 Yurou Xing
Yurou Xing Kejia Zhao2†
Kejia Zhao2† Yongsheng Wang
Yongsheng Wang- 1Thoracic Oncology Ward, Cancer Center, West China Hospital, Sichuan University, Chengdu, Sichuan, China
- 2Department of Thoracic Surgery and Institute of Thoracic Oncology, West China Hospital of Sichuan University, Sichuan University, Chengdu, Sichuan, China
The prognosis of primary central nervous system lymphoma (PCNSL) patients is relatively poor, and there is currently no standard treatment plan. Most patients choose high-dose chemotherapy based on methotrexate. While traditional chemotherapy combined with biological therapy has achieved limited results, some patients still do not respond to treatment or cannot tolerate its toxicity and side effects. Bruton’s tyrosine kinase (BTK) is a key enzyme in B-cell receptor signaling, and its activation is critical for B-cell survival and proliferation. In recent years, BTK inhibitors have shown great potential in treating lymphomas derived from various B cells because of their strong targeting ability and relatively few side effects. They may also be a reasonable treatment choice for PCNSL. This article reviews the mechanism of action, clinical research, adverse reactions, and other issues of BTK inhibitors in treating PCNSL to provide a reference for individualized treatment of patients with PCNSL.
1 Introduction
Primary central nervous system lymphoma (PCNSL) accounts for 4% of primary central nervous system (CNS) tumors and 4% to 6% of extranodal lymphomas (1) and has a relatively worse prognosis than other extranodal diffuse large B-cell lymphomas. High-dose methotrexate (HD-MTX) has improved blood−brain barrier permeability, and immunochemotherapy regimens based on HD-MTX are currently the first-line treatment for PCNSL patients. However, recurrence still occurs in more than 50% of patients. The prognosis of relapsed/refractory (R/R) PCNSL remains extremely poor, and there is currently no unified salvage treatment plan. Diffuse large B-cell lymphoma can be divided into two subtypes: the germinal center B-cell-like (GCB) subtype and the activated B-cell-like (ABC) subtype. Most PCNSLs are activated B-cell-like (ABC) cells. This subtype selectively acquires mutations that target the B-cell receptor (BCR) and promote BCR signaling, which supports the development of BTK inhibitors in PCNSL (2).
The pathophysiological process of PCNSL involves numerous distinct genetic mutations, the most important of which are functional mutations in the genes encoding the CD79b molecule (CD79B), a subunit of the B-cell receptor (BCR), and the MYD88 innate immune signal transduction adaptor (MYD88), which is involved in Toll-like receptor (TLR) and interleukin receptor signaling (3). In normal B cells, the TLR and BCR signaling pathways cooperate to activate the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). Mutated MYD88 and CD79B activate the NF-κB signaling pathway, promoting B-cell survival and proliferation (4). The frequency of MYD88 mutations in PCNSL patients is 76% (5). This high frequency underscores the pivotal role of MYD88 in the pathogenesis and progression of PCNSL, influencing therapeutic strategies targeting the B-cell receptor signaling pathway. In contrast, mutations leading to the activation of the NF-κB signaling pathway are present in more than 90% of patients with PCNSL (6). These findings show that the activation of the NF-κB signaling pathway is the key driver of the occurrence of PCNSL, providing an important theoretical reference for the use of Bruton’s tyrosine kinase (BTK) inhibitors to treat PCNSL.
PCNSL also have the same immune evasion mechanism: gene inactivation of major histocompatibility complex (MHC) classes I and II and β2-microglobulin (B2M), leading to the loss of their synthesized proteins (7). By systematically reviewing the preclinical and clinical data reported for BTK inhibitors, this review focuses on the mechanism of action of BTK inhibitors in PCNSL and the results of existing clinical trials and discusses their therapeutic efficacy, challenges, and future development directions, aiming to provide new perspectives and broader possibilities for treating PCNSL.
2 BTK and BTK inhibitors
2.1 BTK
The BCR pathway is essential for the proliferation and survival of cancer cells in various B-cell malignancies, with BTK being a key component. BTK is a nonreceptor protein kinase belonging to the Tec family (Figure 1) (8). Originally identified as the faulty protein in X-linked agammaglobulinemia by Vetrie et al. in 1993, BTK is also referred to as agammaglobulinemia tyrosine kinase (9, 10). This kinase is expressed predominantly in cells of the hematopoietic lineage, including B cells, mast cells, and macrophages, but not in T cells, natural killer cells, or plasma cells (11). BTK is instrumental in multiple processes, such as B-cell lymphangiogenesis, as well as the development, maturation, and differentiation of immature B cells and their subsequent proliferation and survival (12, 13).
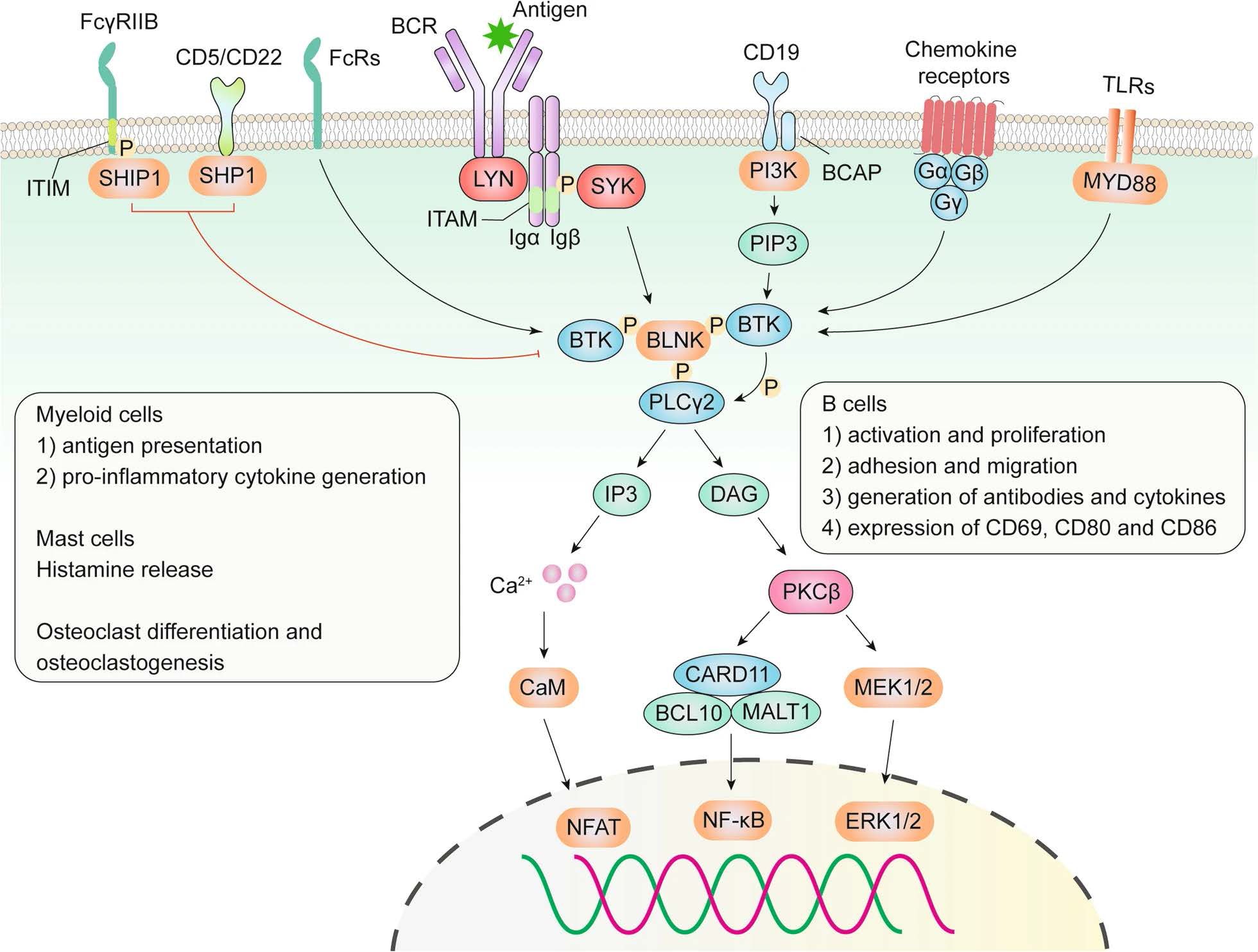
Figure 1. Role of BTK in the BCR, TLR, chemokine receptor, and Fc receptor (FcR) signaling pathways (8).
BTK is a key element within various B-cell receptors (BCRs), serving as a regulator of multiple intracellular signaling pathways mediated by cell surface molecules such as phosphoinositide 3-kinase (PI3K), mitogen-activated protein kinase (MAPK), and NF-κB. This regulation is crucial for the activation, proliferation, and differentiation of antibody-producing plasma cells (14, 15). As a result, the abnormal activation of BTK can lead to several B-cell malignancies, including various types of leukemias and lymphomas (16), as well as autoimmune disorders such as rheumatoid arthritis (RA) and multiple sclerosis (MS) (17).
BTK is composed of five distinct regions: the pleckstrin homology (PH) domain, the Tec homology (TH) domain, the SRC homology 2 (SH2) and 3 (SH3) domains, and a C-terminal catalytic domain (18, 19) (Figure 2) (8). The PH domain is crucial for mediating interactions with phospholipids and proteins. The TH domain, which contains two proline-rich regions (PRRs), plays a role in the autoregulation of BTK. The SH2 and SH3 domains are involved in binding to phosphorylated tyrosine residues and PRRs, respectively, with the SH3 domain also hosting a vital autophosphorylation site at tyrosine 223. The catalytic domain at the C-terminus, featuring the kinase activity center at tyrosine 551, is key for the initial activation of BTK (20). Additionally, within this catalytic domain, the residue Cys481 is a critical covalent binding site targeted by BTK inhibitors.
