 Yosuke Nishio
Yosuke Nishio Kohji Kato1,2,3
Kohji Kato1,2,3 Hisashi Oishi
Hisashi Oishi Shinji Saitoh
Shinji Saitoh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 31 May 2024
Sec. Molecular and Cellular Oncology
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1417607
This article is part of the Research Topic Molecular Mechanisms and Treatment of MYCN-driven Tumors - Volume II View all articles
Somatic mutations in MYCN have been identified across various tumors, playing pivotal roles in tumorigenesis, tumor progression, and unfavorable prognoses. Despite its established notoriety as an oncogenic driver, there is a growing interest in exploring the involvement of MYCN in human development. While MYCN variants have traditionally been associated with Feingold syndrome type 1, recent discoveries highlight gain-of-function variants, specifically p.(Thr58Met) and p.(Pro60Leu), as the cause for megalencephaly-polydactyly syndrome. The elucidation of cellular and murine analytical data from both loss-of-function (Feingold syndrome model) and gain-of-function models (megalencephaly-polydactyly syndrome model) is significantly contributing to a comprehensive understanding of the physiological role of MYCN in human development and pathogenesis. This review discusses the MYCN’s functional implications for human development by reviewing the clinical characteristics of these distinct syndromes, Feingold syndrome, and megalencephaly-polydactyly syndrome, providing valuable insights into the understanding of pathophysiological backgrounds of other syndromes associated with the MYCN pathway and the overall comprehension of MYCN’s role in human development.
MYCN belongs to the MYC proto-oncogene family, alongside MYC and MYCL (1). As transcription factors, MYC proteins govern the diverse genes’ expression pivotal in fundamental cellular biological functions such as proliferation, differentiation, and apoptosis (2, 3). Initially identified as a proto-oncogene amplified in neuroblastoma, MYCN amplification occurs in 20%-30% of neuroblastoma cases, serving as a critical prognostic marker associated with poorer outcomes (4). Furthermore, MYCN amplification and single nucleotide variants have been identified in various other tumors, encompassing Wilms’ tumor, rhabdomyosarcomas, and lung cancers (5–8).
MYCN and other members of the MYC protein family are expressed in various developing human fetal tissues, including the brain, limbs, heart, kidney, and lungs as well as within tumors (9–12). Notably, consistent with these expression patterns, heterozygous loss-of-function (LoF) MYCN variants have harmful effects on fetal development, giving rise to a genetic syndrome marked by multiple congenital anomalies known as Feingold syndrome type 1 (FS1, OMIM #164280) (13).
In addition to the direct MYCN variants, variations in genes associated with the physiological upstream pathways of MYC family, Wnt/β-catenin (14) and sonic hedgehog (SHH) pathways (15–17), contribute to various human developmental diseases (18–24). Furthermore, genes associated with receptor tyrosine kinase (RTK) signaling pathway (25, 26) and F-box and WD repeat domain containing 7 (FBXW7) (27) that contribute to the modulation of stability and activity of MYCN, cyclin D, one of the downstream transcribed genes of MYCN, and MYC associated factor X (MAX), a component of the MYC/MAX complex are documented as the causes of human developmental diseases (28–33). Though these increasing evidence for the association of human developmental diseases with MYCN is being established, the direct functional roles of MYCN on human development remain to be understood comprehensively.
Recently, we established the megalencephaly-polydactyly syndrome (OMIM #620748) in three individuals harboring heterozygous gain-of-function (GoF) MYCN variants, namely p.(Thr58Met) and p.(Pro60Leu), that helped understanding the MYCN’s roles in human development. Intriguingly, this syndrome manifested mirror phenotypes of FS1, featuring megalencephaly and postaxial polydactyly, complicated with neuroblastoma detected in two out of the three patients (34, 35). Functional analysis unveiled excessive stability and reduced phosphorylation at the 58th threonine residue, a crucial locus for MYCN degradation. Insights from the GoF mouse model highlighted the overproliferative tendencies of neuronal progenitors and potentially various tissues, resulting in megalencephaly and the involvement of several organs (34, 35).
Our review mainly focuses on the disorders directly related to MYCN to unravel MYCN’s functional roles in human development through reviewing cellular and murine models representing both LoF and GoF scenarios, along with the concise clinical descriptions of other syndromes associated with the MYCN pathway for the overall comprehension of MYCN’s roles in human development.
Dysregulation of MYCN is observed in various tumors in both pediatric and adult settings, encompassing neuroblastoma, Wilms’ tumor, rhabdomyosarcoma, lung cancer, medulloblastoma, retinoblastoma and basal cell carcinoma (5–8, 36–41). The oncogenic variants include amplification and somatic single nucleotide mutations that stabilize or activate MYCN.
Firstly, MYCN amplification is well established in neuroblastoma accounting for 20%-30% of cases (4), but also observed in 25% of alveolar rhabdomyosarcoma cases (4, 41, 42), 5%-10% of medulloblastomas associated with poor prognosis (36, 40). Among those tumors, medulloblastoma is grouped into four distinct molecular subgroups in terms of gene expression patterns, one of which is SHH group (43), in which the amplifications of MYCN and MYCL are most frequently observed, defining its pathological significance in medulloblastoma (44). In terms of molecular background, the amplification drives tumorigenesis in neural crest cells by maintaining or re-establishing embryonic characteristics such as self-renewal, apoptotic resistance or metabolic flexibility (45). From a murine study with a transgenic mouse expressing MYCN targeted to the neural crest (TH-MYCN mice), the mouse develops neuroblastoma that begins with hyperplastic lesions in paravertebral ganglia within the first weeks after birth, escaping from the normal physiological process of cell death (46). This explains one of the etiologies of tumorigenesis due to amplification, though it should be kept in mind that not all gene amplification result in high level of gene expression (47, 48).
Secondly, somatic single nucleotide mutations, most frequently p.(Pro44Leu) (P44L), are identified in varieties of tumors such as neuroblastoma, medulloblastoma, and Wilms’ tumor (49–51). Among those, it is noteworthy that the P44L mutation is observed as frequently as 1.7% of high-risk neuroblastoma without MYCN amplification (49). Although the functional consequences of the P44L mutation remained unclear until recently, the significantly slower decay of MYCN was observed in OP9-DL1 cells transduced with lentiviral vectors expressing MYCN-P44L (52). Although the 44th proline is located adjacent to the conserved phosphordegron site by FBXW7, the interaction with FBWX7 is not altered in MYCN-P44L, that keeps the molecular background of the active MYCN function under investigations (53). In addition to the P44L variant, the mutationally stabilized variant, p.(Thr58Ala) (T58A), is analyzed using neural stem cells (NSCs) transduced with the variant. Transplantation of N-mycT58A embryonic cerebellar NSCs develops SHH-dependent medulloblastoma (54). Collectively, MYCN functions as a key oncogenic driver in various tumors.
For understanding the essential roles of MYCN in human developmental diseases as well as oncogenesis, the spatiotemporal expression patterns during development should be emphasized. In terms of the temporal aspect, Mycn is highly expressed at embryonic day (E) 13.5, then gradually decreased to its lowest level by postnatal day (P) 15 in mice, suggesting its important roles during embryo- and organogenesis (34). Expanding the focus on the spatial aspect, RNA in situ hybridization and Northern blot analysis of murine embryos suggest the extensive expression profiles of Mycn. Although Myc transcripts are expressed in various tissues at lower levels, Mycn expression indicates the tissue- and cell-specific patterns (55), preferring epithelial tissues, highest in the central nervous system, cranial and spinal ganglia in the peripheral nervous system, and heart (55). In addition, the transcripts are found in the developing gut, kidney, and lung. Interestingly, Myc transcripts are restricted to the mesenchymal compartments, rather than the epithelium, indicating interconnected regulatory mechanisms governing embryogenesis (55). Overall, these spatiotemporal expression profiles during development suggest its pivotal role in normal developmental processes beyond the context of tumorigenesis and progression.
MYCN and other MYC family are regulated by various upstream pathways (Figure 1). Among those are Wnt/β-catenin and SHH pathways that directly target MYCN (14–17), whereas Notch and IL6-JAK-STAT3 pathways are established for the upstream pathways in the context of MYC but not yet of MYCN (56, 57). Wnt signaling pathway initiates with the binding of Wnt ligand to the frizzled receptor and LDL receptor-related protein (LRP) co-receptor. Though GSK-3, CK-1α, Axin, and APC complex with and degrade β-catenin in the absence of the ligand, the binding of the ligand initiates the disruption of the protein complex. Then, dissociated from the complex, accumulates in cytoplasm, subsequently translocating into the nucleus, where it couples with T cell factor/lymphoid enhancer factor, initiating the transcription of targets genes, including MYC and MYCN (58). Speaking of SHH pathway, the sonic hedgehog signaling molecule (SHH) serves as the principal inductive ligand, crucial for shaping the ventral neural tube, determining the anterior-posterior axis of limbs, and influencing the patterning of ventral somites (59, 60). Briefly, SHH initiates a cascade of events by binding to its receptor, patched 1 (PTCH1), relieving its inhibition on smoothened, frizzled class receptor (SMO) and leading to the release of sufu negative regulator of hedgehog signaling (SUFU) from GLI family zinc finger 1, 2, and 3 (GLI1, 2, and 3) (61, 62). This process mediates the intranuclear transition of GLI1 and 2 and prevents GLI3 cleaved into the repressor form, GLI3R, together promoting the GLI1, 2 and GLI3A (the active form) mediated-expression of SHH target genes, including MYCN (15, 16).

