 Gary A. Piazza
Gary A. Piazza Preethi Chandrasekaran
Preethi Chandrasekaran Yulia Y. Maxuitenko
Yulia Y. Maxuitenko Karim I. Budhwani
Karim I. Budhwani- 1Department of Drug Discovery and Development, Harrison College of Pharmacy, Auburn University, Auburn, AL, United States
- 2University of Texas (UT) Southwestern Medical Center, Dallas, TX, United States
- 3CerFlux, Birmingham, AL, United States
- 4University of Alabama at Birmingham, Birmingham, AL, United States
Colorectal cancer (CRC) is a highly prevalent and lethal cancer worldwide. Approximately 45% of CRC patients harbor a gain-in-function mutation in KRAS. KRAS is the most frequently mutated oncogene accounting for approximately 25% of all human cancers. Gene mutations in KRAS cause constitutive activation of the KRAS protein and MAPK/AKT signaling, resulting in unregulated proliferation and survival of cancer cells and other aspects of malignant transformation, progression, and metastasis. While KRAS has long been considered undruggable, the FDA recently approved two direct acting KRAS inhibitors, Sotorasib and Adagrasib, that covalently bind and inactivate KRASG12C. Both drugs showed efficacy for patients with non-small cell lung cancer (NSCLC) diagnosed with a KRASG12C mutation, but for reasons not well understood, were considerably less efficacious for CRC patients diagnosed with the same mutation. Thus, it is imperative to understand the basis for resistance to KRASG12C inhibitors, which will likely be the same limitations for other mutant specific KRAS inhibitors in development. This review provides an update on clinical trials involving CRC patients treated with KRASG12C inhibitors as a monotherapy or combined with other drugs. Mechanisms that contribute to resistance to KRASG12C inhibitors and the development of novel RAS inhibitors with potential to escape such mechanisms of resistance are also discussed.
Introduction
Colorectal cancer (CRC) is the third most prevalent cancer and the second leading cause of cancer related mortality worldwide, according to Global Cancer Statistics 2018 (1). CRC is recognized as a heterogenous malignancy with a complex mutational landscape in which over 45% of cases harbor KRAS mutations but with additional mutations, for example, in components of the APC/β-catenin pathway. While only 3% of CRC patients are diagnosed with the KRASG12C mutation, this type of CRC is often associated with rapid progression and shorter overall survival rate compared to patients diagnosed with non-KRASG12C mutations (2–4). KRASG12C mutations result from a glycine-to-cysteine substitution at position 12 of KRAS protein leading to constitutive activation of KRAS (5).
Under physiological conditions, wild-type (WT) RAS functions enzymatically as a GTPase to regulate normal cell proliferation, for example, in the colonic mucosa to regenerate surface epithelium. RAS is often described as a molecular switch that is “off” when bound to GDP or “on” when GTP is bound, whereby “off” and “on” refer to different conformations of RAS that regulate its capacity to bind effectors such as RAF or PI3K that activate downstream signaling. Upstream of RAS, endogenous mitogens such as epidermal growth factor (EGF), that are enriched in the tumor microenvironment, bind to cell surface receptor tyrosine kinases (RTKs) and activate a cascade of events, starting with removal of GDP from WT RAS isozymes by guanine nucleotide exchange factors (GEFs). When in a nucleotide-free conformation, high intracellular concentrations of GTP rapidly bind and switch RAS “on”, to stimulate MAPK/AKT signaling, culminating in the transcription of genes that encode for proteins essential for normal cell turnover and replacement (6). RAS mutations result in appreciably slower rates of GDP/GTP exchange caused by preventing GTPase activating proteins (GAPs) from removing GTP to turn RAS “off”, resulting in hyperactivation of downstream MAPK/AKT signaling (6). WT RAS isozymes, NRAS and HRAS, are co-expressed in KRAS mutant cancer cells whereby their proliferation can be driven not only by mutant KRAS, but also by extracellular mitogens that activate WT RAS isozymes. Nonetheless, KRASG12C inhibitors have been shown to have exquisite selectively in inhibiting the growth of tumors harboring KRASG12C and, consequently, would not be expected to affect the growth of tumors with other KRAS mutations (or other RAS isozymes). However, unchecked activity from WT RAS isozymes might contribute to intrinsic or acquired resistance. Further, because KRASG12C mutations only account for 3% of CRC cases, there is an urgent medical need to treat CRC patients harboring other KRAS mutations, including patients with G12D (30.1%), G12V (24.2%), G12R (2.1%), or other (19.6%) mutations (7). Thus, a pan-KRAS inhibitor would be expected to have broader use for CRC and other RAS driven cancers given its additional potential to circumvent resistance from unchecked activity of WT RAS isozymes. Nonetheless, both approaches would require a mechanism to selectively inhibit mutant RAS in cancer cells without affecting the activity of WT RAS in normal cells, essential for turnover and replacement in rapidly dividing tissues.
