 Howard S. Oster
Howard S. Oster Moshe Mittelman
Moshe Mittelman- 1Department of Medicine, Tel Aviv Sourasky Medical Center, Tel Aviv University School of Medicine, Tel Aviv, Israel
- 2Department of Hematology, Tel Aviv Sourasky Medical Center, Tel Aviv University School of Medicine, Tel Aviv, Israel
The Myelodysplastic syndromes (MDS) are a heterogenous group of clonal bone marrow (BM) stem cell myeloid neoplasms, characterized by ineffective hematopoiesis that results in dysplasia in hematopoietic cells and peripheral cytopenias, especially anemia, and a propensity to leukemic transformation. The suspicion of MDS is raised by a typical but not specific clinical picture and routine laboratory findings, but the gold standard for MDS diagnosis is still BM examination with the presence of uni-or multi-lineage dysplasia and increased blast percentage, together with exclusion of other reasons. Cytogenetics is also an essential part of the diagnostic and prognostic processes. Flow cytometry and full genetic characterization are helpful but not mandatory for MDS diagnosis. This review summarizes the current steps of diagnostic approach for a patient suspected of having MDS. We also express our hopes that within the near future, non-invasive technologies, especially digital and peripheral blood genetics, will mature and be introduced into practice.
Introduction
The Myelodysplastic syndromes (MDS) are a heterogenous group of clonal bone marrow (BM) stem cell myeloid neoplasms, characterized by ineffective hematopoiesis that results in dysplasia in hematopoietic cells and peripheral cytopenias, especially anemia, and a propensity to leukemic transformation (1–4). MDS incidence increases with aging and is approximately 5 cases per 100,000 people per year in the general population, on average, with a median age of onset of above 70 (3, 5, 6). Once patients are diagnosed with MDS, they are categorized using one of the classifications (7–11). Practically, most patients are assigned to the lower-(LR) or higher-risk (HR) groups. These classifications assist in diagnosis but serve mainly as a prognostic tool and to direct management.
Here we focus on the diagnosis of MDS, both the classic, standard approach as well as some modern modalities which are being tested as diagnostic tools.
Unfortunately, since we are dealing with a heterogenous group of disorders, in contrast with many other diseases, there is no single specific diagnostic test or definitive diagnostic criteria for MDS. The diagnostic process is based on various clinical and laboratory features with exclusion of other diseases.
When to suspect MDS
The suspicion is raised by the clinical and laboratory picture in the elderly. Clinically, MDS symptoms are non-specific. Patients may be asymptomatic, or may have non-specific complaints such as weakness and fatigue. They may also have cardiac complaints, due to the common anemia (Table 1A) (1, 3, 5, 12, 13). Decreased neutrophil count might be associated with recurrent infections and patients might have epistaxis, gingival bleeding or easy bruising if their platelets are low in number or dysfunctional (14).
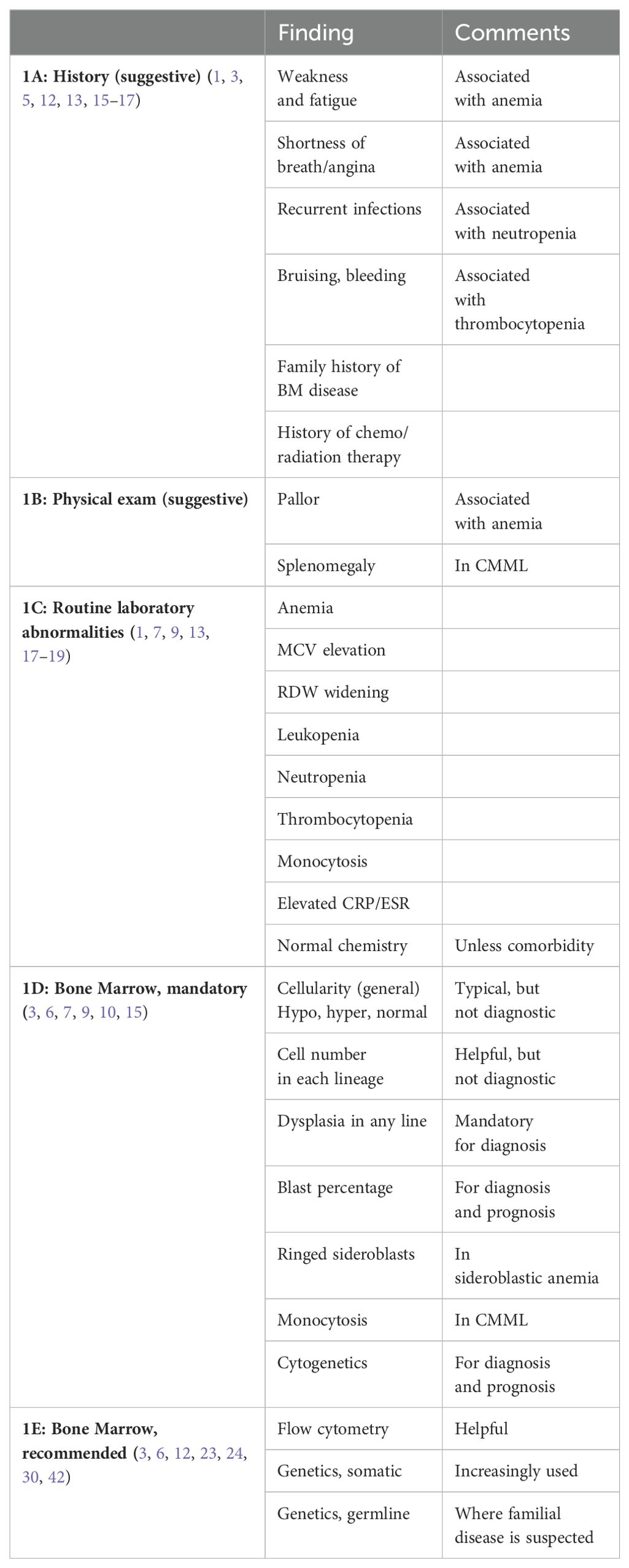
Table 1. Making the diagnosis of MDS.
