 Guenda Di Benedetto1
Guenda Di Benedetto1 Ignazio Barca1Laura De Gregorio2Claudia Scollo3
Ignazio Barca1Laura De Gregorio2Claudia Scollo3 Fiorenza Gianì1
Fiorenza Gianì1 Federica Martorana4
Federica Martorana4 Marco Russo1
Marco Russo1 Francesco Frasca1
Francesco Frasca1 Gabriella Pellegriti1,4*†
Gabriella Pellegriti1,4*† Giulia Sapuppo1
Giulia Sapuppo1- 1Endocrinology Unit, Garibaldi-Nesima Hospital, Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy
- 2Molecular Biology Service, Multi-diagnostic Health Services Centre, Catania, Italy
- 3Endocrinology Service, Department of Internal Medicine, Maggiore Hospital, Modica, Italy
- 4Medical Oncology, Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy
Background: Multiple endocrine neoplasia type 2 syndrome (MEN2) is a hereditary disease resulting from mutations of the rearranged during transfection (RET) protooncogene subclassified into MEN2A [medullary thyroid carcinoma (MTC), pheochromocytoma, and primary hyperparathyroidism] and MEN2B (MTC, pheochromocytoma, Marfanoid habitus, mucous neuromas, and intestinal ganglioneuromatosis). Prophylactic thyroidectomy is recommended in RET-mutated patients. The age at which it should be performed depends on the type and aggressiveness of the mutation.
Aim of the study: This study aimed to evaluate the genotype/phenotype correlation and outcome in pediatric/adolescent carriers of MEN2 RET mutation.
Patients and methods: In a retrospective series of 23 carriers of RET MEN2 mutation who were ≤19 years old at diagnosis and had undergone total thyroidectomy ± lymphadenectomy, the following were analyzed: 1) specific RET mutation, 2) clinical and histopathological characteristics, 3) genotype/phenotype correlation, and 4) outcome at last follow-up.
Results: In our series, the female gender was more prevalent (F/M ratio 2.8/1), and the median age was 14.9 years [interquartile range (IQR) 12.6–17.2]. RET mutations were at very high risk in 4.3% of patients (M918T), high risk in 43.5% (C634), and moderate risk in 52.2% (47.8% C618 and 4.3% C620). All patients underwent surgery: at histology, MTC was found in 19/23 (82.6%) patients, C-cell hyperplasia in 2/23 (8.7%), and benign histology in 2/23 (8.7%). Ten patients (52.6%) had a disease event during the follow-up: 2/19 (10.5%) showed biochemical disease, 6/19 (31.6%) lymph node recurrences, and 2/19 (10.5%) distant metastases (50% liver, 50% bone). At the last follow-up, nine MTCs were not cured. One patient died after 9 years of follow-up at 21 years old (M918T RET+).
Conclusions: From these data, it is clear to see the importance of genetic counseling and RET screening in all first-degree relatives of patients with proven MEN2. The goal should be to subject patients to surgery for prophylactic and not curative purposes, i.e., before the onset of MTC, given the high risk of persistent or recurrent disease also in pediatric/adolescent patients.
1 Introduction
Medullary thyroid carcinoma (MTC) is a rare neoplasm (1%–2% of all thyroid tumors) that originates from the parafollicular cells of the thyroid. At diagnosis, all patients with MTC should be screened for rearranged during transfection (RET) mutations due to the fact that 20%–25% of cases have germline mutation being part of multiple endocrine neoplasia type 2 syndrome (MEN2) (1, 2).
MEN2 is a hereditary disease resulting from mutations of the RET protooncogene subclassified into MEN2A (MTC, pheochromocytoma, and primary hyperparathyroidism) and MEN2B (MTC, pheochromocytoma, Marfanoid habitus, mucous neuromas, and intestinal ganglioneuromatosis).
Hereditary MTC occurs at a younger age, is typically preceded by hyperplasia, and is often multifocal and/or bilateral (3). The age-related penetrance of hyperplasia and MTC depends on specific RET mutations.
Almost all patients with MEN2A (85%) are asymptomatic at diagnosis. The third decade is the age of the peak incidence in index patients.
Genetic counseling must be provided to all first-degree relatives of patients with proven MEN2.
At present, according to the American Thyroid Association (ATA) guidelines, patients are classified based on their phenotype (depending on the specific RET mutations) into three groups: highest, high, and moderate risk with progressive increases in aggressiveness (in terms of development at early age, often with local or distant metastases) (4–6). The ATA’s highest risk includes RET codon M918T mutation, high-risk RET codon C634 and A883F mutation, and moderate risk of all the other mutations.
The optimal timing of prophylactic thyroidectomy is recommended in RET-mutated patients depending on the aggressiveness of the mutation. Children in the highest ATA risk category should undergo total thyroidectomy within the first year of life, regardless of calcitonin level, which is naturally high in the first month of life. Children in the ATA high-risk category should undergo total thyroidectomy before the age of 5 or earlier based on their calcitonin values. Children in the ATA moderate-risk category generally develop a less aggressive tumor at an older age, so they should be screened every 6–12 months from the age of 5 and should undergo thyroidectomy in childhood or early adulthood, or sooner if elevated calcitonin values.