Figure 1 Overview of MYCN-upstream pathway. The Sonic Hedgehog (SHH) and Wnt/β-catenin pathway boosts MYCN transcription, whereas Notch and IL6/JAK/STAT3 pathway regulate MYC transcription. DLL, delta-like; LRP, LDL receptor-related protein. The figure was created with BioRender.com.
MYCN itself regulates cellular proliferation and differentiation by functioning as a transcription factor (Figure 2). Specifically, MYCN, coupled with MAX, controls these processes by activating or inhibiting the transcription of specific genes (63–66). This dimerization processes are competed by DNA-binding counterparts (e.g., MAX dimerization protein (MGA) and zinc finger- and BTB domain-containing protein 17 (ZBTB17, or MIZ-1)) (67, 68), which are essential for early development demonstrated by embryonic lethality of biallelic inactivation alleles though they have not been established as the cause of human diseases (69, 70). For transcriptional targets, POU5F1 is a critical gene involved in cellular self-renewal and differentiation. Studies have shown that MYCN directly binds to the enhancer region of the POU5F1 and enhances its transcription (27, 71). Consequently, POU5F1 engages with the promoter regions of CCND1 and CCND2, critical transcription factors controlling the transition from the G1 phase to the S phase, thereby facilitating cellular growth and proliferation (72, 73). Collectively, MYCN plays a significant role in modulating cells’ gene expression profile through such mechanisms, thereby regulating cellular proliferation and differentiation.
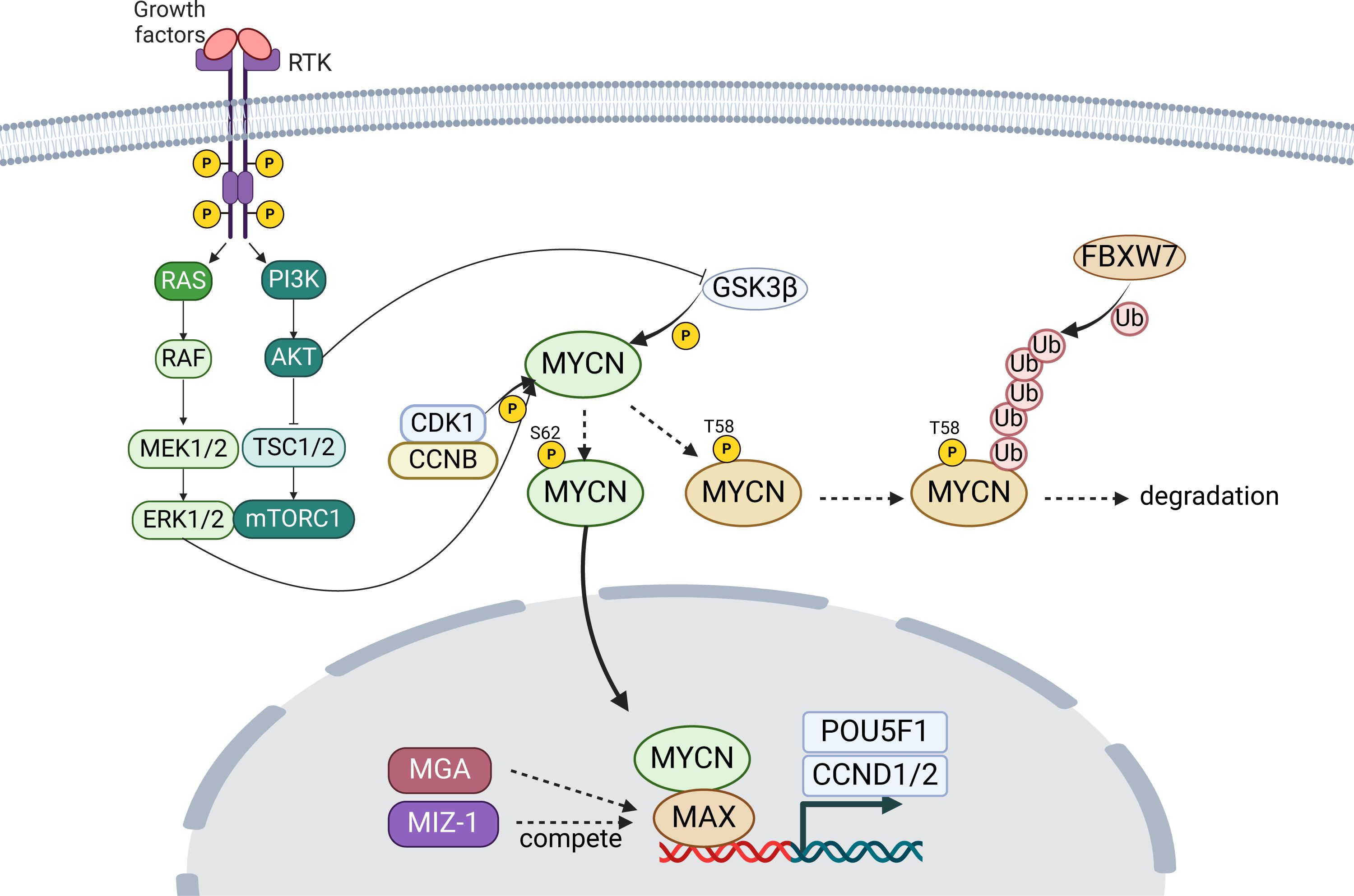
Figure 2 Overview of MYCN stabilization and activity. MYCN targets genes like POU5F1, essential for cell renewal, and promoting proliferation through genes CCND1 and CCND2. Stability of MYCN is modulated by phosphorylation, affecting its degradation or stabilization. RTK, receptor tyrosine kinase; P, phosphate; Ub, ubiquitin. The figure was created with BioRender.com.
In addition to the canonical targets, dysregulated MYCN expression also promotes binding to noncanonical (CANNTG) E-boxes clustered in enhancers, so called, “enhancer invasion” (74). When the expression of MYCN is physiological, it binds predominantly to canonical promoter E-boxes, but the excess MYCN spreads out, binding weaker E-box enhancer sites. The expression of genes with MYCN-invaded enhancers depends on the tissue-specific transcription factor, such as TWIST1 in neuroblastoma, defining highly tumor-specific ‘MYC target gene signatures’, expanding from the canonical targets’ origined signature (74).
In terms of modulating MYCN stability, the phosphorylation at Thr58 (T58) by glycogen synthase kinase three beta (GSK3β) that is regulated by PI3K/AKT pathway, following the phosphorylation at Ser62 (S62) by cyclin-dependent kinase 1 (CDK1) and ERK1/2, plays a crucial role (25, 26, 75, 76) (Figure 2). The phosphorylation at T58 destabilizes the MYCN protein, whereas that of S62 stabilizes it. Accordingly, dephosphorylation at T58 stabilizes the protein, preventing its degradation, following FBXW7-mediated ubiquitination (75, 76).
Collectively, the dynamic interaction between MYCN and associated pathways highlights the sophisticated regulatory networks governing cellular development, underscoring the complexity of developmental biology and the potential implications for understanding and treating developmental disorders.
Human disorders, including developmental diseases and tumors, associated with MYCN upstream pathways, SHH and WNT/β-catenin pathways, are well established so far. Although the contribution of MYCN, their downstream target, to the pathogenesis cannot be ignored to discuss the overall pathophysiology, it remains unclear because of their varieties of effects with multiple targets aside from MYCN. For imagining the potential MYCN’s roles in clinical consequences of those disorders through this review, we first introduce disorders associated with those two important upstream pathways (summarized in Table 1).