RAS signaling
As a membrane bound small guanine nucleotide binding protein, RAS can readily switch between an active GTP-bound state and an inactive GDP-bound conformation under normal physiological conditions. This cascade is modulated by RTKs whereby dimerization is induced by ligand binding. Receptor dimerization leads to the activation of intrinsic tyrosine kinase and autophosphorylation of tyrosine residues. The phosphorylated receptor interacts with GRB2 (growth factor receptor bound protein 2) and GEFs such as SOS (Son of Sevenless), that catalyze GDP/GTP exchange, leading to the active conformation of RAS. RAS-GTP activates several pathways such as RAF-MEK-ERK and PI3K-AKT-mTOR promoting cell proliferation and survival. In normal cells, RAS is switched off by GAPs inducing GTP hydrolysis and forming inactive RAS-GDP. But this is impeded in cancer cells by the inability of GAPs to bind RAS, thereby reducing the hydrolysis of GTP, favoring RAS to be in a constitutively activated conformation (8).
The KRAS protein has a molecular weight of 21 KD and is composed of six beta strands and 5 alpha helices, which form 2 major domains, referred to as the G-domain and the C-terminal (9). The G-domain, which is highly conserved contains the switch I and switch II loops that are responsible for GDP/GTP exchange (10).
Figure 1 illustrates the RAS signaling pathways. The upstream and downstream signaling mechanism of RAS are depicted in Figures 2, 3 respectively.
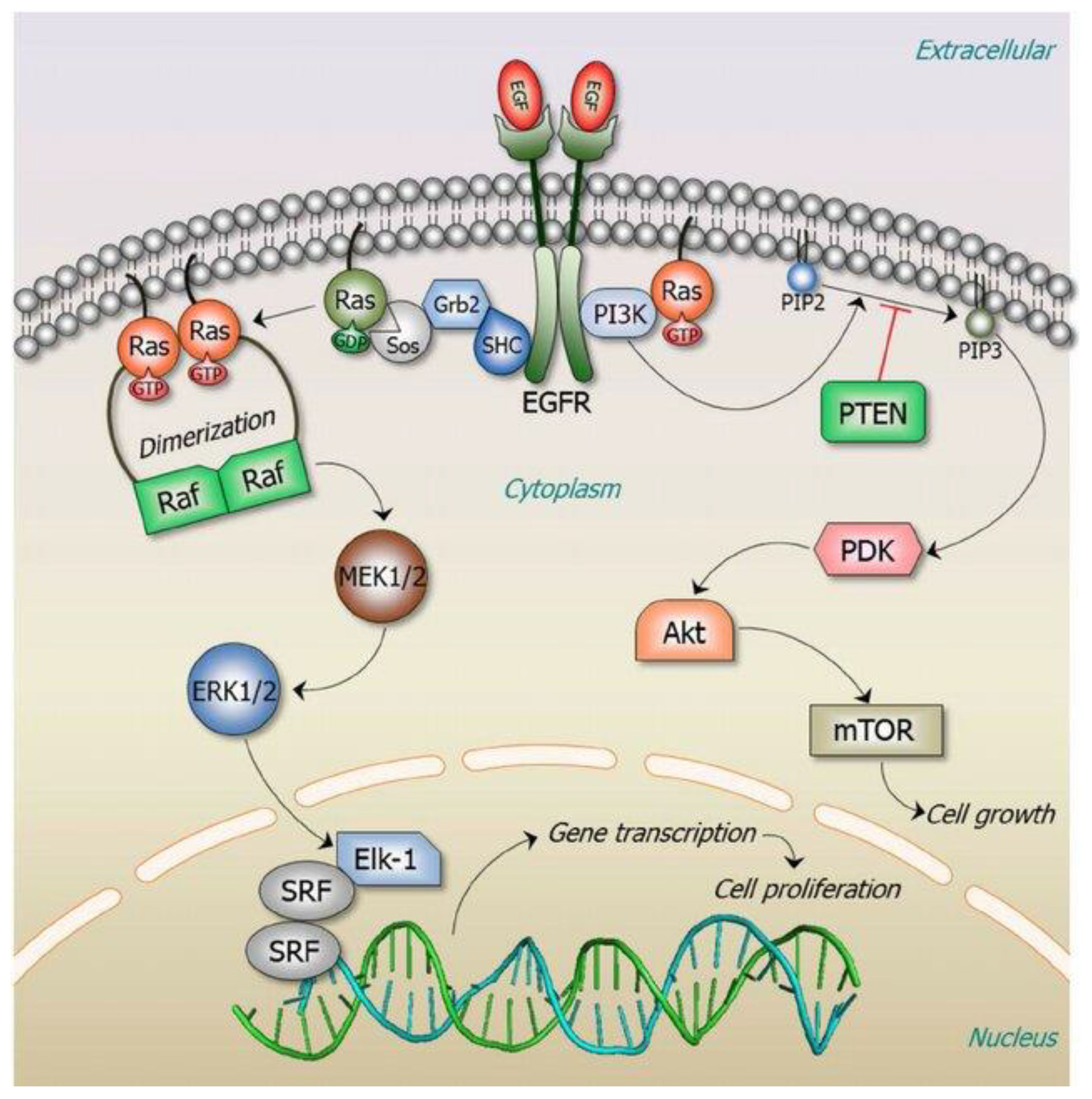
Figure 1 RAS signaling is versatile as it involves numerous cellular functions. The key RAS effector pathway is the mitogen-activated protein kinase (MAPK), Raf-MEK-ERK pathway (11). Reproduced here from Oncotarget (Ruth Nussinov et al., 2014) under Creative Commons Attribution license.
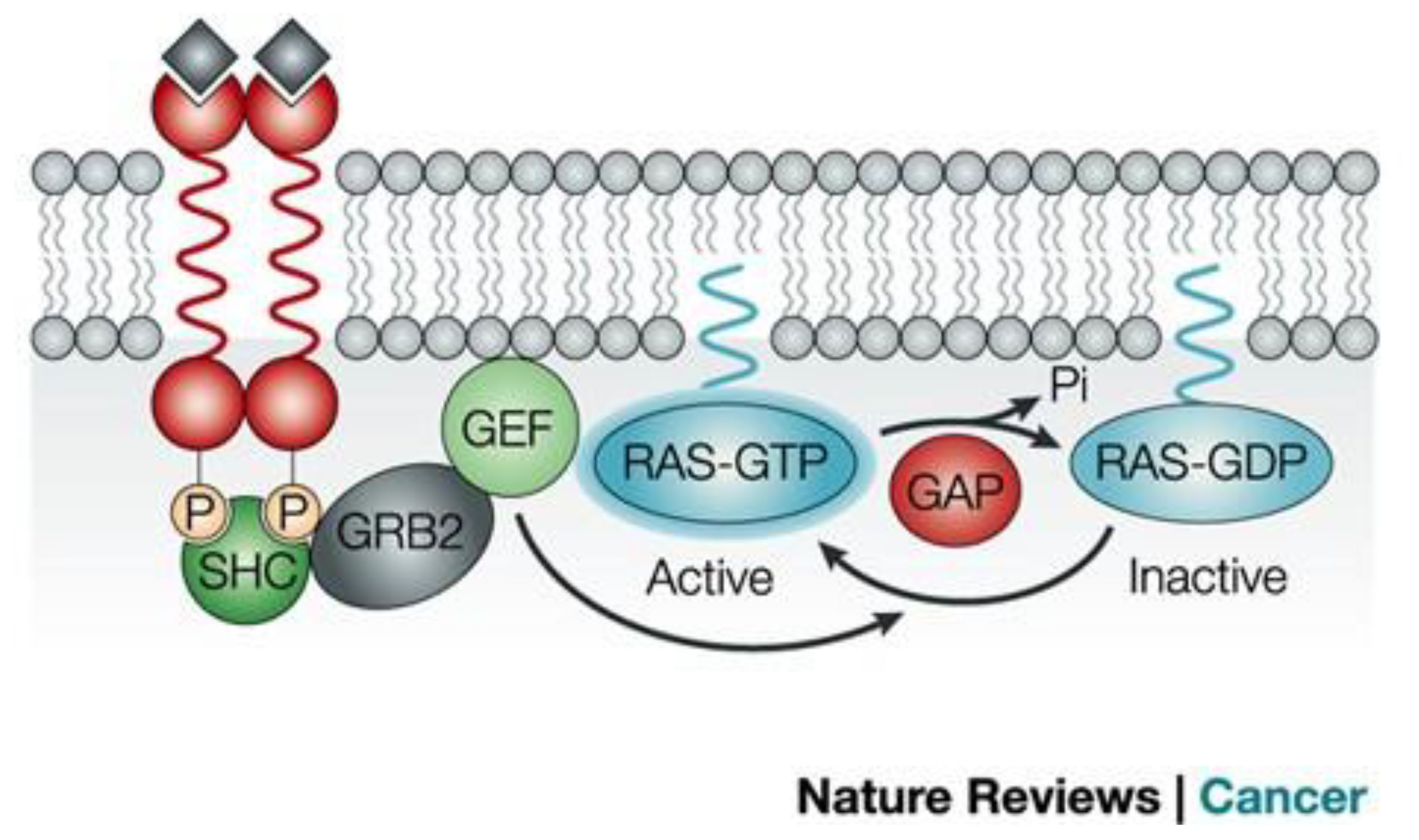
Figure 2 Signaling upstream of RAS (12). The RAS activation is controlled by the cycle of hydrolysis of bound GTP, catalyzed by GTPase activating proteins and the replacement of bound GDP with fresh GTP, which is catalyzed by guanine nucleotide exchange factors. Reproduced here from Nature Reviews Cancer (Julian Downward 2003) under Creative Commons Attribution license.
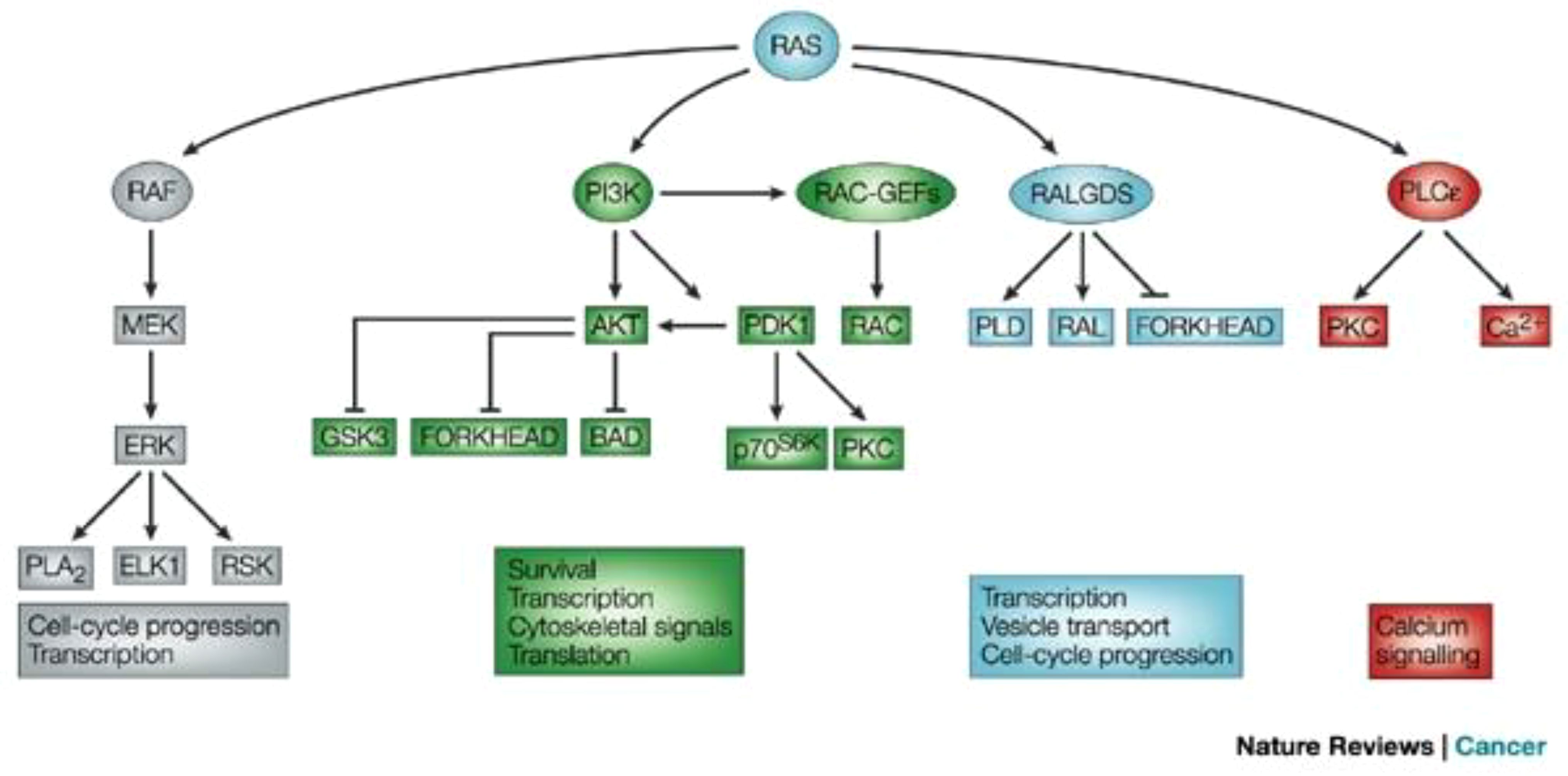
