 Maria Danielsen1
Maria Danielsen1 Thomas Emmanuel1
Thomas Emmanuel1 Morten Muhlig Nielsen2
Morten Muhlig Nielsen2 Lise Maria Lindahl1
Lise Maria Lindahl1 Maria Gluud3
Maria Gluud3 Niels Ødum3
Niels Ødum3 Line Raaby2,4Torben Steiniche4
Line Raaby2,4Torben Steiniche4 Lars Iversen1Rikke Bech1
Lars Iversen1Rikke Bech1 Terkild Brink Buus3Claus Johansen1*
Terkild Brink Buus3Claus Johansen1*- 1Department of Dermatology, Aarhus University Hospital, Aarhus, Denmark
- 2Department of Molecular Medicine (MOMA), Aarhus University Hospital, Aarhus, Denmark
- 3Skin Immunology Research Center, Department of Immunology & Microbiology, University of Copenhagen, Copenhagen, Denmark
- 4Department of Pathology, Aarhus University Hospital, Aarhus, Denmark
Introduction: The majority of patients with mycosis fungoides (MF) have an indolent disease course, but a substantial fraction (20-30%) of patients progress to advanced stages – usually with a grave prognosis. Early differentiation between indolent and aggressive types of MF is important for the choice of treatment regimen and monitoring of the individual patient. Good biomarkers are therefore desired.
Methods: Here, we used spatial transcriptomics on skin samples at time-of-diagnosis to enable prediction of patients who later progressed to advanced stages of MF. Formalin-fixed, paraffin-embedded skin biopsies at time of diagnosis from six patients with MF who progressed to advanced stages of disease within 4 months to 12 years after diagnosis, and nine patients who remained in early-stage disease over 9 to 27 years were analyzed using the GeoMx Digital Spatial Profiler to capture spatially resolved high-plex RNA gene expression data. Five different regions of interest (the epidermis, the basal layer of epidermis, CD4+ T-cells and neighboring cells, and Pautrier’s microabscesses) were profiled for further assessment.
Results and discussion: Interestingly, RUNX2, SHMT2, and MCM7 were upregulated in the enriched population of malignant T-cells in Pautrier’s microabscesses in patients who later developed advanced stages of disease. Expression of RUNX2, SHMT2 and MCM7 in malignant T-cells was confirmed in a subset of patients in MF skin using scRNA-seq datasets across multiple studies and correlating with stage of disease. Taken together, we provide first evidence that RUNX2 has potential as a biomarker to identify MF patients progressing to advanced stage disease. As RUNX2 has not previously been linked to MF, our data also shows the analytical strength of combining spatial transcriptomics with scRNA-seq analysis.
1 Introduction
Mycosis fungoides (MF) is the most common form of cutaneous T-cell lymphoma (CTCL), which is defined as non-Hodgkin lymphoma that presents in the skin without evidence of extracutaneous disease at the time of diagnosis (1). MF is histopathologically characterized by expansion of malignant skin homing CD4+ T-cells in the epidermal layer. However, a histopathologic diagnosis of MF can be difficult to obtain, as the histology of early MF can be non-specific and look like eczema or other inflammatory skin diseases. The clinical manifestations of MF can differ from patient to patient and rely to some extent on the malignant CD4+ T-cell clone and on the cellular and molecular composition of the surrounding microenvironment. Clinically, the diversity manifests in patients with respect to response to therapy and prognosis. MF ranges from localized early-stage disease (IA-IIA) to advanced stages (IIB-IV) with extracutaneous spread and distant metastases (2). In most patients, the disease evolves from few erythematous patches (patch stage) which can develop into more infiltrated plaques (plaque stage). In the patch and limited plaque stage, the prognosis is favorable. However, in a subset of patients, large and often ulcerated tumors (tumor stage) or erythroderma develop. Advanced stage MF is aggressive and associated with increased mortality (3).
MF associated mortality is highest during the first five years after diagnosis, indicating that a subset of patients has an aggressive form of MF (4). Five-year overall survival rates drop significantly with advanced stage disease from 85.8% and 62.2% in stage IB and IIB, respectively, to 23.3% in stage IVB (5). In recent years, improvements in diagnostic tools and treatment modalities have led to better outcomes for patients with MF, and it is evident that age at diagnosis, disease-propagation, treatment, and prolongation of time-to-progression plays a significant role in outcome for these patients (3). Despite prior emerging prognostic markers in MF (4, 6–12), it is still not possible to risk-stratify patients with newly-diagnosed MF in a clinical setting, nor to differentiate the subset of patients at increased risk of developing advanced stage disease. Conventional diagnostic tools, including immunohistochemistry (IHC), bulk DNA sequencing, or RNA expression analysis, do not measure intra-patients and intra-tumor heterogeneity to its full extent, either because of limited plex or lack of spatial resolution. Results from digital spatial profiling can potentially suggest a biologic difference and potentially pinpoint a valuable marker of more indolent vs. aggressive behavior in MF.
Thus, the aim of this study was to investigate the applicability of spatial transcriptome profiling to detect differences in gene expression in the malignant CD4+ T-cells at time-of-diagnosis and compare data from two distinct groups: skin samples from MF patients who progressed to advanced stage disease versus MF patients who remained in early-stage disease.
2 Materials and methods
2.1 Patient cohort
Fifteen patients with MF, who were followed and treated from initial diagnosis (between 1994-2012) and until May 2022 (Table 1) at the Department of Dermatology, Aarhus University Hospital, were included in this study. The study was approved by the local ethical committee (1–10–72–91–13) and the Data Protection Agency (Datatilsynet 1-16-02-478-15).