When MDS is suspected, a careful history must be taken to search for other causes of anemia, including nutritional (folic acid, iron and vitamin B12, especially in vegetarians), medications, alcohol, tobacco, or viral infection. The patient’s history can also determine if there had been exposure to radiation or chemotherapy, or if there is a familial predisposition to bone marrow disease (15, 16).
Other BM diseases, e.g. paroxysmal nocturnal hemoglobinuria, aplastic anemia and myeloproliferative neoplasm (MPN), must also be excluded by history (3, 15, 16).
Physical examination (Table 1B) is often unremarkable or non-specific, with no abnormal findings or only pallor. Bruising, and other bleeding evidence may be found in thrombocytopenic patients. Hepatomegaly and mainly splenomegaly is common in the MDS chronic myelomonocytic leukemia (CMML) subtype (1, 3, 7).
Laboratory findings: MDS laboratory findings are not specific (Table 1C). Erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) are often elevated (17). More than 90% of MDS patients will have anemia; approximately 50% of these patients will be anemic with hemoglogin (Hb) less than 10g/dl (9, 13). The anemia is typically macrocytic with a mean corpuscular volume (MCV) that tends to be high or high normal (1, 3, 7), although not as high as in B12 deficiency. Red cell distribution width (RDW) may be widened, especially in more severe disease (18, 19). Patients with MDS tend to lack the increase in reticulocyte count in contrast with hemolytic anemia (15). About 50% of MDS patients are pancytopenic. Leukopenia and neutropenia with absolute neutrophil count (ANC) less than 800x109/L is found in 18% of patients and low platelet count (< 100x109/L) is observed in 40% of MDS patients (9, 13). Platelet function may be defective as well (14). Monocytosis is the hallmark of CMML, but the recent 2022 classifications lowered the threshold to 0.5x109/L (10, 11). This now includes what used to be referred to as oligomonocytic CMML.
Typically, routine blood chemistry is expected to be normal in MDS, unless there is a comorbidity associated with anemia. Serum iron and iron saturation as well as serum ferritin might be elevated in the MDS sideroblastic subtype.
Since MDS diagnosis is established by exclusion, blood chemistry should exclude nutritional deficiencies, like iron and especially folic acid and vitamin B12, both can cause macrocytic anemia. Blood chemistries should rule out underlying liver or kidney disease, and serology for hepatitis viruses B and C, and CMV, as well as HIV and parvovirus B19 should also be performed.
The peripheral blood (PB) smear might be typical and helpful, but not specific or diagnostic. The red blood cells (RBC) tend to demonstrate anisocytosis or poikylocytosis (7). Occasionally nucleated RBC are observed. The white blood cells (WBC) can show an increased number of immature myeloid cells (“left shift”) with hypolobulation (“Pelger”-like cells) and hypogranulation. PB platelets might be distorted, clumped, large (megaplatelet), or the number might be low. Persistent monocytosis would suggest CMML (7, 15), assuming that other etiologies for monocytosis have been excluded. Often, the PB smear helps to diagnose another hematologic disease and rule out MDS. For example, thrombocytosis or leukocytosis, would suggest an MPN, or at least an overlap MDS/MPN (see below).
Imaging is not a part of the MDS diagnostic process, unless hepatosplenomegaly is evaluated.
In summary, the combination of symptoms and laboratory findings with exclusion of other reasons for anemia or pancytopenia, might raise the suspicion of MDS (Tables 1A–C), however, none of these findings or even several of them, are enough to establish MDS diagnosis.
Bone marrow examination – the gold standard for MDS diagnosis
Once MDS is suspected, other causes of anemia or cytopenia are ruled out, and the patient is defined as having an unexplained anemia (or cytopenia), the next and definitive step in the diagnostic workup is a bone marrow (BM) evaluation, still the gold standard for the diagnosis of MDS (Table 1D). As the organ of blood cell production, BM examination is used to assess abnormalities of blood cells and their hematopoietic precursors, reflecting MDS pathogenesis and establishing the diagnosis. A full BM examination consists of both an aspirate and a biopsy. The main component to establish MDS diagnosis is morphology. Often additional tests are performed, including special staining, cytogenetics, immunophenotyping and more recently, genetics.
BM Morphology: Using May-Grunwald-Giemsa staining, BM aspirate is essential to assess the morphology of single cells. BM cellularity can be estimated but it is not accurate enough, in contrast with that of the biopsy. Typically, the aspirate is characterized by abnormal BM cellularity (hypocellular or hypercellular), but mainly by dysplasia, which can be found in any cell lineage. Dysplasia is considered significant if more than 10% of the nucleated cells of a given line have such changes (3). In the erythroid compartment, the dyserythropoietic changes may include megaloblastosis (similar to B12 deficiency), binuclearity or fractured nuclei (karyorhexis), irregular nuclear edges or internuclear bridging, cytoplasmic inclusions or bridging, vacuolization, fringed cytoplasm or incomplete hemoglobinization (7, 20, 21). The myeloid line may show an increased number of young immature cells, anisocytosis or changes in the shape of the nucleus, including hypolobulation or hypersegmentation. There may also be pseudo granules or cytoplasmic hypogranulation/degranulation. We specifically search for blasts (myeloid in MDS), which are identified by their high nuclear/cytoplasmic ratio, nucleoli, fine nuclear chromatin and cyoplasmic basophilia. Blasts may have granules or Auer rods. Blasts are counted and reported as percentage of nucleated BM cells. The megakaryocytic lineage can have large monolobular forms, small binucleated elements, dispersed nuclei, micromegakaryocytes and degranulation (3, 7). The smears are also stained for iron (Prussian blue) to assess for the presence of ringed sideroblasts (RS). The presence of RS > 15% of BM nucleated RBC diagnoses the MDS subtype of refractory anemia with ringed sideroblasts (RARS) (7). BM aspirate also serves for special tests, especially for MDS exclusion and to establish other hematologic disorders.