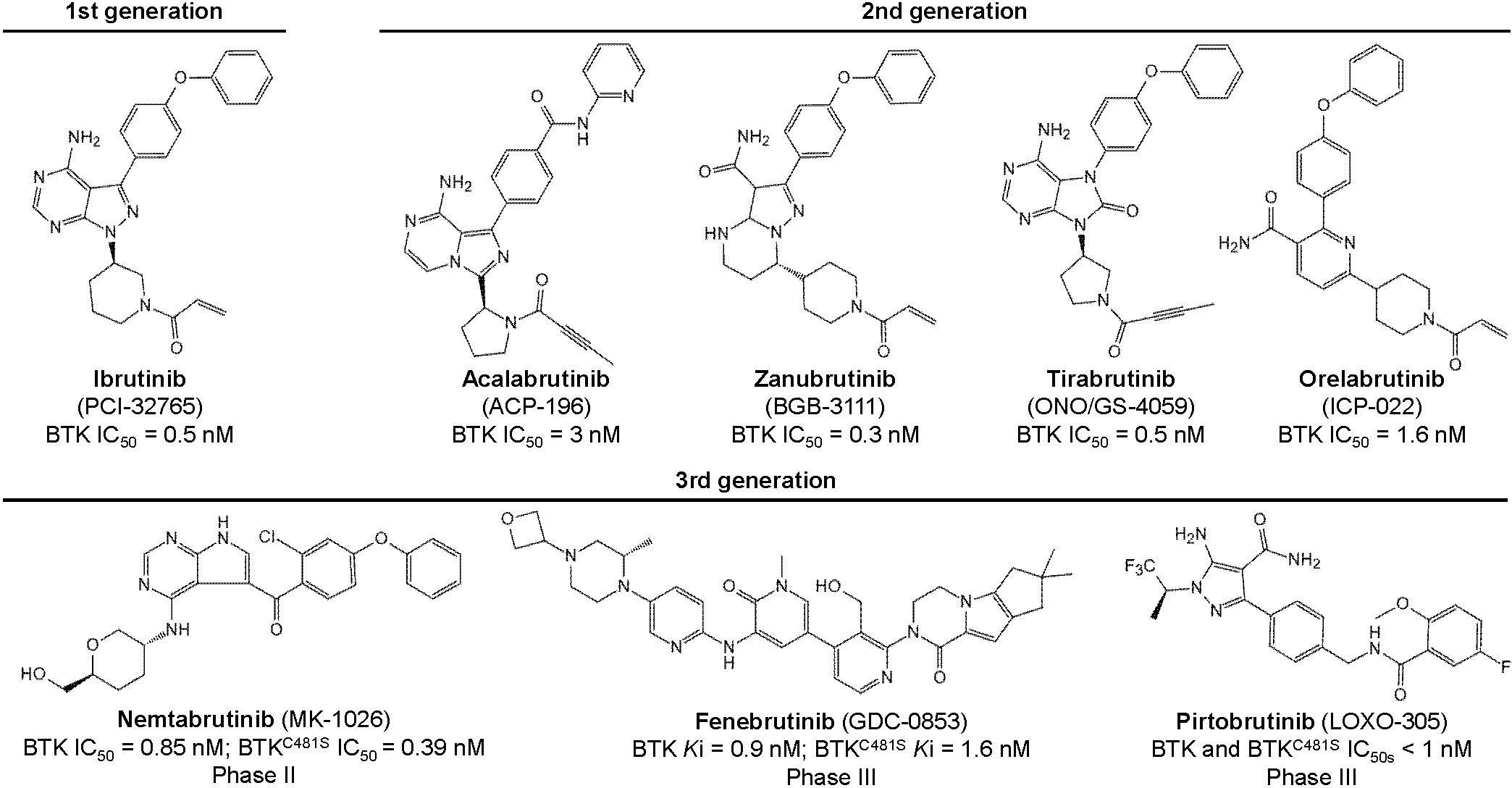
Figure 2. The structure and interactions of BTK (8).
BTK activation occurs through the spleen tyrosine kinase (SYK), which itself is activated by the B-cell receptor (BCR) (20). Once activated, BTK phosphorylates residues Y753 and Y759 on phospholipase C beta 2 (PLCβ2), leading to the formation of inositol trisphosphate (IP3) and diacylglycerol (DAG). These secondary messengers then activate protein kinase C beta (PRKCB/PKCβ) rather than producing it. This results in elevated calcium levels, which then activate the MAPK/extracellular signal-regulated kinase (ERK) pathway, influencing the expression of genes related to cell proliferation, survival, and cytokine secretion. Concurrently, BTK can also activate the protein kinase B (AKT)/NF-κB signaling pathway (21, 22). Additionally, active BTK plays a role in mediating proinflammatory signals, including the production of inflammatory cytokines such as tumor necrosis factor (TNF)-α and interleukin 1b (IL1B), linking it to inflammatory responses (23).
2.2 Mechanisms of action of BTK inhibitors
As previously discussed, Cys481 within BTK is a nucleophilic site that forms a covalent bond with electrophilic inhibitors. Sequence analysis revealed that this cysteine is analogous to Cys773 in the epidermal growth factor receptor (EGFR) family, which has been targeted by several irreversible kinase inhibitors in clinical trials. This similarity inspired the development of a selective BTK inhibitor aimed at irreversibly deactivating this particular site. Through extensive research, scientists synthesized a compound that significantly reduced the phosphorylation of phospholipase C gamma 1 (PLCG1) associated with BTK and inhibited the LYN proto-oncogene Src family tyrosine kinase (LYN)- and spleen tyrosine kinase (SYK)-dependent phosphorylation at Tyr551 on BTK. This compound demonstrated high selectivity for BTK, with a marked preference over LYN or SYK, effectively and irreversibly inhibiting BTK-dependent pathways in the BCR signaling cascade. Consequently, these BTK inhibitors have attracted considerable attention (20).
BTK inhibitors are categorized into two types on the basis of their binding modes and mechanisms of action: irreversible and reversible. Irreversible inhibitors function through a Michael acceptor component that forms a covalent bond with the conserved Cys481 residue within the ATP-binding site, thereby permanently disabling the kinase. Conversely, some reversible inhibitors interact with specific pockets in the SH3 structural domain through noncovalent interactions such as hydrogen bonds or hydrophobic interactions, leading to an inactive kinase conformation (24). Some drugs such as pirtobrutinib blocks the ATP-binding site of BTK through non-covalent, non-C481-dependent binding, thereby overcoming acquired resistance to covalent BTK inhibitors (25). Most of the BTK inhibitors currently approved for use are irreversible (18). However, the clinical efficacy of these irreversible inhibitors, such as ibrutinib—the first of its kind to be marketed—has been compromised by the emergence of drug-resistant mutations. Notably, a mutation in which Cys481 is replaced by serine (C481S) diminishes the reactivity of BTK to ibrutinib and other covalent inhibitors, substantially reducing its effectiveness. For example, the inhibitory potency of ibrutinib is reduced sixfold against the C481S mutant (half-maximal inhibitory concentration = 4.6 nM). Other mutations at Cys481, including C481R, C481F, and C481Y, as well as mutations at the gatekeeper residue Thr474 (T474I, T474S, T474M), have been identified and pose similar challenges (26, 27). While ibrutinib can still bind noncovalently to the C481S mutant, this reversible interaction does not ensure sustained efficacy in patients harboring this mutation (28, 29).
In this context, noncovalent inhibitors that do not rely on interactions with Cys481 have been shown to effectively inhibit BTK mutants such as C481R, T474I, and T474M, representing promising therapeutic alternatives (26). Additionally, reversible inhibitors have demonstrated increased efficacy in the treatment of autoimmune diseases, including rheumatoid arthritis (RA), various forms of multiple sclerosis (MS), chronic graft-versus-host disease, and systemic lupus erythematosus (30, 31). Moreover, recent advancements have led to the development of protein hydrolysis-targeted chimeric molecules, introducing a novel strategy for reducing BTK activity (32).
2.3 Approved BTK inhibitors
Six BTK inhibitors are currently approved and marketed worldwide: first-generation ibrutinib; second-generation acalabrutinib, zanubrutinib, tirabrutinib, and orelabrutinib; and third-generation molecules, including the newly marketed pirtobrutinib and the not-yet-marketed nemtabrutinib and fenebrutinib (Figure 3; Table 1). First- and second-generation BTK inhibitors irreversibly inhibit BTK activity by covalently binding to the Cys481 site in BTK. The third-generation BTK inhibitors are noncovalent inhibitors that do not depend on binding to Cys481 and still have a better inhibitory effect on BTK mutated at this site, with better efficacy and safety.
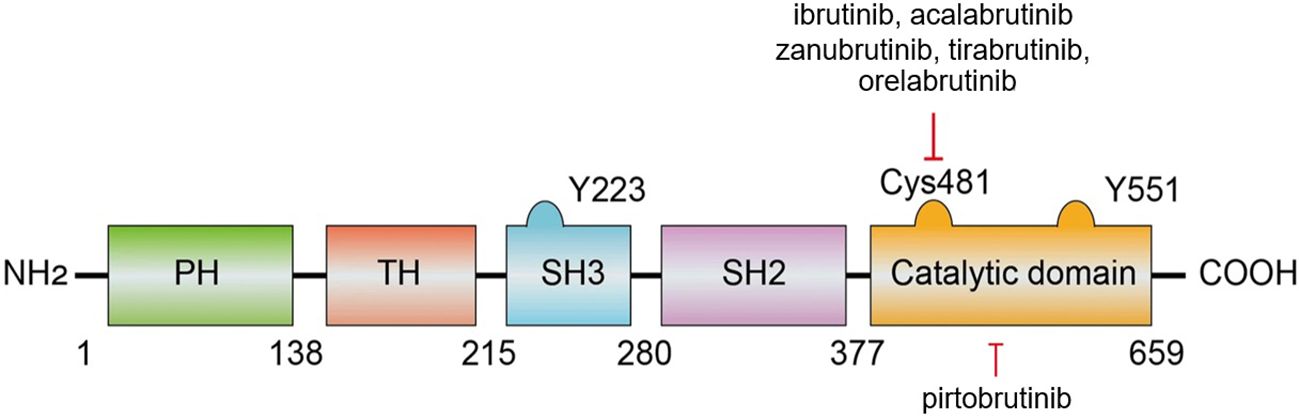
Figure 3. Chemical structures of the BTK inhibitors.
