 Petra Bašová1†Lubomír Minařík1,2†Silvia Carina Magalhaes-Novais3
Petra Bašová1†Lubomír Minařík1,2†Silvia Carina Magalhaes-Novais3 Jana Balounová3Zuzana Zemanová4
Jana Balounová3Zuzana Zemanová4 Tatiana Aghová4Martin Špaček4
Tatiana Aghová4Martin Špaček4 Anna Jonášová2
Anna Jonášová2 Kristýna Gloc Pimková1
Kristýna Gloc Pimková1 Jan Procházka3
Jan Procházka3 Radislav Sedláček3
Radislav Sedláček3 Tomáš Stopka1,2*
Tomáš Stopka1,2*- 1BIOCEV, First Faculty of Medicine, Charles University, Prague, Czechia
- 2Department of Hematology, General Faculty Hospital and Charles University, Prague, Czechia
- 3Czech Centre for Phenogenomics, Institute of Molecular Genetics of the Czech Academy of Sciences, Prague, Czechia
- 4Department of Biochemistry and Laboratory Diagnostics, General Faculty Hospital and Charles University, Prague, Czechia
Introduction: Progressing myelodysplastic syndrome (MDS) into acute myeloid leukemia (AML) is an indication for hypomethylating therapy (HMA, 5-Azacytidine (AZA)) and a BCL2 inhibitor (Venetoclax, VEN) for intensive chemotherapy ineligible patients. Mouse models that engraft primary AML samples may further advance VEN + AZA resistance research.
Methods: We generated a set of transplantable murine PDX models from MDS/AML patients who developed resistance to VEN + AZA and compared the differences in hematopoiesis of the PDX models with primary bone marrow samples at the genetic level. PDX were created in NSGS mice via intraosseal injection of luciferase-encoding Lentivirus-infected MDS/AML primary cells from patient bone marrow. We validated the resistance of PDX-leukemia to VEN and AZA and further tested candidate agents that inhibit the growth of VEN/AZA-resistant AML.
Results and discussion: Transplantable PDX models for MDS/AML arise with 31 % frequency. The lower frequency of transplantable PDX models is not related to peritransplant lethality of the graft, but rather to the loss of the ability of short-term proliferation of leukemic progenitors after 10 weeks of engraftment. There exist subtle genetic and cytological changes between primary and PDX-AML samples however, the PDX models retain therapy resistance observed in patients. Based on in vitro testing and in vivo validation in PDX models, Panobinostat and Dinaciclib are very promising candidate agents that overcome dual VEN + AZA resistance.
Introduction
Although myelodysplastic syndrome (MDS) during progression to acute myeloid leukemia (AML) is indicated for hypomethylating therapy (HMA, 5-Azacytidine (AZA)), the addition of a BCL2 inhibitor (Venetoclax, VEN) yields significantly better responses in patients unsuitable for intensive chemotherapy. Genetic predictors of a favorable response include IDH1/2 or DDX41 mutations, while negative predictors include monosomy 7, EZH2 mutations or N/KRAS activating mutations. Although AZA and VEN have different modes of action, most patients develop dual resistance leading to relapse with an expected survival of 2.4 months (1). Recently, cellular models of stable dual resistance to VEN + AZA have been generated without altering their proliferation properties. Resistance to VEN + AZA leads to an increase in the anti-apoptotic protein MCL1 and a decrease in the pro-apoptotic protein BAX, which has been experimentally validated as a mechanism of resistance in these models (2). However, further research has shown that only co-targeting of BCL2 and MCL1 is effective in AML cell lines with intrinsic or acquired resistance to BH3 mimetics or engineered to genetically overexpress BCL2 or BCL2A1 or downregulate BAX. Thus, it is rather the co-dependence on multiple anti-apoptotic BCL2 proteins and BAX suppression that can be considered as mechanisms of AML resistance to individual BH3 mimetics (3). Genetic screens based on CRISPR-Cas9 have successfully identified several targets in AML, specifically MARCH5, which prevents apoptosis in AML and whose repression enhanced the efficacy of BCL2 inhibitors such as VEN (4), not only in established AML cell lines, but also in PDX models, which are considered to be the most faithful to the primary human disease.
Current patient derived xenograft (PDX) models were established by Wunderlich et al. who generated a mouse strain that transgenically expresses the human cytokines: stem cell factor (SCF), GM-CSF, and IL-3 (SGM3) in a NOD/SCID background, termed NOD/LtSz-scid IL2RG-SGM3 (NSGS), which resulted in improved engraftment of AML cells from patients with various aberrations even compared to the original NSG model (5). PDX exhibit functional heterogeneity, specifically, up to twelve AML patient-derived subclones were generated, each derived from a single stem cell. These clones exhibited different transcriptional and proteomic profiles with a molecular signature of resistance (6). Interestingly, for PDX modelling of AML, reproducible human AML engraftment is mainly limited to high-risk cases. A recent study showed that the majority (85-94%) of mice were engrafted into bone marrow (BM) regardless of risk group, but serial transplantation and long-term cell culture (LTC-IC) assays revealed long-term engraftment in patients with high-risk AML but also with high leukemia-initiating cell counts. This study provided a comprehensive in vivo characterization of human AML in NSGS-type mice and reveals distinct intrinsic characteristics for each risk group with respect to engraftment capacity, LIC, and stromal interactions (7).
PDX models are used to test new therapies such as HDAC inhibitors (8), PPAR alpha inhibitors that specifically inhibit leukemia stem cells (9), FLT3 inhibitors (10, 11), kinase inhibitors (12), or SF3B1 inhibitors (13). In our research, we wanted to build on these previous studies and asked how efficiently NSGS-based PDX models can be created and, more importantly, whether they can be successfully used to test new compounds overcoming resistance to VEN + AZA.
Materials and methods
PDX acceptor mouse strain as a model to engraft and serve for testing of MDS/AML
NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ is also known as NSG-SGM3. The NSG-SGM3 (NSGS) transgenic mouse expresses the human cytokines IL3, GM-CSF (CSF2) and SCF (KITLG), thus combining the properties of the highly immunodeficient NOD scid gamma (NSG, The Jackson Laboratory) mouse, which ideally promotes stable adhesion of myeloid lineages (14) and regulatory T-cell populations through cytokine expression. However, T cell engraftment may paradoxically reduce tumor cell engraftment through the so-called graft vs leukemia effect. Nevertheless, it can be concluded that these NSG-SGM3 mice are almost ideal for implantation of primary AML samples (5) than other models and are useful for immuno-oncology, immunology and other research.
Patient samples and ethics
BM samples (representing residua of those which were used for routine diagnostics) were cryopreserved in liquid nitrogen with patients’ agreement to further analysis. Patient samples were collected in year 2023 following the written informed consent based on the Helsinki declaration and approved by the Ethics Committee of the General Hospital Prague. On 14 June 2018, the Ethics Committee of the General University Hospital discussed and approved the grant project No. 48/18 entitled Development of mouse models of hematological tumors for the purpose of cancer research and experimental preclinical treatment, which was to be carried out in the years 2019-2023. Furthermore, on 16 June 2022, the Ethics Committee approved the grant project No. 38/22 entitled Research on resistance to immunomodulatory drugs (IMiDs) - design of new therapies.
Mouse handling and ethics
Animals were maintained under SPF conditions in individually ventilated cages with controlled temperature (22 ± 2°C) and humidity under a 12 h light/12 h dark cycle and with food and drink ad libitum. Body condition score and body weight were accessed twice a week until the end of the experiment. Mice were sacrificed when signs of disease were visible, such as: lethargy, excessive weight loss (> 20%), change in fur quality, apathy, and hind leg paralysis. The license number for the CCP projects including the ethics was entitled Study of targeted therapy in vivo in PDX model of chemoresistant myeloid malignancies is PDX 63/2020 later modified AVCR 253/2023 SOVII.