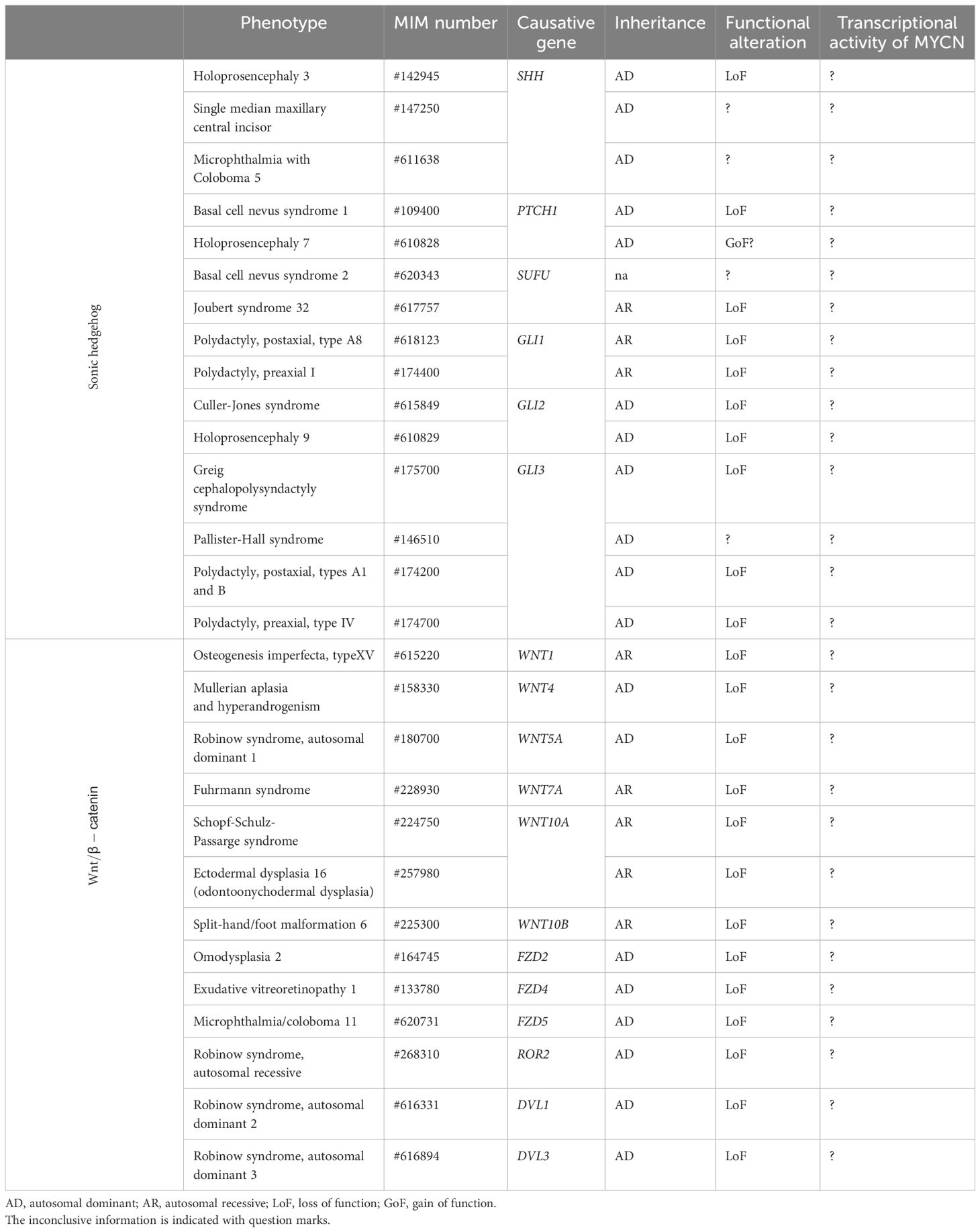
Table 1 MYCN upstream pathway and human developmental diseases.
SHH is associated with three distinct disorders: holoprosencephaly 3 (HPE3, OMIM #142945), single median maxillary central incisor (SMMCI, OMIM #147250) and microphthalmia with coloboma 5 (OMIM #611638). HPE3 is a severer spectrum of the failure for the developing forebrain to correctly segregate into distinct right and left hemispheres (77). In contrast, SMMCI manifests with the milder form, presenting only one deciduous and one permanent maxillary central incisor, often accompanied by short stature (78, 79). Furthermore, microphthalmia with coloboma 5 involves bilateral microphthalmia with inferonasal chorioretinal coloboma (78–80). Though the genotype-phenotype relationship has not been established, mutations in a long-range enhancer located upstream of SHH disrupt limb patterning, leading to the development of preaxial polydactyly (81).
Heterozygous mutations in PTCH1 (patched 1) have been identified in patients with Gorlin syndrome, also known as basal cell nevus syndrome 1 (BCNS1, OMIM #109400), and holoprosencephaly 7 (HPE7, OMIM #610828). BCNS1 is characterized by basal cell carcinomas, epidermal cysts, calcified dural folds, jaw keratocysts, palmar and plantar pits, ovarian fibromas, medulloblastomas, lymphomesenteric cysts, fetal rhabdomyoma, and various signs of maldevelopment, including macrocephaly (20, 21). HPE7, characterized by holoprosencephaly with incomplete penetrance, seems to be caused by the variants, acquiring enhanced repressive activity on the SHH pathway (19, 82).
Homozygous LoF variants within SUFU (SUFU negative regulator of hedgehog signaling) are identified in patients diagnosed with Joubert syndrome 32 (OMIM #617757), characterized by global developmental delay, intellectual disability, dysmorphic facial features, and minor anomalies such as postaxial polydactyly (22). In addition, germline variants in SUFU are found among patients affected with basal cell nevus syndrome 2 (BCNS2, OMIM #620343) (23, 83).
GLIs (GLI family zing fingers) are critical transcription factors, categorized into GLI1, 2, and 3. The GLI1 variants have been identified as the causes of polydactyly, preaxial I (OMIM #174400) and polydactyly, postaxial, type A8 (OMIM #618123) with an autosomal recessive manner (84, 85). Furthermore, mutations or dysregulation of GLI2 are linked to multiple human developmental diseases, such as Culler-Jones syndrome (OMIM #615849), characterized by hypopituitarism, abnormalities of the external genitalia, and postaxial polydactyly (86) and holoprosencephaly 9 (HPE9, OMIM #610829) (87). In addition, the variants in GLI3 have been identified in patients of Greig cephalopolysyndactyly syndrome (GCPS, OMIM #175700) and Pallister-Hall syndrome (PHS, OMIM #146510). The former is characterized by frontal bossing, scaphocephaly, and hypertelorism, alongside pre- and postaxial polydactyly with syndactyly (88), whereas, the latter manifests with hypothalamic hamartoma, pituitary dysfunction, central polydactyly, and visceral malformations (89). GLI3 mutations also cause isolated pre- and postaxial polydactyly (OMIM #174200) (90, 91).
WNTs (Wingless-type MMTB integration site family), a family of secreted glycoproteins, are associated with varieties of developmental diseases (24): WNT1 for osteogenesis imperfecta, type XV characterized by bone fragility and low bone mass with developmental delay and brain malformation (OMIM#615220), WNT4 for mullerian aplasia and hyperandrogenism by aplasia of mullerian duct derivatives (OMIM#158330), WNT5A for Robinow syndrome, autosomal dominant 1 by dysmorphic features, mesomelic limb shortening, hypoplastic external genitalia in males, and renal and vertebral anomalies (OMIM#180700), WNT7A for Fuhrmann syndrome by skeletal anomalies including polydactyly (OMIM#601570), WNT10A for Schopf-Schulz-Passarge syndrome (OMIM#224750), ectodermal dysplasia 16 (odontoonychodermal dysplasia) (OMIM#257980), and WNT10B for split-hand/foot malformation 6 (OMIM#225300).
The receptors for the WNT ligand (FZD, frizzled class receptor) are also associated with various developmental diseases: FZD2 for omodysplasia 2 (OMIM#164745), FZD4 for exudative vitreoretinopathy 1 (OMIM#133780), FZD5 for microphthalmia/coloboma 11 (OMIM#620731), and ROR2 for Robinow syndrome, autosomal recessive (OMIM#268310). Furthermore, DVL1 and 3, encoding dishevelled, an intracellular scaffolding protein that act downstream of transmembrane WNT receptors, have been identified in the patients with Robinow syndrome 2 and 3 (OMIM#616331 and #616894).
MYCN variants have been linked to two distinct human developmental diseases: FS1 and megalencephaly-polydactyly syndrome (summarized in Table 2).
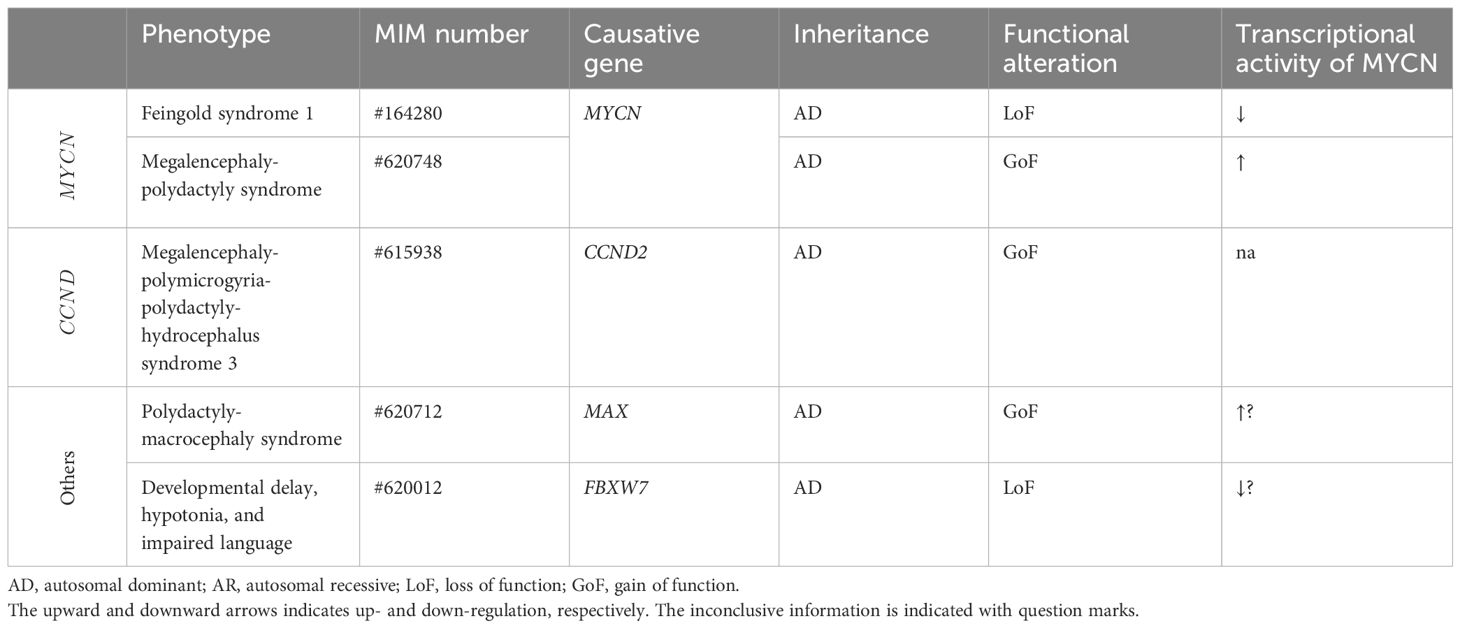
Table 2 MYCN, CCND2, and associated genes and human developmental diseases.
