 Yanyan Ling
Yanyan Ling Huaiqiang Hu2
Huaiqiang Hu2- 1Department of Neurology, Shandong Second Provincial General Hospital, Jinan, China
- 2Department of Neurology, The 960th Hospital of Joint Logistics Force, PLA, Jinan, China
- 3Department of Traumatic Orthopedics, Shandong Second Provincial General Hospital, Jinan, China
- 4Department of Emergency, Shandong Second Provincial General Hospital, Jinan, China
- 5Department of Neurology, The 970th Hospital of Joint Logistics Force, PLA, Yantai, China
Background: Juvenile dermatomyositis (JDM) is a rare autoimmune myopathy whose main clinical manifestations include a characteristic rash, symmetrical proximal muscle weakness, and elevated muscle enzymes. While approximately one-third of adult patients with dermatomyositis (DM) develop malignancies, typically within a year of diagnosis, this phenomenon is not commonly observed in patients with JDM. In this study, we present a rare case of both JDM and Hodgkin’s lymphoma (HL) diagnosed in an adolescent female patient.
Case description: A 14-year-old girl with proximal muscle weakness and myalgia for 8 weeks was admitted to the hospital and ultimately received a diagnosis of DM. A thorough physical examination revealed enlarged lymph nodes on both sides of the cervical, and a lymph node biopsy was performed to diagnose HL. After she underwent radiotherapy and chemotherapy, her symptoms of both HL and DM were alleviated.
Conclusion: The phenomenon of JDM as a paraneoplastic syndrome associated with HL is very rare. Thus, routine cancer screening for DM in adolescents is currently not recommended. The diagnosis of JDM requires a detailed physical examination, and further tumor screening is necessary for patients with unusual physical findings, such as atypical rashes, enlarged lymph nodes, and enlargement of the spleen and/or liver. Even if no malignancy is detected when JDM is diagnosed, long-term follow-up is necessary.
Introduction
Juvenile dermatomyositis (JDM)—an immune-mediated multisystem disease characterized by acute and chronic non-suppurative inflammation affecting the skin, striated muscles, lungs, and gastrointestinal tract (1)—is the most common idiopathic inflammatory myopathy (IIM) in children. Typical clinical manifestations include a lilac rash on the eyelids, Gottron’s sign, and symmetrical proximal muscle weakness of the limbs. Additionally, significant elevation of muscle enzymes, such as creatine kinase (CK), is observed (2, 3). Epidemiological data indicate an annual incidence of 2 to 4 cases per million children (4), with the onset typically peaking between the ages of 5 and 10 years and girls being more commonly affected than boys, with an estimated ratio of 2-5:1 (5). In adults, the association between dermatomyositis (DM) and malignancy is well known, with a reported prevalence of 7 to 60%, especially in those demonstrating disease onset after the age of 50 (6, 7). Therefore, extensive screening for occult cancers is common for adults with DM. In contrast, an association between DM and malignancy has rarely been noted in children. Here, we present a case of JDM in a patient with Hodgkin’s lymphoma (HL) as a paraneoplastic manifestation. Additionally, we conducted a PubMed search and retrieved reports of four cases of HL combined with JDM. Our case report clarifies whether routine evaluation of malignant tumors is recommended for patients with JDM and the circumstances under which further screening for malignant tumors is required.
Case presentation
A 14-year-old girl with obesity experiencing symmetrical proximal limb muscle weakness and myalgia for 8 weeks was admitted to the 960th Hospital of Joint Logistics Force, PLA. Cutaneous manifestations included a lilac rash around both eyes and a flaky V-shaped rash on the chest. The patient did not have speaking or breathing difficulties, a family history of autoimmune disease or malignancy, or a history of fever before the onset of muscle pain. Her development was otherwise normal.
During the physical examination, the patient was found to experience pain in the extremities upon pressing. She also exhibited generalized edema, including periorbital swelling and bilateral non-pitting edema of the upper and lower limbs. Bilateral cervical lymph node enlargement was detected on palpation; however, there was no evidence of organomegaly. The Manual Muscle Testing (MMT-8) score was 76, with the following specific muscle group scores: 8 for elbow flexors, 8 for hip flexors, 10 for shoulder abductors, 10 for wrist extensors, 10 for ankle dorsiflexors, 10 for neck flexors, 10 for hip abductors, and 10 for hip extensors (8). Electromyography (EMG) findings were consistent with the diagnosis of inflammatory muscle disease. To further evaluate the bilateral cervical lymphadenopathy noted during the physical examination, a chest computed tomography (CT) scan was performed. CT revealed an anterior mediastinal mass, bilateral subclavian lymph node enlargement, and bilateral axillary lymph node enlargement (Figure 1). Biopsy of the left brachii muscle showed evidence consistent with the presence of DM (Figure 2), and cervical lymph node biopsy indicated nodular sclerosing HL (nsHL) (Figure 3). F-fluorodeoxyglucose positron emission tomography (FDG-PET) revealed mild diffuse increases in subcutaneous FDG metabolism in the extremities, along with anterior diaphragmatic lymphadenopathy and involvement of the sternal stalk (Figure 4). Bone marrow aspiration results showed no abnormalities.