Intraosseal transplantation technique
Three days before transplantation mice were fed with high caloric diet and 2% of gentamycin (G1397, Merck) was added in their drinking water. The gentamycin was kept for 7 days from the day of transplantation and changed every 3 days. On the day of transplantation mice were irradiated with sublethal dose of 120 cGy using XRAD320 X-ray system.
For the intraosseal cell application, after anesthetized with 20% Zoletil (Biopharm), mice hind limbs were shaved and cleaned with scrubs of betadine. Mice were placed in a supine position on a sterile surgical platform and hind limbs were fixed at 90 degrees from the platform. Using 1 ml syringe with 27 gauge needle the femur was slowly perfused with 20 µl of cell suspension. Aspiration was performed to confirm proper needle placement. Post-operative analgesics were administered (Bupaq, Richter Pharma AG), and mice were kept in a warm environment during the recovery period. Secondary (F2) and tertiary (F3) transplantation: hematopoietic tissue from F1 PDX mice were applicated into sublethal irradiated mice (BM: intraosseal; LIV, SPL: intravenous) (procedure shown above).
Bioluminescence-based engraftment monitoring
In vivo whole-body imaging of mice was performed every 4 weeks using the LagoX machine (Spectral Imaging Instruments) with Aura software. The mice were anaesthetized by 3% isoflurane inhalation and injected intraperitoneally with 300 µl of 15 mg/ml Xeno-light D-luciferine substrate (Perkin Elmer, 122799) to visualize the tumor cells. The luminescence exposure time was 300 s, and the X-ray image was taken subsequently. Data for the calculation of radiance were obtained from Spectral Instruments Imaging by Aura Imaging Software. Radiance is a calibrated absolute measurement of photon emission from the subject (photons/second/cm2/steradian). Mean Rad is defined as Total radiance/number of pixels in the ROI (region of interest) defined as the image area quantified.
Flow cytometry analysis
Mouse blood samples (40-60 μl) were collected into EDTA-coated tubes. Red blood cells were lysed twice using ACK buffer. Following lysis, cells were washed with FACS buffer (2% FCS, 10 mM HEPES, 2 mM EDTA in HBSS without Ca2+ and Mg2+) and subsequently stained with a cocktail of fluorescently labeled antibodies supplemented with anti-mouse and human Fc block (Becton Dickinson 553142, 564220, both 1:200) in FACS buffer. The following antibodies were used: anti-hCD3-BV786 (Becton Dickinson 566781, 1:200), anti-hCD45-FITC (Biolegend 368508, 1:150), anti-hCD33-PECy7 (Biolegend 366618, 1:100), anti-hCD56-APC-Cy7 (Biolegend 362512, 1:200), and anti-mCD45-AF700 (Thermo Fisher Scientific 56-0451-82, 1:400). Following a washing step in FACS buffer, blood cells were resuspended in 195 μl of FACS buffer and mixed with 5 μl of CountBright™ Absolute Counting Beads (Thermo Fisher Scientific) and 0.5 μl of Hoechst 33258 (10 μg/ml). The entire sample was acquired on a Cytec Aurora cytometer. Data analysis was performed using FlowJo software, and absolute cell counts per 1 μl of blood were calculated.
WST-1 assay and specific chemotherapeutic agents
The WST1 is a cell proliferation colorimetric assay (Roche, Basel, Switzerland) to obtain IC50 (concentration of drug required for 50% inhibition). WST-1 assay performed on primary AML samples, PDX samples and cell line OCI-M2-AZA-R. Cells were seeded (50 000 live cells/well) on 96 or 384-well flat bottom plate with freshly prepared inhibitors (concentration scale 1 nM – 10 µM; diluted in primary cells medium (RPMI1640 + 25% FBS + 10 ng/ml SCF, G-CSF, GM-CSF); total volume with cells 40 - 100 µL). Outer wells were filled with PBS to prevent evaporation. After 48 hours incubation period (in 37°C 5% CO2) WST-1 reagent was added to each well and absorbancy at 440 nm and 660 nm (reference value) was measured 60 min after addition using Tecan Reader. Each treatment condition was normalized to vehicle treatment and IC50 was calculated using Prism Graphpad software. Panel of specific inhibitors included: 5-Azacytidine (AZA), Venetoclax (VEN), Panobinostat (PAN), Dinaciclib (DIN) and Sorafenib (SOR). All inhibitors were purchased from MedChemExpress (Monmouth Junction, NJ, USA) or Selleckchem (Houston, TX, USA). These inhibitors were administered i.p. in human-equivalent dosages three times a week followed by a 7-day pause. AZA=2.14 mg/kg, VEN=5.7 mg/kg, PANhigh = 10 mg/kg, PANlow = 5 mg/kg; DINhigh = 40 mg/kg, DINlow = 20 mg/kg.
Statistics
The data sets were compared using: t-test, Unpaired, two-tailed, confidence intervals 95%: *≤ 0.05, **≤ 0.001, ***≤ 0.0001, ****≤ 0.00001. Mean and error bars (SD, SEM) are shown.
Results
Investigating engraftment efficacy of PDX MDS/AML models
MDS/AML models can be derived from human leukemia cell lines (CDX models) or patients (PDX models) and are used to assess therapeutic efficacy and toxicity. Various models are currently available in commercial databases, but very little is known about how these models were generated and with what frequency. We set to determine whether the development of AML in PDX mice is a common or rare event. After xenotransplantation into irradiated NSGS recipients (as described in M&M and Supplementary Figure 1A), we monitored tumor tissue growth every two weeks by bioluminescence. We found that all 13 primary MDS/AML patient samples (Table 1) (using 34 NSGS mice, see Supplementary Figure 1B) were engrafted at 5 weeks after intraosseal injection. However, after 10 weeks, there was a sudden drop in increasing bioluminescence in some of the samples that led to complete loss of bioluminescent signal (i.e., 50% of the patient samples). This suggests that after a relatively long phase of proliferating engrafted tumor cells, either the proliferating capacity is diminished or external events such as tumor-specific lymphocytes may induce MDS/AML rejection.