FS1 is characterized by a range of congenital anomalies, including digital anomalies (such as absent or hypoplastic phalanges), microcephaly, dysmorphic facial features, gastrointestinal atresia, and intellectual disability (13). Occasional findings may also include renal and cardiac abnormalities, along with hearing impairment (13). LoF variants in MYCN have been identified as the cause for this syndrome (92, 93).
In contrast to FS1 phenotypes, a de novo MYCN missense variant, p.(Thr58Met), is identified in a 15-year-old patient with megalencephaly, postaxial polydactyly, and neuroblastoma (34). The variant is located at the phosphorylation site in the MYC box 1, highly conserved, and absent in any population database (eg, gnomAD), although present in the database of somatic mutations in human cancers (eg, COSMIC). Functional analysis demonstrated the GoF properties of the identified variant (discussed later). Additionally, two additional cases involving a fetus and an 8-month-old patient, both of whom presented with remarkably similar phenotypes, including megalencephaly and postaxial polydactyly, with neuroblastoma observed in the latter, is documented to have heterozygous GoF MYCN variants, namely p.(Thr58Met) and p.(Pro60Leu) (35). The novel variant, p.(Pro60Leu), is located just near the phosphorylation site (the 58th threonine) in the MYC box 1, highly conserved with a damaging in silico prediction, and absent in any population database (eg, gnomAD), despite present in the database of somatic mutations in human cancers (eg, COSMIC). Notably, these patients exhibited a reduced amount of white matter and ventriculomegaly, differentiating this condition from other megalencephalic syndromes, especially those linked to disturbances in the mTOR pathway (31–33). Interestingly, the phenotypic characteristics of this novel syndrome mirrored those of FS1.
CCND2 GoF variants have been associated with megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome 3 (MPPH3, OMIM#615938), a developmental brain disorder characterized by megalencephaly, bilateral perisylvian polymicrogyria, and postaxial polydactyly (28).
There are several MYCN-related genes established as the causative genes for human developmental diseases. GoF mutations in MAX are implicated in polydactyly-macrocephaly syndrome (PDMCS, OMIM#620712), characterized by postaxial polydactyly, progressive macrocephaly, ocular anomalies, and neurodevelopmental issues (29). The FBXW7 is identified as the causative gene for developmental delay, hypotonia, and impaired language (OMIM #606278), characterized by global developmental delay, delayed speech, and distinctive facial features (30).
A germline knockout mouse is embryonic lethal, dying around E11.5, accompanied by notable organ abnormalities in the nervous system, mesonephros, lung, and gut, consistent with its expression pattern (55). Expanding the focus on neural crest-specific Mycn conditional knockout mice, the targeted removal of Mycn impaired the proliferation of granule neuron progenitors (GNPs), disrupted foliation, and resulted in a diminished cerebellar mass (94–96). As suggested in conventional knockout embryos, the increased Myc mRNA levels in Mycn-null GNPs were observed, and the simultaneous deletion of both Myc and Mycn worsened the impairment in cerebellar development (94–96). Furthermore, Mycn deficiency induced the premature expression of two cyclin-dependent kinase inhibitors, Kip1 and Ink4c, within the cerebellar primordium (94). Disrupting the Kip1 and Ink4c genes in Mycn-null cerebella partially restores GNPs proliferation and cerebellar foliation, providing conclusive genetic evidence supporting the assertion that Mycn expression, along with the concurrent down-regulation of Ink4c and Kip1, significantly contributes to the proper developmental trajectory of the cerebellum. In addition to its extensive analyses of the central nervous system, skeletal mesenchymal stem cell-specific Mycn conditional knockout mice were generated as models for Feingold syndrome (97). These mice, recapitulating the human phenotypes of limb abnormalities, revealed another dimension of significance during embryo- and organogenesis. Although the expression of miR-17-92, which is coded by MIR17HG, the causative gene of Feingold syndrome type 2, is controlled with transcriptional regulation by MYCN, the study demonstrated the distinct molecular mechanisms between Feingold syndrome type 1 and 2 (97). Interestingly, Mir17-92 deficiency leads to the upregulation of TGF-β signaling, while Mycn deficiency induces the downregulation of PI3K signaling in limb mesenchymal cells. In addition, skeletal anomalies arising from Mir17-92 deficiency can be effectively ameliorated through genetic or pharmacological inhibition of TGF-β signaling, indicating a crucial role for elevated TGF-β signaling in the skeletal abnormalities of Feingold syndrome type 2, but the skeletal phenotype associated with Mycn-deficiency experiences only partial mitigation through Pten heterozygosity and fails to respond to TGF-β inhibition (97). These findings strongly emphasize that, despite phenotypic similarities, distinct and complicated molecular mechanisms govern the maldevelopment of Feingold syndrome types 1 and 2 (97).
We previously established the GoF MYCN-induced megalencephaly-polydactyly syndrome (34, 35) (summarized in Figure 3). For the functional aspect, HEK293T cells transfected with plasmids expressing MYCN-WT, MYCN-T58M, and MYCN-P60L revealed significantly lower T58 phosphorylation levels in those transfected with both MYCN-T58M and -P60L compared to MYCN-WT (34, 35). Additionally, when plasmids expressing MYCN-WT, MYCN-T58M, and MYCN-P60L were co-transfected with Fbw7, the ubiquitin ligase targeting MYCN, both MYCN-T58M and -P60L displayed increased stability and resistance to degradation (34, 35). Crucially, the mutant MYCN proteins retained their ability to activate transcription of downstream genes (CCND1 and CCND2), affirming their sustained canonical activity as transcription factors, revealing GoF characteristics of both variants (34, 35).
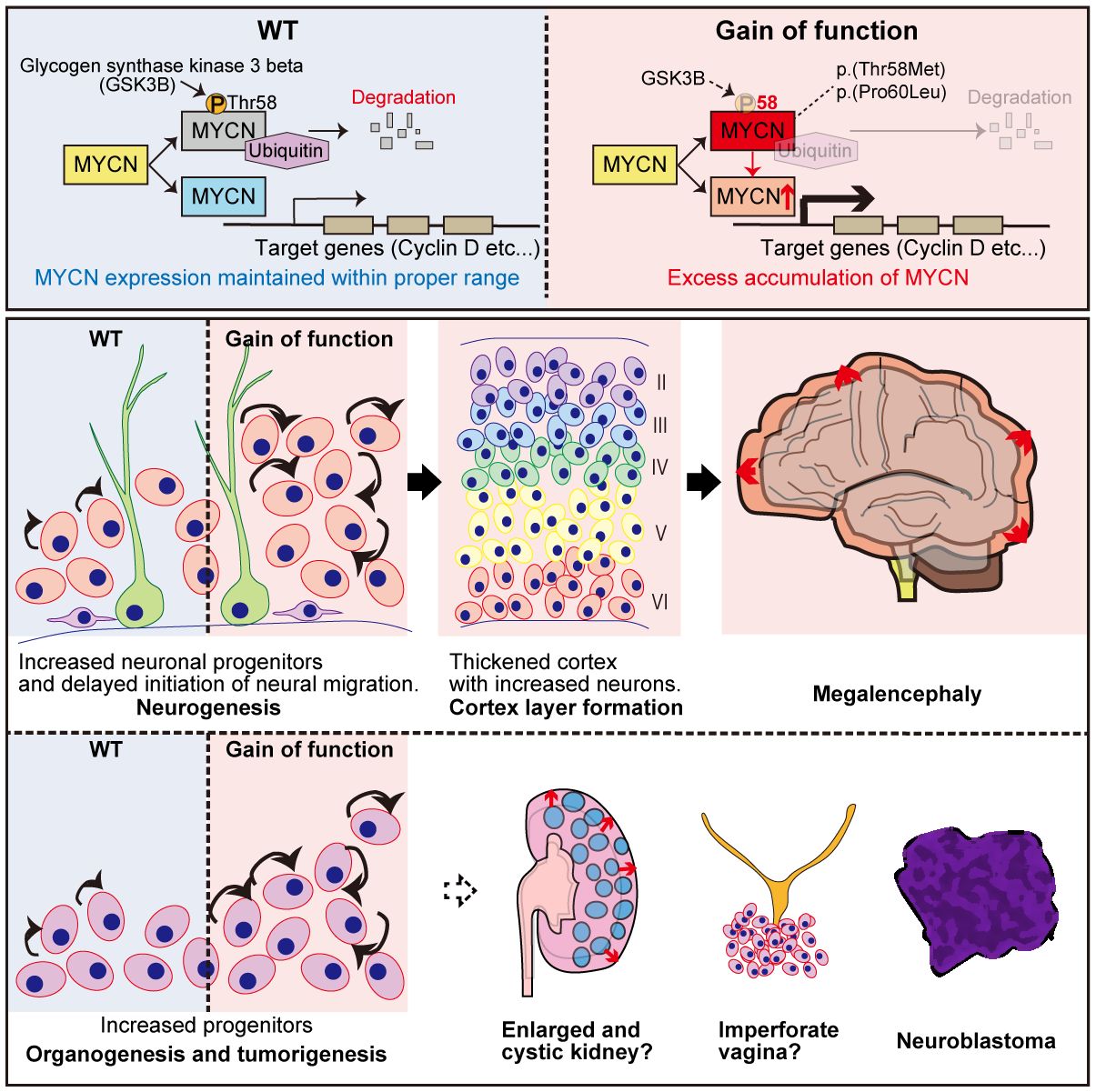
Figure 3 Graphical overview of MYCN gain-of-function induced megalencephaly-polydactyly syndrome. This figure presents findings on MYCN’s role in megalencephaly-polydactyly syndrome, highlighting gain-of-function MYCN variants linked to decreased protein degradation and increased cell proliferation. Through functional analyses and mouse models, the study reveals that these gain-of-function mutations lead to over-proliferation of neural precursors and morphological anomalies in kidneys and the female reproductive tract. The findings confirm MYCN’s significant proliferative role in both organogenesis and tumorigenesis, establishing a syndrome that contrasts with Feingold syndrome, which is associated with MYCN loss-of-function mutations.