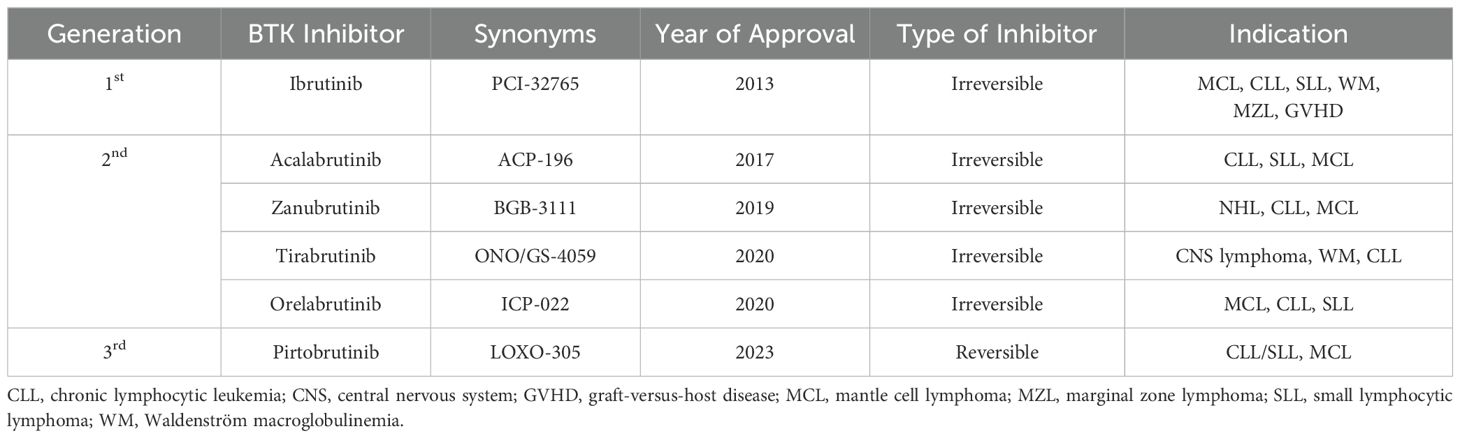
Table 1. Summary of approved BTK inhibitors.
Ibrutinib, a pioneering and selective BTK inhibitor, was recognized as a breakthrough therapy by the U.S. Food and Drug Administration (FDA) in 2013, marking a significant milestone in medical treatment. It effectively inhibits tumor cell proliferation and impacts the adhesion and migration of malignant cells and the tumor microenvironment, which includes T and mesenchymal cells (33–37). In November 2013, the FDA initially approved ibrutinib for the treatment of mantle cell lymphoma (MCL). It was later approved to address various B-cell lymphomas, including chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), Waldenström macroglobulinemia (WM), and marginal zone lymphoma (MZL). Despite its efficacy, resistance has developed in some patients who have relapsed following treatment with ibrutinib.
Research has indicated that certain baseline molecular and cytogenetic characteristics, such as del(17p)/tumor protein p53 (TP53) mutations and complex karyotypes (23 chromosomal abnormalities), increase the risk of disease progression in patients with primary chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) undergoing treatment with ibrutinib. Patients carrying the del(17p)/TP53 mutation are particularly prone to recurrence, which is often linked to BTK mutations (38, 39). Specifically, in relapsed/refractory (R/R) patients, the C481 residue of BTK is most commonly mutated to serine (C481S), which impedes the covalent attachment of ibrutinib to the structural domain of BTK (29, 40–42). Additionally, cells with the BTK T316A mutation exhibit resistance to ibrutinib, both at the cellular and molecular levels, comparable to the resistance observed with the C481S mutation (43). Moreover, missense mutations in PLCG2, a target of BTK, have emerged as another resistance mechanism. The activation of PLCG2 enables CLL cells to proliferate independently of BTK control, often cooccurring with BTK mutations to foster ibrutinib resistance (43). Another contributing factor to CLL resistance is del(8p), which results in the loss of TRAIL-R and significant epigenetic alterations (44). In response to these resistance issues and to mitigate side effects associated with ibrutinib, such as skin and dermatologic issues (45), bleeding, infections (46), headaches, and atrial fibrillation, newer generation BTK inhibitors, such as acalabrutinib and zanubrutinib, were developed and received FDA approval in 2017 and 2019, respectively (47). These advancements aim to offer more effective and tolerable treatment options for patients with CLL and related conditions.
The three approved BTK inhibitors—ibrutinib, zanubrutinib, and acalabrutinib—share similarities and differences in their action and usage. All three bind irreversibly and covalently to the Cys481 residue in the ATP-binding pocket of BTK. In terms of biochemical binding kinetics, ibrutinib is recognized as the most potent, followed by zanubrutinib and then acalabrutinib. Acalabrutinib stands out for having the highest selectivity and the lowest rate of off-target effects, followed by zanubrutinib, with ibrutinib having the least selectivity (48). Pharmacodynamic and pharmacokinetic variations among these inhibitors influence dosing, efficacy, and the profile of adverse effects observed in clinical practice. For example, acalabrutinib, with its shorter half-life and higher rate of BTK occupancy, is administered twice daily compared with ibrutinib’s once-daily regimen (95.3% vs. 87.6% BTK occupancy) (49). For zanubrutinib, studies have shown that a dosage of 160 mg twice daily achieves sustained complete suppression and more than 95% BTK occupancy in lymph nodes, making it more effective than a single daily dose of 320 mg. Thus, 160 mg twice daily has been recommended for further clinical studies (50). The interplay between rapid absorption and elimination in drug kinetics can lead to swift target inhibition while minimizing the risk of off-target effects and drug interactions. The selective properties and short half-life of acalabrutinib ensure thorough and continuous BTK inhibition, avoiding the toxicity associated with the inhibition of other kinases. Achieving full target engagement helps mitigate drug resistance arising from BTK mutations and lowers the incidence of Richter transformation (51).
Tirabrutinib is a highly selective and highly active BTK inhibitor developed in Japan for the treatment of recurrent or refractory primary central nervous system lymphoma (52). A phase 1/2 clinical study confirmed an overall response rate of 63.6% in patients with relapsed/refractory PCNSL treated with tirabrutinib. Common adverse reactions are rash, neutropenia, leukopenia, and lymphocytopenia (53). Orelabrutinib is a highly selective BTK inhibitor that is currently approved only for MCL and CLL in China (54). A phase I/II clinical study reported an overall response rate of 81.1% in patients with relapsed or refractory (r/r) mantle cell lymphoma (MCL). PMID: 37078706 Another phase 2 clinical study evaluating orelabrutinib (150 mg once daily) in patients with refractory or recurrent chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL). The total effective rate was 92.5% (74/80). Orelabrutinib is well tolerated, and the most common adverse reactions are decreases in leukocytes, hemoglobin and platelets and infection (55).
Recently, researchers have introduced pirtobrutinib, a reversible, noncovalent BTK inhibitor that is highly selective and effectively inhibits BTK phosphorylation, cellular proliferation, and tumor growth (56, 57). Unlike previous inhibitors, pirtobrutinib binds to BTK without Cys481, offering a potential solution to overcome the resistance observed with ibrutinib, acalabrutinib, and zanubrutinib. Nemtabrutinib is a multitarget inhibitor that can strongly inhibit the transmission of BCR signals. It can competitively occupy the ATP-binding pocket of BTK without interacting with C481, overcoming the resistance caused by this mutation. A phase I study demonstrated the initial efficacy of nemtabrutinib in patients with relapsed/refractory B-cell malignancies (58). The most common side effect of the drug is neutropenia. These new developments may represent significant advancements in the treatment of B-cell malignancies. Fenebrutinib is also a reversible BTK inhibitor that can both inhibit B-cell activation and reduce MS-induced inflammation. At present, related research in the field of multiple sclerosis is being carried out.
3 Clinical applications of BTK inhibitors for treating PCNSL
In the context of PCNSL, numerous studies have documented significant genomic alterations. Common genetic anomalies in PCNSL include single nucleotide and copy number variations, with the most frequently mutated genes being MYD88, CD79B, caspase recruitment domain family member 11 (CARD11), and TNF-α-induced protein 3 (TNFAIP3) (59). MYD88 serves as a critical adaptor molecule in the Toll-like receptor (TLR) and interleukin 1 receptor type 1 (IL1R1) signaling pathways (60). Research by Ngo et al. indicated that activated B-cell-like diffuse large B-cell lymphoma (ABC-DLBCL) relies on MYD88, with the MYD88 L265P mutation being particularly prevalent in systemic DLBCL (61). This mutation is also commonly observed in PCNSL, making it the predominant subtype (62). CD79B, the second most frequently mutated gene in PCNSL, enhances B-cell receptor (BCR) signaling and NF-κB activation, thus providing survival signals to tumor cells (5, 63–65). Mutations in MYD88 L265P and CD79B are also utilized to classify ABC-DLBCL into distinct categories (66). Additionally, CARD11, which is a downstream component of the BCR pathway, has mutations that potentially activate NF-κB, contributing to the pathogenesis of PCNSL (67). Notably, mutations in CARD11 have been linked to resistance against the BTK inhibitor ibrutinib in several human B-cell malignancies (2, 29).
