 Giuseppe Di Grazia1Chiara Conti1Sabrina Nucera1Gianmarco Motta2,3Federica Martorana2,3*Stefania Stella2,4Michele Massimino4,5Mario Giuliano6Paolo Vigneri2,3
Giuseppe Di Grazia1Chiara Conti1Sabrina Nucera1Gianmarco Motta2,3Federica Martorana2,3*Stefania Stella2,4Michele Massimino4,5Mario Giuliano6Paolo Vigneri2,3- 1Department of Human Pathology “G. Barresi”, University of Messina, Messina, Italy
- 2Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy
- 3University Oncology Department, Humanitas Istituto Clinico Catanese, Catania, Italy
- 4Center of Experimental Oncology and Hematology, Azienda Ospedaliera Universitaria (A.O.U.) Policlinico “G. Rodolico - S. Marco”, Catania, Italy
- 5Department of General Surgery and Medical-Surgical Specialties, University of Catania, Catania, Italy
- 6Department of Clinical Medicine and Surgery, University of Naples Federico II, Naples, Italy
The REarranged during Transfection (RET) receptor tyrosine kinase plays a crucial role in the development of various anatomical structures during embryogenesis and it is involved in many physiological cellular processes. This protein is also associated with the initiation of various cancer types, such as thyroid cancer, non-small cell lung cancer, and multiple endocrine neoplasms. In breast cancer, and especially in the estrogen receptor-positive (ER+) subtype, the activity of RET is of notable importance. Indeed, RET seems to be involved in tumor progression, resistance to therapies, and cellular proliferation. Nevertheless, the ways RET alterations could impact the prognosis of breast cancer and its response to treatment remain only partially elucidated. Several inhibitors of RET kinase have been developed thus far, with various degrees of selectivity toward RET inhibition. These molecules showed notable efficacy in the treatment of RET-driven tumors, including some breast cancer cases. Despite these encouraging results, further investigation is needed to fully understand the potential role RET inhibition in breast cancer. This review aims to recapitulate the existing evidence about the role of RET oncogene in breast cancer, from its pathogenic and potentially prognostic role, to the clinical applications of RET inhibitors.
1 Introduction
The REarranged during Transfection (RET) oncogene was first discovered in 1980 and subsequently identified as a pivotal cancerogenic determinant for papillary and medullary thyroid carcinomas and other malignancies, paving the way for new diagnostic and therapeutic strategies (1).
The RET oncogene is involved in the regulation of cell growth and differentiation, playing a crucial role in embryonic development and in the maintenance of tissue homeostasis in adults (2).
RET rearrangements or mutations determine aberrant activation of its catalytic activity, contributing to tumor formation and progression (3). In particular, dysfunctional activation of RET signaling induces uncontrolled activity of the MAPK and PI3K/AKT/mTOR pathways, ultimately leading to cell proliferation (4).
Given the key role of the RET oncogene in cancer, its inhibition has represented an attractive therapeutic strategy. Indeed, several multi-tyrosine kinase inhibitors have been developed over the years and primarily tested in different tumor types, including thyroid carcinoma, renal cancer and hepatocellular carcinoma, where they currently represent the standard of care (5–7). More recently, selective RET inhibitors showed remarkable activity in multiple cancer types sharing RET rearrangements (8). Hence, selective RET inhibitors currently represent an agnostic therapeutic option for patients whose tumor harbor a RET fusion (9, 10).
With more than 2.3 million new diagnoses and 685.000 deaths in 2020, breast cancer (BC) represents a major health concern worldwide (11). Despite the significant advances achieved in the treatment of this disease, new and effective therapeutic options are continuously investigated and tested, in search for further improvement of disease outcome (12). Indeed, RET inhibition may represent an exploitable target across different BC subtypes.
Aim of this review is to provide an overview of the physiological and pathological functions of the RET oncogene, focusing on its role in BC biology. We reviewed the available pre-clinical and clinical evidence concerning RET pharmacological inhibition in this disease, also highlighting future challenges and perspectives for targeting this oncogene in the era of personalized medicine.
2 RET physiological and pathological functions
The REarranged during Transfection proto-oncogene is located on chromosome 10q11.2 and encodes for a transmembrane tyrosine kinase receptor (RTK) with an extracellular, a transmembrane and an intracellular domain (13). The extracellular region has four cadherin-like domains, a calcium binding site and a cysteine-rich domain. The intracellular region consists of the active tyrosine kinase domain flanked by two regulatory regions: the juxta-membrane domain and a C-tail (13).
RET activation is triggered by glial cell line-derived neurotrophic factor (GDNF) family ligands (GFLs) such as GDNF, neurturin (NRTN), artemin (ARTN), or persephin (PSPN) that bind a glycosylphosphatidylinositol-anchored cell surface protein: GDNF family receptor-alfa (GFR-α) 1-4 (13). In turn, the GDNF-GFR α1 complex induces RET homodimerization, leading to auto-phosphorylation of its intracellular tyrosine residues (14, 15). These phosphorylated residues recruit several adaptor proteins, inducing the pleiotropic propagation of external stimuli. It has been clearly demonstrated that different residues activate distinct downstream pathways. For instance, RET-Y687 binds the SHP2 phosphatase, activating the PI3K/AKT pathway, while RET-Y752 and Y928 promote STAT3 phosphorylation, nuclear translocation and signaling. Moreover, RET-Y905 is critical for binding adaptor proteins Grb7/10 and RET-Y981 contributes to activation of the SRC kinase. Additionally, RET-Y1015 recruits phospholipase C-gamma (PLC-γ) thereby activating the protein kinase C (PKC) pathway, while RET-Y1062 recruits adaptor proteins which activate both the PI3K/AKT and MAPK pathways. Finally, RET-Y1096 binds Grb2 and activates the MAPK pathway (15) (Figure 1).
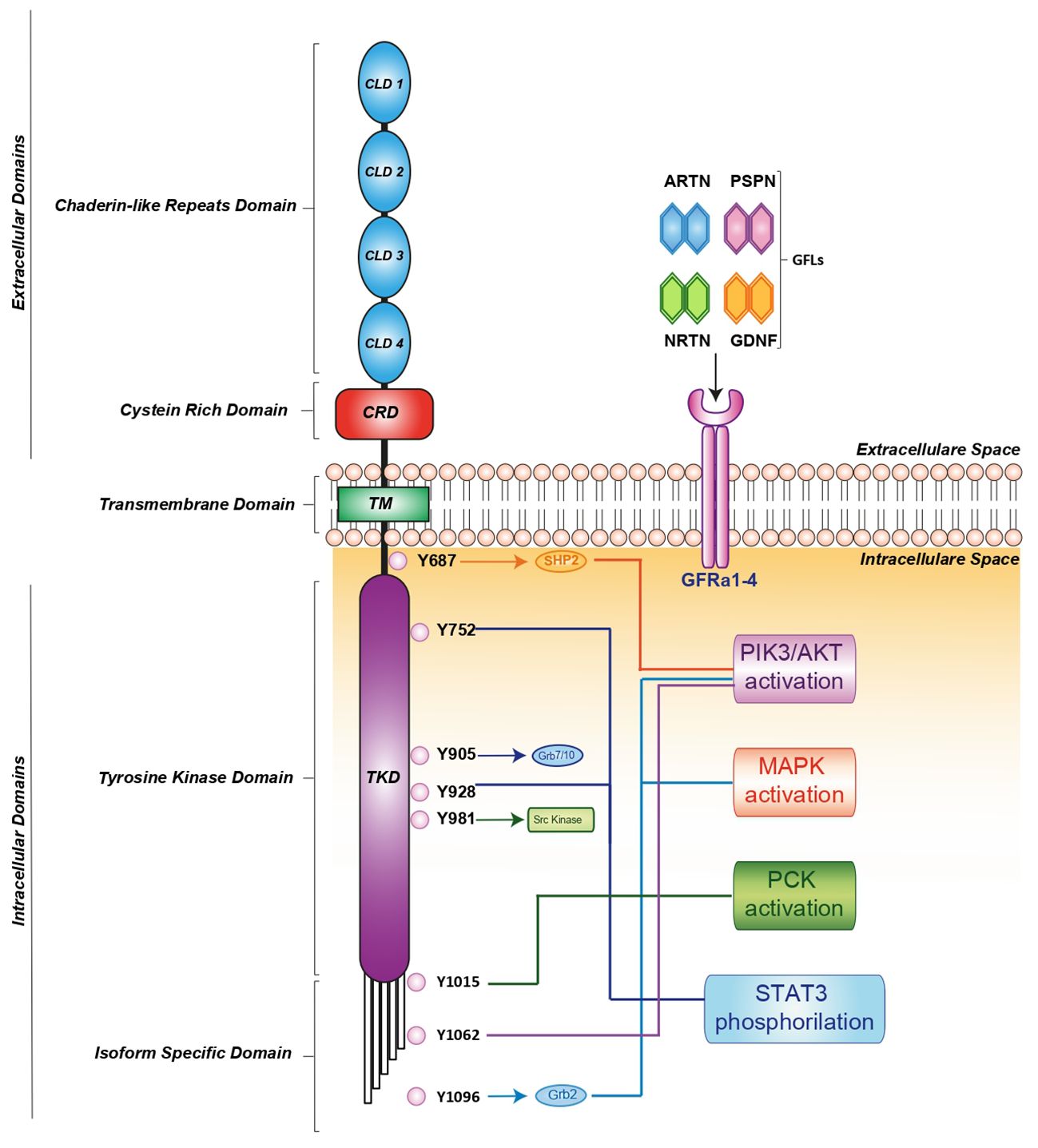
Figure 1 RET receptor structure, co-receptors, corresponding ligands and intracellular signaling pathway. RET activation is mediated by indirect interaction with one of four ligands: GDNF, ARTN, NRTN and PSPN linked to one of four GFRa1-4 co-receptors. The formation of the ligand/co-receptor/RET complex induces RET dimerization and triggers autophosphorylation at intracellular tyrosine residues leading the activation of downstream signaling pathways essential for cell growth, proliferation, survival, differentiation, or appetite control. The orange, light blue, blue and green arrows indicate the binding of adapter proteins. The light blue, blue, red, orange, green and violet lines indicate the activation of downstream signaling pathways. ARTN, Artemin; GDNF, glial cel-linederived neurotrophic factor; GFRa, GDNF family receptor-alfa; NRTN, Neurturin; PSPN, Persephin; RET, Rearranged during Transfection.