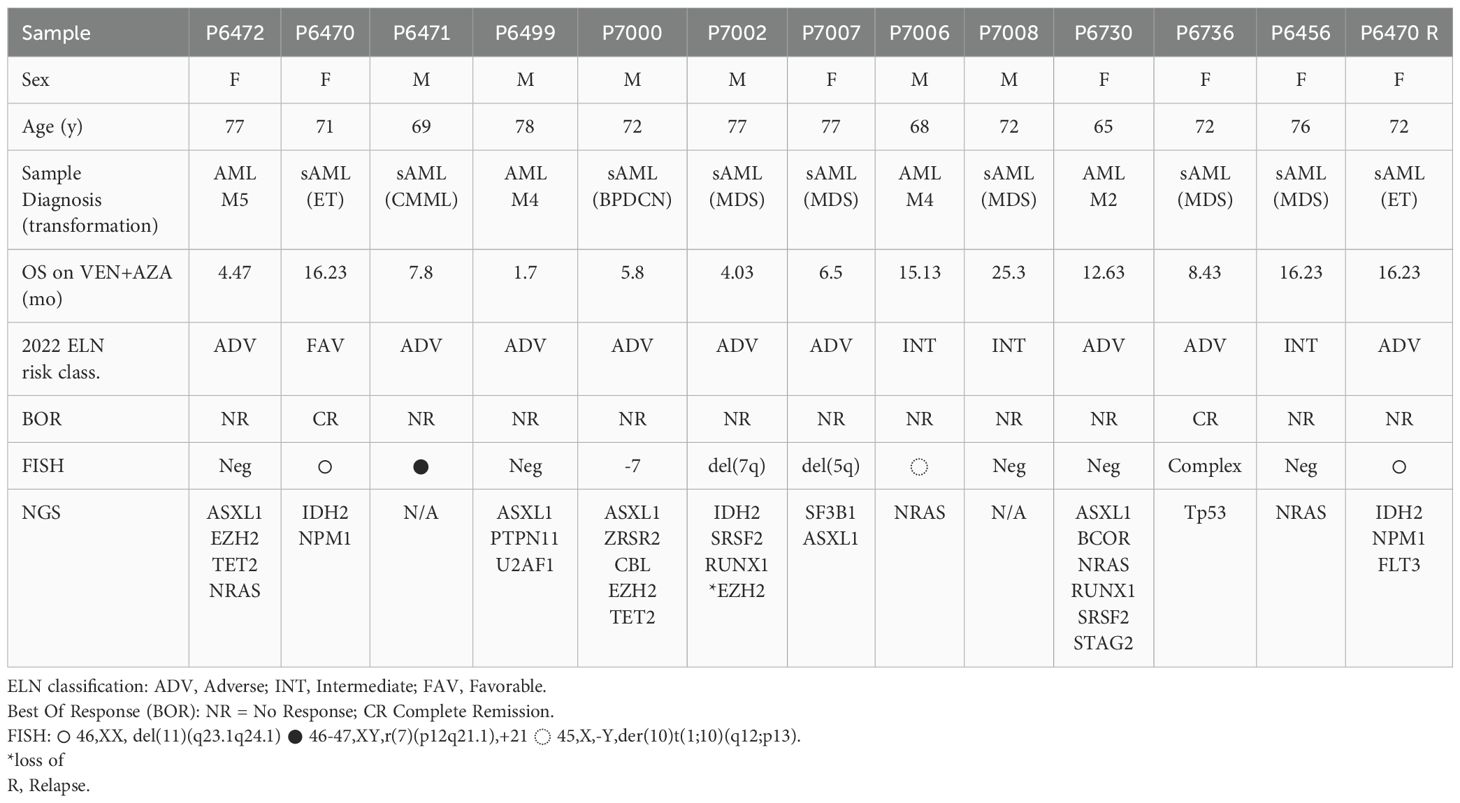
Table 1. Characteristics of primary 13 MDS/AML patients for xenotransplantation into NSGS mice.
In terms of tumor engraftment, the question is whether there is a repopulation of tumor-specific stem cells or whether the tumor growth in the first months consists only of short-lived leukemic cells, both of which are testable by the transplantation approach. To confirm the translatability of proliferating bioluminescent PDX-AML tumors, we performed a set of subsequent secondary and tertiary transplantations. Of the total number of transplanted samples, the number of those that were able to proliferate in secondary and tertiary recipients was reduced to 31% (i.e., 4 patient PDX models/out of 13 patients, Supplementary Figure 1B). These results suggest that the proliferative capacity of MDS/AML in the PDX model is exhausted between 5 and 10 weeks after transplantation, and approximately 1 out of 3 of MDS/AML samples retain repopulating stem-like activity.
Molecular differences between MDS/AML sample and PDX model
PDX models are widely used in research; their main advantage is that they are derived from patients, so they should faithfully recapitulate the context of the disease. On the other hand, it is unclear at this point whether a PDX model is completely identical to a patient sample. Therefore, we have taken a very detailed look at our generated PDX models using in depth methods (see Figure 1). All newly derived PDX models were assessed for tumor growth by bioluminescence and flow cytometry. Bioluminescence showed tumor growth from the primary site of inoculation (Figure 1B) and expansion of growth between weeks 1 and 9-12 of follow-up (Figure 1C). Analysis of the peritoneal cavity unveiled noticeable hepatosplenomegaly in PDX mice, which was notably more pronounced compared to the control NSGS mice (Figure 1B). Tumor analysis by flow cytometry showed accumulation of human CD45+ and CD33+ cells (Figures 1D, E). Supplementary Figures 2A-B show flow cytometry analysis after engraftment in blood and all hematopoietic tissue. Supplementary Figure 2C shows flow cytometry analysis after engraftment after the 1st, 2nd, and 3rd transplantation. Taken together, these data show that the newly created PDX models are unequivocally from leukemia stem cells, thus transplantable, and lead to significant tumor outgrowth in the recipients, resulting in overwhelming with tumor tissue.
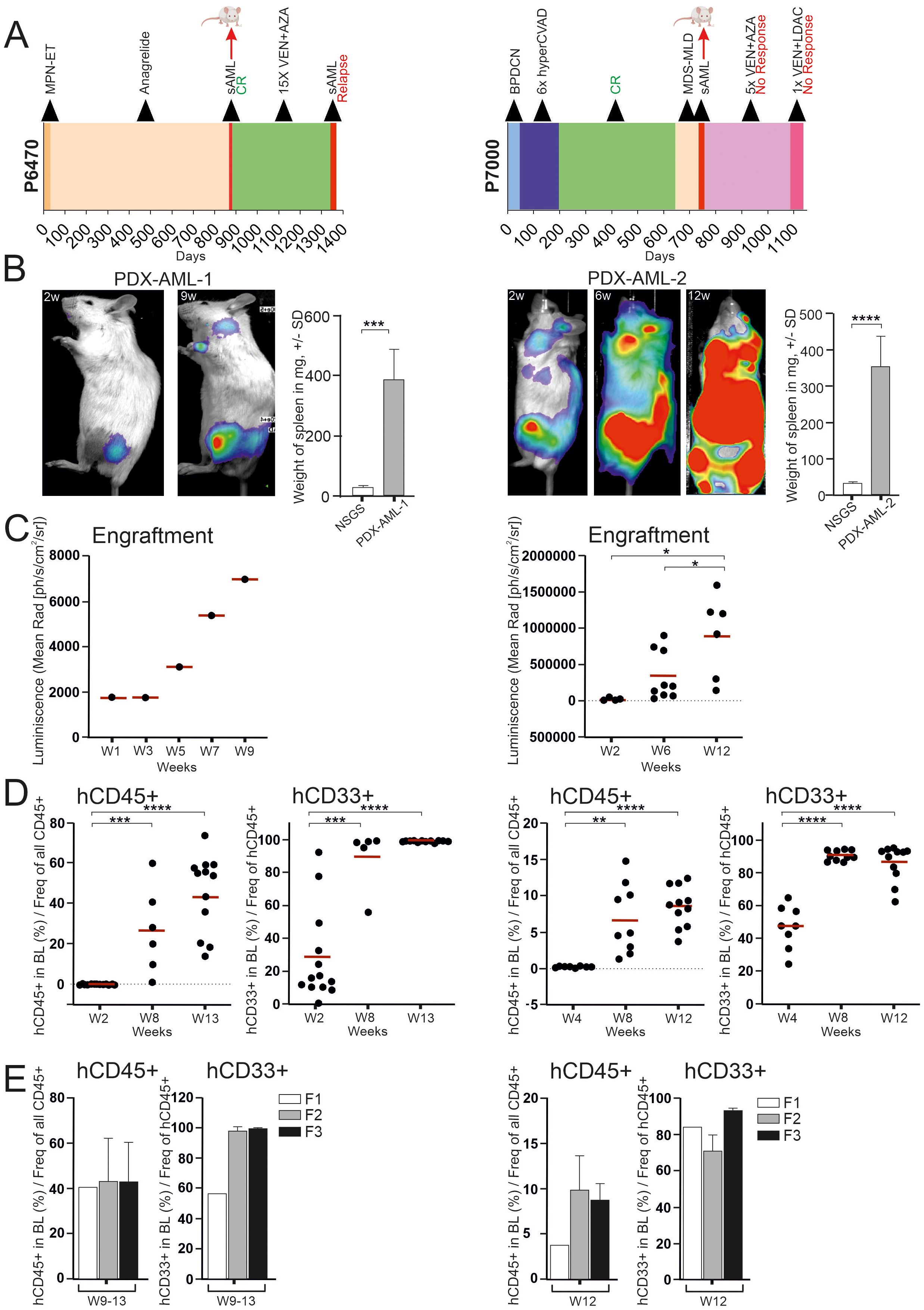
Figure 1. PDX model containing progressed MDS/AML with response to VEN (left side) and with resistance to VEN + AZA (right side). (A) Swimmer plots of two patients: P6470 with response to VEN (left) and P7000 with resistance to VEN + AZA (right). Graph includes a bar showing the length of treatment duration for each patient, the sample collection time for mouse transplantation is indicated by the red arrow. (B) Luminescence under general anesthesia (isoflurane) at indicated weeks after transplantation. Weight differences of NSGS spleens (N = 5) and PDX (N = 11, left; N = 8, right) in weeks 12-15. Mean ± SD, p-values (t-test, Unpaired, two-tailed). (C) Analysis of luciferase activity in PDX mouse (N = 1, during 9 weeks) (left) and in PDX mice at timepoints 2, 6, 12 weeks [W2-6 (N = 9), W12 (N = 6)] (right). Mean is shown, (t-test, Unpaired, two-tailed). (D) PB-FACS analysis of hCD45 or hCD33 (y-axis) at 2, 8 and 13 weeks (x-axis) [W2 (N = 13), W8 (N = 6), W13 (N = 12)] (left) and also PB-FACS analysis of indicated surface markers (y-axis) at 4, 8 and 12 weeks [W4 (N = 8), W8 (N = 10), W12 (N = 11)] (right). The data sets were compared using: t-test, Unpaired, two-tailed, confidence intervals 95%: *≤ 0.05, **≤ 0.001, ***≤ 0.0001, ****≤ 0.00001. Mean is shown. (E) PB-FACS analysis of indicated surface markers (y-axis) at terminal week in individual transplantation [F1 (N = 1), F2 (N = 8), F3 (N = 5)] (left) and [F1 (N = 1), F2 (N = 3), F3 (N = 7)] (right).