The BM trephine biopsy is important for evaluating the BM cells in their milieu, and provides information on cellularity, although this parameter has not been found to be critical for MDS diagnosis or prognosis (3, 7, 22). BM biopsy might also identify possible fibrosis. BM biopsy is less reliable in evaluating the morphology of single cells or counting blasts. Stains include hematoxylin/eosin as well as Giemsa. Specimens also undergo immunohistochemistry stains for Glycophorin A or C for the erythroid line, CD34 and CD117 for blasts, CD61 or CD42b for the megakaryocytes, KP1/CD68 or PGM1/CD68R for monocytes, CD20 for the B-cell line, and CD3 for the T-cell line (3). Reticulin stain or Gomori’s silver impregnation are used to evaluate for BM fibrosis (3, 15). Occasionally, other cells can be identified that may lead to other diagnoses like cellular metastases from other malignancies.
In summary, BM examination, especially the dysplastic features and blast percentage are mandatory in establishing MDS diagnosis (Table 1D). Moreover, once diagnosed, these BM findings, and especially the blast percentage further assist in categorizing and predicting the prognosis of the patient, according to the various classifications (4, 7, 8, 10). Finally, the blast % also distinguishes between HR-MDS and acute leukemia, although the line between these two entities declines over the years from 30% to 10% (depending on genetic signatures) (4, 7, 10, 11).
Flow cytometry: Immunophenotyping by flow cytometry (FC) is an adjunct diagnostic tool assisting in establishing MDS diagnosis and might also serve for follow up (Table 1E) (3, 12). The technology is based on detection of multiple aberrancies on a particular cell, as opposed to any single marker. The combined profile of several markers, can distinguish MDS from other cytopenias (23, 24). For example, a score that identifies at least two of the following criteria – increased CD34+ progenitors in nucleated BM cells, a decrease in B cell progenitors among CD34+ cells, change in CD45 expression on myeloid progenitor cells as compared with lymphocytes, and a decrease in sideward light scatter (SCC) of neutrophils compared with lymphocytes – demonstrated a specificity of over 90% in patients with low risk MDS (25, 26). Other markers can increase sensitivity and even identify cellular dysplasia. Unfortunately, due to high cost and the need for special facilities, this method has not been widely adopted globally, and is regarded as rather helpful but not mandatory.
Cytogenetics: Cytogenetics is performed with a combination of G-banding and FISH techniques. While it may not be required to establish MDS diagnosis, no diagnostic workup is complete without performing it (3, 6, 9). (Table 1D). At least 20 cells in metaphase should be examined. Thus, applying cytogenetics with the typical chromosomal abnormalities assists in the diagnosis. The common MDS cytogenetic findings are chromosomes 5 (deletion or monosomy), chromosome 7, + 8 (27). Cytotogenetics is even more important in predicting prognosis (8, 9). In the WHO 2016 classification of MDS, the use of cytogenetics was important for diagnosis especially where the existence of dysplasia is not seen at all, is less than 10% in all cell lineages, or is equivocal. Such patients were then seen as MDS-unclassifiable (4). In the current classification systems, that has been replaced by incorporation of clonal cytopenia of undetermined significance (CCUS) (10, 11), but the principle is the same.
Genetics: Over the last couple of decades, it has become clear that like other malignancies, genetic mutations are responsible for the development of the malignant clone(s) in MDS and these genetic signatures control the disease course (Table 1E) (28). We know today that 90% of MDS patients do harbor myeloid mutations (3, 29–31), (Table 2) with 2-3 mutations per patient at MDS diagnosis, on the average. Many mutations were described in MDS, but six are found in at least 10% of MDS patients: SF3B1, TET2, SRSF2, ASXL1, DNMT3A and RUNX1 (6, 31–41). However, in contrast with other hematologic neoplasms, CML or CLL for example, introduction of genetics into clinical practice, both for diagnosis and prognosis (42) is still in its infancy. Several tough hurdles still prevent broad genetic application (6, 30, 31, 43–46). We learned that not all mutation were born equal - there are driver mutations, obviously more important clinically while other are just passengers. Often there are not only founding driver mutations, but also sub-clonal driver mutations which generate a new clone with both the newly acquired as well as the original founding mutations. Certain mutations are associated with favorable (isolated SF3B1 (32, 33)) or poor (TP53 (31, 47), RUNX1(40, 41)) prognosis. The variant allele frequency (VAF) and hotspot of the mutation appear to be important. The function of mutations as well as occurrence of co-mutations and gene-gene interaction is still not elucidated. We have also learned that mono-allelic mutation, for example TP53, is associated with a better prognostic phenotype as opposed to bi-allelic mutation (48). Finally, no mutation has been found to be unique or diagnostic for MDS, with the only exception of SF3B1 mutation which is recognized today as an MDS defining signature (10, 11, 15). Moreover, these mutations have been found in healthy aging people too, and most of them will never become sick, the phenomenon defined as age-related clonal hematopoiesis (ARCH) (49, 50), or clonal hematopoiesis of indeterminate potential (CHIP) (51, 52). Table 2 lists other mutations (with references) that have an association with MDS.