Recent research has highlighted that innovations targeting the BCR and TLR signaling pathways have been pivotal in advancing the treatment of PCNSL. These pathways offer various therapeutic targets. Upstream of the BCR signaling pathway, PI3K can be downregulated to suppress signaling. Some previously studied PI3K inhibitors have limited clinical use due to gastrointestinal toxicity. Amdizalisib, a novel highly selective PI3Kδ inhibitor, is currently undergoing clinical exploration for the treatment of hematologic malignancies (68). The drug has shown good safety and efficacy in patients with R/R lymphoma. Amdizalisib is not currently used in PCNSL, but owing to its good therapeutic effect and safety in other lymphomas, it may provide a new treatment option for patients with PCNSL. Another PI3K inhibitor, Linperlisib, a small-molecule inhibitor with high blood−brain barrier permeability, can effectively treat PCNSL and improve patient survival (69–71). Downstream, immunomodulatory drugs such as lenalidomide can inhibit interferon regulatory factor 4 (IRF4), thereby impacting NF-κB functionality. Proteasome inhibitors can also be used to prevent NF-κB from entering the nucleus, altering gene expression. However, a significant limitation of proteasome inhibitors is their inability to cross the blood−brain barrier, complicating their use in treating PCNSL (59). At the core of this pathway lies BTK, which is targeted effectively by BTK inhibitors.
3.1 First-generation BTK inhibitors
Ibrutinib is a first-generation BTK inhibitor. A phase I clinical trial by Gromes et al. (3) used ibrutinib monotherapy to treat 20 patients with R/R PCNSL, with an objective response rate (ORR) of 77%, including five patients with a complete response (CR), a PFS of 4.6 months, and an mOS of 15 months. In a further phase II clinical trial of single-agent ibrutinib in the treatment of R/R PCNSL and secondary CNS lymphoma (SCNSL), the ORR was 81%, the median PFS (mPFS) was four months, and the mOS was 19.5 months (72). In a phase II clinical trial (73) in which ibrutinib monotherapy was used in patients with R/R PCNSL or primary vitreoretinal lymphoma (PVSL), the ORR was 52%, the mPFS was 4.8 months, and the mOS was 19.2 months. These three studies demonstrated that ibrutinib monotherapy had a good clinical response in treating PCNSL.
Interestingly, the efficacy of BTK inhibitors in PCNSL is distinct from their performance in systemic lymphoma, where the single agent ibrutinib achieves only a 10% CR rate and a 15% PR rate (2). These findings underscore the limited impact of ibrutinib outside the CNS, in contrast with its more favorable outcomes in PCNSL, where despite a modest median PFS, the ORR is higher. This disparity may be attributed to the increased prevalence of BCR/TLR pathway alterations, such as mutations in MYD88, in PCNSL (73). Notably, even PCNSL patients without significant genomic alterations in the BCR signaling pathway respond to ibrutinib (73). However, the co-occurrence of CD79B and MYD88 mutations, while enhancing the sensitivity of systemic lymphomas to ibrutinib (2), does not seem to confer the same level of responsiveness in CNS disorders, potentially owing to a lesser reliance on the BCR signaling pathway in these conditions (3). These mutations are present in approximately 37% of PCNSL patients (64). Additionally, mutations in CARD11 and TNFAIP3, which operate downstream of BTK, have been identified as potential sources of resistance to ibrutinib in both systemic lymphoma (74) and PCNSL treated with ibrutinib monotherapy (3). This resistance is particularly relevant when considering the mechanisms of resistance that may arise when ibrutinib is used in combination with cytotoxic chemotherapy (65).
Since the efficacy of ibrutinib alone is often transient or incomplete, exploring its efficacy in combination with other antineoplastic drugs is meaningful. Gromes et al. (3) conducted three clinical trials. The first trial treated patients with PCNSL or SCNSL with ibrutinib, HD-MTX, and rituximab and reported an ORR of 89% (65). The second trial combined ibrutinib with copanisib to treat patients with R/R PCNSL and reported an ORR of 67% (75). The third trial combined 560–840 mg of ibrutinib with rituximab and lenalidomide to treat patients with R/R PCNSL or SCNSL and reported an mPFS of 3.03 months (76).
In 2017, Lionakis et al. (64) treated patients with R/R PCNSL via a regimen of ibrutinib with rituximab, liposomal adriamycin, temozolomide, etoposide, and dexamethasone (DA-TEDDi-R), with 86% achieving a CR and an overall efficacy rate of 94% and a PFS of 15.3 months. In 2020, Mark et al. (77) conducted a similar trial using ibrutinib in combination with temozolomide, etoposide, liposomal adriamycin, dexamethasone, and rituximab (TEDDi-R) to treat patients with R/R PCNSL, with one-year PFS and OS rates of 60.0% and 100%, respectively. In the same year, another study retrospectively analyzed 22 patients with R/R PCNSL treated with ibrutinib in combination with temozolomide and reported an ORR of 55% and a PFS of 11.7 months (78).
Despite high response rates to treatment, PFS with ibrutinib monotherapy is less than five months, suggesting the early emergence of resistance (79–81). Ibrutinib combination therapy extended PFS in pretreated patients to approximately nine months (65). Currently, a number of prospective studies are being conducted to combine ibrutinib with drugs such as lenalidomide, copanlisib, checkpoint inhibitors, and traditional chemotherapy. These results have not yet been published, but they provide new ideas and expectations for the application of BTK inhibitors in PCNSL. In conclusion, ibrutinib alone or in combination with chemotherapy has clear efficacy for the treatment of PCNSL. Combination chemotherapy is more efficacious than ibrutinib alone but also increases the risk of adverse effects, and the optimal combination approach still warrants further exploration.
3.2 Second-generation BTK inhibitors
Acalabrutinib, zanubrutinib, tirabrutinib, and orelabrutinib are second-generation BTK inhibitors, all of which are covalent. Zanubrutinib has greater target selectivity and fewer off-target effects than does ibrutinib. Zhang et al. (82) administered a zanubrutinib-containing regimen to four patients with newly diagnosed PCNSL and four patients with R/R PCNSL, and all patients with primary PCNSL and 75% of patients with R/R PCNSL achieved CR. Song et al. (83) combined rituximab, zanubrutinib, lenalidomide, and temozolomide with or without MTX (RLZT ± MTX) to treat PCNSL, achieving an ORR of 79.2%, making it a promising regimen for elderly patients who are intolerant of high-dose radiotherapy. A clinical study enrolled two groups of patients. One group included young patients in the RLZT+MTX group who received rituximab, lenalidomide, zanubrutinib, temozolomide, or high-dose methotrexate. The ORR of this group was 86.7% (CR rate 40%), and the other group of elderly patients was the RLZT group. The ORR of the RLZT group was 76.5% (CR rate 35.3%). This suggests that RLZT (without methotrexate) may be an option for older patients who cannot be treated with methotrexate (84). Another prospective study combined zanubrutinib with high-dose cytarabine to treat patients with R/R PCNSL, achieving an ORR of 75%, a median follow-up of 12 months, a PFS of 5.6 months, and an unmet mOS (85). All four studies used regimens combining zanubrutinib with other agents with favorable clinical responses. However, the results of zanubrutinib monotherapy have not been clarified, and an ongoing trial (NCT05117814) aimed to evaluate the efficacy and safety of zanubrutinib monotherapy in treating R/R PCNSL and SCNSL.
Four ongoing studies are examining the efficacy of acalabrutinib in treating PCNSL. One (NCT04688151) is a dose-escalation trial combining acalabrutinib with durvalumab to treat R/R PCNSL or SCNSL. The second is a trial combining acalabrutinib with rituximab and durvalumab to treat R/R PCNSL. The remaining two trials (NCT04906902 and NCT04548648) are designed to evaluate the safety and efficacy of acalabrutinib monotherapy for treating R/R PCNSL.
Tirabrutinib was the first BTK approved globally to treat R/R PCNSL. One study reported that elderly patients with PCNSL were switched to maintenance therapy with tirabrutinib after being unable to continue HD-MTX treatment, with their tumors almost disappearing and their cognitive function improving after two months (86). A phase I/II clinical trial in which single-agent tirabrutinib was used to treat patients with R/R PCNSL achieved an ORR of 64% and an mPFS of 2.9 months (53). Kawasaki et al. (87) conducted a prospective study evaluating tirabrutinib for R/R PCNSL, achieving an ORR of 63.0%. While the above studies suggest that tirabrutinib has a favorable clinical response in treating R/R PCNSL alone, the efficacy of combining it with other chemotherapeutic agents remains unknown. An ongoing trial (NCT04947319) is combining tirabrutinib with two different HD-MTX regimens (MTX, temozolomide, and rituximab or MTX, rituximab, procarbazine, and vincristine) to treat PCNSL and evaluate the safety and efficacy of tirabrutinib in combination with other chemotherapeutic agents.