The PDX-AML-1 model was generated from a sample of female patient with secondary AML following ET who completed 15 cycles of VEN + AZA and achieved complete remission (CR) and progressed after 15 cycles of therapy (Table 1, Figure 1A left). NGS panel, flow cytometry and cytological analyses were used to compare tumor and PDX samples. Compared to the primary sample, the PDX-AML-1 model showed a loss of AML-associated monocytes (Figure 2A). The pathogenic mutations in DNMT3A, GATA2 and IDH2 persisted, however, unlike transient loss of NPM1 bearing clone in the time of remission (Figure 2B). During the disease progression, we also observed the positivity of FLT3-ITD. FACS profile confirmed the loss of monocytes (Figure 2C; Supplementary Figure 3A). Interestingly, the flow cytometry results for PDX-AML-1 are very similar to those seen in this patient P6470 at relapse. Incidentally, the relapse of this patient was also transplanted into NSGS mice and successfully generated a PDX model, designated PDX-AML-3 (Supplementary Figures 4A-D; Table 1 in red). Of note, the survival of the PDX-AML-3 mice is markedly shorter, which can be attributed to the selection of more aggressive clone/s (Supplementary Figure 4D).
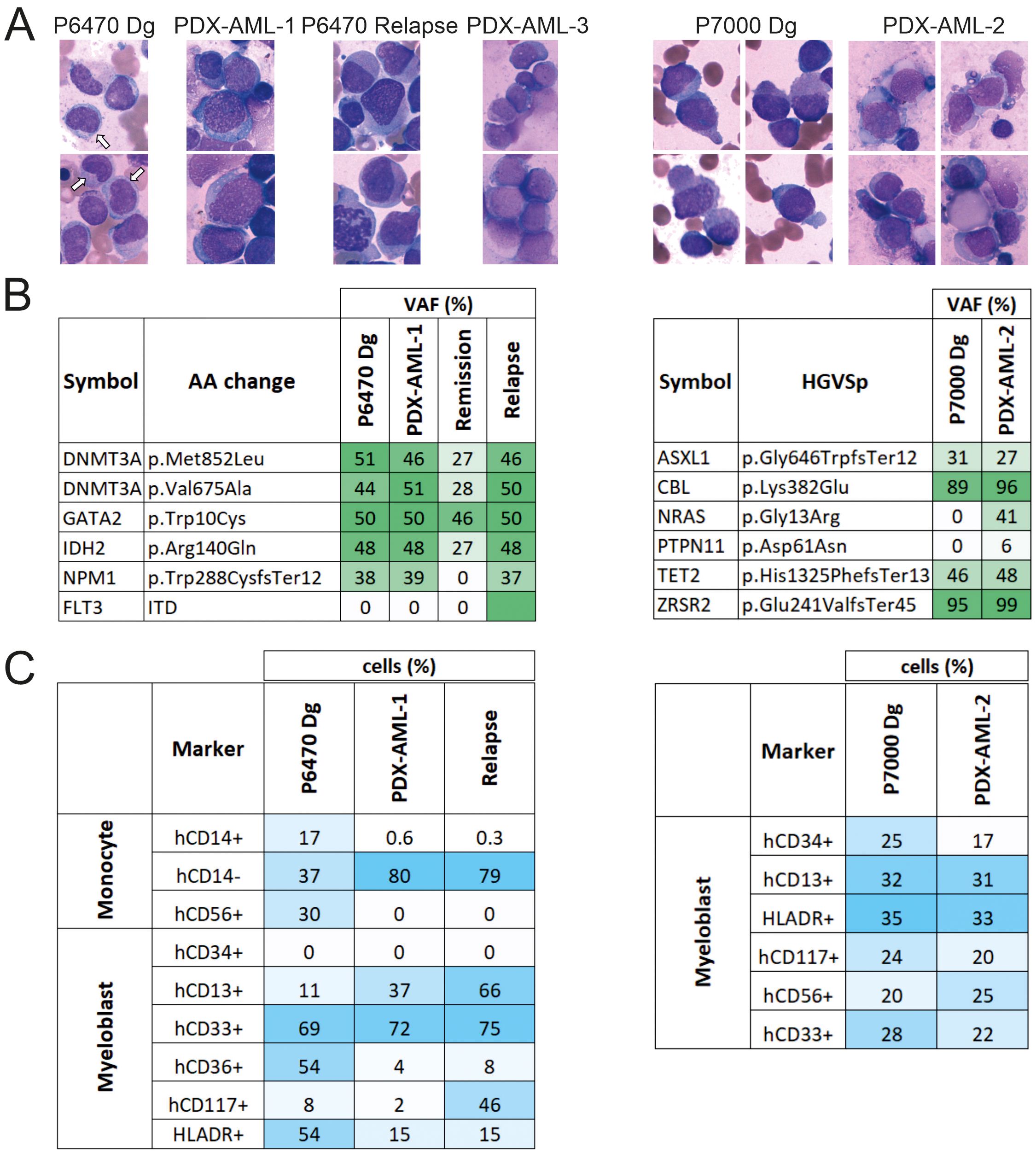
Figure 2. Comparison of the primary AML patient samples and the PDX-AML model. (A) Cytology of primary cells from diagnosis sample in comparison with cells from PDX-AML model stained via Giemsa-May-Grunwald protocol (white arrow = monocyte). (B) Mutation profile of primary samples in comparison with cells from PDX-AML models. (C) FACS analysis of indicated surface markers of primary samples in comparison with cells from PDX-AML model.