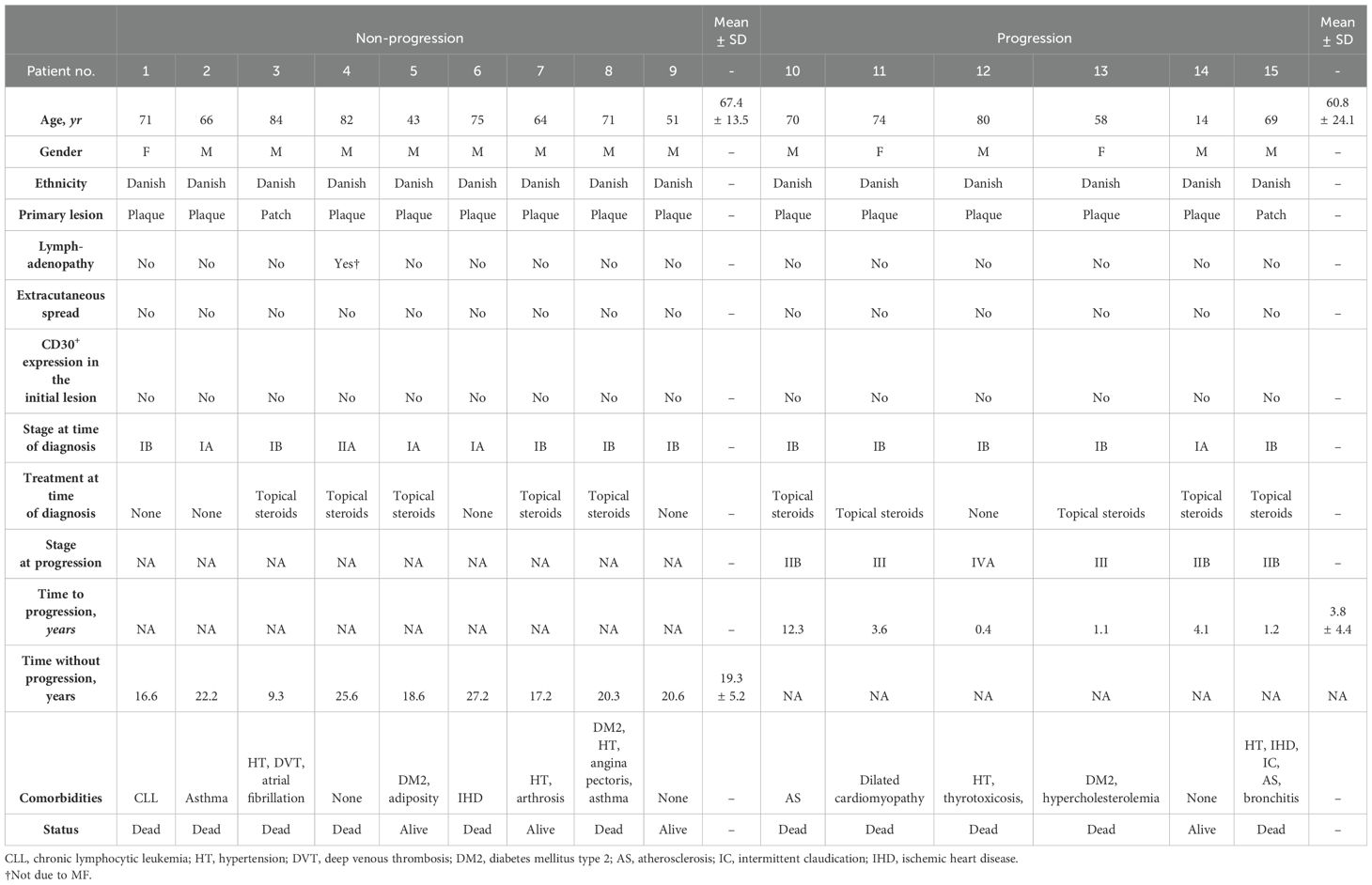
Table 1. Demographic and clinical characteristics of the patient cohort.
By individual-level linkage to the medical records, we identified and acquired the specific skin biopsy that first established the diagnosis of early-stage MF. There had to be visible Pautrier’s microabscesses (a cluster enriched with malignant CD4+ T-cells in the epidermis) as assessed by the pathologist at the time of diagnosis. From the patient files, we extracted important clinical variables with complete and long-term follow-up such as age, gender, ethnicity, clinical stage at time of diagnosis and at progression, treatment at time of diagnosis, comorbidities, CD30 status and extracutaneous spread (Table 1). The early MF diagnosis (stage IA-IIA) was confirmed by clinical as well as immuno- and histopathological findings referenced in the patient files. The patients were staged according to the International Society for Cutaneous Lymphomas/European Organization of Research and Treatment of Cancer proposal (13). In total, seventeen formalin-fixed, paraffin-embedded (FFPE) skin biopsies were acquired. Two patients had to be excluded due to exhaustion of the biopsy core. Disease progression was defined as progression from the early stages (IA-IIA) to the advanced stages of MF (stage IIB-IVB), because progression to advanced stages of MF significantly affects the prognosis (14). The patients were thus subdivided into two groups according to whether their disease progressed to advanced stages (stage ≥ IIB) or remained in the early stages (stage < IIB) during follow-up. Some patients were treated with topical steroids and none of the patients received phototherapy at the time of diagnosis.
2.2 Sample tissue features and selecting visualization markers
Pathological archives were searched for the initial biopsy which confirmed the diagnosis of MF and a medical record following treatment and status of progression. Serial sections of five µm thickness derived from FFPE tissue blocks were sectioned. One section was stained with hematoxylin and eosin (H&E) as previously described (15). Furthermore, a serial unstained section was mounted on a positively charged histology slide (Superfrost Plus microscope slides cat: 12-550-15, Fisher Scientific, Waltham, MA, USA) for analyses using the GeoMx Digital Spatial Profiling (DSP) technology (NanoString Technologies Inc., Seattle, USA) in order to perform spatial transcriptomics. H&E-stained slides were digitized for image analysis using the whole slide digital pathology scanner NanoZoomer 2.0-HT (RRID: SCR_021658, Hamamatsu Photonics K.K, Hamamatsu City, Japan) with a 20×objective. Identification of epidermis, dermis, Pautrier’s microabscesses and basal layer was scored with 100% consensus agreement by three observers (M.D., T.E., and L.I.).
2.3 Preparation of samples for DSP
The process began with the deparaffinization of slides, followed by antigen retrieval. Subsequently, the samples underwent treatment with proteinase K (1 ug/ml for 15 minutes). Next, the slides were incubated overnight with DSP Barcoded RNA (indexing RNA oligos) prior to being stained with fluorescently conjugated antibodies to CD4, PanCK and DNA (Supplementary Table S1) for one hour. The slides were then washed in 2x saline-sodium citrate. Slides were introduced in the DSP instrument and submerged/washed in PBS with 0.1% Tween 20 as described in the NanoString Protocol.
2.4 ROI placement and compartmentalization/segmentation strategies
Fluorescence scans (x20) were performed to obtain a high-resolution image of the tissue (Figure 1). Fluorescence scans were also imported into QuPath (RRID: SCR_018257, version 0.3.2) (16) which allowed for image analysis and cell counting. Regions-of-interest (ROIs) were selected in the DSP by matching with the previous selected regions on the H&E-stained serial section. Up to four ROIs were chosen per tissue consisting of the dermis, Pautrier’s microabscesses, the basal layer and a part of epidermis (Figures 1E–H). ROIs in the dermis were segmented into CD4+ T-cells and the surrounding tumor microenvironment.