Mouse models were generated to delve deeper into Mycn gain- and loss-of-function roles in murine development (35). The GoF (T58M/WT) mice recapitulated megalencephaly-polydactyly syndrome, while the haploinsufficient model (frameshift/WT) manifested microcephaly, which is one of the significant characteristics of Feingold syndrome (35). Semi-quantitative analysis unveiled decreased T58 phosphorylation and increased Mycn accumulation in GoF mice. Further investigations into the central nervous system at P0 and P7 elucidated a thickened cerebral cortex with increased neurons in GoF mice, contributing to megalencephaly, indicating that the megalencephalic phenotype in GoF mice was already determined prenatally (35).
The study then analyzed back neurogenesis in GoF mice, uncovering delayed neuronal migration and an elevated number of intermediate neural precursors at E14.5, contributing to the pathophysiological background of megalencephaly (35). The increased number of intermediate neural precursors (IPCs) and delayed neuronal migration were consistent with prior research indicating that Mycn overexpression leads to delayed cell cycle exit and migration. More precisely, MYCN has been reported to modulate the equilibrium between symmetrical and asymmetrical cell division, with overexpression promoting symmetrical cell division (98, 99).
Beyond the central nervous system manifestations, morphological alterations in various organs of GoF mice were observed, including digits, female reproductive system, and kidneys, which was consistent with its almost ubiquitous expression pattern (35). In the digits, over 90% of the mice exhibited postaxial polydactyly, which could be explained by the prior research that has elucidated the involvement of Shh signaling in limb development, influencing the establishment of the anterior-posterior axis for digit specification and the proliferation of limb mesenchymal cell (100–103).
Within the female reproductive system, imperforate vagina and hydrometra, arising from the vaginal closure with over-proliferative epithelium, were identified (35). Shh signaling is recognized for its role in activating the proliferation of Müllerian epithelial cell in the uterus and vagina (104), potentially contributing to the histological alterations observed in the female reproductive tract in our mouse model. Furthermore, the Shh signal is implicated in the progression of polycystic kidney disease (105), and its inhibition has demonstrated preventive effects on excessive proliferation and microcyst formation in the disease (106).
The triple knockout mice of Ccnd1, 2, and 3 were generated to investigate the role of cyclin Ds during development (107). The mice were viable until mid or late gestation but died due to a heart defect associated with severe hematologic abnormality, anemia, suggesting important roles in expanding hematopoietic stem cells. Fibroblasts deficient of Ccnds proliferated almost normally but required increased mitogenic stimulation in cell cycle reentry (107). The GoF CCND2 variants, the cause of MPPH3, were clustered around a residue that is phosphorylated by glycogen synthase kinase 3β (GSK3β) (28). Mutant CCND2 prevents itself from proteasomal degradation compared to wildtype CCND2. In utero electroporation of mutant CCND2 into embryonic mouse brains resulted in the transfected progenitors proliferating more and exhibited delayed cell cycle exit compared to cells transfected with wildtype CCND2, suggesting important functions in regulating cell cycle progression (28).
The GoF MAX variants, the cause of polydactyly-macrocephaly syndrome, are present in the b-HLH-LZ domain, where the mutant MAX binds its target E-box sequence with a lower apparent affinity, leading to a more efficient heterodimerization with Myc and an increase in transcriptional activity of Myc (29).
Speaking of FBXW7, Fbw7 knockout mice exhibited hematopoietic and vascular development deficiencies, leading to early embryonic lethality (108, 109). The absence of Fbw7 resulted in elevated levels of cyclin E and increased notch signaling, emphasizing its regulatory functions in cell cycle control and notch pathway modulation (109). Consistently, cellular analysis with HEK293T cells transfected with the pathogenic variants of FBXW7 showed an impaired ability to ubiquitinate and degrade substrates such as CCNE1 and CCNE2 (30).
Our comprehensive review sheds new light on the dual roles of MYCN in developmental processes as well as well-established roles in the context of oncogenesis. It should be emphasized that the intricate balance of MYCN expression is fundamental for normal development. The reduced expression leads to FS1 (92, 93) and excessive stability results in megalencephaly-polydactyly syndrome in developmental scenarios (34, 35). The importance of this intricately controlled balance is reinforced by the partially overlapped phenotypes of MYCN-related disorders and from the fact in the oncogenic field that dysfunctions of all the MYCN-related genes introduced in this review are associated with tumorigenesis and progression: hedgehog signaling (110–113), cyclin Ds (114–116), FBXW7 (117–119), and MAX (6).
The proper spatiotemporal expression of MYCN, both quantitatively and geographically, accomplished both by the precisely regulated transcription by the upstream pathways and intricately adjusted phosphorylation followed by ubiquitination by FBXW7 is key to normal developmental processes (120). Needless to mention the direct aberration affecting the quantity of MYCN expression, the dysregulation of its pathway component exerts adverse effects enough to compromise the normal developmental processes.
The strength of this review is that the MYCN’s direct contributions to the pathogenesis of human developmental diseases are made clear. This could result in the detailed understanding of the disorders associated with MYCN, such as SHH or Wnt/β-catenin pathway, contributing to the development of the therapeutic approaches targeting this specific pathway. Noteworthy, the growing evidence of the therapeutic approaches is proposed in terms of the treatment for MYCN-associated tumors, which targets MYCN transcription, stability, MYCN cofactors/coregulators and MYCN downstream targets (121). In addition, the investigations targeting hedgehog signaling, cyclin D, and FBXW7 are also under development (122–125). Though its relative difficulty for application to developmental disease due to the more intricate adjustment during development, not only quantitatively but also geographically, this oncologic knowledge might contribute to the development of therapeutic approaches even to the developmental disorders discussed in this review.
This review has potential limitations. Firstly, the disorder with GoF MYCN was just discovered that the clinical and molecular information is limited so far. For explaining the MYCN-related pathogenesis in associated disorders, it should be based not only on clinical information but also on molecular data. The future study would enrich the molecular understanding of the diseases, resulting in the comprehensive understanding of associated disorders. Secondly, we lack the data regarding MYCN’s transcriptional signature for the GoF model. It is unclear whether “enhancer invasion” would be present in the model in our situation where the expression level would be intermediate compared with MYCN amplification, The comprehensive understanding of the transcriptional signature is required to understand the pathophysiology more specifically.
The dual roles of MYCN in development and oncogenesis underscore the significance in cellular proliferation, differentiation, and apoptosis. The intricate network of interactions and signaling pathways involving MYCN and its associated genes highlights the complexity of regulatory mechanisms governing normal development and their potential disruption in diseases. Continuing exploration of these pathways holds promise for unraveling the molecular backgrounds of developmental disorders and cancers, offering hope for new therapeutic strategies. Our review advances our understanding of MYCN’s role in human biology and opens new avenues for research into the prevention and treatment of the conditions associated with its dysregulation.
YN: Writing – original draft, Writing – review & editing. KK: Writing – original draft, Writing – review & editing. HO: Writing – review & editing. YT: Writing – review & editing. SS: Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was partially supported by JSPS KAKENHI Grant Number JP24K02424 (SS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. DePinho R, Mitsock L, Hatton K, Ferrier P, Zimmerman K, Legouy E, et al. Myc family of cellular oncogenes. J Cell Biochem. (1987) 33:257–66. doi: 10.1002/jcb.240330404
2. Bretones G, Delgado MD, León J. Myc and cell cycle control. Biochim Biophys Acta. (2015) 1849:506–16. doi: 10.1016/j.bbagrm.2014.03.013
4. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. (2003) 3:203–16. doi: 10.1038/nrc1014
5. Rickman DS, Schulte JH, Eilers M. The expanding world of N-MYC-driven tumors. Cancer Discovery. (2018) 8:150–63. doi: 10.1158/2159-8290.CD-17-0273
6. Jiménez Martín O, Schlosser A, Furtwängler R, Wegert J, Gessler M. MYCN and MAX alterations in Wilms tumor and identification of novel N-MYC interaction partners as biomarker candidates. Cancer Cell Int. (2021) 21:555. doi: 10.1186/s12935-021-02259-2
7. Driman D, Thorner PS, Greenberg ML, Chilton-MacNeill S, Squire J. MYCN gene amplification in rhabdomyosarcoma. Cancer. (1994) 73:2231–7. doi: 10.1002/(ISSN)1097-0142
8. Massó-Vallés D, Beaulieu ME, Soucek LMYC. MYCL, and MYCN as therapeutic targets in lung cancer. Expert Opin Ther Targets. (2020) 24:101–14. doi: 10.1080/14728222.2020.1723548
9. Ota S, Zhou ZQ, Keene DR, Knoepfler P, Hurlin PJ. Activities of N-Myc in the developing limb link control of skeletal size with digit separation. Development. (2007) 134:1583–92. doi: 10.1242/dev.000703
10. Moens CB, Stanton BR, Parada LF, Rossant J. Defects in heart and lung development in compound heterozygotes for two different targeted mutations at the N-myc locus. Development. (1993) 119:485–99. doi: 10.1242/dev.119.2.485
11. Grady EF, Schwab M, Rosenau W. Expression of N-myc and c-src during the development of fetal human brain. Cancer Res. (1987) 47:2931–6.