The PDX-AML-2 model was derived from a male-patient with secondary/therapy-related AML following Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN) who underwent 5 cycles of VEN + AZA achieving ‘No Response’ which was further managed with single cycle of VEN + LowDose AraC (Table 1, Figure 1A right); at this point the patient died due to robust progression and infection complication. Compared to the patient sample, the cytological (Figure 2A) and flow cytometry profiles (Figure 2C; Supplementary Figure 3B) were preserved in the PDX-AML-2 model, and pathogenic mutations of ASXL1, CBL, TET2 and ZRSR2 persisted alongside deletion of a chromosome 7. Importantly, additional mutations of NRAS and PTPN11 were also found in the PDX sample (Figure 2B). Comparison of the primary sample with PDX MDS/AML shows that these are clearly very similar, but not identical, and that differences exist in cell surface markers that are formed or lost, cytological composition, and this is also reflected by the additional pathogenic mutations. These changes may arise from a limited number of stem cells that engraft MDS/AML within the mouse PDX. It was subsequently during the relapse that we noticed that the mutational profile of the PDX sample and the relapse showed some similarities, and the same can be said for the flow cytometry and cytology profile (see Figures 2A-C). Another PDX model designated as PDX-AML-4 was recently generated from patient P6730: 65-year-old women with AML M2 marked with mutations in ASXL1, BCOR, NRAS, RUNX1, SRSF2, and STAG2 (see Table 1 in red).
Validating the therapeutic efficacy using PDX-derived cells and in a CDX model
One way to assess the similarity or dissimilarity of a primary MDS/AML sample to PDX is to test for sensitivity to specific chemotherapeutic agents. Specifically, we validated resistance (seen in patient) to AZA (in the PDX-AML-1 model) and dual resistance to VEN + AZA (in the PDX-AML-2 model). To do so, we used the WST-1 assay in PDX-derived cells (Figure 3A). Our data show that the sensitivity to AZA and VEN in both PDX models is identical to the patient response to these drugs (Figure 3B). Considering this, we further tested other candidate agents. Specifically, we found shared resistance to Sorafenib (SOR) with which neither patient was treated. Conversely, we also found significant sensitivity to Dinaciclib (DIN; inhibitor of CDK 1/2/5/9) and Panobinostat (PAN; HDAC inhibitor). These results show that in the case of resistance to AZA or VEN + AZA, there is not an induction of multidrug resistance, but on the contrary, it seems that cells retained sensitivity to some other drugs, which can be used as a salvage option in case of resistance to AZA or AZA+VEN.
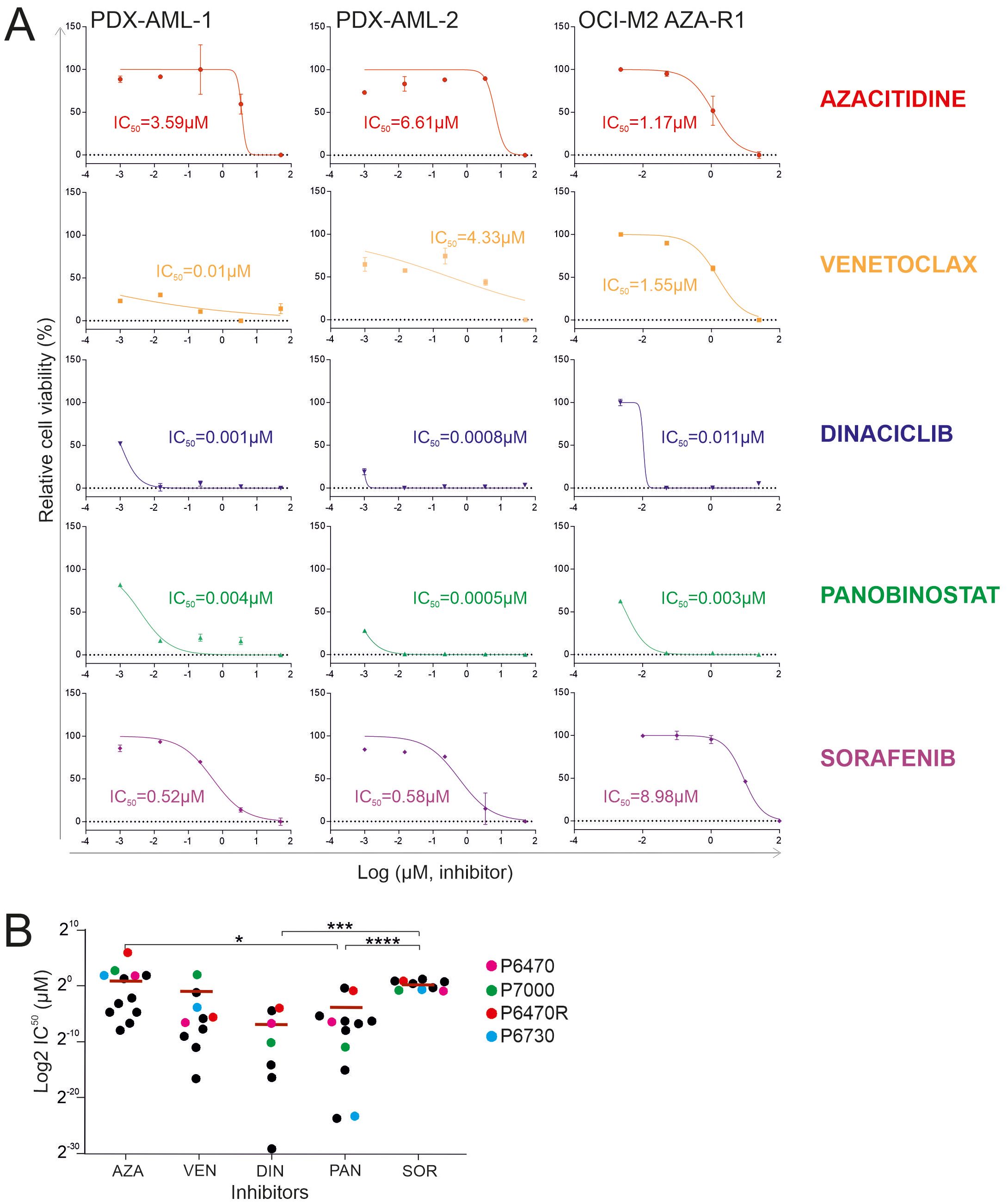
Figure 3. WST-1 proliferation essay. (A) Comparison of the effect of 5-Azacytidine (AZA), Venetoclax (VEN), Panobinostat (PAN), Dinaciclib (DIN) and Sorafenib (SOR) in primary MDS/AML cells and the AZA-resistant OCI-M2 cell line. (B) Comparison of IC50 values of following inhibitors: 5-Azacytidine (AZA), hypomethylating agent; Venetoclax (VEN), BCL2 inhibitor; Dinaciclib (DIN), CDK1/2/5/9 inhibitor; Panobinostat (PAN), HDAC inhibitor; Sorafenib (SOR) kinase inhibitor in AZA-resistant primary AML cells (N = 12). Data from established PDX models are shown in violet (P6470), red (P6470R), green (P7000), and blue (P6730). The data sets were compared using: t-test, Unpaired, two-tailed, confidence intervals 95%: *≤ 0.05, ***≤ 0.0001, ****≤ 0.00001. Mean is shown.
To further validate our assumptions derived from the experiment described in Figure 3A, which indicate the significant utility of Dinaciclib and Panobinostat for patients relapsing on the VEN + AZA regimen, we looked at the sensitivity of primary refractory progenitors following the VEN + AZA regimen to Dinaciclib and Panobinostat. Specifically, we used a cohort of patients treated with VEN + AZA. These patient samples (N=13) also include the samples that were used to successfully generate a PDX model. We used the WST1 method and measured the IC50 for the candidate drugs. We also further validated the IC50 for AZA and VEN. Figure 3B shows that Dinaciclib and Panobinostat are very effective alternatives for administration in patients with resistance to the VEN + AZA regimen, which cannot be stated for Sorafenib.
To test the tumor-specific effect of PAN and DIN in vivo, we used CDX models that we have generated from the MDS/AML cell line OCI-M2 in the past and that are resistant to AZA and VEN. After i.v. injection of tumor cells, we first assessed whether the tumor cells had engrafted in the NSGS mice. Subsequently, after 2 weeks, we initiated therapy (placebo, VEN + AZA, PAN and DIN in two doses) and assessed their effect on tumor growth by bioluminescence. These data clearly showed that while control therapy (PBS) and treatment with VEN + AZA were unable to block rapid tumor accumulation in the mouse, tumor growth was more gradual in the case of PAN and DIN therapy (Figure 4A). Bioluminescence trends of the treated PDX models are shown in the Figure 4B. Tumor growth affects the survival of treated animals, specifically the DIN therapy (unlike of PAN) led to a significant prolongation of survival (Figure 4C). The evolution of bioluminescence over time (weeks 0, 2, 8, and 10) indicates that while all tumors have a roughly similar tumor signal at baseline, PAN and DIN signals are still visible at week 8 and only DIN signal is visible at week 10 (Figure 4D; Supplementary Figure 5). However, DIN appears to be relatively more effective to clear VEN + AZA resistant cells compared to PAN. Thus, in summary, we suggest that based on in vitro and in vivo modeling in CDX/PDX systems, novel and previously unconsidered drugs such as DIN or PAN can be considered for the treatment of VEN + AZA resistant MDS/AML patients in the future.
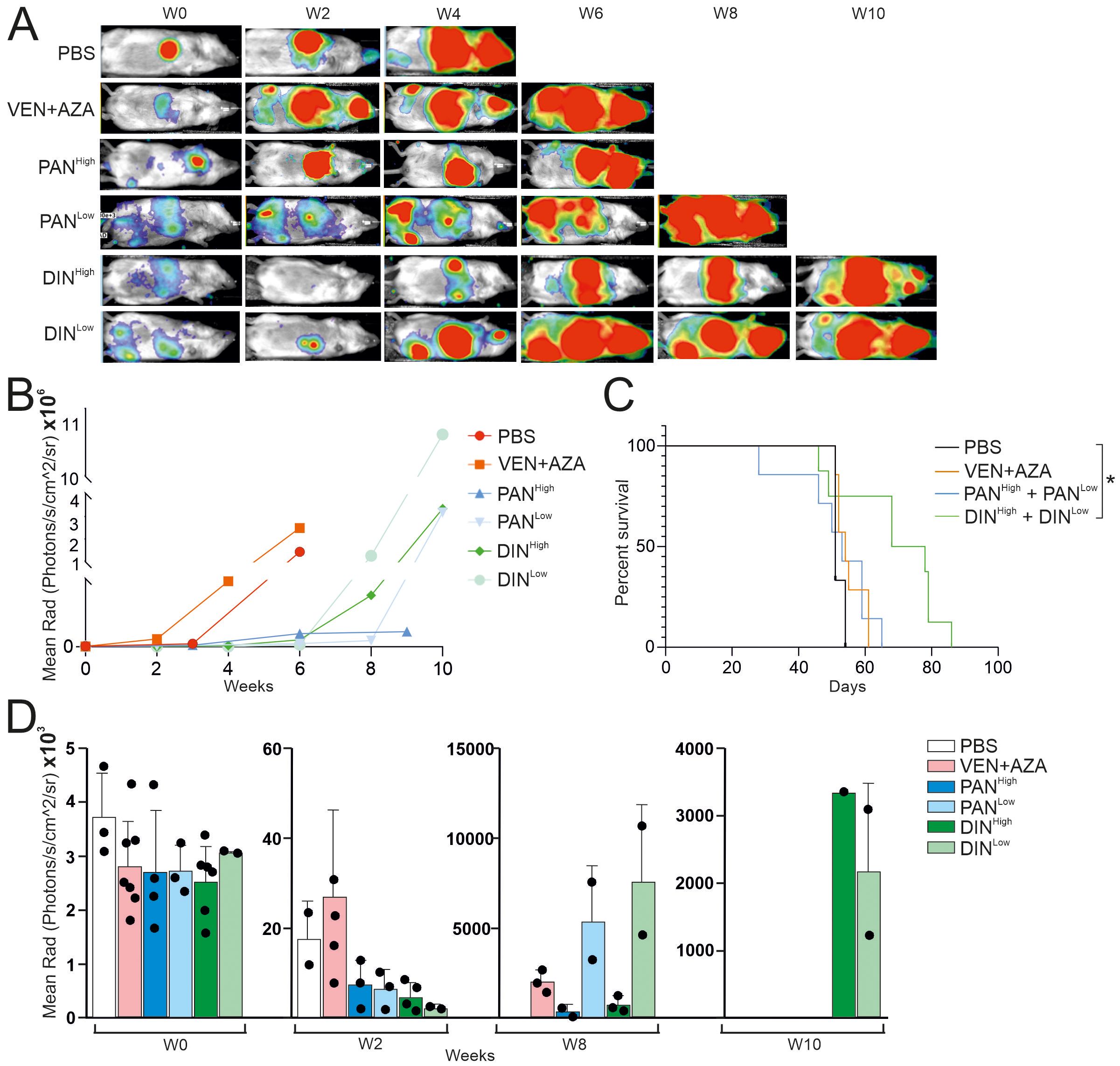
Figure 4. Testing the effect of PAN and DIN inhibitors on the survival of CDX mice containing the OCI-M2-AZA-R cell line. (A) Luminescence under general anesthesia (isoflurane) at indicated weeks after starting treatment with the inhibitors. (B) Analysis of luciferase activity in treated CDX mice (N=1 for each inhibitor treatment, during 10 weeks). (C) Survival curve of CDX treated mice [PBS (N=3), AZA/VEN (N=7), PANHigh (N=4), PANLow (N=3), DINHigh (N=6), DINLow (N=2)]. The data sets were compared using: t-test, Unpaired, two-tailed, confidence intervals 95%: *≤ 0.05. (D) Analysis of luciferase activity in treated CDX mice at 0, 2, 8 and 10 weeks after start of treatment.
Discussion
Our study showed that transplantable PDX models for MDS/AML resistant to VEN + AZA+ can be generated with a probability of 31% (~1 in 3) in NSGS mice. Lower engraftment efficiency as well as molecular signatures between PDX and primary AML have recently been also described, and it is more likely the leukemic initiating cells that determine this (7). In addition to the intrinsic nature of AML, there are also factors at the T cell level that can influence engraftment. Recently, a study of transgenic expression of interleukin IL15 improved T cell efficiency and thus survival in a PDX model. IL15 led to T cells remaining in a less differentiated state, thus preventing their exhaustion (15). In our PDX modeling, we also observed that in rare cases after loss AML engraftment, T cells accumulated at the engraftment site (data not shown), suggesting that the role of autologous lymphocytes is very important and possibly actionable. Previous studies have also shown a clonal discrepancy between PDX and primary cells. A recent study has shown that by investigating the clonal dynamics of somatic mutations in the PDX system, Resistant/Refractory (R/R) AMLs dominate in PDX mice generated from multi-clonal AML cells at diagnosis, although R/R clones have low allelic frequency in a diagnosis sample, indicating their engraftment potential (16).
Our study showed that resistance to VEN + AZA can be overcome with some already FDA approved drugs. Specifically, DIN and to lesser extent also PAN are two effective inhibitors that can be considered in patients with developed VEN + AZA resistance who are at risk of very short survival upon VEN + AZA failure. However, clinical validation of this preclinical result remains one of the long-term goals resulting from this work. Although several mechanisms of VEN resistance in AML have been identified, recent research has shown that heterogeneity in resistance mechanisms across patient populations exists also at the transcriptomic and cell signaling levels, specifically with activation of the PI3K-AKT-mTOR signaling axis and energy metabolism pathways. Other transcriptional features included transcriptional repression of HOX expression, activation of JAK-STAT signaling, or overexpression of interferon signaling (17). Given the significant transcriptional differences in VEN resistance, the efficacy of HDAC inhibitors is understandable. Similar and detailed results were also reported by another study, which investigated the mechanisms by which the anti-AML activity of VEN could be potentiated by dual mTORC1/TORC2 inhibition. Among others, for example, constitutive AKT activation counteracted the synergism between Venetoclax and PI3K or AKT inhibitors (18). We previously showed for the CDX AZA-resistant model (based on the OCI-M2 MDS/AML cell line) that AKT signaling plays an important and actionable role (19). Recent research has shown that a VEN in combination with the PPARα agonist chiglitazar led to synergistic suppression of AML progression in PDX models. Mechanistically, chiglitazar-mediated PPARα activation inhibited the transcriptional activity of the PIK3AP1 gene promoter and down-regulated the PI3K/AKT signaling pathway and anti-apoptotic BCL2 proteins, leading to inhibition of cell proliferation and induction of apoptosis, which was synergized with VEN (20). Another work suggested another unique mechanism that via AXL/MERTK inhibition effectively blocks VEN-resistant FLT3-ITD AML cells including those overexpressing MCL1 (21). Another recent study showed that increased expression of the ubiquitin ligase RNF5 contributes to the development and survival of AML, while inhibition of RNF5 causes transcriptional changes that overlap with those observed with histone deacetylase (HDAC) inhibition. RNF5 induces the formation of K29 ubiquitin chains on the histone-binding protein RBBP4, thereby promoting its recruitment and subsequent epigenetic regulation of genes involved in AML maintenance. Therefore, knockdown of RNF5 or RBBP4 increases the sensitivity of AML cells to HDAC inhibitors (22). Considering these results, it is likely that susceptibility to HDAC inhibitors in AML may be potentiated or may arise because of clonal selection during development of VEN + AZA resistance.
Like what we have attempted, there are laboratories developing new AML PDX models (23). There are even already PDX models with a particular pattern of mutations, specifically e.g. DNMT3A mutants, on which specific therapies can be tested (24). In addition, a methodological article to support the development of new PDX models has recently also been published (25). PDX research has led to the introduction of new therapies, specifically some agents such as CD47 inhibitors can sensitize to VEN + AZA treatment (26). The stress sensor GADD45A in AML is also associated with treatment resistance in AML and PDX models have been utilized during this research (27). PDX models have also been used to understand the role of “multiple drug resistance” mechanisms in AML (28) or to test novel CAR-T therapies (29). Thus, the role of PDX models in modern AML pharmacotherapy is beyond dispute and in this respect our work represents a step towards testing new approaches to VEN + AZA resistance and introducing new therapies.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethical Committee of the General Hospital, Prague #38/22. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. The animal studies were approved by Ethical Committe at the Institute of Molecular Genetics, #AVCR 253/2023 SOVII. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
PB: Data curation, Formal analysis, Writing – original draft, Writing – review & editing, Validation. LM: Validation, Writing – original draft, Writing – review & editing, Conceptualization, Investigation, Methodology, Supervision. SM-N: Investigation, Methodology, Writing – review & editing. JB: Methodology, Writing – review & editing, Validation. ZZ: Methodology, Writing – review & editing, Investigation. TA: Investigation, Methodology, Writing – review & editing, Validation. MŠ: Investigation, Methodology, Writing – review & editing. AJ: Investigation, Methodology, Writing – review & editing, Data curation. KP: Investigation, Methodology, Writing – review & editing. JP: Methodology, Supervision, Writing – review & editing. RS: Funding acquisition, Investigation, Supervision, Writing – review & editing. TS: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Grants: TS, LM, PB, KP were supported by the GAČR (24-10353S), which covered the cost of flow cytometry and sequencing. In addition, AZV CR (NU22-05-00374, NU21-08-00312) provided funding for personnel and research, specifically labeling of mouse stem cells with retroviruses, culture experiments, and other analyses. National Cancer Research Institute (reg. no. LX22NPO5102) - funded by the European Union – Next Generation EU, the EXCELES programme provided funding for staff and research support, specifically genetic analyses of cancer cells as well as biobanking. TS team was also sponsored by CZ.01.01.01/01/22_002/0000832 Development of technology for the production of biomedical preparations for advanced therapy in the field of haematooncology. ZZ, TA, MŠ and AJ were funded through RVO (MoH DRO-VFN-64165), which supported staff costs and materials for cytogenetic analyses and DNA sequencing. Mouse experiments were supported by the Czech Academy of Sciences RVO 68378050, LM2018126 and LM2023036 Czech Centre for Phenogenomics provided by MEYS and by CZ.02.1.01/0.0/0.0/18_046/0015861 CCP Infrastructure Upgrade II by MEYS and ESIF. SM-N, JB designed and performed the in vivo studies. JP and RS supervised the in vivo studies.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2024.1414950/full#supplementary-material
References
1. Maiti A, Rausch CR, Cortes JE, Pemmaraju N, Daver NG, Ravandi F, et al. Outcomes of relapsed or refractory acute myeloid leukemia after frontline hypomethylating agent and venetoclax regimens. Haematologica. (2021) 106:894–8. doi: 10.3324/haematol.2020.252569
2. Carter JL, Su Y, Qiao X, Zhao J, Wang G, Howard M, et al. Acquired resistance to venetoclax plus azacitidine in acute myeloid leukemia: In vitro models and mechanisms. Biochem Pharmacol. (2023) 216:115759. doi: 10.1016/j.bcp.2023.115759
3. Carter BZ, Mak PY, Tao W, Zhang Q, Ruvolo V, Kuruvilla VM, et al. Maximal activation of apoptosis signaling by cotargeting antiapoptotic proteins in BH3 mimetic-resistant AML and AML stem cells. Mol Cancer Ther. (2022) 21:879–89. doi: 10.1158/1535-7163.MCT-21-0690
4. Lin S, Larrue C, Scheidegger NK, Seong BKA, Dharia NV, Kuljanin M, et al. An in vivo CRISPR screening platform for prioritizing therapeutic targets in AML. Cancer Discovery. (2022) 12:432–49. doi: 10.1158/2159-8290.CD-20-1851
5. Wunderlich M, Chou FS, Link KA, Mizukawa B, Perry RL, Carroll M, et al. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia. (2010) 24:1785–8. doi: 10.1038/leu.2010.158
6. Zeller C, Richter D, Jurinovic V, Valtierra-Gutierrez IA, Jayavelu AK, Mann M, et al. Adverse stem cell clones within a single patient’s tumor predict clinical outcome in AML patients. J Hematol Oncol. (2022) 15:25. doi: 10.1186/s13045-022-01232-4
7. Diaz de la Guardia R, Velasco-Hernandez T, Gutierrez-Aguera F, Roca-Ho H, Molina O, Nombela-Arrieta C, et al. Engraftment characterization of risk-stratified AML in NSGS mice. Blood Adv. (2021) 5:4842–54. doi: 10.1182/bloodadvances.2020003958
8. Zhou H, Qin D, Xie C, Zhou J, Jia S, Zhou Z, et al. Combinations of HDAC inhibitor and PPAR agonist induce ferroptosis of leukemic stem cell-like cells in acute myeloid leukemia. Clin Cancer Res. (2024) 30(23):5430–44. doi: 10.1158/1078-0432.c.7565502
9. Zhou H, Jiang Y, Huang Y, Zhong M, Qin D, Xie C, et al. Therapeutic inhibition of PPARalpha-HIF1alpha-PGK1 signaling targets leukemia stem and progenitor cells in acute myeloid leukemia. Cancer Lett. (2023) 554:215997. doi: 10.1016/j.canlet.2022.215997
10. Zhang Y, Wang P, Wang Y, Shen Y. Sitravatinib as a potent FLT3 inhibitor can overcome gilteritinib resistance in acute myeloid leukemia. biomark Res. (2023) 11:8. doi: 10.1186/s40364-022-00447-4
11. Tan Y, Xin L, Wang Q, Xu R, Tong X, Chen G, et al. FLT3-selective PROTAC: Enhanced safety and increased synergy with Venetoclax in FLT3-ITD mutated acute myeloid leukemia. Cancer Lett. (2024) 592:216933. doi: 10.1016/j.canlet.2024.216933
12. Yang T, Ke H, Liu J, An X, Xue J, Ning J, et al. Narazaciclib, a novel multi-kinase inhibitor with potent activity against CSF1R, FLT3 and CDK6, shows strong anti-AML activity in defined preclinical models. Sci Rep. (2024) 14:9032. doi: 10.1038/s41598-024-59650-y
13. Wheeler EC, Martin BJE, Doyle WC, Neaher S, Conway CA, Pitton CN, et al. Splicing modulators impair DNA damage response and induce killing of cohesin-mutant MDS and AML. Sci Transl Med. (2024) 16:eade2774. doi: 10.1126/scitranslmed.ade2774
14. Coughlan AM, Harmon C, Whelan S, O’Brien EC, O’Reilly VP, Crotty P, et al. Myeloid engraftment in humanized mice: impact of granulocyte-colony stimulating factor treatment and transgenic mouse strain. Stem Cells Dev. (2016) 25:530–41. doi: 10.1089/scd.2015.0289
15. Mu-Mosley H, Ostermann L, Muftuoglu M, Vaidya A, Bonifant CL, Velasquez MP, et al. Transgenic expression of IL15 retains CD123-redirected T cells in a less differentiated state resulting in improved anti-AML activity in autologous AML PDX models. Front Immunol. (2022) 13:880108. doi: 10.3389/fimmu.2022.880108
16. Kawashima N, Ishikawa Y, Kim JH, Ushijima Y, Akashi A, Yamaguchi Y, et al. Comparison of clonal architecture between primary and immunodeficient mouse-engrafted acute myeloid leukemia cells. Nat Commun. (2022) 13:1624. doi: 10.1038/s41467-022-29304-6
17. Mohanty V, Baran N, Huang Y, Ramage CL, Cooper LM, He S, et al. Transcriptional and phenotypic heterogeneity underpinning venetoclax resistance in AML. bioRxiv. (2024). doi: 10.1101/2024.01.27.577579
18. Satta T, Li L, Chalasani SL, Hu X, Nkwocha J, Sharma K, et al. Dual mTORC1/2 inhibition synergistically enhances AML cell death in combination with the BCL2 antagonist venetoclax. Clin Cancer Res. (2023) 29:1332–43. doi: 10.1158/1078-0432.CCR-22-2729
19. Minarik L, Pimkova K, Kokavec J, Schaffartzikova A, Vellieux F, Kulvait V, et al. Analysis of 5-azacytidine resistance models reveals a set of targetable pathways. Cells. (2022) 11(2)223. doi: 10.3390/cells11020223
20. Xie C, Zhou H, Qin D, Zheng H, Tang Y, Li W, et al. Bcl-2 inhibition combined with PPARalpha activation synergistically targets leukemic stem cell-like cells in acute myeloid leukemia. Cell Death Dis. (2023) 14:573. doi: 10.1038/s41419-023-06075-6
21. Post SM, Ma H, Malaney P, Zhang X, Aitken MJL, Mak PY, et al. AXL/MERTK inhibitor ONO-7475 potently synergizes with venetoclax and overcomes venetoclax resistance to kill FLT3-ITD acute myeloid leukemia. Haematologica. (2022) 107:1311–22. doi: 10.3324/haematol.2021.278369
22. Khateb A, Deshpande A, Feng Y, Finlay D, Lee JS, Lazar I, et al. The ubiquitin ligase RNF5 determines acute myeloid leukemia growth and susceptibility to histone deacetylase inhibitors. Nat Commun. (2021) 12:5397. doi: 10.1038/s41467-021-25664-7
23. Stevens AM, Terrell M, Rashid R, Fisher KE, Marcogliese AN, Gaikwad A, et al. Addressing a Pre-Clinical Pipeline Gap: Development of the Pediatric Acute Myeloid Leukemia Patient-Derived Xenograft Program at Texas Children’s Hospital at Baylor College of Medicine. Biomedicines. (2024) 12(2):394. doi: 10.3390/biomedicines12020394
24. Palam LR, Ramdas B, Pickerell K, Pasupuleti SK, Kanumuri R, Cesarano A, et al. Loss of Dnmt3a impairs hematopoietic homeostasis and myeloid cell skewing via the PI3Kinase pathway. JCI Insight. (2023) 8(9):e163864. doi: 10.1172/jci.insight.163864
25. Brown FC, Carmichael CL. Patient-derived xenograft models for leukemias. Methods Mol Biol. (2024) 2806:31–40. doi: 10.1007/978-1-0716-3858-3_4
26. Ramsey HE, Gorska AE, Smith BN, Monteith AJ, Fuller L, Arrate MP, et al. TLR3 agonism augments CD47 inhibition in acute myeloid leukemia. Haematologica. (2024) 109(7):2111–21. doi: 10.3324/haematol.2023.283850
27. Hassan N, Yi H, Malik B, Gaspard-Boulinc L, Samaraweera SE, Casolari DA, et al. Loss of the stress sensor GADD45A promotes stem cell activity and ferroptosis resistance in LGR4/HOXA9-dependent AML. Blood. (2024) 144(1):84–98. doi: 10.1182/blood.2024024072
28. Olesinski EA, Bhatia KS, Wang C, Pioso MS, Lin XX, Mamdouh AM, et al. Acquired multidrug resistance in AML is caused by low apoptotic priming in relapsed myeloblasts. Blood Cancer Discovery. (2024) 5(3):180–201. doi: 10.1158/2643-3230.BCD-24-0001
Keywords: myelodysplastic syndrome, PDX (patient derived xenograft), 5-Azacytidine, Venetoclax (BCL2 inhibitor), therapeutic targets
Citation: Bašová P, Minařík L, Magalhaes-Novais SC, Balounová J, Zemanová Z, Aghová T, Špaček M, Jonášová A, Pimková KG, Procházka J, Sedláček R and Stopka T (2025) Investigating resistance to 5-Azacytidine and Venetoclax in PDX models of MDS/AML. Front. Oncol. 14:1414950. doi: 10.3389/fonc.2024.1414950
Received: 09 April 2024; Accepted: 06 December 2024;
Published: 07 January 2025.
Edited by:
Ulrich Germing, Heinrich Heine University of Düsseldorf, GermanyReviewed by:
Valentina Giudice, University of Salerno, ItalyLucia Gozzo, Gaspare Rodolico Hospital, Italy
Copyright © 2025 Bašová, Minařík, Magalhaes-Novais, Balounová, Zemanová, Aghová, Špaček, Jonášová, Pimková, Procházka, Sedláček and Stopka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomáš Stopka, dHN0b3BrYUBsZjEuY3VuaS5jeg==
†These authors have contributed equally to this work and share first authorship