RET mRNA is overexpressed during the earliest phases of embryological development declining in the later phases of pregnancy. Indeed, a functional RET is necessary for renal embryogenesis and for the development of the sympathetic, parasympathetic and enteric nervous systems, allowing proper anatomical distribution of entero-endocrine and entero-chromaffin cells. Thus, intra-uterine RET loss may determine several malformations, including renal agenesia, severe kidney abnormalities, aberrant spermatogenesis or Hirschprung’s disease (16).
In adult life, high levels of RET can be found in the salivary glands, in the dopaminergic neurons of the substantia nigra and in the smooth muscle of the arterial wall, where they provide for proper functioning of the sympathetic nervous system. Lower RET expression can be observed in the heart, spleen, liver, kidneys, lungs, ovaries and testis (16).
Oncogenic RET activation mostly occurs because of chromosomal rearrangements or gene mutations that directly or indirectly activate its kinase domain (17).
Chromosomal rearrangements take place through the juxtaposition of 3’ RET sequences - encoding for the catalytic domain - with the 5’ sequences of other genes displaying a protein dimerization domain. Indeed, most RET fusions lack the transmembrane domain coding regions and give rise to a constitutively active protein. The most frequent rearrangements involve the coiled-coil domain containing 6 gene (CCDC6-RET), the nuclear receptor co-activator 4 gene (NCOA4-RET) and the kinesin family member 5B gene (KIF5B-RET) (15). RET fusions can be found in approximately 20% of papillary thyroid cancers, 1-2% of non-small-cell lung cancers (NSCLCs) and <1% of many other solid tumors, including ovarian, pancreatic, salivary and colorectal malignancies (18).
Single nucleotide variants in the RET sequence can be either inherited or somatic (Figure 2).
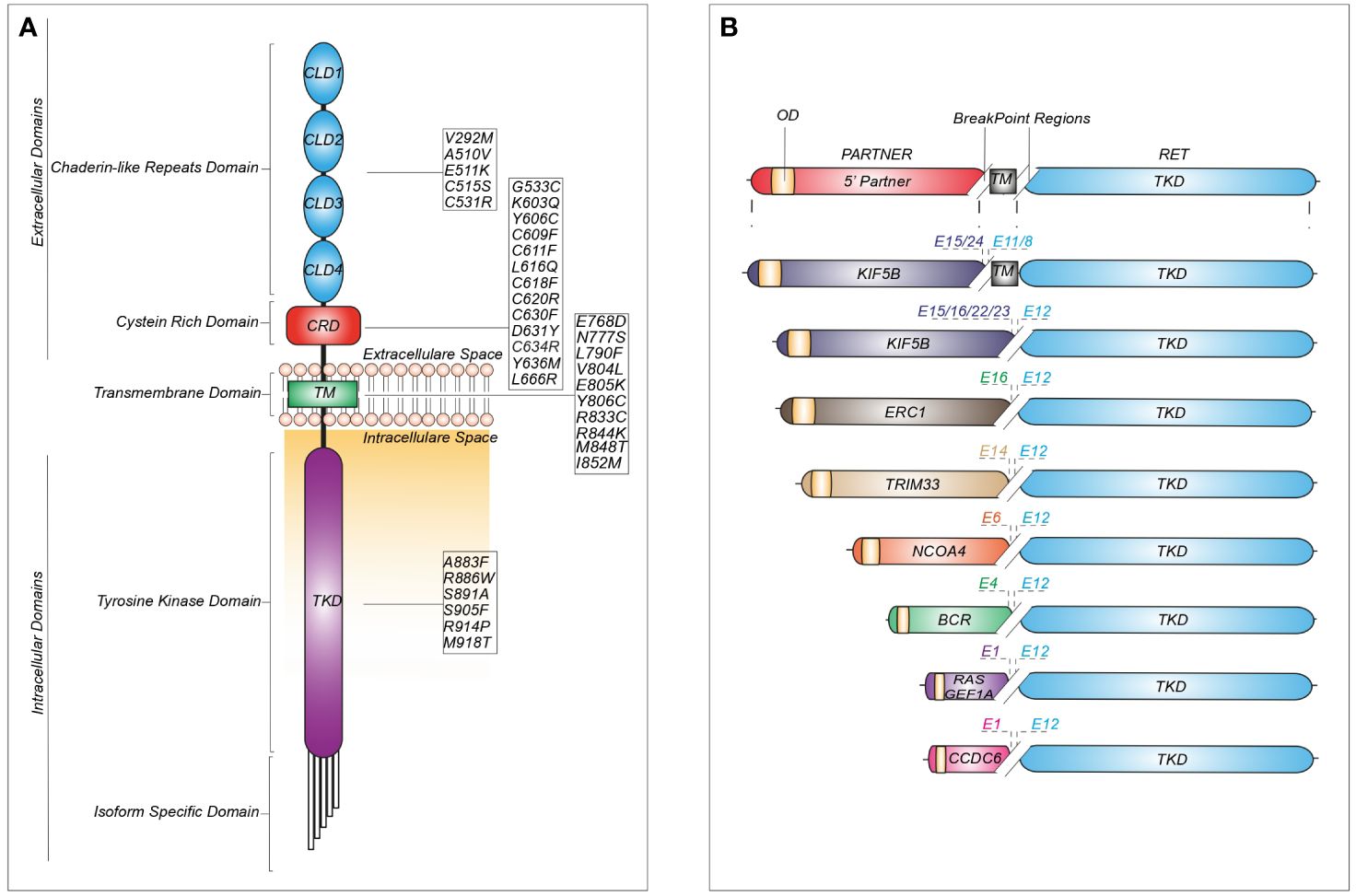
Figure 2 Schematic representation of recurrent RET mutations and rearrangements in cancer. (A) Structure of the RET protein reporting the most common mutations identified in cadherin-like repeats domains (CLDs), cystein-rich domain (CRD), transmembrane domain (TM) and tyrosine kinase domain (TKD). (B) RET fusions containing the most common upstream gene partners characterized by an oligomerization domain (OD) and different break-point regions dependent on the exon (E) involved in the generation of the chimeric junction.
Germline missense mutations are linked to autosomal dominant multiple endocrine neoplasia type 2 (MEN2), which is associated with an increased risk of medullary thyroid carcinoma (MTC), pheochromocytoma and other tumors (18). More than 95% of individuals with MEN2A display a germline mutation in RET exons 10 or 11, involving the cysteine-rich area of the extracellular domain. Indeed, these regions are prone to mutations which cause cysteine replacement with other amino acids. These modifications reduce the likelihood of generating intra-molecular disulphide-bonds, promoting instead inter-molecular covalent disulphide bonds between free cysteine residues of RET monomers, thereby increasing receptor dimerization and activation (19). Unlike MEN2A, MEN2B is associated with germline mutations in the kinase domain, such as the commonly reported substitutions M918T or A883F. Interestingly, the protein encoded by the RET-M918T variant can signal as a monomer, owing to increased ATP-binding affinity and altered protein conformation, leading to loss of kinase autoinhibition (20).
Somatic RET mutations are found in 65% of all sporadic MTCs. The majority of these tumors present the RET M918T mutation, although additional somatic substitutions, such as E768D and A883F, have also been described. Less frequently, RET mutations can be found in sporadic pheochromocytoma (15%) (20).