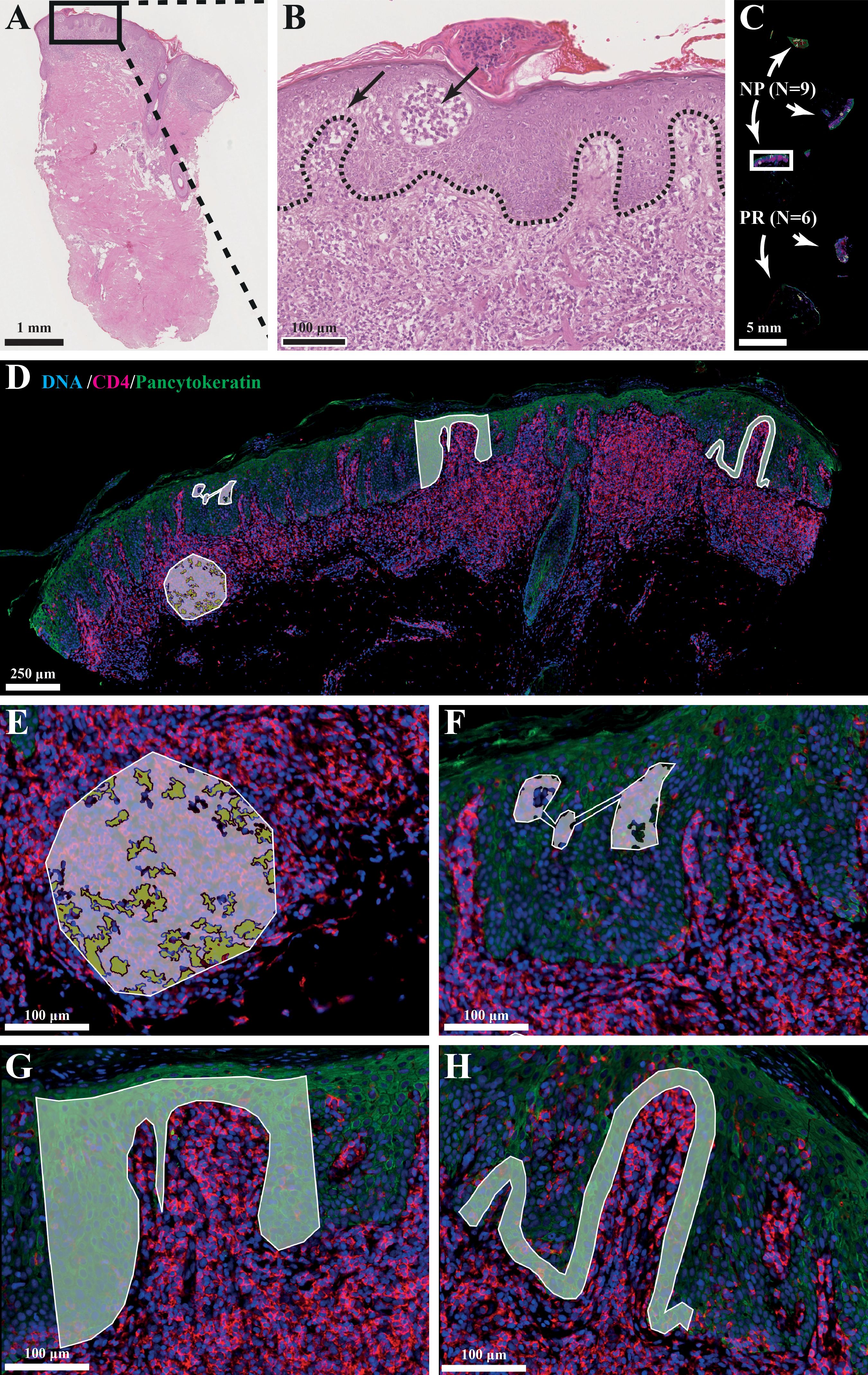
Figure 1. (A, B) Histological hallmarks of MF: intraepidermal vesicles (Pautrier’s microabscesses) and epidermotropism (atypical lymphocytes palisade along dermal-epidermal junction). (C) Slide setup with number of biopsies and type (NP=non-progression, PR=progression). (D) Photo of selected ROI/AOIs from one tissue sample. (E) ROI: dermal infiltrate, AOI: segmented into +/- CD4+ cells. (F) ROI: Pautrier’s microabscess. (G) ROI: Block of epidermis. (H) ROI: Basal layer.
2.5 Library preparation and sequencing
Indexing RNA oligos (probes), marked with a DSP barcode, were released from specific ROIs using UV light. Subsequently, 10 µl of liquid from above the ROI was gathered via a microcapillary tip and deposited into a 96-well plate. The probes underwent quantification through Next-Generation-Sequencing (NGS), wherein the resulting read numbers for each probe were then associated with the expression level of their corresponding gene targets.
To preserve ROI identity, indexing RNA oligos from each ROI were amplified via PCR using primers that hybridized to constant regions and contained distinct dual-indexed barcoding sequences. The PCR products underwent pooling and double purification using Ampure XP beads. The concentration and purity of the library were determined using a high-sensitivity DNA Bioanalyzer chip. Sequencing, involving paired-end reads (2x75 bp), was conducted on an Illumina MiSeq instrument.
2.6 Data processing
Count data files (dcc files) were produced from NGS fastq files using GeoMx HGS Pipeline software from NanoString. Subsequent data analysis including graphical presentations were made using the R statistical programming language and the packages NanoStringNCTools, GeomxTools and GeoWorkflows (17–19). Fraction of trimmed, stitched, and aligned reads were above 80% for all segments. Sequencing saturation was above 90% for all segments. A segment cutoff range based on biological diversity of 2% was applied, leaving 7,712 genes above the threshold.
2.7 Normalization
Third quartile (Q3) normalization was used prior to downstream visualization and differential expression analysis. Data were stratified according to cell type and five separate linear mixed models for the basal layer, cd4_dermis, cd4_dermis_micro, cd4_microabscess and epidermis were applied to contrast the progression variable with the scan id variable as a random factor. In addition, two models were made to combine evidence across different cell types. For both models, cell types were used as random effect factor.
Volcano plots were used for interpreting differential gene expression results, and the top five up- and downregulated genes for all models were reviewed in the literature. In addition, a non-exhaustive list of MF defining genes was added (Supplementary Table S2).
2.8 Relevance of spatial RNA profiling using single-cell RNA sequencing
Count matrices from six studies were obtained or generated from the Gene Expression Omnibus [accession numbers GSE182861 (20), GSE165623 (21), GSE173205 (22), and GSE128531 (23)], Sequence Read Archive (SRP332550 (24)), or Genome Sequence Archive (HRA000166 (25)). Upregulated genes from patients with progression in the Pautrier’s microabscess model were compared to findings in these scRNA-seq studies. Details on processing, integration, and analysis of scRNA-seq data can be found in Supplementary Methods.