12. Hirvonen H, Mäkelä TP, Sandberg M, Kalimo H, Vuorio E, Alitalo K. Expression of the myc proto-oncogenes in developing human fetal brain. Oncogene. (1990) 5:1787–97.
13. Celli J, van Bokhoven H, Brunner HG. Feingold syndrome: clinical review and genetic mapping. Am J Med Genet A. (2003) 122a:294–300. doi: 10.1002/ajmg.a.20471
14. Kuwahara A, Hirabayashi Y, Knoepfler PS, Taketo MM, Sakai J, Kodama T, et al. Wnt signaling and its downstream target N-myc regulate basal progenitors in the developing neocortex. Development. (2010) 137:1035–44. doi: 10.1242/dev.046417
15. Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. (2008) 22:2454–72. doi: 10.1101/gad.1693608
16. Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. (2010) 24:670–82. doi: 10.1101/gad.1902910
17. Hatton BA, Knoepfler PS, Kenney AM, Rowitch DH, de Alborán IM, Olson JM, et al. N-myc is an essential downstream effector of Shh signaling during both normal and neoplastic cerebellar growth. Cancer Res. (2006) 66:8655–61. doi: 10.1158/0008-5472.CAN-06-1621
18. Nanni L, Ming JE, Bocian M, Steinhaus K, Bianchi DW, De Die-Smulders C, et al. The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum Mol Genet. (1999) 8:2479–88. doi: 10.1093/hmg/8.13.2479
19. Ming JE, Kaupas ME, Roessler E, Brunner HG, Golabi M, Tekin M, et al. Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum Genet. (2002) 110:297–301. doi: 10.1007/s00439-002-0695-5
20. Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. (1996) 272:1668–71. doi: 10.1126/science.272.5268.1668
21. Hahn H, Wicking C, Zaphiropoulos PG, Gailani MR, Shanley S, Chidambaram A, et al. Mutations of the human homolog of drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. (1996) 85:841–51. doi: 10.1016/S0092-8674(00)81268-4
22. De Mori R, Romani M, D’Arrigo S, Zaki MS, Lorefice E, Tardivo S, et al. Hypomorphic recessive variants in SUFU impair the sonic hedgehog pathway and cause Joubert syndrome with Cranio-facial and skeletal defects. Am J Hum Genet. (2017) 101:552–63. doi: 10.1016/j.ajhg.2017.08.017
23. Pastorino L, Ghiorzo P, Nasti S, Battistuzzi L, Cusano R, Marzocchi C, et al. Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A. (2009) 149a:1539–43. doi: 10.1002/ajmg.a.32944
24. Ng LF, Kaur P, Bunnag N, Suresh J, Sung ICH, Tan QH, et al. WNT signaling in disease. Cells. (2019) 8:826. doi: 10.3390/cells8080826
25. Carter JL, Hege K, Yang J, Kalpage HA, Su Y, Edwards H, et al. Targeting multiple signaling pathways: the new approach to acute myeloid leukemia therapy. Signal Transduction Targeted Ther. (2020) 5:288. doi: 10.1038/s41392-020-00361-x
26. Gustafson WC, Weiss WA. Myc proteins as therapeutic targets. Oncogene. (2010) 29:1249–59. doi: 10.1038/onc.2009.512
27. Otto T, Horn S, Brockmann M, Eilers U, Schüttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. (2009) 15:67–78. doi: 10.1016/j.ccr.2008.12.005
28. Mirzaa G, Parry DA, Fry AE, Giamanco KA, Schwartzentruber J, Vanstone M, et al. De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nat Genet. (2014) 46:510–5. doi: 10.1038/ng.2948
29. Harris EL, Roy V, Montagne M, Rose AMS, Livesey H, Reijnders MRF, et al. A recurrent de novo MAX p.Arg60Gln variant causes a syndromic overgrowth disorder through differential expression of c-Myc target genes. Am J Hum Genet. (2024) 111:119–32. doi: 10.1016/j.ajhg.2023.11.010
30. Stephenson SEM, Costain G, Blok LER, Silk MA, Nguyen TB, Dong X, et al. Germline variants in tumor suppressor FBXW7 lead to impaired ubiquitination and a neurodevelopmental syndrome. Am J Hum Genet. (2022) 109:601–17. doi: 10.1016/j.ajhg.2022.03.002
31. Dragoo DD, Taher A, Wong VK, Elsaiey A, Consul N, Mahmoud HS, et al. PTEN hamartoma tumor syndrome/cowden syndrome: genomics, oncogenesis, and imaging review for associated lesions and Malignancy. Cancers. (2021) 13:3120. doi: 10.3390/cancers13133120
32. Shiohama T, Levman J, Vasung L, Takahashi E. Brain morphological analysis in PTEN hamartoma tumor syndrome. Am J Med Genet A. (2020) 182:1117–29. doi: 10.1002/ajmg.a.61532
33. Alcantara D, Timms AE, Gripp K, Baker L, Park K, Collins S, et al. Mutations of AKT3 are associated with a wide spectrum of developmental disorders including extreme megalencephaly. Brain. (2017) 140:2610–22. doi: 10.1093/brain/awx203
34. Kato K, Miya F, Hamada N, Negishi Y, Narumi-Kishimoto Y, Ozawa H, et al. MYCN de novo gain-of-function mutation in a patient with a novel megalencephaly syndrome. J Med Genet. (2019) 56:388–95. doi: 10.1136/jmedgenet-2018-105487
35. Nishio Y, Kato K, Tran Mau-Them F, Futagawa H, Quélin C, Masuda S, et al. Gain-of-function MYCN causes a megalencephaly-polydactyly syndrome manifesting mirror phenotypes of Feingold syndrome. Hum Genet Genomics Advances. (2023) 4:100238. doi: 10.1016/j.xhgg.2023.100238
36. Aldosari N, Bigner SH, Burger PC, Becker L, Kepner JL, Friedman HS, et al. MYCC and MYCN oncogene amplification in medulloblastoma. A fluorescence in situ hybridization study on paraffin sections from the Children's Oncology Group. Arch Pathol Lab Med. (2002) 126:540–4. doi: 10.5858/2002-126-0540-MAMOAI
37. Freier K, Flechtenmacher C, Devens F, Hartschuh W, Hofele C, Lichter P, et al. Recurrent NMYC copy number gain and high protein expression in basal cell carcinoma. Oncol Rep. (2006) 15:1141–5. doi: 10.3892/or
38. Lee WH, Murphree AL, Benedict WF. Expression and amplification of the N-myc gene in primary retinoblastoma. Nature. (1984) 309:458–60. doi: 10.1038/309458a0
39. Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, et al. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med. (1985) 313:1111–6. doi: 10.1056/NEJM198510313131802
40. Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan QW, Goldenberg DD, et al. Pleiotropic role for MYCN in medulloblastoma. Genes Dev. (2010) 24:1059–72. doi: 10.1101/gad.1907510
41. Tonelli R, McIntyre A, Camerin C, Walters ZS, Di Leo K, Selfe J, et al. Antitumor activity of sustained N-myc reduction in rhabdomyosarcomas and transcriptional block by antigene therapy. Clin Cancer Res. (2012) 18:796–807. doi: 10.1158/1078-0432.CCR-11-1981
42. Williamson D, Lu YJ, Gordon T, Sciot R, Kelsey A, Fisher C, et al. Relationship between MYCN copy number and expression in rhabdomyosarcomas and correlation with adverse prognosis in the alveolar subtype. J Clin Oncol. (2005) 23:880–8. doi: 10.1200/JCO.2005.11.078
43. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. (2012) 123:465–72. doi: 10.1007/s00401-011-0922-z
44. Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. (2012) 488:49–56. doi: 10.1038/nature11327
45. Otte J, Dyberg C, Pepich A, Johnsen JI. MYCN function in neuroblastoma development. Front Oncol. (2020) 10:624079. doi: 10.3389/fonc.2020.624079
46. Hansford LM, Thomas WD, Keating JM, Burkhart CA, Peaston AE, Norris MD, et al. Mechanisms of embryonal tumor initiation: distinct roles for MycN expression and MYCN amplification. Proc Natl Acad Sci U.S.A. (2004) 101:12664–9. doi: 10.1073/pnas.0401083101
47. Chang HH, Tseng YF, Lu MY, Yang YL, Chou SW, Lin DT, et al. MYCN RNA levels determined by quantitative in situ hybridization is better than MYCN gene dosages in predicting the prognosis of neuroblastoma patients. Mod Pathol. (2020) 33:531–40. doi: 10.1038/s41379-019-0410-x
48. Cohn SL, London WB, Huang D, Katzenstein HM, Salwen HR, Reinhart T, et al. MYCN expression is not prognostic of adverse outcome in advanced-stage neuroblastoma with nonamplified MYCN. J Clin Oncol. (2000) 18:3604–13. doi: 10.1200/JCO.2000.18.21.3604
49. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. (2013) 45:279–84. doi: 10.1038/ng.2529
50. Jones DT, Jäger N, Kool M, Zichner T, Hutter B, Sultan M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. (2012) 488:100–5. doi: 10.1038/nature11284
51. Williams RD, Chagtai T, Alcaide-German M, Apps J, Wegert J, Popov S, et al. Multiple mechanisms of MYCN dysregulation in Wilms tumour. Oncotarget. (2015) 6:7232–43. doi: 10.18632/oncotarget.v6i9
52. Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. (2017) 49:1211–8. doi: 10.1038/ng.3909
53. Bonilla X, Parmentier L, King B, Bezrukov F, Kaya G, Zoete V, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Genet. (2016) 48:398–406. doi: 10.1038/ng.3525
54. Swrtling F, Savov V, Persson A, Chen J, Hackett C, Northcott P, et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell. (2012) 21:601–13. doi: 10.1016/j.ccr.2012.04.012
55. Stanton BR, Perkins AS, Tessarollo L, Sassoon DA, Parada LF. Loss of N-myc function results in embryonic lethality and failure of the epithelial component of the embryo to develop. Genes Dev. (1992) 6:2235–47. doi: 10.1101/gad.6.12a.2235
56. Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. (2006) 20:2096–109. doi: 10.1101/gad.1450406
57. Subramaniam KS, Omar IS, Kwong SC, Mohamed Z, Woo YL, Mat Adenan NA, et al. Cancer-associated fibroblasts promote endometrial cancer growth via activation of interleukin-6/STAT-3/c-Myc pathway. Am J Cancer Res. (2016) 6:200–13.
58. Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang X, et al. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther. (2022) 7:3. doi: 10.1038/s41392-021-00762-6
59. Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, et al. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. (1993) 75:1417–30. doi: 10.1016/0092-8674(93)90627-3
60. Roelink H, Augsburger A, Heemskerk J, Korzh V, Norlin S, Ruiz i Altaba A, et al. Floor plate and motor neuron induction by vhh-1, a vertebrate homolog of hedgehog expressed by the notochord. Cell. (1994) 76:761–75. doi: 10.1016/0092-8674(94)90514-2
61. Choudhry Z, Rikani AA, Choudhry AM, Tariq S, Zakaria F, Asghar MW, et al. Sonic hedgehog signalling pathway: a complex network. Ann Neurosci. (2014) 21:28–31. doi: 10.5214/ans.0972.7531
62. Carballo GB, Honorato JR, De Lopes GPF. Spohr TCLDSE. A highlight on Sonic hedgehog pathway. Cell Communication Signaling. (2018) 16):11. doi: 10.1186/s12964-018-0220-7
63. Blackwood EM, Eisenman RN. Regulation of Myc: Max complex formation and its potential role in cell proliferation. Tohoku J Exp Med. (1992) 168:195–202. doi: 10.1620/tjem.168.195
64. Nair SK, Burley SK. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell. (2003) 112:193–205. doi: 10.1016/S0092-8674(02)01284-9
65. Wenzel A, Schwab M. The mycN/max protein complex in neuroblastoma. Short review. Eur J Cancer. (1995) 31a:516–9. doi: 10.1016/0959-8049(95)00060-V
66. Cheung L, J E, Haber M, M D. “The MYCN Oncogene,” in Oncogene and Cancer - From Bench to Clinic, ed. Siregar Y. (IntechOpen) (2013) 437–454.
67. Hurlin PJ, Steingrìmsson E, Copeland NG, Jenkins NA, Eisenman RN. Mga, a dual-specificity transcription factor that interacts with Max and contains a T-domain DNA-binding motif. EMBO J. (1999) 18:7019–28. doi: 10.1093/emboj/18.24.7019
68. Bédard M, Maltais L, Montagne M, Lavigne P. Miz-1 and Max compete to engage c-Myc: implication for the mechanism of inhibition of c-Myc transcriptional activity by Miz-1. Proteins. (2017) 85:199–206. doi: 10.1002/prot.25214
69. Adhikary S, Peukert K, Karsunky H, Beuger V, Lutz W, Elsässer HP, et al. Miz1 is required for early embryonic development during gastrulation. Mol Cell Biol. (2003) 23:7648–57. doi: 10.1128/MCB.23.21.7648-7657.2003
70. Qin J, Wang C, Zhu Y, Su T, Dong L, Huang Y, et al. Mga safeguards embryonic stem cells from acquiring extraembryonic endoderm fates. Sci Adv. (2021) 7:eabe5689. doi: 10.1126/sciadv.abe5689
71. Suenaga Y, Nakatani K, Nakagawara A. De novo evolved gene product NCYM in the pathogenesis and clinical outcome of human neuroblastomas and other cancers. Japanese J Clin Oncol. (2020) 50:839–46. doi: 10.1093/jjco/hyaa097
72. Su C. Survivin in survival of hepatocellular carcinoma. Cancer Lett. (2016) 379:184–90. doi: 10.1016/j.canlet.2015.06.016
73. Bai M, Yuan M, Liao H, Chen J, Xie B, Yan D, et al. OCT4 pseudogene 5 upregulates OCT4 expression to promote proliferation by competing with miR-145 in endometrial carcinoma. Oncol Rep. (2015) 33:1745–52. doi: 10.3892/or.2015.3763
74. Zeid R, Lawlor MA, Poon E, Reyes JM, Fulciniti M, Lopez MA, et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat Genet. (2018) 50:515–23. doi: 10.1038/s41588-018-0044-9
75. Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. (2000) 14:2501–14. doi: 10.1101/gad.836800
76. Sjostrom SK, Finn G, Hahn WC, Rowitch DH, Kenney AM. The Cdk1 complex plays a prime role in regulating N-myc phosphorylation and turnover in neural precursors. Dev Cell. (2005) 9:327–38. doi: 10.1016/j.devcel.2005.07.014
77. Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, et al. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. (1996) 14:357–60. doi: 10.1038/ng1196-357
78. Nanni L, Ming JE, Du Y, Hall RK, Aldred M, Bankier A, et al. SHH mutation is associated with solitary median maxillary central incisor: a study of 13 patients and review of the literature. Am J Med Genet. (2001) 102:1–10. doi: 10.1002/(ISSN)1096-8628
79. Marini M, Cusano R, De Biasio P, Caroli F, Lerone M, Silengo M, et al. Previously undescribed nonsense mutation in SHH caused autosomal dominant holoprosencephaly with wide intrafamilial variability. Am J Med Genet A. (2003) 117a:112–5. doi: 10.1002/ajmg.a.10163
80. Schimmenti LA, de la Cruz J, Lewis RA, Karkera JD, Manligas GS, Roessler E, et al. Novel mutation in sonic hedgehog in non-syndromic colobomatous microphthalmia. Am J Med Genet A. (2003) 116a:215–21. doi: 10.1002/ajmg.a.10884
81. Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, Oostra BA, et al. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet. (2003) 12:1725–35. doi: 10.1093/hmg/ddg180
82. Ribeiro LA, Murray JC, Richieri-Costa A. PTCH mutations in four Brazilian patients with holoprosencephaly and in one with holoprosencephaly-like features and normal MRI. Am J Med Genet A. (2006) 140:2584–6. doi: 10.1002/ajmg.a.31369
83. Álvarez-Salafranca M, García-García M, Montes-Torres A, Rivera-Fuertes I, López-Giménez MT, Ara M. SUFU-associated Gorlin syndrome: Expanding the spectrum between classic nevoid basal cell carcinoma syndrome and multiple hereditary infundibulocystic basal cell carcinoma. Australas J Dermatol. (2023) 64:249–54. doi: 10.1111/ajd.14014
84. Palencia-Campos A, Ullah A, Nevado J, Yildirim R, Unal E, Ciorraga M, et al. GLI1 inactivation is associated with developmental phenotypes overlapping with Ellis-van Creveld syndrome. Hum Mol Genet. (2017) 26:4556–71. doi: 10.1093/hmg/ddx335
85. Ullah A, Umair M, Majeed AI, Abdullah, Jan A, Ahmad W. A novel homozygous sequence variant in GLI1 underlies first case of autosomal recessive pre-axial polydactyly. Clin Genet. (2019) 95:540–1. doi: 10.1111/cge.13495
86. França MM, Jorge AA, Carvalho LR, Costalonga EF, Vasques GA, Leite CC, et al. Novel heterozygous nonsense GLI2 mutations in patients with hypopituitarism and ectopic posterior pituitary lobe without holoprosencephaly. J Clin Endocrinol Metab. (2010) 95:E384–91. doi: 10.1210/jc.2010-1050
87. Roessler E, Du YZ, Mullor JL, Casas E, Allen WP, Gillessen-Kaesbach G, et al. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc Natl Acad Sci U.S.A. (2003) 100:13424–9. doi: 10.1073/pnas.2235734100
88. Biesecker LG. The Greig cephalopolysyndactyly syndrome. Orphanet J Rare Dis. (2008) 3:10. doi: 10.1186/1750-1172-3-10
89. Démurger F, Ichkou A, Mougou-Zerelli S, Le Merrer M, Goudefroye G, Delezoide AL, et al. New insights into genotype-phenotype correlation for GLI3 mutations. Eur J Hum Genet. (2015) 23:92–102. doi: 10.1038/ejhg.2014.62
90. Fujioka H, Ariga T, Horiuchi K, Otsu M, Igawa H, Kawashima K, et al. Molecular analysis of non-syndromic preaxial polydactyly: preaxial polydactyly type-IV and preaxial polydactyly type-I. Clin Genet. (2005) 67:429–33. doi: 10.1111/j.1399-0004.2005.00431.x
91. Al-Qattan MM. A novel frameshift mutation of the GLI3 gene in a family with broad thumbs with/without big toes, postaxial polydactyly and variable syndactyly of the hands/feet. Clin Genet. (2012) 82:502–4. doi: 10.1111/j.1399-0004.2012.01866.x