The increasing use of NGS platforms has uncovered both classical and novel RET mutations in other cancers, including RET-C634R in breast carcinoma, RET-E511K in endometrial and Merkel-cell carcinomas, RET-M918T in a paraganglioma and atypical lung carcinoid and RET-V804M in colorectal carcinoma, meningioma, gastrointestinal stromal tumors (GIST) and hepatoma (9) (Figure 2).
3 RET as an oncogene in breast cancer
In the last years, the potential role of RET in BC development and progression has been extensively investigated (Figure 3).
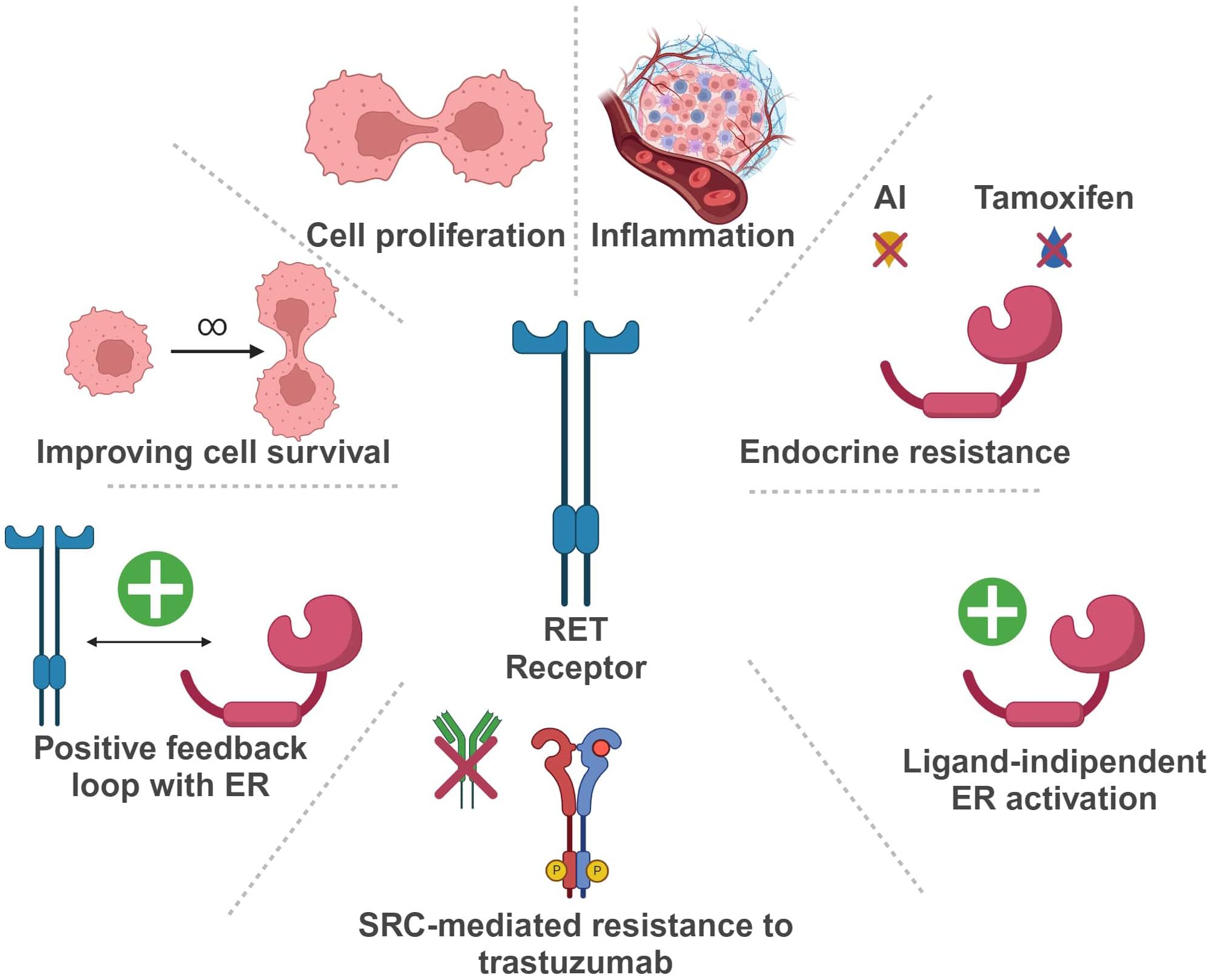
Figure 3 Schematic representation of RET oncogene role in Breast Cancer (Created with BioRender.com). RET, Rearranged during Transfection; ER, estrogen receptor; AI, aromatase inhibitor.
In BC, RET is more frequently overexpressed than rearranged or mutated. Indeed, high levels of RET RNA, with or without gene amplification, can be found in up to 40-60% of breast tumors, usually correlating with increased protein expression (21–25).
More in detail, in ER positive (ER+) BC, RET overexpression seems to be involved in tumorigenesis and resistance to endocrine therapy (26–30). In a study by Gattelli et al., chronic overexpression of wild-type RET (isoform 51) in the mammary gland promoted the development of ER+ BC in a transgenic mouse model (22). These tumors harbored intense MAPK and PI3K/AKT/mTOR signaling, underlying the relevance of these pathways in RET-driven BC, as reported in additional studies (24, 27, 30, 31). According to Wang et al., exposure of BC cells to estrogen enhances RET expression through ER and FOXA1 transcriptional activity, leading to the activation of downstream pathways and eventually promoting cell proliferation (32).
RET hyperexpression may also determine estrogen-independent activation of the estrogen receptor (ER)-α, leading to an endocrine resistant phenotype (26, 27, 33). Moreover, autocrine production of RET ligands, such as GDNF, by ER+ BC cells may be responsible for RET-mediated endocrine resistance, rather than RET overexpression itself (34). However, high levels of RET and GDNF in ER+ BC cells seem to be associated with estrogen-independent signaling activation via NTRK, KRAS and PI3K, and these features seemingly correlate with inferior survival outcomes (35). In turn, ER activation can upregulate both RET and GFR-α1, due to the presence of estrogen-responsive elements (EREs) in their promoters (36, 37). Other evidence supports a role for RET in the development of endocrine resistance. In an in-vitro endocrine resistant BC model, the GDNF/RET axis was strongly active and promoted cell survival (33, 38). Hyperactivation of this pathway may determine estrogen-independent ER functioning and promote the transcription of genes related to the immune response, such as STAT1 and STAT3 (22). Additionally, GDNF/RET signaling seems to induce a positive feedback loop with early growth response-1 (EGR1) that can cause endocrine resistance through the activation of cyclin D1 (39).
Zheng and colleagues described another positive feedback loop involving BRD4/ER-α-RET-ER-α in ER+ BC cell lines (40). In detail, estrogen receptor super-enhancers (ERSEs) seem to promote ER-α-induced carcinogenesis, regulating transcription of ER target genes, such as RET. Moreover, the bromodomain protein BRD4, which is a massive regulator of ERSEs, controls RET activation through ERSEs itself. On the other hand, RET activation induces the up-regulation of the RAS/RAF/MEK2/ERK/p90RSK/ER-α cascade, leading to a loop activation of ER-α, a crucial event for ER-α-induced gene transcription and development of a malignant phenotype (40).
On a different note, in ER+ BC cells endocrine therapy may exert a paradoxical effect on the GDNF/RET axis, increasing RET expression at both the RNA and protein level, a mechanism that seemingly relies on pro-inflammatory mediators (24, 41). Indeed, it has been reported that both fulvestrant and aromatase inhibitors can increase inflammatory cytokines levels (e.g. IL-6) (24, 41). Furthermore, the RET promoter displays binding sites for STAT and NFKB, that can stimulate IL-6 expression. In line with these observations, adding an anti-IL-6 antibody in BC cell culture reduces RET expression. These data reinforce the hypothesis that RET signaling can mediate endocrine resistance through inflammation mechanisms (31, 33).
Given its involvement in endocrine resistance, RET overexpression has controversially been proposed as a negative prognostic biomarker in ER+ BC. In a cohort of 93 ER+ BC patients, individuals carrying a RET polymorphism associated with lower RET expression (rs2435357C>T) displayed longer survival (42). Consistently, several reports have described an association between RET overexpression and a worse prognosis (31, 32). However, RET hyperexpression by immunohistochemistry did not predict inferior survival in two large cohorts of ER+ BC (32, 43).
RET overexpression can also be present in HER2-enriched (HER2+ve) and triple negative (TN) BC (6, 38). In HER2+ tumors, RET can cause resistance to anti-HER2 therapies, such as trastuzumab. Indeed, GDNF/RET signaling can reduce trastuzumab-induced apoptosis, eventually promoting cell survival and treatment resistance. In a xenograft model, the addition of recombinant GDNF (rGDNF) in cultures stimulates tumor growth in both trastuzumab-sensitive and -resistant cell lines (38). This effect is reversible upon SRC inhibition, suggesting a potential role for SRC in RET-driven resistance to trastuzumab (38).