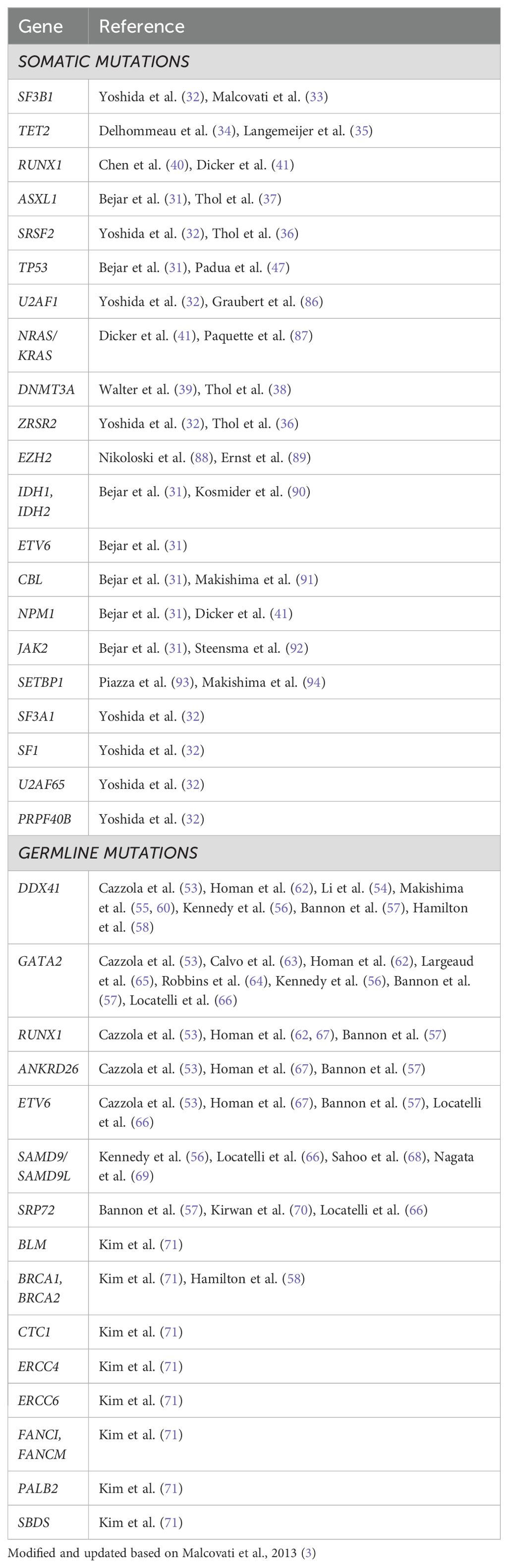
Table 2. Genetic mutations in MDS.
A relatively new area is the germline mutation in MDS. Until several years ago we looked at germline mutations as a pediatric problem. Over the last couple of decades several such mutations are not only observed but also result in a clinical phenotype, which might be detected only at an adult age. Examples are DDX41 (53–62), GATA-2 (53, 56, 57, 62–66), and others (53, 56–58, 66–71) (Table 2).
The challenges we face now is how to detect these individuals, how to follow and manage them, and most importantly who of the family members to screen. We expect to have some of the answers within the next few years.
In summary, one cannot underestimate the role of genetics in diagnosis, as well as in the pathogenesis and prognosis (31, 42), but in 2024 we are still in the beginning of this era, and the genetic profile, although routinely performed in many parts of the world, is still not a mandatory tool in the diagnostic workup. Of course, the high cost and lack of professional skills to perform this analysis further prevents its wide application.
It is noted that some of these mutations found (not only but also) in MDS already serve as targets for treatment. Examples are the APR-246 targeting TP53 mutations (72) and the IDH1/2 inhibitors (73).
MDS/MPN overlap syndrome
When a suspected patient undergoes a workup diagnostic procedure, one has to consider both disorders with overlapping features, as well as diseases that should be excluded. Patients with myeloproliferative neoplasms (MPN) share several clinical, laboratory and imaging features with MDS. Both the WHO and the ICC guidelines devote attention to such patients (10, 11, 74). The debate whether the MDS subgroup CMML is indeed MDS or should be re-classified with MPN has been with us since the first classification (7). MDS/MPN overlap is suggested when there is splenomegaly and/or elevated WBC (>13k) with or without monocytosis, now > 0.5x109/L (CMML) and/or PLT count (>450k). It can also be associated with ring sideroblasts (with SF3B1) (11, 15).
Pre-MDS states
Several pieces of evidence suggest that MDS develops over time (75) in which the malignant clone evolves before the clinical disease is diagnosed. The occurrence of myeloid mutations in healthy individuals with a higher tendency to further evolve into full blown myeloid diseases, especially MDS, further supports this concept (49, 50). Like other hematologic neoplasms such as multiple myeloma (monoclonal gammopathy of undetermined significance) and chronic lymphocytic leukemia (monoclonal B-cell lymphocytosis), pre-MDS states are recognized too. These entities include idiopathic cytopenia of undetermined/unknown significance (ICUS), many of which end up being clonal cytopenia of unknown significance (CCUS). ICUS is characterized by cytopenia without a known cause and without the minimal criteria to establish MDS diagnosis (76–78). In CCUS, a clonal myeloid mutation is observed, with some overlap with ARCH and CHIP (51), however, it cannot be (still) defined as MDS. There may also be dysplasia without cytopenia (IDUS, idiopathic dysplasia of unknown significance) (76, 79), and BM clonal changes without cytopenia. Does it make sense to diagnose these pre-MDS states? Probably yes. Although no current therapeutic policy justifies intervention in these disease states, one can foresee such in the future, especially considering the coming biological less-invasive technologies. How to diagnose these conditions? It is likely that genetic studies and identification of individuals at risk (e.g. germline mutations) might assist. However, one cannot ignore the social, ethical and financial considerations making it still very difficult at present.
Novel approaches to diagnose MDS
As stated above BM examination and its morphology is still the gold standard tool to diagnose MDS. Many still believe that the information obtained, including the morphological findings and the blast percentage cannot be replaced by any other method. However, since this examination is invasive and painful, and morphological evaluation is somewhat subjective with high interobserver variations (80), one would want to avoid it. Work toward this goal has progressed along two lines and is still under investigation but quite promising.
