC assay, Wei et al. demonstrated that tumor angiogenesis is inhibited after treatment with verteporfin, which downregulates angiopoietin-2 (Ang2) by suppressing YAP activity (213). The correlation between Ang2 and YAP has also been clarified in previous studies, where Ang2 was reported as an important biomarker of vasculogenic events (214), and YAP also functions to promote angiogenesis through regulating angiogenic germination and remodeling of HUVECs (215). Verteporfin can also repress VM by downregulating MMP2, VE-cadherin, and a-SMA expression (216). In addition, RSK and TTK were identified as novel modulators of angiogenesis and potential targets for anti- angiogenic therapy (217). In accordance with the findings of these studies, resistance of tumors to anti-angiogenic drugs is a complex process involving multiple genes, factors, mechanisms, and the tumor microenvironment. Changing the expression of certain key transcripts in angiogenesis and activating or inhibiting specific signaling pathways may enhance the sensitivity of tumors to treatment. Hence, identification of new targets for anti-angiogenesis treatment and combining them with other drugs could provide new avenues for exploration.
Tumor anti-angiogenesis therapy has not achieved optimal therapeutic effects, due to the development of resistance over time, raising concerns regarding whether the combination of m6A and anti-angiogenic drugs will contribute to improved antitumor therapy. Some recent studies have discussed mechanisms involving m6A-regulated tumor angiogenesis or VM in drug treatment and resistance, and suggested that targeting m6A-regulated tumor angiogenesis also has therapeutic potential. VEGFR-targeted treatment is a common method of inhibiting tumor angiogenesis. The IGFL2-AS1/AR signaling axis is clinically associated with VM formation, and is strongly increased during pazopanib resistance of metastatic ccRCC. Mechanistically, the lncRNA, IGFL2-AS1, interacts with the 5’UTR of AR mRNA to regulate the activity of the upstream open reading frame and enhance translation of AR mRNA, which was demethylated by METTL3/METTL14, during pazopanib resistance (158).Chen et al. manifested that a novel pharmaceutical intervention strategy for the treatment of patients with sunitinib might be targeting TRAF1 mRNA and its pathways in the near future. Mechanistically, METTL14-mediated m6A modification of TRAF1 enhances its stability in an IGF2BP2-dependent manner, upregulating TRAF1 expression in sunitinib resistant cells. In addition, TRAF1 overexpression significantly stimulates AKT/mTOR/HIF-1α/VEGFA signaling, while silencing TRAF1 increases sunitinib-induced anti-apoptotic and anti-angiogenic effects (162). Further, Lin et al. found that the methyltransferase, METTL3, is dramatically down-regulated in human sorafenib-resistant HCC. METTL3 depletion decreases the stability of its downstream target, FOXO3, in a YTHDF1-dependent manner and reduces FOXO3 expression levels, ultimately promoting sorafenib resistance (148). hence, targeting METTL3 expression may enhance treatment response to sorafenib. In many types of cancer, tumor angiogenesis is also accompanied by increased YAP mRNA expression levels and activity. The m6A demethylase, ALKBH5, decreases YTHDF-mediated YAP expression to suppress tumor growth and metastasis in NSCLC cells (171). Further, IGF2BP2 recognizes m6A sites on YAP mRNA and facilitates its translation efficiency in CRC cells. The IGF2BP2/YAP/ErbB2 axis promotes CRC cells proliferation, invasion, and migration and represses CRC cell apoptosis (218). Therefore, targeting m6A enzymes which mediate YAP expression and activity could be a promising therapeutic strategy. This suggests the new idea that m6A can increase the sensitivity of anti-angiogenic drugs through targeted regulation; therefore, combination therapy using m6A inhibitors or activators together with angiogenesis inhibitors, is expected to improve anti-tumor efficacy.
Nevertheless, there has been relatively little research to date on the mechanism underlying targeting of m6A combined with anti-angiogenesis approaches, and further in-depth research is needed to determine the clinical value of such methods, in terms of anti-tumor therapy and survival improvements for patients with cancer. Overall, m6A regulatory factors are of great significance in tumor prognosis and diagnosis. Some m6A-related molecules are currently in clinical trials, and m6A is expected to become a drug target for clinical anti-cancer treatment in the future (Table 3).
Table 3. Clinical trials of m6A regulators related to cancer. (Data from ClinicalTrials.gov).
The connections between tumors and angiogenesis have been explored for more than five decades, since they were first proposed by Professor Folkman. The rapid development of high-throughput sequencing technologies, popularization of bioinformatics, and emergence of highly specific antibodies, have led to verification that m6A methylation is involved in various tumor malignant processes, including proliferation, invasion, metastasis, and immune escape, as well as angiogenesis. At present, there is an increasing research focus on the relationship between m6A and tumor angiogenesis, and m6A modification has been found to directly or indirectly regulate tumor cell angiogenesis. In different types of tumor, m6A modification influences biological behaviors through affecting the stability and expression of target mRNAs and activating or repressing angiogenesis-related signaling pathways, to regulate tumor angiogenesis. Further, m6A modification also occurs in ncRNAs, including lncRNAs, miRNAs, and circRNAs, and m6A-modified ncRNAs can impact their downstream signaling axes, thereby regulating tumor angiogenesis in an m6A-dependent manner. There is a growing body of evidence suggesting that m6A modification regulates tumor angiogenesis and malignant phenotypes through an extremely complex interaction network, resulting in influences on the occurrence, development, treatment, and prognosis of cancer. Further study of the potential mechanisms underlying the relationships between m6A methylation and tumor angiogenesis will improve understanding and provide novel possibilities for tumor diagnostic methods and therapeutic strategies in the near future. At present, the effectiveness of anti-angiogenic drugs for treating patients with cancer is suboptimal. The strategy of targeting m6A in combination with anti-angiogenic drugs or vascular mimicry inhibitors is predicted to open a promising new frontier for future tumor treatment; however, enormous research challenges remain. According to existing research, it is clear that only a proportion of m6A regulators have been investigated, and the relationships between other m6A regulators and tumor angiogenesis remains unclear. Further exploration is needed to determine whether other factors that promote or suppress m6A deposition contribute to tumor angiogenesis. Additionally, m6A regulators have a dual regulatory effect on tumor angiogenesis in different cancers; and even within the same cancer, different researchers hold opposite views. Differences among tumor microenvironments and upstream and downstream genes may be important factors affecting the expression and function of m6A regulators. Therefore, multi-center, large-scale research could provide deeper and more comprehensive understanding of mechanisms involving m6A in tumor angiogenesis, and facilitate screening and development of specific inhibitors or activators targeting m6A. Given the extensive research on the mechanisms underlying the role of m6A modification in tumor angiogenesis, the development of specific m6A-targeted drugs will be of great significance for achieving personalized and precise treatment and, combined with other approaches, such drugs may improve malignant tumor therapy sensitivity, reduce drug resistance and side effects, and achieve superior therapeutic effects. Although there have been numerous reports indicating that various m6A regulators have potential diagnostic, prognostic, and therapeutic value in the context of anti-tumor angiogenesis, research on the regulation of tumor angiogenesis by m6A remains in its infancy. The development of specific m6A-targeted activators or inhibitors suitable for clinical application and the translation of scientific research into clinical practice will require considerable further efforts.