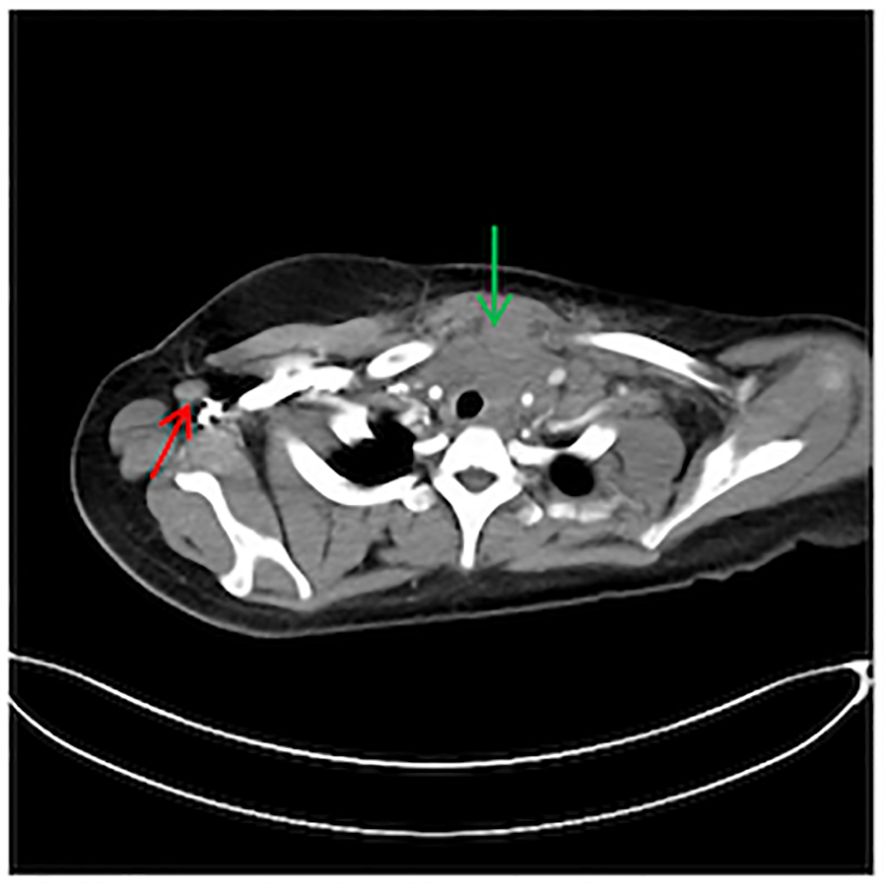
Figure 1 Chest computed tomography (CT) showing an anterior mediastinal mass (Green) and right axillary lymph node enlargement (Red).
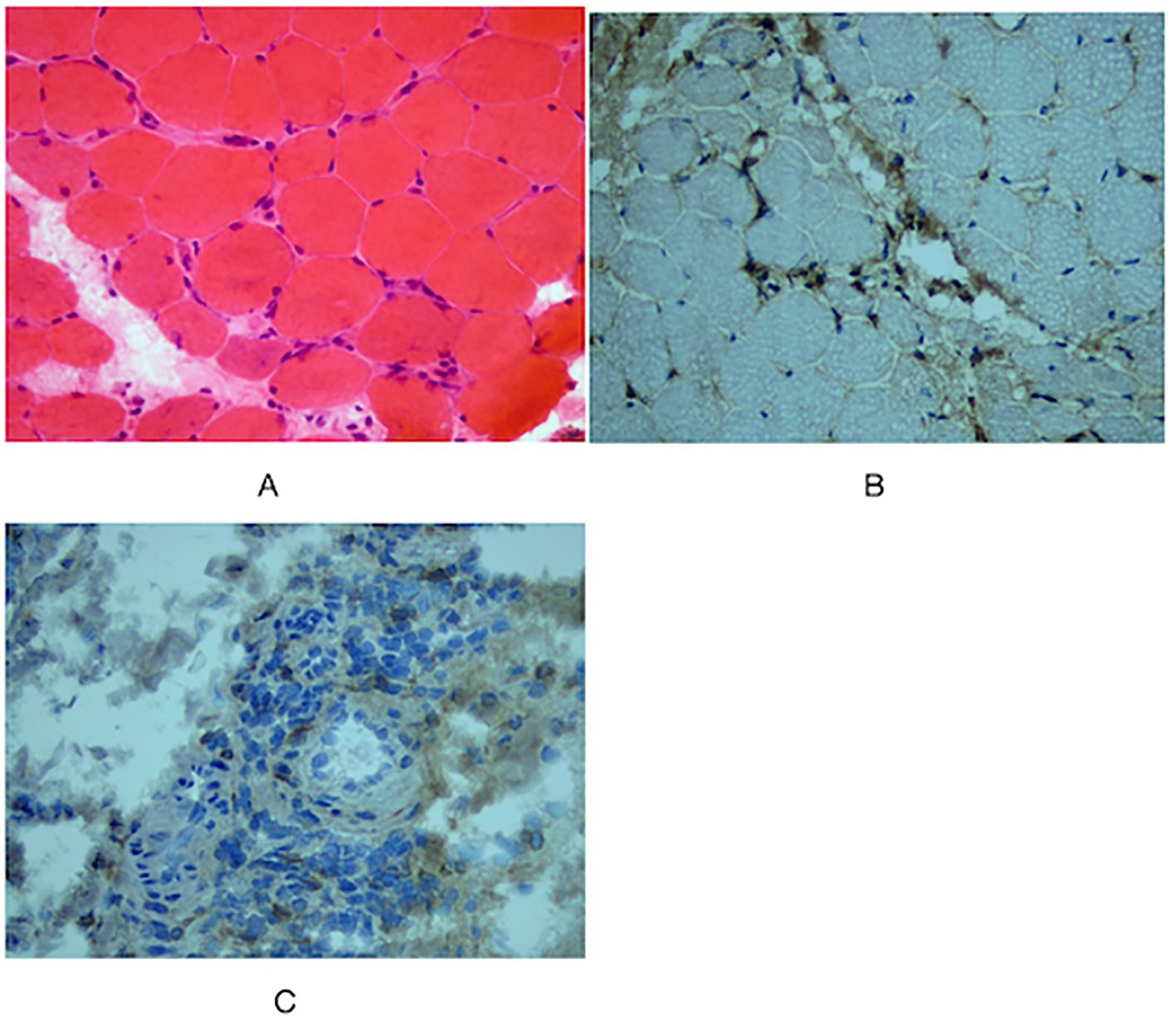
Figure 2 Muscle biopsy of left musculus biceps brachii. (A) Hematoxylin and Eosin (HE) staining showed perifascicular atrophy of muscle fibers, and perivascular inflammatory infiltrates (x40). No regenerating muscle fibers are