The first approach applies digital tools comparing numerous data collected from large numbers of patients, to data obtained from healthy subjects. Our group, in collaboration with the European MDS group, analyzed such data collected from 501 MDS patients and compared them to 501 controls. We developed a simple diagnostic model applying 10 simple parameters that are easily available (age, sex, hemoglobin, white blood cells, platelets, mean corpuscular volume, neutrophils, monocytes, glucose, and creatinine) (81, 82). These variables of a suspected individual can be applied and the model was designed to provide one of three possible predictive conclusions: probably MDS (pMDS), probably not MDS (pnMDS) and indeterminate. We found that we could predict or rule out MDS in over 80% of patients with unexplained anemia with an area under the receiver operator characteristics (ROC) curve (AUC) of 0.96. We recently validated the model using data from patients and controls who had not been included in the development of the model (83). Also, external validation was performed by the Dusseldorf group, using data from a different center and found the model especially useful in ruling out MDS (84).
The second approach to skip BM examination in the diagnostic workup is based on the assumption that most relevant information, especially genetics, can be found in the PB if we have the right methodology to use it. One example is the work recently presented, identifying PB CD34+ hematopoietic stem and progenitor cells (HSPC) and performing single cell RNAseq, which can potentially diagnose MDS or pre-MDS states (85). These approaches are still investigational and not the standard, however, it is likely that such non-invasive methods will obviate the need for BM evaluations in many patients for diagnosing MDS.
Summary
In 2024 we are still conservative regarding the diagnosis of MDS. To make the diagnosis of MDS, some tests are mandatory, especially BM examination (aspirate and/or biopsy) identifying dysplasia in one or more and enumeration of blasts, as well as exclusion of other reasons for anemia (or cytopenia). Cytogenetics is also an essential part of the diagnostic process. Suspicious clinical picture, macroctyic anemia (or cytopenia), peripheral blood abnormalities, presence of BM ringed sideroblasts, flow cytometry and myeloid somatic mutations as well as other genetic assays are helpful and recommended but not critical for MDS diagnosis. We express our hopes that within the near future, non-invasive technologies, such as those described (digital and PB genetics) or others, will mature and be introduced into practice.
Author contributions
HO: Conceptualization, Writing – original draft, Writing – review & editing. MM: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mittelman M. The myelodysplastic syndromes–1990. Isr J Med Sci. (1990) 26:468–78. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=2205597.
2. Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med. (2009) 361:1872–85. doi: 10.1056/NEJMra0902908
3. Malcovati L, Hellstrom-Lindberg E, Bowen D, Ades L, Cermak J, Del Canizo C, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. (2013) 122:2943–64. doi: 10.1182/blood-2013-03-492884
4. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
5. de Swart L, Smith A, Johnston TW, Haase D, Droste J, Fenaux P, et al. Validation of the revised international prognostic scoring system (IPSS-R) in patients with lower-risk myelodysplastic syndromes: a report from the prospective European LeukaemiaNet MDS (EUMDS) registry. Br J Haematol. (2015) 170:372–83. doi: 10.1111/bjh.13450
6. Cazzola M. Myelodysplastic syndromes. N Engl J Med. (2020) 383:1358–74. doi: 10.1056/NEJMra1904794
7. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. (1982) 51:189–99. http://www.ncbi.nlm.nih.gov/pubmed/6952920.
8. Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. (1997) 89:2079–88. http://www.ncbi.nlm.nih.gov/pubmed/9058730.
9. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. (2012) 120:2454–65. doi: 10.1182/blood-2012-03-420489
10. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. (2022) 36:1703–19. doi: 10.1038/s41375-022-01613-1
11. Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. (2022) 140:1200–28. doi: 10.1182/blood.2022015850
12. Garcia-Manero G, Chien KS, Montalban-Bravo G. Myelodysplastic syndromes: 2021 update on diagnosis, risk stratification and management. Am J Hematol. (2020) 95:1399–420. doi: 10.1002/ajh.25950
13. Sekeres MA, Taylor J. Diagnosis and treatment of myelodysplastic syndromes: A review. JAMA. (2022) 328:872–80. doi: 10.1001/jama.2022.14578
14. Mittelman M, Zeidman A. Platelet function in the myelodysplastic syndromes. Int J Hematol. (2000) 71:95–8. https://www.ncbi.nlm.nih.gov/pubmed/10745619.
15. Hasserjian RP, Germing U, Malcovati L. Diagnosis and classification of myelodysplastic syndromes. Blood. (2023) 142:2247–57. doi: 10.1182/blood.2023020078
16. Ades L, Itzykson R, Fenaux P. Myelodysplastic syndromes. Lancet. (2014) 383:2239–52. doi: 10.1016/S0140-6736(13)61901-7
17. Oster HS, Sklyar E, Goldshmidt N, Mittelman M. Routine inflammatory markers are elevated in myelodysplastic syndromes at presentation. Mediterr J Hematol Infect Dis. (2023) 15:e2023044. doi: 10.4084/MJHID.2023.044
18. Luis TC, Wilkinson AC, Beerman I, Jaiswal S, Shlush LI. Biological implications of clonal hematopoiesis. Exp Hematol. (2019) 77:1–5. doi: 10.1016/j.exphem.2019.08.004
19. Shi Z, Li B, Huang H, Qin T, Xu Z, Zhang H, et al. Prognostic impact of red blood cell distribution width in myelodysplastic syndromes. Br J Haematol. (2019) 186:352–5. doi: 10.1111/bjh.15830
20. Mittelman M, Weinstein T, Pick AI, Djaldetti M. Intercellular bridges between erythroblasts in a patient with preleukemia. Haematologica. (1987) 72:533–5. https://www.ncbi.nlm.nih.gov/pubmed/3126111.
21. Linman JW, Bagby GC Jr. The preleukemic syndrome (hemopoietic dysplasia). Cancer. (1978) 42:854–64. doi: 10.1002/1097-0142(197808)42:2+<854::aid-cncr2820420707>3.0.co;2-w
22. Greenbaum U, Joffe E, Filanovsky K, Oster HS, Kirgner I, Levi I, et al. Can bone marrow cellularity help in predicting prognosis in myelodysplastic syndromes? Eur J Haematol. (2018) 101:502–7. doi: 10.1111/ejh.13134
23. van de Loosdrecht AA, Westers TM. Cutting edge: flow cytometry in myelodysplastic syndromes. J Natl Compr Canc Netw. (2013) 11:892–902. doi: 10.6004/jnccn.2013.0106
24. Ogata K, Della Porta MG, Malcovati L, Picone C, Yokose N, Matsuda A, et al. Diagnostic utility of flow cytometry in low-grade myelodysplastic syndromes: a prospective validation study. Haematologica. (2009) 94:1066–74. doi: 10.3324/haematol.2009.008532
25. Della Porta MG, Picone C, Pascutto C, Malcovati L, Tamura H, Handa H, et al. Multicenter validation of a reproducible flow cytometric score for the diagnosis of low-grade myelodysplastic syndromes: results of a European LeukemiaNET study. Haematologica. (2012) 97:1209–17. doi: 10.3324/haematol.2011.048421
26. Porwit A, Bene MC, Duetz C, Matarraz S, Oelschlaegel U, Westers TM, et al. Multiparameter flow cytometry in the evaluation of myelodysplasia: Analytical issues: Recommendations from the European LeukemiaNet/International Myelodysplastic Syndrome Flow Cytometry Working Group. Cytometry B Clin Cytom. (2023) 104:27–50. doi: 10.1002/cyto.b.22108
27. Schanz J, Tuchler H, Sole F, Mallo M, Luno E, Cervera J, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. (2012) 30:820–9. doi: 10.1200/JCO.2011.35.6394
29. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. (2014) 28:241–7. doi: 10.1038/leu.2013.336
30. Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. (2013) 122:3616–27. doi: 10.1182/blood-2013-08-518886
31. Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. (2011) 364:2496–506. doi: 10.1056/NEJMoa1013343
32. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. (2011) 478:64–9. doi: 10.1038/nature10496
33. Malcovati L, Papaemmanuil E, Bowen DT, Boultwood J, Della Porta MG, Pascutto C, et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood. (2011) 118:6239–46. doi: 10.1182/blood-2011-09-377275
34. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. (2009) 360:2289–301. doi: 10.1056/NEJMoa0810069
35. Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. (2009) 41:838–42. doi: 10.1038/ng.391
36. Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood. (2012) 119:3578–84. doi: 10.1182/blood-2011-12-399337
37. Thol F, Friesen I, Damm F, Yun H, Weissinger EM, Krauter J, et al. Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J Clin Oncol. (2011) 29:2499–506. doi: 10.1200/JCO.2010.33.4938
38. Thol F, Winschel C, Ludeking A, Yun H, Friesen I, Damm F, et al. Rare occurrence of DNMT3A mutations in myelodysplastic syndromes. Haematologica. (2011) 96:1870–3. doi: 10.3324/haematol.2011.045559
39. Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. (2011) 25:1153–8. doi: 10.1038/leu.2011.44
40. Chen CY, Lin LI, Tang JL, Ko BS, Tsay W, Chou WC, et al. RUNX1 gene mutation in primary myelodysplastic syndrome–the mutation can be detected early at diagnosis or acquired during disease progression and is associated with poor outcome. Br J Haematol. (2007) 139:405–14. doi: 10.1111/j.1365-2141.2007.06811.x
41. Dicker F, Haferlach C, Sundermann J, Wendland N, Weiss T, Kern W, et al. Mutation analysis for RUNX1, MLL-PTD, FLT3-ITD, NPM1 and NRAS in 269 patients with MDS or secondary AML. Leukemia. (2010) 24:1528–32. doi: 10.1038/leu.2010.124
42. Bernard E, Tuechler H, Greenberg PL, Hasserjian RP, Ossa JEA, Nannya Y, et al. Molecular international prognostic scoring system for myelodysplastic syndromes. NEJM Evidence. (2022) 1:EVIDoa2200008. doi: 10.1056/EVIDoa2200008
43. Sallman DA, Komrokji R, Vaupel C, Cluzeau T, Geyer SM, McGraw KL, et al. Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes. Leukemia. (2016) 30:666–73. doi: 10.1038/leu.2015.304
44. Nazha A, Komrokji R, Meggendorfer M, Jia X, Radakovich N, Shreve J, et al. Personalized prediction model to risk stratify patients with myelodysplastic syndromes. J Clin Oncol. (2021) 39:3737–46. doi: 10.1200/JCO.20.02810
45. Dalton WB, Helmenstine E, Pieterse L, Li B, Gocke CD, Donaldson J, et al. The K666N mutation in SF3B1 is associated with increased progression of MDS and distinct RNA splicing. Blood Adv. (2020) 4:1192–6. doi: 10.1182/bloodadvances.2019001127
46. Jiang L, Wang L, Shen C, Zhu S, Lang W, Luo Y, et al. Impact of mutational variant allele frequency on prognosis in myelodysplastic syndromes. Am J Cancer Res. (2020) 10:4476–87. https://www.ncbi.nlm.nih.gov/pubmed/33415012.
47. Padua RA, Guinn BA, Al-Sabah AI, Smith M, Taylor C, Pettersson T, et al. RAS, FMS and p53 mutations and poor clinical outcome in myelodysplasias: a 10-year follow-up. Leukemia. (1998) 12:887–92. doi: 10.1038/sj.leu.2401044
48. Bernard E, Nannya Y, Hasserjian RP, Devlin SM, Tuechler H, Medina-Martinez JS, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. (2020) 26:1549–56. doi: 10.1038/s41591-020-1008-z
49. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. (2014) 371:2488–98. doi: 10.1056/NEJMoa1408617
50. Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. (2014) 371:2477–87. doi: 10.1056/NEJMoa1409405
51. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. (2015) 126:9–16. doi: 10.1182/blood-2015-03-631747
52. Cacic AM, Schulz FI, Germing U, Dietrich S, Gattermann N. Molecular and clinical aspects relevant for counseling individuals with clonal hematopoiesis of indeterminate potential. Front Oncol. (2023) 13:1303785. doi: 10.3389/fonc.2023.1303785
53. Cazzola M. Introduction to a review series on germ line predisposition to hematologic Malignancies: time to consider germ line testing. Blood. (2023) 141:1509–12. doi: 10.1182/blood.2023019846
54. Li P, Brown S, Williams M, White T, Xie W, Cui W, et al. The genetic landscape of germline DDX41 variants predisposing to myeloid neoplasms. Blood. (2022) 140:716–55. doi: 10.1182/blood.2021015135
55. Makishima H, Saiki R, Nannya Y, Korotev S, Gurnari C, Takeda J, et al. Germ line DDX41 mutations define a unique subtype of myeloid neoplasms. Blood. (2023) 141:534–49. doi: 10.1182/blood.2022018221
56. Kennedy AL, Shimamura A. Genetic predisposition to MDS: clinical features and clonal evolution. Blood. (2019) 133:1071–85. doi: 10.1182/blood-2018-10-844662
57. Bannon SA, DiNardo CD. Hereditary predispositions to myelodysplastic syndrome. Int J Mol Sci. (2016) 17(6):838. doi: 10.3390/ijms17060838
58. Hamilton KV, Fox LC, Nichols KE. How I communicate with patients and families about germ line genetic information. Blood. (2023) 141:3143–52. doi: 10.1182/blood.2022017379
59. Badar T, Nanaa A, Foran JM, Viswanatha D, Al-Kali A, Lasho T, et al. Clinical and molecular correlates of somatic and germline DDX41 variants in patients and families with myeloid neoplasms. Haematologica. (2023) 108:3033–43. doi: 10.3324/haematol.2023.282867
60. Makishima H, Bowman TV, Godley LA. DDX41-associated susceptibility to myeloid neoplasms. Blood. (2023) 141:1544–52. doi: 10.1182/blood.2022017715
61. Maierhofer A, Mehta N, Chisholm RA, Hutter S, Baer C, Nadarajah N, et al. The clinical and genomic landscape of patients with DDX41 variants identified during diagnostic sequencing. Blood Adv. (2023) 7:7346–57. doi: 10.1182/bloodadvances.2023011389
62. Homan CC, Drazer MW, Yu K, Lawrence DM, Feng J, Arriola-Martinez L, et al. Somatic mutational landscape of hereditary hematopoietic Malignancies caused by germline variants in RUNX1, GATA2, and DDX41. Blood Adv. (2023) 7:6092–107. doi: 10.1182/bloodadvances.2023010045
63. Calvo KR, Hickstein DD. The spectrum of GATA2 deficiency syndrome. Blood. (2023) 141:1524–32. doi: 10.1182/blood.2022017764
64. Robbins DJ, Pavletich TS, Patil AT, Pahopos D, Lasarev M, Polaki US, et al. Linking GATA2 to myeloid dysplasia and complex cytogenetics in adult myelodysplastic neoplasm and acute myeloid leukemia. Blood Adv. (2024) 8:80–92. doi: 10.1182/bloodadvances.2023011554
65. Largeaud L, Collin M, Monselet N, Vergez F, Fregona V, Larcher L, et al. Somatic genetic alterations predict hematological progression in GATA2 deficiency. Haematologica. (2023) 108:1515–29. doi: 10.3324/haematol.2022.282250
66. Locatelli F, Strahm B. How I treat myelodysplastic syndromes of childhood. Blood. (2018) 131:1406–14. doi: 10.1182/blood-2017-09-765214
67. Homan CC, Scott HS, Brown AL. Hereditary platelet disorders associated with germ line variants in RUNX1, ETV6, and ANKRD26. Blood. (2023) 141:1533–43. doi: 10.1182/blood.2022017735
68. Sahoo SS, Pastor VB, Goodings C, Voss RK, Kozyra EJ, Szvetnik A, et al. Clinical evolution, genetic landscape and trajectories of clonal hematopoiesis in SAMD9/SAMD9L syndromes. Nat Med. (2021) 27:1806–17. doi: 10.1038/s41591-021-01511-6
69. Nagata Y, Narumi S, Guan Y, Przychodzen BP, Hirsch CM, Makishima H, et al. Germline loss-of-function SAMD9 and SAMD9L alterations in adult myelodysplastic syndromes. Blood. (2018) 132:2309–13. doi: 10.1182/blood-2017-05-787390
70. Kirwan M, Walne AJ, Plagnol V, Velangi M, Ho A, Hossain U, et al. Exome sequencing identifies autosomal-dominant SRP72 mutations associated with familial aplasia and myelodysplasia. Am J Hum Genet. (2012) 90:888–92. doi: 10.1016/j.ajhg.2012.03.020
71. Kim B, Yun W, Lee S-T, Choi JR, Yoo KH, Koo HH, et al. Prevalence and clinical implications of germline predisposition gene mutations in patients with acute myeloid leukemia. Sci Rep. (2020) 10:14297. doi: 10.1038/s41598-020-71386-z
72. Sallman DA, DeZern AE, Garcia-Manero G, Steensma DP, Roboz GJ, Sekeres MA, et al. Eprenetapopt (APR-246) and azacitidine in TP53-mutant myelodysplastic syndromes. J Clin Oncol. (2021) 39:1584–94. doi: 10.1200/JCO.20.02341
73. Komrokji R, Al Ali N, Chan O, Sweet K, Kuykendall A, Lancet J, et al. IDH mutations are enriched in myelodysplastic syndrome patients with severe neutropenia and can be a potential for targeted therapy. Haematologica. (2023) 108:1168–72. doi: 10.3324/haematol.2022.281607
74. Symeonidis A, Germing U. Editorial: Hybrid or mixed myelodyplastic/myeloproliferative disorders: Current trends in diagnosis and treatment. Front Oncol. (2023) 13:1166163. doi: 10.3389/fonc.2023.1166163
75. Joffe E, Greenbaum U, Man-El G, Filanovsky K, Oster HS, Levi I, et al. Kinetics of pre-myelodysplastic syndromes blood values correlate with disease risk and survival. Hematol Oncol. (2020) 38:782–91. doi: 10.1002/hon.2777
76. Valent P, Bain BJ, Bennett JM, Wimazal F, Sperr WR, Mufti G, et al. Idiopathic cytopenia of undetermined significance (ICUS) and idiopathic dysplasia of uncertain significance (IDUS), and their distinction from low risk MDS. Leuk Res. (2012) 36:1–5. doi: 10.1016/j.leukres.2011.08.016
77. Malcovati L, Galli A, Travaglino E, Ambaglio I, Rizzo E, Molteni E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. (2017) 129:3371–8. doi: 10.1182/blood-2017-01-763425
78. Galli A, Todisco G, Catamo E, Sala C, Elena C, Pozzi S, et al. Relationship between clone metrics and clinical outcome in clonal cytopenia. Blood. (2021) 138:965–76. doi: 10.1182/blood.2021011323
79. Valent P. ICUS, IDUS, CHIP and CCUS: diagnostic criteria, separation from MDS and clinical implications. Pathobiology. (2019) 86:30–8. doi: 10.1159/000489042
80. Gorak EJ, Otterstatter M, Al Baghdadi T, Gillis N, Foran JM, Liu JJ, et al. Discordant pathologic diagnoses of myelodysplastic neoplasms and their implications for registries and therapies. Blood Adv. (2023) 7:6120–9. doi: 10.1182/bloodadvances.2023010061
81. Oster HS, Carmi G, Kolomansky A, Joffe E, Kaye I, Kirgner I, et al. Is bone marrow examination always necessary to establish the diagnosis of myelodysplastic syndromes? A proposed non-invasive diagnostic model. Leuk Lymphoma. (2018) 59:2227–32. doi: 10.1080/10428194.2017.1416363
82. Oster HS, Crouch S, Smith A, Yu G, Abu Shrkihe B, Baruch S, et al. A predictive algorithm using clinical and laboratory parameters may assist in ruling out and in diagnosing MDS. Blood Adv. (2021) 5:3066–75. doi: 10.1182/bloodadvances.2020004055
83. Polakow A, Oster H, Golsdshmidt N, Mittelman M. P004 - topic: AS01-diagnosis/AS01a-cytomorphology: NON-INVASIVE WEB-BASED DIAGNOSTIC ALGORITHM FOR MDS – MODEL PERFORMANCE AND VALIDATION. Leukemia Res. (2023) 128:107139. doi: 10.1016/j.leukres.2023.107139
84. Schulz F, Nachtkamp K, Oster HS, Mittelman M, Gattermann N, Schweier S, et al. Validation of a novel algorithm with a high specificity in ruling out MDS. Int J Lab Hematol. (2024) 46(3):510–4. doi: 10.1111/ijlh.14234
85. Furer N, Rappoport N, Tanay A, Shlush L. Natural and age-related variation in circulating human hematopoietic stem cells. Eur Hematol Association HemaSphere. (2023) 7:e6115724. doi: 10.1097/01.HS9.0000967320.61157.24
86. Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. (2011) 44:53–7. doi: 10.1038/ng.1031
87. Paquette RL, Landaw EM, Pierre RV, Kahan J, Lubbert M, Lazcano O, et al. N-ras mutations are associated with poor prognosis and increased risk of leukemia in myelodysplastic syndrome. Blood. (1993) 82:590–9. https://www.ncbi.nlm.nih.gov/pubmed/8329714.
88. Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. (2010) 42:665–7. doi: 10.1038/ng.620
89. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. (2010) 42:722–6. doi: 10.1038/ng.621
90. Kosmider O, Gelsi-Boyer V, Slama L, Dreyfus F, Beyne-Rauzy O, Quesnel B, et al. Mutations of IDH1 and IDH2 genes in early and accelerated phases of myelodysplastic syndromes and MDS/myeloproliferative neoplasms. Leukemia. (2010) 24:1094–6. doi: 10.1038/leu.2010.52
91. Makishima H, Yoshida K, Nguyen N, Przychodzen B, Sanada M, Okuno Y, et al. Somatic SETBP1 mutations in myeloid Malignancies. Nat Genet. (2013) 45:942–6. doi: 10.1038/ng.2696
92. Steensma DP, Dewald GW, Lasho TL, Powell HL, McClure RF, Levine RL, et al. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both "atypical" myeloproliferative disorders and myelodysplastic syndromes. Blood. (2005) 106:1207–9. doi: 10.1182/blood-2005-03-1183
93. Piazza R, Valletta S, Winkelmann N, Redaelli S, Spinelli R, Pirola A, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet. (2013) 45:18–24. doi: 10.1038/ng.2495
Keywords: Myelodysplastic syndromes, cytogenetics, computer model, peripheral blood (PB), bone marrow examination, genetic mutations
Citation: Oster HS and Mittelman M (2024) How we diagnose Myelodysplastic syndromes. Front. Oncol. 14:1415101. doi: 10.3389/fonc.2024.1415101
Received: 09 April 2024; Accepted: 22 August 2024;
Published: 13 September 2024.
Edited by:
Sabine Blum, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandReviewed by:
Eleftheria Hatzimichael, University of Ioannina, GreeceAnnika Kasprzak, University Hospital of Düsseldorf, Germany
Copyright © 2024 Oster and Mittelman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Moshe Mittelman, bW9zaGVtdEBnbWFpbC5jb20=