Orelabrutinib has higher cerebrospinal fluid concentrations than ibrutinib does. A retrospective study evaluated the efficacy of orelabrutinib monotherapy in treating PCNSL, achieving a six-month OS rate of 100% (88). A clinical study enrolled patients with PCNSL who were treated with rituximab plus high-dose methotrexate and orelabrutinib. On day 15 of orelibrutinib treatment, the patients underwent a lumbar puncture, and the concentration of orelabrutinib in the cerebrospinal fluid was measured. One study confirmed that the cerebrospinal fluid concentration of orelabrutinib was much greater than that of drugs such as ibrutinib (89). Several recent clinical studies have examined the combination of orelabrutinib with other chemotherapy drugs to treat PCNSL. Yang et al. (90) treated 15 R/R PCNSL patients with orelabrutinib combined with rituximab, HD-MTX, temozolomide, and lenalidomide, achieving an ORR of 86.7% and a CR rate of 73.3%. Another phase II clinical trial achieved an ORR of 100% when orelabrutinib was combined with HD-MTX and rituximab to treat patients with newly diagnosed PCNSL (91). A retrospective study by Zeng et al. (92) evaluated the efficacy and safety of a regimen of tiotropium, orelabrutinib, and MTX with or without rituximab (TOM ± R) in treating patients with PCNSL, achieving an ORR of 92.3%, a CR rate of 53.9%, and a six-month PFS of 63.6%. Zhao et al. (93) administered orelabrutinib, rituximab, and HD-MTX to 34 patients newly diagnosed with PCNSL, achieving an ORR of 94.4% and a CR rate of 88.9%. A phase I/II clinical trial by Zhang et al. (94) evaluated the safety and efficacy of combining orelabrutinib, an anti-programmed cell death 1 (PDCD1/PD-1) antibody, and formostatin to treat patients with newly diagnosed PCNSL, achieving an ORR of 88.9% in a phase I study, with a phase II study still ongoing.
The five BTK inhibitors currently on the market are all covalent inhibitors that are prone to drug-resistant mutations and adverse reactions caused by off-target effects. The most commonly reported adverse reactions to ibrutinib include diarrhea, bleeding, atrial fibrillation, and infection, which may lead to treatment discontinuation in severe cases (Table 2). One study reported that 56% of patients treated with the single agent ibrutinib and 52% of patients treated with combination therapy developed infections (46). Among more than 500 patients who received ibrutinib for malignancy between 2009 and mid-2016, more than 75% developed new or worsening hypertension within a median of 30 months (46). Second-generation covalent BTK inhibitors mostly have higher target selectivity and less off-target toxicity, but they can still cause adverse reactions, such as headache, diarrhea, and infection. The risk of adverse reactions increases when some chemotherapy drugs are used together.

Table 2. Summary of efficacy and common adverse reactions of 5 BTK inhibitors.
There are a number of other BTK inhibitors that have been reported. TL-895 is a very potent and highly selective BTK inhibitor (95). Another BTK inhibitor, M7583, has been shown to be effective in treating human B-cell malignancies (96). In addition, a Phase I clinical study demonstrated that the BTK inhibitor DTRMWXHS-12 in combination with everolimus and pomadomide was tolerated and clinically active (97). These drugs are expected to be further studied in the future. The compound RSH-7 strongly inhibits BTK and FLT3 signaling pathways by up-regulating proapoptotic protein and down-regulating Bcl-2 levels, and effectively inhibits the proliferation of various hematologic malignant tumor cells. RSH-7 may be a promising compound for the treatment of hematological malignancies (98). Preclinical studies have confirmed that BGB-8035 is highly selective and a promising preclinical candidate compound for the treatment of autoimmune diseases and B-cell lymphoma (99). JNJ-64264681 is a covalent, irreversible BTK inhibitor that has shown good oral efficacy in both cancer and autoimmune models and has entered human clinical studies (100). Studies have confirmed that AS-1763 has a significant effect on the in vivo xenogenic tumor model, and has entered the phase I clinical study (101). BTK inhibitors such as NX-2127, HPCL-760, and CG-806 can destroy B-cell receptors and inhibit B-cell pathways, which is expected to be further explored in the future (102, 103). Currently ongoing clinical trails of BTK inhibitors are shown in Table 3.
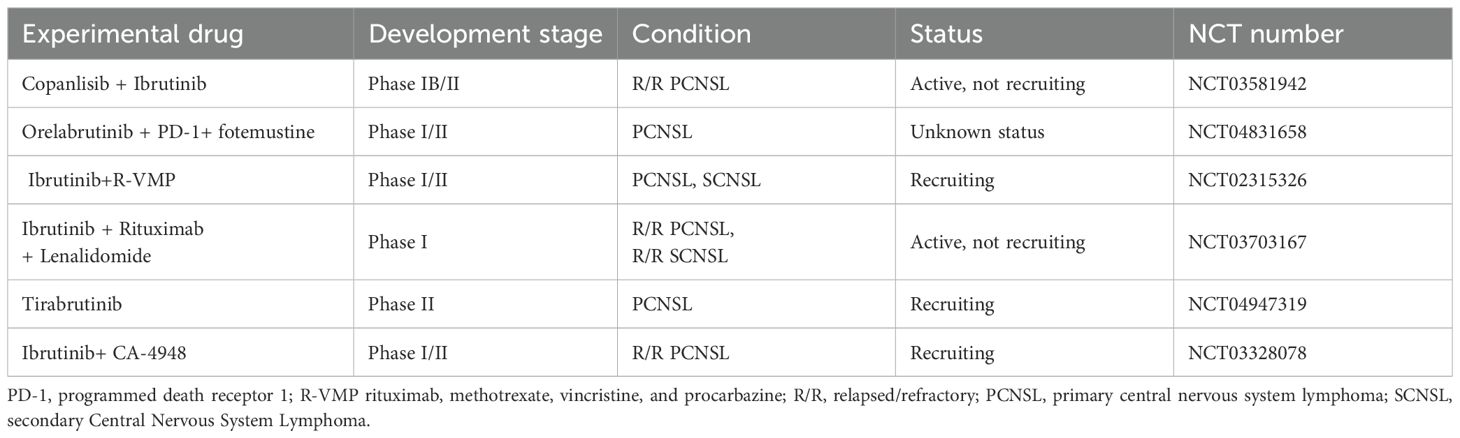
Table 3. Summary of ongoing clinical trials of BTK inhibitors.
Another concern is the issue of resistance. First- and second-generation BTK inhibitors inhibit BTK kinase activity by binding to its ATP-binding site and then covalently modifying Cys481. Most often, mutations convert this active cysteine to serine (C481S) and, less frequently, to phenylalanine (C481F), tyrosine (C481Y), or arginine (C481R), and the L528W mutation inactivates BTK. The growth and survival of DLBCL cells with kinase-naïve BTK are dependent on Toll-like receptor 9 (TLR9), leading to resistance to BTK inhibitors (104). While second-generation BTK inhibitors have shown better BTK selectivity and less off-target toxicity, they cannot reverse the resistance of tumor cells to ibrutinib. In conclusion, the role of BTK inhibitors in PCNSL is still in the clinical trial stage. While some clinical data are promising, they remain limited due to small sample sizes and a lack of blinding, randomized control, and comparison. Most phase III clinical trials on BTK inhibitors in PCNSL are ongoing, and we expect that more evidence will be available to confirm the efficacy, safety, and resistance of BTK inhibitors in PCNSL, with the goal of providing a basis for individualized treatment of patients with PCNSL in the real world.
4 Conclusion
Since PCNSL have similar biological properties and therapeutic responses, BTK inhibitors provide a new option for their treatment. Compared with first-generation BTK inhibitors, second-generation BTK inhibitors have significantly fewer off-target effects and fewer adverse effects, further improving patient prognosis. However, drug-resistant mutations in tumor cells remain a challenge. Pirtobrutinib, the only approved third-generation noncovalent BTK inhibitor, is used to treat R/R lymphomas after at least two lines of prior systemic therapy, and the effectiveness and efficacy of noncovalent BTK inhibitors in treating PCNSL remain unknown. Further clinical studies examining the efficacy of BTK inhibitors in treating PCNSL are needed. The therapeutic efficacy of BTK inhibitors as monotherapies is usually short-lived and incomplete, and the combination of BTK inhibitors with other treatments may increase their adverse effects while further improving patient prognosis. However, combining BTK inhibitors may solve the problem of drug resistance, and exploring the development of BTK inhibitor-based combination therapy to further improve the efficacy and safety of treating PCNSL is worthwhile.
5 Future directions
Novel BTK inhibitors, immunomodulators, anti-PD-1 drugs, CAR-T cells, etc., have provided new options for the treatment of PCNSL patients. However, more clinical studies on long-term efficacy and survival are needed. Promising therapeutic strategies should be actively developed, and more drug combinations should be explored to provide further reference for clinical practice. Increased expression or activity of BTK has been associated with increased blood-brain barrier permeability in several studies, and orelabrutinib has shown potential in the treatment of central system lymphoma. In the development of a new generation of BTK inhibitors, improving the blood-brain barrier permeability of BTK inhibitors is an important direction.