3 Results
To establish the similarity between the histopathology of the non-progression group and the progression group we performed manual cell assessments in the investigated compartments in the samples used for DSP (Supplementary Figure S1). No significant difference between the two groups for any of the assessments was observed.
3.1 Upregulation of specific genes in CD4+ T-cells from Pautrier’s microabscesses in MF patients with progression
Next, we assessed the five different investigated compartments using DSP. The top five up- or downregulated genes for all five models and known genes-of-interest (Supplementary Table S2) in the progression vs. non-progression groups were labeled separately (Figure 2). None of the genes met the threshold of false detection rate (FDR) <0.1, nor were any genes differentially expressed when pooling all data and comparing progression vs. non-progression. All top five up- or downregulated genes were reviewed in the literature to identify candidate markers of progression. Comparison within the Pautrier’s microabscesses ensured expression data from clusters of cells enriched with malignant CD4+ T-cells (Figure 2B). Comparing this compartment in MF patients with progression vs. non-progression, we found upregulation of RUNX2 (p=.002, FDR=.91), ATP5ME (p=.000, FDR=.99), CD37 (p=.003, FDR=.99), TIMM13 (p=.004, FDR=.99), LPIN3 (p=.006, FDR=.99), SHMT2 (p=.014, FDR=.99), SP1 (p=.004, FDR=.99), UPF2 (p=.008, FDR=.99), AKAP1 (p=.009, FDR=.99), SLC16A3 (p=.009, FDR=.99), MCM7 (p=.013, FDR=.99), PSTPIP1 (p=.014, FDR=.99), PRPF31 (p=.016, FDR=.99) and ZNF419 (p=.007, FDR=.99) in malignant CD4+ T-cells in Pautrier’s microabscesses in MF patients with progression compared with the non-progression group (Figure 2B).
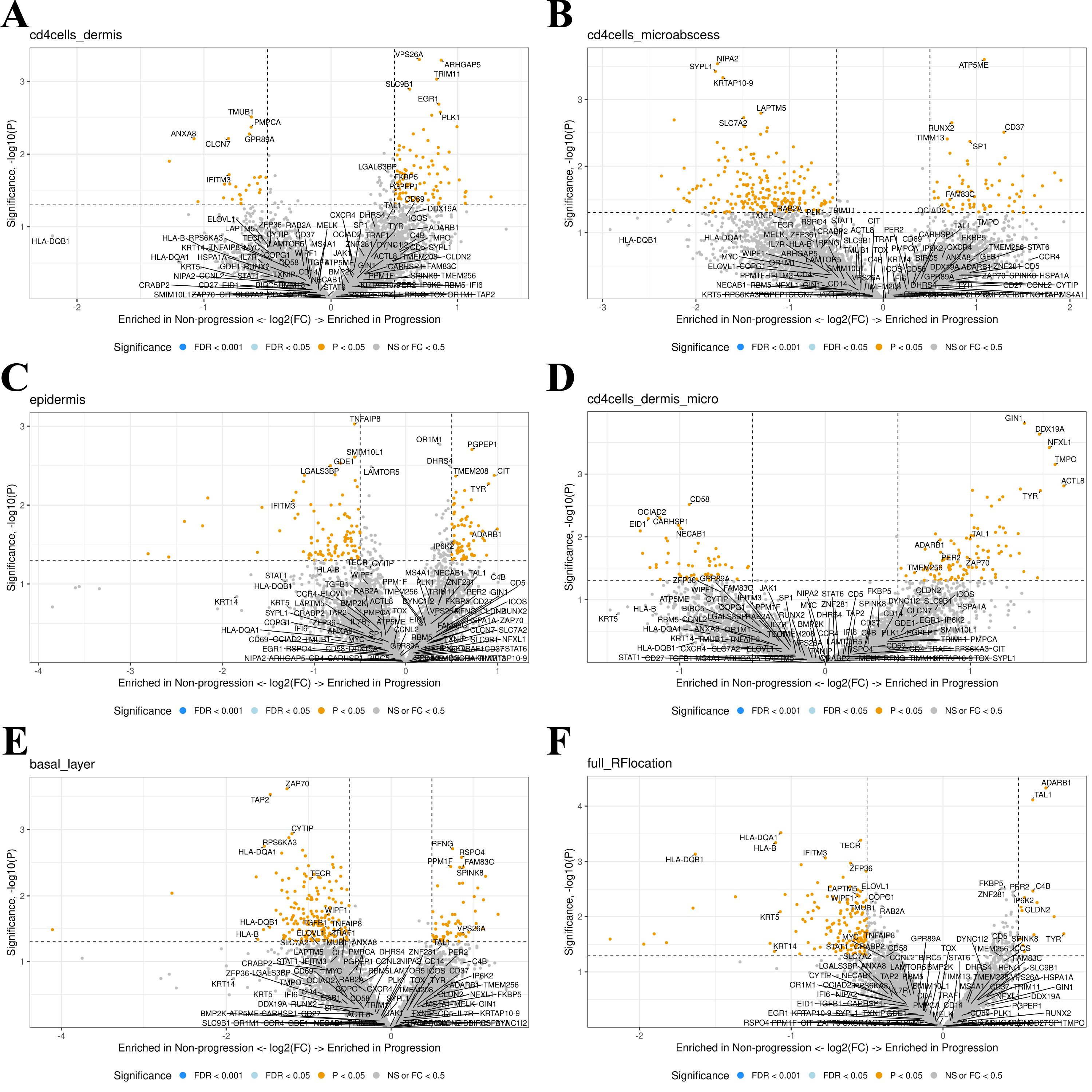
Figure 2. (A–E) A canonical visualization of the top five up- or downregulated genes for all five comparison models and known genes-of-interest (p<0.5) in progression vs. non-progression. None of the genes fell under the false detection rate (FDR) of 0.1. (F) All comparison models were combined in a total model showing the up- or downregulated genes enriched in progression vs. non-progression.
To validate expression of the progression-related genes within malignant T-cells from MF patients, we integrated and reanalyzed a collection of published scRNA-seq data sets containing a total of 64 skin samples (non-lesional/patch/plaque/tumor) from 40 patients with MF (stage I: six patients, stage II: 22 patients III: two patients, stage IV: eight patients, and two patients with unknown stage) and 12 healthy controls across six studies (20–25) (Figure 3). T-cell receptor clonality and the expression of malignant-associated genes were used to distinguish between malignant and nonmalignant T-cells (Figures 3D, E). For use as progression biomarkers, we focused on RUNX2, MCM7 and SHMT2 that all exhibited high expression within malignant cells and limited expression among other abundant cell types (Figure 3F). Interestingly, RUNX2 was also highly expressed in type 2 innate lymphoid cells (Figure 3F). Supporting that RUNX2 expression within Pautrier’s microabscesses in patients that progress was derived from malignant T-cells, we found that RUNX2 was abundantly and almost exclusively found within malignant T-cells from MF patients and largely absent in other cell types and healthy controls (Figure 3G). MCM7 and SHMT2 transcripts were enriched within malignant T-cells, but also found among other cell types in both MF patients and healthy donors (Figures 3H, I). Supporting the potential utility as an early marker of MF patients that will progress, which only occurs in a subset of patients, we found heterogeneous expression of RUNX2 in malignant T-cells between different patients at the early (I-II) stages as well as an inverse correlation with overall CTCL stage (Figure 3J). Similar patterns were less clear with MCM7 and SHMT2.