In some studies of children with MEN2 undergoing prophylactic surgery, no lymph node metastases or postoperative residual disease occurred when baseline serum calcitonin (Ctn) levels were <30–40 pg/mL (7–9).
A risk/benefit assessment between the risks of thyroidectomy and the possibility that MTC in patients with MEN2 will not be cured if thyroidectomy is delayed must be taken into account.
In the present study, we retrospectively evaluated a series of 23 pediatric/adolescent RET-mutated patients by analyzing clinical and histopathological characteristics, the specific RET mutation, the genotype/phenotype correlation, and the outcome at the last follow-up.
2 Patients and methods
A consecutive series of 23 patients with germline mutation of the RET protooncogene (MEN2), with an age at diagnosis ≤19 years, all followed up at Endocrinology Thyroid Clinic, Garibaldi-Nesima Medical Center, in Catania, from 1980 to 2018 were retrospectively analyzed. The median follow-up was 9.7 years, with a median of 4.1 and a range of 2–36 years. Two patients were index cases, and the other patients were identified during family screening.
Patients underwent total thyroidectomy (TT) ± lymphadenectomy (unilateral or bilateral) according to pre-surgery evaluation (mutation aggressiveness, ultrasound, and Ctn value). The criteria for lymph node (LN) central and/or lateral dissection were as follows: a) Ctn value >20 pg/mL (central) and >50 pg/mL (lateral), b) pre-surgery evidence of neck LN involvement either clinical or at ultrasound examination, and c) intra-surgery suspicion of metastatic LN.
The following data were collected: clinical (gender, age at diagnosis, and date of follow-up), histological (size of the primary tumor, multifocality, and number of metastatic lymph nodes at diagnosis), biochemical [Ctn and carcinoembryonic antigen (CEA) values at diagnosis and during follow-up], and morphological (computed tomography (CT), PET, magnetic resonance imaging (MRI), etc.) data and mutation status. Regarding genetic analysis, genomic DNA was purified from peripheral blood lymphocytes of the index case using the High Purification PCR Template Preparation Kit (Roche Diagnostic GmbH, Mannheim, Germany). RET gene exons 10, 11, 13, 14, 15, and 16 were analyzed using PCR and DNA direct sequencing. Exon 11 mutation was confirmed on an independent sample of the index case, and mutation analysis of his relatives was performed using specific amplification primers (5′-CCTCTGGCGGTGCCAAGCCTC-3′; 5′-CCTCGTCTGCCCAGCGTTG-3′) and exon 11 direct sequencing.
Tumors were staged according to the 8th TNM edition (10): T (primary tumor’s maximum size) and N (LN metastases) were assessed at pathological examination. The N status was indicated as N0 when all excised nodes were negative, N1a when positive nodes were only in the central compartment (levels VI–VII), and N1b when laterocervical nodes (levels I–V) were involved. M (distant metastases) was evaluated using total body imaging techniques [CT, MRI, whole-body bone scan (WBS), 18-fluorodeoxyglucose positron emission tomography (FDG-PET), and positron emission tomography with receptor tracers (for example, gallium, 68GA-PET)]. Patients were staged according to the TNM VIII edition.
All patients were periodically followed up by Ctn and CEA measurements, neck ultrasound, and further imaging methods when necessary. The intensity of subsequent examinations, every 6–12 months, was modulated on the basis of the initial risk assessment after surgery. At the last follow-up visit, persistence/recurrence of disease was defined by measurable values of Ctn and CEA or structural evidence of disease on ultrasound of the neck or other imaging methods. In particular, biochemical incomplete response is defined as detectable serum calcitonin and abnormal CEA with no evidence of structural disease. All patients who had persistent/relapsed disease during follow-up underwent additional diagnostic imaging procedures (CT, MRI, FDG-PET, 68GA-PET, and WBS), additional surgeries, and/or other therapies when required.
2.1 Statistical analysis
Categorical variables were expressed as frequencies and percentages. Normally distributed quantitative variables were expressed as mean ± standard deviation (SD), while non-normally distributed variables were expressed as median and interquartile range (IQR). The normality of quantitative variables was assessed using the Kolmogorov–Smirnov test. Categorical variables were analyzed using the chi-square test with Yates’s correction or Fisher’s exact test. A p-value <0.05 was considered statistically significant for all analyses. Data analysis was conducted using SPSS statistic software version 13.0 for Windows.
3 Results
3.1 Characteristics of the patients
The clinical and histopathological characteristics of the 23 pediatric/adolescent patients, six male and 17 female, carriers of RET MEN2 mutation, included in the present study are shown in Table 1.