Figure 3 Signaling downstream of RAS (12). The main effector proteins with which RAS interacts, once in its active GTP-bound state, is shown. Reproduced here from Nature Reviews Cancer (Julian Downward 2003) under Creative Commons Attribution license.
Development of KRASG12C inhibitors for colorectal cancer
Until recently, KRAS was considered undruggable as the protein apparently lacked deep pockets for small molecule binding, apart from the nucleotide binding domain (13). In addition, the high affinity of KRAS for GTP makes it difficult to develop competitive small molecule inhibitors to block GTP activation of RAS. Despite these challenges, multiple attempts have been made to discover small molecules to directly inhibit RAS. Early clinical trials of Sotorasib (the first FDA approved KRASG12C inhibitor) in CRC patients with a KRASG12C mutation resulted in lack of response as well as in non-small cell lung cancer (NSCLC) patients with the same mutation. For example, Sotorasib monotherapy in CRC KRASG12C patients previously treated with fluoropyrimidine, oxaliplatin, and irinotecan, demonstrated 9.7% objective response rate (ORR) in 62 patients (14). Another study evaluated the effect of Adagrasib (the second FDA approved KRASG12C inhibitor) in 43 CRC patients in KRYSTAL-1 (NCT03785249) trial (Table 1) which demonstrated 19% response rates (15). The reasons underlying the lower response to the drugs in CRC targeting G12C may be the rapid development of treatment related adaptive signaling resistance as these tumors undergo significant rebound in ERK phosphorylation (16). Another hypothesis for this contradictory response is the presence of higher levels of upstream receptor tyrosine phosphorylation compared to NSCLC, specifically in EGFR (16). Thus, combination therapies with small molecule KRAS inhibitors and anti-EGFR monoclonal antibodies have gained traction recently. In line with this, CodeBreak 101 (NCT04185883) reported an overall survival of 30% in 40 metastatic CRC patients (Table 1) treated with Panitumumab and Sotorasib combination therapy (17). Similarly, Adagrasib in combination with Cetuximab demonstrated an ORR of 46% compared to Adagrasib monotherapy with ORR of 19% (18).