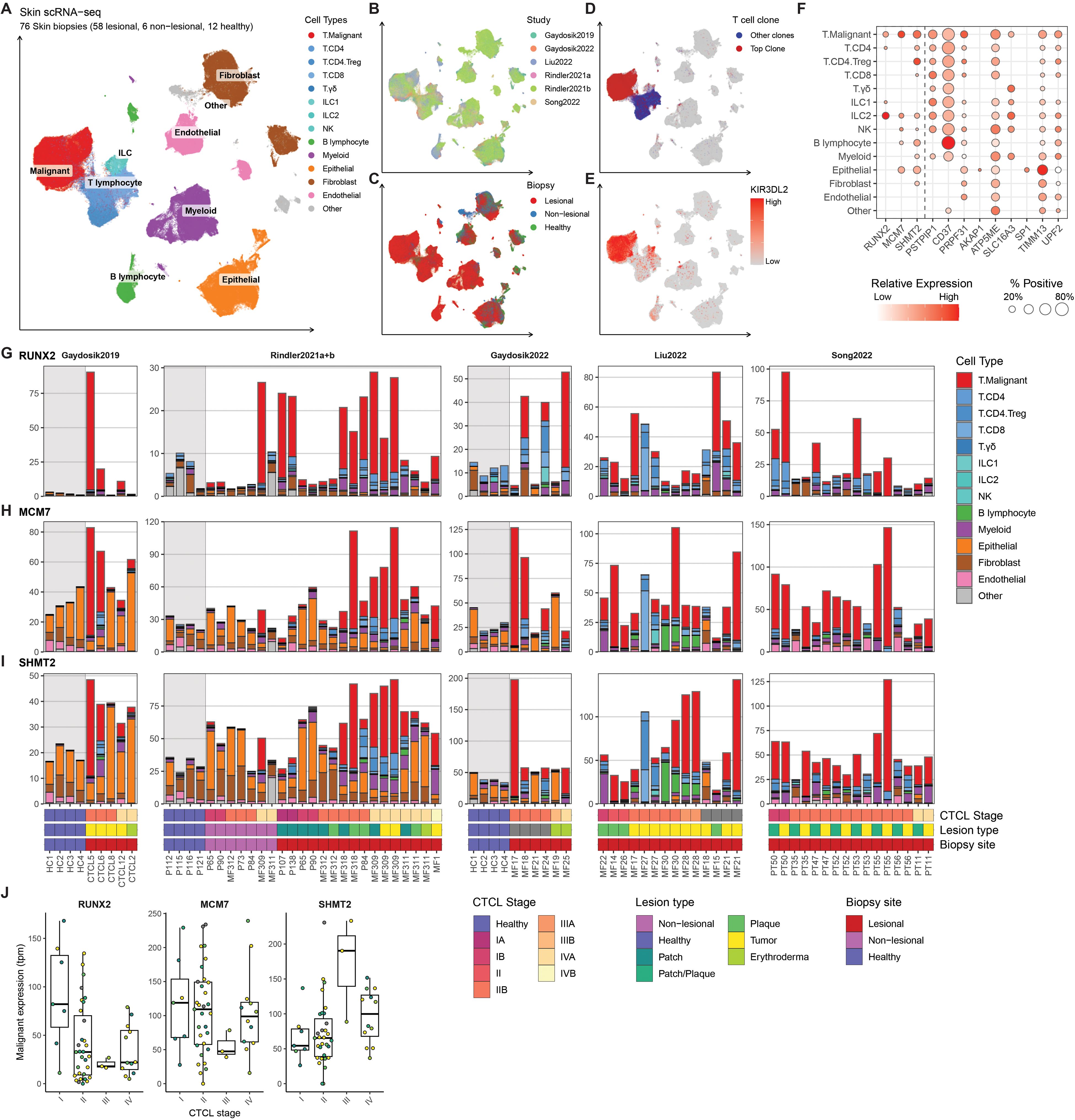
Figure 3. (A–E) Single-cell RNA sequencing (scRNA-seq) data from 76 skin biopsies (57 lesional, 7 non-lesional, 12 healthy) from 40 MF patients (stage I: six patients, stage II: 22 patients III: two patients, stage IV: eight patients, and two patients with unknown stage) and 12 healthy donors across six studies (20–25) were integrated using scANVI and visualized using Uniform Manifold Approximation and Projection (UMAP) colored by (A) cell types, (B) Study or origin, (C) Biopsy site, (D) Expression of top T cell receptor (TCR)-clone marking malignant cells and (E) expression of KIR3DL2 known to be high in malignant cells in CTCL. (F) scRNA-seq expression of genes upregulated in patients with progression in the Pautrier’s microabscess model shown as percent positive cells and mean expression across identified cell types. (G–I). Transcript expression of the three selected genes (RUNX2, MCM7 and SHMT2) across each of the 76 Skin biopsies shown as transcripts per million (tpm) by sample. Bar height and colors indicate transcript contribution of each cell type population. (J) Mean malignant expression (as tpm) of RUNX2, MCM7 and SHMT2 divided into CTCL stage. Each dot represents mean malignant expression from one biopsy colored by lesion type. Bold horizontal line depicts median, box edges show 25th and 75th percentiles. The expression values indicated are gated on malignant T-cells only.
Taken together, these data confirm that RUNX2 is highly expressed by malignant CD4+ T-cells in a subset of MF patients at early CTCL stages, and higher RUNX2 expression was detected in Pautrier’s microabscesses in early-stage disease in patients who progressed to advanced stages.
4 Discussion
The prognosis of patients with advanced MF is poor. This calls for a biologic marker which preferably at time of diagnosis, can identify patients progressing to advanced stage MF.
In the current study, gene expression data from spatial profiling of skin biopsies from patients who progressed to advanced stage disease were compared with patients who remained in early-stage disease. Current methods often rely on analyzing bulk samples which may dilute spatial compartments in the skin. However, spatial transcriptomic methods could help elucidate areas with known enriched pathological cells, discovering disease specific differences that so far have been hidden (26). Interestingly, our analyses provided multiple genes of interest. From the comparative model of Pautrier’s microabscesses, the gene expression of the transcription factor RUNX2 was found to be upregulated differentially at time of diagnosis in patients who progressed to advanced stage disease. RUNX2 is a member of the RUNX family of transcription factors and encodes a nuclear protein with a Runt DNA-binding domain. This protein is essential for osteoblastic differentiation and skeletal morphogenesis and acts as a scaffold for nucleic acids and regulatory factors involved in skeletal gene expression (27, 28). Interestingly, RUNX2 is connected to the RUNX3 gene which is already known to be dysregulated in cutaneous T-cell lymphoma (29), emphasizing the necessity of looking further into this gene as a potential biomarker. Prior experimental studies have found an association between RUNX2 and other T-cell cancers (30–32). In other malignancies, RUNX2 has been shown to drive expression of genes and pathways involved in invasion, angiogenesis, and metastasis such as MMP9, VEGF, IL-8, TGFβ, and AKT/PI3K (33), all of which have been reported as overexpressed by malignant T-cells from MF patients (34). Of notice, ectopic RUNX2 expression has also been linked to genomic instability and drug resistance in cancer (35–37). Thus, it seems likely that an increased RUNX2 expression by malignant T-cells at the time of diagnosis – as reported here – is not only an important novel biomarker but also a putative molecular driver of disease progression in MF. While RUNX2 may be a driver of malignant transformation at the time of diagnosis it may act differently at more advanced stages as we observed a trend towards a lower concentration. Accordingly, the upregulation of RUNX2 needs to be validated in a bigger cohort. One of the weaknesses in this study is the relatively high FDR values which thereby indicate a higher proportion of false positives among the genes identified as significantly differentially expressed. One possible explanation for the high FDR values in our samples could be due to poor RNA quality of our FFPE samples, which may be attributed to their age, with some samples being 20-25 years old. Lastly, from the assumption that a marker of progression will be greatly expressed in later stages of disease as compared to early-stage, MCM7 involved in DNA replication, and SHMT2 involved in metabolism are also interesting candidates for further investigation (38). Both genes have a lower expression in scRNA-seq in healthy skin as compared with malignant T-cells and MCM7 expression exhibited a clear correlation with stage of disease. Moreover, increased levels of MCM7 or SHMT2 expression have been associated with poor prognosis in various cancers, including breast and lung cancers (39–41). It is notable that scRNA-seq analysis revealed high expression of RUNX2 in type 2 innate lymphoid cells, indicating the need for further investigation of this cell type in MF.
MF can progress to high-grade large cell lymphoma which may be characterized by the increased expression of CD30 (42). A recent study utilized DSP to investigate CD30+ transformed MF and primary cutaneous anaplastic large-cell lymphoma (43). It was demonstrated that CD30+ tumor areas in MF with large-cell transformation had an increased concentration of macrophages and fibroblasts and a decreased concentration of regulatory T-cells compared with primary cutaneous anaplastic large-cell lymphoma. MMP9, a marker of cell-cell membrane interaction, was posited as a potential novel target in advanced MF (43). Classic Hodgkin lymphoma is also characterized by CD30+ malignant cells in the lymph nodes. In a recent study CD4+ T-cells close to and at 20 μm distance from CD30+ cells were profiled using DSP. PD-1 and CTLA-4 were significantly upregulated in cells close to CD30+ cells compared with cells at a distance from CD30+ cells (44). Both studies included relatively few patients, and none of the patients in our cohort were CD30+. However, both studies illustrate the good potential of DSP in biomarker identification in cutaneous lymphomas.
In conclusion, the present study identifies putative novel biomarkers for early detection of disease progression in patients with cutaneous lymphoma.
Data availability statement
The datasets used and analyzed during the current study are available at hosted at GEO repository (https://www.ncbi.nlm.nih.gov/geo/, accession number GSE275677) and the corresponding author on reasonable request.
Ethics statement
The studies involving humans were approved by Danish Ethics Committee (1–10–72–91–13) and The Danish Data Protection Agency (Datatilsynet 1-16-02-478-15). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
MD: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing – original draft, Writing – review & editing, Funding acquisition. TE: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Methodology, Resources, Validation, Visualization, Writing – original draft, Writing – review & editing. MN: Data curation, Formal analysis, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. LL: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Writing – original draft, Writing – review & editing. MG: Formal analysis, Investigation, Writing – original draft, Writing – review & editing. NØ: Funding acquisition, Resources, Supervision, Writing – original draft, Writing – review & editing. LR: Methodology, Resources, Supervision, Writing – original draft, Writing – review & editing. TS: Validation, Writing – original draft, Writing – review & editing, Resources, Supervision. LI: Writing – original draft, Writing – review & editing, Conceptualization, Funding acquisition, Methodology, Resources, Supervision. RB: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. TB: Data curation, Formal analysis, Validation, Visualization, Writing – original draft, Writing – review & editing. CJ: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Part of the funding for the present study was provided by a Nordic WTA Grant provided by NanoString to LI. MD was supported by funds designated for DSP by Aage Bangs Fond (Grant number: 900600), TE was supported by funds designated for DSP by Aage Bangs Fond (Grant number: Not specified), Fonden til Lægevidenskabens Fremme (Grant number: 19-L-0131), Danish Psoriasis Association (Grant number: Not specified), Danske Lægers Forsikringsforening (Grant number: Not specified), Lægeforeningen (Grant number: 2019-3780/40), Robert Wehnerts og Kirsten Wehnerts Fond (Grant number: 18978), Carl og Ellen Hertz´ Videnskabslegat (Grant number: 7179-2), and Grosserer L. F. Foghts Fond (Grant number: 20.010). Novo Nordic Foundation Tandem Program (Grant number: NNF210C0066950). The funding bodies did not have any role in the study design; in the collection, analysis, and interpretation of data; in the writing of the manuscript; or in the decision to submit the article for publication.
Acknowledgments
We thank Susanne Lorenz and Marie Kristine Gillstrøm at Radium Hospital, Oslo University, for the technical assistance. We also thank Kristine Møller and Annette Blak Rasmussen for the excellent laboratory aid.
Conflict of interest
Author TE has participated in investigator-initiated clinical trials sponsored by Janssen and Leo Pharma and has served as paid speaker for BMS. Author CJ has served as a consultant and/or paid speaker for Eli Lilly, Abbvie, Leo Pharma and L’Oréal. Author LI has served as a consultant and/or paid speaker for and/or participated in clinical trials sponsored by: AbbVie, Almirall, Amgen, AstraZeneca, BMS, Boehringer Ingelheim, Celgene, Centocor, Eli Lilly, Janssen Cilag, Kyowa, Leo Pharma, MSD, Novartis, Pfizer, Regranion, Samsung, Union Therapeutics and UCB. Author LI was employed by the company MC2 Therapeutics A/S.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2024.1421443/full#supplementary-material
Abbreviations
CD, cluster of differentiation; CTCL, cutaneous T-cell lymphoma; DNA, deoxyribonucleic acid; DSP, digital spatial profiling; FDR, false detection rate; FFPE, formalin-fixed, paraffin-embedded; H&E, hematoxylin and eosin; IHC, immunohistochemistry; LOQ, limit of quantification; MF, mycosis fungoides; NGS, next-generation-sequencing; PanCK, pancytokeratin; PBS, phosphate buffered saline; PCR, polymerase chain reaction; RNA, ribonucleic acid; ROI, regions of interest; UV, ultraviolet; WTA, whole transcriptome assay.
References
1. Willemze R, Hodak E, Zinzani PL, Specht L, Ladetto M. Primary cutaneous lymphomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2013) 24:vi149–54. doi: 10.1093/annonc/mdt242
2. Willemze R, Cerroni L, Kempf W, Berti E, Facchetti F, Swerdlow SH, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood. (2019) 133:1703–14. doi: 10.1182/blood-2018-11-881268
3. Kim YH, Liu HL, Mraz-Gernhard S, Varghese A, Hoppe RT. Long-term outcome of 525 patients with mycosis fungoides and sézary syndrome. Arch Dermatol. (2003) 139(7):857–866. doi: 10.1001/archderm.139.7.857
4. Lindahl LM, Besenbacher S, Rittig AH, Celis P, Willerslev-Olsen A, Gjerdrum LMR, et al. Prognostic miRNA classifier in early-stage mycosis fungoides: development and validation in a Danish nationwide study. Blood. (2018) 131:759–70. doi: 10.1182/blood-2017-06-788950
5. Mourad A, Gniadecki R. Overall survival in mycosis fungoides: A systematic review and meta-analysis. J Invest Dermatol. (2020) 140:495–7. doi: 10.1016/j.jid.2019.07.712
6. Mizuno Y, Shibata S, Miyagaki T, Ito Y, Taira H, Omori I, et al. Serum cell-free DNA as a new biomarker in cutaneous T-cell lymphoma. J Dermatol. (2022) 49:1124–30. doi: 10.1111/1346-8138.16520
7. Gluud M, Willerslev-Olsen A, Gjerdrum LMR, Lindahl LM, Buus TB, Andersen MH, et al. MicroRNAs in the pathogenesis, diagnosis, prognosis and targeted treatment of cutaneous T-cell lymphomas. Cancers (Basel). (2020) 12:1229. doi: 10.3390/cancers12051229
8. Walia R, Yeung CCS. An update on molecular biology of cutaneous T cell lymphoma. Front Oncol. (2020) 9. doi: 10.3389/fonc.2019.01558
9. Gambichler T, Salveridou K, Schmitz L, Käfferlein HU, Brüning T, Stockfleth E, et al. Low Drosha protein expression in cutaneous T-cell lymphoma is associated with worse disease outcome. J Eur Acad Dermatol Venereology. (2019) 33:1695–9. doi: 10.1111/jdv.15652
10. Barata MC, Teixeira SP, Batista MD, Tomimori J, Landman G, Silva MRR. [amp]]alpha;Eβ7 expression increases with the extent of cutaneous involvement in mycosis fungoides. Am J Dermatopathol. (2019) 41:630–6. doi: 10.1097/DAD.0000000000001397
11. Mascolo M, Romano MF, Ilardi G, Romano S, Baldo A, Scalvenzi M, et al. Expression of FK506-binding protein 51 (FKBP51) in Mycosis fungoides. J Eur Acad Dermatol Venereology. (2018) 32:735–44. doi: 10.1111/jdv.14614
12. Argyropoulos KV, Pulitzer M, Perez S, Korkolopoulou P, Angelopoulou M, Baxevanis C, et al. Tumor-infiltrating and circulating granulocytic myeloid-derived suppressor cells correlate with disease activity and adverse clinical outcomes in mycosis fungoides. Clin Trans Oncol. (2020) 22:1059–66. doi: 10.1007/s12094-019-02231-7
13. Olsen E, Vonderheid E, Pimpinelli N, Willemze R, Kim Y, Knobler R, et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Ca. Blood. (2007) 110:1713–22. doi: 10.1182/blood-2007-03-055749
14. Agar NS, Wedgeworth E, Crichton S, Mitchell TJ, Cox M, Ferreira S, et al. Survival outcomes and prognostic factors in mycosis fungoides/sézary syndrome: validation of the revised international society for cutaneous lymphomas/european organisation for research and treatment of cancer staging proposal. J Clin Oncol. (2010) 28:4730–9. doi: 10.1200/JCO.2009.27.7665
15. Raaby L, Rosada C, Langkilde A, Lauridsen KL, Vinter H, Ommen P, et al. Langerhans cell markers CD1a and CD207 are the most rapidly responding genes in lesional psoriatic skin following adalimumab treatment. Exp Dermatol. (2017) 26:804–10. doi: 10.1111/exd.13304
16. Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep. (2017) 7:16878. doi: 10.1038/s41598-017-17204-5
17. Griswold M, Ortogero N, Yang Z, Vitancol R, Henderson D. GeomxTools: nanoString geoMx tools. (2024). R package version 3.8.0.
18. Griswold M, Reeves J, Divakar P, Yang Z, Zimmerman S, Vitancol R, et al. GeoMxWorkflows: GeoMx Digital Spatial Profiler (DSP) data analysis workflows. (2024). R package version 1.10.0.