LQ: Writing – original draft, Writing – review & editing. XZ: Writing – review & editing. XQ: Writing – review & editing. XC: Writing – review & editing. SL: Resources, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Natural Science Foundation of China (82260579) and the Natural Science Foundation of Guangxi, China, (2020GXNSFAA159056)
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kretschmer M, Rüdiger D, Zahler S. Mechanical aspects of angiogenesis. Cancers. (2021) 13:4987. doi: 10.3390/cancers13194987
2. Kumari R, Syeda S, Shrivastava A. Nature’s elixir for cancer treatment: targeting tumor-induced neovascularization. Curr Med Chem. (2024) 31:5281–304. doi: 10.2174/0109298673282525240222050051
3. Zhou Z, Lv J, Yu H, Han J, Yang X, Feng D, et al. Mechanism of RNA modification N6-methyladenosine in human cancer. Mol Cancer. (2020) 19:104. doi: 10.1186/s12943-020-01216-3
4. Shi H, Chai P, Jia R, Fan X. Novel insight into the regulatory roles of diverse RNA modifications: Re-defining the bridge between transcription and translation. Mol Cancer. (2020) 19:78. doi: 10.1186/s12943-020-01194-6
5. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell. (2012) 149:1635–46. doi: 10.1016/j.cell.2012.05.003
6. Nombela P, Miguel-López B. Blanco S.The role of m6A, m5C and Ψ RNA modifications in cancer: Novel therapeutic opportunities. Mol Cancer. (2021) 20:18. doi: 10.1186/s12943-020-01263-w
7. Zhou X, Li C, Chen T, Li W, Wang X, Yang Q. Targeting RNA N6-methyladenosine to synergize with immune checkpoint therapy. Mol Cancer. (2023) 22:36. doi: 10.1186/s12943-023-01746-6
8. Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. (2019) 20:608–24. doi: 10.1038/s41580-019-0168-5
9. Wang X, Ma R, Zhang X, Cui L, Ding Y, Shi W, et al. Crosstalk between N6-methyladenosine modification and circular RNAs: current understanding and future directions. Mol Cancer. (2021) 20:121. doi: 10.1186/s12943-021-01415-6
10. Li H, Wu H, Wang Q, Ning S, Xu S, Pang D. Dual effects of N6-methyladenosine on cancer progression and immunotherapy. Mol Ther Nucleic Acids. (2021) 24::25–39. doi: 10.1016/j.omtn.2021.02.001
11. Hu B, Wang X, Gu X, Zou C, Gao Z, Zhang H, et al. N6-methyladenosine (m6A) RNA modification in gastrointestinal tract cancers: roles, mechanisms, and applications. Mol Cancer. (2019) 18:178. doi: 10.1186/s12943-019-1099-7
12. Cao X, Geng Q, Fan D, Wang Q, Wang X, Zhang M, et al. m6A methylation: a process reshaping the tumour immune microenvironment and regulating immune evasion. Mol Cancer. (2023) 22:42. doi: 10.1186/s12943-022-01704-8
13. Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. (2015) 12:767–72. doi: 10.1038/nmeth.3453
14. Shi B, Liu W, Yang K, Jiang G, Wang H. The role, mechanism, and application of RNA methyltransferase METTL14 in gastrointestinal cancer. Mol Cancer. (2022) 21:163. doi: 10.1186/s12943-022-01634-5
15. Yang X, Liu Q, Xu W, Zhang Y, Yang Y, Ju L, et al. m6A promotes R-loop formation to facilitate transcription termination. Cell Res. (2019) 29:1035–8. doi: 10.1038/s41422-019-0235-7
16. Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson SC, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature. (2017) 552:126–31. doi: 10.1038/nature24678
17. Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N6-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature. (2015) 518:560–4. doi: 10.1038/nature14234
18. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. (2012) 485:201–6. doi: 10.1038/nature11112
19. Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell. (2015) 162:1299–308. doi: 10.1016/j.cell.2015.08.011
20. Wu Y, Yang X, Chen Z, Tian L, Jiang G, Chen F, et al. m6A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol Cancer. (2019) 18:87. doi: 10.1186/s12943-019-1014-2
21. Chen R, Chen X, Xia L, Zhang J, Pan Z, Ma X, et al. N6-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat Commun. (2019) 10:4695. doi: 10.1038/s41467-019-12651-2
22. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell. (2015) 161:1388–99. doi: 10.1016/j.cell.2015.05.014
23. Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. (2017) 27:315–28. doi: 10.1038/cr.2017.15
24. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. (2014) 505:117–20. doi: 10.1038/nature12730
25. Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, et al. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. (2015) 347:1002–6. doi: 10.1126/science.1261417
26. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. (2018) 20:285–95. doi: 10.1038/s41556-018-0045-z
27. Yang D, Qiao J, Wang G, Lan Y, Li G, Guo X, et al. N6-Methyladenosine modification of lincRNA 1281 is critically required for mESC differentiation potential. Nucleic Acids Res. (2018) 46:3906–20. doi: 10.1093/nar/gky130
28. Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet. (2014) 15:293–306. doi: 10.1038/nrg3724
29. Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, Mele A, et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. (2015) 29:2037–53. doi: 10.1101/gad.269415.115
30. Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5′ UTR m6A promotes cap-independent translation. Cell. (2015) 163:999–1010. doi: 10.1016/j.cell.2015.10.012
31. Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian S. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature. (2015) 526:591–4. doi: 10.1038/nature15377
32. Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. (2021) 6:74. doi: 10.1038/s41392-020-00450-x
33. Lou X, Wang J, Wei Y, Sun J. Emerging role of RNA modification N6-methyladenosine in immune evasion. Cell Death Dis. (2021) 12:300. doi: 10.1038/s41419-021-03585-z
34. Li W, Hao Y, Zhang X, Xu S. Pang D.Targeting RNA N6-methyladenosine modification: a precise weapon in overcoming tumor immune escape. Mol Cancer. (2022) 21:176. doi: 10.1186/s12943-022-01652-3
35. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. (2014) 10:93–5. doi: 10.1038/nchembio.1432
36. Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. (1997) 3:1233–47.