observed, and there is no apparent nuclear migration of muscle fibers. Immunohistochemical staining showed (B) CD4+, (C) CD68+ (×40).
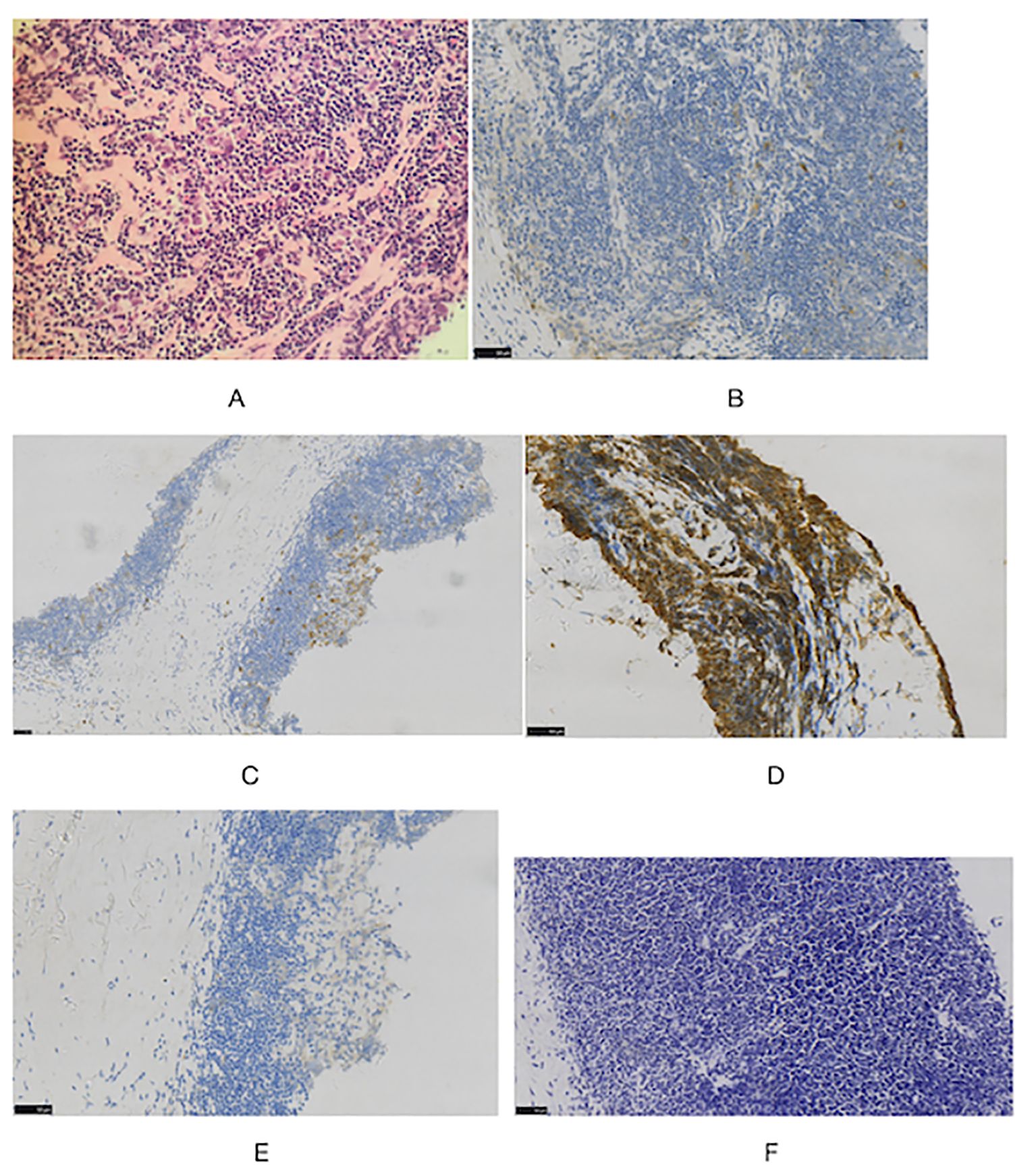
Figure 3 Histologic images in cervical lymph node (A) staining showed typical Hodgkin Reed-Sternberg (H-RS) cells in a variable inflammatory background. (HE, x40). Immunohistochemical staining also showed (B) CD30(+), (C) CD15(+), (D) CD20(+), (E) ALK(-) (F) EBER(-) (×40).
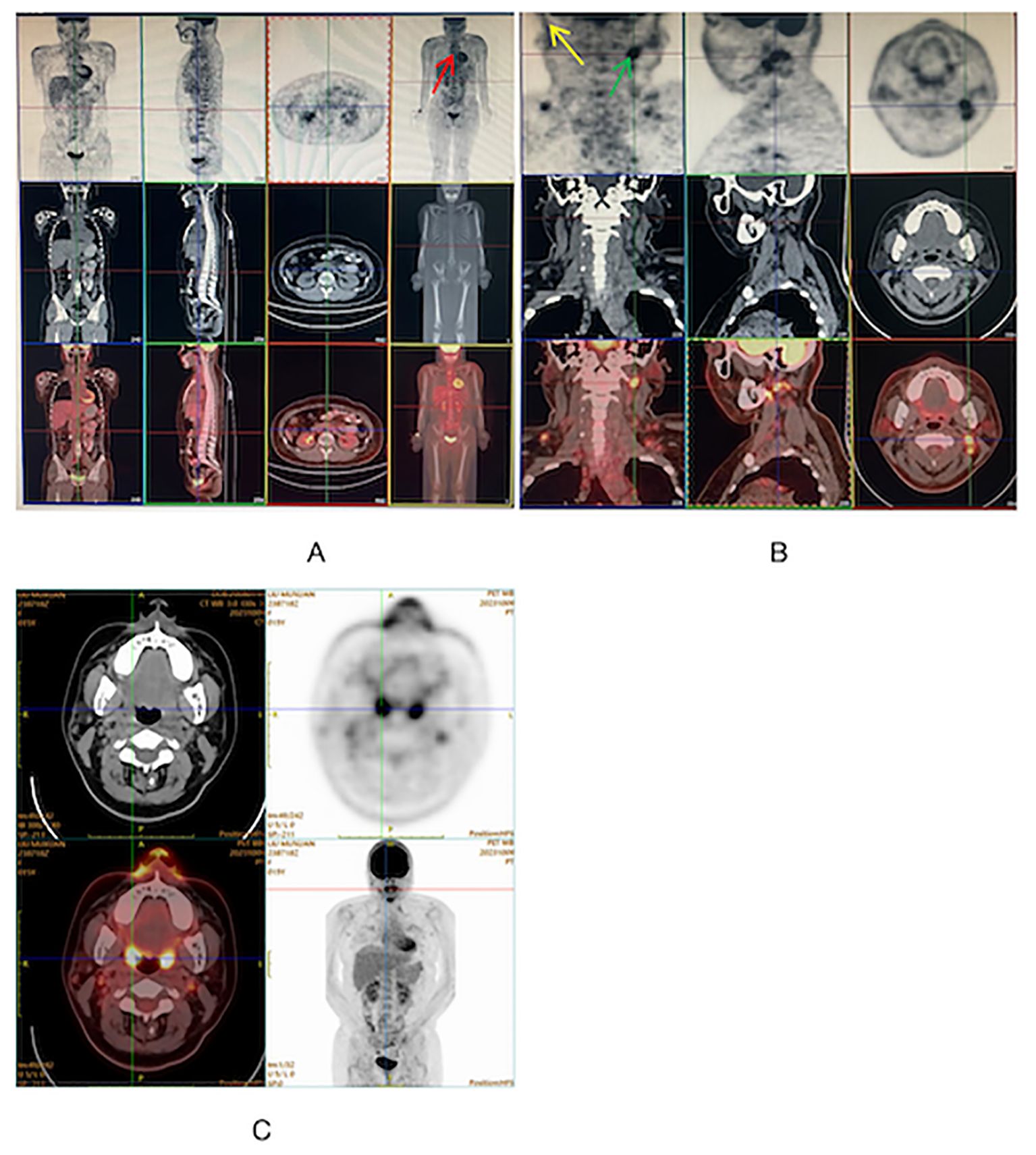
Figure 4 (A, B) FDG-PET on admission showing diffuse uptake in mediastinum supradiaphragmatic lymph nodes (Red), left supraclavicular lymph node (Yellow) and right cervical lymph nodes (Green). (C) FDG-PET at 11 months after diagnosis showing disappearance of abnormal signals.
Laboratory investigations revealed an elevated C-reactive protein (CRP) level of 12.40 mg/L (normal range: 0–5 mg/L). Tests for anti-nuclear antibodies (ANAs), anti-double-stranded DNA (anti-dsDNA), and anti-extractable nuclear antigen antibodies (including anti-Jo-1, anti-PM-Scl 70, anti-PM-Scl 100, anti-Ku, anti-Ro-52, anti-SSA, anti-SSB, and anti-Sm) were negative, as were serology tests for myositis-specific antibodies (MSAs) (anti-Mi-2, -Jo1, -EJ, -PL-12, -PL-7, -SAE 1/2, -MDA5, -SRP, -HMGCR, -TIF-1γ, -SSA/Ro52, and -NXP-2). Line-blot for MSAs detection in JDM serum. Serum samples were tested by the commercial line blot Simcere Diagnostics autoimmune myositis 12 Ag (MYO12D-12) following the manufacturers’ instructions. Test strips were scanned and evaluated for band intensity using the LineScan software (Dr DOT4). Following the manufacturer’s recommendations, classified as negative (-) or positive (+). Negative: <5 AU (arbitrary units); gray zone: 5-10 AU (retest after 8-12 weeks); positive: >10 AU. CK, lactate dehydrogenase (LDH), alanine transaminase (ALT), and aspartate transaminase (AST) levels; white blood cell (WBC), red blood cell (RBC), and platelet (PLT) counts; and erythrocyte sedimentation rate (ESR) were within the normal ranges.
Ultimately, stage IV A nsHL concurrent with JDM was diagnosed. Chemotherapy was initiated according to the protocol for stage IV A, which comprised a regimen of adriamycin, bleomycin, vinblastine, and dacarbazine (ABVD). Prednisolone (2 mg/kg/day, orally) was administered for 2 months to treat JDM. The patient underwent six courses of chemotherapy, along with 15 sessions of local radiation therapy, to treat HL. Eleven months after the diagnosis, HL and JDM were completely in remission (Figure 4).
Discussion
JDM was diagnosed in our patient who met four of the five Bohan and Peter criteria: typical skin changes, proximal muscle weakness, myopathic changes detected by EMG, and abnormal muscle biopsy findings (9). She also met the diagnostic criteria of the European League Against Rheumatism/American College of Rheumatology (EULAR/ACR) with a score of 10.7, thus falling in the “definite IIM” category (aggregate score ≥ 8.7) (2). However, only a few cases of patients with JDM either having DM as a paraneoplastic manifestation or developing a lymphoma after diagnosis of JDM have been reported. While the link between DM and lymphoma is well documented in adults, it has rarely been observed in adolescent patients with JDM.
We conducted a search on PubMed for case reports and review literature using the following terms: juvenile dermatomyositis, myositis, malignancies, cancer, tumor, children, adolescents, and pediatrics, either individually or in combination. This search yielded the case reports of four pediatric patients with JDM and lymphoma (10–13). Table 1 shows the characteristics of these four cases and ours (patient 5). Three of these patients were girls, and the remaining two were boys, with an average age of 13.8 years. JDM and HL were diagnosed in three patients (patients 1, 4, and 5) simultaneously, all of whom demonstrated unusual physical findings at the time of JDM diagnosis, including enlarged lymph nodes. The other two patients (patients 2 and 3) exhibited no atypical rash, lymphadenopathy, or splenomegaly and/or hepatomegaly as unusual physical findings at the time of JDM diagnosis. The patient 2 was found to have a right supraclavicular mass 13 months after being diagnosed with JDM, and was diagnosed HL IIA. The patient 3 was found to have a painless left supraclavicular lymph node 5 months after being diagnosed with JDM. The biopsy result of this supraclavicular lymph node was consistent with HL.
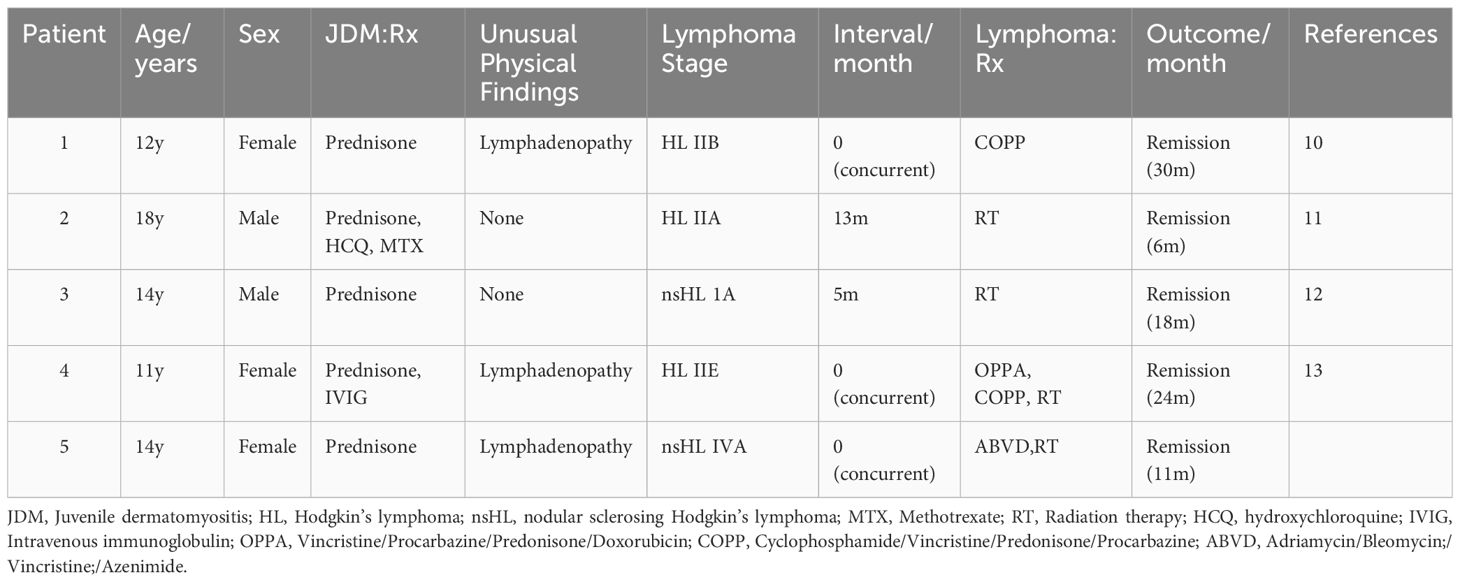
Table 1 Presentation of lymphoma and JDM in pediatric patients.
The pathogenesis of JDM and lymphoma has not been fully elucidated, and multiple mechanisms may be attributed to the link between the two. First, long-term medication-induced immunosuppression (14) has been suggested as a contributing factor. Stübgen described five patients whose lymphoma regressed only upon withdrawal of such drugs and when combined with locoregional radiation, supporting the notion of treatment-associated lymphoma (15). Second, high inflammatory activity in patients with autoimmune diseases is considered a major risk factor for developing lymphoma (16). Additionally, the occurrence of JDM as a paraneoplastic phenomenon associated with lymphoma has rarely been reported (17). Evidence indicating a potential association between JDM and malignancy is limited, which is in contrast with that demonstrated in the adult population (14). A majority of the studies regarding the association between JDM and malignant tumors consist of isolated case reports. In a comprehensive review of the literature spanning 45 years, 12 cases establishing an association between JDM/polymyositis (PM) and malignancy were identified. It showed that the occurrence of a paraneoplastic syndrome can be concluded if a malignant tumor occurs within 12 months of diagnosing DM and/or successful tumor treatment can improve the symptoms of DM (18). Among the five cases described in Table 1, three (patients 1, 4, and 5) support the hypothesis that JDM is a paraneoplastic phenomenon (13). This is evidenced by the simultaneous diagnosis of JDM and HL, with the lymphoma already at stage II or IV when diagnosed. Subsequent radiotherapy and chemotherapy treatment alleviated symptoms of DM, such as muscle weakness, pain, and rash. Patient 3 was diagnosed with HL within 12 months of being diagnosed with JDM, suggesting a paraneoplastic phenomenon. However, patient 2 had a malignancy that was less clearly temporally related to the JDM diagnosis than it was in the other three patients (12, 18).
The etiology of JDM as a paraneoplastic phenomenon of lymphoma remains unknown; however, some mechanisms have been proposed. First, environmental factors can trigger both cancer and myositis in genetically susceptible hosts (15). Second, malignant cells in HL secrete cytokines excessively. Activated cytokines enable T cell-mediated myocyte toxicity or complement-mediated microangiopathy, and the tumor products cause muscle and skin inflammation (19). Third, cross-reactions between tumors and muscle or skin antigens can cause dysregulation of the immune system (20–22).
Previous studies have shown that anti-TIF-1γ autoantibodies are one of the most common MSAs in adult DM and JDM patients (23, 24). DM patients, adult or adolescent, with positive anti-TIF-1γ autoantibodies often present with extensive and severe skin lesions, including photosensitive rashes, skin ulcers, and muscle atrophy (25). Importantly, in adults with DM, anti-TIF-1γ autoantibodies are associated with an increased risk of cancer (23). According to previous studies, the positivity rate of anti-TIF-1γ antibodies in patients with DM-associated tumors is over 50%, confirming their reliability as a predictor of malignancy in DM (26). However, no association between anti-TIF-1γ autoantibodies and malignant tumors has been found in adolescent patients with JDM (27). Research indicates that patients, whether adults or adolescents, who test positive for anti-TIF-1γ may also exhibit other autoantibodies, such as anti-Sp4, anti-CCAR1, and anti-TBL1XR1 autoantibodies (28, 29). Patients with JDM who present with anti-TIF-1γ antibodies and associated autoantibodies exhibit less muscle weakness, less frequent skin ulcers, and lower anti-TIF-1γ autoantibody titers (29) perhaps because, in this subgroup of patients, the titer of anti-TIF-1γ autoantibodies appears to decrease due to other autoantibodies targeting antigens that form complexes with TIF-1γ (28). Therefore, the potential myotoxicity effect of anti-TIF1-γ autoantibodies may be weakened. Results reported in different studies show that autoantibodies against Sp4 and CCAR1 are associated with a reduced risk of cancer in adults with anti-TIF-1γ -positive DM. The specific mechanism by which the presence of associated antibodies in such patients reduces tumor risk is unclear, but it may be that they are eradicated more efficiently in those who mount an immune response against both TIF-1γ and the associated autoantigens (28).
JDM is a rare paraneoplastic syndrome, and large-scale data regarding the relationship between MSAs and malignancy are currently lacking. However, unusual physical findings during diagnosis of JDM, such as atypical rash, organomegaly, or lymphadenopathy, may necessitate a thorough evaluation to exclude malignancy. We observed unusual features, including lymphadenopathy, in our patient. Patients 2 and 3 had no unusual physical findings at the time of diagnosis of JDM, while HL was detected 13 and 5 months later, respectively (11). Hence, even if a malignancy is not detected when JDM is diagnosed, follow-up is necessary. Early identification is critical in such patients because steroid treatment can partially inhibit tumor growth, potentially delaying the establishment of a correct diagnosis, which could negatively impact the patient’s prognosis.
Conclusion
Here, we described a rare case of JDM as a paraneoplastic syndrome of HL, but only a few cases of JDM as a paraneoplastic manifestation have been reported, and thus, routine screening for lymphoma in patients with JDM does not seem to be warranted. Unusual physical findings during the diagnosis of JDM may indicate the presence of a malignant tumor, necessitating further evaluation to screen for malignancy. Even if no malignant tumor is found at the time of JDM diagnosis, follow-up is necessary. Regular follow-up of JDM patients should not solely focus on the characteristics of myositis but should also encompass a systematic and comprehensive physical examination. Comprehensive malignancy screening is required if any unusual physical findings are observed during follow-up. Further study of paraneoplastic syndromes in children, focusing on pathophysiology and treatment strategies, is required.
Data availability statement
The original contributions presented in the study are included in the article supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by The 960 Hospital of the PLA Joint Logistic Support Force. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
YL: Formal Analysis, Writing – original draft. HH: Software, Writing – review & editing. XX: Methodology, Supervision, Writing – review & editing. JF: Investigation, Supervision, Writing – review & editing. ML: Formal Analysis, Methodology, Writing – review & editing. HL: Formal Analysis, Methodology, Writing – review & editing. MC: Formal Analysis, Investigation, Supervision, Writing – review & editing. XW: Formal Analysis, Investigation, Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We thank the participant and all the authors for their support and contributions to this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mendez EP, Lipton R, Ramsey-Goldman R, Phil R, Susan B, Alan D, et al. US incidence of juvenile dermatomyositis, 1995-1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheumatol. (2003) 49:300–5. doi: 10.1002/art.11122
2. Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, Visser M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. (2017) 76:1955–64. doi: 10.1136/annrheumdis-2017-211468
3. Barut K, Aydin PO, Adrovic A, Sahin S, Kasapcopur O. Juvenile dermatomyositis: a tertiary center experience. Clin Rheumatol. (2017) 36:361–6. doi: 10.1007/s10067-016-3530-4
4. Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet. (2002) 360:1197–202. doi: 10.1016/S0140-6736(02)11279-7
5. Pachman LM, Abbott K, Sinacore JM, Amoruso L, Dyer A, Lipton R, et al. Duration of illness is an important variable for untreated children with juvenile dermatomyositis. J Pediatr. (2006) 148:247–53. doi: 10.1016/j.jpeds.2005.10.032
6. Hill CL, Zhang Y, Sigurgeirsson B, Pukkala E, Mellemkjaer L, Airio A, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. (2001) 357:96–100. doi: 10.1016/S0140-6736(00)03540-6
7. Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and Malignancy risk: a metaanalysis study. J Rheumatol. (2015) 42:282–91. doi: 10.3899/jrheum.140566
8. Rosina S, Varnier GC, Pistorio A, Pilkington C, Maillard S, Civino A, et al. Development and testing of reduced versions of the manual muscle test-8 in juvenile dermatomyositis. J Rheumatol. (2021) 48:898–906. doi: 10.3899/jrheum.200543
9. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. (1975) 292:344–7. doi: 10.1056/NEJM197502132920706
10. Döring E, Grulich M, Dörffel W. [Lymphogranulomatosis and dermatomyositis]. Padiatr Grenzgeb. (1983) 22:141–7.
11. Bittar B, Rose CD. Early development of Hodgkin’s lymphoma in association with the use of methotrexate for the treatment of dermatomyositis. Ann Rheum Dis. (1995) 54:607–8. doi: 10.1136/ard.54.7.607-b
12. Shay M, Braester A, Cohen I. Dermatomyositis as presenting symptom of Hodgkin’s disease. Ann Hematol. (1991) 63:116–8. doi: 10.1007/BF01707284
13. Yoshimoto Suzuki Y, Koto M, Tanaka M, Yamanaka J, Shichino H. Paraneoplastic dermatomyositis with pediatric Hodgkin lymphoma: A case report. Pediatr Int. (2020) 62:1297–9. doi: 10.1111/ped.14334
14. Stübgen JP. Juvenile dermatomyositis/polymyositis and lymphoma. J Neurol Sci. (2017) 377:19–24. doi: 10.1016/j.jns.2017.03.033
15. Stübgen JP. Inflammatory myopathies and lymphoma. J Neurol Sci. (2016) 369:377–89. doi: 10.1016/j.jns.2016.08.060
16. Pasquet F, Pavic M, Ninet J, Hot A. [Autoimmune diseases and cancers. Part I: cancers complicating autoimmune diseases and their treatment]. Rev Med Interne. (2014) 35:310–6. doi: 10.1016/j.revmed.2013.10.336
17. Sherry DD, Haas JE, Milstein JM. Childhood polymyositis as a paraneoplastic phenomenon. Pediatr Neurol. (1993) 9:155–6. doi: 10.1016/0887-8994(93)90055-H
18. Morris P, Dare J. Juvenile dermatomyositis as a paraneoplastic phenomenon: an update. J Pediatr Hematol Oncol. (2010) 32:189–91. doi: 10.1097/MPH.0b013e3181bf29a2
19. Hsu SM, Waldron JW Jr, Hsu PL, Hough AJ Jr. Cytokines in Malignant lymphomas: review and prospective evaluation. Hum Pathol. (1993) 24:1040–57. doi: 10.1016/0046-8177(93)90183-H
20. Bernard P, Bonnetblanc JM. Dermatomyositis and Malignancy. J Invest Dermatol. (1993) 100:128S–32S. doi: 10.1111/1523-1747.ep12356956
21. Casciola-Rosen L, Nagaraju K, Plotz P, Wang K, Levine S, Gabrielson E, et al. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J Exp Med. (2005) 201:591–601. doi: 10.1084/jem.20041367
22. Yang H, Peng Q, Yin L, Li S, Shi J, Zhang Y, et al. Identification of multiple cancer-associated myositis-specific autoantibodies in idiopathic inflammatory myopathies: a large longitudinal cohort study. Arthritis Res Ther. (2017) 19:259. doi: 10.1186/s13075-017-1469-8
23. Lundberg IE, Fujimoto M, Vencovsky J, Aggarwal R, Holmqvist M, Christopher-Stine L, et al. Idiopathic inflammatory myopathies. Nat Rev Dis Primers. (2021) 7:86. doi: 10.1038/s41572-021-00321-x
24. Best M, Molinari N, Chasset F, Vincent T, Cordel N, Bessis D. Use of anti-transcriptional intermediary factor-1 gamma autoantibody in identifying adult dermatomyositis patients with cancer: A systematic review and meta-analysis. Acta Derm Venereol. (2019) 99:256–62. doi: 10.2340/00015555-3091
25. Kishi T, Warren-Hicks W, Bayat N, Targoff IN, Huber AM, Ward MM, et al. Corticosteroid discontinuation, complete clinical response and remission in juvenile dermatomyositis. Rheumatol (Oxford). (2021) 60:2134–45. doi: 10.1093/rheumatology/keaa371
26. Venalis P, Selickaja S, Lundberg K, Rugiene R, Lundberg IE. Association of anti-transcription intermediary factor 1γ Antibodies with paraneoplastic rheumatic syndromes other than dermatomyositis. Arthritis Care Res (Hoboken). (2018) 70:648–51. doi: 10.1002/acr.23325
27. Gunawardena H, Wedderburn LR, North J, Betteridge Z, Dunphy J, Chinoy H, et al. Clinical associations of autoantibodies to a p155/140 kDa doublet protein in juvenile dermatomyositis. Rheumatol (Oxford). (2008) 47:324–8. doi: 10.1093/rheumatology/kem359
28. Sherman MA, Yang Q, Gutierrez-Alamillo L, Pak K, Flegel WA, Mammen AL, et al. Clinical features and immunogenetic risk factors associated with additional autoantibodies in anti-transcriptional intermediary factor 1γ Juvenile-onset dermatomyositis. Arthritis Rheumatol. (2024) 76:631–7. doi: 10.1002/art.42768
29. Sherman MA, Pak K, Pinal-Fernandez I, Flegel WA, Targoff IN, Miller FW, et al. Autoantibodies recognizing specificity protein 4 co-occur with anti-transcription intermediary factor 1 and are associated with distinct clinical features and immunogenetic risk factors in juvenile myositis. Arthritis Rheumatol. (2023) 75:1668–77. doi: 10.1002/art.42512
Keywords: juvenile dermatomyositis, Hodgkin’s lymphoma, paraneoplastic syndrome, malignancy, nodular sclerosing Hodgkin’s lymphoma
Citation: Ling Y, Hu H, Xu X, Feng J, Li M, Li H, Cheng M and Wang X (2024) Paraneoplastic dermatomyositis and Hodgkin’s lymphoma in a 14-year-old girl: a case report and literature review. Front. Oncol. 14:1416083. doi: 10.3389/fonc.2024.1416083
Received: 11 April 2024; Accepted: 23 July 2024;
Published: 07 August 2024.
Edited by:
Angela Mastronuzzi, Bambino Gesù Children’s Hospital (IRCCS), ItalyReviewed by:
Ozgur Kasapcopur, Istanbul University-Cerrahpasa, TürkiyeSara Sabbagh, Medical College of Wisconsin, United States
Copyright © 2024 Ling, Hu, Xu, Feng, Li, Li, Cheng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Cheng, eHd4eTEyMTIxM0AxNjMuY29t; Xiaoling Wang, Q01DTUxZWTE5OTBAMTYzLmNvbQ==
†First author