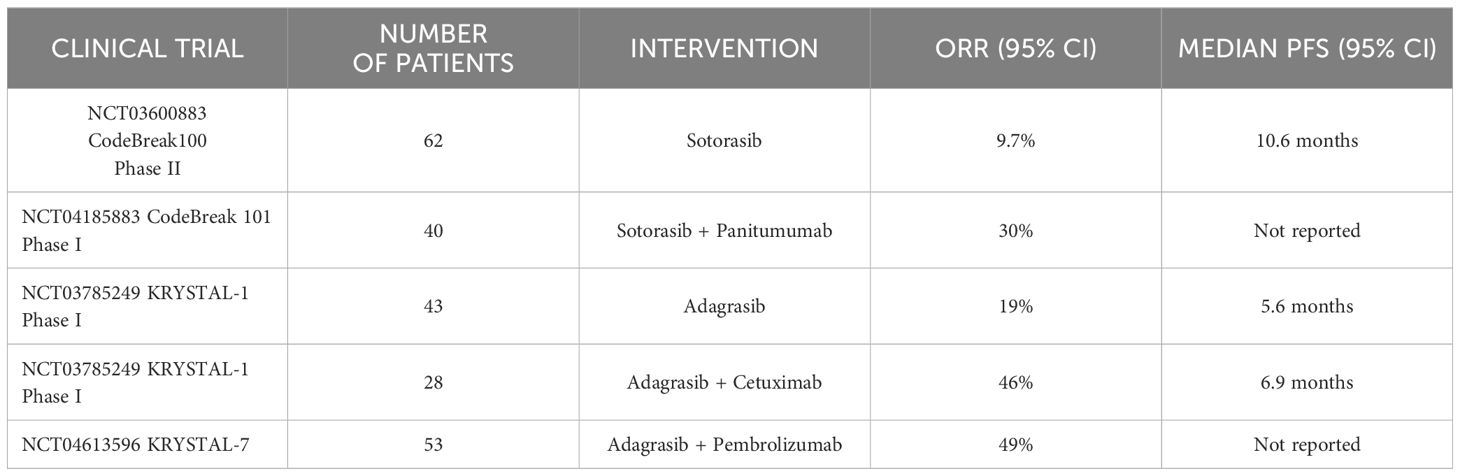
Table 1 Summary of key clinical trials of KRASG12C inhibitors in CRC, advanced solid tumors, and NSCLC patients.
Mechanisms of resistance to KRASG12C inhibitors
The limited response of CRC patients to KRASG12C inhibitors may be attributed to multiple mechanisms of resistance, both upstream and downstream of KRAS as well as co-lateral pathways (e.g., Wnt/β-catenin) that can compensate for the effects of a mutant specific KRAS inhibitor. In addition, co-occurring mutations such as G13D, R68M, and A59S/T confer resistance selectively to Sotorasib, while Q99L alteration is selective to Adagrasib (19). The most common X96D/S mutation confers strongest resistance to both drugs (20). A frequently identified mechanism of resistance that diminishes the therapeutic efficacy of KRAS inhibitors is the induction of bypass MAPK signaling to overcome KRAS blockade. Initial studies revealed significant suppression of negative regulators of MAPK signaling and that ERK dependent signaling is reactivated to bypass KRASG12C treatment. Further insights into resistance mechanism suggest that only the cells with KRASG12C in the inactive confirmation are strongly inhibited by novel KRASG12C inhibitors. This leads to non-uniform rates of inactive to active KRASG12C cycling. Subsequently, these cells with KRASG12C preferentially held in active confirmation could be insensitive to treatment and could mediate reactivation of MAPK signaling (21, 22).
Furthermore, CRC cells show early development of adaptive resistance to KRASG12C inhibitors by rapid upregulation of p-MEK and p-ERK and increased basal phosphorylation and activation of EGFR. CRC cells respond to EGF stimulation by activating RAS-MAPK signaling even in the presence of an activating KRASG12C mutation, which contrasts with NSCLC cells. In line with this, preclinical studies suggest that primary resistance to KRAS inhibition is less likely, and the predominant issue appears to be drug-induced (acquired) resistance. This contrasts with NSCLC where the key issue is primary (intrinsic) resistance. In summary, EGFR specifically mediates adaptive resistance response in CRC cells. Finally, resistance is also observed by induction of epithelial to mesenchymal transition in conjunction with increased PI3K/AKT signaling due to upregulated EGFR signaling and subsequently leading to increased MAPK signaling via FGFR (23).
Thus, increased RTK signaling coupled with other mechanisms such as increased GTP-bound KRASG12C leads to tumor progression and triggers further downstream signaling. Alternate pathways such as Wnt/β-catenin signaling is activated interacting with mutant KRAS signaling further promoting oncogenic signaling and increased resistance (24).
Primary resistance also plays a role in lack of efficacy of KRAS inhibitors driven by multiple mechanisms. This includes rapid adaptive feedback RTK-RAS-MAPK reactivation signaling upon deficit host immune system. The formation of active GTP-bound KRASG12C from non-uniform cycling between GTP-bound active and GDP-bound inactive states driven by EGF and persistent upstream RTK activity with signaling through alternative wild-type RAS forms in CRC. The induction of EMT and disinhibition of cell-cycle transition by co-occurring alterations in CDKN2A also contribute to low efficacy of KRAS inhibitors. The differences in pharmacokinetic properties of different KRAS inhibitors also contribute to low efficacy of some of these inhibitors (25).
Early phase clinical trials of KRASG12C inhibitors
The first human phase 1 trial on Sotorasib at a daily dose of 960 mg in 42 CRC patients (CodeBreak 100, NCT03600883) demonstrated modest clinical activity (ORR of 7.1%) compared to NSCLC patients with ORR of 32.3% (26). The phase 2 CodeBreak 100 trial with the same dose of Sotorasib demonstrated an ORR of 9.7% in patients with metastatic KRASG12C mutant CRC with prior fluoropyrimidine, oxaliplatin, and irinotecan treatment (14). The KRYSTAL-1 phase 1/2 trial (NCT03785249) investigating Adagrasib in patients previously treated with chemotherapy or anti-PD1 showed ORR of 22% and disease control rate (DCR) of 87% in 45 CRC patients (15).
Current clinical trials involving KRASG12C inhibitors
There are 12 different KRASG12C inhibitors currently under clinical investigation. In total, there are 76 entries for KRASG12C trials, out of which 39 were trials in CRC patients, while 33 were trials involving lung cancer patients. (Two trials were excluded as they are not specific to KRASG12C mutant tumors, and 2 others were excluded as they are only diagnostic studies).
Noteworthy, Novartis JDQ443 binds KRAS without involving H95 residue and maintains activity among tumors with a dual G12C and H95 KRAS mutation. This non-selectivity may reduce or alleviate acquired resistance (27, 28). On a similar note, JNJ-74699157 binds near the switch II pocket through a different cysteine residue interaction and may also mitigate resistance (29). More recently, a striking finding on Eli Lilly’s LY3537982, demonstrated that this novel KRAS inhibitor in combination with Cetuximab showed 45% ORR in 11 CRC patients (30). As would be expected, combinations of KRASG12C inhibitors and inhibitors of other downstream components of RAS/MAPK pathways such as BRAF or MEK offer promising approaches as well (31). Another combination strategy was based on KRAS inhibition triggering pro-inflammatory changes in the tumor microenvironment. This was shown by combining anti-PD1 therapy and Sotorasib, which demonstrated increased CD8+ T-cell infiltration in the tumor microenvironment and promising efficacy (32).
Figure 4 shows the structure of KRAS surfaces targeted by KRAS inhibitors. a) switch II pocket of KRASG12c bound to AMG510 b) MRTX1133 with KRASG12D/GDP.