The role of RET hyperexpression in TNBC is not fully elucidated. However, RET-directed TKIs seem to inhibit TNBC growth, providing preclinical evidence for the use of RET inhibitors in this subtype of breast carcinomas (6).
Besides RET overexpression, other genomic aberrations have been reported in BC. RET fusions are exceedingly rare (0.1%) but represent a potential therapeutic target for BC patients (44). The most common rearrangements are CCDC6-RET, NCOA4-RET and RasGEF domain family member 1A (RASFGEF1A)-RET (45). Additionally, new fusion partners have been recently identified, such as ELKS/RAB6-interacting/CAST family member 1 (ERC1)-RET, zinc finger protein 485 (ZNF485)-RET and Sperm Antigen with Calponin Homology and Coiled-Coil Domains 1 Like (SPECC1L)-RET (45, 46). Similarly, RET SNVs have been anecdotally described in breast tumors (0.2%) (17). They are usually missense mutations, such as C634R and M918T (responsible for MEN2A and MEN2B, respectively). Other substitutions, such as E511K, C611R, C620F, L633V, C634F and T636M, involve the extracellular domain, while V804M occurs in the kinase domain (45). These SNVs have been found more frequently in metastatic sites, rather than in primary tumors. Several in vitro studies have defined the pathways activated by these alterations that include MAPK, PI3K/AKT, mTOR, FAK and JAK/STAT (47). Other mutations, such as M918T, can maintain STAT3 constitutively active (48). Lastly, alterations involving non-coding RET sequences (e.g., promoter regions) that may influence the expression of TK domain coding exons have also been reported in BC (49–51), but their biological significance is still uncertain.
4 Targeting RET in breast cancer
4.1 Pre-clinical evidence
According to preclinical evidence, RET represents an actionable target in many solid tumors, including BC, and different RET inhibitors have been tested both in vitro and in xenograft models. However, most of the evidence available so far, regards mainly multi-kinase inhibitors, whose anti-RET activity cannot be easily assessed.
Among these, sunitinib (N-[2-(diethylamino)ethyl]-5-[(Z)-(5-fluoro-2-oxo-1H-indol-3-ylidene)methyl]-2,4-dimethyl-1H-pyrrole-3-carboxamide) reduced MAPK/ERK pathway activity in RET-expressing BC cell lines (7). Similarly, treatment with vandetanib reduced RET phosphorylation and activation, promoting tumor regression in BC patient-derived xenografts (PDXs) (6).
More recently, cabozantinib (1-N-[4-(6,7-dimethoxyquinolin-4-yl)oxyphenyl]-1-N’-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide) has been tested in RET overexpressing cell cultures and PDXs from resected brain metastases, determining a reduction of RET phosphorylation and inhibiting tumor growth (52).
RET inhibitors have also been tested in association with endocrine therapy in ER+ BC, with controversial results. The combination of both sunitinib or vandetanib and tamoxifen significantly reduced tumor growth in vivo (28, 30, 53). On the other hand, the addition of tamoxifen, letrozole or fulvestrant to NVP-AST487 (a multi-kinase RET-targeting inhibitor) did not show an additional benefit in xenograft models (24). However, these combinations seem to decrease disease metastatic index and tumor dissemination (24).
Limited preclinical evidence has been generated in BC models harboring RET rearrangements with the available evidence indicating that cabozantinib reduces the proliferative potential of breast cancer cells displaying the NCOA4-RET fusion (52).
Finally, an antibody-drug conjugate (ADC) encompassing a fully human anti-RET antibody (Y087) and the microtubule inhibitor maytansine (DM1) has been developed and tested both in vitro and in vivo. The compound showed signs of activity in BC xenograft models, with preliminary safety studies in primates suggesting neuropathy as the main on-target toxicity (54).
4.2 Clinical evidence
Given the encouraging preclinical results, RET inhibitors have been tested in BC patients, mostly in early phase trials (Table 1). Results are already available from studies with either multi-kinase inhibitors (anlotinib, cabozantinib, lenvatinib, vandetanib) or RET-specific inhibitors (selpercatinib), with several other trials still ongoing (Table 2).
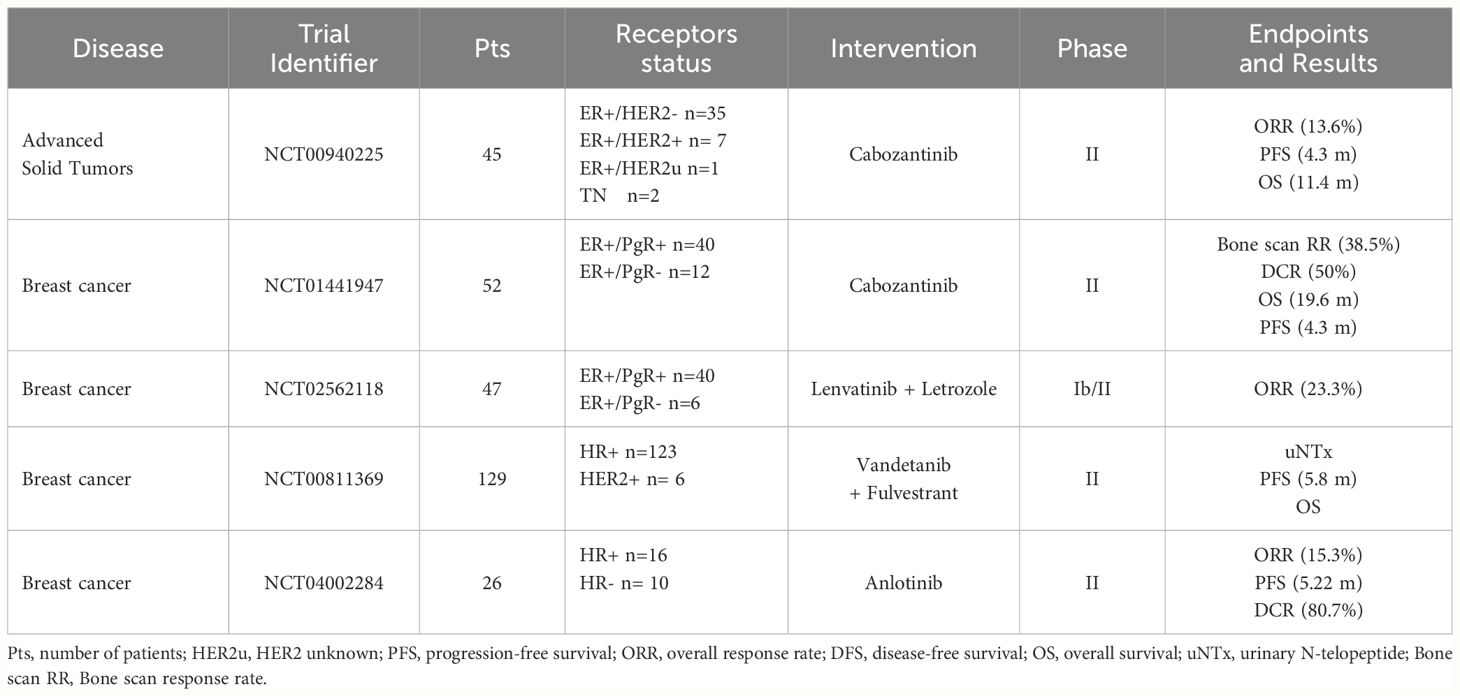
Table 1 Published clinical trials with RET-inhibitors including breast cancer patients.
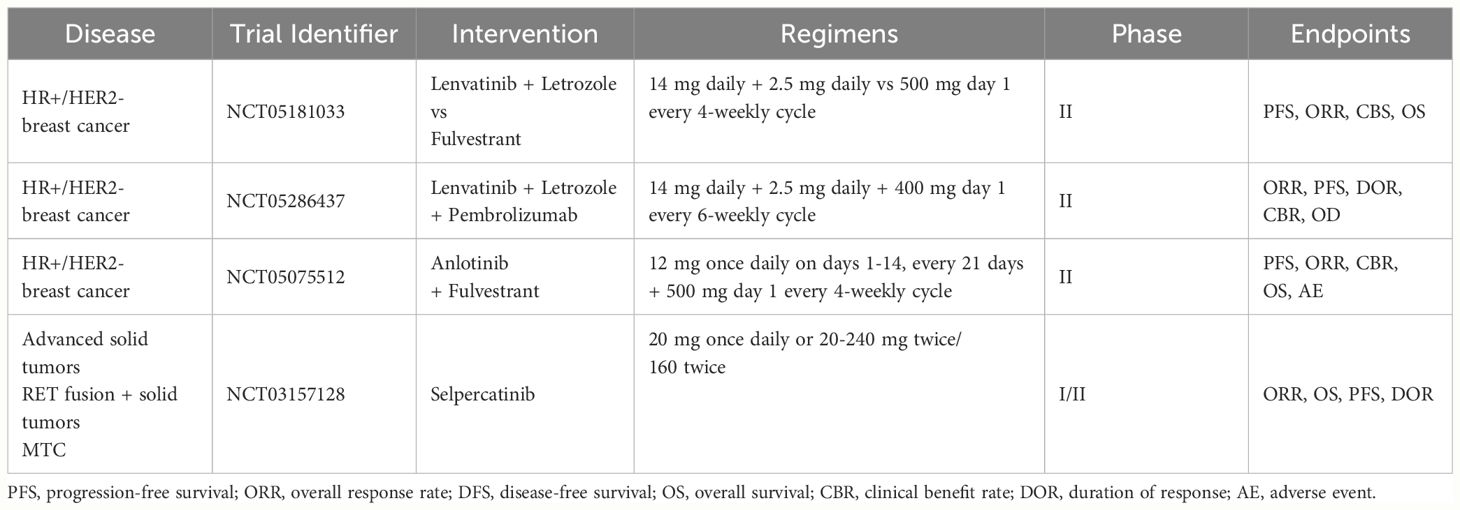
Table 2 Ongoing clinical trials with RET-inhibitors enrolling breast cancer patients.
4.2.1 Non-selective multi-kinase inhibitors
Anlotinib (1-[[4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxyquinolin-7-yl]oxymethyl]cyclopropan-1-amine) is a tyrosine kinase inhibitor with several targets, including RET, FGFR, c-KIT, PDGFR, and VEGFR (55, 56). The efficacy and safety of this inhibitor has been evaluated in a Chinese population with advanced solid tumors (55). In a single-arm phase II trial, 26 HER2- metastatic BC patients, unselected for the presence of RET alterations, received anlotinib after failing standard treatment options (56). Objective response rate (ORR), the primary endpoint, was 15.4%, while median PFS was 5.22 months. Overall, anlotinib activity was superimposable in ER+ and TNBC patients. Treatment was well tolerated with grade 3-4 adverse events (AEs) mostly ascribed to hypertension (26.9%) and hand-foot syndrome (3.8%) (56).
The multi-kinase inhibitor cabozantinib targets RET, MET, VEGFR2 and other receptor tyrosine kinases such as FLT3 and c-KIT (5). In a single-arm phase II trial, cabozantinib, at the dose of 100 mg or 60 mg once daily, showed clinical activity in 52 ER+ metastatic pre-treaded BC patients with bone metastases (with or without extra-osseous metastases) (57). Bone scan response rate, the primary endpoint, was 38.5%, while disease control rate (DCR) was 50%. Median PFS and OS were 4.3 and 19.6 months, respectively. No difference emerged between patients with bone-only and extra-osseous disease. Approximately 80% of patients receiving cabozantinib at 100 mg required a dose reduction. Most common adverse events were fatigue, hypertension, diarrhea and nausea (57). In another phase II trial conducted in different tumor types, cabozantinib was administered for a 12-week, open-label lead-in period. Patients with partial response at week 12 were kept on open label cabozantinib, while those with stable disease were randomized to cabozantinib or placebo. At the time of disease progression, treatment was unblinded and patients on cabozantinib discontinued it, while those on placebo resumed the TKI until a new progression occurred. Objective response rate (ORR) for the 12-week lead-in stage and PFS after randomization were the co-primary endpoints. Among the 45, heavily pretreated, BC patients enrolled (43 ER+ve, 2 TN) ORR at 12 weeks was 13.6% and DCR 46.7%. Median PFS was 4.3 months and median OS was 11.4 months. Most frequent adverse events of all grades were fatigue, palmar-plantar erythrodysesthesia, nausea and diarrhea (58). A published report describes the case of a 63-year-old woman with recurrent ER+/HER2+ BC and a NCOA4-RET rearrangement that achieved a clinical and radiological response to the combination of cabozantinib, trastuzumab and exemestane (45).
The multi-kinase inhibitor foretinib (1-N’-[3-fluoro-4-[6-methoxy-7-(3-morpholin-4-ylpropoxy)quinolin-4-yl]oxyphenyl]-1-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide) has been tested, in combination with lapatinib, in a phase Ib trial that enrolled 19 patients with advanced HER2+ BC. However, the TKI association showed limited activity (median PFS 3.2 months), and foretinib development has been halted (59).
Lenvatinib (4-[3-chloro-4-(cyclopropylcarbamoylamino)phenoxy]-7-methoxyquinoline-6-carboxamide) is another multi-kinase inhibitor with several targets, such as RET, FGFR, c-KIT, PDGFR, and VEGFR (60–62). In a phase Ib/II trial the combination of lenvatinib with letrozole was evaluated in 47 post-menopausal women with pre-treated advanced ER+/HER2- BC (63). Twenty-three percent of patients presented an objective response, with a median duration of 6.9 months and a median time-to-progression of 6.2 months. The potential correlation between RET expression by immunohistochemistry and clinical benefit was investigated. Despite a trend toward an increased efficacy in patients with higher RET expression, the association was not statistically significant.
Vandetanib (N-(4-bromo-2-fluorophenyl)-6-methoxy-7-[(1-methylpiperidin-4-yl)methoxy]quinazolin-4-amine) is a multi-kinase inhibitor with activity against and RET, EGFR1 and VEGFR2. A randomized phase II trial investigated the use of vandetanib or placebo with fulvestrant in 129 postmenopausal ER+ metastatic BC patients, with bone only or bone-predominant disease (64). Reduction of urinary N-telopeptide of type 1 collagen (uNTx) was the primary outcome of this study. Secondary outcomes included PFS, OS, pain response and number of skeletal-related events (SREs). The trial failed to show any statistically significant difference between treatment arms for both the primary and secondary endpoints, indicating no advantage of adding vandetanib to fulvestrant in this population (64).
4.2.2 Selective inhibitors
Selpercatinib (6-(2-hydroxy-2-methylpropoxy)-4-[6-[6-[(6-methoxypyridin-3-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]pyrazolo[1,5-a]pyridine-3-carbonitrile) is a selective inhibitor of wild-type, mutant and re-arranged RET. The efficacy of this drug has been explored in a phase I/II, multicenter, open-label, multicohort clinical trial (LIBRETTO-001) in patients with tumors harboring RET alterations (65, 66). Two patients with BC and RET fusions were enrolled in this trial, experiencing a partial and a complete response, respectively. The patient achieving a complete response was a 46-year-old pre-menopausal Japanese woman, with multiple nodal and lung metastases from ER+ BC at baseline, progressing to first line therapy with tamoxifen and goserelin (67). Next generation sequencing analysis performed on tumor tissue detected the CCDC6-RET fusion. Upon selpercatinib initiation, the patient experienced a rapid clinical improvement, along with a partial response, followed by a complete response after 3 months of treatment (67).
Another report described the case of a 36 years-old Chinese woman with heavily pretreated TNBC, displaying a CCDC6-RET fusion and achieving a partial response with the RET selective inhibitor pralsetinib (N-[(1S)-1-[6-(4-fluoropyrazol-1-yl)pyridin-3-yl]ethyl]-1-methoxy-4-[4-methyl-6-[(5-methyl-1H-pyrazol-3-yl)amino]pyrimidin-2-yl]cyclohexane-1-carboxamide) (68).
Several clinical trials are evaluating the efficacy of RET inhibitors in monotherapy or in combination with other agents in BC patients. Most of these early phase trials are testing a combination of multi-kinase inhibitors and hormonal therapy in patients with metastatic ER+/HER2- BC, progressing after a previous line of endocrine therapy.
5 Conclusions and future perspectives
The intricate role of the RET oncogene in BC biology has been extensively explored since the early nineties, with most findings generated in the ER+ subtype (3, 33, 34). As this body of evidence shows, in BC RET is frequently overexpressed rather than rearranged or mutated (21, 35, 69). Still, thus far a prognostic or predictive role for RET hyperexpression in breast malignancies has not been demonstrated.
In ER+ BC patients, alterations of RET signaling contribute to the onset and maintenance of endocrine resistance (29, 34, 39). Whether these alterations are also implicated in lack of response to other therapies, such as cyclin dependent kinase 4/6 (CDK4/6) inhibitors or ADCs, is unknown, and could represent a field for future investigations.
Data from clinical trials testing non-selective RET inhibitors in BC, either as monotherapy or in combination with endocrine therapy, are conflicting and results from ongoing trials are awaited (57, 60, 64, 70).. Among these, a phase II study is comparing the combination of lenvatinib and letrozole to fulvestrant in advanced ER+ BC patients who progressed to first line endocrine therapy plus a CDK4/6 inhibitor (NCT05181033). Another phase II trial is testing lenvatinib, pembrolizumab and letrozole in endocrine-resistant advanced BC (NCT05286437). The association of lenvatinib and pembrolizumab has proven effective in multiple cancer types, including some scarcely sensitive to immune-checkpoint inhibitors monotherapy (e.g., mismatch repair-proficient endometrial cancer) (71). Hence, a rationale exists to test this combination in ER+ BC. In general, a major pitfall of past and ongoing studies of non-selective RET inhibitors in BC is the lack of a predictive biomarker that could inform patient selection.
In the last years, the search for actionable molecular drivers has led to major breakthroughs in the treatment of several malignancies and to the definition of so-called agnostic targets, which offer the opportunity to tailor patient treatment to the disease molecular profile rather than to the tumor site of origin (72, 73). This is the case for NTRK rearrangements that can be addressed by specific inhibitors, and for tumor mutational burden (TMB) or mismatch repair deficiency (MMRd), that are predictive of immunotherapy efficacy (74–76). More recently, RET rearrangements have also been claimed as agnostic biomarkers and in 2022 the Food and Drug Administration approved the selective RET inhibitor selpercatinib for the treatment of patients with advanced solid tumors with RET gene fusions after the results of the phase I/II LIBRETTO trial (10).
Moreover, ADCs targeting RET or GFRA1, may represent another innovative approach in the future, although only preclinical evidence is available so far (54, 77).
Despite being exceedingly rare, RET rearrangements represent an invaluable therapeutic possibility for BC patients. Published reports confirm the activity of selective RET inhibitors across different BC subtypes in patients with advanced disease, progressing to several lines of standard treatments (9, 10). RET point mutations may also predict response to selective inhibitors, as already demonstrated in medullar and papillary thyroid cancer (65). However, whether the same activity is retained in BC displaying RET mutations has yet to be demonstrated.
Importantly, RET inhibitors, especially multi-kinase compounds, may induce different and potentially severe toxicities, such as hypertension and hemorrhagic or thrombotic events (7, 9, 10, 55, 56, 58, 59, 63, 64). Future studies evaluating these drugs must take into account strategies to mitigate and manage toxicity, including dose reduction or intermittent schedules, cardiological monitoring, evaluation of concurrent medications and comorbidity.
Overall, clinical application of RET inhibitors in BC is still in an early stage of development. Most of the existing evidence derive from small studies, and no recommendation exists about the RET testing and its timing in BC patients. Since RET inhibitors represent an off-label option for BC patients in most countries, with the exception of the United States where selpercatinib has received an agnostic indication, RET alterations could be tested in patients with advanced BC after progression to standard options, to check for potential eligibility in clinical trials.
Future researches should focus on: i) mechanistic studies elucidating the precise molecular mechanisms by which RET alterations drive BC progression and therapy resistance; ii) combinations between RET inhibitors and other compounds such as hormonal treatments and immunotherapies; iii) identification of reliable predictive biomarkers,to select patients who are most likely to benefit from RET-targeted therapies.
Understanding the role of the RET oncogene in BC biology may increase our ability to exploit its therapeutic potential in the era of personalized medicine. Future studies should elucidate the prognostic and predictive role of RET alterations in BC, with the aim of expanding the proportion of patients who may benefit from RET inhibition, eventually integrating them into the therapeutic armamentarium for BC patients.
Author contributions
GDG: Conceptualization, Writing – original draft. CC: Writing – original draft. SN: Writing – original draft. GM: Methodology, Writing – original draft. FM: Conceptualization, Methodology, Writing – review & editing. SS: Writing – review & editing. MM: Writing – review & editing. MG: Supervision, Writing – review & editing. PV: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Research Plan of the University of Catania PIA.CE.RI. - linea di intervento 2 (MD_RESETT_GLIO).
Conflict of interest
FM advisory role and honoraria from Amgen; Daiichi-Sankyo; Eli-Lilly; Gilead; GSK; Novartis; Pfizer; Roche. Support for travels and accommodations from Eli-Lilly; Gilead; Pfizer; Roche; Sophos. MG advisory role and honoraria from AstraZeneca, Daichii Sankyo, Eisai, Exact Sciences, Gilead, Lilly, Menarini Stemline; MSD, Novartis, Pfizer, Roche, Seagen. Support for travels and accommodations from Lilly, Pfizer, AstraZeneca. Research funding to the institution from AstraZeneca. PV advisory role and honoraria from Astra-Zeneca; Daiichi-Sankyo; Eli-Lilly; Gilead; Incyte; Istituto Gentili; Novartis; Pfizer; Roche; Seagen; Teva. Support for travel and accommodation from Daiichi-Sankyo; Eli-Lilly; Novartis. Research funding to the institution from Novartis and AstraZeneca.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, et al. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell. (1990) 60:557–63. doi: 10.1016/0092-8674(90)90659-3
2. Sawada H, Ibi M, Kihara T, Urushitani M, Nakanishi M, Akaike A, et al. Neuroprotective mechanism of glial cell line-derived neurotrophic factor in mesencephalic neurons. J Neurochem. (2000) 74:1175–84. doi: 10.1046/j.1471-4159.2000.741175.x
3. Pecar G, Liu S, Hooda J, Atkinson JM, Oesterreich S, Lee AV. RET signaling in breast cancer therapeutic resistance and metastasis. Breast Cancer Res. (2023) 25:26. doi: 10.1186/s13058-023-01622-7
4. Gild ML, Landa I, Ryder M, Ghossein RA, Knauf JA, Fagin JA. Targeting mTOR in RET mutant medullary and differentiated thyroid cancer cells. Endocr Relat Cancer. (2013) 20:659–67. doi: 10.1530/ERC-13-0085
5. Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. (2011) 10:2298–308. doi: 10.1158/1535-7163.MCT-11-0264
6. Hatem R, Labiod D, Château-Joubert S, de Plater L, El Botty R, Vacher S, et al. Vandetanib as a potential new treatment for estrogen receptor-negative breast cancers. Int J Cancer. (2016) 138:2510–21. doi: 10.1002/ijc.29974
7. Spanheimer PM, Lorenzen AW, De Andrade JP, Kulak MV, Carr JC, Woodfield GW, et al. Receptor tyrosine kinase expression predicts response to sunitinib in breast cancer. Ann Surg Oncol. (2015) 22:4287–94. doi: 10.1245/s10434-015-4597-x
8. Subbiah V, Gainor JF, Rahal R, Brubaker JD, Kim JL, Maynard M, et al. Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discovery. (2018) 8:836–49. doi: 10.1158/2159-8290.CD-18-0338
9. Subbiah V, Cassier PA, Siena S, Garralda E, Paz-Ares L, Garrido P, et al. Pan-cancer efficacy of pralsetinib in patients with RET fusion–positive solid tumors from the phase 1/2 ARROW trial. Nat Med. (2022) 28:1640–5. doi: 10.1038/s41591-022-01931-y
10. Subbiah V, Wolf J, Konda B, Kang H, Spira A, Weiss J, et al. Tumour-agnostic efficacy and safety of selpercatinib in patients with RET fusion-positive solid tumours other than lung or thyroid tumours (LIBRETTO-001): a phase 1/2, open-label, basket trial. Lancet Oncol. (2022) 23:1261–73. doi: 10.1016/S1470-2045(22)00541-1
11. Arnold M, Morgan E, Rumgay H, Mafra A, Singh D, Laversanne M, et al. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast. (2022) 66:15–23. doi: 10.1016/j.breast.2022.08.010
12. Gradishar WJ. Advances in the management of metastatic breast cancer. J Natl Compr Cancer Network. (2023) 21:1–4. doi: 10.6004/jnccn.2023.5006
13. Plaza-Menacho I, Mologni L, McDonald NQ. Mechanisms of RET signaling in cancer: Current and future implications for targeted therapy. Cell Signal. (2014) 26:1743–52. doi: 10.1016/j.cellsig.2014.03.032
14. Li AY, McCusker MG, Russo A, Scilla KA, Gittens A, Arensmeyer K, et al. RET fusions in solid tumors. Cancer Treat Rev. (2019) 81:101911. doi: 10.1016/j.ctrv.2019.101911
15. Regua AT, Najjar M, Lo H-W. RET signaling pathway and RET inhibitors in human cancer. Front Oncol. (2022) 12:932353. doi: 10.3389/fonc.2022.932353
16. Takahashi M, Kawai K, Asai N. Roles of the RET proto-oncogene in cancer and development. JMA J. (2020) 3:175–81. doi: 10.31662/jmaj.2020-0021
17. Kato S, Subbiah V, Marchlik E, Elkin SK, Carter JL, Kurzrock R. RET aberrations in diverse cancers: Next-generation sequencing of 4,871 patients. Clin Cancer Res. (2017) 23:1988–97. doi: 10.1158/1078-0432.CCR-16-1679
18. Drilon A, Hu ZI, Lai GGY, Tan DSW. Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat Rev Clin Oncol. (2018) 15:151–67. doi: 10.1038/nrclinonc.2017.175
19. Zhao L, Wang N, Zhang D, Jia Y, Kong F. A comprehensive overview of the relationship between RET gene and tumor occurrence. Front Oncol. (2023) 13:1090757. doi: 10.3389/fonc.2023.1090757
20. Lodish MB, Stratakis CA. RET oncogene in MEN2, MEN2B, MTC and other forms of thyroid cancer. Expert Rev Anticancer Ther. (2008) 8:625–32. doi: 10.1586/14737140.8.4.625
21. Gattelli A, Hynes NE, Schor IE, Vallone SA. Ret receptor has distinct alterations and functions in breast cancer. J Mammary Gland Biol Neoplasia. (2020) 25:13–26. doi: 10.1007/s10911-020-09445-4
22. Gattelli A, García Solá ME, Roloff TC, Cardiff RD, Kordon EC, Chodosh LA, et al. Chronic expression of wild-type Ret receptor in the mammary gland induces luminal tumors that are sensitive to Ret inhibition. Oncogene. (2018) 37:4046–54. doi: 10.1038/s41388-018-0235-y
23. Lo Nigro C, Rusmini M, Ceccherini I. RET in breast cancer: pathogenic implications and mechanisms of drug resistance. Cancer Drug Resistance. (2019) 2:1136–52. doi: 10.20517/cdr.2019.66
24. Gattelli A, Nalvarte I, Boulay A, Roloff TC, Schreiber M, Carragher N, et al. Ret inhibition decreases growth and metastatic potential of estrogen receptor positive breast cancer cells. EMBO Mol Med. (2013) 5:1335–50. doi: 10.1002/emmm.201302625
25. Zhou L, Li J, Zhang X, Xu Z, Yan Y, Hu K. An integrative pan cancer analysis of RET aberrations and their potential clinical implications. Sci Rep. (2022) 12:13913. doi: 10.1038/s41598-022-17791-y
26. Boulay A, Breuleux M, Stephan C, Fux C, Brisken C, Fiche M, et al. The ret receptor tyrosine kinase pathway functionally interacts with the ERα Pathway in breast cancer. Cancer Res. (2008) 68:3743–51. doi: 10.1158/0008-5472.CAN-07-5100
27. Spanheimer PM, Woodfield GW, Cyr AR, Kulak MV, White-Baer LS, Bair TB, et al. Expression of the RET proto-oncogene is regulated by TFAP2C in breast cancer independent of the estrogen receptor. Ann Surg Oncol. (2013) 20:2204–12. doi: 10.1245/s10434-012-2570-5
28. Spanheimer PM, Park J-M, Askeland RW, Kulak MV, Woodfield GW, De Andrade JP, et al. Inhibition of RET increases the efficacy of antiestrogen and is a novel treatment strategy for luminal breast cancer. Clin Cancer Res. (2014) 20:2115–25. doi: 10.1158/1078-0432.CCR-13-2221
29. Plaza-Menacho I, Morandi A, Robertson D, Pancholi S, Drury S, Dowsett M, et al. Targeting the receptor tyrosine kinase RET sensitizes breast cancer cells to tamoxifen treatment and reveals a role for RET in endocrine resistance. Oncogene. (2010) 29:4648–57. doi: 10.1038/onc.2010.209
30. Spanheimer PM, Cyr AR, Gillum MP, Woodfield GW, Askeland RW, Weigel RJ. Distinct pathways regulated by RET and estrogen receptor in luminal breast cancer demonstrate the biological basis for combination therapy. Ann Surg. (2014) 259:793–9. doi: 10.1097/SLA.0b013e3182a6f552
31. Esseghir S, Todd SK, Hunt T, Poulsom R, Plaza-Menacho I, Reis-Filho JS, et al. A role for glial cell-derived neurotrophic factor-induced expression by inflammatory cytokines and RET/GFRα1 receptor up-regulation in breast cancer. Cancer Res. (2007) 67:11732–41. doi: 10.1158/0008-5472.CAN-07-2343
32. Wang C, Mayer JA, Mazumdar A, Brown PH. The rearranged during transfection/papillary thyroid carcinoma tyrosine kinase is an estrogen-dependent gene required for the growth of estrogen receptor positive breast cancer cells. Breast Cancer Res Treat. (2012) 133:487–500. doi: 10.1007/s10549-011-1775-9
33. Morandi A, Martin LA, Gao Q, Pancholi S, Mackay A, Robertson D, et al. GDNF-RET signaling in ER-positive breast cancers is a key determinant of response and resistance to aromatase inhibitors. Cancer Res. (2013) 73:3783–95. doi: 10.1158/0008-5472.CAN-12-4265
34. Horibata S, Rice EJ, Mukai C, Marks BA, Sams K, Zheng H, et al. ER-positive breast cancer cells are poised for RET-mediated endocrine resistance. PLoS One. (2018) 13:e0194023. doi: 10.1371/journal.pone.0194023
35. Kakati RT, Kim H, Whitman A, Spanheimer PM. High expression of the RET receptor tyrosine kinase and its ligand GDNF identifies a high-risk subset of estrogen receptor positive breast cancer. Breast Cancer Res Treat. (2023) 199:589–601. doi: 10.1007/s10549-023-06937-9
36. Stine ZE, McGaughey DM, Bessling SL, Li S, McCallion AS. Steroid hormone modulation of RET through two estrogen responsive enhancers in breast cancer. Hum Mol Genet. (2011) 20:3746–56. doi: 10.1093/hmg/ddr291
37. Lin C-Y, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M, et al. Whole-genome cartography of estrogen receptor α Binding sites. PLoS Genet. (2007) 3:e87. doi: 10.1371/journal.pgen.0030087
38. Gardaneh M, Shojaei S, Kaviani A, Behnam B. GDNF induces RET–SRC–HER2-dependent growth in trastuzumab-sensitive but SRC-independent growth in resistant breast tumor cells. Breast Cancer Res Treat. (2017) 162:231–41. doi: 10.1007/s10549-016-4078-3
39. Marks BA, Pipia IM, Mukai C, Horibata S, Rice EJ, Danko CG, et al. GDNF-RET signaling and EGR1 form a positive feedback loop that promotes tamoxifen resistance via cyclin D1. BMC Cancer. (2023) 23:138. doi: 10.1186/s12885-023-10559-1
40. Zheng ZZ, Xia L, Hu GS, Liu JY, Hu YH, Chen YJ, et al. Super-enhancer-controlled positive feedback loop BRD4/ERα-RET-ERα promotes ERα-positive breast cancer. Nucleic Acids Res. (2022) 50:10230–48. doi: 10.1093/nar/gkac778
41. Morandi A, Isacke CM. Targeting RET–interleukin-6 crosstalk to impair metastatic dissemination in breast cancer. Breast Cancer Res. (2014) 16:301. doi: 10.1186/bcr3608
42. Griseri P, Garrone O, Lo Sardo A, Monteverde M, Rusmini M, Tonissi F, et al. Genetic and epigenetic factors affect RET gene expression in breast cancer cell lines and influence survival in patients. Oncotarget. (2016) 7:26465–79. doi: 10.18632/oncotarget.v7i18
43. Pavanelli AC, Mangone FR, Yoganathan P, Bessa SA, Nonogaki S, de Toledo Osório CAB, et al. Comprehensive immunohistochemical analysis of RET, BCAR1, and BCAR3 expression in patients with Luminal A and B breast cancer subtypes. Breast Cancer Res Treat. (2022) 192:43–52. doi: 10.1007/s10549-021-06452-9
44. Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. (2014) 5:4846. doi: 10.1038/ncomms5846
45. Paratala BS, Chung JH, Williams CB, Yilmazel B, Petrosky W, Williams K, et al. RET rearrangements are actionable alterations in breast cancer. Nat Commun. (2018) 9:4821. doi: 10.1038/s41467-018-07341-4
46. Ross DS, Liu B, Schram AM, Razavi P, Lagana SM, Zhang Y, et al. Enrichment of kinase fusions in ESR1 wild-type, metastatic breast cancer revealed by a systematic analysis of 4854 patients. Ann Oncol. (2020) 31:991–1000. doi: 10.1016/j.annonc.2020.04.008
47. Plaza-Menacho I, Morandi A, Mologni L, Boender P, Gambacorti-Passerini C, Magee AI, et al. Focal adhesion kinase (FAK) binds RET kinase via its FERM domain, priming a direct and reciprocal RET-FAK transactivation mechanism. J Biol Chem. (2011) 286:17292–302. doi: 10.1074/jbc.M110.168500
48. Plaza Menacho I, Koster R, van der Sloot AM, Quax WJ, Osinga J, van der Sluis T, et al. RET-familial medullary thyroid carcinoma mutants Y791F and S891A activate a src/JAK/STAT3 pathway, independent of glial cell line–derived neurotrophic factor. Cancer Res. (2005) 65:1729–37. doi: 10.1158/0008-5472.CAN-04-2363
49. Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. (2017) 45:D777–83. doi: 10.1093/nar/gkw1121
50. Rheinbay E, Parasuraman P, Grimsby J, Tiao G, Engreitz JM, Kim J, et al. Recurrent and functional regulatory mutations in breast cancer. Nature. (2017) 547:55–60. doi: 10.1038/nature22992
51. Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day INM, et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. (2015) 31:1536–43. doi: 10.1093/bioinformatics/btv009
52. Varešlija D, Priedigkeit N, Fagan A, Purcell S, Cosgrove N, O’Halloran PJ, et al. Transcriptome characterization of matched primary breast and brain metastatic tumors to detect novel actionable targets. JNCI: J Natl Cancer Institute. (2019) 111:388–98. doi: 10.1093/jnci/djy110
53. Andreucci E, Francica P, Fearns A, Martin L-A, Chiarugi P, Isacke CM, et al. Targeting the receptor tyrosine kinase RET in combination with aromatase inhibitors in ER positive breast cancer xenografts. Oncotarget. (2016) 7:80543–53. doi: 10.18632/oncotarget.v7i49
54. Nguyen M, Miyakawa S, Kato J, Mori T, Arai T, Armanini M, et al. Preclinical efficacy and safety assessment of an antibody–drug conjugate targeting the c-RET proto-oncogene for breast carcinoma. Clin Cancer Res. (2015) 21:5552–62. doi: 10.1158/1078-0432.CCR-15-0468
55. Sun Y, Niu W, Du F, Du C, Li S, Wang J, et al. Safety, pharmacokinetics, and antitumor properties of anlotinib, an oral multi-target tyrosine kinase inhibitor, in patients with advanced refractory solid tumors. J Hematol Oncol. (2016) 9:105. doi: 10.1186/s13045-016-0332-8
56. Hu N, Si Y, Yue J, Sun T, Wang X, Jia Z, et al. Anlotinib has good efficacy and low toxicity: a phase II study of anlotinib in pre-treated HER-2 negative metastatic breast cancer. Cancer Biol Med. (2021) 18:849–59. doi: 10.20892/j.issn.2095-3941.2020.0463
57. Xu J, Higgins MJ, Tolaney SM, Come SE, Smith MR, Fornier M, et al. A phase II trial of cabozantinib in hormone receptor-positive breast cancer with bone metastases. Oncologist. (2020) 25:652–60. doi: 10.1634/theoncologist.2020-0127
58. Tolaney SM, Nechushtan H, Ron I-G, Schöffski P, Awada A, Yasenchak CA, et al. Cabozantinib for metastatic breast carcinoma: results of a phase II placebo-controlled randomized discontinuation study. Breast Cancer Res Treat. (2016) 160:305–12. doi: 10.1007/s10549-016-4001-y
59. Chia SK, Ellard SL, Mates M, Welch S, Mihalcioiu C, Miller WH, et al. A phase-I study of lapatinib in combination with foretinib, a c-MET, AXL and vascular endothelial growth factor receptor inhibitor, in human epidermal growth factor receptor 2 (HER-2)-positive metastatic breast cancer. Breast Cancer Res. (2017) 19:54. doi: 10.1186/s13058-017-0836-3
60. Kudo M, Finn RS, Qin S, Han K-H, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. (2018) 391:1163–73. doi: 10.1016/S0140-6736(18)30207-1
61. Schlumberger M, Tahara M, Wirth LJ, Robinson B, Brose MS, Elisei R, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. New Engl J Med. (2015) 372:621–30. doi: 10.1056/NEJMoa1406470
62. Motzer R, Alekseev B, Rha S-Y, Porta C, Eto M, Powles T, et al. Lenvatinib plus pembrolizumab or everolimus for advanced renal cell carcinoma. New Engl J Med. (2021) 384:1289–300. doi: 10.1056/NEJMoa2035716
63. Lim JSJ, Wong ALA, Ow SGW, Ngoi NYL, Chan GHJ, Ang YLE, et al. Phase ib/II dose expansion study of lenvatinib combined with letrozole in postmenopausal women with hormone receptor–positive breast cancer. Clin Cancer Res. (2022) 28:2248–56. doi: 10.1158/1078-0432.CCR-21-4179
64. Clemons MJ, Cochrane B, Pond GR, Califaretti N, Chia SKL, Dent RA, et al. Randomised, phase II, placebo-controlled, trial of fulvestrant plus vandetanib in postmenopausal women with bone only or bone predominant, hormone-receptor-positive metastatic breast cancer (MBC): the OCOG ZAMBONEY study. Breast Cancer Res Treat. (2014) 146:153–62. doi: 10.1007/s10549-014-3015-6
65. Wirth LJ, Sherman E, Robinson B, Solomon B, Kang H, Lorch J, et al. Efficacy of selpercatinib in RET-altered thyroid cancers. New Engl J Med. (2020) 383:825–35. doi: 10.1056/NEJMoa2005651
66. Drilon A, Oxnard GR, Tan DSW, Loong HHF, Johnson M, Gainor J, et al. Efficacy of selpercatinib in RET fusion–positive non–small-cell lung cancer. New Engl J Med. (2020) 383:813–24. doi: 10.1056/NEJMoa2005653
67. Watanabe S, Takeda M, Otani T, Yoshida T, Sakai K, Olek E, et al. Complete response to selective RET inhibition with selpercatinib (LOXO-292) in a patient with RET fusion–positive breast cancer. JCO Precis Oncol. (2021) 5:103–6. doi: 10.1200/PO.20.00282
68. Zhao J, Xu W, Zhuo X, Liu L, Zhang J, Jiang F, et al. Response to pralsetinib in multi-drug-resistant breast cancer with CCDC6-RET mutation. Oncologist. (2023) 28:e416–24. doi: 10.1093/oncolo/oyad115
69. Mechera R, Soysal SD, Piscuoglio S, Ng CKY, Zeindler J, Mujagic E, et al. Expression of RET is associated with Oestrogen receptor expression but lacks prognostic significance in breast cancer 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis. BMC Cancer. (2019) 19:41. doi: 10.1186/s12885-018-5262-0
70. Gordon MS, Vogelzang NJ, Schoffski P, Daud A, Spira AI, O’Keeffe BA, et al. Activity of cabozantinib (XL184) in soft tissue and bone: Results of a phase II randomized discontinuation trial (RDT) in patients (pts) with advanced solid tumors. J Clin Oncol. (2011) 29:3010–0. doi: 10.1200/jco.2011.29.15_suppl.3010
71. Makker V, Colombo N, Casado Herráez A, Santin AD, Colomba E, Miller DS, et al. Lenvatinib plus pembrolizumab for advanced endometrial cancer. New Engl J Med. (2022) 386:437–48. doi: 10.1056/NEJMoa2108330
72. Gouda MA, Nelson BE, Buschhorn L, Wahida A, Subbiah V. Tumor-agnostic precision medicine from the AACR GENIE database: Clinical implications. Clin Cancer Res. (2023) 29:2753–60. doi: 10.1158/1078-0432.CCR-23-0090
73. Tateo V, Marchese PV, Mollica V, Massari F, Kurzrock R, Adashek JJ. Agnostic approvals in oncology: getting the right drug to the right patient with the right genomics. Pharmaceuticals. (2023) 16:614. doi: 10.3390/ph16040614
74. Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. (2018) 15:731–47. doi: 10.1038/s41571-018-0113-0
75. Strickler JH, Hanks BA, Khasraw M. Tumor mutational burden as a predictor of immunotherapy response: Is more always better? Clin Cancer Res. (2021) 27:1236–41. doi: 10.1158/1078-0432.CCR-20-3054
76. Zhao P, Li L, Jiang X, Li Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J Hematol Oncol. (2019) 12:54. doi: 10.1186/s13045-019-0738-1
Keywords: breast cancer, RET oncogene, targeted therapy, clinical trials, TKI - tyrosine kinase inhibitor
Citation: Di Grazia G, Conti C, Nucera S, Motta G, Martorana F, Stella S, Massimino M, Giuliano M and Vigneri P (2024) REThinking the role of the RET oncogene in breast cancer. Front. Oncol. 14:1427228. doi: 10.3389/fonc.2024.1427228
Received: 03 May 2024; Accepted: 19 July 2024;
Published: 01 August 2024.
Edited by:
Isabella Castellano, University of Turin, ItalyReviewed by:
Shiva Prasad Kollur, Amrita Vishwa Vidyapeetham, IndiaDharmendra Kumar Yadav, Gachon University, Republic of Korea
Copyright © 2024 Di Grazia, Conti, Nucera, Motta, Martorana, Stella, Massimino, Giuliano and Vigneri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Federica Martorana, federica.martorana@unict.it