Author contributions
YX: Writing – original draft. KZ: Writing – review & editing. YZ: Writing – review & editing. YW: Conceptualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Calimeri T, Steffanoni S, Gagliardi F, Chiara A, Ferreri A. How we treat primary central nervous system lymphoma. ESMO Open. (2021) 6:100213. doi: 10.1016/j.esmoop.2021.100213
2. Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. (2015) 21:922–6. doi: 10.1038/nm.3884
3. Grommes C, Pastore A, Palaskas N, Tang SS, Campos C, Schartz D, et al. Ibrutinib unmasks critical role of Bruton tyrosine kinase in primary CNS lymphoma. Cancer Discovery. (2017) 7:1018–29. doi: 10.1158/2159-8290.CD-17-0613
4. Nepal G, Khurana M, Bucheli DH, Bhandari S, Joshi U, Bhagat R, et al. Ibrutinib in refractory or relapsing primary central nervous system lymphoma: a systematic review. Neurol Int. (2022) 14:99–108. doi: 10.3390/neurolint14010009
5. Nakamura T, Tateishi K, Niwa T, Matsushita Y, Tamura K, Kinoshita M, et al. Recurrent mutations of CD79B and MYD88 are the hallmark of primary central nervous system lymphomas. Neuropathol Appl Neurobiol. (2016) 42:279–90. doi: 10.1111/nan.2016.42.issue-3
6. Chapuy B, Roemer MG, Stewart C, Tan Y, Abo RP, Zhang L, et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood J Am Soc Hematol. (2016) 127:869–81. doi: 10.1182/blood-2015-10-673236
7. Riemersma SA, Jordanova ES, Schop RF, Philippo K, Looijenga LH, Schuuring E, et al. Extensive genetic alterations of the HLA region, including homozygous deletions of HLA class II genes in B-cell lymphomas arising in immune-privileged sites. Blood J Am Soc Hematol. (2000) 96:3569–77. doi: 10.1182/bloodv96.10.3569
8. Alu SA, Lei ES, Han RF, Wei K, Wei LH. BTK inhibitors in the treatment of hematological Malignancies and inflammatory diseases: mechanisms and clinical studies. J Hematol Oncol. (2022) 15:138. doi: 10.1186/s13045-022-01353-w
9. Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell Malignancy. Int Rev Immunol. (2012) 31:119–32. doi: 10.3109/08830185.2012.664797
10. Vetrie D, Vorechovský I, Sideras P, Holland J, Davies A, Flinter F, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. (1993) 361:226–33. doi: 10.1038/361226a0
11. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. New Engl J Med. (2013) 369:32–42. doi: 10.1056/NEJMoa1215637
12. Kil LP, De Bruijn MJ, Van Nimwegen M, Corneth OB, Van Hamburg JP, Dingjan GM, et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood J Am Soc Hematol. (2012) 119:3744–56. doi: 10.1182/blood-2011-12-397919
13. Hendriks R, De Bruijn M, Maas A, Dingjan GM, Karis A, Grosveld F, et al. Inactivation of Btk by insertion of lacZ reveals defects in B cell development only past the pretl cell stage. EMBO J. (1996) 15:4862–72. doi: 10.1002/j.1460-2075.1996.tb00867.x
14. Dingjan GM, Middendorp S, Dahlenborg K, Maas A, Grosveld F, Hendriks RW. Bruton's tyrosine kinase regulates the activation of gene rearrangements at the λ light chain locus in precursor B cells in the mouse. J Exp Med. (2001) 193:1169–78. doi: 10.1084/jem.193.10.1169
15. Liang C, Tian D, Ren X, Ding S, Jia M, Xin M, et al. The development of Bruton's tyrosine kinase (BTK) inhibitors from 2012 to 2017: A mini-review. Eur J medicinal Chem. (2018) 151:315–26. doi: 10.1016/j.ejmech.2018.03.062
16. Ysebaert L, Michallet A-S. Bruton's tyrosine kinase inhibitors: lessons learned from bench-to-bedside (first) studies. Curr Opin Oncol. (2014) 26:463–8. doi: 10.1097/CCO.0000000000000114
17. Carnero Contentti E, Correale J. Bruton’s tyrosine kinase inhibitors: a promising emerging treatment option for multiple sclerosis. Expert Opin emerging Drugs. (2020) 25:377–81. doi: 10.1080/14728214.2020.1822817
18. Wang X, Wong J, Sevinsky CJ, Kokabee L, Khan F, Sun Y, et al. Bruton's tyrosine kinase inhibitors prevent therapeutic escape in breast cancer cells. Mol Cancer Ther. (2016) 15:2198–208. doi: 10.1158/1535-7163.MCT-15-0813
19. Yoon Y. Small chemicals with inhibitory effects on PtdIns (3, 4, 5) P3 binding of Btk PH domain. Bioorganic Medicinal Chem Lett. (2014) 24:2334–9. doi: 10.1016/j.bmcl.2014.03.068
20. Pan Z, Scheerens H, Li S-J, Schultz BE, Sprengeler PA, Burrill LC, et al. Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem. (2007) 2:58–61. doi: 10.1002/cmdc.200600221
21. Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton’s tyrosine kinase in B cells and Malignancies. Mol Cancer. (2018) 17:57. doi: 10.1186/s12943-018-0779-z
22. Zuo Y, Pan Z. Small-Molecule Inhibitors of bruton’s tyrosine kinase. Cancer II. (2018) 28:75–104. doi: 10.1007/7355_2017_6
23. Burger JA, Wiestner A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat Rev Cancer. (2018) 18:148–67. doi: 10.1038/nrc.2017.121
24. Brullo C, Villa C, Tasso B, Russo E, Spallarossa A. Btk inhibitors: a medicinal chemistry and drug delivery perspective. Int J Mol Sci. (2021) 22:7641. doi: 10.3390/ijms22147641
25. Wierda WG, Shah NN, Cheah CY, Lewis D, Hoffmann MS, Coombs CC, et al. Pirtobrutinib, a highly selective, non-covalent (reversible) BTK inhibitor in patients with B-cell Malignancies: analysis of the Richter transformation subgroup from the multicentre, open-label, phase 1/2 BRUIN study. Lancet Haematol. (2024) 11:e682–92. doi: 10.1016/S2352-3026(24)00172-8
26. Johnson AR, Kohli PB, Katewa A, Gogol E, Belmont LD, Choy R, et al. Battling Btk mutants with noncovalent inhibitors that overcome Cys481 and Thr474 mutations. ACS Chem Biol. (2016) 11:2897–907. doi: 10.1021/acschembio.6b00480
27. Liu L, Shi B, Wang X, Xiang H. Strategies to overcome resistance mutations of Bruton's tyrosine kinase inhibitor ibrutinib. Future Medicinal Chem. (2018) 10:343–56. doi: 10.4155/fmc-2017-0145
28. Furman RR, Chen S, Lu P, Setty M, Perez AR, Guo A, et al. Ibrutinib resistance in chronic lymphocytic leukemia. New Engl J Med. (2014) 370:2352–4. doi: 10.1056/NEJMc1402716
29. Woyach JA, Furman RR, Liu T-M, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. New Engl J Med. (2014) 370:2286–94. doi: 10.1056/NEJMoa1400029
30. Feng Y, Duan W, Cu X, Liang C, Xin M. Bruton’s tyrosine kinase (BTK) inhibitors in treating cancer: a patent review (2010-2018). Expert Opin Ther Patents. (2019) 29:217–41. doi: 10.1080/13543776.2019.1594777
31. Norman P. Investigational Bruton’s tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Expert Opin Investigational Drugs. (2016) 25:891–9. doi: 10.1080/13543784.2016.1182499
32. Tinworth CP, Lithgow H, Dittus L, Bassi ZI, Hughes SE, Muelbaier M, et al. PROTAC-mediated degradation of Bruton’s tyrosine kinase is inhibited by covalent binding. ACS Chem Biol. (2019) 14:342–7. doi: 10.1021/acschembio.8b01094
33. Ponader S, Chen S-S, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood J Am Soc Hematol. (2012) 119:1182–9. doi: 10.1182/blood-2011-10-386417
34. de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor–and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood J Am Soc Hematol. (2012) 119:2590–4. doi: 10.1182/blood-2011-11-390989
35. Niemann CU, Herman SE, Maric I, Gomez-Rodriguez J, Biancotto A, Chang BY, et al. Disruption of in vivo chronic lymphocytic leukemia tumor–microenvironment interactions by ibrutinib–findings from an investigator-initiated phase II study. Clin Cancer Res. (2016) 22:1572–82. doi: 10.1158/1078-0432.CCR-15-1965
36. Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood J Am Soc Hematol. (2013) 122:2539–49. doi: 10.1182/blood-2013-06-507947
37. Herman SE, Mustafa RZ, Jones J, Wong DH, Farooqui M, Wiestner A. Treatment with ibrutinib inhibits BTK-and VLA-4–dependent adhesion of chronic lymphocytic leukemia cells in vivo. Clin Cancer Res. (2015) 21:4642–51. doi: 10.1158/1078-0432.CCR-15-0781
38. Kadri S, Lee J, Fitzpatrick C, Galanina N, Sukhanova M, Venkataraman G, et al. Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv. (2017) 1:715–27. doi: 10.1182/bloodadvances.2016003632
39. Campo E, Cymbalista F, Ghia P, Jäger U, Pospisilova S, Rosenquist R, et al. TP53 aberrations in chronic lymphocytic leukemia: an overview of the clinical implications of improved diagnostics. Haematologica. (2018) 103:1956. doi: 10.3324/haematol.2018.187583
40. Woyach JA, Ruppert AS, Guinn D, Lehman A, Blachly JS, Lozanski A, et al. BTKC481S-mediated resistance to ibrutinib in chronic lymphocytic leukemia. J Clin Oncol. (2017) 35:1437. doi: 10.1200/JCO.2016.70.2282