19. Aboyoun P, Ortogero N. NanoStringNCTools: nanoString nCounter tools. (2024). R package version 1.12.0.
20. Gaydosik AM, Stonesifer CJ, Khaleel AE, Geskin LJ, Fuschiotti P. Single-cell RNA sequencing unveils the clonal and transcriptional landscape of cutaneous T-cell lymphomas. Clin Cancer Res. (2022) 28:2610–22. doi: 10.1158/1078-0432.CCR-21-4437
21. Rindler K, Bauer WM, Jonak C, Wielscher M, Shaw LE, Rojahn TB, et al. Single-cell RNA sequencing reveals tissue compartment-specific plasticity of mycosis fungoides tumor cells. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.666935
22. Rindler K, Jonak C, Alkon N, Thaler FM, Kurz H, Shaw LE, et al. Single-cell RNA sequencing reveals markers of disease progression in primary cutaneous T-cell lymphoma. Mol Cancer. (2021) 20:124. doi: 10.1186/s12943-021-01419-2
23. Gaydosik AM, Tabib T, Geskin LJ, Bayan CA, Conway JF, Lafyatis R, et al. Single-cell lymphocyte heterogeneity in advanced cutaneous T-cell lymphoma skin tumors. Clin Cancer Res. (2019) 25:4443–54. doi: 10.1158/1078-0432.CCR-19-0148
24. Song XX, Chang S, Seminario-Vidal L, de Mingo Pulido A, Tordesillas L, Song XX, et al. Genomic and single-cell landscape reveals novel drivers and therapeutic vulnerabilities of transformed cutaneous T-cell lymphoma. Cancer Discovery. (2022) 12:1294–313. doi: 10.1158/2159-8290.CD-21-1207
25. Liu X, Jin S, Hu S, Li R, Pan H, Liu Y, et al. Single-cell transcriptomics links Malignant T cells to the tumor immune landscape in cutaneous T cell lymphoma. Nat Commun. (2022) 13:1158. doi: 10.1038/s41467-022-28799-3
26. Piñeiro AJ, Houser AE, Ji AL. Research techniques made simple: spatial transcriptomics. J Invest Dermatol. (2022) 142:993–1001. doi: 10.1016/j.jid.2021.12.014
27. Lian J, Stein G. Runx2/cbfa1: A multifunctional regulator of bone formation. Curr Pharm Des. (2003) 9:2677–85. doi: 10.2174/1381612033453659
28. Komori T. Requisite roles of Runx2 and Cbfb in skeletal development. J Bone Miner Metab. (2003) 21:193–7. doi: 10.1007/s00774-002-0408-0
29. Dulmage BO, Akilov O, Vu JR, Falo LD, Geskin LJ. Dysregulation of the TOX-RUNX3 pathway in cutaneous T-cell lymphoma. Oncotarget. (2019) 10:3104–13. doi: 10.18632%2Foncotarget.5742
30. Blyth K, Vaillant F, Hanlon L, Mackay N, Bell M, Jenkins A, et al. Runx2 and MYC collaborate in lymphoma development by suppressing apoptotic and growth arrest pathways in vivo. Cancer Res. (2006) 66:2195–201. doi: 10.1158/0008-5472.CAN-05-3558
31. Cohen-Solal KA, Boregowda RK, Lasfar A. RUNX2 and the PI3K/AKT axis reciprocal activation as a driving force for tumor progression. Mol Cancer. (2015) 14:137. doi: 10.1186/s12943-015-0404-3
32. Zitti B, Hoffer E, Zheng W, Pandey RV, Schlums H, Perinetti Casoni G, et al. Human skin-resident CD8+ T cells require RUNX2 and RUNX3 for induction of cytotoxicity and expression of the integrin CD49a. Immunity. (2023) 56:1285–302. doi: 10.1016/j.immuni.2023.05.003
34. Krejsgaard T, Lindahl LM, Mongan NP, Wasik MA, Litvinov IV, Iversen L, et al. Malignant inflammation in cutaneous T-cell lymphoma—a hostile takeover. Semin Immunopathol. (2017) 39:269–82. doi: 10.1007/s00281-016-0594-9
35. Zhang PP, Wang YC, Cheng C, Zhang F, Ding DZ, Chen DK. Runt-related transcription factor 2 influences cell adhesion-mediated drug resistance and cell proliferation in B-cell non-Hodgkin’s lymphoma and multiple myeloma. Leuk Res. (2020) 92:106340. doi: 10.1016/j.leukres.2020.106340
36. Samarakkody AS, Shin NY, Cantor AB. Role of RUNX family transcription factors in DNA damage response. Molecules Cells. (2020) 43:99–106. Korean Society for Molecular and Cellular Biology.
37. Hay J, Gilroy K, Huser C, Kilbey A, Mcdonald A, MacCallum A, et al. Collaboration of MYC and RUNX2 in lymphoma simulates T-cell receptor signaling and attenuates p53 pathway activity. J Cell Biochem. (2019) 120:18332–45. doi: 10.1002/jcb.29143
38. Springer PS, Holding avid R, Groover A, Yordan C, Martienssen RA. The essential Mcm7 protein PROLIFERA is localized to the nucleus of dividing cells during the G1 phase and is required maternally for early Arabidopsis development. Development. (2000) 127:1815–22. doi: 10.1242/dev.127.9.1815
39. Huang TH, Huo L, Wang YN, Xia W, Wei Y, Chang SS, et al. Epidermal growth factor receptor potentiates MCM7-mediated DNA replication through tyrosine phosphorylation of lyn kinase in human cancers. Cancer Cell. (2013) 23:796–810. doi: 10.1016/j.ccr.2013.04.027
40. Zhong X, Guan X, Dong Q, Yang S, Liu W, Zhang L. Examining Nek2 as a better proliferation marker in non-small cell lung cancer prognosis. Tumor Biol. (2014) 35:7155–62. doi: 10.1007/s13277-014-1935-8
41. Bernhardt S, Bayerlová M, Vetter M, Wachter A, Mitra D, Hanf V, et al. Proteomic profiling of breast cancer metabolism identifies SHMT2 and ASCT2 as prognostic factors. Breast Cancer Res. (2017) 19:112. doi: 10.1186/s13058-017-0905-7
42. Wu H, Telang GH, Lessin SR, Vonderheid EC. Mycosis fungoides with CD30-positive cells in the epidermis. Am J Dermatopathol. (2000) 22:212–6. doi: 10.1097/00000372-200006000-00002
43. Choi ME, Lee MY, Won CH, Chang SE, Lee MW, Lee WJ. Spatially resolved transcriptomes of CD30+-transformed mycosis fungoides and cutaneous anaplastic large-cell lymphoma. J Invest Dermatol. (2023) 144(2):331–340.e2. doi: 10.1016/j.jid.2023.05.030
Keywords: mycosis fungoides, digital spatial profiling, biomarkers, RNA profiling, spatial transcriptomics
Citation: Danielsen M, Emmanuel T, Nielsen MM, Lindahl LM, Gluud M, Ødum N, Raaby L, Steiniche T, Iversen L, Bech R, Buus TB and Johansen C (2024) RUNX2 as a novel biomarker for early identification of patients progressing to advanced-stage mycosis fungoides. Front. Oncol. 14:1421443. doi: 10.3389/fonc.2024.1421443
Received: 22 April 2024; Accepted: 04 September 2024;
Published: 07 October 2024.
Edited by:
Hsin-Sheng Yang, University of Kentucky, United StatesReviewed by:
Chalid Assaf, Helios Hospital Krefeld, GermanyAnna Jarzab, Technical University of Munich, Germany
Eiji Kiyohara, Osaka University, Japan
Alan Zhou, Northwestern Medicine, United States
Copyright © 2024 Danielsen, Emmanuel, Nielsen, Lindahl, Gluud, Ødum, Raaby, Steiniche, Iversen, Bech, Buus and Johansen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claus Johansen, Y2xhdXMuam9oYW5zZW5AY2xpbi5hdS5kaw==