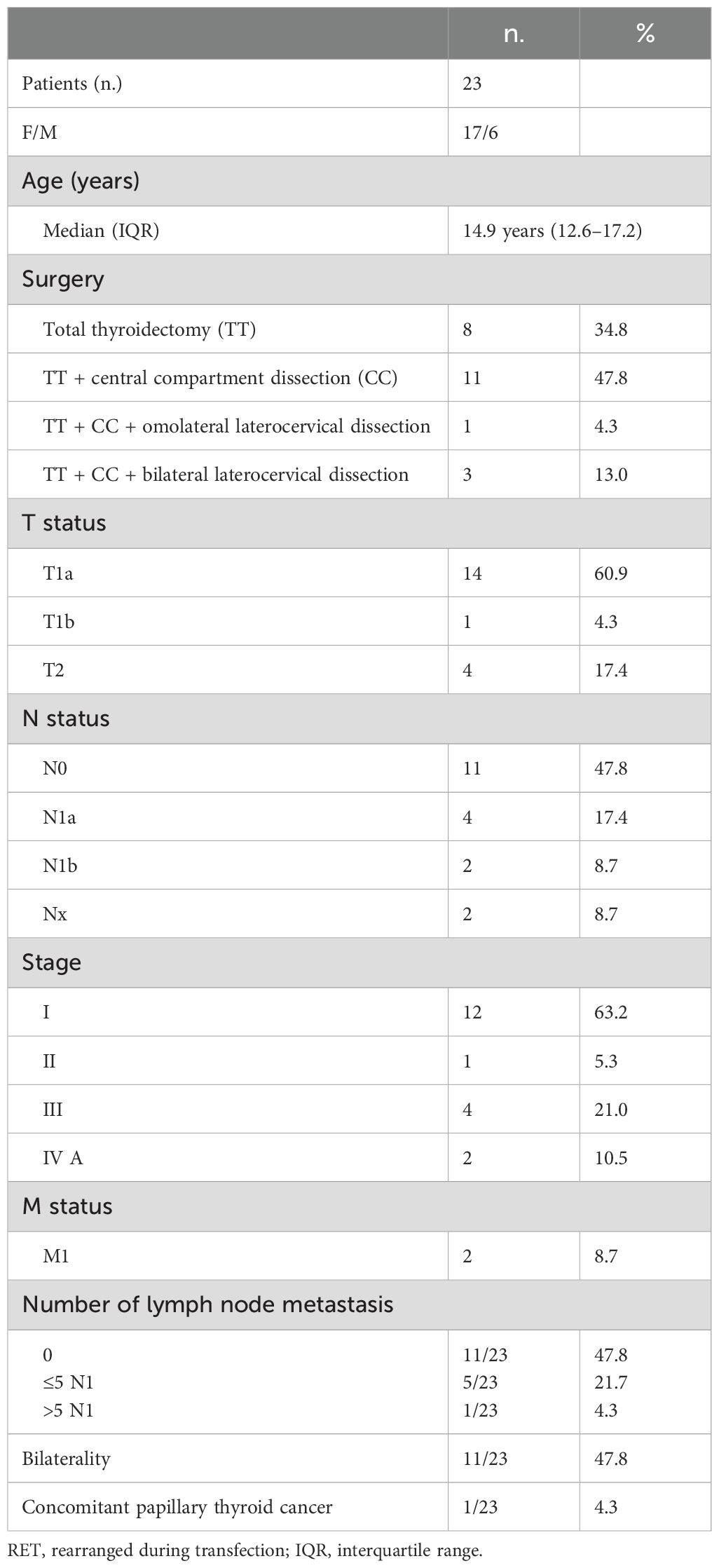
Table 1. Clinical and histopathological characteristics of 23 RET-mutated patients at diagnosis.
The differences in baseline characteristics between patients with high- and moderate-risk mutations are shown in Supplementary Table S1.
The median age at diagnosis was 14.9 years (IQR 12.6–17.2), and most patients were female (73.9%, F/M ratio 2.8/1). Total thyroidectomy was performed in eight patients (34.8%). LN surgery was performed in 15 (65.2%) patients: central compartment LN dissection in 11 and central and lateral LN dissection in four patients, of which three were bilateral. In 14 (60.9%), tumor size was ≤1 cm, and lymph node metastases were found in six (40.0%) patients who underwent lymph node dissection. At histology, MTC was found in 19/23 (82.6%) patients, C-cell hyperplasia in 2/23 (8.7%), and benign histology in 2/23 (8.7%). Two patients (8.7%) had distant metastasis at diagnosis (one liver and one bone).
Regarding the other manifestations of multiple endocrine neoplasia, only one patient was diagnosed with pheochromocytoma at 27 years old, and no patient developed hyperparathyroidism. The presence of the other manifestations of the syndrome in only a few cases is due to their young age and the later onset of pheochromocytoma or hyperparathyroidism.
Figure 1 shows the genealogical tree of a family with a 618 CYS TYR mutation.
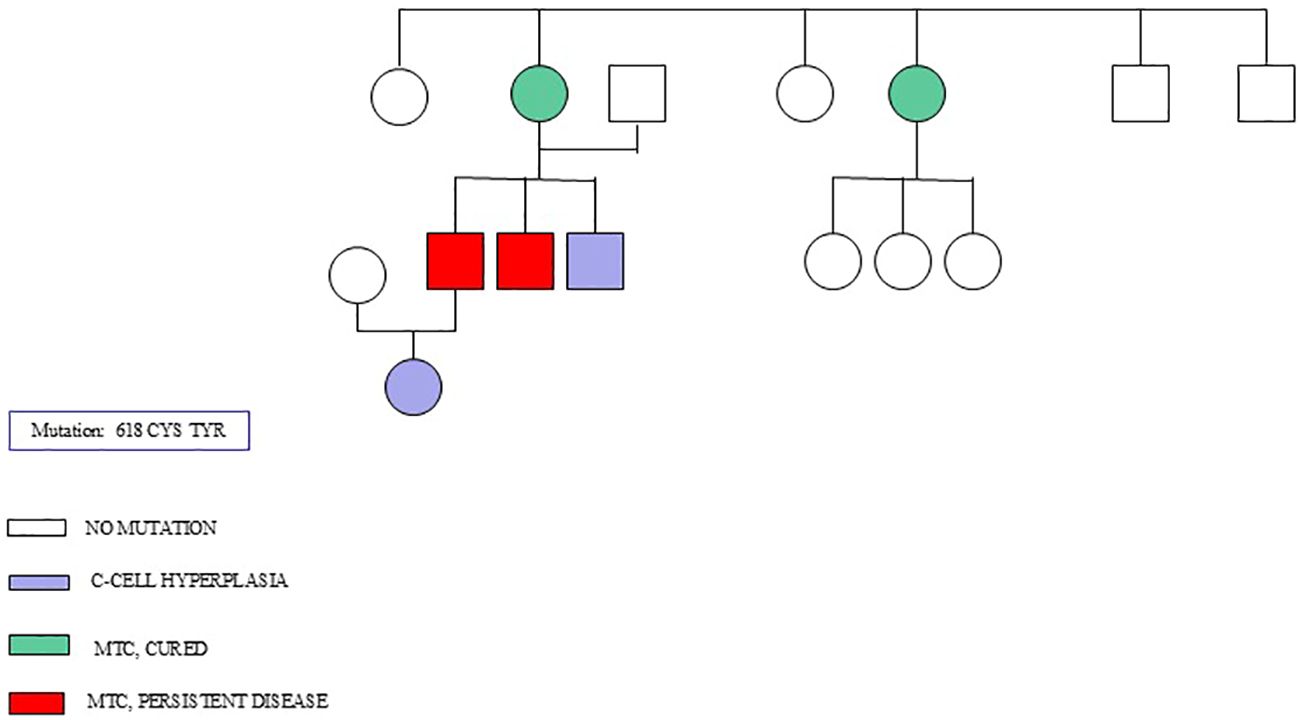
Figure 1. A genealogical tree of a family with 618 CYS TYR mutation.
3.2 RET mutation
The mutations were at very high risk in 4.3% of patients (M918T), high risk in 43.5% (C634), and moderate risk in 52.2% (47.8% C618 and 4.3% C620).
Of patients with C618 mutation, 4/11 (36.4%) had C-cell hyperplasia/negative histology; instead, in all patients with other mutations, MTC was already diagnosed.
3.3 Disease events and response to therapy at last follow-up
Ten of 19 (52.6%) had a disease event related to MTC during the follow-up: 2/19 (10.5%) biochemical disease, 6/19 (31.6%) lymph node recurrences, and 2/19 (10.5%) distant metastases (50% liver and 50% bone).
During follow-up, due to disease persistence, 5/6 patients (26.3%) underwent reoperation with subsequent histological diagnosis of lymph node metastasis.
At the last follow-up, nine MTCs were not cured (55.6%% with RET C634 mutation, 33.3% with C618, and 11.1% with M918T). All patients had positive calcitonin values and lymph node metastases, and two patients had distant metastases. Analyzing the rate of persistent disease according to the mutation, 5/10 (50%) had RET C634 mutation, 3/11 (27.3%) had C618, and 1/1 (100%) had M918T.
We analyzed the outcome in the two main groups of patients according to the mutation (618 vs. 634 mutated patients), and there was no significant difference (p = 0.38), probably due to the sample size.
One patient died after 9 years of follow-up at 21 years old (M918T). Among N1 patients, the number of lymph node metastases ranged from 1 to 8 (median 3). Most N1 patients 83.3% (5/6) were not cured at the last follow-up.
3.4 Outcome at last follow-up according to RET mutation and the age of carrier relatives
We evaluated whether the carrier’s age at diagnosis could have influenced the late diagnosis and therefore the surgery in our patients, resulting in a high percentage of medullary carcinoma diagnoses and disease persistence (Table 2).
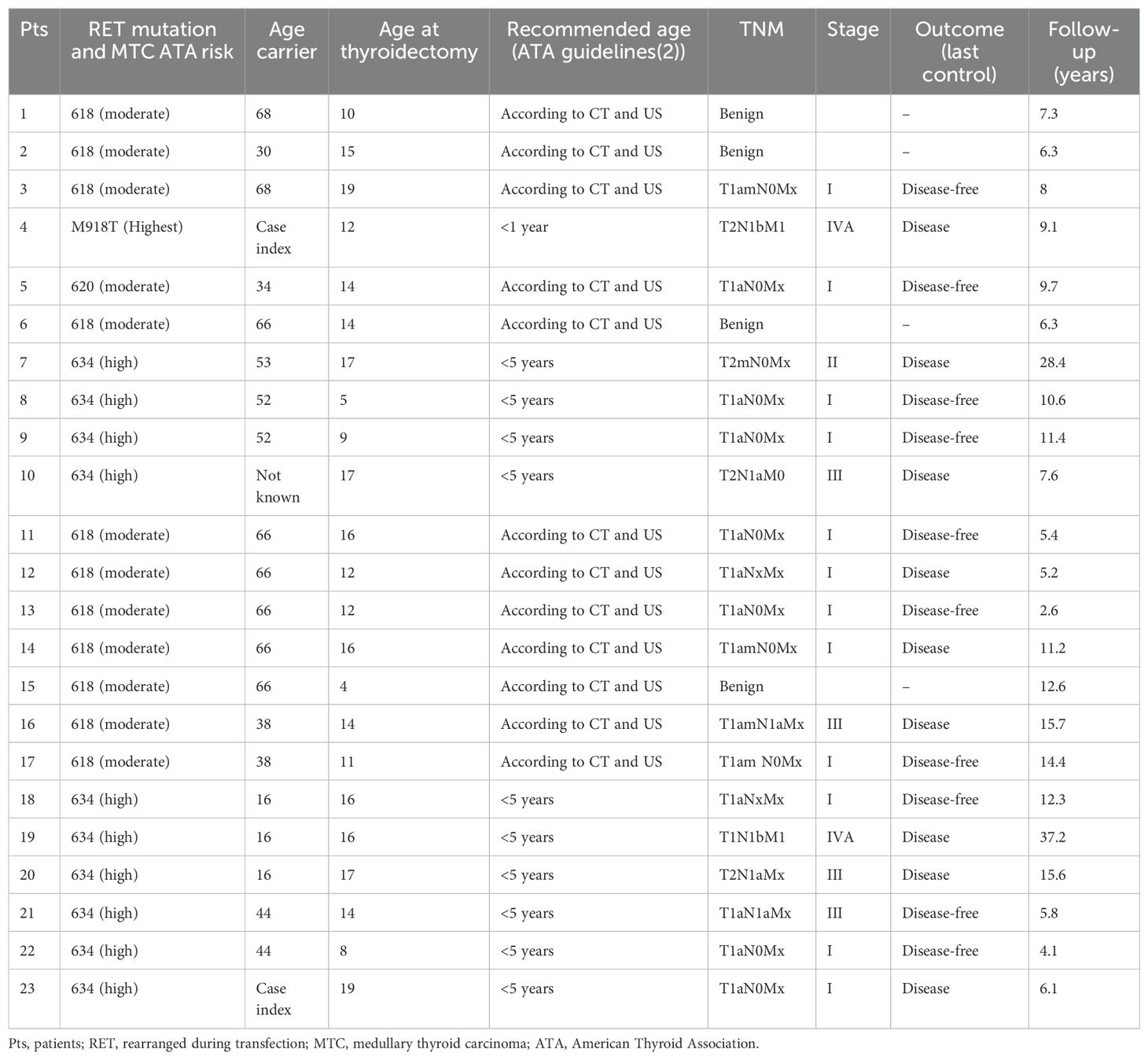
Table 2. Outcome at last follow-up according to RET mutation and the age of carrier relatives.
All four patients with benign histology were thyroidectomized at a young age (4, 10, 14, and 15 years old) and had a moderate risk of RET mutation.
Evaluating the median age at thyroidectomy and the outcome (disease or disease-free) according to the RET mutation risk, we found the following:
1. no difference in children with moderate risk RET mutations (median, 14 years old in both groups, p = 1), and
2. significant difference in children with high-risk RET mutations (disease-free 9 years old and 17 years old in the other group, p = 0.04).
4 Discussion
MTC is a rare tumor accounting for approximately 1–2% of all thyroid tumors. Approximately 20%–25% of cases have germline mutation of RET being included in MEN2, so at diagnosis, genetic counseling and RET screening must be provided to all first-degree relatives of patients with proven MEN2.
Hereditary MTC is usually preceded by hyperplasia. The age of onset of hyperplasia and MTC depends on the specific RET mutation.
At present, three groups of risk, depending on the specific RET mutations, have been identified (2): highest (including RET codon M918T mutation), high (including RET codon C634 and A883F mutations), and moderate risk (including all the others RET mutations) with progressive increases in aggressiveness in terms of onset and the stage, local, or distant disease (4, 6).
The optimal timing of the thyroid screening (with ultrasound and calcitonin determination) and the prophylactic thyroidectomy is recommended in RET-mutated patients according to the risk category. For the highest-risk category, total thyroidectomy is recommended within the first year. For the high-risk category, it is recommended that patients undergo a physical examination, serum calcitonin level test, and neck ultrasound from the age of 3 years and total thyroidectomy before 5 years old. For the moderate-risk category generally, patients should be screened every 6–12 months from the age of 5 and undergo surgery in childhood/early adulthood (earlier if calcitonin value is elevated) (10, 11).
The goal in relatives of patients having MEN2 syndromes is the presymptomatic detection of the RET mutation and consequently the correct timing of follow-up and/or thyroidectomy before the onset of MTC (12–16).
The hereditary form of MTC, being part of MEN2 syndrome, is subclassified into two forms: MEN2A (95%) and MEN2B (5%). In our series, only one patient (4.3%) had M918T RET mutation (MEN2B), similar to literature data.
MEN2A is characterized by the presence of MTC associated with pheochromocytoma hyperparathyroidism or both, with a different frequency depending on the specific RET mutation.
In our series, only one patient had the diagnosis of pheochromocytoma at 27 years old, and no patient developed hyperparathyroidism; instead, in their relatives, seven developed pheochromocytoma and two hyperparathyroidism. Therefore, the presence of the other manifestations of the syndrome in only a few cases is due to their young age and the later onset of pheochromocytoma or hyperparathyroidism.
The median age in our series was 14.9 years, older than 10 years reported in a paper by Sanso et al., which included 60 patients aged 6 months to 21 years, with 18 thyroidectomized (17).
Histologically, 21/23 (91.3%) patients had parafollicular cell hyperplasia, 19/23 (82.6%) had MTC, and 2/23 (8.7%) had benign histology. These data are similar to those shown in the paper of Sanso (17): 18/18 (100%) had parafollicular cell hyperplasia and 15/18 (83.3%) had MTC.
Regarding lymph node metastases in our series, 6/19 (31.6%) had N1a or N1b involvement, higher than in the Sanso series (3/15, 20%).
Of patients with C618 mutation, 4/11 (36.4%) had C-cell hyperplasia/negative histology; instead, in all patients with other mutations, MTC was already diagnosed.
Our data are different from the data reported in the literature by Romei et al. (66), who found a significantly higher frequency for the Val804met mutation of the non-cysteine region in exon 14 of the RET gene.
In a recent paper by Elisei et al. (18), 2,031 patients (1,264 with sporadic MTC, 117 with hereditary MTC, and 650 relatives) were evaluated. They found that in the group of clinically familial cases, Cys634 mutations were the most prevalent (31.6%), following the V804M RET mutation (19.6%).
In our series, 1/23 patients (4.3%) presented an association between MTC and papillary/follicular thyroid carcinoma. The coexistence of MTC and papillary/follicular thyroid carcinoma is a rare phenomenon that occurs in a range of 1% to 19% of all thyroid tumors in relation to the different series and must be considered incidental, as one cannot, however, exclude the possibility of a shared pathogenetic mechanism of both tumors (19, 20). Progression and prognosis are similar to the predominant component of the tumor. Radioactive iodine therapy can be used for follicular components.
As reported by several authors, the presence of lymph node metastases, the localization of N1, and the number of lymph nodes involved at diagnosis represent negative prognostic factors in patients with MTC (21, 22).
After the initial surgical treatment, in our series, the disease at the last follow-up was 39%, 23.5% in N0 patients, and 83.3% in N1 patients (75% in N1a and 100% in N1b).
During the follow-up, 10/19 patients (52.6%) had a disease event: 10.5% biochemical disease, 31.6% lymph node recurrences, and 10.5% distant metastases (liver and bone). During follow-up, due to disease persistence, five (26.3%) underwent reoperation with a histological diagnosis of lymph node metastasis.
At the last follow-up, nine MTCs were not cured. All patients had positive calcitonin values and lymph node metastases, and two patients had distant metastases. Analyzing the rate of persistent disease according to the mutation, 5/10 (50%) had RET C634 mutation, 3/11 (27.3%) C618, and 1/1 (100%) M918T. The only patient who died after 9 years of follow-up at 21 years old had M918T, confirming the literature data of the greater aggressiveness of MEN2B.
The limitations of the present analysis were that it is a retrospective study with possible enrollment bias and that the total number of patients was not numerous, depending on both the fact that it was a single-center study and the rarity of the syndrome.
5 Conclusions
From these data, it is clear to see the importance of genetic counseling and RET screening in all first-degree relatives of patients with proven MEN2. The objective should be to identify the correct timing of screening (calcitonin and ultrasound) and thyroidectomy before the onset of MTC develops or while it is clinically not evident and confined to the gland.
It is clear that the late diagnosis of MTC, understood as an advanced stage of the disease (stages III–IV), is associated with a high risk of persistent or recurrent disease also in pediatric/adolescent patients.
Data availability statement
The data supporting the findings of this study are derived from clinical records and known RET proto-oncogene mutations, which are publicly available in databases such as ClinVar. Due to ethical and privacy constraints, individual patient data cannot be shared publicly. De-identified data may be made available upon reasonable request to the corresponding author, in compliance with applicable legal and ethical standards.
Ethics statement
The studies involving humans were approved by Comitato Etico Catania 2, Catania, Italy. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. All procedures involving human participants were inaccordance with the ethical standards of the institutional researchcommittee and with the Declaration of Helsinki as revised in 2013.
Author contributions
GD: Conceptualization, Data curation, Investigation, Writing – review & editing. IB: Data curation, Writing – review & editing, Validation. LD: Writing – review & editing, Investigation. CS: Investigation, Writing – review & editing, Conceptualization, Data curation, Validation, Visualization. FG: Writing – review & editing. FM: Writing – review & editing. MR: Writing – review & editing, Conceptualization, Investigation, Validation, Visualization. FF: Validation, Writing – review & editing, Supervision. GP: Supervision, Validation, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Visualization. GS: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Supervision, Validation, Visualization, Writing – review & editing, Software, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2024.1464890/full#supplementary-material
Supplementary Table 1 | Differences in baseline characteristics between patients with high and moderate risk mutations.
References
1. Miranda-Filho A, Lortet-Tieulent J, Bray F, Cao B, Franceschi S, Vaccarella S, et al. Thyroid cancer incidence trends by histology in 25 countries: a population-based study. Lancet Diabetes Endocrinol. (2021) 9:225–34. doi: 10.1016/S2213-8587(21)00027-9
2. Wells SA, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. (2015) 25:567–610. doi: 10.1089/thy.2014.0335
3. Wolfe Hubert J, Melvin Kenneth EW, Cervi-Skinner Sergio J, AL Saadi Abdul A, Juliar Joseph F, Jackson Charles E, et al. C-cell hyperplasia preceding medullary thyroid carcinoma. New Engl J Med. (1973) 289:437–41. doi: 10.1056/NEJM197308302890901
4. Wells. Cancer of the Thyroid Invasive: Trends in SEER Incidence and U.S. Mortality Using the Joinpoint Regression Program, 1975–2011(SEER) Stat version 8.1.2 Rate Session. (Access the SEER 18 database at www.seer.cancer.gov). Incidence - SEER 18 Regs Research Data + Hurricane Katrina Impacted Louisiana Cases, Nov 596 WELLS ET AL. 2012 Sub (2000–2010) < Katrina/Rita Population Adjustment > - Linked To County Attributes - Total U.S., 1969– 2011 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, Surveillance Systems Branch, released April 2013, based on the November 2012 submission. (2012). Available at: https://seer.cancer.gov/archive/csr/1975_2011/results_merged/sect_26_thyroid.pdf
5. Tuttle RM, Ball DW, Byrd D, Daniels GH, Dilawari RA, Doherty GM, et al. Medullary carcinoma. J Natl Compr Canc Netw. (2010) 8:512–30. doi: 10.6004/jnccn.2010.0040
6. Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. (2001) 86:5658–71. doi: 10.1210/jcem.86.12.8070
7. Rohmer V, Vidal-Trecan G, Bourdelot A, Niccoli P, Murat A, Wemeau JL, et al. Prognostic factors of disease-free survival after thyroidectomy in 170 young patients with a RET germline mutation: a multicenter study of the Groupe Francais d’Etude des Tumeurs Endocrines. J Clin Endocrinol Metab. (2011) 96:E509–518. doi: 10.1210/jc.2010-1234
8. Niccoli-Sire P, Murat A, Rohmer V, Gibelin H, Chabrier G, Conte-Devolx B, et al. When should thyroidectomy be performed in familial medullary thyroid carcinoma gene carriers with non-cysteine RET mutations? Surgery. (2003) 134:1029–36. doi: 10.1016/j.surg.2003.07.019
9. MachensA ,Schneyer U, Holzhausen H-J, Dralle H. Prospects of remission in medullary thyroid carcinoma according to basal calcitonin level. J Clin Endocrinol Metab. (2005) 90(4):2029–34. doi: 10.1210/jc.2004-1836
10. Zordan P, Tavella S, Brizzolara A, Biticchi R, Ceccherini I, Garofalo S, et al. The immediate upstream sequence of the mouse Ret gene controls tissue-specific expression in transgenic mice(2006). Available online at: https://iris.unimol.it/handle/11695/2967. doi: 10.3892/ijmm.18.4.601
11. PDQ Pediatric Treatment Editorial Board. Childhood multiple endocrine neoplasia (MEN) syndromes treatment (PDQ®): health professional version. In: PDQ Cancer Information Summaries. National Cancer Institute (US, Bethesda (MD (2002). Available at: http://www.ncbi.nlm.nih.gov/books/NBK552292/.
12. Skinner MA, Moley JA, Dilley WG, Owzar K, Debenedetti MK, Wells SA. Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. N Engl J Med. (2005) 353:1105–13. doi: 10.1056/NEJMoa043999
13. Skinner MA. Management of hereditary thyroid cancer in children. Surg Oncol. (2003) 12:101–4. doi: 10.1016/S0960-7404(03)00033-1
14. Fitze G. Management of patients with hereditary medullary thyroid carcinoma. Eur J Pediatr Surg. (2004) 14:375–83. doi: 10.1055/s-2004-821209
15. Learoyd DL, Gosnell J, Elston MS, Saurine TJ, Richardson AL, Delbridge LW, et al. Experience of prophylactic thyroidectomy in multiple endocrine neoplasia type 2A kindreds with RET codon 804 mutations. Clin Endocrinol (Oxf). (2005) 63:636–41. doi: 10.1111/j.1365-2265.2005.02394.x
16. Haddad RI, Bischoff L, Ball D, Bernet V, Blomain E, Busaidy NL, et al. Thyroid carcinoma, version 2.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2022) 20:925–51. doi: 10.6004/jnccn.2022.0040
17. Sanso GE, Domene HM, Garcia R, Pusiol E, de M, Roque M, et al. Very early detection of RET proto-oncogene mutation is crucial for preventive thyroidectomy in multiple endocrine neoplasia type 2 children: presence of C-cell Malignant disease in asymptomatic carriers. Cancer. (2002) 94:323–30. doi: 10.1002/cncr.10228
18. Elisei R, Tacito A, Ramone T, Ciampi R, Bottici V, Cappagli V, et al. Twenty-five years experience on RET genetic screening on hereditary MTC: an update on the prevalence of germline RET mutations. Genes (Basel). (2019) 10:698. doi: 10.3390/genes10090698
19. Calapkulu M, Gul OO, Cander S, Sagiroglu MF, Saraydaroglu O, Erturk E, et al. Co-existence of papillary and medullary thyroid carcinoma: reports of three cases. J Coll Physicians Surg Pak. (2022) 32:S156–8.
20. Kim WG, Gong G, Kim EY, Kim TY, Hong SJ, Kim WB, et al. Concurrent occurrence of medullary thyroid carcinoma and papillary thyroid carcinoma in the same thyroid should be considered as coincidental. Clin Endocrinol (Oxf). (2010) 72:256–63. doi: 10.1111/j.1365-2265.2009.03622.x
21. Scollo C, Baudin E, Travagli JP, Caillou B, Bellon N, Leboulleux S, et al. Rationale for central and bilateral lymph node dissection in sporadic and hereditary medullary thyroid cancer. J Clin Endocrinol Metab. (2003) 88:2070–5. doi: 10.1210/jc.2002-021713
22. Modigliani E, Cohen R, Campos JM, Conte-Devolx B, Maes B, Boneu A, et al. Prognostic factors for survival and for biochemical cure in medullary thyroid carcinoma: results in 899 patients. The GETC Study Group. Groupe d’étude des tumeurs à calcitonine. Clin Endocrinol (Oxf). (1998) 48:265–73. doi: 10.1046/j.1365-2265.1998.00392.x
Keywords: pediatric and adolescent MEN 2, childhood medullary thyroid cancer, RET mutation, response to treatment, outcome, persistent disease, thyroid cancer
Citation: Di Benedetto G, Barca I, De Gregorio L, Scollo C, Gianì F, Martorana F, Russo M, Frasca F, Pellegriti G and Sapuppo G (2025) Medullary thyroid cancer in MEN2 pediatric/adolescent carriers of RET mutation: genotype/phenotype correlation and outcome in a retrospective series of 23 patients. Front. Oncol. 14:1464890. doi: 10.3389/fonc.2024.1464890
Received: 15 July 2024; Accepted: 04 November 2024;
Published: 07 January 2025.
Edited by:
Davide Leardini, University of Bologna, ItalyReviewed by:
Daniele Zama, IRCCS University Hospital of Bologna Sant Orsola Polyclinic, ItalyFrancesco Pegoraro, University of Florence, Italy
Copyright © 2025 Di Benedetto, Barca, De Gregorio, Scollo, Gianì, Martorana, Russo, Frasca, Pellegriti and Sapuppo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriella Pellegriti, Z2FicmllbGxhLnBlbGxlZ3JpdGlAdW5pY3QuaXQ=
†ORCID: Gabriella Pellegriti, orcid.org/0000-0001-6102-379X