41. Cheng S, Guo A, Lu P, Ma J, Coleman M, Wang Y. Functional characterization of BTKC481S mutation that confers ibrutinib resistance: exploration of alternative kinase inhibitors. Leukemia. (2015) 29:895–900. doi: 10.1038/leu.2014.263
42. Quinquenel A, Fornecker L-M, Letestu R, Ysebaert L, Fleury C, Lazarian G, et al. Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: a FILO group study. Blood J Am Soc Hematol. (2019) 134:641–4. doi: 10.1182/blood.2019000854
43. Liu T-M, Woyach JA, Zhong Y, Lozanski A, Lozanski G, Dong S, et al. Hypermorphic mutation of phospholipase C, γ2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation. Blood J Am Soc Hematol. (2015) 126:61–8. doi: 10.1182/blood-2015-02-626846
44. Shaffer III AL, Phelan JD, Wang JQ, Huang D, Wright GW, Kasbekar M, et al. Overcoming acquired epigenetic resistance to BTK inhibitors. Blood Cancer Discovery. (2021) 2:630–47. doi: 10.1158/2643-3230.BCD-21-0063
45. Sibaud V, Beylot-Barr M, Protin C, Vigarios E, Recher C, Ysebaert L. Dermatological toxicities of Bruton’s tyrosine kinase inhibitors. Am J Clin Dermatol. (2020) 21:799–812. doi: 10.1007/s40257-020-00535-x
46. Tillman BF, Pauff JM, Satyanarayana G, Talbott M, Warner JL. Systematic review of infectious events with the Bruton tyrosine kinase inhibitor ibrutinib in the treatment of hematologic Malignancies. Eur J Haematology. (2018) 100:325–34. doi: 10.1111/ejh.2018.100.issue-4
47. Wen T, Wang J, Shi Y, Qian H, Liu P. Inhibitors targeting Bruton’s tyrosine kinase in cancers: drug development advances. Leukemia. (2021) 35:312–32. doi: 10.1038/s41375-020-01072-6
48. Kaptein A, de Bruin G, Emmelot-van Hoek M, van de Kar B, de Jong A, Gulrajani M, et al. Potency and selectivity of BTK inhibitors in clinical development for B-cell Malignancies. Blood. (2018) 132:1871. doi: 10.1182/blood-2018-99-109973
49. Alsadhan A, Cheung J, Gulrajani M, Gaglione EM, Nierman P, Hamdy A, et al. Pharmacodynamic analysis of BTK inhibition in patients with chronic lymphocytic leukemia treated with acalabrutinib. Clin Cancer Res. (2020) 26:2800–9. doi: 10.1158/1078-0432.CCR-19-3505
50. Tam CS, Trotman J, Opat S, Burger JA, Cull G, Gottlieb D, et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell Malignancies and safety and efficacy evaluation in CLL. Blood J Am Soc Hematol. (2019) 134:851–9. doi: 10.1182/blood.2019001160
51. Wu J, Zhang M, Liu D. Acalabrutinib (ACP-196): a selective second-generation BTK inhibitor. J Hematol Oncol. (2016) 9:1–4. doi: 10.1186/s13045-016-0250-9
52. Dhillon S. Tirabrutinib: first approval. Drugs. (2020) 80:835–40. doi: 10.1007/s40265-020-01318-8
53. Narita Y, Nagane M, Mishima K, Terui Y, Arakawa Y, Yonezawa H, et al. Phase I/II study of tirabrutinib, a second-generation Bruton's tyrosine kinase inhibitor, in relapsed/refractory primary central nervous system lymphoma. Neuro Oncol. (2021) 23:122–33. doi: 10.1093/neuonc/noaa145
54. Dhillon S. Orelabrutinib: first approval. Drugs. (2021) 81:503–7. doi: 10.1007/s40265-021-01482-5
55. Xu W, Zhou K, Wang T, Yang S, Liu L, Hu Y, et al. Orelabrutinib in relapsed or refractory chronic lymphocytic leukemia/small lymphocytic lymphoma patients: Multi-center, single-arm, open-label, phase 2 study. Am J Hematol. (2023) 98:571–9. doi: 10.1002/ajh.26826
56. Jiang VC, Liu Y, Lian J, Huang S, Jordan A, Cai Q, et al. Cotargeting of BTK and MALT1 overcomes resistance to BTK inhibitors in mantle cell lymphoma. J Clin Invest. (2023) 133:e165694. doi: 10.1172/JCI165694
57. Mato AR, Shah NN, Jurczak W, Cheah CY, Pagel JM, Woyach JA, et al. Pirtobrutinib in relapsed or refractory B-cell Malignancies (BRUIN): a phase 1/2 study. Lancet. (2021) 397:892–901. doi: 10.1016/S0140-6736(21)00224-5
58. Woyach JA, Stephens DM, Flinn IW, Bhat SA, Savage RE, Chai F, et al. First-in-human study of the reversible BTK inhibitor nemtabrutinib in patients with relapsed/refractory chronic lymphocytic leukemia and B-cell non-hodgkin lymphoma. Cancer Discovery. (2024) 14:66–75. doi: 10.1158/2159-8290.CD-23-0670
59. Schaff LR, Grommes C. Update on novel therapeutics for primary CNS lymphoma. Cancers. (2021) 13:5372. doi: 10.3390/cancers13215372
60. Jeelall YS, Horikawa K. Oncogenic MYD88 mutation drives Toll pathway to lymphoma. Immunol Cell Biol. (2011) 89:659. doi: 10.1038/icb.2011.31
61. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim K-H, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. (2011) 470:115–9. doi: 10.1038/nature09671
62. Hilal T, Maguire A, Kosiorek HE, Rimsza LM, Rosenthal AC. Clinical features and cell of origin subtyping using gene expression profiling in HIV-negative patients with primary central nervous system lymphoma. Blood. (2018) 132. doi: 10.1182/blood-2018-99-110660
63. Braggio E, Van Wier S, Ojha J, McPhail E, Asmann YW, Egan J, et al. Genome-wide analysis uncovers novel recurrent alterations in primary central nervous system lymphomas. Clin Cancer Res. (2015) 21:3986–94. doi: 10.1158/1078-0432.CCR-14-2116
64. Lionakis MS, Dunleavy K, Roschewski M, Widemann BC, Butman JA, Schmitz R, et al. Inhibition of B cell receptor signaling by ibrutinib in primary CNS lymphoma. Cancer Cell. (2017) 31:833–843.e5. doi: 10.1016/j.ccell.2017.04.012
65. Grommes C, Tang SS, Wolfe J, Kaley TJ, Daras M, Pentsova EI, et al. Phase 1b trial of an ibrutinib-based combination therapy in recurrent/refractory CNS lymphoma. Blood J Am Soc Hematol. (2019) 133:436–45. doi: 10.1182/blood-2018-09-875732
66. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. New Engl J Med. (2018) 378:1396–407. doi: 10.1056/NEJMoa1801445
67. Montesinos-Rongen M, Schmitz R, Brunn A, Gesk S, Richter J, Hong K, et al. Mutations of CARD11 but not TNFAIP3 may activate the NF-κB pathway in primary CNS lymphoma. Acta neuropathologica. (2010) 120:529–35. doi: 10.1007/s00401-010-0709-7
68. Jiang S, Li X, Xue W, Xia S, Wang J, Sai Y, et al. Preclinical pharmacokinetic characterization of amdizalisib, a novel PI3Kdelta inhibitor for the treatment of hematological Malignancies. Front Pharmacol. (2024) 15:1392209. doi: 10.3389/fphar.2024.1392209
69. Grommes C, Nayak L, Tun HW, Batchelor TT. Introduction of novel agents in the treatment of primary CNS lymphoma. Neuro Oncol. (2019) 21:306–13. doi: 10.1093/neuonc/noy193
70. Hoang-Xuan K, Deckert M, Ferreri AJM, Furtner J, Gallego Perez-Larraya J, Henriksson R, et al. European Association of Neuro-Oncology (EANO) guidelines for treatment of primary central nervous system lymphoma (PCNSL). Neuro Oncol. (2023) 25:37–53. doi: 10.1093/neuonc/noac196
71. Zou L, Qi Y, Tang L, Du Y, Xiang M, Chen X, et al. Clinical review considerations of class I PI3K inhibitors in hematolymphatic Malignancies by Center for Drug Evaluation. Chin J Cancer Res. (2022) 34:415–21. doi: 10.21147/j.issn.1000-9604.2022.04.09
72. Grommes C, Kaley TJ, Nolan C, Omuro AMP, Wolfe J, Mellinghoff IK, et al. Phase I study of single agent ibrutinib in recurrent/refractory primary (PCNSL) and secondary CNS lymphoma (SCNSL). American. Clin Oncol. (2016) 34(15). doi: 10.1200/JCO.2016.34.15_suppl.2046
73. Soussain C, Choquet S, Blonski M, Leclercq D, Houillier C, Rezai K, et al. Ibrutinib monotherapy for relapse or refractory primary CNS lymphoma and primary vitreoretinal lymphoma: Final analysis of the phase II ‘proof-of-concept’iLOC study by the Lymphoma study association (LYSA) and the French oculo-cerebral lymphoma (LOC) network. Eur J Cancer. (2019) 117:121–30. doi: 10.1016/j.ejca.2019.05.024
74. Wu C, de Miranda NF, Chen L, Wasik AM, Mansouri L, Jurczak W, et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: Impact of recurrent CARD11 mutations. Oncotarget. (2016) 7:38180. doi: 10.18632/oncotarget.v7i25
75. Grommes C, Gavrilovic I, Miller AM, Stone JB, Kaley T, Madzsar JT, et al. Phase Ib of copanlisib in combination with ibrutinib in recurrent/refractory primary CNS lymphoma (PCNSL). Blood. (2019) 134:1598. doi: 10.1182/blood-2019-126214
76. Grommes C, Piotrowski A, Pentsova E, Nolan C, Francis J, DeAngelis L, et al. Phase ib trial with dose expansion of the bruton's tyrosine kinase (BTK) inhibitor, ibrutinib, in combination with rituximab and lenalidomide in patients with Refractory/Recurrent primary central nervous system lymphoma (PCNSL) and Refractory/Recurrent secondary central nervous system lymphoma (SCNSL). Blood. (2020) 136:48. doi: 10.1182/blood-2020-143075