37. Wang T, Kong S, Tao M, Ju S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer. (2022) 19:88. doi: 10.1186/s12943-020-01204-7
38. Yoshida A, Oyoshi T, Suda A, Futaki S, Imanishi M. Recognition of G-quadruplex RNA by a crucial RNA methyltransferase component, METTL14. Nucleic Acids Res. (2022) 50:449–57. doi: 10.1093/nar/gkab1211
39. Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m6A Modification. Cell Stem Cell. (2018) 22:191–205.e9. doi: 10.1016/j.stem.2017.11.016
40. Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of mettl3 and mettl14 methyltransferases. Mol Cell. (2016) 63:306–17. doi: 10.1016/j.molcel.2016.05.041
41. Zeng Z, Pan Q, Sun Y, Huang H, Chen X, Chen T, et al. METTL3 protects METTL14 from STUB1-mediated degradation to maintain m6A homeostasis. EMBO Rep. (2023) 24:e55762. doi: 10.15252/embr.202255762
42. Ping X, Sun B, Wang L, Xiao W, Yang X, Wang W, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. (2014) 24:177–89. doi: 10.1038/cr.2014.3
43. Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m6A machinery component Wtap/Fl(2)d. Genes Dev. (2018) 32:415–29. doi: 10.1101/gad.309146.117
44. Zolotukhin AS, Uranishi H, Lindtner S, Bear J, Pavlakis GN, Felber BK. Nuclear export factor RBM15 facilitates the access of DBP5 to mRNA. Nucleic Acids Res. (2009) 37:7151–62. doi: 10.1093/nar/gkp782
45. Zhang S. Mechanism of N6-methyladenosine modification and its emerging role in cancer. Pharmacol Ther. (2018) 189:173–83. doi: 10.1016/j.pharmthera.2018.04.011
46. Qian J, Gao J, Sun X, Cao M, Shi L, Xia T, et al. KIAA1429 acts as an oncogenic factor in breast cancer by regulating CDK1 in an N6-methyladenosine-independent manner. Oncogene. (2019) 38:6123–41. doi: 10.1038/s41388-019-0861-z
47. Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, et al. VIRMA mediates preferential m6A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discovery. (2018) 4:10. doi: 10.1038/s41421-018-0019-0
48. Wen J, Lv R, Ma H, Shen H, He C, Wang J, et al. Zc3h13 regulates nuclear RNA m6A methylation and mouse embryonic stem cell self-renewal. Mol Cell. (2018) 69:1028–1038.e6. doi: 10.1016/j.molcel.2018.02.015
49. Růžička K, Zhang M, Campilho A, Bodi Z, Kashif M, Saleh M, et al. Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol. (2017) 215:157–72. doi: 10.1111/nph.14586
50. Ma H, Wang X, Cai J, Dai Q, Natchiar SK, Lv R, et al. N6-Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol. (2019) 15:88–94. doi: 10.1038/s41589-018-0184-3
51. Pinto R, Vågbø CB, Jakobsson ME, Kim Y, Baltissen MP, O’Donohue M, et al. The human methyltransferase ZCCHC4 catalyses N6-methyladenosine modification of 28S ribosomal RNA. Nucleic Acids Res. (2022) 48:830–46. doi: 10.1093/nar/gkz1147
52. Sepich-Poore C, Zheng Z, Schmitt E, Wen K, Zhang ZS, Cui X, et al. The METTL5-TRMT112 N6-methyladenosine methyltransferase complex regulates mRNA translation via 18S rRNA methylation. J Biol Chem. (2022) 298:101590. doi: 10.1016/j.jbc.2022.101590
53. van Tran N, Ernst FGM, Hawley BR, Zorbas C, Ulryck N, Hackert P, et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. (2019) 47:7719–33. doi: 10.1093/nar/gkz619
54. Ruszkowska A. METTL16, methyltransferase-like protein 16: current insights into structure and function. Int J Mol Sci. (2021) 22:2176. doi: 10.3390/ijms22042176
55. Sun L, Zhang Y, Yang B, Sun S, Zhang P, Luo Z, et al. Lactylation of METTL16 promotes cuproptosis via m6A-modification on FDX1 mRNA in gastric cancer. Nat Commun. (2023) 14:6523. doi: 10.1038/s41467-023-42025-8
56. Ma Q, Gui Y, Ma X, Zhang B, Xiong W, Yang S, et al. N6-methyladenosine writer METTL16-mediated alternative splicing and translation control are essential for murine spermatogenesis. Genome Biol. (2024) 25:193. doi: 10.1186/s13059-024-03332-5
57. Barone S, Cerchia C, Summa V, Brindisi M. Methyl-transferase-like protein 16 (METTL16): the intriguing journey of a key epitranscriptomic player becoming an emerging biological target. J Med Chem. (2024) 67:14786–806. doi: 10.1021/acs.jmedchem.4c01247
58. Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, et al. The U6 snRNA m 6 A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell. (2017) 169:824–835.e14. doi: 10.1016/j.cell.2017.05.003
59. Zhang H, Yin M, Huang H, Zhao G, Lu M. METTL16 in human diseases: What should we do next? Open Med (Wars). (2023) 18:20230856. doi: 10.1515/med-2023-0856
60. Yue S, Liu H, Su H, Luo C, Liang H, Zhang B, et al. m6A-regulated tumor glycolysis: new advances in epigenetics and metabolism. Mol Cancer. (2023) 22:137. doi: 10.1186/s12943-023-01841-8
61. Zhao X, Yang Y, Sun B, Shi Y, Yang X, Xiao W, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. (2014) 24:1403–19. doi: 10.1038/cr.2014.151
62. Fu Y, Jia G, Pang X, Wang RN, Wang X, Li CJ, et al. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat Commun. (2013) 4:1798. doi: 10.1038/ncomms2822
63. Tang C, Klukovich R, Peng H, Wang Z, Yu T, Zhang Y, et al. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3′-UTR mRNAs in male germ cells. Proc Natl Acad Sci U S A. (2018) 115:E325–33. doi: 10.1073/pnas.1717794115
64. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. (2013) 49:18–29. doi: 10.1016/j.molcel.2012.10.015
65. Yang G, Sun Z, Zhang N. Reshaping the role of m6A modification in cancer transcriptome: a review. Cancer Cell Int. (2021) 20:353. doi: 10.1186/s12935-020-01445-y
66. Ueda Y, Ooshio I, Fusamae Y, Kitae K, Kawaguchi M, Jingushi K, et al. AlkB homolog 3-mediated tRNA demethylation promotes protein synthesis in cancer cells. Sci Rep. (2017) 7:42271. doi: 10.1038/srep42271
67. An Y, Duan H. The role of m6A RNA methylation in cancer metabolism. Mol Cancer. (2022) 21:14. doi: 10.1186/s12943-022-01500-4
68. Meyer KD, Jaffrey SR. Rethinking m6A readers, writers, and erasers. Annu Rev Cell Dev Biol. (2017) 33:319–42. doi: 10.1146/annurev-cellbio-100616-060758
69. Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4–NOT deadenylase complex. Nat Commun. (2016) 7:12626. doi: 10.1038/ncomms12626
70. Lee Y, Choe J, Park OH, Kim YK. Molecular Mechanisms Driving mRNA Degradation by m6A Modification. Trends Genet. (2020) 36:177–88. doi: 10.1016/j.tig.2019.12.007
71. Zou Z, Sepich-Poore C, Zhou X, Wei J, He C. The mechanism underlying redundant functions of the YTHDF proteins. Genome Biol. (2023) 24:17. doi: 10.1186/s13059-023-02862-8
72. Xiao W, Adhikari S, Dahal U, Chen Y, Hao Y, Sun B, et al. Nuclear m 6 A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell. (2016) 61:507–19. doi: 10.1016/j.molcel.2016.01.012
73. Roundtree IA, Luo G, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. Elife. (2017) 6:e31311. doi: 10.7554/eLife.31311
74. Lesbirel S, Viphakone N, Parker M, Parker J, Heath C, Sudbery I, et al. The m6A-methylase complex recruits TREX and regulates mRNA export. Sci Rep. (2018) 8:13827. doi: 10.1038/s41598-018-32310-8
75. Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. (2017) 27:1115–27. doi: 10.1038/cr.2017.99
76. Wang J, Tan L, Jia B, Yu X, Yao R, OUYang N, et al. Downregulation of m6A Reader YTHDC2 Promotes the Proliferation and Migration of Malignant Lung Cells via CYLD/NF-κB Pathway. Int J Biol Sci. (2021) 17:2633–51. doi: 10.7150/ijbs.58514
77. Liu N, Pan T. N6-methyladenosine–encoded epitranscriptomics. Nat Struct Mol Biol. (2016) 23:98–102. doi: 10.1038/nsmb.3162
78. Lewis CJT, Pan T, Kalsotra A. RNA modifications and structures cooperate to guide RNA–protein interactions. Nat Rev Mol Cell Biol. (2017) 18:202–10. doi: 10.1038/nrm.2016.163
79. Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. (2017) 45:6051–63. doi: 10.1093/nar/gkx141
80. Sun L, Fazal FM, Li P, Broughton JP, Lee B, Tang L, et al. RNA structure maps across mammalian cellular compartments. Nat Struct Mol Biol. (2019) 26:322–30. doi: 10.1038/s41594-019-0200-7
81. Edens BM, Vissers C, Su J, Arumugam S, Xu Z, Shi H, et al. FMRP Modulates Neural Differentiation through m6A-Dependent mRNA Nuclear Export. Cell Rep. (2019) 28:845–854.e5. doi: 10.1016/j.celrep.2019.06.072
82. Zhang F, Kang Y, Wang M, Li Y, Xu T, Yang W, et al. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum Mol Genet. (2018) 27:3936–50. doi: 10.1093/hmg/ddy292
83. Wu R, Li A, Sun B, Sun J, Zhang J, Zhang T, et al. A novel m6A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. (2019) 29:23–41. doi: 10.1038/s41422-018-0113-8
84. Wang Y, Wang Y, Patel H, Chen J, Wang J, Chen Z, et al. Epigenetic modification of m6A regulator proteins in cancer. Mol Cancer. (2023) 22:102. doi: 10.1186/s12943-023-01810-1
85. Sun S, Gao T, Pang B, Su X, Guo C, Zhang R, et al. RNA binding protein NKAP protects glioblastoma cells from ferroptosis by promoting SLC7A11 mRNA splicing in an m6A-dependent manner. Cell Death Dis. (2022) 13:73. doi: 10.1038/s41419-022-04524-2
86. Wang H, Tang A, Cui Y, Gong H, Li H. LRPPRC facilitates tumor progression and immune evasion through upregulation of m6A modification of PD-L1 mRNA in hepatocellular carcinoma. Front Immunol. (2023) 14:1144774. doi: 10.3389/fimmu.2023.1144774
87. Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, et al. Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature. (2019) 567:414–9. doi: 10.1038/s41586-019-1016-7
88. Shim S, Lee HG, Lee H, Seo PJ. H3K36me2 is highly correlated with m6A modifications in plants. J Integr Plant Biol. (2020) 62:1455–60. doi: 10.1111/jipb.12917
89. Fernández-Justel JM, Santa-María C, Martín-Vírgala S, Ramesh S, Ferrera-Lagoa A, Salinas-Pena M, et al. Histone H1 regulates non-coding RNA turnover on chromatin in a m6A-dependent manner. Cell Rep. (2022) 40:111329. doi: 10.1016/j.celrep.2022.111329
90. Gallego A, Fernández-Justel JM, Martín-Vírgala S, Maslon MM, Gómez M. Slow RNAPII Transcription Elongation Rate, Low Levels of RNAPII Pausing, and Elevated Histone H1 Content at Promoters Associate with Higher m6A Deposition on Nascent mRNAs. Genes. (2022) 13:1652. doi: 10.3390/genes13091652
91. Fish L, Navickas A, Culbertson B, Xu Y, Nguyen HCB, Zhang S, et al. Nuclear TARBP2 drives oncogenic dysregulation of RNA splicing and decay. Mol Cell. (2019) 75:967–81. doi: 10.1016/j.molcel.2019.06.001
92. He PC, Wei J, Dou X, Harada BT, Zhang Z, Ge R, et al. Exon architecture controls mRNA m6A suppression and gene expression. Science. (2023) 379:677–82. doi: 10.1126/science.abj9090
93. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. (1971) 285:1182–6. doi: 10.1056/NEJM197111182852108
94. Li X, Zhou J, Wang X, Li C, Ma Z, Wan Q, et al. New advances in the research of clinical treatment and novel anticancer agents in tumor angiogenesis. BioMed Pharmacother. (2023) 163:114806. doi: 10.1016/j.biopha.2023.114806
95. Dianat-Moghadam H, Nedaeinia R, Keshavarz M, Azizi M, Kazemi M, Salehi R. Immunotherapies targeting tumor vasculature: challenges and opportunities. Front Immunol. (2023) 14:1226360. doi: 10.3389/fimmu.2023.1226360
96. Zhang R, Yao Y, Gao H, Hu X. Mechanisms of angiogenesis in tumour. Front Oncol. (2024) 14:1359069. doi: 10.3389/fonc.2024.1359069
97. Ahluwalia A, Jones MK, Matysiak-Budnik T, Tarnawski AS. VEGF and colon cancer growth beyond angiogenesis: does VEGF directly mediate colon cancer growth via a non-angiogenic mechanism? Curr Pharm Des. (2014) 20:1041–4. doi: 10.2174/1381612819999131218175905
98. Wicks EE, Semenza GL. Hypoxia-inducible factors: cancer progression and clinical translation. J Clin Invest. (2022) 132:e159839. doi: 10.1172/jci159839
99. Qi S, Deng S, Lian Z, Yu K. Novel drugs with high efficacy against tumor angiogenesis. Int J Mol Sci. (2022) 23:6934. doi: 10.3390/ijms23136934
100. Liao D, Johnson RS. Hypoxia: A key regulator of angiogenesis in cancer. Cancer Metastasis Rev. (2007) 26:281–90. doi: 10.1007/s10555-007-9066-y
101. Li T, Kang G, Wang T, Huang H. Tumor angiogenesis and anti−angiogenic gene therapy for cancer (Review). Oncol Lett. (2018) 16:687–702. doi: 10.3892/ol.2018.8733
102. Ribatti D, Nico B, Crivellato E, Roccaro AM, Vacca A. The history of the angiogenic switch concept. Leukemia. (2007) 21:44–52. doi: 10.1038/sj.leu.2404402
103. Pathak A, Pal AK, Roy S, Nandave M, Jain K, Yuan S. Role of angiogenesis and its biomarkers in development of targeted tumor therapies. Stem Cells Int. (2024) 2024:9077926. doi: 10.1155/2024/9077926
104. Rajabi M, Mousa S. The role of angiogenesis in cancer treatment. Biomedicines. (2017) 5:34. doi: 10.3390/biomedicines5020034
105. Venkatakrishnan G, Parvathi VD. Decoding the mechanism of vascular morphogenesis to explore future prospects in targeted tumor therapy. Med Oncol. (2022) 39:178. doi: 10.1007/s12032-022-01810-z
106. Vimalraj S. A concise review of VEGF, PDGF, FGF, Notch, angiopoietin, and HGF signalling in tumor angiogenesis with a focus on alternative approaches and future directions. Int J Biol Macromol. (2022) 221:1428–38. doi: 10.1016/j.ijbiomac.2022.09.129
107. Ribatti D. The chick embryo chorioallantoic membrane as an experimental model to study in vivo angiogenesis in glioblastoma multiforme. Brain Res Bull. (2022) 182:26–9. doi: 10.1016/j.brainresbull.2022.02.005
108. Parmar D, Apte M. Angiopoietin inhibitors: A review on targeting tumor angiogenesis. Eur J Pharmacol. (2021) 899:174021. doi: 10.1016/j.ejphar.2021.174021
109. Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe’er J, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. (1999) 155:739–52. doi: 10.1016/S0002-9440(10)65173-5
110. Wei X, Chen Y, Jiang X, Peng M, Liu Y, Mo Y, et al. Mechanisms of vasculogenic mimicry in hypoxic tumor microenvironments. Mol Cancer. (2021) 20:7. doi: 10.1186/s12943-020-01288-1
111. Treps L, Clere N. Vasculogenic mimicry, a complex and devious process favoring tumorigenesis – Interest in making it a therapeutic target. Pharmacol Ther. (2021) 223:107805. doi: 10.1016/j.pharmthera.2021.107805
112. Luo Q, Wang J, Zhao W, Peng Z, Liu X, Li B, et al. Vasculogenic mimicry in carcinogenesis and clinical applications. J Hematol Oncol. (2020) 13:19. doi: 10.1186/s13045-020-00858-6
113. Zhang Y, Bai J, Cheng R, Zhang D, Qiu Z, Liu T, et al. TAZ promotes vasculogenic mimicry in gastric cancer through the upregulation of TEAD4. J Gastroenterol Hepatol. (2020) 37:714–26. doi: 10.1111/jgh.15779
114. Li W, Zong S, Shi Q, Li H, Xu J, Hou F. Hypoxia-induced vasculogenic mimicry formation in human colorectal cancer cells: Involvement of HIF-1a, Claudin-4, and E-cadherin and Vimentin. Sci Rep. (2016) 6:37534. doi: 10.1038/srep37534
115. Liu W, Xu G, Jia W, Li J, Ma J, Chen K, et al. Prognostic significance and mechanisms of patterned matrix vasculogenic mimicry in hepatocellular carcinoma. Med Oncol. (2011) 28:S228–38. doi: 10.1007/s12032-010-9706-x
116. Mao X, Xue X, Wang L, Zhang X, Yan M, Tu Y, et al. CDH5 is specifically activated in glioblastoma stemlike cells and contributes to vasculogenic mimicry induced by hypoxia. Neuro Oncol. (2013) 15:865–79. doi: 10.1093/neuonc/not029
117. Wu S, Yu L, Wang D, Zhou L, Cheng Z, Chai D, et al. Aberrant expression of CD133 in non-small cell lung cancer and its relationship to vasculogenic mimicry. BMC Cancer. (2012) 12:535. doi: 10.1186/1471-2407-12-535
118. Nisar MA, Zheng Q, Saleem MZ, Ahmmed B, Ramzan MN, Din SRU, et al. IL-1β Promotes vasculogenic mimicry of breast cancer cells through p38/MAPK and PI3K/akt signaling pathways. Front Oncol. (2021) 11:618839. doi: 10.3389/fonc.2021.618839
119. Ayala-Domínguez L, Olmedo-Nieva L, Muñoz-Bello JO, Contreras-Paredes A, Manzo-Merino J, Martínez-Ramírez I, et al. Mechanisms of vasculogenic mimicry in ovarian cancer. Front Oncol. (2019) 9:998. doi: 10.3389/fonc.2019.00998
120. Haiaty S, Rashidi M, Akbarzadeh M, Maroufi NF, Yousefi B, Nouri M. Targeting vasculogenic mimicry by phytochemicals: A potential opportunity for cancer therapy. IUBMB Life. (2020) 72:825–41. doi: 10.1002/iub.2233
121. Pezzella F, Ribatti D. Vascular co-option and vasculogenic mimicry mediate resistance to antiangiogenic strategies. Cancer Rep. (2022) 5:e1318. doi: 10.1002/cnr2.1318
122. Zhang J, Qiao L, Liang N, Xie J, Luo H, Deng G, et al. Vasculogenic mimicry and tumor metastasis. J BUON. (2016) 21:533–41.
123. Zhang S, Zhang D, Sun B. Vasculogenic mimicry: Current status and future prospects. Cancer Lett. (2007) 254:157–64. doi: 10.1016/j.canlet.2006.12.036
124. Lu X, Sun W, Ge C, Zhang W, Fan Y. Contribution of the PI3K/MMPs/Ln-5γ2 and EphA2/FAK/Paxillin signaling pathways to tumor growth and vasculogenic mimicry of gallbladder carcinomas. Int J Oncol. (2013) 42:2103–15. doi: 10.3892/ijo.2013.1897
125. Zhang J, Zhou H, Zhang X, Mu W, Hu J, Liu G, et al. Hypoxic induction of vasculogenic mimicry in hepatocellular carcinoma: role of HIF-1 α, RhoA/ROCK and Rac1/PAK signaling. BMC Cancer. (2020) 20:32. doi: 10.1186/s12885-019-6501-8
126. Fang T, Lin L, Ye ZJ, Fang L, Shi S, Yu KD, et al. Dexmedetomidine promotes angiogenesis and vasculogenic mimicry in human hepatocellular carcinoma through α 2-AR/HIF-1α/VEGFA pathway. BioMed Environ Sci. (2022) 35:931–42. doi: 10.3967/bes2022.120
127. Shah AA, Kamal MA, Akhtar S. Tumor angiogenesis and VEGFR-2: mechanism, pathways and current biological therapeutic interventions. Curr Drug Metab. (2021) 22:50–9. doi: 10.2174/1389200221666201019143252
128. Brat DJ, Van Meir EG. Glomeruloid microvascular proliferation orchestrated by VPF/VEGF. Am J Pathol. (2001) 158:789–96. doi: 10.1016/S0002-9440(10)64025-4
129. Straume O, Akslen LA. Increased expression of VEGF-receptors (FLT-1, KDR, NRP-1) and thrombospondin-1 is associated with glomeruloid microvascular proliferation, an aggressive angiogenic phenotype, in Malignant melanoma. Angiogenesis. (2003) 6:295–301. doi: 10.1023/B:AGEN.0000029408.08638.aa
130. Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. (2010) 468:829–33. doi: 10.1038/nature09624
131. Akslen LA, Straume O, Geisler S, Sørlie T, Chi J, Aas T, et al. Glomeruloid microvascular proliferation is associated with lack of response to chemotherapy in breast cancer. Br J Cancer. (2011) 105:9–12. doi: 10.1038/bjc.2011.203
132. Eguchi R, Wakabayashi I. HDGF enhances VEGF−dependent angiogenesis and FGF−2 is a VEGF−independent angiogenic factor in non−small cell lung cancer. Oncol Rep. (2022) 44:14–28. doi: 10.3892/or.2020.7580
133. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. (2023) 73:17–48. doi: 10.3322/caac.21763
134. Liu X, He H, Zhang F, Hu X, Bi F, Li K, et al. m6A methylated EphA2 and VEGFA through IGF2BP2/3 regulation promotes vasculogenic mimicry in colorectal cancer via PI3K/AKT and ERK1/2 signaling. Cell Death Dis. (2022) 13:483. doi: 10.1038/s41419-022-04950-2
135. Gao Y, Ma X, Lu H, Xu P, Xu C. PLAU is associated with cell migration and invasion and is regulated by transcription factor YY1 in cervical cancer. Oncol Rep. (2023) 49:25. doi: 10.3892/or.2022.8462
136. Lin M, Zhang Z, Gao M, Yu H, Sheng H, Huang J. MicroRNA-193a-3p suppresses the colorectal cancer cell proliferation and progression through downregulating the PLAU expression. Cancer Manag Res. (2019) 11:5353–63. doi: 10.2147/CMAR.S208233
137. Yu T, Liu J, Wang Y, Chen W, Liu Z, Zhu L, et al. METTL3 promotes colorectal cancer metastasis by stabilizing PLAU mRNA in an m6A-dependent manner. Biochem Biophys Res Commun. (2022) 23:614. doi: 10.1016/j.bbrc.2022.04.141
138. Zhang G, Wang T, Huang Z, Chen Y, Sun L, Xia X, et al. METTL3 dual regulation of the stability of LINC00662 and VEGFA RNAs promotes colorectal cancer angiogenesis. Discovery Oncol. (2022) 13:89. doi: 10.1007/s12672-022-00557-3
139. Bian Y, Wang Y, Xu S, Gao Z, Li C, Fan Z, et al. m6A Modification of Long Non-Coding RNA HNF1A-AS1 Facilitates Cell Cycle Progression in Colorectal Cancer via IGF2BP2-Mediated CCND1 mRNA Stabilization. Cells. (2022) 11:3008. doi: 10.3390/cells11193008
140. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
141. Bao C, Wang J, Ma W, Wang X, Cheng Y. HDGF: a novel jack-of-all-trades in cancer. Future Oncol. (2014) 10:2675–85. doi: 10.2217/fon.14.194
142. Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, et al. METTL3-mediated m6A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. (2020) 69:1193–205. doi: 10.1136/gutjnl-2019-319639
143. Liu M, Li J, Huang Z, Li Y. Gastric cancer risk-scoring system based on analysis of a competing endogenous RNA network. Transl Cancer Res. (2020) 9:3889–902. doi: 10.21037/tcr-19-2977
144. Wang N, Huo X, Zhang B, Chen X, Zhao S, Shi X, et al. METTL3-mediated ADAMTS9 suppression facilitates angiogenesis and carcinogenesis in gastric cancer. Front Oncol. (2022) 12:861807. doi: 10.3389/fonc.2022.861807
145. Xu P, Yang J, Chen Z, Zhang X, Xia Y, Wang S, et al. N6-methyladenosine modification of CENPF mRNA facilitates gastric cancer metastasis via regulating FAK nuclear export. Cancer Commun (Lond). (2023) 43:685–705. doi: 10.1002/cac2.12443
146. Hooglugt A, van der Stoel MM, Boon RA, Huveneers S. Endothelial YAP/TAZ signaling in angiogenesis and tumor vasculature. Front Oncol. (2021) 10:612802. doi: 10.3389/fonc.2020.612802
147. Qiao K, Liu Y, Xu Z, Zhang H, Zhang H, Zhang C, et al. RNA m6A methylation promotes the formation of vasculogenic mimicry in hepatocellular carcinoma via Hippo pathway. Angiogenesis. (2021) 24:83–96. doi: 10.1007/s10456-020-09744-8
148. Lin Z, Niu Y, Wan A, Chen D, Liang H, Chen X, et al. RNA m6A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy. EMBO J. (2020) 39:e103181. doi: 10.15252/embj.2019103181
149. Lin W, Zhang T, Ding G, Hao L, Zhang B, Yu J, et al. Circular RNA circ−CCT3 promotes hepatocellular carcinoma progression by regulating the miR−1287−5p/TEAD1/PTCH1/LOX axis. Mol Med Rep. (2021) 23:375. doi: 10.3892/mmr.2021.12014
150. Takenaka K, Katakura H, Chen F, Ogawa E, Adachi M, Wada H, et al. The ratio of membrane-bound form Flt-1 mRNA to VEGF mRNA correlates with tumor angiogenesis and prognosis in non-small cell lung cancer. Cancer Lett. (2007) 246:34–40. doi: 10.1016/j.canlet.2006.01.022
151. Liu H, Jiang Y, Lu J, Peng C, Ling Z, Chen Y, et al. m6A-modification regulated circ-CCT3 acts as the sponge of miR-378a-3p to promote hepatocellular carcinoma progression. Epigenetics. (2023) 18:2204772. doi: 10.1080/15592294.2023.2204772
152. Bertrand M, Szeremeta1 F, Hervouet-Coste1 N, Sarou-Kanian V, Landon C, Morisset-Lopez S, et al. An adult Drosophila glioma model to highlight metabolic dysfunctions and evaluate the role of the serotonin 5-HT7 receptor as a potential therapeutic target. FASEB J. (2023) 37:e23230. doi: 10.1096/fj.202300783RR
153. Wu Z, Lin Y, Wei N. N6-methyladenosine-modified HOTAIRM1 promotes vasculogenic mimicry formation in glioma. Cancer Sci. (2023) 114:129–41. doi: 10.1111/cas.15578
154. Tao M, Li X, He L, Rong X, Wang H, Pan J, et al. Decreased RNA m6A methylation enhances the process of the epithelial mesenchymal transition and vasculogenic mimicry in glioblastoma. Am J Cancer Res. (2022) 12:893–906.
155. Liu M, Ruan X, Liu X, Dong W, Wang D, Yang C, et al. The mechanism of BUD13 m6A methylation mediated MBNL1-phosphorylation by CDK12 regulating the vasculogenic mimicry in glioblastoma cells. Cell Death Dis. (2022) 13:1017. doi: 10.1038/s41419-022-05426-z
156. Feng Y, Wu F, Wu Y, Guo Z, Ji X. LncRNA DGUOK-AS1 facilitates non-small cell lung cancer growth and metastasis through increasing TRPM7 stability via m6A modification. Transl Oncol. (2023) 32:101661. doi: 10.1016/j.tranon.2023.101661
157. Mao X, Wang G, Wang Z, Duan C, Wu X, Xu H. Theranostic lipid nanoparticles for renal cell carcinoma. Adv Mater. (2023) 25:e2306246. doi: 10.1002/adma.202306246
158. Cheng B, Xie M, Zhou Y, Li T, Liu W, Yu W, et al. Vascular mimicry induced by m6A mediated IGFL2-AS1/AR axis contributes to pazopanib resistance in clear cell renal cell carcinoma. Cell Death Discovery. (2023) 9:121. doi: 10.1038/s41420-023-01423-z
159. Guo Y, Wang Q, Wang J, Gu Y, Song P, Wang S, et al. METTL3 modulates m6A modification of CDC25B and promotes head and neck squamous cell carcinoma Malignant progression. Exp Hematol Oncol. (2022) 11:14. doi: 10.1186/s40164-022-00256-3
160. Wang G, Dai Y, Li K, Cheng M, Xiong G, Wang X, et al. Deficiency of mettl3 in bladder cancer stem cells inhibits bladder cancer progression and angiogenesis. Front Cell Dev Biol. (2021) 9:627706. doi: 10.3389/fcell.2021.627706
161. Liu H, Gu J, Huang Z, Han Z, Xin J, Yuan L, et al. Fine particulate matter induces METTL3-mediated m6A modification of BIRC5 mRNA in bladder cancer. J Hazard Mater. (2022) 437:129310. doi: 10.1016/j.jhazmat.2022.129310
162. Chen Y, Lu Z, Qi C, Yu C, Li Y, Huan W, et al. N6-methyladenosine-modified TRAF1 promotes sunitinib resistance by regulating apoptosis and angiogenesis in a METTL14-dependent manner in renal cell carcinoma. Mol Cancer. (2022) 21:111. doi: 10.1186/s12943-022-01549-1
163. Xie J, Huang X, Chen Q, Ma Y, Zhao Y, Liuet L, et al. m6A modification-mediated BATF2 acts as a tumor suppressor in gastric cancer through inhibition of ERK signaling. Mol Cancer. (2020) 19:114. doi: 10.1186/s12943-020-01223-4
164. Wen H, Tang J, Cui Y, Hou M, Zhou J. m6A modification-mediated BATF2 suppresses metastasis and angiogenesis of tongue squamous cell carcinoma through inhibiting VEGFA. Cell Cycle. (2023) 22:100–16. doi: 10.1080/15384101.2022.2109897
165. Panneerdoss S, Eedunuri VK, Yadav P, Timilsina S, Rajamanickam S, Viswanadhapalli S, et al. Cross-talk among writers, readers, and erasers of m6A regulates cancer growth and progression. Sci Adv. (2018) 4:eaar8263. doi: 10.1126/sciadv.aar8263
166. Ye M, Chen J, Yu P, Hu C, Wang B, Baoet J, et al. WTAP activates MAPK signaling through m6A methylation in VEGFA mRNA-mediated by YTHDC1 to promote colorectal cancer development. FASEB J. (2023) 37:e23090. doi: 10.1096/fj.202300344RRR
167. Tan A, Dang Y, Chen G, Mo Z. Overexpression of the fat mass and obesity associated gene (FTO) in breast cancer and its clinical implications. Int J Clin Exp Pathol. (2015) 8:13405–10.
168. Rong Z, Li Z, He J, Liu L, Ren X, Gao J, et al. Downregulation of fat mass and obesity associated (FTO) promotes the progression of intrahepatic cholangiocarcinoma. Front Oncol. (2019) 9:369. doi: 10.3389/fonc.2019.00369
169. Shen W, Pu J, Zuo Z, Gu S, Sun J, Tan B, et al. The RNA demethylase ALKBH5 promotes the progression and angiogenesis of lung cancer by regulating the stability of the LncRNA PVT1. Cancer Cell Int. (2022) 22:353. doi: 10.1186/s12935-022-02770-0
170. Zhang H, Zhou J, Li J, Wang Z, Chen Z, Lv Z, et al. N6-methyladenosine promotes translation of VEGFA to accelerate angiogenesis in lung cancer. Cancer Res. (2023) 83:2208–25. doi: 10.1158/0008-5472.CAN-22-2449
171. Jin D, Guo J, Wu Y, Yang L, Wang X, Du J, et al. m6A demethylase ALKBH5 inhibits tumor growth and metastasis by reducing YTHDFs-mediated YAP expression and inhibiting miR-107/LATS2–mediated YAP activity in NSCLC. Mol Cancer. (2020) 19:40. doi: 10.1186/s12943-020-01161-1
172. Guo Y, Guo Y, Chen C, Fan D, Wu X, Zhao L, et al. Circ3823 contributes to growth, metastasis and angiogenesis of colorectal cancer: involvement of miR-30c-5p/TCF7 axis. Mol Cancer. (2021) 20:93. doi: 10.1186/s12943-021-01372-0
173. Ou C, Sun Z, He X, Li X, Fan S, Zheng X, et al. Targeting YAP1/LINC00152/FSCN1 signaling axis prevents the progression of colorectal cancer. Adv Sci (Weinh). (2019) 7:1901380. doi: 10.1002/advs.201901380
174. Li T, Xie J, Shen C, Cheng D, Shi Y, Wu Z, et al. Amplification of long noncoding RNA ZFAS1 promotes metastasis in hepatocellular carcinoma. Cancer Res. (2015) 75:3181–91. doi: 10.1158/0008-5472.CAN-14-3721
175. Yu T, Yao L, Yin H, Teng Y, Hong M, Wu Q, et al. ALKBH5 Promotes Multiple Myeloma Tumorigenicity through inducing m6A-demethylation of SAV1 mRNA and Myeloma Stem Cell Phenotype. Int J Biol Sci. (2022) 8:2235–48. doi: 10.7150/ijbs.64943
176. Wang J, Tan L, Yu X, Cao X, Jia B, Chen R, et al. lncRNA ZNRD1-AS1 promotes Malignant lung cell proliferation, migration, and angiogenesis via the miR-942/TNS1 axis and is positively regulated by the m6A reader YTHDC2. Mol Cancer. (2022) 21:229. doi: 10.1186/s12943-022-01705-7
177. Yuan W, Chen S, Li B, Han X, Meng B, Zou Y, et al. The N6-methyladenosine reader protein YTHDC2 promotes gastric cancer progression via enhancing YAP mRNA translation. Transl Oncol. (2022) 16:101308. doi: 10.1016/j.tranon.2021.101308
178. Rashid M, Zadeh LR, Baradaran B, Molavi O, Ghesmati Z, Sabzichi M, et al. Up-down regulation of HIF-1α in cancer progression. Gene. (2021) 798:145796. doi: 10.1016/j.gene.2021.145796
179. Liang L, Zhu Y, Li J, Zeng J, Wu L. ALKBH5-mediated m6A modification of circCCDC134 facilitates cervical cancer metastasis by enhancing HIF1A transcription. J Exp Clin Cancer Res. (2022) 41:261. doi: 10.1186/s13046-022-02462-7
180. Tiwari A, Tashiro K, Dixit A, Soni A, Vogel K, Hall B, et al. Loss of HIF1A from pancreatic cancer cells increases expression of PPP1R1B and degradation of p53 to promote invasion and metastasis. Gastroenterology. (2020) 159:1882–1897.e5. doi: 10.1053/j.gastro.2020.07.046
181. Bai R, Sun M, Chen Y, Zhuo S, Song G, Wang T, et al. H19 recruited N6-methyladenosine (m6A) reader YTHDF1 to promote SCARB1 translation and facilitate angiogenesis in gastric cancer. Chin Med J (Engl). (2023) 136:1719–31. doi: 10.1097/CM9.0000000000002722
182. Hou J, Zhang H, Liu J, Zhao Z, Wang J, Lu Z, et al. YTHDF2 reduction fuels inflammation and vascular abnormalization in hepatocellular carcinoma. Mol Cancer. (2019) 18:163. doi: 10.1186/s12943-019-1082-3
183. Sasahira T, Kurihara-Shimomura M, Shimomura H, Kirita T. SERPINE2 is an oral cancer-promoting factor that induces angiogenesis and lymphangiogenesis. Int J Clin Oncol. (2021) 26:1831–9. doi: 10.1007/s10147-021-01970-4
184. Langer EM, Kendsersky ND, Daniel CJ, Kuziel GM, Pelz C, Murphy KM, et al. ZEB1-repressed microRNAs inhibit autocrine signaling that promotes vascular mimicry of breast cancer cells. Oncogene. (2018) 37:1005–19. doi: 10.1038/onc.2017.356
185. Dixit D, Prager BC, Gimple RC, Poh HX, Wang Y, Wu Q, et al. The RNA m6A reader YTHDF2 maintains oncogene expression and is a targetable dependency in glioblastoma stem cells. Cancer Discovery. (2021) 11:480–99. doi: 10.1158/2159-8290.CD-20-0331
186. Xu Z, Chen S, Liu R, Chen H, Xu B, Xu W, et al. Circular RNA circPOLR2A promotes clear cell renal cell carcinoma progression by facilitating the UBE3C-induced ubiquitination of PEBP1 and, thereby, activating the ERK signaling pathway. Mol Cancer. (2022) 21:146. doi: 10.1186/s12943-022-01607-8
187. Chang G, Shi L, Ye Y, Shi H, Zeng L, Tiwary S, et al. YTHDF3 induces the translation of m6A-enriched gene transcripts to promote breast cancer brain metastasis. Cancer Cell. (2020) 38:857–871.e7. doi: 10.1016/j.ccell.2020.10.004
188. Yang Z, Wang T, Wu D, Min Z, Tan J, Yu B. RNA N6-methyladenosine reader IGF2BP3 regulates cell cycle and angiogenesis in colon cancer. J Exp Clin Cancer Res. (2020) 39:203. doi: 10.1186/s13046-020-01714-8
189. Fang H, Sun Q, Zhou J, Zhang H, Song Q, Zhang H, et al. m6A methylation reader IGF2BP2 activates endothelial cells to promote angiogenesis and metastasis of lung adenocarcinoma. Mol Cancer. (2023) 22:99. doi: 10.1186/s12943-023-01791-1
190. Claesson-Welsh L, Welsh M. VEGFA and tumour angiogenesis. J Intern Med. (2013) 273:114–27. doi: 10.1111/joim.12019
191. Zhang J, Li W, Wang W, Chen Q, Xu Z, Deng M, et al. Dual roles of FAK in tumor angiogenesis: A review focused on pericyte FAK. Eur J Pharmacol. (2023) 947:175694. doi: 10.1016/j.ejphar.2023.175694
192. Yao H, Li J, Liu Z, Ouyang C, Qiu Y, Zheng X, et al. Ablation of endothelial Atg7 inhibits ischemia-induced angiogenesis by upregulating Stat1 that suppresses Hif1a expression. Autophagy. (2023) 19:1491–511. doi: 10.1080/15548627.2022.2139920
193. Uemura1 A, Fukushima Y. Rho GTPases in retinal vascular diseases. Int J Mol Sci. (2021) 22:3684. doi: 10.3390/ijms22073684
194. Ye H, Wang RY, Yu XZ, Wu YK, Yang BW, Ao MY, et al. Exosomal circNFIX promotes angiogenesis in ovarian cancer via miR-518a-3p/TRIM44 axis. Kaohsiung J Med Sci. (2023) 39:26–39. doi: 10.1002/kjm2.12615
195. Wang R, Ye H, Yang B, Ao M, Yu X, Wu Y, et al. m6A-modified circNFIX promotes ovarian cancer progression and immune escape via activating IL-6R/JAK1/STAT3 signaling by sponging miR-647. Int Immunopharmacol. (2023) 124:110879. doi: 10.1016/j.intimp.2023.110879
196. Zeng H, Hou Y, Zhou X, Lang L, Luo H, Sun Y, et al. Cancer-associated fibroblasts facilitate premetastatic niche formation through lncRNA SNHG5-mediated angiogenesis and vascular permeability in breast cancer. Theranostics. (2022) 12:7351–70. doi: 10.7150/thno.74753
197. Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discovery. (2018) 18:99–115. doi: 10.1038/s41573-018-0004-1
198. Liu Y, Cao X. Characteristics and significance of the pre-metastatic niche. Cancer Cell. (2016) 30:668–81. doi: 10.1016/j.ccell.2016.09.011
199. Jiang L, Li Y, He Y, Wei D, Yan L, Wen H. Knockdown of m6A reader IGF2BP3 inhibited hypoxia-induced cell migration and angiogenesis by regulating hypoxia inducible factor-1α in stomach cancer. Front Oncol. (2021) 11:711207. doi: 10.3389/fonc.2021.711207
200. Tang E, Wang Y, Liu T, Yan B. Gastrin promotes angiogenesis by activating HIF-1α/β-catenin/VEGF signaling in gastric cancer. Gene. (2019) 704:42–8. doi: 10.1016/j.gene.2019.04.029
201. Ma Y, Shi B, Guo J, Liu J, Yang X, Xin R, et al. microRNA-320b suppresses HNF4G and IGF2BP2 expression to inhibit angiogenesis and tumor growth of lung cancer. Carcinogenesis. (2021) 42:762–71. doi: 10.1093/carcin/bgab023
202. Gu Z, Yang Yang Y, Ma Q, Wang H, Zhao S, Qi Y, et al. HNRNPC, a predictor of prognosis and immunotherapy response based on bioinformatics analysis, is related to proliferation and invasion of NSCLC cells. Respir Res. (2022) 23:362. doi: 10.1186/s12931-022-02227-y
203. He X, Tan L, Ni J, Shen G. Expression pattern of m6A regulators is significantly correlated with Malignancy and antitumor immune response of breast cancer. Cancer Gene Ther. (2020) 28:188–96. doi: 10.1038/s41417-020-00208-1
204. Li B, Wang F, Wang N, Hou K, Du J. Identification of implications of angiogenesis and m6A modification on immunosuppression and therapeutic sensitivity in low-grade glioma by network computational analysis of subtypes and signatures. Front Immunol. (2022) 13:871564. doi: 10.3389/fimmu.2022.871564
205. Lu X, Friedrich LJ, Efferth T. Natural products targeting tumour angiogenesis. Br J Pharmacol. (2023). doi: 10.1111/bph.16232
206. Atkins MB, Gravis G, Drosik K, Demkow T, Tomczak P, Wong SS, et al. Trebananib (AMG 386) in combination with sunitinib in patients with metastatic renal cell cancer: an open-label, multicenter, phase II study. J Clin Oncol. (2015) 33:3431–8. doi: 10.1200/JCO.2014.60.6012
207. Aldin A, Besiroglu B, Adams A, Monsef I, Piechotta V, Tomlinson E, et al. First-line therapy for adults with advanced renal cell carcinoma: a systematic review and network meta-analysis. Cochrane Database Syst Rev. (2023) 5:CD013798. doi: 10.1002/14651858.CD013798.pub2
208. He M, Yang H, Shi H, Hu Y, Chang C, Liu S, et al. Sunitinib increases the cancer stem cells and vasculogenic mimicry formation via modulating the lncRNA-ECVSR/ERβ/Hif2-α signaling. Cancer Lett. (2022) 524:15–28. doi: 10.1016/j.canlet.2021.08.028
209. Duran I, Lambea J, Maroto P, González-Larriba JL, Flores L, Granados-Principal S, et al. Resistance to targeted therapies in renal cancer: the importance of changing the mechanism of action. Target Oncol. (2016) 12:19–35. doi: 10.1007/s11523-016-0463-4
210. Molina AM, Lin X, Korytowsky B, Matczak E, Lechuga MJ, Wiltshire R, et al. Sunitinib objective response in metastatic renal cell carcinoma: Analysis of 1059 patients treated on clinical trials. Eur J Cancer. (2014) 50:351–8. doi: 10.1016/j.ejca.2013.08.021
211. Makhov P, Joshi S, Ghatalia P, Kutikov A, Uzzo RG, Kolenko VM, et al. Resistance to systemic therapies in clear cell renal cell carcinoma: mechanisms and management strategies. Mol Cancer Ther. (2018) 17:1355–64. doi: 10.1158/1535-7163
212. Soria1 JC, Mauguen A, Reck M, AB S, Saijo N, DH J, et al. Systematic review and meta-analysis of randomised, phase II/III trials adding bevacizumab to platinum-based chemotherapy as first-line treatment in patients with advanced non-small-cell lung cancer. Ann Oncol. (2013) 24:20–30. doi: 10.1093/annonc/mds590
213. Wei H, Wang F, Wang Y, Li T, Xiu P, Zhong J, et al. Verteporfin suppresses cell survival, angiogenesis and vasculogenic mimicry of pancreatic ductal adenocarcinoma via disrupting the YAP-TEAD complex. . Cancer Sci. (2017) 108:478–87. doi: 10.1111/cas.13138
214. Wang F, Dong X, Xiu P, Zhong J, Wei H, Xu Z, et al. T7 peptide inhibits angiogenesis via downregulation of angiopoietin-2 and autophagy. Oncol Rep. (2015) 33:675–84. doi: 10.3892/or.2014.3653
215. Choi HJ, Zhang H, Park H, Choi KS, Lee HW, Agrawal V, et al. Yes-associated protein regulates endothelial cell contact-mediated expression of angiopoietin-2. Nat Commun. (2015) 6:6943. doi: 10.1038/ncomms7943
216. Oura K, Morishita A, Tani J, Masaki T. Tumor immune microenvironment and immunosuppressive therapy in hepatocellular carcinoma: A review. Int J Mol Sci. (2021) 22:5801. doi: 10.3390/ijms22115801
217. Hammoud L, Adams JR, Loch AJ, Marcellus RC, Uehling DE, Aman A, et al. Identification of RSK and TTK as modulators of blood vessel morphogenesis using an embryonic stem cell-based vascular differentiation assay. Stem Cell Rep. (2016) 7:787–801. doi: 10.1016/j.stemcr.2016.08.004
Keywords: N6-methyladenosine, writer, eraser, reader, tumor angiogenesis, tumor therapy
Citation: Qin L, Zeng X, Qiu X, Chen X and Liu S (2024) The role of N6-methyladenosine modification in tumor angiogenesis. Front. Oncol. 14:1467850. doi: 10.3389/fonc.2024.1467850
Received: 21 July 2024; Accepted: 11 November 2024;
Published: 03 December 2024.
Edited by:
Jinjun Wu, Guangzhou University of Chinese Medicine, ChinaCopyright © 2024 Qin, Zeng, Qiu, Chen and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shiquan Liu, cG9lbXBvd2VyQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.