92. van Bokhoven H, Celli J, van Reeuwijk J, Rinne T, Glaudemans B, van Beusekom E, et al. MYCN haploinsufficiency is associated with reduced brain size and intestinal atresias in Feingold syndrome. Nat Genet. (2005) 37:465–7. doi: 10.1038/ng1546
93. Marcelis CL, Hol FA, Graham GE, Rieu PN, Kellermayer R, Meijer RP, et al. Genotype-phenotype correlations in MYCN-related Feingold syndrome. Hum Mutat. (2008) 29:1125–32. doi: 10.1002/humu.v29:9
94. Zindy F, Knoepfler PS, Xie S, Sherr CJ, Eisenman RN, Roussel MF. N-Myc and the cyclin-dependent kinase inhibitors p18Ink4c and p27Kip1 coordinately regulate cerebellar development. Proc Natl Acad Sci U.S.A. (2006) 103:11579–83. doi: 10.1073/pnas.0604727103
95. Kenney AM, Cole MD, Rowitch DH. Nmycupregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. (2003) 130:15–28. doi: 10.1242/dev.00182
96. Corrales JD, Rocco GL, Blaess S, Guo Q, Joyner AL. Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development. (2004) 131:5581–90. doi: 10.1242/dev.01438
97. Mirzamohammadi F, Kozlova A, Papaioannou G, Paltrinieri E, Ayturk UM, Kobayashi T. Distinct molecular pathways mediate Mycn and Myc-regulated miR-17-92 microRNA action in Feingold syndrome mouse models. Nat Commun. (2018) 9:1352. doi: 10.1038/s41467-018-03788-7
98. Huttner WB, Kosodo Y. Symmetric versus asymmetric cell division during neurogenesis in the developing vertebrate central nervous system. Curr Opin Cell Biol. (2005) 17:648–57. doi: 10.1016/j.ceb.2005.10.005
99. Izumi H, Kaneko Y, Nakagawara A. The role of MYCN in symmetric vs. Asymmetric cell division of human neuroblastoma cells. Front Oncol. (2020) 10:570815. doi: 10.3389/fonc.2020.570815
100. Riddle RD, Johnson RL, Laufer E, Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. (1993) 75:1401–16. doi: 10.1016/0092-8674(93)90626-2
101. Ahn S, Joyner AL. Dynamic changes in the response of cells to positive hedgehog signaling during mouse limb patterning. Cell. (2004) 118:505–16. doi: 10.1016/j.cell.2004.07.023
102. Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, Tabin CJ. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell. (2004) 118:517–28. doi: 10.1016/j.cell.2004.07.024
103. Francis-West P, Hill R. Uncoupling the role of sonic hedgehog in limb development: growth and specification. Sci Signal. (2008) 1:pe34. doi: 10.1126/scisignal.126pe34
104. Nakajima T, Iguchi T, Sato T. Hedgehog signaling plays roles in epithelial cell proliferation in neonatal mouse uterus and vagina. Cell Tissue Res. (2012) 348:239–47. doi: 10.1007/s00441-012-1350-7
105. Hsieh CL, Jerman SJ, Sun Z. Non-cell-autonomous activation of hedgehog signaling contributes to disease progression in a mouse model of renal cystic ciliopathy. Hum Mol Genet. (2022) 31:4228–40. doi: 10.1093/hmg/ddac175
106. Silva LM, Jacobs DT, Allard BA, Fields TA, Sharma M, Wallace DP, et al. Inhibition of Hedgehog signaling suppresses proliferation and microcyst formation of human Autosomal Dominant Polycystic Kidney Disease cells. Sci Rep. (2018) 8:4985. doi: 10.1038/s41598-018-23341-2
107. Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell. (2004) 118:477–91. doi: 10.1016/j.cell.2004.07.025
108. Thompson BJ, Jankovic V, Gao J, Buonamici S, Vest A, Lee JM, et al. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J Exp Med. (2008) 205:1395–408. doi: 10.1084/jem.20080277
109. Tetzlaff MT, Yu W, Li M, Zhang P, Finegold M, Mahon K, et al. Defective cardiovascular development and elevated cyclin E and Notch proteins in mice lacking the Fbw7 F-box protein. Proc Natl Acad Sci U.S.A. (2004) 101:3338–45. doi: 10.1073/pnas.0307875101
110. Liao X, Siu MK, Au CW, Wong ES, Chan HY, Ip PP, et al. Aberrant activation of hedgehog signaling pathway in ovarian cancers: effect on prognosis, cell invasion and differentiation. Carcinogenesis. (2009) 30:131–40. doi: 10.1093/carcin/bgn230
111. Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. (2007) 67:2187–96. doi: 10.1158/0008-5472.CAN-06-3281
112. Souzaki M, Kubo M, Kai M, Kameda C, Tanaka H, Taguchi T, et al. Hedgehog signaling pathway mediates the progression of non-invasive breast cancer to invasive breast cancer. Cancer Sci. (2011) 102:373–81. doi: 10.1111/j.1349-7006.2010.01779.x
113. Huaitong X, Yuanyong F, Yueqin T, Peng Z, Wei S, Kai S. Microvesicles releasing by oral cancer cells enhance endothelial cell angiogenesis via Shh/RhoA signaling pathway. Cancer Biol Ther. (2017) 18:783–91. doi: 10.1080/15384047.2017.1373213
114. Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: a perspective. Oncogene. (2005) 24:2909–15. doi: 10.1038/sj.onc.1208618
115. Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. (2011) 11:558–72. doi: 10.1038/nrc3090
116. Takano Y, Kato Y, van Diest PJ, Masuda M, Mitomi H, Okayasu I. Cyclin D2 overexpression and lack of p27 correlate positively and cyclin E inversely with a poor prognosis in gastric cancer cases. Am J Pathol. (2000) 156:585–94. doi: 10.1016/S0002-9440(10)64763-3
117. Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. (2008) 8:83–93. doi: 10.1038/nrc2290
118. Grim JE, Knoblaugh SE, Guthrie KA, Hagar A, Swanger J, Hespelt J, et al. Fbw7 and p53 cooperatively suppress advanced and chromosomally unstable intestinal cancer. Mol Cell Biol. (2012) 32:2160–7. doi: 10.1128/MCB.00305-12
119. Sancho R, Jandke A, Davis H, Diefenbacher ME, Tomlinson I, Behrens A. F-box and WD repeat domain-containing 7 regulates intestinal cell lineage commitment and is a haploinsufficient tumor suppressor. Gastroenterology. (2010) 139:929–41. doi: 10.1053/j.gastro.2010.05.078
120. Kenney AM, Rowitch DH. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol. (2000) 20:9055–67. doi: 10.1128/MCB.20.23.9055-9067.2000
121. Liu Z, Chen SS, Clarke S, Veschi V, Thiele CJ. Targeting MYCN in pediatric and adult cancers. Front Oncol. (2020) 10:623679. doi: 10.3389/fonc.2020.623679
122. Nguyen NM, Cho J. Hedgehog pathway inhibitors as targeted cancer therapy and strategies to overcome drug resistance. Int J Mol Sci. (2022) 23:1733. doi: 10.3390/ijms23031733
123. Chen S, Leng P, Guo J, Zhou H. FBXW7 in breast cancer: mechanism of action and therapeutic potential. J Exp Clin Cancer Res. (2023) 42:226. doi: 10.1186/s13046-023-02767-1
124. Chen S, Lin J, Zhao J, Lin Q, Liu J, Wang Q, et al. FBXW7 attenuates tumor drug resistance and enhances the efficacy of immunotherapy. Front Oncol. (2023) 13. doi: 10.3389/fonc.2023.1147239
Keywords: MYCN, megalencephaly, polydactyly, Feingold syndrome, human development
Citation: Nishio Y, Kato K, Oishi H, Takahashi Y and Saitoh S (2024) MYCN in human development and diseases. Front. Oncol. 14:1417607. doi: 10.3389/fonc.2024.1417607
Received: 15 April 2024; Accepted: 15 May 2024;
Published: 31 May 2024.
Edited by:
Yusuke Suenaga, Chiba Cancer Center, JapanCopyright © 2024 Nishio, Kato, Oishi, Takahashi and Saitoh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shinji Saitoh, c3MxMUBtZWQubmFnb3lhLWN1LmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.