77. Roschewski M, Melani C, Lakhotia R, Pittaluga S, Phelan JD, Peer C, et al. Phase 1 study of escalating doses of ibrutinib and temozolomide, etoposide, liposomal doxorubicin, dexamethasone, rituximab (TEDDI-R) with isavuconazole for relapsed and refractory primary CNS lymphoma. Blood. (2020) 136:12–3. doi: 10.1182/blood-2020-137637
78. Renaud L, Bossard JB, Terriou L, Cambier N, Chanteau G, Carpentier B, et al. Treatment with temozolomide and ibrutinib in recurrent/refractory primary (PCNSL) and secondary CNS lymphoma (SCNSL). Blood (2020) 136. doi: 10.1182/blood-2020-138764
79. Ghesquieres H, Chevrier M, Laadhari M, Chinot O, Choquet S, Molucon-Chabrot C, et al. Lenalidomide in combination with intravenous rituximab (REVRI) in relapsed/refractory primary CNS lymphoma or primary intraocular lymphoma: a multicenter prospective ‘proof of concept’phase II study of the French Oculo-Cerebral lymphoma (LOC) Network and the Lymphoma Study Association (LYSA). Ann Oncol. (2019) 30:621–8. doi: 10.1093/annonc/mdz032
80. Rubenstein JL, Geng H, Fraser EJ, Formaker P, Chen L, Sharma J, et al. Phase 1 investigation of lenalidomide/rituximab plus outcomes of lenalidomide maintenance in relapsed CNS lymphoma. Blood Adv. (2018) 2:1595–607. doi: 10.1182/bloodadvances.2017014845
81. Tun HW, Johnston PB, DeAngelis LM, Atherton PJ, Pederson LD, Koenig PA, et al. Phase 1 study of pomalidomide and dexamethasone for relapsed/refractory primary CNS or vitreoretinal lymphoma. Blood J Am Soc Hematol. (2018) 132:2240–8. doi: 10.1182/blood-2018-02-835496
82. Zhang Y, Li Y, Zhuang Z, Wang W, Wei C, Zhao D, et al. Preliminary evaluation of zanubrutinib-containing regimens in DLBCL and the cerebrospinal fluid distribution of zanubrutinib: a 13-case series. Front Oncol. (2021) 11:760405. doi: 10.3389/fonc.2021.760405
83. Song J, Liu H, Jiao Z, Gao D, Ding K, Wang Y, et al. Zanubrutinib, lenalidomide, rituximab, temozolomide and methotrexate (RLZT ± MTX) as first-line treatment for newly diagnosed PCNSL: a prospective, open-lable, multicenter clinical trial. Blood. (2022) 140:3739–40. doi: 10.1182/blood-2022-164677
84. Ferreri AJM, Illerhaus G, Doorduijn JK, Auer DP, Bromberg JEC, Calimeri T, et al. Primary central nervous system lymphomas: EHA-ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. (2024) 35:491–507. doi: 10.1002/hem3.v8.6
85. Lin Z, Chen B, Ma J, Ma Y, Kang H. Investigation of high dose cytarabine plus zenubrutinib in relapsed/refractory primary central nervous system lymphoma. Blood. (2022) 140:12070–0. doi: 10.1182/blood-2022-158730
86. Okamura T, Hosoi H, Matsufusa T, Akagi Y, Iwamoto R, Kosako H, et al. Tirabrutinib maintenance therapy for a patient with high-dose methotrexate-ineligible primary central nervous system lymphoma. Ann Hematol. (2022) 101:1379–81. doi: 10.1007/s00277-021-04744-0
87. Kawasaki A, Matsushita M, Yoshida S, Matsukura T, Izutsu K, Nishikawa R, et al. Real-world evidence for the safety and effectiveness of tirabrutinib in 140 Japanese patients with recurrent or refractory primary central nervous system lymphoma: interim report of postmarketing surveillance. Blood. (2022) 140:6685–6. doi: 10.1182/blood-2022-156472
88. Wu J-J, Wang W-H, Dong M, Ma S-S, Zhang X-D, Zhu L-N, et al. Orelabrutinib-bruton tyrosine kinase inhibitor-based regimens in the treatment of central nervous system lymphoma: a retrospective study. Investigational New Drugs. (2022) 40:650–9. doi: 10.1007/s10637-022-01219-5
89. Cookson E, Smith LL. 2022 ASH annual meeting. Lancet Haematol. (2023) 10:e87–8. doi: 10.1016/S2352-3026(22)00401-X
90. Yang C, Cui Y, Ren X, Li M, Yu K, Shen S, et al. Orelabrutinib combined with lenalidomide and immunochemotherapy for relapsed/refractory primary central nervous system lymphoma: a retrospective analysis of case series. Front Oncol. (2022) 12:901797. doi: 10.3389/fonc.2022.901797
91. Ma J, He Q, Lu K, Wang L. A multicenter, single-arm, prospective phase II study of orelabrutinib combined with high-dose methotrexate and rituximab sequential autologous hematopoietic stem cell transplantation in newly-dignosed primary central nervous system lymphoma. Blood. (2022) 140:3744–5. doi: 10.1182/blood-2022-165720
92. Zeng R, Sun Z, Xiao L, Hui Z. Thiotepa, orelabrutinib, and methotrexate combined with or without the rituximab regimens in the treatment of patients with central nervous system lymphoma. Blood. (2022) 140:9536–7. doi: 10.1182/blood-2022-165970
93. Zhao S, Liu Y, Zhu Z, He J, Liu L, Wang X, et al. Orelabrutinib, rituximab, and high-dose methotrexate (HD-MTX) in newly diagnosed primary central nervous system lymphoma (PCNSL): a retrospective analysis on efficacy, safety, and biomarker. Blood. (2022) 140:1338–9. doi: 10.1182/blood-2022-165710
94. Zhang X, Wang W, Ma S, Fan M, Zhu L, Zhang L, et al. A phase I/II study of orelabrutinib combined with anti-programmed cell death protein-1 antibody and fotemustine for patients with newly diagnosed primary central nervous system lymphoma (PCNSL). Blood. (2022) 140:12075–6. doi: 10.1182/blood-2022-159383
95. Goodstal SM, Lin J, Crandall T, Crowley L, Bender AT, Pereira A, et al. Preclinical evidence for the effective use of TL-895, a highly selective and potent second-generation BTK inhibitor, for the treatment of B-cell Malignancies. Sci Rep. (2023) 13:20412. doi: 10.1038/s41598-023-47735-z
96. Jurczak W, Rule S, Townsend W, Tucker D, Sarholz B, Scheele J, et al. Phase I, first-in-human trial of Bruton's tyrosine kinase inhibitor M7583 in patients with B-cell Malignancies. Leuk Lymphoma. (2021) 62:2392–9. doi: 10.1080/10428194.2021.1913139
97. Huntington SF, Schuster SJ, Ding W, Koehler AB, Brander DM, Rosenthal AC, et al. DTRMWXHS-12, a novel Bruton tyrosine kinase inhibitor, in combination with everolimus and pomalidomide in patients with relapsed/refractory lymphomas: An open-label, multicenter, phase 1a/1b study. Am J Hematol. (2023) 98:739–49. doi: 10.1002/ajh.26888
98. Zhang D, Xu G, Zhao J, Wang Y, Wu X, He X, et al. Structure-activity relationship investigation for imidazopyrazole-3-carboxamide derivatives as novel selective inhibitors of Bruton's tyrosine kinase. Eur J Med Chem. (2021) 225:113724. doi: 10.1016/j.ejmech.2021.113724
99. Guo Y, Hu N, Liu Y, Zhang W, Yu D, Shi G, et al. Discovery of BGB-8035, a highly selective covalent inhibitor of bruton's tyrosine kinase for B-cell Malignancies and autoimmune diseases. J Med Chem. (2023) 66:4025–44. doi: 10.1021/acs.jmedchem.2c01938
100. Tichenor MS, Wiener JJM, Rao NL, Bacani GM, Wei J, Pooley Deckhut C, et al. Discovery of JNJ-64264681: A potent and selective covalent inhibitor of bruton's tyrosine kinase. J Med Chem. (2022) 65:14326–36. doi: 10.1021/acs.jmedchem.2c01026
101. Kawahata W, Asami T, Kiyoi T, Irie T, Kashimoto S, Furuichi H, et al. Discovery of AS-1763: A potent, selective, noncovalent, and orally available inhibitor of bruton's tyrosine kinase. J Med Chem. (2021) 64:14129–41. doi: 10.1021/acs.jmedchem.1c01279
102. Robbins DW, Noviski MA, Tan YS, Konst ZA, Kelly A, Auger P, et al. Discovery and preclinical pharmacology of NX-2127, an orally bioavailable degrader of bruton's tyrosine kinase with immunomodulatory activity for the treatment of patients with B cell Malignancies. J Med Chem. (2024) 67:2321–36. doi: 10.1021/acs.jmedchem.3c01007
103. Thieme E, Liu T, Bruss N, Roleder C, Lam V, Wang X, et al. Dual BTK/SYK inhibition with CG-806 (luxeptinib) disrupts B-cell receptor and Bcl-2 signaling networks in mantle cell lymphoma. Cell Death Dis. (2022) 13:246. doi: 10.1038/s41419-022-04684-1
Keywords: B-cell receptor signaling, Bruton’s tyrosine kinase (BTK) inhibitors, primary central nervous system lymphoma (PCNSL), mechanism, lymphoma
Citation: Xing Y, Zhao K, Zhang Y and Wang Y (2024) BTK inhibition in primary central nervous system lymphoma: mechanisms, clinical efficacy, and future perspectives. Front. Oncol. 14:1463505. doi: 10.3389/fonc.2024.1463505
Received: 12 July 2024; Accepted: 29 November 2024;
Published: 24 December 2024.
Edited by:
Mohamed A. Yassin, Qatar University, QatarReviewed by:
Walter Hanel, The Ohio State University, United StatesChunhua Ma, Henan Normal University, China
Copyright © 2024 Xing, Zhao, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongsheng Wang, d2FuZ3lzQHNjdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship