 Elisabeth A. Pedersen1†
Elisabeth A. Pedersen1† Paul W. Harms
Paul W. Harms
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 29 July 2024
Sec. Skin Cancer
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1413793
This article is part of the Research TopicManagement of Rare Oncological CasesView all 62 articles
Merkel cell carcinoma (MCC) is an aggressive cutaneous neuroendocrine carcinoma thought to arise via either viral (Merkel cell polyomavirus) or ultraviolet-associated pathways. Surgery and radiotherapy have historically been mainstays of management, and immunotherapy has improved outcomes for advanced disease. However, there remains a lack of effective therapy for those patients who fail to respond to these established approaches, underscoring a critical need to better understand MCC biology for more effective prognosis and treatment. Here, we review the fundamental aspects of MCC biology and the recent advances which have had profound impact on management. The first genetically-engineered mouse models for MCC tumorigenesis provide opportunities to understand the potential MCC cell of origin and may prove useful for preclinical investigation of novel therapeutics. The MCC cell of origin debate has also been advanced by recent observations of MCC arising in association with a clonally related hair follicle tumor or squamous cell carcinoma in situ. These studies also suggested a role for epigenetics in the origin of MCC, highlighting a potential utility for this therapeutic avenue in MCC. These and other therapeutic targets form the basis for a wealth of ongoing clinical trials to improve MCC management. Here, we review these recent advances in the context of the existing literature and implications for future investigations.
Merkel cell carcinoma (MCC) is a poorly differentiated primary cutaneous neuroendocrine carcinoma. It is among the most aggressive cutaneous solid tumors, with significant rates of disease-associated morbidity and mortality. MCC is now known to consist of two molecular subclasses: Merkel cell polyomavirus-associated and UV-associated, which are denoted as virus-positive MCC (VP-MCC) and virus-negative MCC (VN-MCC) throughout this review. Recently, immunotherapy has dramatically improved prognosis in advanced disease, but is not universally effective. Thus, there is continued need for better molecular understanding, prognostication, and therapeutic options for MCC.
In this review, we discuss the current state of MCC treatment and how it relates to the cellular and molecular pathogenesis of MCC. We also review the emerging therapeutic strategies and the related need for improved preclinical models of MCC.
MCC arises most frequently in fair-skinned patients, with 89.9% of MCC cases identified in White, 5.7% in Hispanic, 2.3% in Asian American or Pacific Islander, and 1.5% in Black patients (1). The greatest incidence of MCC has been noted in Australia (1.6 cases per 100,000 individuals) (2), and is in line with the observed greater MCC incidence with geographic proximity to the equator (3). In 2013, the incidence in the United States was approximately 0.7 per 100,000, which was a 95% increase since 2000 (4). MCC is more common in males compared to females (3:2, respectively) (4–6), and the median age of diagnosis is >75 years, with very rare incidence in patients < 40 years (5). For MCC, the incidence continues to rise with each additional decade of life, whereas in most other malignancies, the incidence peaks at a mid-/later decade of life and then declines with further age (2). The incidence of MCC is greater in patients with impaired immune functioning such as organ transplantation, hematologic malignancy, immunosuppressive medications, and other causes of decreased immunity (3).
MCC most often arises as a red-to-violaceous papule or nodule on sun-exposed skin. It may also arise as a subcutaneous nodule with no overlying skin changes. Tumors often grow rapidly over the course of 2-3 months. MCC has a high propensity for metastasis; up to 35% of patients will have either regional lymph node or distant metastases (7) at the time of diagnosis. Frequent sites of metastatic involvement are adjacent skin (satellite or in-transit metastases); regional lymph nodes; and distant sites including bone, visceral organs (liver, lungs), distant skin, and distant lymph nodes.
MCC is a poorly-differentiated neuroendocrine carcinoma with round cell morphology (Figure 1) (2, 7–9). In most cases, the tumor is centered in the dermis, possibly with extension into the subcutis (Figure 1A) (7). In a minority of cases, there is pagetoid scatter in the overlying epidermis. Purely intraepidermal cases are exceptionally rare. There can be squamous metaplasia, eccrine differentiation, or rarely sarcomatoid change. A minority of VN-MCC is accompanied by SCCIS in the overlying epidermis, or less frequently invasive SCC (7, 10). Rarely, VP-MCC can be associated with other tumor types including trichoblastoma or poroma (11, 12).
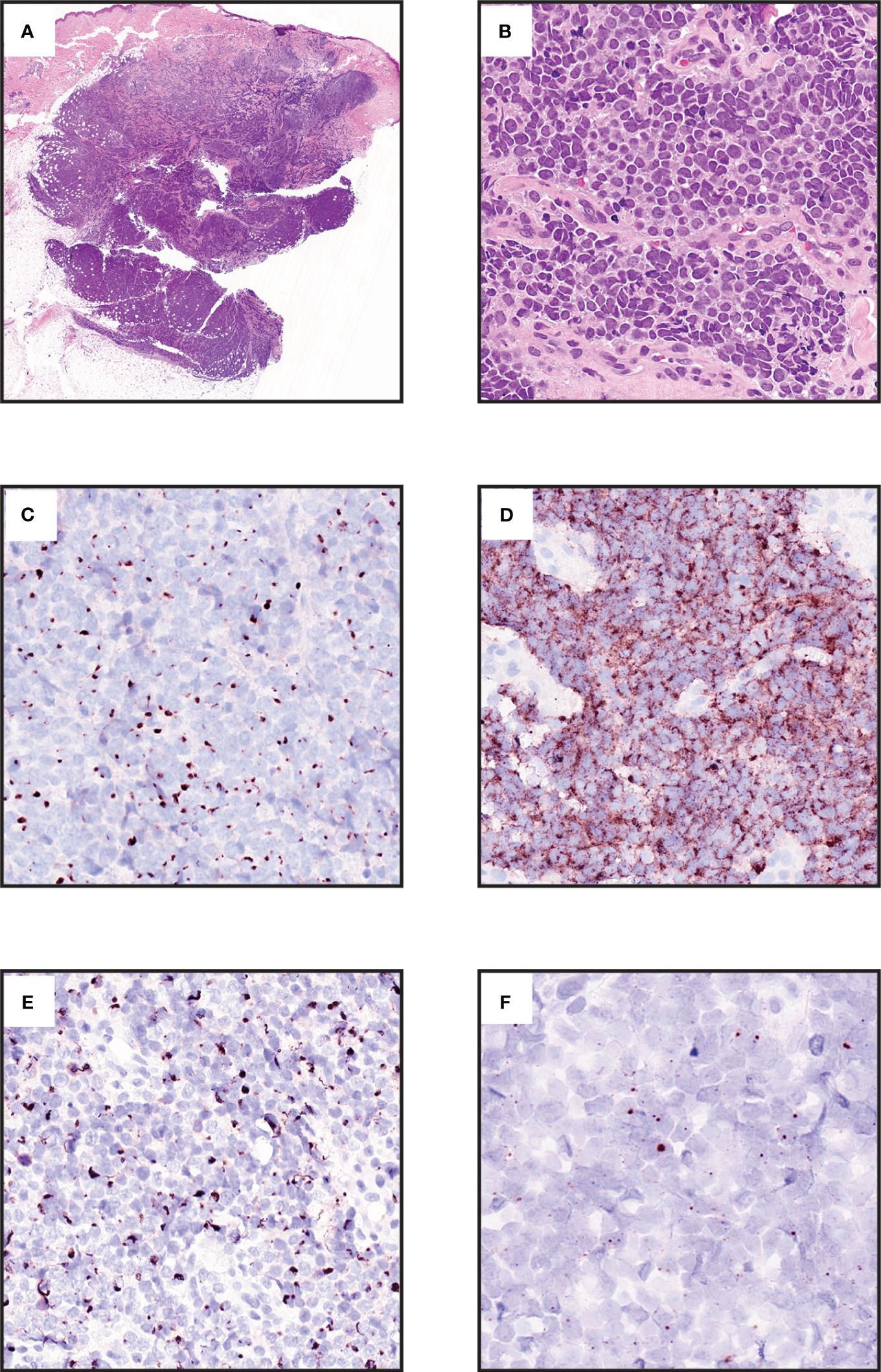
Figure 1 Morphologic and immunophenotypic features of Merkel cell carcinoma. (A) Expansile blue cell tumor occupying dermis with extension into subcutis (hematoxylin and eosin [H&E], 0.7x. (B) Small round cell tumor with neuroendocrine chromatin, mitotic activity, and crush artifact (H&E, 40x). (C) Cytokeratin 20 displays distinctive paranuclear dot labelling. (Chromogen DAB, counterstain hematoxylin, 40x). (D) Neuroendocrine marker Chromogranin A is immunohistochemically expressed in most cases (Chromogen DAB, counterstain hematoxylin, 40x). (E ) Neurofilament immunohistochemistry displays paranuclear dot labelling and provides a more specific marker than cytokeratin 20 for MCC (Chromogen DAB, counterstain hematoxylin, 40x). (F) RNA in situ hybridization for the MCPyV T-antigen transcript in a virus-positive MCC (Chromogen DAB, counterstain hematoxylin, 80x).
MCC, like other poorly-differentiated tumors, can be challenging to distinguish from histopathologic mimics. The differential diagnosis for MCC depends on site of presentation and tumor morphology (7, 9). Extracutaneous small cell carcinoma metastatic to the skin is morphologically identical to MCC. Other small round cell tumors, such as lymphoma, small cell melanoma, or Ewing sarcoma, might also enter the differential diagnosis (7, 9). For MCC with more prominent pleomorphism, other poorly differentiated cutaneous carcinomas might also be considered (7).
Several other neuroendocrine tumors arise in the skin or subcutis and must be distinguished from MCC. Endocrine mucin-producing sweat gland carcinoma, and the related neuroendocrine subtype of mucinous carcinoma, are adenocarcinomas of the eyelid that may resemble MCC but lack round cell morphology (13). Primary cutaneous neuroendocrine tumors (carcinoids) display morphologic features of neuroendocrine tumors but are histologically lower grade than MCC (14). Recently, another category of primary cutaneous neuroendocrine carcinoma, termed sweat gland carcinoma with neuroendocrine differentiation (SCAND), has been proposed; however, these tumors also lack the round cell morphology of MCC (15).
Immunohistochemistry is critical for confirming the diagnosis of MCC and excluding histopathologic mimics (7–9). It should be noted that no single marker is completely sensitive for MCC, thus diagnosis often requires multiple immunohistochemical stains, expert evaluation, and clinical correlation to confirm the diagnosis. Commonly used markers include CK20 expression in a paranuclear dot pattern (Figure 1C), which is distinctive for MCC, but may not always be present (7). Neuroendocrine markers such as chromogranin A or synaptophysin are typically expressed (Figure 1D). Paranuclear dot labeling for neurofilament is also highly characteristic (Figure 1E) (7, 16, 17). Thyroid transcription factor 1 (TTF-1) and cytokeratin 7 are typically negative. Detection of MCPyV by immunohistochemistry or RNA in-situ hybridization supports a diagnosis of MCC (Figure 1F) (7, 10, 18). INSM1 is a highly sensitive marker for MCC, although specificity is limited relative to other neuroendocrine carcinomas (19–21). SATB2 has more limited sensitivity, but provides added specificity in that strong diffuse expression favors MCC over metastatic small cell carcinoma (22–24). SOX2 expression is highly sensitive for MCC (92%), and is highly specific for MCC compared to other cutaneous carcinomas, as well as 70% specific for MCC compared to other neuroendocrine carcinomas (25, 26). ATOH1 is expressed in MCC, however there is mixed data regarding the specificity of this marker for excluding other non-MCC neuroendocrine carcinomas (23, 27–30). VN-MCC has a higher rate of immunophenotypic aberrancy such as CK20 negativity (7, 31–33).
Morphology alone is usually sufficient to distinguish MCC from malignancies other than small cell carcinomas. However, several pitfalls merit mention. Neuroendocrine marker expression can be seen in some cases of other cutaneous carcinoma including basal cell carcinoma. Brisk obscuring inflammation, and/or expression of certain lymphoid markers (TdT, PAX5, SOX11, immunoglobulins) in a subset of MCC (7, 34, 35), can raise confusion with a lymphoid process. S100 expression has been described for MCC (7), and conversely can be lacking in small cell melanoma (36), potentially raising diagnostic difficulty. However, nuclear SOX10 expression is exceedingly rare in MCC, and other melanocytic markers will be negative, thus allowing reliable distinction from small cell melanoma (7, 37–40). MCC can express CD99 similar to small round cell sarcomas such as Ewing sarcoma, but will lack EWSR1 rearrangements (2, 7, 41).
In challenging cases such as neuroendocrine carcinomas presenting in the parotid, or carcinomas of unknown primary, analyses demonstrating MCPyV or UV-signature mutations can also be useful for distinguishing MCC from extracutaneous neuroendocrine carcinomas (2, 7, 23, 42–45). Of note, the potential for primary parotid neuroendocrine carcinomas to be MCPyV-positive has been debated (44, 46–48), and thus currently clinicopathologic correlation remains essential for classifying MCPyV-positive neuroendocrine carcinomas in the parotid.
The AJCC 8th edition defines staging guidelines for MCC based upon tumor size and the presence/pattern of metastatic spread (5). In addition, it is well established that advanced age, male sex, immunosuppression, and patients with a known primary lesion with clinically evident nodal metastases are associated with a higher risk of recurrence (49, 50). Additional prognostic factors are a subject of ongoing research. Many potential biomarkers for predicting MCC outcome have been proposed (2, 7, 51). Although data from smaller studies is mixed, several large studies have found that the viral status of MCC is prognostic: specifically, the presence of MCPyV is a favorable prognostic finding, with improved disease-specific and progression-free survival in VP-MCC compared to VN-MCC (2, 7, 10, 52–54). Tumor expression of p63 has been proposed to be predictive of outcomes (7, 55–58), however the clinical utility of this marker has been questioned (58). Many studies have found that immune infiltration patterns or marker expression can be informative of outcome for MCC tumors (2, 7, 10, 59, 60), especially CD8+ T-cells (7, 61–63). The predictive value of immune markers might vary based upon tumor MCPyV status (10), which was often not considered in such studies. Notably, the great majority of these studies were based upon outcomes data collected prior to the approval of immunotherapy for MCC, and thus reappraisal of these findings in the immunotherapy era will be essential.
Clinical trials and the results of single- and multi-institution cohort studies provide the best evidence to support current practices for MCC treatment. Given the rarity of MCC, high-powered randomized controlled trials to identify optimal treatment approaches have not been feasible. The National Comprehensive Cancer Network (NCCN) Guidelines for MCC provides an annually updated evidence-based, and consensus-driven algorithmic approach for management of all stages of MCC (64). In addition, the European consensus-based interdisciplinary guideline was published in 2022 (65). Importantly, for patients with any stage of MCC, a multidisciplinary team is recommended as management requires the involvement of multiple specialties. Please refer to the NCCN and European Consensus guidelines for the most comprehensive and up-to-date algorithms for management (64, 65).
Initial evaluation and work-up of a patient newly diagnosed with MCC includes a thorough history with special attention directed towards immunosuppression. Immunosuppression is a known adverse prognostic factor, and it is recommended that immunosuppressive regimens are reduced by the prescribing physician when medically feasible. The initial physical exam should focus on identifying clinical staging parameters including the clinical size of the primary tumor, clinical in-transit metastases, and clinically detectable regional lymph node metastases. For patients without clinical lymph node metastases, baseline imaging with PET-CT is often recommended, as it has been shown to find additional asymptomatic sites of disease and impact staging (66). Suspected metastases should be confirmed by biopsy. Subsequent management is based upon stage of disease (64).
Surgical treatment with or without radiotherapy is the current standard of care. Due to the lack of randomized controlled trials, the optimal surgical margin is not well defined. The current NCCN recommendation is for excision with 1-2 cm margins, with Mohs micrographic excision (excision done by stages with frozen section evaluation of margins) designated as useful in certain circumstances (64). The risk of occult metastasis is high in patients presenting without clinically detected regional lymph node metastasis, and sentinel lymph node biopsy (SLNB) is recommended at the time of surgical removal of the primary tumor. Studies have demonstrated SLNB positivity rates as high as 26 - 45% (67–69) in institution based-cohorts. Observation is recommended for patients with a negative SLNB result, with evidence supporting a low risk of regional recurrence after a negative SLNB (70). For management of SLNB-positive disease, current guidelines recommend further treatment including lymph node dissection or radiation therapy (64), which have both demonstrated association with low rates of regional recurrence (71, 72).
MCC is highly sensitive to radiation therapy, and adjuvant radiation is recommended in cases of positive surgical margins (versus re-excision) and in the presence of adverse risk factors, such as primary tumors > 1 cm in size, immunosuppression, head/neck site, or lymphovascular invasion. For patients who are not surgical candidates, the recommendation is for multidisciplinary discussion and consideration of definitive radiation therapy or systemic therapy (64).
For patients with clinically detected lymph node metastases prior to surgery, a lymph node biopsy via fine needle aspiration or core needle biopsy is recommended to confirm the presence of metastatic MCC. For treatment of biopsy-confirmed clinically detectable lymph node metastases, current NCCN guidelines recommend clinical trial, or combination of lymph node dissection and radiation therapy. In addition, neoadjuvant therapy (e.g. treatment prior to surgery) with immunotherapy is emerging as a potential option for treatment.
Reports of immune-mediated spontaneous regression in some cases of MCC suggested the potential for therapeutic harnessing of antitumor immunity for advanced disease (2). Immunotherapy, also known as immune checkpoint inhibitor therapy, has become widely used in management of metastatic MCC and preferred over traditional chemotherapy due to improved responses and more favorable side effect profiles. Three immune checkpoint inhibitors are currently FDA-approved for the management of metastatic or advanced MCC. Avelumab, a monoclonal antibody targeted against programmed cell death ligand 1 (anti-PD-L1) was FDA-approved in 2017 for metastatic MCC. The JAVELIN Merkel 200 trial part A showed a 32% objective response rate to second-line avelumab (73); long-term follow up demonstrated a median overall survival (OS) of 12.6 months and a 5-year OS of 26% (74). Results of JAVELIN Merkel 200 part B showed a 62% objective response rate (ORR) to first-line avelumab (75). More recently, a retrospective review of patients with advanced MCC initiated on avelumab reported durable response rates as high as 73% (76). Pembrolizumab and retifanlimab, monoclonal antibody therapies targeting programmed cell death protein -1 (PD-1), were FDA-approved in 2018 and 2023, respectively, for recurrent, locally advanced and metastatic MCC (77). In the Keynote-017 trial, pembrolizumab demonstrated 56% overall response rate (ORR) (78); long-term follow-up demonstrated a 24-month overall survival rate of 68.7% (79). The PODIUM-201 trial demonstrated a 46.2% ORR, and 62% of patients had a response of ≥ 12 months (80). Nivolumab, an anti-PD-1 antibody, is also showing promise in the treatment of MCC, with numerous ongoing studies evaluating the efficacy of single and multi-agent therapy (2, 81).
Emerging neoadjuvant studies have shown promising results and are being recommended more frequently. The Checkmate 358 trial demonstrated that half of patients with clinically detectable lymph node metastases who received neoadjuvant nivolumab responded with tumor reductions of > 30% (82). NCCN guidelines currently recommend multidisciplinary discussion for consideration of neoadjuvant immunotherapy (64).
Overall, evidence across studies suggests that immunotherapy can achieve durable response in a significant proportion of patients with advanced MCC, especially when used as first-line therapy (83). However, patient factors including advanced age often seen in MCC patients and morbidity from treatment may alter treatment course or deem immunotherapy a less favorable option. Additional investigations with longer follow-up times in larger cohorts will be essential for more definitive assessment of durable response rates across different patient groups.
Management of metastatic MCC has historically included chemotherapy with etoposide and carboplatin/cisplatin-based regimens; however, the response to chemotherapy is not durable and most patients experience relapse. In a retrospective study of patients with metastatic MCC, first-line chemotherapy had a response rate was 55%, but also had a median progression free survival of 94 days (84). For second-line chemotherapy, the response rate was 23% with a median progression free survival of 61 days. Given initial response rates, chemotherapy may be useful in certain circumstances, such as for those who cannot undergo immune checkpoint inhibition due to immunocompromised status or the presence of a comorbid autoimmune condition (2, 85). It may also serve as a palliative therapy to slow the rate of rapidly growing symptomatic tumors or metastases.
As immunotherapy is not universally effective in MCC, predicting treatment response could be helpful to avoid the risk of immunotherapy toxicity in patients who will not benefit from therapy, and to consider alternative treatments if those become available. However, predictors of MCC response to immunotherapy have been elusive. Responses have not correlated with tumor PD-L1 expression (73, 78, 82, 86, 87), MCPyV status (73, 82, 86–89), tumor mutational burden (82, 86, 87), or UV mutation signatures (87). A retrospective study showed that single nucleotide variants of ARID2 and NTRK1 were associated with a better response to immunotherapy (87). More sophisticated approaches may be informative, such as characterizing immune populations (especially T-cells) (88, 90–95) or measuring spatial associations of cell populations in the tumor microenvironment (88, 96). However, to date none of these approaches have reached routine clinical use for decisions on immunotherapy eligibility.
After definitive treatment with surgery, radiation, and/or systemic therapy, close surveillance is typically recommended. Due to the significant risk of recurrence, current management for MCC includes imaging (such as by CT) every 3-6 months for several years following diagnosis (64). Alternative and emerging methods for disease surveillance include circulating burden of antibodies against MCPyV T-antigens (64, 77, 97, 98), and circulating tumor DNA (77, 99).
The above average prevalence of MCC in immune-compromised transplant and HIV patients (100) alluded to a possible viral etiology. This possibility was confirmed when analysis of MCC tumors via digital transcriptome subtraction methodology identified viral transcripts from a previously unknown human polyomavirus, which the authors thus named the Merkel cell polyomavirus (MCPyV) (101). The initial finding that only about 80% of tumors had integrated viral sequences suggested that a fraction of MCCs arise by a distinct mechanism. Following additional studies, two often histologically indistinguishable molecular subclasses have now been defined: MCPyV associated (virus-positive MCC; VP) and UV-associated (virus-negative MCC; VN) tumors (2, 43, 45, 102).
The MCPyV belongs to the Polyomaviridae (PyV) family of small non-enveloped, circular, double-stranded DNA viruses, of which to date, 15 have been isolated from human samples with 13 identified as genuine human viruses (103). Several polyomaviruses are associated with cutaneous infections (trichodysplasia spinulosa polyomavirus, human polyomavirus 6, and human polyomavirus 7) and other human diseases (JC Polyomavirus, BK polyomavirus). However, MCPyV is the sole family member classified as a Group 2A carcinogen (probably carcinogenic to humans) by the International Agency for Research on Cancer (104). Polyomaviruses are widely prevalent in the human population, and MCPyV can be detected in the skin of most healthy individuals and at low levels in a wide variety of tissues. Serological studies indicate primary infection occurs most likely in early childhood and seroprevalence in adults is typically 60–80% (105).
During the MCPyV infectious cycle, the MCPyV helicase (C-terminal domain of LTAg) mediates replication of viral DNA by recruiting the host cell machinery. The viral DNA of MCPyV can be repeatedly and rapidly amplified during a single cell cycle, referred to as unlicensed replication (106). The host cell factors that contribute to MCPyV replication are poorly understood. In healthy populations, antiviral responses induced by MCPyV, such as IFN production, may inhibit early transcription and restrict propagation, whereas skin injury or UV irradiation may induce growth factors promoting viral propagation, thus providing a way to support persistent MCPyV infection (107, 108). Degradation of LTAg by ubiquitin ligases, which may be abrogated by cellular stresses, is another proposed mechanism for latency and reactivation of MCPyV (109–112), although this mechanism has been debated (113).
Despite a lifelong history of MCPyV exposure and infection in most individuals, development of MCC tumors remains remarkably rare. Productive viral infection is associated with host cell death (114), precluding the possibility of cell transformation during the normal viral life cycle. Two critical events are thus hypothesized to precede MCPyV-driven transformation: accidental integration of the viral genome into the host genome, and loss of viral replicative capacity due to LTAg mutations which yield a non-replicative truncated LTAg (tLTAg) (101, 115). The clonal pattern of viral integration suggests infection and integration occurs before tumor expansion, providing evidence for MCPyV being causal for tumorigenesis (101).
Like other polyomaviruses, the MCPyV genome consists of an early region (ER) coding for regulatory proteins involved in replication and transcription, a late region (LR) coding for structural proteins, and a non-coding control region (NCCR) containing the origin of DNA replication, regulatory elements, and transcription promoters (116). Four spliced transcripts are produced by the ER that code for four proteins: the large T antigen (LTAg) and the alternative splicing product 57kT; the small T antigen (sTAg); and alternate frame of the LTAg open reading frame (ALTO, as analogous to middle T antigen or MT)(Table 1) (101, 129, 131). The LR encodes two capsid proteins (VP1 and VP2) required for viral assembly, and a microRNA targeting the TAg transcripts (132–134).
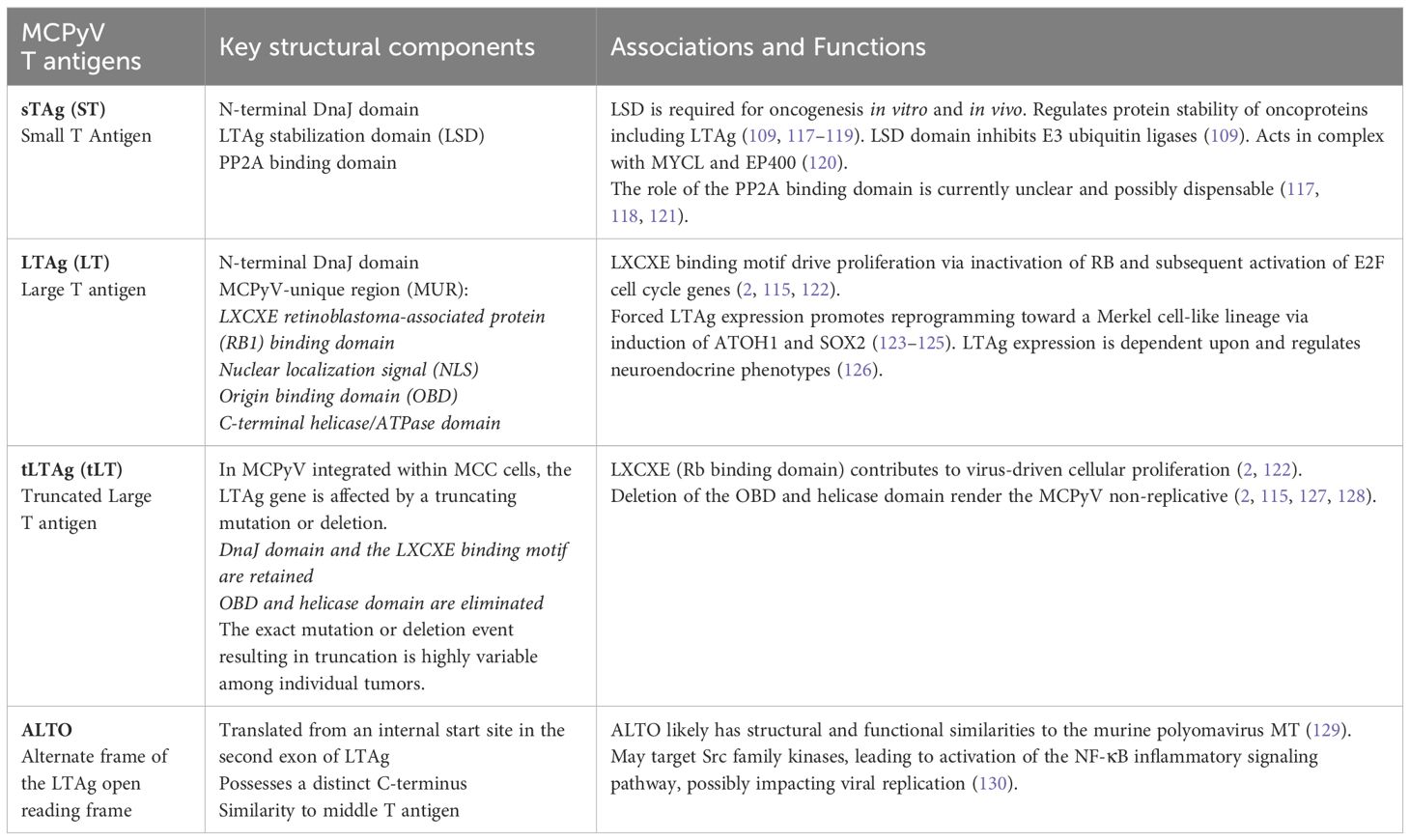
Table 1 MCPyV T antigen gene products, domains, and functions.
Evidence suggests that tLTAg and sTAg both contribute to the oncogenic potential of MCPyV (117, 118, 135–140). Expression of these oncoproteins is required for VP-MCC cell growth and survival (141, 142). Interestingly, while LTAgs from most other polyomaviruses such as SV40 typically demonstrate transforming ability (143), the MCPyV tLTAg is not sufficient to drive transformation alone in vitro (135) or in vivo (118), but rather cooperates with sTAg to drive transformation (137).
The LTAg contains several functional domains, most notably the LXCXE retinoblastoma-associated protein (RB1) binding domain, and C-terminal region responsible for mediating viral replication [reviewed in (144)]. In MCPyV integrated within MCC cells, the LTAg gene is affected by a truncating mutation or deletion to generate tLTAg, eliminating the C-terminus and rendering the virus non-replicative (101, 115, 145) while sparing the LXCXE binding motif (2, 146), which plays a critical role in LTAg-driven proliferation via inactivation of RB and subsequent activation of E2F cell cycle genes (2).
In contrast to sTAgs of other polyomaviruses, the MCPyV sTAg displays transformation capability in culture (135) and in vivo (117, 118, 139). Several potential mechanisms for this activity have been investigated, including promoting activity of the mTOR pathway (135), regulation of protein stability via a unique LTAg stabilization domain (LSD) of sTAg proposed to inhibit E3 ubiquitin ligases and stabilize oncoproteins including LTAg (109, 117–119), cell surface localization of proteins involved in tumor cell recognition and tumor microenvironment regulation such as CD47 (147), and interacting with protein phosphatases and the NF-kB essential modulator (NEMO) to achieve inhibition of NF-kB signaling, a potential mechanism for repressing inflammatory signaling (148).
One of the most recently described roles for sTAg may have an even more profound influence on cellular functions. MCPyV sTAg can complex with the MYC homolog, L-MYC and its binding partner MAX, which in turn recruit the chromatin remodeling complex EP400 (120). The sTAg-MYCL-EP400 complex transcriptionally activates hundreds of downstream targets involved in ribosomal biogenesis, splicing, glycolysis, and other metabolic functions. This complex also increases levels of the mouse double minute 2 homolog (MDM2) E3 ubiquitin-protein ligase, which binds p53 and promotes its ubiquitination and proteasomal degradation, as well as CK1α, an activator of MDM4 (120, 149). Thus, although VP-MCC typically harbor wild-type TP53 (43), p53 is indirectly inactivated by MCPyV sTAg via this mechanism. Additional activated targets of the sTAg-MYCL-EP400 complex include LSD1, RCOR2 and INSM1, which form part of a transcriptional repressor complex that opposes the ncBAF complex involved in differentiation, and the ATOH1 transcription factor which governs Merkel cell fate (150). The sTAg complex has also been shown to upregulate genes involved in cell motility, and downregulate genes involved with cell adhesion properties, revealing possible sTAg roles in invasion and metastasis (120, 140, 151–153).
Analogous to other sTAgs, the MCPyV sTAg contains a PP2A binding domain. However, the role of the PP2A binding domain in sTAg-mediated transformation is currently unclear and possibly dispensable (117, 118, 121).
To date, the functional role of MCPyV ALTO has not been well recognized, but emerging evidence now suggests that ALTO has structural and functional similarities to the murine polyomavirus MT, targeting Src family kinases, leading to activation of the NF- κB inflammatory signaling pathway, and possibly impacting viral replication (130).
While not yet functionally understood, significant levels of intrinsic protein structure disorder have recently been described in sTAg, LTAg, ALTO, 57kT, and VP1, characterized by the lack of a consistent three-dimensional structure (154). These disordered structures may present MCPyV viral proteins with unique functional properties including exceptional binding promiscuity and interactions with unexpected partners (154). This feature might contribute to the high diversity of functions that have been attributed to MCPyV T antigens.
Initial focused sequencing studies on MCC identified recurrent mutations including TP53 and RB1 in a subset of tumors but did not correlate these findings with MCPyV or global mutation changes (54, 155–159). However, later studies that broadly examined the MCC mutational landscape in the context of MCPyV immediately identified two molecular subclasses with significant differences in mutational burden (43, 45, 102). Virus-negative tumors displayed a high UV-signature mutational burden with recurrent inactivation of tumor suppressor genes TP53 and RB1, while virus-positive tumors had an extremely low mutational burden (43, 45, 102). VN-MCC can present with a mutational burden of 40 mutations per megabase, which is higher than any cancer sequenced by The Cancer Genome Atlas and more than 100-fold higher than VP-MCC (43, 102). Metastatic VN-MCC tumors exhibit additional genomic complexity (160). In contrast, VP-MCC typically have relatively normal diploid genomes with few somatic mutations (43, 45, 102), and those present are often subclonal, suggesting they occurred after tumor initiating events (161). Mutations in UV-associated VN-MCC often result in inactivation of genes involved in several signaling pathways, including NOTCH (NOTCH1, NOTCH2), DNA damage repair (KMT2A, KMT2C KMT2D, ASXL1, ARID1A, ARID1B, SMARCA4) and chromatin-modifying pathways (ATM, MSH2, BRCA1, BRCA2, and BCOR) (10, 43, 45, 86, 102, 146).
Oncogene activation events tend to be detected with greater frequency in VN-MCC than VP-MCC and are overall heterogeneous (10, 86). The most highly recurrent oncogene activation events include MYCL amplification and PIK3CA activating point mutation. Other hotspot activating mutations reported in MCC include TERT promoter, KNSTRN, RAC1, HRAS, KRAS, NRAS, AKT1, CTNNB1, IDH1, IDH2 and EZH2, among others (2, 42, 43, 45, 86, 102, 157, 162, 163).
At the transcriptional level, VN-MCC shows distinct patterns of gene expression from VP-MCC (10, 164–168), including upregulation of genes involved in the epithelial-to-mesenchymal transition and developmental processes, and downregulation of genes involved in neuroendocrine and epidermal differentiation, which may contribute to its more aggressive nature (165, 167).
Epigenetic modifications encompass a spectrum of reversible covalent modifications, especially to histone proteins and DNA, that exert powerful effects on chromatin accessibility and hence gene expression (169, 170). Alterations in epigenetic patterning can contribute to the hallmarks of cancer, and are frequently observed in malignancies (169–171).
A consistent model for the contribution of epigenetic dysregulation to MCC tumor biology has remained elusive. Although many sequencing studies have identified mutations involving chromatin modifier genes in MCC, the genes involved are heterogeneous with no highly recurrent mutational driver of epigenetic dysregulation (10, 42, 43, 45, 86, 102, 161, 172, 173). Until recently there have been relatively few studies establishing patterns and significance of epigenetic alterations in MCC. However, recent studies have provided new insights into the biological importance of the tumor epigenome in MCC.
Methylation of DNA occurs on cytosine residues and is a major regulator of gene expression. Methylation of promoter CpG islands and adjacent shores is associated with silencing, whereas methylation of gene body sites is associated with increased expression (174–176). Patterns of DNA methylation can yield insights into tumor classification and cell of origin (177). DNA methylation is perturbed in tumorigenesis, including global methylation changes and specific silencing of tumor suppressor genes (171). DNA hypomethylation agents have been approved as antitumor therapy for some myeloid neoplasms and are under investigation as therapeutics for solid tumors (178, 179), underscoring the mechanistic importance of altered DNA methylation in tumor biology.
Until recently, studies into epigenetic patterning in MCC have been limited to specific genes. Promoter hypermethylation has been described for tumor suppressor genes including CDKN2A, RASSF1, RB1, DUSP2, and MGMT (180–184), although protein expression studies have not shown that these genes are consistently silenced in MCC tumors (53, 164, 182, 184, 185). Hypomethylation of the PTCH1 tumor suppressor has been described in MCC, but this did not consistently correlate with gene expression (186). Thus, the biological significance of these methylation events has remained unclear. The cell surface protein PD-1 (encoded by PDCD1), long known to be associated with immune exhaustion when expressed on T-cells, has recently been proposed to also be a tumor suppressor expressed on some tumor cells (187). PDCD1 promoter hypermethylation has been described in MCC, in which context it may correlate with reduced immune response and worse prognosis (188).
Recent studies have provided greater detail on patterns of DNA methylation in MCC (189–192). Based on global genomic patterns, MCC assorts into a distinct cluster from other tumor types, with closest similarity to small cell lung carcinoma, and relative similarity to squamous cell carcinoma (189). Comparison to other tumor types by a score-based approach also demonstrated the epithelial and neuroendocrine-type patterning of DNA methylation in MCC (190). VP-MCC displays distinctive genome-wide DNA methylation patterns as compared to VN-MCC, although the magnitude and significance of such differences is unclear (189–191). In particular, VP-MCC displays globally decreased methylation compared to VN-MCC, as indicated by direct measurement of genomic methylation, and by expression of the hypomethylation marker long interspersed nuclear element-1 (189, 193). The mechanism for DNA methylation dysregulation in MCC remains unclear, as DNA methyltransferase genes (DNMTs) are not consistently overexpressed or mutated in MCC (43, 45, 102, 189). Nonetheless, promoter methylation might provide a mechanistic explanation for expression patterns of many diagnostic and prognostic markers in MCC (189, 191). There is also evidence that DNA methylation patterns change with MCC progression (191). DNA methylation may also coordinate or promote other epigenetic changes in MCC such as histone methylation and acetylation (189, 191). In support of a biological role for DNA methylation in MCC biology, treatment with the DNA hypomethylating agent decitabine is associated with antiproliferative effects in VN-MCC and cell death in VP-MCC (189).
In addition to this proposed role in cell proliferation and survival, DNA methylation also appears to be a mechanism of immune evasion for MCC (189). HLA presentation of “non-self” antigens, either viral antigens for VP-MCC or UV-mutational neoantigens for VN-MCC (2), represents a potentially powerful trigger for antitumor immunity against MCC. HLA presentation can be downregulated by direct silencing of HLA genes and/or by repressing expression of the antigen presenting machinery responsible for mediating cell-surface localization and antigen presentation by HLA. In MCC, promoter hypermethylation is observed for HLA and antigen presenting machinery genes (189). This is likely functionally significant, as treatment by a hypomethylating agent resulted in de-repression and restored membrane localization of HLA (189). Therefore, hypomethylating agents might increase the antigenicity of MCC to promote antitumor immunity.
The reversal of DNA methylation is achieved via an intermediate step in which TET enzymes convert 5-methylcytosine to 5-hydroxymethylcytosine (5-hmc). The dysregulation of TET activity plays a significant role in driving some hematopoietic neoplasms (194). Inactivating mutations of TET2, and activating mutations of IDH1/IDH2 that inhibit TET activity, can occur in MCC but are uncommon relative to other potential drivers (10, 42, 86). Global loss of 5-hmc has been reported in aggressive cases of MCC (195), although the mechanism for TET dysregulation in such cases remains unclear.
Histone modifications, especially methylation, phosphorylation, and acetylation, can play roles in either promoting or silencing gene expression (170, 196). Histone acetylation on lysine residues promotes chromatin relaxation to facilitate gene expression, and is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs). Histone methylation (either mono-, di-, or tri-methylation) on lysine and arginine residues represents a complex multistep regulatory process that generally promotes closed chromatin and transcriptional repression (170). However, some histone methylation marks such as H3K4 trimethylation are associated with active transcription. Histone methylation is regulated by histone methyltransferases (including SET domain proteins such as the MLL family) and demethylases such as LSD1. Analogous to DNA methylation, histone modification can be a mechanism for tumor suppressor silencing or oncogene overexpression, and thus histone modifiers such as histone deacetylases represent therapeutic targets in tumors (170).
The Polycomb repressor complex 2 (PRC2) silences genes via repressive trimethylation of Histone 3 lysine 27 (H3K27Me3) (197). Dysregulated activity of PRC2, especially the catalytic subunit EZH2, has been implicated in many malignancies (197). EZH2 inhibitors display antitumor activity in epithelioid sarcoma and follicular lymphoma (198, 199). The role of PRC2 activity in MCC may be more complex. During embryogenesis, PRC2 acts to oppose normal Merkel cell development via silencing of the master Merkel cell fate regulator SOX2 (200). Global H3K27Me3 loss can be observed in many MCC consistent with loss of PRC2 activity (173, 201–204). H3K27Me3 loss occurs in tandem with the transition from a squamous progenitor population into MCC in at least some cases (173), suggesting similar roles for PRC2 in antagonizing both normal Merkel cell development and the emergence of MCC.
Given this evidence that PRC2 opposes Merkel cell formation, it is perhaps surprising that the PRC2 catalytic subunit EZH2 has been shown to be expressed in some MCC tumors (205–208), in which context it has been correlated with worse prognosis (206, 207). Activating EZH2 mutations, however, are rare in MCC (10, 42, 43, 86). EZH2 expression can be promoted by E2F transcription factors (209), suggesting a mechanism by which this gene might be upregulated in Rb-deficient tumors such as MCC. Thus far, there is conflicting data on the potential efficacy and mechanism of EZH2 inhibitors in MCC, with 2 reports finding that inhibition of the EZH2 methyltransferase activity had cytotoxic effects on VP-MCC cells and/or xenografts (208, 210), whereas another study found that protein degraders of EZH2 impaired VP-MCC cell line viability independently of the methyltransferase function (211). The antitumor effect of EZH2 inhibition on MCC might be mediated by reversing EZH2-mediated repression of inner ear differentiation genes including SIX1 (210). Further investigations are needed to better clarify the complex roles for PRC2 and the EZH2 subunit in MCC tumor biology.
The Myc oncoprotein exerts broad effects on transcription patterns in tumors via multiple mechanisms, including epigenetic modification. In keeping with a role for Myc family dysregulation in MCC, MYCL (and to a lesser extent MYC) can be amplified in a subset of MCC tumors (10, 212). More recently, evidence has shown that MCPyV small T antigen also acts in complex with MYCL and EP400 to promote LSD1 expression. In turn, LSD1 regulates methylation of H3K4 and H3K9 (associated with transcriptional activation and repression, respectively), resulting in gene expression changes that promote tumor proliferation and survival (150, 213). Importantly, Myc overexpression in MCC has been linked to the activating epigenetic mark H3K27Ac in the MYC promoter region (214). This represents a potential therapeutic vulnerability, as the H3K27Ac is bound by BRD4, which can be targeted by BET inhibitors to result in antitumor effects in MCC (214–216); however, the Myc-dependency of this effect has been debated (215).
Other histone methylation marks that may be altered in MCC include H3K9 methylation via PRDM8 (217), and induction of histone changes by sTAg (including H3K4Me2, H4K20me2, and the DNA damage associated phosphorylation of H2AX) (218).
Alongside DNA methylation, histone modifications also appear to be an epigenetic mechanism for immune evasion by MCC. Silencing of antigen-presenting machinery in MCC via histone deacetylation results in loss of surface HLA expression, promoting immune evasion (219). Thus, HDAC inhibitors can restore HLA expression and promote immunogenicity of MCC (189, 219–221). H3K9 deacetylation has been associated with silencing of MHC class I chain-related protein, thus removing an activating signal for NK cell targeting of tumor cells (222). Histone methylation may also play a role in HLA silencing in MCC. Specifically, HLA silencing correlates with loss of global H3K27Me3 in MCC, both in fully developed tumors (208, 223), and during the transition of precursor squamous cells into MCC (173); this silencing can be reversed by EZH2 inhibition (223).
Of small regulatory RNAs, evidence thus far supports roles for microRNAs in MCC tumor biology (224). MCCs display distinct patterns of microRNA expression relative to normal tissues and other skin tumor types (225). In addition, microRNA expression patterns may differ between VP-MCC and VN-MCC (226, 227), possibly due in part to microRNA regulation by MCPyV T-antigen(s) (228). A highly expressed microRNA in a subset of MCC is miR-375 (226, 229). Studies have found that miR-375 may contribute to tumor biology in MCC via intracellular and intercellular mechanisms, although reports are mixed regarding the significance and mechanism of this effect (123, 228, 230–234). In addition to these cellular microRNAs, MCPyV encodes a microRNA, MCV-mir-M1-5p, that may suppress LTAg expression and contribute to viral latency and episomal persistence (134, 228, 235, 236).
MCC was originally named “trabecular carcinoma”, but further investigation soon revealed a high degree of similarity with normal Merkel cells (MCs), prompting a name change to Merkel cell carcinoma (237, 238), and also suggesting these tumors were derived from their namesake. However, multiple lines of evidence now suggest that MCs are not the cell of origin for MCC, including tissue localization, post-mitotic and terminal differentiation status of MCs, differential expression of multiple markers including keratins, as well as the failure of mouse modeling using MC specific drivers (239). Considerable debate about the MCC cell of origin persists, with proposed cells of origin including epithelial, dermal, and lymphoid cell populations (Figure 2) (34, 240, 241).
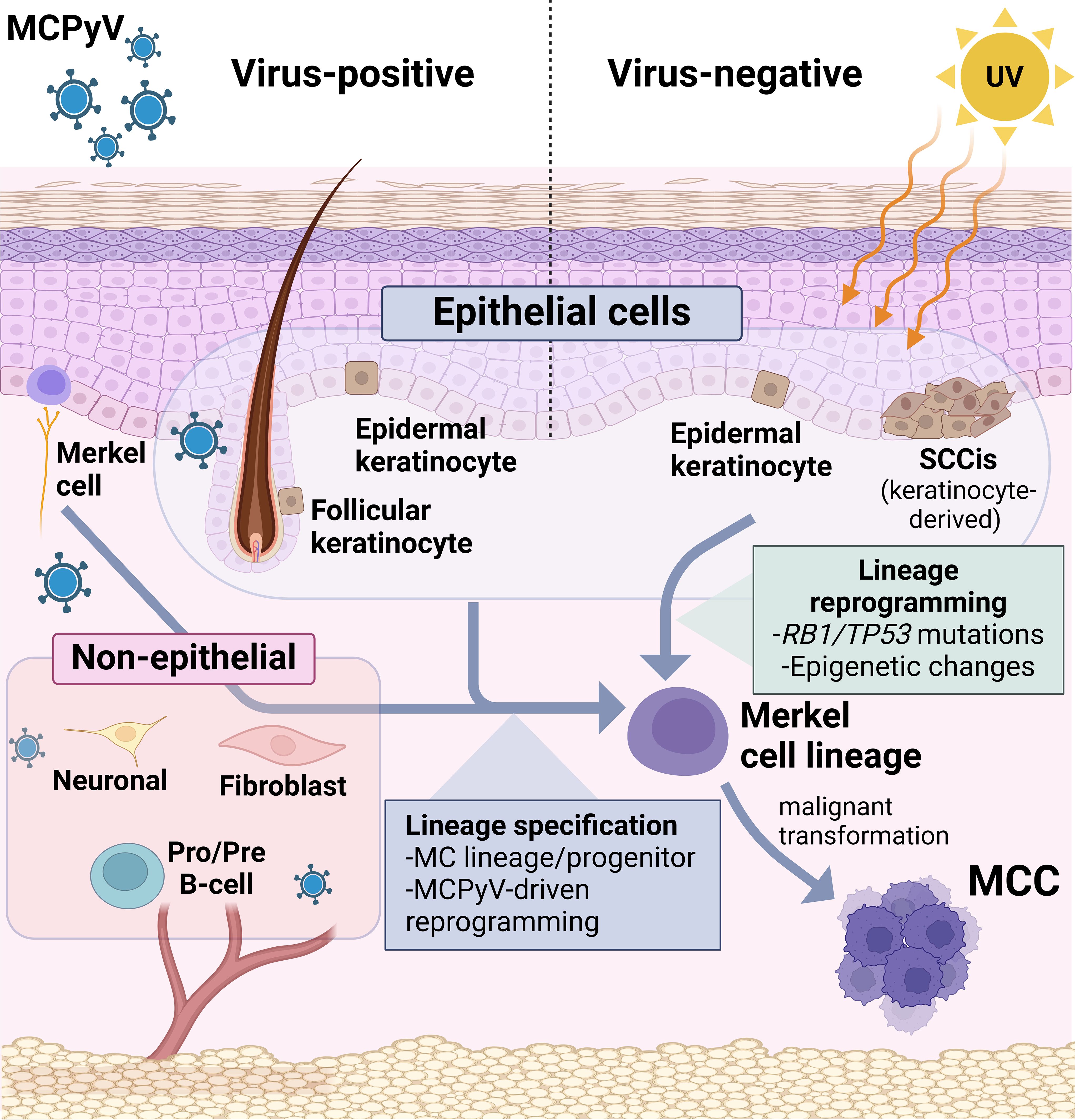
Figure 2 Candidates for MCC cell of origin. (Left) Proposed candidates for VP-MCC include many diverse cell types, especially UV-shielded dermal populations such as follicular epithelial cells and precursors, and non-epithelial lineages including neuronal, fibroblast, and pre-pro B-cells. By contrast, direct observations from human tumors strongly suggest epidermal keratinocytes to be the precursors for VN-MCC. In either scenario, lineage reprogramming (with an as-yet unknown trigger) would be required for a shift to the Merkel cell phenotype in addition to malignant transformation.
Furthermore, current genomic data suggests VP- and VN-MCC arise from distinct progenitor populations since VN tumors carry a high UV-signature mutational burden (43, 45, 102), which is characteristic of tumors originating in the epidermis. In contrast, VP tumors harbor few UV-mutations, suggesting they are not derived from sun-damaged epidermal cells, and possibly reside at deeper locations (Figure 2). This is in keeping with the nascent tumors arising in mice with epidermal-driven expression of TAgs and ATOH1, which appeared spatially restricted to the outer root sheath of the hair follicle, near the bulge compartment (242). The lack of UV-mutations, and the dermal location characteristic of MCC, could also be explained by a non-epithelial cell of origin. Interestingly, dermal fibroblasts have a similar mutational burden as VP-MCCs (239, 243) and thus far are the only host cell that has been shown to support the MCPyV viral life cycle by in vitro assays (241). Another non-epithelial candidate cell type with dermal localization is the B-cell lineage, proposed mainly due to MCC expression of early B-cell lineage markers and immunoglobulins (34, 244). Neuronal cells are another possible candidate, since TAg knockdown in VP-MCC cells converts them to a differentiated neuronal phenotype (124).
Although lymphoid or mesenchymal populations have been proposed for the VP-MCC cell of origin, currently, most mouse modeling studies support epithelial populations for this role, since MCPyV TAgs were shown to drive transformation or tumor development in mice from epidermal drivers (117, 118, 138, 242). While murine MCCs only developed upon exogenous ATOH1 expression driving an epidermal-neuroendocrine fate switch, this lineage was also maintained following transient ATOH1 activation (118). This suggests that MCCs may therefore arise from epithelial cells with transitory ATOH1 expression if a neuroendocrine cell fate feed forward loop is established (200, 245, 246). These findings support the concept that commitment to the MC lineage may be a fundamental characteristic of the MCC cell of origin.
Recent data showing mutational overlap between a trichoblastoma and a VP-MCC in a rare combined tumor implies that viral integration in a follicular epithelial cell triggered MCC development (11). Although rare, this case demonstrates the potential for follicular epithelial cells to give rise to human VP-MCC tumors. This aligns with reports of MCPyV protein expression in human follicular epithelial cells, consistent with infection of that cell population by wild-type MCPyV in human skin (247). Furthermore, the appearance of nascent tumors spatially restricted to the hair follicle outer root sheath, in mice with epidermal-driven expression of TAgs and ATOH1, also supports these cells as a potential cell of origin (242).
VN-MCC can display squamous differentiation, or coexist with invasive squamous cell carcinoma or SCCIS (7, 10). Squamous and neuroendocrine areas of differentiation in combined tumors share mutations and copy number abnormalities (159, 172); however, it is not clear to what extent this similarity reflects squamous metaplasia and/or bidirectional tumor differentiation, rather than evidence for MCC arising within a squamous precursor. Recent studies more specifically comparing SCCIS associated with VN-MCC found that both components share key driver mutations in TP53 and RB1 (173, 248, 249) and copy number variations (173, 250). Together, these findings suggest that SCCIS is clonally related to the underlying MCC and thus may be a progenitor lesion. However, these studies did not identify a mutational event responsible for the transition from SCCIS to MCC, rather uncovering a shift associated with epigenetic changes (173, 248).
Comprehensive analysis of DNA-methylation patterns in classical VP- and VN-MCC cell lines suggests an epithelial origin for both MCC subtypes (189, 190). Taken together, these findings provide strong support for an epithelial origin for MCC, and further suggest that VP-MCC and VN-MCC might arise from distinct follicular epithelial and epidermal progenitors, respectively.
Immune therapy has improved the prognosis of advanced and metastatic MCC, but is not universally effective or tolerated. Thus, there is continued need for additional therapeutic options that are effective either alone or that work in combination with immunotherapy. Furthermore, the potential benefit of adjuvant or neoadjuvant immunotherapy in MCC must be defined. Here, we review the current status of therapeutic strategies that are being studied in the clinical setting, based upon the fundamental observations of MCC tumor biology described above.
Adjuvant immunotherapy trials are underway to determine the efficacy of immunotherapy to prevent disease relapse in treated patients. The ADMEC-O trial (NCT02196961) is evaluating adjuvant nivolumab in treated stage I-IV MCC patients (251). Interim analysis showed a 9% risk reduction at 1 year and 10% at 2 years (252). The STAMP (Surgically Treated Adjuvant Merkel Cell Carcinoma with Pembrolizumab) trial (NCT03712605) is evaluating pembrolizumab as adjuvant therapy in patients with resected stage I-IIIB MCC versus current standard-of-care observation (253). Adjuvant avelumab is being studied in stage III MCC patients in the ADAM (Adjuvant Avelumab in Merkel) trial (NCT03271372) (251). Clinical trial NCT03798639 is studying the effect of nivolumab and radiation or nivolumab and ipilimumab for patients with resected stage IIIA or IIIB MCC (251).
Neoadjuvant therapy trials are underway for MCC. The Checkmate-358 trial (NCT02488759) evaluated neoadjuvant nivolumab in stage IIA-IV MCC (82). Additional trials are ongoing to evaluate the efficacy of the neoadjuvant approach with immunotherapies including pembrolizumab (NCT05496036), pembrolizumab with lenvatinib (NCT04869137), cemiplimab (NCT04975152), and retifanlimab with cisplatin (NCT05594290) (77). Neoadjuvant intratumoral injection with L19IL2/L19TNF (NCT05329792) and PH-762, an RNAi targeting PD-1 (NCT06014086) is also under evaluation (251).
Combination therapy with nivolumab and the CTLA-4 inhibitor ipilimumab is under evaluation as first-line therapy and in checkpoint inhibitor-refractory disease. A randomized, open label phase 2 trial (NCT03071406) evaluated the combination of ipilimumab and nivolumab with or without stereotactic body radiation therapy (SBRT) as first line or following PD-1/PD-L1 monotherapy (254). This study found ORR in both the first-line and second-line treatment groups. A retrospective study of 14 patients from the German-based multi-center prospective skin cancer registry ADOREG demonstrated a response to combined ipilimumab and nivolumab in Avelumab-refractory patients, demonstrating a progression-free survival rate of 42.9% at 12 months and 26.8% at 24 months (255). Conversely, a retrospective study of 13 patients refractory of checkpoint inhibitor inhibition found only a limited benefit from treatment with combination of ipilimumab and nivolumab (256). Another retrospective study, of patients who received rescue therapy following unsuccessful anti-PD1 therapy, observed objective responses in 4/13 patients treated with anti-CTLA4 in combination with immunotherapy, although some responses were not durable (257). The Checkmate-358 trial (NCT02488759) evaluated ipilimumab in combination with nivolumab for metastatic MCC (251); the results have not been published.
Given the high immunogenicity of MCC, continued investigations into immune-based approaches other than PD-1/PD-L1 inhibition have substantial promise for identifying effective new therapies. Therapeutic vaccines are an active avenue of investigation for cancer therapy, including MCC (77). In melanoma, a trial evaluating the role of a vaccine against telomerase in combination with pembrolizumab was shown to have preliminary positive results (258). For MCC, vaccines might potentially target either MCPyV epitopes for VP-MCC, or patient-specific tumor neoantigens for VN-MCC (259). Mouse modeling experiments with DNA vaccines based upon LTAg, sTAg, and VP1 peptides have shown promising results (260–263). A phase I DNA vaccine trial utilizing a modified MCPyV LTAg is underway in MCPyV-positive MCC patients (NCT05422781), with results pending (251).
Talimogene laherparepvec (T-VEC) is an injectable engineered oncolytic virus that has been shown to induce an immunologic anti-tumor response in melanoma (264). The efficacy of T-VEC injection is currently in clinical trials as a single agent in locally advanced MCC (NCT03458117), with or without radiotherapy in metastatic lesions of MCC (NCT02819843), and with nivolumab in refractory disease (NCT02978625) (251).
CD47 is a cell surface ligand that opposes antitumor immunity by preventing immune-mediated phagocytosis. In vitro expression of CD47 has been shown to be influenced by the sTAg of MCPyV (147). Anti-CD47 therapy has been investigated in a clinical trial for CTCL which also included MCC (NCT02890368) (147).
Other immune-based therapeutic strategies under investigation for MCC include natural killer cell immunotherapy, stimulating tumor MHC expression (via approaches including interferons or epigenetic modifiers), and adoptive T-cell transfer including chimeric antigen receptor (CAR) T-cells (251, 259).
Epigenetic modulation can normalize cell cycle and apoptosis regulation, thus facilitating removal of aberrant cells. Histone deacetylases (HDACs) are a class of enzymes that play an important role in gene expression. Abnormal regulation and expression of HDACs has been identified in several malignancies. Domatinostat is a HDAC class I inhibitor that has shown anti-tumor effects and upregulation of MHC class 1 molecules in MCC cells (221). Domatinostat in combination with Avelumab is currently being evaluated in the MERKLIN 2 trial for patients with advanced MCC who have progressed on anti-checkpoint inhibitor therapy (NCT04393753) (251). The Bromodomains and Extra-terminal Domains (BET) family of proteins also play a role in gene expression. Both a BET inhibitor and BET degrader, JQ1, and BETd-246, respectively, have demonstrated in vitro anti-tumor effect in MCC cell lines (215, 216). Similarly, the hypomethylating agent decitabine has shown anti-tumor effects in MCC cell lines and xenograft tumors (189). Finally, as noted above, activity of the histone demethylase LSD1 (KDM1A) may represent a therapeutic vulnerability for MCC (150, 213). These promising early results underscore the need for further investigation of epigenetic modifiers as therapeutics for MCC.
As the underlying mechanisms of MCC tumor biology are more precisely characterized, distinct therapeutic targets will emerge. These might be useful as single agents, in combination with immunotherapy, or for immunotherapy-refractory disease. There have been many trials of investigational agents in both humans and preclinical mouse models based upon biomarker expression patterns and therapeutic performance in other tumor types, as summarized in Table 2 (89, 149, 197, 205–208, 210, 211, 223, 259, 265–274, 277–279, 283–285, 288–294, 304).
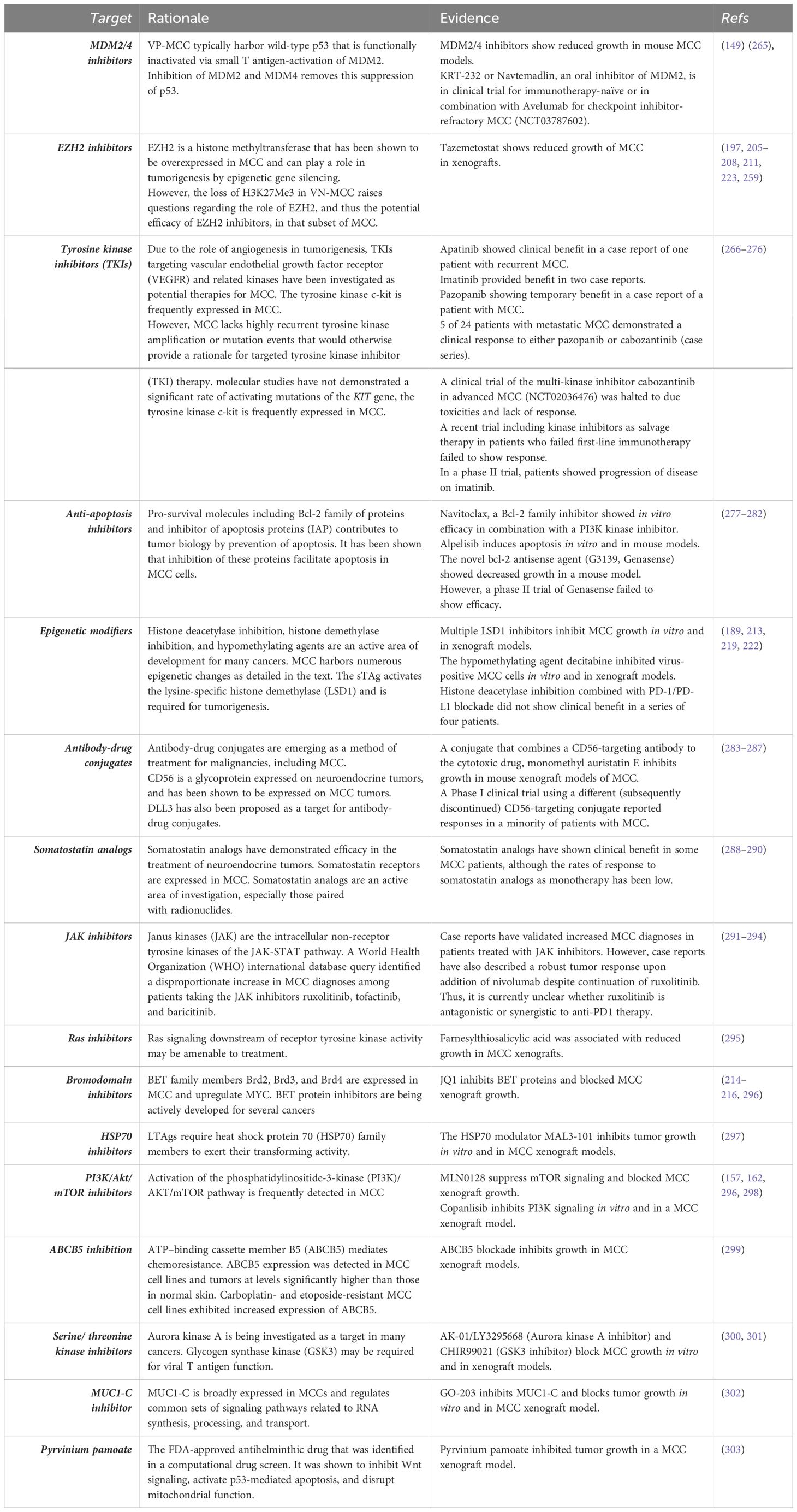
Table 2 Emerging targets in clinical and preclinical studies.
Accurate and reliable preclinical models are invaluable for testing new therapeutic strategies prior to testing in humans. While a better understanding of the molecular pathogenesis of MCC has led to improve in vitro and in vivo models since the discovery of the MCPyV [reviewed in (305)], multiple questions remain that could be addressed by mouse models. Types of murine models used in cancer research include cell line xenografts (CLX), patient-derived xenografts (PDX), syngeneic models, and genetically engineered mouse models (GEMMs)(Figure 3). Each provides different opportunities or poses specific challenges for investigating tumor initiation, progression, and metastases as well as possible prevention and therapeutic strategies. Successful development of these models is critical to advance understanding of both viral and UV-driven MCC tumors.
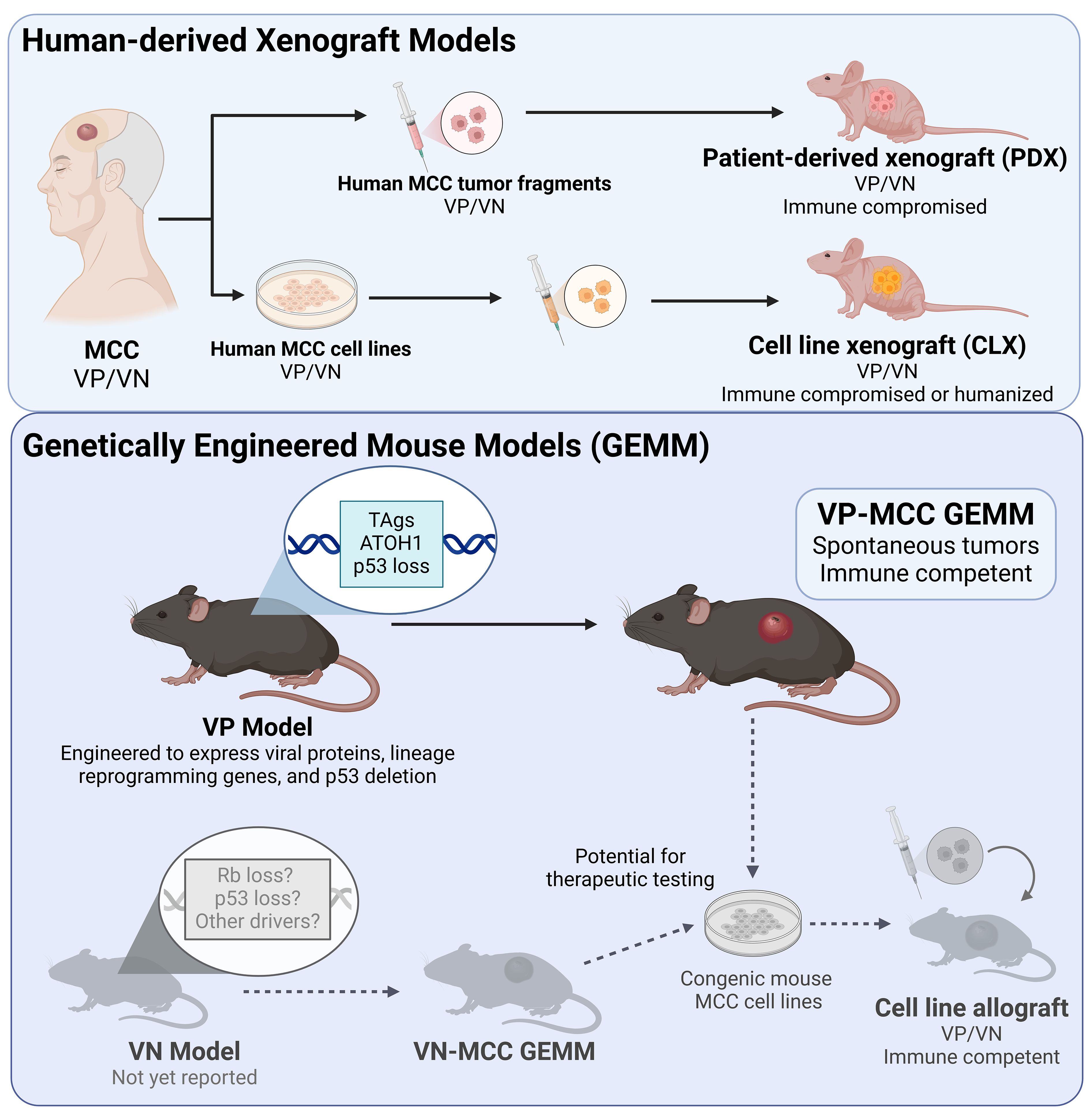
Figure 3 Mouse models of MCC. (Top panel) Mouse models of MCC utilizing xenografts of human MCC include patient-derived xenograft models in immune compromised mice, and cell line xenografts in immune compromised or humanized mice. Of these, only humanized mice allow for assessment of antitumor immune response. (Bottom panel) Generation of a murine virus positive (VP) MCC tumor required genetically engineered mice expressing MCC oncoproteins (TAgs) accompanied by ATOH1 for neuroendocrine reprogramming, and inactivation of p53. The resulting tumors can be studied in immune competent mice, allowing for studies of antitumor immunity (such as for immunotherapy). There is currently no virus negative (VN) MCC mouse model, however such a model would presumably require inactivation of the Trp53 and RB1 tumor suppressor genes similar to human VN-MCC tumors. Generation of congenic mouse cell lines from VP and VN GEMMs would allow pre-clinical therapeutic studies in an immune competent syngeneic model. Examples in gray have not yet been reported.
Establishment of cancer cell lines from human tumors has traditionally allowed for the development of CLX models in immunocompromised mice, which have classically been powerful tools for drug efficacy and toxicity studies, including in MCC (150, 189, 213, 215, 216, 219, 222, 265, 275, 277, 280–282, 284, 295–303).
While the rarity of MCC may in part be responsible for the lack of PDX models to date, several recent studies now provide compelling preclinical data for the use of targeted monotherapies or potential combinatorial regimens in select MCC patients. The PI3K/mTOR/AKT pathway is commonly activated in MCC, and use of the PI3K inhibitor copanlisib, specifically targeting PI3K-α and PI3K-δ isoforms, showed antitumor effects in multiple MCC PDX models compared to other PI3K inhibitors (298). CDK4/6 inhibition with palbociclib increases PD-L1 expression, and when combined with HIF2alpha inhibition results in growth inhibition using both CDX models and a PDX-derived cell line (306). Additionally, PDX models have proven valuable for assessing MDM2 inhibition via the small molecule inhibitor, milademetan, in MCCs with wild-type p53 (265).
Though classically used for therapeutic strategizing, CLX and PDX models have additionally provided opportunities for establishing and characterizing MCC cell lines (277, 307–309). They have been valuable for observing the appearance of circulating MCC tumor cells and metastasis formation (310), determining the requirement of TAg expression in MCC cells (136), as well as for studying vaccine development (260, 261), vasculogenic mimicry as a novel MCC biomarker (311), and circulating microRNA miR-375 as a surrogate biomarker (230). Xenograft models have also helped establish the role of cancer-associated fibroblasts in promoting MCC progression (312).
Undoubtedly, transplantable mouse models can be powerful in specific settings. However, a GEMM that develops spontaneous MCC tumors recapitulating the human cancer will provide the best insight into tumor development and reveal possibilities for prevention and treatment. The clonal integration of MCPyV sequences expressing TAgs in MCC supports this virus as the driver of tumorigenesis. However, despite multiple attempts to capitalize on MCPyV TAg expression in mice over the years (117, 118, 138, 139, 242, 313, 314), generation of a faithful, accessible MCC GEMM has been complicated and hindered by the unknown cell of origin.
The in vitro transforming potential of sTAg (135) was subsequently corroborated in transgenic mice expressing sTAg (117). Although this study did not yield MCC tumors, it was the first evidence in a mouse model that sTAg expression is sufficient for epithelial transformation. Using a Keratin 5 (K5) promoter to drive expression of wild-type or mutant sTAgs in epidermis, this model revealed that the transformation phenotype was dependent on the sTAg LSD-binding domain. These sTAg transforming studies, initially performed in a preterm embryo model, were validated in an adult model that rapidly developed epidermal hyperplasia and advanced skin lesions with histological changes that collectively recapitulated human squamous cell carcinoma in situ (SCCIS), also known as Bowen’s disease (117).
That same year, two additional studies implicated the TAgs in cancer formation (138, 139). Use of an epithelial tissue-specific conditional mouse model further demonstrated that a K14-driven MCPyV early region expressing TAgs has tumorigenic activity in vivo. These mice also developed epidermal hyperplasia, with some advancing to cutaneous papillomas (138). Another mouse model generated to conditionally express sTAg from the ROSA26 locus revealed ubiquitous sTAg was lethal at high levels, while lower expression could induce a hyperplastic skin phenotype (139). Expression of ubiquitous sTAg in the context of a homozygous p53 deletion in this model generated poorly differentiated malignancies in spleen and liver, however, MCC development was not observed.
Taken together, these initial mouse models demonstrated the capability of sTAg to drive undifferentiated malignancy, epidermal hyperplasia, and epidermal dysplasia, but did not culminate in a neuroendocrine tumor. The lack of MCC development suggests the TAgs might not have been targeted to the appropriate cell population or required the contribution of additional factors. An early theory of MCC origin (no longer considered likely) posited that these represent malignant transformation of Merkel cells (MCs), due to their shared structural and immunohistochemical characteristics (239). However, sTAg-targeted to embryonic or adult Merkel cells showed no sustained proliferative capacity and no transforming potential, even in the p53 null setting (139, 239). This is consistent with most MCs being post-mitotic or terminally differentiated, and thus unlikely to generate a neoplastic proliferation (315).
Merkel cells themselves have been shown to be derived from an epidermal progenitor in an ATOH1 dependent manner (316, 317), and epidermal expression of ATOH1 in embryonic and adult mice drives formation of ectopic Merkel cells (245). MCC tumors express ATOH1 (27, 28, 30), suggesting that these tumors might arise from epidermal progenitors in a process that recapitulates the differentiation pathway of normal Merkel cells. This insight led to a key advance in MCC mouse modeling. When keratinocytes were reprogrammed to the MC lineage by ATOH1, sTAg expression was sufficient to initiate small blue cell tumors resembling human intraepidermal MCC in late-stage mouse embryos (118). These MCC-like tumors expressed MC markers and K8 and K20 in a clumped or dot-like pattern typically detected in MCC tumor cells (7, 318).
This finding was followed up by a study designed to ascertain whether TAg-expressing epidermal cells reprogrammed to the MC lineage via ATOH1 would develop murine MCC tumors in adult mice (242). These conditional mice expressing sTAg, tLTAg and ATOH1 in Krt5-expressing cells and their progeny, yielded microscopic collections of proliferating MCC-like cells, but no gross tumors.
An important modification of this mouse model was additional targeted deletion of Trp53 (Figure 3). This change, which ensured the loss of p53 activity known to be critical in human MCC tumors, was the final step to yield macroscopic skin tumors with classic MCC histology including a monomorphous small blue cell phenotype, finely stippled chromatin, prominent mitoses, and nuclear molding. Furthermore, they mimicked human MCC tumors at the marker expression level, and cross-species transcriptomic analysis revealed close similarity to human tumors. Thus, this model which relies on epidermal expression of MCPyV TAgs, ectopic expression of ATOH1 to drive epidermal reprogramming, and loss of p53, is the first GEMM to successfully develop murine MCC (242).
Despite the initial promise raised by CLX and PDX studies, targeted approaches for MCC have performed poorly in clinical trials (267, 270, 274, 279, 289), and there is currently no approved targeted therapy for advanced MCC. Since CLX models do not necessarily accurately recapitulate the phenotypic and molecular heterogeneity of MCC tumors, use of PDX models utilizing patient tumor material may be more representative and better predictors of clinical responses to anticancer agents (319–321). Importantly, both CLX and PDX models require immune depleted mice, thus they are not informative for immunotherapy strategies. Preclinical immunotherapy studies are best tested in a mouse model where an intact immune system is present.
For these reasons, “humanized” mouse models, consisting of immune deficient mice with various human immune cell engraftments, used in conjunction with CLXs or PDXs have become fundamentally important in onco-immunology studies (322, 323). Use of a humanized mouse model has suggested the potential application for T-cell-mediated immunotherapy directed against MCPyV TAgs (324). Although not yet extensively used for MCC studies, such humanized mice may provide a powerful platform for testing responsiveness to novel immunotherapies or combination regimens, as well as better predicting clinical responses.
Another alternative strategy is to develop syngeneic models utilizing immune-competent animals. Syngeneic mouse models utilize immune-competent mice that develop tumors derived from murine cancer cells that have been engrafted from genetically identical congenic mouse strains. Syngeneic models are typically considered the workhorse for investigating immunotherapies and studying the immune surveillance of cancer development (322, 325, 326). Since the only current FDA-approved MCC therapies target the PD-1/PD-L1 (programmed cell death protein 1/PD1 ligand) immune checkpoint pathway, which benefits ~50% of MCC patients (73, 75, 76, 78, 327), a syngeneic model would be a tremendous benefit and provide a much-needed immunotherapy pre-clinical model. However, currently there are no syngeneic MCC mouse models available.
To date, MCC viral-driven mouse modeling has focused on expression of individual or TAg combinations, however the role of each has not been fully dissected. sTAg in the absence of LTAg was shown to be the primary oncogenic driver in several mouse models (117, 118, 139). The precise contribution of tLTAg in the latest murine MCC model (242) is currently unknown. A recent study utilizing the previously developed K14-driven transgenic mouse expressing both sTAg and tLTAg (138) indicates the resulting epithelial phenotypes are dependent upon the LTAg-pRb interaction (313). The functional role of TAgs was additionally investigated in a multi-stage chemically-induced mouse model of skin carcinogenesis, revealing that expression of TAgs in stratified epithelia synergize with the tumor-inducing agent dimethylbenz(a)anthracene (DMBA), but not the tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA), suggesting TAgs operate predominantly as tumor promoters (314). Exactly how the MCPyV TAgs contribute to MCC tumorigenesis remains an open question, and poorly understood or disparate functions of TAgs in vivo may be a consequence of experimental design and modeling strategies which do not truly recapitulate human MCCs.
The unexpected requirement for the loss of p53 to drive full blown virus-positive murine tumors remains perplexing (242). While TP53 mutations are common in UV-driven MCCs, they are rare in viral-driven MCC (10, 43, 45, 102) conceivably because sTAg upregulates MDM2 and the MDM4 activator CK1α to repress p53, thus abrogating the need for mutational inactivation (149). Interestingly, the requirement for the loss of p53 in sTAg-induced transformation was seen previously in mouse embryonic fibroblasts and in mice, and it was hypothesized that sTAg induces mitotic catastrophe and p53 activation leading to apoptosis (139). The presence of activated p53 and apoptotic cells in microscopic lesions of adult mice expressing sTAg, tLTAg and ATOH1 potentially supports this theory (242). However, the differential requirement for the loss of Trp53 in human and mouse TAg-driven tumors may be a result of divergence in p53-regulated targets between the species (328). Regardless of mechanism, abrogating p53 activity in viral-driven MCC appears to be critical.
The development of murine MCCs established a fundamental role for the TAgs in viral-driven MCC tumorigenesis. However this model is dependent on the shift of TAg-expressing epidermal cells into a Merkel cell fate via ectopic ATOH1 expression (242). While this highlights the novelty of manipulating cell fate to generate tumors of unknown origin, it does not accurately reflect MCC development in humans. Although expression of MCPyV TAgs in Sox9-positive epidermal cells has recently been shown to drive hyperproliferative lesions and reprogram cells to express neuroendocrine markers, full blown MCC tumors were not observed (125). Determining whether other types of skin cells can be ATOH1-reprogrammed to yield MCCs, or whether different candidate cells of origin are competent to form MCCs without exogenous ATOH1 or p53 deficiency, remains an active area of research.
Effectively modeling VN-MCC in mice remains challenging. Although transplantable VN-MCC mouse models have been successfully generated and utilized (277), there are currently no GEMMs reported in the peer-reviewed literature for this tumor subtype. While the frequent inactivation of TP53 and RB1 genes in human VN-MCC tumors indicates this will likely be a prerequisite for development of a VN-MCC GEMM, additional drivers or pathway perturbations that might be required are unclear. Furthermore, the unknown cell of origin and speculation, that VP- and VN-MCCs arise from distinct progenitor populations (2, 173, 239, 248), suggest this mouse model may be particularly challenging to develop.
The pathogenesis of MCC has remained poorly understood. Recent studies have fundamentally advanced our understanding regarding potential cells of origin for MCC, the role of epigenetic modifications in tumor biology, and potential therapeutic options. Remaining challenges include more conclusive evidence for MCC origins, mouse models for VN-MCC, syngeneic mouse models suitable for immunotherapy research, and effective therapies for patients who fail to respond to, or tolerate, immunotherapy.
EP: Conceptualization, Supervision, Visualization, Writing – original draft, Writing – review & editing. MV: Conceptualization, Data curation, Methodology, Project administration, Visualization, Writing – original draft, Writing – review & editing. MJ: Writing – original draft, Writing – review & editing. KH: Writing – original draft, Writing – review & editing. PH: Conceptualization, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. PH receives support from the National Institute of Arthritis and Musculoskeletal and Skin Diseases P30 AR075043. MV receives support from the National Cancer Institute R01 CA241947.
Figures 2 and 3 were created with Biorender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mohsin N, Martin MR, Reed DJ, Vilasi SM, Miao L, Hill NT, et al. Differences in merkel cell carcinoma presentation and outcomes among racial and ethnic groups. JAMA Dermatol. (2023) 159:536–40. doi: 10.1001/jamadermatol.2023.0061
2. Harms PW, Harms KL, Moore PS, DeCaprio JA, Nghiem P, Wong MKK, et al. The biology and treatment of Merkel cell carcinoma: current understanding and research priorities. Nat Rev Clin Oncol. (2018) 15:763–76. doi: 10.1038/s41571-018-0103-2
3. SChadendorf D, Lebbé C, Zur Hausen A, Avril MF, Hariharan S, Bharmal M, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. Eur J Cancer. (2017) 71:53–69. doi: 10.1016/j.ejca.2016.10.022
4. Paulson KG, Park SY, Vandeven NA, Lachance K, Thomas H, Chapuis AG, et al. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J Am Acad Dermatol. (2018) 78:457–463.e2. doi: 10.1016/j.jaad.2017.10.028
5. Harms KL, Healy MA, Nghiem P, Sober AJ, Johnson TM, Bichakjian CK, et al. Analysis of prognostic factors from 9387 merkel cell carcinoma cases forms the basis for the new 8th edition AJCC staging system. Ann Surg Oncol. (2016) 23:3564–71. doi: 10.1245/s10434-016-5266-4
6. Silling S, Kreuter A, Gambichler T, Meyer T, Stockfleth E, Wieland U. Epidemiology of merkel cell polyomavirus infection and merkel cell carcinoma. Cancers. (2022) 14(24):6176. doi: 10.3390/cancers14246176
7. Tetzlaff MT, Harms PW. Danger is only skin deep: aggressive epidermal carcinomas. An overview of the diagnosis, demographics, molecular-genetics, staging, prognostic biomarkers, and therapeutic advances in Merkel cell carcinoma. Modern Pathol. (2020) 33:42–55. doi: 10.1038/s41379-019-0394-6
8. Harms PW. Update on merkel cell carcinoma. Clin Lab Med. (2017) 37:485–501. doi: 10.1016/j.cll.2017.05.004
9. Pulitzer M. Merkel cell carcinoma. Surg Pathol Clin. (2017) 10:399–408. doi: 10.1016/j.path.2017.01.013
10. Harms KL, Zhao L, Johnson B, Wang X, Carskadon S, Palanisamy N, et al. Virus-positive merkel cell carcinoma is an independent prognostic group with distinct predictive biomarkers. Clin Cancer res: an Off J Am Assoc Cancer Res. (2021) 27:2494–504. doi: 10.1158/1078-0432.CCR-20-0864
11. Kervarrec T, Aljundi M, Appenzeller S, Samimi M, Maubec E, Cribier B, et al. Polyomavirus-positive merkel cell carcinoma derived from a trichoblastoma suggests an epithelial origin of this merkel cell carcinoma. J Invest Dermatol. (2020) 140:976–85. doi: 10.1016/j.jid.2019.09.026
12. Kervarrec T, Appenzeller S, Tallet A, Jullie ML, Sohier P, Guillonneau F, et al. Detection of wildtype Merkel cell polyomavirus genomic sequence and VP1 transcription in a subset of Merkel cell carcinoma. Histopathology. (2024) 84:356–68. doi: 10.1111/his.15068
13. Au RTM, Bundele MM. Endocrine mucin-producing sweat gland carcinoma and associated primary cutaneous mucinous carcinoma: Review of the literature. J cutaneous Pathol. (2021) 48:1156–65. doi: 10.1111/cup.13983
14. Jedrych J, Pulitzer M. Primary carcinoid tumor of the skin: a literature review. Int J Surg Pathol. (2014) 22:129–35. doi: 10.1177/1066896913516672
15. Goto K, Kukita Y, Honma K, Ohike N, Komori T, Ishida Y, et al. Sweat-gland carcinoma with neuroendocrine differentiation (SCAND): a clinicopathologic study of 13 cases with genetic analysis. Modern Pathol. (2022) 35:33–43. doi: 10.1038/s41379-021-00921-8
16. McCalmont TH. Paranuclear dots of neurofilament reliably identify Merkel cell carcinoma. J cutaneous Pathol. (2010) 37:821–3. doi: 10.1111/j.1600-0560.2010.01567_1.x
17. Stanoszek LM, Chan MP, Palanisamy N, Carskadon S, Siddiqui J, Patel RM, et al. Neurofilament is superior to cytokeratin 20 in supporting cutaneous origin for neuroendocrine carcinoma. Histopathology. (2019) 74:504–13. doi: 10.1111/his.13758
18. Wang L, Harms PW, Palanisamy N, Carskadon S, Cao X, Siddiqui J, et al. Age and gender associations of virus positivity in merkel cell carcinoma characterized using a novel RNA in situ hybridization assay. Clin Cancer Res. (2017) 23:5622–30. doi: 10.1158/1078-0432.CCR-17-0299
19. Rush PS, Rosenbaum JN, Roy M, Baus RM, Bennett DD, Lloyd RV. Insulinoma-associated 1: A novel nuclear marker in Merkel cell carcinoma (cutaneous neuroendocrine carcinoma). J cutaneous Pathol. (2018) 45:129–35. doi: 10.1111/cup.13079
20. Leblebici C, Yeni B, Savli TC, Aydin O, Gunes P, Cinel L, et al. A new immunohistochemical marker, insulinoma-associated protein 1 (INSM1), for Merkel cell carcinoma: Evaluation of 24 cases. Ann Diagn Pathol. (2019) 40:53–8. doi: 10.1016/j.anndiagpath.2019.04.002
21. Lilo MT, Chen Y, LeBlanc RE. INSM1 is more sensitive and interpretable than conventional immunohistochemical stains used to diagnose merkel cell carcinoma. Am J Surg Pathol. (2018) 42:1541–8. doi: 10.1097/PAS.0000000000001136
22. Fukuhara M, Agnarsdottir M, Edqvist PH, Coter A, Ponten F. SATB2 is expressed in Merkel cell carcinoma. Arch Dermatol Res. (2016) 308:449–54. doi: 10.1007/s00403-016-1655-6
23. Kervarrec T, Tallet A, Miquelestorena-Standley E, Houben R, Schrama D, Gambichler T, et al. Diagnostic accuracy of a panel of immunohistochemical and molecular markers to distinguish Merkel cell carcinoma from other neuroendocrine carcinomas. Modern Pathol. (2019) 32:499–510. doi: 10.1038/s41379-018-0155-y
24. Bellizzi AM. SATB2 in neuroendocrine neoplasms: strong expression is restricted to well-differentiated tumours of lower gastrointestinal tract origin and is most frequent in Merkel cell carcinoma among poorly differentiated carcinomas. Histopathology. (2020) 76:251–64. doi: 10.1111/his.13943
25. Laga AC, Lai CY, Zhan Q, Huang SJ, Velazquez EF, Yang Q, et al. Expression of the embryonic stem cell transcription factor SOX2 in human skin: relevance to melanocyte and merkel cell biology. Am J Pathol. (2010) 176:903–13. doi: 10.2353/ajpath.2010.090495
26. Thanguturi S, Tallet A, Miquelestorena-Standley E, Coco C, Le Corre Y, Hainaut-Wierzbicka E, et al. Investigation of the RB1-SOX2 axis constitutes a tool for viral status determination and diagnosis in Merkel cell carcinoma. Virchows Arch. (2022) 480:1239–54. doi: 10.1007/s00428-022-03315-6
27. Heiskala K, Arola J, Heiskala M, Andersson LC. Expression of Reg IV and Hath1 in neuroendocrine neoplasms. Histol histopathol. (2010) 25:63–72. doi: 10.14670/HH-25.63
28. Gambichler T, Mohtezebsade S, Wieland U, Silling S, Hoh AK, Dreissigacker M, et al. Prognostic relevance of high atonal homolog-1 expression in Merkel cell carcinoma. J Cancer Res Clin Oncol. (2017) 143:43–9. doi: 10.1007/s00432-016-2257-6
29. Vilasi SM, Nguyen J, Wang CJ, Miao L, Daily K, Eid M, et al. ATOH1, TFAP2B, and CEACAM6 as immunohistochemical markers to distinguish merkel cell carcinoma and small cell lung cancer. Cancers. (2024) 16(4):788. doi: 10.3390/cancers16040788
30. Leonard JH, Cook AL, Van Gele M, Boyle GM, Inglis KJ, Speleman F, et al. Proneural and proneuroendocrine transcription factor expression in cutaneous mechanoreceptor (Merkel) cells and Merkel cell carcinoma. Int J Cancer J Int du Cancer. (2002) 101:103–10. doi: 10.1002/ijc.10554
31. Miner AG, Patel RM, Wilson DA, Procop GW, Minca EC, Fullen DR, et al. Cytokeratin 20-negative Merkel cell carcinoma is infrequently associated with the Merkel cell polyomavirus. Modern Pathol. (2014) 28(4):498–504. doi: 10.1038/modpathol.2014.148
32. Pasternak S, Carter MD, Ly TY, Doucette S, Walsh NM. Immunohistochemical profiles of different subsets of Merkel cell carcinoma. Hum Pathol. (2018) 82:232–8. doi: 10.1016/j.humpath.2018.07.022
33. Kervarrec T, Tallet A, Miquelestorena-Standley E, et al. Morphologic and immunophenotypical features distinguishing Merkel cell polyomavirus-positive and negative Merkel cell carcinoma. Modern Pathol. (2019) 32:1605–16. doi: 10.1038/s41379-019-0288-7
34. Zur Hausen A, Rennspiess D, Winnepenninckx V, Speel EJ, Kurz AK. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry. Cancer Res. (2013) 73:4982–7. doi: 10.1158/0008-5472.CAN-13-0616
35. Murakami I, Takata K, Matsushita M, Nonaka D, Iwasaki T, Kuwamoto S, et al. Immunoglobulin expressions are only associated with MCPyV-positive Merkel cell carcinomas but not with MCPyV-negative ones: comparison of prognosis. Am J Surg Pathol. (2014) 38:1627–35. doi: 10.1097/PAS.0000000000000279
36. Kirkham N. Small cell melanoma. Histopathology. (2002) 40:196–8. doi: 10.1046/j.1365-2559.2002.01317.x
37. Wong AS, du Plessis JJ, Jackett LA. Merkel cell carcinoma: an unusual case with SOX10 nuclear staining. Pathology. (2023) 55:718–21. doi: 10.1016/j.pathol.2022.12.352
38. Jackson CR, Linos K. SOX10 dot-like paranuclear positivity in merkel cell carcinoma: report of 2 cases. Am J dermatopathol. (2019) 41:694–5. doi: 10.1097/DAD.0000000000001230
39. Miettinen M, McCue PA, Sarlomo-Rikala M, Biernat W, Czapiewski P, Kopczynski J, et al. Sox10–a marker for not only schwannian and melanocytic neoplasms but also myoepithelial cell tumors of soft tissue: a systematic analysis of 5134 tumors. Am J Surg Pathol. (2015) 39:826–35. doi: 10.1097/PAS.0000000000000398
40. Nonaka D, Chiriboga L, Rubin BP. Sox10: a pan-schwannian and melanocytic marker. Am J Surg Pathol. (2008) 32:1291–8. doi: 10.1097/PAS.0b013e3181658c14
41. Fernandez-Flores A, Suarez-Penaranda JM, Alonso S. Study of EWS/FLI-1 rearrangement in 18 cases of CK20+/CM2B4+ Merkel cell carcinoma using FISH and correlation to the differential diagnosis of Ewing sarcoma/peripheral neuroectodermal tumor. Appl immunohistochem Mol morphol. (2013) 21:379–85. doi: 10.1097/PAI.0b013e318273a9e0
42. Harms PW, Collie AM, Hovelson DH, Cani AK, Verhaegen ME, Patel RM, et al. Next generation sequencing of Cytokeratin 20-negative Merkel cell carcinoma reveals ultraviolet-signature mutations and recurrent TP53 and RB1 inactivation. (2016) 29(3):240–8. doi: 10.1038/modpathol.2015.154
43. Harms PW, Vats P, Verhaegen ME, Robinson DR, Wu YM, Dhanasekaran SM, et al. The distinctive mutational spectra of polyomavirus-negative merkel cell carcinoma. Cancer Res. (2015) 75:3720–7. doi: 10.1158/0008-5472.CAN-15-0702
44. Sun L, Cliften PF, Duncavage EJ, Lewis JS Jr., Chernock RD. UV signature mutations reclassify salivary high-grade neuroendocrine carcinomas as occult metastatic cutaneous merkel cell carcinomas. Am J Surg Pathol. (2019) 43:682–7. doi: 10.1097/PAS.0000000000001231
45. Wong SQ, Waldeck K, Vergara IA, Schroder J, Madore J, Wilmott JS, et al. UV-associated mutations underlie the etiology of MCV-negative merkel cell carcinomas. Cancer Res. (2015) 75:5228–34. doi: 10.1158/0008-5472.CAN-15-1877
46. Fisher CA, Harms PW, McHugh JB, Edwards PC, Siddiqui J, Palanisamy N, et al. Small cell carcinoma in the parotid harboring Merkel cell polyomavirus. Oral surgery Oral med Oral Pathol Oral Radiol. (2014) 118:703–12. doi: 10.1016/j.oooo.2014.09.012
47. Chernock RD, Duncavage EJ, Gnepp DR, El-Mofty SK, Lewis JS Jr. Absence of Merkel cell polyomavirus in primary parotid high-grade neuroendocrine carcinomas regardless of cytokeratin 20 immunophenotype. Am J Surg Pathol. (2011) 35:1806–11. doi: 10.1097/PAS.0b013e318236a9b0
48. de Biase D, Ragazzi M, Asioli S, Eusebi V. Extracutaneous Merkel cell carcinomas harbor polyomavirus DNA. Hum Pathol. (2012) 43:980–5. doi: 10.1016/j.humpath.2011.08.014
49. McEvoy AM, Lachance K, Hippe DS, Cahill K, Moshiri Y, Lewis CW, et al. Recurrence and mortality risk of merkel cell carcinoma by cancer stage and time from diagnosis. JAMA Dermatol. (2022) 158:382–9. doi: 10.1001/jamadermatol.2021.6096
50. McEvoy AM, Hippe DS, Lachance K, Park S, Cahill K, Redman M, et al. Merkel cell carcinoma recurrence risk estimation is improved by integrating factors beyond cancer stage: A multivariable model and web-based calculator. J Am Acad Dermatol. (2024) 90:569–76. doi: 10.1016/j.jaad.2023.11.020
51. Hoang MP, Donizy P, Wu CL, Kopczynski J, Pieniazek M, Miller DM, et al. TdT expression is a marker of better survival in merkel cell carcinoma, and expression of B-cell markers is associated with merkel cell polyomavirus. Am J Clin Pathol. (2020) 154:38–47. doi: 10.1093/ajcp/aqaa017
52. Moshiri AS, Doumani R, Yelistratova L, Blom A, Lachance K, Shinohara MM, et al. Polyomavirus-negative merkel cell carcinoma: A more aggressive subtype based on analysis of 282 cases using multimodal tumor virus detection. J Invest Dermatol. (2016) 137(4):819–27. doi: 10.1016/j.jid.2016.10.028
53. Sihto H, Kukko H, Koljonen V, Sankila R, Bohling T, Joensuu H. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in Merkel cell carcinoma. Clin Cancer Res. (2011) 17:4806–13. doi: 10.1158/1078-0432.CCR-10-3363
54. Higaki-Mori H, Kuwamoto S, Iwasaki T, Kato M, Murakami I, Nagata K, et al. Association of Merkel cell polyomavirus infection with clinicopathological differences in Merkel cell carcinoma. Hum Pathol. (2012) 43:2282–91. doi: 10.1016/j.humpath.2012.04.002
55. Portilla N, Alzate JP, Sierra FA, Parra-Medina R. A Systematic review and Meta-Analysis of the survival and clinicopathological features of p63 expression in Merkel cell carcinoma. Australas J Dermatol. (2020) 61:e276–82. doi: 10.1111/ajd.13211
56. Hall BJ, Pincus LB, Yu SS, Oh DH, Wilson AR, McCalmont TH. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J cutaneous Pathol. (2012) 39:911–7. doi: 10.1111/j.1600-0560.2012.01964.x
57. Asioli S, Righi A, Volante M, Eusebi V, Bussolati G. p63 expression as a new prognostic marker in Merkel cell carcinoma. Cancer. (2007) 110:640–7. doi: 10.1002/cncr.22828
58. Stetsenko GY, Malekirad J, Paulson KG, et al. p63 expression in Merkel cell carcinoma predicts poorer survival yet may have limited clinical utility. Am J Clin Pathol. (2013) 140:838–44. doi: 10.1309/AJCPE4PK6CTBNQJY
59. Feldmeyer L, Hudgens CW, Ray-Lyons G, Nagarajan P, Aung PP, Curry JL, et al. Density, distribution, and composition of immune infiltrates correlate with survival in merkel cell carcinoma. Clin Cancer Res. (2016) 22(22):5553–63. doi: 10.1158/1078-0432.CCR-16-0392
60. Sihto H, Bohling T, Kavola H, Koljonen V, Salmi M, Jalkanen S, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res. (2012) 18:2872–81. doi: 10.1158/1078-0432.CCR-11-3020
61. Paulson KG, Iyer JG, Simonson WT, Blom A, Thibodeau RM, Schmidt M, et al. CD8+ lymphocyte intratumoral infiltration as a stage-independent predictor of Merkel cell carcinoma survival: a population-based study. Am J Clin Pathol. (2014) 142:452–8. doi: 10.1309/AJCPIKDZM39CRPNC
62. Paulson KG, Iyer JG, Tegeder AR, Thibodeau R, Schelter J, Koba S, et al. Transcriptome-wide studies of merkel cell carcinoma and validation of intratumoral CD8+ lymphocyte invasion as an independent predictor of survival. J Clin Oncol. (2011) 29:1539–46. doi: 10.1200/JCO.2010.30.6308
63. Donizy P, Wu CL, Kopczynski J, Pieniazek M, Biecek P, Rys J, et al. Prognostic role of tumoral PD-L1 and IDO1 expression, and intratumoral CD8+ and foxP3+ Lymphocyte infiltrates in 132 primary cutaneous merkel cell carcinomas. Int J Mol Sci. (2021) 22(11):5489. doi: 10.3390/ijms22115489
64. Schmults CD, Blitzblau R, Aasi SZ, Alam M, Amini A, Bibee K, et al. NCCN guidelines(R) insights: merkel cell carcinoma, version 1.2024. J Natl Compr Canc Netw. (2024) 22(1D):e240002. doi: 10.6004/jnccn.2024.0002
65. Gauci ML, Aristei C, Becker JC, Blom A, Bataille V, Dreno B, et al. Diagnosis and treatment of Merkel cell carcinoma: European consensus-based interdisciplinary guideline - Update 2022. Eur J Cancer. (2022) 171:203–31. doi: 10.1016/j.ejca.2022.03.043
66. Singh N, Alexander NA, Lachance K, Lewis CW, McEvoy A, Akaike G, et al. Clinical benefit of baseline imaging in Merkel cell carcinoma: Analysis of 584 patients. J Am Acad Dermatol. (2021) 84:330–9. doi: 10.1016/j.jaad.2020.07.065
67. Sims JR, Grotz TE, Pockaj BA, Joseph RW, Foote RL, Otley CC, et al. Sentinel lymph node biopsy in Merkel cell carcinoma: The Mayo Clinic experience of 150 patients. Surg Oncol. (2018) 27:11–7. doi: 10.1016/j.suronc.2017.10.005
68. Gupta SG, Wang LC, Penas PF, Gellenthin M, Lee SJ, Nghiem P. Sentinel lymph node biopsy for evaluation and treatment of patients with Merkel cell carcinoma: The Dana-Farber experience and meta-analysis of the literature. Arch Dermatol. (2006) 142:685–90. doi: 10.1001/archderm.142.6.685
69. Schwartz JL, Griffith KA, Lowe L, Wong SL, McLean SA, Fullen DR, et al. Features predicting sentinel lymph node positivity in Merkel cell carcinoma. J Clin Oncol. (2011) 29:1036–41. doi: 10.1200/JCO.2010.33.4136
70. Grotz TE, Joseph RW, Pockaj BA, Foote RL, Otley CC, Bagaria SP, et al. Negative sentinel lymph node biopsy in merkel cell carcinoma is associated with a low risk of same-nodal-basin recurrences. Ann Surg Oncol. (2015) 22:4060–6. doi: 10.1245/s10434-015-4421-7
71. Lee JS, Durham AB, Bichakjian CK, Harms PW, Hayman JA, McLean SA, et al. Completion lymph node dissection or radiation therapy for sentinel node metastasis in merkel cell carcinoma. Ann Surg Oncol. (2019) 26:386–94. doi: 10.1245/s10434-018-7072-7
72. Perez MC, Oliver DE, Weitman ES, Boulware D, Messina JL, Torres-Roca J, et al. Management of sentinel lymph node metastasis in merkel cell carcinoma: completion lymphadenectomy, radiation, or both? Ann Surg Oncol. (2019) 26:379–85. doi: 10.1245/s10434-018-6810-1
73. Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D'Angelo SP, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. (2016) 17:1374–85. doi: 10.1016/S1470-2045(16)30364-3
74. D’Angelo SP, Bhatia S, Brohl AS, Hamid O, Mehnert JM, Terheyden P, et al. Avelumab in patients with previously treated metastatic Merkel cell carcinoma (JAVELIN Merkel 200): updated overall survival data after >5 years of follow-up. ESMO Open. (2021) 6:100290. doi: 10.1016/j.esmoop.2021.100290
75. D’Angelo SP, Russell J, Lebbe C, Chmielowski B, Gambichler T, Grob JJ, et al. Efficacy and safety of first-line avelumab treatment in patients with stage IV metastatic merkel cell carcinoma: A preplanned interim analysis of a clinical trial. JAMA Oncol. (2018) 4:e180077. doi: 10.1001/jamaoncol.2018.0077
76. Bhatia S, Nghiem P, Veeranki SP, Vanegas A, Lachance K, Tachiki L, et al. Real-world clinical outcomes with avelumab in patients with Merkel cell carcinoma treated in the USA: a multicenter chart review study. J Immunother Cancer. (2022) 10. doi: 10.1136/jitc-2022-004904
77. Singh N, McClure EM, Akaike T, Park SY, Huynh ET, Goff PH, et al. The evolving treatment landscape of merkel cell carcinoma. Curr Treat Options Oncol. (2023) 24:1231–58. doi: 10.1007/s11864-023-01118-8
78. Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD-1 blockade with pembrolizumab in advanced merkel-cell carcinoma. N Engl J Med. (2016) 374:2542–52. doi: 10.1056/NEJMoa1603702
79. Nghiem P, Bhatia S, Lipson EJ, Sharfman WH, Kudchadkar RR, Brohl AS, et al. Durable tumor regression and overall survival in patients with advanced merkel cell carcinoma receiving pembrolizumab as first-line therapy. J Clin Oncol. (2019) 37:693–702. doi: 10.1200/JCO.18.01896
80. In brief: Retifanlimab (Zynyz) for Merkel cell carcinoma. Med Lett Drugs Ther. (2023) 65:e73–4. doi: 10.58347/tml.2023.1674h
81. Uchi H. Merkel cell carcinoma: an update and immunotherapy. Front Oncol. (2018) 8:48. doi: 10.3389/fonc.2018.00048
82. Topalian SL, Bhatia S, Amin A, Kudchadkar RR, Sharfman WH, Lebbe C, et al. Neoadjuvant nivolumab for patients with resectable merkel cell carcinoma in the checkMate 358 trial. J Clin Oncol. (2020) 38:2476–87. doi: 10.1200/JCO.20.00201
83. Angeles CV, Sabel MS. Immunotherapy for Merkel cell carcinoma. J Surg Oncol. (2021) 123:775–81. doi: 10.1002/jso.26319
84. Iyer JG, Blom A, Doumani R, Lewis C, Tarabadkar ES, Anderson A. Response rates and durability of chemotherapy among 62 patients with metastatic Merkel cell carcinoma. Cancer Med. (2016) 5:2294–301. doi: 10.1002/cam4.815
85. Nghiem P, Kaufman HL, Bharmal M, Mahnke L, Phatak H, Becker JC. Systematic literature review of efficacy, safety and tolerability outcomes of chemotherapy regimens in patients with metastatic Merkel cell carcinoma. Future Oncol. (2017) 13:1263–79. doi: 10.2217/fon-2017-0072
86. Knepper TC, Montesion M, Russell JS, Sokol ES, Frampton GM, Miller VA, et al. The genomic landscape of merkel cell carcinoma and clinicogenomic biomarkers of response to immune checkpoint inhibitor therapy. Clin Cancer Res. (2019) 25:5961–71. doi: 10.1158/1078-0432.CCR-18-4159
87. Kacew AJ, Dharaneeswaran H, Starrett GJ, Thakuria M, LeBoeuf NR, Silk AW, et al. Predictors of immunotherapy benefit in Merkel cell carcinoma. Oncotarget. (2020) 11:4401–10. doi: 10.18632/oncotarget.v11i47
88. Spassova I, Ugurel S, Kubat L, Zimmer L, Terheyden P, Mohr A, et al. Clinical and molecular characteristics associated with response to therapeutic PD-1/PD-L1 inhibition in advanced Merkel cell carcinoma. J Immunother Cancer. (2022) 10(1):e003198. doi: 10.1136/jitc-2021-003198
89. Nghiem P, Bhatia S, Lipson EJ, Sharfman WH, Kudchadkar RR, Brohl AS, et al. Three-year survival, correlates and salvage therapies in patients receiving first-line pembrolizumab for advanced Merkel cell carcinoma. J Immunother Cancer. (2021) 9(4):e002478. doi: 10.1136/jitc-2021-002478
90. Pulliam T, Jani S, Jing L, Ryu H, Jojic A, Shasha C, et al. Circulating cancer-specific CD8 T cell frequency is associated with response to PD-1 blockade in Merkel cell carcinoma. Cell Rep Med. (2024) 5:101412. doi: 10.1016/j.xcrm.2024.101412
91. Ryu H, Bi TM, Pulliam TH, Sarkar K, Church CD, Kumar N, et al. Merkel cell polyomavirus-specific and CD39(+)CLA(+) CD8 T cells as blood-based predictive biomarkers for PD-1 blockade in Merkel cell carcinoma. Cell Rep Med. (2024) 5:101390. doi: 10.1016/j.xcrm.2023.101390
92. Church C, Pulliam T, Longino N, Park SY, Smythe KS, Makarov V, et al. Transcriptional and functional analyses of neoantigen-specific CD4 T cells during a profound response to anti-PD-L1 in metastatic Merkel cell carcinoma. J Immunother Cancer. (2022) 10(9):e005328. doi: 10.1136/jitc-2022-005328
93. Spassova I, Ugurel S, Terheyden P, Sucker A, Hassel JC, Ritter C, et al. Predominance of central memory T cells with high T-cell receptor repertoire diversity is associated with response to PD-1/PD-L1 inhibition in merkel cell carcinoma. Clin Cancer Res. (2020) 26:2257–67. doi: 10.1158/1078-0432.CCR-19-2244
94. Tao Q, Du JX, Zhang S, Lin W, Luo Y, Liu Y, et al. Longitudinal multi-functional analysis identified responses of T cells, B cells, and monocytes as hallmarks of immunotherapy tolerance in patients with merkel cell carcinoma. PloS One. (2023) 18:e0293922. doi: 10.1371/journal.pone.0293922
95. Tabachnick-Cherny S, Pulliam T, Rodriguez HJ, Fan X, Hippe DS, Jones DC, et al. Characterization of immunosuppressive myeloid cells in Merkel cell carcinoma: correlation with resistance to PD-1 pathway blockade. Clin Cancer Res. (2023) 30(6):1189–99. doi: 10.1158/1078-0432.CCR-23-1957
96. Giraldo NA, Nguyen P, Engle EL, Kaunitz GJ, Cottrell TR, Berry S, et al. Multidimensional, quantitative assessment of PD-1/PD-L1 expression in patients with Merkel cell carcinoma and association with response to pembrolizumab. J Immunother Cancer. (2018) 6:99. doi: 10.1186/s40425-018-0404-0
97. Samimi M, Molet L, Fleury M, Laude H, Carlotti A, Gardair C, et al. Prognostic value of antibodies to Merkel cell polyomavirus T antigens and VP1 protein in patients with Merkel cell carcinoma. Br J Dermatol. (2016) 174:813–22. doi: 10.1111/bjd.2016.174.issue-4
98. Paulson KG, Lewis CW, Redman MW, Simonson WT, Lisberg A, Ritter D, et al. Viral oncoprotein antibodies as a marker for recurrence of Merkel cell carcinoma: A prospective validation study. Cancer. (2017) 123:1464–74. doi: 10.1002/cncr.30475
99. Akaike T, Hippe DS, So N, Maloney N, Gunnell L, Hall E, et al. Circulating tumor DNA reflects tumor burden and detects early recurrence in patients with Merkel cell carcinoma. presented at: International Society of Investigative Dermatology. Tokyo, Japan J Invest Dermatol. (2023) 143(5):S265. doi: 10.26226/m.64199a16337ccc00143861ee
100. Engels EA, Frisch M, Goedert JJ, Biggar RJ, Miller RW. Merkel cell carcinoma and HIV infection. Lancet. (2002) 359:497–8. doi: 10.1016/S0140-6736(02)07668-7
101. Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. (2008) 319:1096–100. doi: 10.1126/science.1152586
102. Goh G, Walradt T, Markarov V, Blom A, Riaz N, Doumani R, et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget. (2016) 7:3403–15. doi: 10.18632/oncotarget.v7i3
103. Moens U, Prezioso C, Pietropaolo V. Genetic diversity of the noncoding control region of the novel human polyomaviruses. Viruses. (2020) 12(12):1406. doi: 10.3390/v12121406
104. Becker JC, Stang A, DeCaprio JA, Cerroni L, Lebbe C, Veness M, et al. Merkel cell carcinoma. Nat Rev Dis Primers. (2017) 3:17077. doi: 10.1038/nrdp.2017.77
105. Yang JF, You J. Merkel cell polyomavirus and associated Merkel cell carcinoma. Tumour Virus Res. (2022) 13:200232. doi: 10.1016/j.tvr.2021.200232
106. Wan L, Toland S, Robinson-McCarthy LR, Lee N, Schaich MA, Hengel SR, et al. Unlicensed origin DNA melting by MCV and SV40 polyomavirus LT proteins is independent of ATP-dependent helicase activity. Proc Natl Acad Sci USA. (2023) 120:e2308010120. doi: 10.1073/pnas.2308010120
107. Wang R, Yang JF, Senay TE, Liu W, You J. Characterization of the impact of merkel cell polyomavirus-induced interferon signaling on viral infection. J Virol. (2023) 97:e0190722. doi: 10.1128/jvi.01907-22
108. Krump NA, Wang R, Liu W, Yang JF, Ma T, You J. Merkel cell polyomavirus infection induces an antiviral innate immune response in human dermal fibroblasts. J Virol. (2021) 95:e0221120. doi: 10.1128/JVI.02211-20
109. Kwun HJ, Shuda M, Feng H, Camacho CJ, Moore PS, Chang Y. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe. (2013) 14:125–35. doi: 10.1016/j.chom.2013.06.008
110. Pham AM, Ortiz LE, Lukacher AE, Kwun HJ. Merkel cell polyomavirus large T antigen induces cellular senescence for host growth arrest and viral genome persistence through its unique domain. Cells. (2023) 12(3):380. doi: 10.3390/cells12030380
111. Ortiz LE, Pham AM, Kwun HJ. Identification of the merkel cell polyomavirus large tumor antigen ubiquitin conjugation residue. Int J Mol Sci. (2021) 22(13):7169. doi: 10.3390/ijms22137169
112. Kwun HJ, Chang Y, Moore PS. Protein-mediated viral latency is a novel mechanism for Merkel cell polyomavirus persistence. Proc Natl Acad Sci USA. (2017) 114:E4040–7. doi: 10.1073/pnas.1703879114
113. Dye KN, Welcker M, Clurman BE, Roman A, Galloway DA. Merkel cell polyomavirus Tumor antigens expressed in Merkel cell carcinoma function independently of the ubiquitin ligases Fbw7 and beta-TrCP. PloS Pathog. (2019) 15:e1007543. doi: 10.1371/journal.ppat.1007543
114. Kwun HJ, Wendzicki JA, Shuda Y, Moore PS, Chang Y. Merkel cell polyomavirus small T antigen induces genome instability by E3 ubiquitin ligase targeting. Oncogene. (2017) 36:6838. doi: 10.1038/onc.2017.389
115. Shuda M, Feng H, Kwun HJ, Rosen ST, Gjoerup O, Moore PS, et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci USA. (2008) 105:16272–7. doi: 10.1073/pnas.0806526105
116. Moens U, Van Ghelue M, Johannessen M. Oncogenic potentials of the human polyomavirus regulatory proteins. Cell Mol Life Sci. (2007) 64:1656–78. doi: 10.1007/s00018-007-7020-3
117. Verhaegen ME, Mangelberger D, Harms PW, Vozheiko TD, Weick JW, Wilbert DM, et al. Merkel cell polyomavirus small T antigen is oncogenic in transgenic mice. J Invest Dermatol. (2015) 135:1415–24. doi: 10.1038/jid.2014.446
118. Verhaegen ME, Mangelberger D, Harms PW, Eberl M, Wilbert DM, Meireles J, et al. Merkel cell polyomavirus small T antigen initiates merkel cell carcinoma-like tumor development in mice. Cancer Res. (2017) 77:3151–7. doi: 10.1158/0008-5472.CAN-17-0035
119. Shuda M, Velasquez C, Cheng E, Cordek DG, Kwun HJ, Chang Y, et al. CDK1 substitutes for mTOR kinase to activate mitotic cap-dependent protein translation. Proc Natl Acad Sci USA. (2015) 112:5875–82. doi: 10.1073/pnas.1505787112
120. Cheng J, Park DE, Berrios C, White EA, Arora R, Yoon R, et al. Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PloS Pathog. (2017) 13:e1006668. doi: 10.1371/journal.ppat.1006668
121. Kwun HJ, Shuda M, Camacho CJ, Gamper AM, Thant M, Chang Y, et al. Restricted protein phosphatase 2A targeting by Merkel cell polyomavirus small T antigen. J Virol. (2015) 89:4191–200. doi: 10.1128/JVI.00157-15
122. Richards KF, Guastafierro A, Shuda M, Toptan T, Moore PS, Chang Y. Merkel cell polyomavirus T antigens promote cell proliferation and inflammatory cytokine gene expression. J Gen Virol. (2015) 96:3532–44. doi: 10.1099/jgv.0.000287
123. Fan K, Gravemeyer J, Ritter C, Rasheed K, Gambichler T, Moens U, et al. MCPyV large T antigen-induced atonal homolog 1 is a lineage-dependency oncogene in merkel cell carcinoma. J Invest Dermatol. (2020) 140:56–65 e3. doi: 10.1016/j.jid.2019.06.135
124. Harold A, Amako Y, Hachisuka J, Bai Y, Li MY, Kubat L, et al. Conversion of Sox2-dependent Merkel cell carcinoma to a differentiated neuron-like phenotype by T antigen inhibition. Proc Natl Acad Sci USA. (2019) 116:20104–14. doi: 10.1073/pnas.1907154116
125. Weber M, Nguyen MB, Li MY, Flora P, Shuda M, Ezhkova E. Merkel cell polyomavirus T antigen-mediated reprogramming in adult merkel cell progenitors. J Invest Dermatol. (2023) 143:2163–2176 e6. doi: 10.1016/j.jid.2023.04.031
126. Frost TC, Gartin AK, Liu M, Cheng J, Dharaneeswaran H, Keskin DB, et al. YAP1 and WWTR1 expression inversely correlates with neuroendocrine markers in Merkel cell carcinoma. J Clin Invest. (2023) 133(5):e157171. doi: 10.1172/JCI157171
127. Kwun HJ, Guastafierro A, Shuda M, Meinke G, Bohm A, Moore PS, et al. The minimum replication origin of merkel cell polyomavirus has a unique large T-antigen loading architecture and requires small T-antigen expression for optimal replication. J Virol. (2009) 83:12118–28. doi: 10.1128/JVI.01336-09
128. Li J, Wang X, Diaz J, Tsang SH, Buck CB, You J. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J Virol. (2013) 87:9173–88. doi: 10.1128/JVI.01216-13
129. Carter JJ, Daugherty MD, Qi X, Bheda-Malge A, Wipf GC, Robinson K, et al. Identification of an overprinting gene in Merkel cell polyomavirus provides evolutionary insight into the birth of viral genes. Proc Natl Acad Sci USA. (2013) 110:12744–9. doi: 10.1073/pnas.1303526110
130. Peng WY, Abere B, Shi H, Toland S, Smithgall TE, Moore PS, et al. Membrane-bound Merkel cell polyomavirus middle T protein constitutively activates PLCgamma1 signaling through Src-family kinases. Proc Natl Acad Sci USA. (2023) 120:e2316467120. doi: 10.1073/pnas.2316467120
131. Gjoerup O, Chang Y. Update on human polyomaviruses and cancer. Adv Cancer Res. (2010) 106:1–51. doi: 10.1016/S0065-230X(10)06001-X
132. Neu U, Hengel H, Blaum BS, Schowalter RM, Macejak D, Gilbert M, et al. Structures of Merkel cell polyomavirus VP1 complexes define a sialic acid binding site required for infection. PloS pathogens. (2012) 8:e1002738. doi: 10.1371/journal.ppat.1002738
133. Schowalter RM, Buck CB. The Merkel cell polyomavirus minor capsid protein. PloS pathogens. (2013) 9:e1003558. doi: 10.1371/journal.ppat.1003558
134. Lee S, Paulson KG, Murchison EP, Afanasiev OK, Alkan C, Leonard JH, et al. Identification and validation of a novel mature microRNA encoded by the Merkel cell polyomavirus in human Merkel cell carcinomas. J Clin Virol. (2011) 52:272–5. doi: 10.1016/j.jcv.2011.08.012
135. Shuda M, Kwun HJ, Feng H, Chang Y, Moore PS. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J Clin Invest. (2011) 121:3623–34. doi: 10.1172/JCI46323
136. Houben R, Adam C, Baeurle A, Hesbacher S, Grimm J, Angermeyer S, et al. An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. Int J Cancer J Int du Cancer. (2012) 130:847–56. doi: 10.1002/ijc.26076
137. Cheng J, Rozenblatt-Rosen O, Paulson KG, Nghiem P, DeCaprio JA. Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. J Virol. (2013) 87:6118–26. doi: 10.1128/JVI.00385-13
138. Spurgeon ME, Cheng J, Bronson RT, Lambert PF, DeCaprio JA. Tumorigenic activity of merkel cell polyomavirus T antigens expressed in the stratified epithelium of mice. Cancer Res. (2015) 75:1068–79. doi: 10.1158/0008-5472.CAN-14-2425
139. Shuda M, Guastafierro A, Geng X, Shuda Y, Ostrowski SM, Lukianov S, et al. Merkel cell polyomavirus small T antigen induces cancer and embryonic merkel cell proliferation in a transgenic mouse model. PloS One. (2015) 10:e014232: doi: 10.1371/journal.pone.0142329
140. Nwogu N, Ortiz LE, Kwun HJ. Surface charge of Merkel cell polyomavirus small T antigen determines cell transformation through allosteric FBW7 WD40 domain targeting. Oncogenesis. (2020) 9:53. doi: 10.1038/s41389-020-0235-y
141. Houben R, Shuda M, Weinkam R, Schrama D, Feng H, Chang Y, et al. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J Virol. (2010) 84:7064–72. doi: 10.1128/JVI.02400-09
142. Shuda M, Chang Y, Moore PS. Merkel cell polyomavirus-positive Merkel cell carcinoma requires viral small T-antigen for cell proliferation. J Invest Dermatol. (2014) 134:1479–81. doi: 10.1038/jid.2013.483
143. Khalili K, Sariyer IK, Safak M. Small tumor antigen of polyomaviruses: role in viral life cycle and cell transformation. J Cell Physiol. (2008) 215:309–19. doi: 10.1002/jcp.21326
144. Ahmed MM, Cushman CH, DeCaprio JA. Merkel cell polyomavirus: oncogenesis in a stable genome. Viruses. (2021) 14(1):58. doi: 10.3390/v14010058
145. Houben R, Angermeyer S, Haferkamp S, Aue A, Goebeler M, Schrama D, et al. Characterization of functional domains in the Merkel cell polyoma virus Large T antigen. Int J Cancer J Int du Cancer. (2015) 136:E290–300. doi: 10.1002/ijc.29200
146. Starrett GJ, Thakuria M, Chen T, Marcelus C, Cheng J, Nomburg J, et al. Clinical and molecular characterization of virus-positive and virus-negative Merkel cell carcinoma. Genome Med Mar 18. (2020) 12:30. doi: 10.1186/s13073-020-00727-4
147. Schlemeyer T, Ohnezeit D, Virdi S, Korner C, Weisselberg S, Starzonek S, et al. Merkel cell carcinoma and immune evasion: merkel cell polyomavirus small T-antigen−Induced surface changes can be reverted by therapeutic intervention. J Invest Dermatol. (2022) 142:3071–3081.e13. doi: 10.1016/j.jid.2022.04.029
148. Griffiths DA, Abdul-Sada H, Knight LM, Jackson BR, Richards K, Prescott EL, et al. Merkel cell polyomavirus small T antigen targets the NEMO adaptor protein to disrupt inflammatory signaling. J Virol. (2013) 87:13853–67. doi: 10.1128/JVI.02159-13
149. Park DE, Cheng J, Berrios C, Montero J, Cortes-Cros M, Ferretti S, et al. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc Natl Acad Sci USA. (2019) 116:1027–32. doi: 10.1073/pnas.1818798116
150. Park DE, Cheng J, McGrath JP, Lim MY, Cushman C, Swanson SK, et al. Merkel cell polyomavirus activates LSD1-mediated blockade of non-canonical BAF to regulate transformation and tumorigenesis. Nat Cell Biol. (2020) 22:603–15. doi: 10.1038/s41556-020-0503-2
151. Knight LM, Stakaityte G, Wood JJ, Abdul-Sada H, Griffiths DA, Howell GJ, et al. Merkel cell polyomavirus small T antigen mediates microtubule destabilization to promote cell motility and migration. J Virol. (2015) 89:35–47. doi: 10.1128/JVI.02317-14
152. Stakaityte G, Nwogu N, Dobson SJ, Knight LM, Wasson CW, Salguero FJ, et al. Merkel cell polyomavirus small T antigen drives cell motility via rho-GTPase-induced filopodium formation. J Virol. (2018) 92. doi: 10.1128/JVI.00940-17
153. Nwogu N, Boyne JR, Dobson SJ, Poterlowicz K, Blair GE, Macdonald A, et al. Cellular sheddases are induced by Merkel cell polyomavirus small tumour antigen to mediate cell dissociation and invasiveness. PloS Pathog. (2018) 14:e1007276. doi: 10.1371/journal.ppat.1007276
154. Lanclos N, Radulovic P, Bland J, Oganisyan V, Radefeld K, Uversky VN. Implications of intrinsic disorder and functional proteomics in the merkel cell polyomavirus life cycle. J Cell Biochem. (2023). doi: 10.1002/jcb.30485
155. Cimino PJ, Robirds DH, Tripp SR, Pfeifer JD, Abel HJ, Duncavage EJ. Retinoblastoma gene mutations detected by whole exome sequencing of Merkel cell carcinoma. Modern Pathol. (2014) 27(8):1073–87. doi: 10.1038/modpathol.2013.235
156. Van Gele M, Kaghad M, Leonard JH, Van Roy N, Naeyaert JM, Geerts ML, et al. Mutation analysis of P73 and TP53 in Merkel cell carcinoma. Br J Cancer. (2000) 82:823–6. doi: 10.1054/bjoc.1999.1006
157. Nardi V, Song Y, Santamaria-Barria JA, Cosper AK, Lam Q, Faber AC, et al. Activation of PI3K signaling in Merkel cell carcinoma. Clin Cancer Res. (2012) 18:1227–36. doi: 10.1158/1078-0432.CCR-11-2308
158. Popp S, Waltering S, Herbst C, Moll I, Boukamp P. UV-B-type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int J Cancer J Int du Cancer. (002) 99:352–60. doi: 10.1002/ijc.10321
159. Pulitzer MP, Brannon AR, Berger MF, Louis P, Scott SN, Jungbluth AA, et al. Cutaneous squamous and neuroendocrine carcinoma: genetically and immunohistochemically different from Merkel cell carcinoma. Modern Pathol. (2015) 28(8):1023–32. doi: 10.1038/modpathol.2015.60
160. Robb TJ, Ward Z, Houseman P, Woodhouse B, Patel R, Fitzgerald S, et al. Chromosomal aberrations accumulate during metastasis of virus-negative merkel cell carcinoma. J Invest Dermatol. (2023) 143:1168–1177 e2. doi: 10.1016/j.jid.2023.01.015
161. Harms KL, Lazo de la Vega L, Hovelson DH, Rahrig S, Cani AK, Liu CJ, et al. Molecular profiling of multiple primary merkel cell carcinoma to distinguish genetically distinct tumors from clonally related metastases. JAMA Dermatol. (2017) 153:505–12. doi: 10.1001/jamadermatol.2017.0507
162. Hafner C, Houben R, Baeurle A, Ritter C, Schrama D, Landthaler M, et al. Activation of the PI3K/AKT pathway in Merkel cell carcinoma. PloS One. (2012) 7:e31255. doi: 10.1371/journal.pone.0031255
163. Veija T, Sarhadi VK, Koljonen V, Bohling T, Knuutila S. Hotspot mutations in polyomavirus positive and negative Merkel cell carcinomas. Cancer Genet. (2016) 209:30–5. doi: 10.1016/j.cancergen.2015.11.006
164. Harms PW, Patel RM, Verhaegen ME, Giordano TJ, Nash KT, Johnson CN, et al. Distinct gene expression profiles of viral- and nonviral-associated merkel cell carcinoma revealed by transcriptome analysis. J Invest Dermatol. (2013) 133:936–45. doi: 10.1038/jid.2012.445
165. Karpinski P, Rosales I, Laczmanski L, Kowalik A, Wenson S, Hoang MP. Expression of genes associated with epithelial-mesenchymal transition in merkel cell polyomavirus-negative merkel cell carcinoma. Lab Invest. (2023) 103:100177. doi: 10.1016/j.labinv.2023.100177
166. Starrett GJ, Marcelus C, Cantalupo PG, Katz JP, Cheng J, Akagi K, et al. Merkel cell polyomavirus exhibits dominant control of the tumor genome and transcriptome in virus-associated merkel cell carcinoma. mBio. (2017) 8(1):e02079-16. doi: 10.1128/mBio.02079-16
167. Sundqvist BZ, Kilpinen SK, Bohling TO, Koljonen VSK, Sihto HJ. Transcriptomic analyses reveal three distinct molecular subgroups of Merkel cell carcinoma with differing prognoses. Int J Cancer J Int du Cancer. (2023) 152:2099–108. doi: 10.1002/ijc.34425
168. Torre-Castro J, Rodriguez M, Alonso-Alonso R, Mendoza Cembranos MD, Diaz-Alejo JF, Rebollo-Gonzalez M, et al. LT and SOX9 expression are associated with gene sets that distinguish Merkel cell polyomavirus-positive and -negative Merkel cell carcinoma. Br J Dermatol. (2024) 190(6):876–84. doi: 10.1093/bjd/ljae033
169. Jones PA, Baylin SB. The epigenomics of cancer. Cell. (2007) 128:683–92. doi: 10.1016/j.cell.2007.01.029
170. Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. (2017) 357(6348):eaal2380. doi: 10.1126/science.aal2380
171. Stirzaker C, Taberlay PC, Statham AL, Clark SJ. Mining cancer methylomes: prospects and challenges. Trends Genet. (2014) 30:75–84. doi: 10.1016/j.tig.2013.11.004
172. Carter MD, Gaston D, Huang WY, Greer WL, Pasternak S, Ly TY, et al. Genetic profiles of different subsets of Merkel cell carcinoma show links between combined and pure MCPyV-negative tumors. Hum Pathol. (2018) 71:117–25. doi: 10.1016/j.humpath.2017.10.014
173. Harms PW, Verhaegen ME, Hu K, Hrycaj SM, Chan MP, Liu CJ, et al. Genomic evidence suggests that cutaneous neuroendocrine carcinomas can arise from squamous dysplastic precursors. Modern Pathol. (2022) 35:506–14. doi: 10.1038/s41379-021-00928-1
174. Lao VV, Grady WM. Epigenetics and colorectal cancer. Nat Rev Gastroenterol Hepatol. (2011) 8:686–700. doi: 10.1038/nrgastro.2011.173
175. Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. (2009) 41:178–86. doi: 10.1038/ng.298
176. Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. (2007) 39:457–66. doi: 10.1038/ng1990
177. Mancarella D, Plass C. Epigenetic signatures in cancer: proper controls, current challenges and the potential for clinical translation. Genome Med. (2021) 13:23. doi: 10.1186/s13073-021-00837-7
178. Nunes SP, Henrique R, Jeronimo C, Paramio JM. DNA methylation as a therapeutic target for bladder cancer. Cells. (2020) 9(8):1850. doi: 10.3390/cells9081850
179. Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol. (2016) 8(9):a019505. doi: 10.1101/cshperspect.a019505
180. Haag T, Richter AM, Schneider MB, Jimenez AP, Dammann RH. The dual specificity phosphatase 2 gene is hypermethylated in human cancer and regulated by epigenetic mechanisms. BMC Cancer. (2016) 16:49. doi: 10.1186/s12885-016-2087-6
181. Richter AM, Haag T, Walesch S, Herrmann-Trost P, Marsch WC, Kutzner H, et al. Aberrant promoter hypermethylation of RASSF family members in merkel cell carcinoma. Cancers. (2013) 5:1566–76. doi: 10.3390/cancers5041566
182. Improta G, Ritter C, Pettinato A, Vasta V, Schrama D, Fraggetta F, et al. MGMT promoter methylation status in Merkel cell carcinoma: in vitro versus in vivo. J Cancer Res Clin Oncol. (2017) 143:1489–97. doi: 10.1007/s00432-017-2413-7
183. Lassacher A, Heitzer E, Kerl H, Wolf P. p14ARF hypermethylation is common but INK4a-ARF locus or p53 mutations are rare in Merkel cell carcinoma. J Invest Dermatol. (2008) 128:1788–96. doi: 10.1038/sj.jid.5701256
184. Sahi H, Savola S, Sihto H, Koljonen V, Bohling T, Knuutila S. RB1 gene in Merkel cell carcinoma: hypermethylation in all tumors and concurrent heterozygous deletions in the polyomavirus-negative subgroup. APMIS: Acta pathologica microbiologica immunologica Scandinavica. (2014) 122:1157–66. doi: 10.1111/apm.2014.122.issue-12
185. Talsma CE, Wang M, Andea AA, Fullen DR, Harms PW, Chan MP. Expression of p16 in Merkel cell carcinoma. J cutaneous Pathol. (2021) 48:455–7. doi: 10.1111/cup.13790
186. Gambichler T, Dreissigacker M, Kasakovski D, Skrygan M, Wieland U, Silling S, et al. Patched 1 expression in Merkel cell carcinoma. J Dermatol. (2021) 48:64–74. doi: 10.1111/1346-8138.15611
187. Wang X, Yang X, Zhang C, Wang Y, Cheng T, Duan L, et al. Tumor cell-intrinsic PD-1 receptor is a tumor suppressor and mediates resistance to PD-1 blockade therapy. Proc Natl Acad Sci USA. (2020) 117:6640–50. doi: 10.1073/pnas.1921445117
188. Ricci C, Morandi L, Righi A, Gibertoni D, Maletta F, Ambrosi F, et al. PD-1 (PDCD1) promoter methylation in Merkel cell carcinoma: prognostic relevance and relationship with clinico-pathological parameters. Modern Pathol. (2019) 32:1359–72. doi: 10.1038/s41379-019-0261-5
189. Harms PW, Verhaegen ME, Vo JN, Tien JC, Pratt D, Su F, et al. Viral status predicts patterns of genome methylation and decitabine response in merkel cell carcinoma. J Invest Dermatol. (2021) 142(3 Pt A):641–52. doi: 10.1016/j.jid.2021.07.173
190. Gravemeyer J, Spassova I, Verhaegen ME, Dlugosz AA, Hoffmann D, Lange A, et al. DNA-methylation patterns imply a common cellular origin of virus- and UV-associated Merkel cell carcinoma. Oncogene. (2022) 41:37–45. doi: 10.1038/s41388-021-02064-1
191. Gujar H, Mehta A, Li HT, Tsai YC, Qiu X, Weisenberger DJ, et al. Characterizing DNA methylation signatures and their potential functional roles in Merkel cell carcinoma. Genome Med. (2021) 13:130. doi: 10.1186/s13073-021-00946-3
192. Chteinberg E, Vogt J, Kolarova J, Bormann F, van den Oord J, Speel EJ, et al. The curious case of Merkel cell carcinoma: epigenetic youth and lack of pluripotency. Epigenetics. (2020) 15:1319–24. doi: 10.1080/15592294.2020.1773096
193. Kuriyama Y, Shimizu A, Kim J, Yasuda M, Motegi SI. The differential expression of long interspersed nuclear elements-1 as a marker for hypomethylation in Merkel cell carcinoma. Clin Exp Dermatol. (2022) 47:1726–8. doi: 10.1111/ced.15265
194. Li W, Xu L. Epigenetic function of TET family, 5-methylcytosine, and 5-hydroxymethylcytosine in hematologic Malignancies. Oncol Res Treat. (2019) 42:309–18. doi: 10.1159/000498947
195. Gambichler T, Schmitt K, Ruddel I, Dreibigacker M, Stockfleth E, Becker JC. Decreased 5-hydroxymethylcytosine immunoreactivity in primary Merkel cell carcinoma is a strong predictor for disease-specific death. Br J Dermatol. (2019) 181:389–90. doi: 10.1111/bjd.17705
196. Mazziotta C, Lanzillotti C, Gafa R, Touze A, Durand MA, Martini F, et al. The role of histone post-translational modifications in merkel cell carcinoma. Front Oncol. (2022) 12:832047. doi: 10.3389/fonc.2022.832047
198. Morschhauser F, Tilly H, Chaidos A, McKay P, Phillips T, Assouline S, et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. (2020) 21:1433–42. doi: 10.1016/S1470-2045(20)30441-1
199. Gounder M, Schoffski P, Jones RL, Agulnik M, Cote GM, Villalobos VM, et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: an international, open-label, phase 2 basket study. Lancet Oncol. (2020) 21:1423–32. doi: 10.1016/S1470-2045(20)30451-4
200. Bardot ES, Valdes VJ, Zhang J, Perdigoto CN, Nicolis S, Hearn SA, et al. Polycomb subunits Ezh1 and Ezh2 regulate the Merkel cell differentiation program in skin stem cells. EMBO J. (2013) 32:1990–2000. doi: 10.1038/emboj.2013.110
201. Busam KJ, Pulitzer MP, Coit DC, Arcila M, Leng D, Jungbluth AA, et al. Reduced H3K27me3 expression in Merkel cell polyoma virus-positive tumors. Modern Pathol. (2017) 30:877–83. doi: 10.1038/modpathol.2017.8
202. Marchione DM, Lisby A, Viaene AN, Santi M, Nasrallah M, Wang LP, et al. Histone H3K27 dimethyl loss is highly specific for Malignant peripheral nerve sheath tumor and distinguishes true PRC2 loss from isolated H3K27 trimethyl loss. Modern Pathol. (2019) 32:1434–46. doi: 10.1038/s41379-019-0287-8
203. Matsushita M, Iwasaki T, Wardhani LO, Kuwamoto S, Nonaka D, Nagata K, et al. Decreased H3K27me3 expression is associated with merkel cell polyomavirus-negative merkel cell carcinoma, especially combined with cutaneous squamous cell carcinoma. Anticancer Res. (2019) 39:5573–9. doi: 10.21873/anticanres.13751
204. Krajisnik A, Rezaee N, Crystal J, Duncan ER, Balzer BL, Frishberg DP, et al. The intricate relationship between H3K27 trimethylation and merkel cell polyomavirus status in merkel cell carcinoma. Am J dermatopathol. (2023) 45:783–5. doi: 10.1097/DAD.0000000000002541
205. Veija T, Koljonen V, Bohling T, Kero M, Knuutila S, Sarhadi VK. Aberrant expression of ALK and EZH2 in Merkel cell carcinoma. BMC Cancer. (2017) 17:236. doi: 10.1186/s12885-017-3233-5
206. Harms KL, Chubb H, Zhao L, Fullen DR, Bichakjian CK, Johnson TM, et al. Increased expression of EZH2 in Merkel cell carcinoma is associated with disease progression and poorer prognosis. Hum Pathol. (2017) 67:78–84. doi: 10.1016/j.humpath.2017.07.009
207. Acikalin A, Bagir E, Paydas S. Prognostic and predictive value of EZH2 expression and the tumor immune microenvironment in Merkel cell carcinoma. Pol J Pathol. (2021) 72:140–7. doi: 10.5114/pjp.2021.109516
208. Durand MA, Drouin A, Mouchard A, Durand L, Esnault C, Berthon P, et al. Distinct regulation of EZH2 and its repressive H3K27me3 mark in polyomavirus-positive and -negative merkel cell carcinoma. J Invest Dermatol. (2023) 143:1937–1946 e7. doi: 10.1016/j.jid.2023.02.038
209. Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. (2003) 22:5323–35. doi: 10.1093/emboj/cdg542
210. Gartin AK, Frost TC, Cushman CH, Leeper BA, Gokhale PC, DeCaprio JA. Merkel cell carcinoma sensitivity to EZH2 inhibition is mediated by SIX1 derepression. J Invest Dermatol. (2022) 142:2783–2792.e15. doi: 10.1016/j.jid.2022.03.008
211. Khattri M, Amako Y, Gibbs JR, Collura JL, Arora R, Harold A, et al. Methyltransferase-independent function of enhancer of zeste homologue 2 maintains tumorigenicity induced by human oncogenic papillomavirus and polyomavirus. Tumour Virus Res. (2023) 16:200264. doi: 10.1016/j.tvr.2023.200264
212. Paulson KG, Lemos BD, Feng B, Jaimes N, Penas PF, Bi X, et al. Array-CGH reveals recurrent genomic changes in Merkel cell carcinoma including amplification of L-Myc. J Invest Dermatol. (2009) 129:1547–55. doi: 10.1038/jid.2008.365
213. Leiendecker L, Jung PS, Krecioch I, Neumann T, Schleiffer A, Mechtler K, et al. LSD1 inhibition induces differentiation and cell death in Merkel cell carcinoma. EMBO Mol Med. (2020) 12:e12525. doi: 10.15252/emmm.202012525
214. Sengupta D, Kannan A, Kern M, Moreno MA, Vural E, Stack B Jr, et al. Disruption of BRD4 at H3K27Ac-enriched enhancer region correlates with decreased c-Myc expression in Merkel cell carcinoma. Epigenetics. (2015) 10:460–6. doi: 10.1080/15592294.2015.1034416
215. Choi JE, Verhaegen ME, Yazdani S, Malik R, Harms PW, Mangelberger D, et al. Characterizing the therapeutic potential of a potent BET degrader in merkel cell carcinoma. Neoplasia. (2019) 21:322–30. doi: 10.1016/j.neo.2019.01.003
216. Shao Q, Kannan A, Lin Z, Stack BC Jr., Suen JY, Gao L. BET protein inhibitor JQ1 attenuates Myc-amplified MCC tumor growth in vivo. Cancer Res. (2014) 74:7090–102. doi: 10.1158/0008-5472.CAN-14-0305
217. Orouji E, Peitsch WK, Orouji A, Houben R, Utikal J. Unique role of histone methyltransferase PRDM8 in the tumorigenesis of virus-negative merkel cell carcinoma. Cancers. (2020) 12(4):1057. doi: 10.3390/cancers12041057
218. Wu JH, Narayanan D, Limmer AL, Simonette RA, Rady PL, Tyring SK. Merkel cell polyomavirus small T antigen induces DNA damage response. Intervirology. (2019) 62:96–100. doi: 10.1159/000501419
219. Ritter C, Fan K, Paschen A, Reker Hardrup S, Ferrone S, Nghiem P, et al. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci Rep. (2017) 7:2290. doi: 10.1038/s41598-017-02608-0
220. Ugurel S, Spassova I, Wohlfarth J, Drusio C, Cherouny A, Melior A, et al. MHC class-I downregulation in PD-1/PD-L1 inhibitor refractory Merkel cell carcinoma and its potential reversal by histone deacetylase inhibition: a case series. Cancer Immunol Immunother. (2019) 68:983–90. doi: 10.1007/s00262-019-02341-9
221. Song L, Bretz AC, Gravemeyer J, Spassova I, Muminova S, Gambichler T, et al. The HDAC inhibitor domatinostat promotes cell-cycle arrest, induces apoptosis, and increases immunogenicity of merkel cell carcinoma cells. J Invest Dermatol. (2021) 141:903–912 e4. doi: 10.1016/j.jid.2020.08.023
222. Ritter C, Fan K, Paulson KG, Nghiem P, Schrama D, Becker JC. Reversal of epigenetic silencing of MHC class I chain-related protein A and B improves immune recognition of Merkel cell carcinoma. Sci Rep. (2016) 6:21678. doi: 10.1038/srep21678
223. Durand MA, Drouin A, Bachiri K, Durand L, Berthon P, Houben R, et al. Enhancer of zeste homolog 2 inhibition induces HLA class I re-expression in merkel cell carcinoma. J Invest Dermatol. (2023) 144(6):1398–401.e1. doi: 10.1016/j.jid.2023.10.036
224. Mazziotta C, Cervellera CF, Lanzillotti C, Touze A, Gaboriaud P, Tognon M, et al. MicroRNA dysregulations in Merkel cell carcinoma: Molecular mechanisms and clinical applications. J Med Virol. (2023) 95:e2837: doi: 10.1002/jmv.28375
225. Ning MS, Kim AS, Prasad N, Levy SE, Zhang H, Andl T. Characterization of the merkel cell carcinoma miRNome. J skin cancer. (2014) 2014:289548. doi: 10.1155/2014/289548
226. Xie H, Lee L, Caramuta S, Hoog A, Browaldh N, Bjornhagen V, et al. MicroRNA expression patterns related to merkel cell polyomavirus infection in human merkel cell carcinoma. J Invest Dermatol. (2014) 134:507–17. doi: 10.1038/jid.2013.355
227. Veija T, Sahi H, Koljonen V, Bohling T, Knuutila S, Mosakhani N. miRNA-34a underexpressed in Merkel cell polyomavirus-negative Merkel cell carcinoma. Virchows Arch. (2015) 466:289–95. doi: 10.1007/s00428-014-1700-9
228. Kumar S, Xie H, Shi H, Gao J, Juhlin CC, Bjornhagen V, et al. Merkel cell polyomavirus oncoproteins induce microRNAs that suppress multiple autophagy genes. Int J Cancer J Int du Cancer. (2020) 146:1652–66. doi: 10.1002/ijc.32503
229. Renwick N, Cekan P, Masry PA, McGeary SE, Miller JB, Hafner M, et al. Multicolor microRNA FISH effectively differentiates tumor types. J Clin Invest. (2013) 123:2694–702. doi: 10.1172/JCI68760
230. Fan K, Ritter C, Nghiem P, Blom A, Verhaegen ME, Dlugosz A, et al. Circulating cell-free miR-375 as surrogate marker of tumor burden in merkel cell carcinoma. Clin Cancer Res. (2018) 24:5873–82. doi: 10.1158/1078-0432.CCR-18-1184
231. Kumar S, Xie H, Scicluna P, Lee L, Bjornhagen V, Hoog A, et al. MiR-375 regulation of LDHB plays distinct roles in polyomavirus-positive and -negative merkel cell carcinoma. Cancers. (2018) 10(11):443. doi: 10.3390/cancers10110443
232. Abraham KJ, Zhang X, Vidal R, Pare GC, Feilotter HE, Tron VA. Roles for miR-375 in Neuroendocrine Differentiation and Tumor Suppression via Notch Pathway Suppression in Merkel Cell Carcinoma. Am J Pathol. (2016) 186:1025–35. doi: 10.1016/j.ajpath.2015.11.020
233. Fan K, Spassova I, Gravemeyer J, Ritter C, Horny K, Lange A, et al. Merkel cell carcinoma-derived exosome-shuttle miR-375 induces fibroblast polarization by inhibition of RBPJ and p53. Oncogene. (2021) 40:980–96. doi: 10.1038/s41388-020-01576-6
234. Fan K, Zebisch A, Horny K, Schrama D, Becker JC. Highly Expressed miR-375 is not an Intracellular Oncogene in Merkel Cell Polyomavirus-Associated Merkel Cell Carcinoma. Cancers. (2020) 12(3):529. doi: 10.3390/cancers12030529
235. Seo GJ, Chen CJ, Sullivan CS. Merkel cell polyomavirus encodes a microRNA with the ability to autoregulate viral gene expression. Virol Jan 20. (2009) 383:183–7. doi: 10.1016/j.virol.2008.11.001
236. Theiss JM, Gunther T, Alawi M, Neumann F, Tessmer U, Fischer N, et al. A comprehensive analysis of replicating merkel cell polyomavirus genomes delineates the viral transcription program and suggests a role for mcv-miR-M1 in episomal persistence. PloS Pathog. (2015) 11:e1004974. doi: 10.1371/journal.ppat.1004974
237. Toker C. Trabecular carcinoma of the skin. A question of title. Am J dermatopathol. (1982) 4:497–500. doi: 10.1097/00000372-198212000-00003
238. Rywlin AM. Malignant Merkel-cell tumor is a more accurate description than trabecular carcinoma. Am J dermatopathol. (1982) 4:513–5. doi: 10.1097/00000372-198212000-00007
239. Sunshine JC, Jahchan NS, Sage J, Choi J. Are there multiple cells of origin of Merkel cell carcinoma? Oncogene. (2018) 37:1409–16. doi: 10.1038/s41388-017-0073-3
240. Tilling T, Moll I. Which are the cells of origin in merkel cell carcinoma? J skin cancer. (2012) 2012:680410. doi: 10.1155/2012/680410
241. Liu W, Yang R, Payne AS, Schowalter RM, Spurgeon ME, Lambert PF, et al. Identifying the target cells and mechanisms of merkel cell polyomavirus infection. Cell Host Microbe. (2016) 19:775–87. doi: 10.1016/j.chom.2016.04.024
242. Verhaegen ME, Harms PW, Van Goor JJ, Arche J, Patrick MT, Wilbert D, et al. Direct cellular reprogramming enables development of viral T antigen-driven Merkel cell carcinoma in mice. J Clin Invest. (2022) 132(7):e152069. doi: 10.1172/JCI152069
243. Milholland B, Dong X, Zhang L, Hao X, Suh Y, Vijg J. Differences between germline and somatic mutation rates in humans and mice. Nat Commun. (2017) 8:15183. doi: 10.1038/ncomms15183
244. Sauer CM, Haugg AM, Chteinberg E, Rennspiess D, Winnepenninckx V, Speel EJ, et al. Reviewing the current evidence supporting early B-cells as the cellular origin of Merkel cell carcinoma. Crit Rev Oncol Hematol. (2017) 116:99–105. doi: 10.1016/j.critrevonc.2017.05.009
245. Ostrowski SM, Wright MC, Bolock AM, Geng X, Maricich SM. Ectopic Atoh1 expression drives Merkel cell production in embryonic, postnatal and adult mouse epidermis. Development. (2015) 142:2533–44. doi: 10.1242/dev.123141
246. Helms AW, Abney AL, Ben-Arie N, Zoghbi HY, Johnson JE. Autoregulation and multiple enhancers control Math1 expression in the developing nervous system. Development. (2000) 127:1185–96. doi: 10.1242/dev.127.6.1185
247. Nemeth K, Gorog A, Mezey E, Pinter D, Kuroli E, Harsing J, et al. Cover Image: Detection of hair follicle-associated Merkel cell polyomavirus in an immunocompromised host with follicular spicules and alopecia. Br J Dermatol. (2016) 175:1409. doi: 10.1111/bjd.15039
248. Kervarrec T, Appenzeller S, Samimi M, Sarma B, Sarosi EM, Berthon P, et al. Merkel cell polyomavirusNegative merkel cell carcinoma originating from in situ squamous cell carcinoma: A keratinocytic tumor with neuroendocrine differentiation. J Invest Dermatol. (2022) 142:516–27. doi: 10.1016/j.jid.2021.07.175
249. DeCoste RC, Walsh NM, Gaston D, Ly TY, Pasternak S, Cutler S, et al. RB1-deficient squamous cell carcinoma: the proposed source of combined Merkel cell carcinoma. Modern Pathol. (2022). doi: 10.1038/s41379-022-01151-2
250. Pacaud M, Kervarrec T, Masliah-Planchon J, Tallet A, Collin C, Guyetant S, et al. Merkel cell carcinoma from renal transplant recipients are mostly MCPyV-negative and are frequently associated with squamous cell carcinomas or precursors. J Eur Acad Dermatol Venereol. (2023). doi: 10.1111/jdv.19076
252. Becker JC, Ugurel S, Leiter U, Meier F, Gutzmer R, Haferkamp S, et al. Adjuvant immunotherapy with nivolumab versus observation in completely resected Merkel cell carcinoma (ADMEC-O): disease-free survival results from a randomised, open-label, phase 2 trial. Lancet. (2023) 402:798–808. doi: 10.1016/S0140-6736(23)00769-9
253. Teke ME, Rossi AJ, Hernandez JM, Gastman B. Pembrolizumab compared with standard-of-care observation in treating patients with completely resected stage I-III merkel cell cancer (STAMP). Ann Surg Oncol. (2022) 6:3379–80. doi: 10.1245/s10434-022-11498-0
254. Kim S, Wuthrick E, Blakaj D, Eroglu Z, Verschraegen C, Thapa R, et al. Combined nivolumab and ipilimumab with or without stereotactic body radiation therapy for advanced Merkel cell carcinoma: a randomised, open label, phase 2 trial. Lancet. (2022) 400:1008–19. doi: 10.1016/S0140-6736(22)01659-2
255. Glutsch V, Schummer P, Kneitz H, Gesierich A, Goebeler M, Klein D, et al. Ipilimumab plus nivolumab in avelumab-refractory Merkel cell carcinoma: a multicenter study of the prospective skin cancer registry ADOREG. J Immunother Cancer. (2022) 10(11):e005930. doi: 10.1136/jitc-2022-005930
256. Shalhout SZ, Emerick KS, Kaufman HL, Silk AW, Thakuria M, Miller DM. A retrospective study of ipilimumab plus nivolumab in anti-PD-L1/PD-1 refractory merkel cell carcinoma. J Immunother. (2022) 45:299–302. doi: 10.1097/CJI.0000000000000432
257. LoPiccolo J, Schollenberger MD, Dakhil S, Rosner S, Ali O, Sharfman WH, et al. Rescue therapy for patients with anti-PD-1-refractory Merkel cell carcinoma: a multicenter, retrospective case series. J Immunother Cancer. (2019) 7:170. doi: 10.1186/s40425-019-0661-6
258. Ellingsen EB, O’Day S, Mezheyeuski A, Gromadka A, Clancy T, Kristedja TS, et al. Clinical activity of combined telomerase vaccination and pembrolizumab in advanced melanoma: results from a phase I trial. Clin Cancer Res. (2023) 29:3026–36. doi: 10.1158/1078-0432.CCR-23-0416
259. Celikdemir B, Houben R, Kervarrec T, Samimi M, Schrama D. Current and preclinical treatment options for Merkel cell carcinoma. Expert Opin Biol Ther. (2023) 23:1015–34. doi: 10.1080/14712598.2023.2257603
260. Gomez B, He L, Tsai YC, Wu TC, Viscidi RP, Hung CF. Creation of a Merkel cell polyomavirus small T antigen-expressing murine tumor model and a DNA vaccine targeting small T antigen. Cell Biosci. (2013) 3:29. doi: 10.1186/2045-3701-3-29
261. Zeng Q, Gomez BP, Viscidi RP, Peng S, He L, Ma B, et al. Development of a DNA vaccine targeting Merkel cell polyomavirus. Vaccine. (2012) 30:1322–9. doi: 10.1016/j.vaccine.2011.12.072
262. Gomez BP, Wang C, Viscidi RP, Peng S, He L, Wu TC, et al. Strategy for eliciting antigen-specific CD8+ T cell-mediated immune response against a cryptic CTL epitope of merkel cell polyomavirus large T antigen. Cell Biosci. (2012) 2:36. doi: 10.1186/2045-3701-2-36
263. Xu D, Jiang S, He Y, Jin X, Zhao G, Wang B. Development of a therapeutic vaccine targeting Merkel cell polyomavirus capsid protein VP1 against Merkel cell carcinoma. NPJ Vaccines. (2021) 6:119. doi: 10.1038/s41541-021-00382-9
264. Shalhout SZ, Miller DM, Emerick KS, Kaufman HL. Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol. (2023) 20:160–77. doi: 10.1038/s41571-022-00719-w
265. Ananthapadmanabhan V, Frost TC, Soroko KM, Knott A, Magliozzi BJ, Gokhale PC, et al. Milademetan is a highly potent MDM2 inhibitor in Merkel cell carcinoma. JCI Insight. (2022) 7(13):e160513. doi: 10.1172/jci.insight.160513
266. Feng Y, Song X, Jia R. Case report: favorable response to the tyrosine kinase inhibitor apatinib in recurrent merkel cell carcinoma. Front Oncol. (2021) 11:625360. doi: 10.3389/fonc.2021.625360
267. Davids MS, Charlton A, Ng SS, Chong ML, Laubscher K, Dar M, et al. Response to a novel multitargeted tyrosine kinase inhibitor pazopanib in metastatic Merkel cell carcinoma. J Clin Oncol. (2009) 27:e97–100. doi: 10.1200/JCO.2009.21.8149
268. Tarabadkar ES, Thomas H, Blom A, Parvathaneni U, Olencki T, Nghiem P, et al. Clinical benefit from tyrosine kinase inhibitors in metastatic merkel cell carcinoma: A case series of 5 patients. Am J Case Rep. (2018) 19:505–11. doi: 10.12659/AJCR.908649
269. Maroto P, Porta C, Capdevila J, Apolo AB, Viteri S, Rodriguez-Antona C, et al. Cabozantinib for the treatment of solid tumors: a systematic review. Ther Adv Med Oncol. (2022) 14:17588359221107112. doi: 10.1177/17588359221107112
270. Rabinowits G, Lezcano C, Catalano PJ, McHugh P, Becker H, Reilly MM, et al. Cabozantinib in patients with advanced merkel cell carcinoma. Oncologist. (2018) 23:814–21. doi: 10.1634/theoncologist.2017-0552
271. Su LD, Fullen DR, Lowe L, Uherova P, Schnitzer B, Valdez R. CD117 (KIT receptor) expression in Merkel cell carcinoma. Am J dermatopathol. (2002) 24:289–93. doi: 10.1097/00000372-200208000-00001
272. Frenard C, Peuvrel L, Brocard A, Saint-Jean M, Moreau A, Dreno B, et al. Dramatic response of an inoperable Merkel cell carcinoma with imatinib. JAAD Case Rep. (2016) 1:16–8. doi: 10.1016/j.jdcr.2015.10.007
273. Loader DE, Feldmann R, Baumgartner M, Breier F, Schrama D, Becker JC, et al. Clinical remission of Merkel cell carcinoma after treatment with imatinib. J Am Acad Dermatol. (2013) 69:e181–3. doi: 10.1016/j.jaad.2013.03.042
274. Samlowski WE, Moon J, Tuthill RJ, Heinrich MC, Balzer-Haas NS, Merl SA, et al. A phase II trial of imatinib mesylate in merkel cell carcinoma (neuroendocrine carcinoma of the skin): A Southwest Oncology Group study (S0331). Am J Clin Oncol. (2010) 33:495–9. doi: 10.1097/COC.0b013e3181b9cf04
275. Knepper TC, Panchaud RA, Muradova E, Cohen L, DeCaprio JA, Khushalani NI, et al. An analysis of the use of targeted therapies in patients with advanced Merkel cell carcinoma and an evaluation of genomic correlates of response. Cancer Med. (2021) 10:5889–96. doi: 10.1002/cam4.4138
276. Swick BL, Ravdel L, Fitzpatrick JE, Robinson WA. Merkel cell carcinoma: evaluation of KIT (CD117) expression and failure to demonstrate activating mutations in the C-KIT proto-oncogene - implications for treatment with imatinib mesylate. J cutaneous Pathol. (2007) 34:324–9. doi: 10.1111/j.1600-0560.2006.00613.x
277. Verhaegen ME, Mangelberger D, Weick JW, Vozheiko TD, Harms PW, Nash KT, et al. Merkel cell carcinoma dependence on bcl-2 family members for survival. J Invest Dermatol. (2014). doi: 10.1038/jid.2014.138
278. Chteinberg E, Wetzels S, Gerritsen W, Temmerman L, van den Oord J, Biessen E, et al. Navitoclax combined with Alpelisib effectively inhibits Merkel cell carcinoma cell growth in vitro. Ther Adv Med Oncol. (2020) 12:1758835920975621. doi: 10.1177/1758835920975621
279. Shah MH, Varker KA, Collamore M, Zwiebel JA, Coit D, Kelsen D, et al. G3139 (Genasense) in patients with advanced merkel cell carcinoma. Am J Clin Oncol. (2009) 32:174–9. doi: 10.1097/COC.0b013e31817eebf8
280. Schlagbauer-Wadl H, Klosner G, Heere-Ress E, Waltering S, Moll I, Wolff K, et al. Bcl-2 antisense oligonucleotides (G3139) inhibit Merkel cell carcinoma growth in SCID mice. J Invest Dermatol. (2000) 114:725–30. doi: 10.1046/j.1523-1747.2000.00937.x
281. Arora R, Shuda M, Guastafierro A, Feng H, Toptan T, Tolstov Y, et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci Trans Med. (2012) 4:133ra56. doi: 10.1126/scitranslmed.3003713
282. Dresang LR, Guastafierro A, Arora R, Normolle D, Chang Y, Moore PS. Response of Merkel cell polyomavirus-positive merkel cell carcinoma xenografts to a survivin inhibitor. PloS One. (2013) 8:e80543. doi: 10.1371/journal.pone.0080543
283. Leiendecker L, Jung PS, Obenauf AC. Targeting CD56 with an antibody-drug conjugate in Merkel cell carcinoma. Br J Dermatol. (2022) 186:209–10. doi: 10.1111/bjd.20894
284. Esnault C, Leblond V, Martin C, Desgranges A, Baltus CB, Aubrey N, et al. Adcitmer(®), a new CD56-targeting monomethyl auristatin E-conjugated antibody, is a potential therapeutic approach in Merkel cell carcinoma. Br J Dermatol. (2022) 186:295–306. doi: 10.1111/bjd.20770
285. Shah MH, Lorigan P, O’Brien ME, Fossella FV, Moore KN, Bhatia S, et al. Phase I study of IMGN901, a CD56-targeting antibody-drug conjugate, in patients with CD56-positive solid tumors. Invest New Drugs. (2016) 34:290–9. doi: 10.1007/s10637-016-0336-9
286. McNiff JM, Cowper SE, Lazova R, Subtil A, Glusac EJ. CD56 staining in Merkel cell carcinoma and natural killer-cell lymphoma: magic bullet, diagnostic pitfall, or both? J cutaneous Pathol. (2005) 32:541–5. doi: 10.1111/j.0303-6987.2005.00378.x
287. Kurokawa M, Nabeshima K, Akiyama Y, Maeda S, Nishida T, Nakayama F, et al. CD56: a useful marker for diagnosing Merkel cell carcinoma. J Dermatol Sci. (2003) 31:219–24. doi: 10.1016/S0923-1811(03)00029-X
288. Stueven AK, Kayser A, Wetz C, Amthauer H, Wree A, Tacke F, et al. Somatostatin analogues in the treatment of neuroendocrine tumors: past, present and future. Int J Mol Sci. (2019) 20(12):3049. doi: 10.3390/ijms20123049
289. Akaike T, Qazi J, Anderson A, Behnia FS, Shinohara MM, Akaike G, et al. High somatostatin receptor expression and efficacy of somatostatin analogues in patients with metastatic Merkel cell carcinoma. Br J Dermatol. (2021) 184:319–27. doi: 10.1111/bjd.19150
290. Askari E, Moghadam SZ, Wild D, Delpassand E, Baldari S, Nilica B, et al. Peptide receptor radionuclide therapy in merkel cell carcinoma: A comprehensive review. J Nucl Med Technol. (2023) 51:22–5. doi: 10.2967/jnmt.122.264904
291. Jalles C, Lepelley M, Mouret S, Charles J, Leccia MT, Trabelsi S. Skin cancers under Janus kinase inhibitors: A World Health Organization drug safety database analysis. Therapie. (2022) 77(6):649–56. doi: 10.1016/j.therap.2022.04.005
292. Rastrelli M, Ferrazzi B, Tropea S, Costa A, Finotto S, Marino D, et al. Aggressive merkel cell carcinoma after janus kinase inhibitor ruxolitinib for polycythemia vera. In vivo. (2019) 33(5):1667–9. doi: 10.21873/invivo.11653
293. Wallis LS, Rakita U, Grushchak S, Asadbeigi SN, Yazdan P, Liu W, et al. Merkel cell carcinoma associated with tofacitinib therapy. JAAD Case Rep. (2021) 14:94–6. doi: 10.1016/j.jdcr.2021.06.011
294. Debureaux PE, Arrondeau J, Bouscary D, Goldwasser F. Nivolumab combined with ruxolitinib: antagonism or synergy? Ann Oncol. (2018) 29:1334–5. doi: 10.1093/annonc/mdy077
295. Jansen B, Heere-Ress E, Schlagbauer-Wadl H, Halaschek-Wiener J, Waltering S, Moll I, et al. Farnesylthiosalicylic acid inhibits the growth of human Merkel cell carcinoma in SCID mice. J Mol Med (Berl). (1999) 77:792–7. doi: 10.1007/s001099900052
296. Kannan A, Lin Z, Shao Q, Zhao S, Fang B, Moreno MA, et al. Dual mTOR inhibitor MLN0128 suppresses Merkel cell carcinoma (MCC) xenograft tumor growth. Oncotarget. (2016) 7:6576–92. doi: 10.18632/oncotarget.v7i6
297. Adam C, Baeurle A, Brodsky JL, Wipf P, Schrama D, Becker JC, et al. The HSP70 modulator MAL3-101 inhibits Merkel cell carcinoma. PloS One. (2014) 9:e92041. doi: 10.1371/journal.pone.0092041
298. Fang B, Kannan A, Zhao S, Nguyen QH, Ejadi S, Yamamoto M, et al. Inhibition of PI3K by copanlisib exerts potent antitumor effects on Merkel cell carcinoma cell lines and mouse xenografts. Sci Rep. (2020) 10:8867. doi: 10.1038/s41598-020-65637-2
299. Kleffel S, Lee N, Lezcano C, Wilson BJ, Sobolewski K, Saab KR, et al. ABCB5-targeted chemoresistance reversal inhibits merkel cell carcinoma growth. J Invest Dermatol. (2016) 136:838–46. doi: 10.1016/j.jid.2015.12.038
300. Das BK, Kannan A, Nguyen Q, Gogoi J, Zhao H, Gao L. Selective inhibition of aurora kinase A by AK-01/LY3295668 attenuates MCC tumor growth by inducing MCC cell cycle arrest and apoptosis. Cancers. (2021) 13(15):3708. doi: 10.3390/cancers13153708
301. Houben R, Hesbacher S, Sarma B, Schulte C, Sarosi EM, Popp S, et al. Inhibition of T-antigen expression promoting glycogen synthase kinase 3 impairs merkel cell carcinoma cell growth. Cancer Lett. (2022) 524:259–67. doi: 10.1016/j.canlet.2021.10.031
302. Morimoto Y, Fushimi A, Yamashita N, Hagiwara M, Bhattacharya A, Cheng J, et al. Addiction of Merkel cell carcinoma to MUC1-C identifies a potential new target for treatment. Oncogene. (2022) 41:3511–23. doi: 10.1038/s41388-022-02361-3
303. Yang J, Lim JT, Victor P, Chen C, Khwaja H, Schnellmann RG, et al. Integrative analysis reveals therapeutic potential of pyrvinium pamoate in Merkel cell carcinoma. bioRxiv. (2023). doi: 10.1101/2023.11.01.565218
304. Xie H, Kaye FJ, Isse K, Sun Y, Ramoth J, French DM, et al. Delta-like protein 3 expression and targeting in merkel cell carcinoma. Oncologist. (2020) 25:810–7. doi: 10.1634/theoncologist.2019-0877
305. Loke ASW, Lambert PF, Spurgeon ME. Current in vitro and in vivo models to study MCPyV-associated MCC. Viruses. (2022) 14(10):2204. doi: 10.3390/v14102204
306. Lee JH, Lee JD, Paulson K, Voillet V, Berndt A, Church C, et al. Enhancing immunogenic responses through CDK4/6 and HIF2alpha inhibition in Merkel cell carcinoma. Heliyon. (2024) 10:e23521. doi: 10.1016/j.heliyon.2023.e23521
307. Leonard JH, Bell JR, Kearsley JH. Characterization of cell lines established from Merkel-cell (“small-cell”) carcinoma of the skin. Int J Cancer J Int du Cancer. (1993) 55:803–10. doi: 10.1002/ijc.2910550519
308. Krasagakis K, Almond-Roesler B, Geilen C, Fimmel S, Krengel S, Chatzaki E, et al. Growth and characterization of a cell line from a human primary neuroendocrine carcinoma of the skin (Merkel cell carcinoma) in culture and as xenograft. J Cell Physiol. (2001) 187:386–91. doi: 10.1002/jcp.1086
309. Guastafierro A, Feng H, Thant M, Kirkwood JM, Chang Y, Moore PS, et al. Characterization of an early passage Merkel cell polyomavirus-positive Merkel cell carcinoma cell line, MS-1, and its growth in NOD scid gamma mice. J Virol Methods. (2013) 187:6–14. doi: 10.1016/j.jviromet.2012.10.001
310. Knips J, Czech-Sioli M, Spohn M, Heiland M, Moll I, Grundhoff A, et al. Spontaneous lung metastasis formation of human Merkel cell carcinoma cell lines transplanted into scid mice. Int J Cancer J Int du Cancer. (2017) 141:160–71. doi: 10.1002/ijc.30723
311. Lezcano C, Kleffel S, Lee N, Larson AR, Zhan Q, DoRosario A, et al. Merkel cell carcinoma expresses vasculogenic mimicry: demonstration in patients and experimental manipulation in xenografts. Lab Invest. (2014) 94:1092–102. doi: 10.1038/labinvest.2014.99
312. Albertini S, Martuscelli L, Borgogna C, Virdi S, Indenbirken D, Lo Cigno I, et al. Cancer-associated fibroblasts exert proangiogenic activity in merkel cell carcinoma. J Invest Dermatol. (2023) 143:965–976 e15. doi: 10.1016/j.jid.2022.12.006
313. Spurgeon ME, Cheng J, Ward-Shaw E, Dick FA, DeCaprio JA, Lambert PF. Merkel cell polyomavirus large T antigen binding to pRb promotes skin hyperplasia and tumor development. PloS Pathog. (2022) 18:e1010551. doi: 10.1371/journal.ppat.1010551
314. Spurgeon ME, Liem A, Buehler D, Cheng J, DeCaprio JA, Lambert PF. The merkel cell polyomavirus T antigens function as tumor promoters in murine skin. Cancers. (2021) 13(2):222. doi: 10.3390/cancers13020222
315. Wright MC, Reed-Geaghan EG, Bolock AM, Fujiyama T, Hoshino M, Maricich SM. Unipotent, Atoh1+ progenitors maintain the Merkel cell population in embryonic and adult mice. J Cell Biol. (2015) 208:367–79. doi: 10.1083/jcb.201407101
316. Morrison KM, Miesegaes GR, Lumpkin EA, Maricich SM. Mammalian Merkel cells are descended from the epidermal lineage. Dev Biol. (2009) 336:76–83. doi: 10.1016/j.ydbio.2009.09.032
317. Van Keymeulen A, Mascre G, Youseff KK, Harel I, Michaux C, De Geest N, et al. Epidermal progenitors give rise to Merkel cells during embryonic development and adult homeostasis. J Cell Biol. (2009) 187:91–100. doi: 10.1083/jcb.200907080
318. Moll R, Lowe A, Laufer J, Franke WW. Cytokeratin 20 in human carcinomas. A new histodiagnostic marker detected by monoclonal antibodies. Am J Pathol. (1992) 140:427–47.
319. Gillet JP, Calcagno AM, Varma S, Marino M, Green LJ, Vora MI, et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc Natl Acad Sci USA. (2011) 108:18708–13. doi: 10.1073/pnas.1111840108
320. Mak IW, Evaniew N, Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. Am J Transl Res. (2014) 6:114–8.
321. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. (2015) 21:1318–25. doi: 10.1038/nm.3954
322. Zitvogel L, Pitt JM, Daillere R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nat Rev Cancer. (2016) 16:759–73. doi: 10.1038/nrc.2016.91
323. Cogels MM, Rouas R, Ghanem GE, Martinive P, Awada A, Van Gestel D, et al. Humanized mice as a valuable pre-clinical model for cancer immunotherapy research. Front Oncol. (2021) 11:784947. doi: 10.3389/fonc.2021.784947
324. Gavvovidis I, Leisegang M, Willimsky G, Miller N, Nghiem P, Blankenstein T. Targeting merkel cell carcinoma by engineered T cells specific to T-antigens of merkel cell polyomavirus. Clin Cancer Res. (2018) 24:3644–55. doi: 10.1158/1078-0432.CCR-17-2661
325. Ostrand-Rosenberg S. Animal models of tumor immunity, immunotherapy and cancer vaccines. Curr Opin Immunol. (2004) 16:143–50. doi: 10.1016/j.coi.2004.01.003
326. Dranoff G. Experimental mouse tumour models: what can be learnt about human cancer immunology? Nat Rev Immunol. (2011) 12:61–6. doi: 10.1038/nri3129
327. Colunga A, Pulliam T, Nghiem P. Merkel cell carcinoma in the age of immunotherapy: facts and hopes. Clin Cancer Res. (2018) 24:2035–43. doi: 10.1158/1078-0432.CCR-17-0439
Keywords: neuroendocrine carcinoma (NEC), immunotherapy, mouse model, Merkel cell carcinoma (MCC), epigenetics, viral tumorigenesis, UV signature, clinical trial
Citation: Pedersen EA, Verhaegen ME, Joseph MK, Harms KL and Harms PW (2024) Merkel cell carcinoma: updates in tumor biology, emerging therapies, and preclinical models. Front. Oncol. 14:1413793. doi: 10.3389/fonc.2024.1413793
Received: 07 April 2024; Accepted: 08 July 2024;
Published: 29 July 2024.
Edited by:
Nikhil I. Khushalani, Moffitt Cancer Center, United StatesReviewed by:
Ilze Strumfa, Riga Stradiņš University, LatviaCopyright © 2024 Pedersen, Verhaegen, Joseph, Harms and Harms. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul W. Harms, cGF1bGhhcm1AbWVkLnVtaWNoLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.