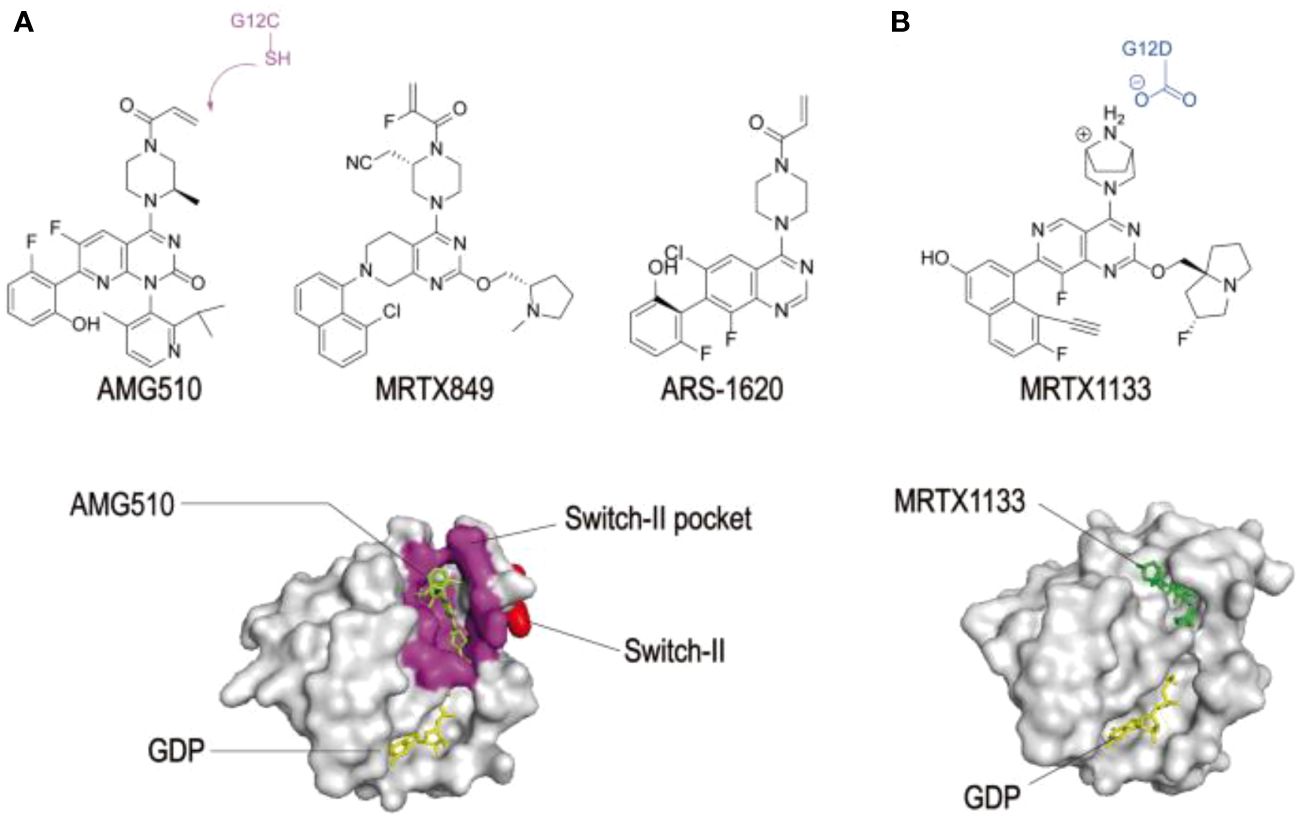
Figure 4 Structures of KRAS surfaces targeted by KRAS mutant inhibitors. a Switch-II pocket (purple) of KRAS (G12C) bound to AMG510 (PDB: 6OIM). b MRTX1133 with KRAS G12D/GDP (PDB: 7RPZ). (33) Reproduced here from Molecular Cancer (Weidong Zhang et al., 2022) under Creative Commons Attribution license.
Combination treatment strategies
Considering mechanisms of resistance upstream of RAS, combined treatment of Sotorasib or Adagrasib with EGFR inhibitors is currently being evaluated in clinical trials. Preclinical studies reported that the EGRF inhibitor, Cetuximab, sensitizes KRASG12C mutated CRC cell lines to Sotorasib, leading to sustained downregulation of phosphorylated MEK and ERK proteins, causing cell proliferation arrest and apoptosis (16). The KRYSTAL-1 (NCT03785249) trial conducted in 28 CRC patients reported ORR of 46% and DCR of 100% in patients treated with Cetuximab and Adagrasib (16).
CodeBreak 101 umbrella trial tested Sotorasib with inhibitors of MEK, CDK4/6, mTOR, or VEGFR in additional cohorts. Similarly, KRYSTAL-1 trial is also exploring similar combination strategies. Combinations of Sotorasib and the MEK inhibitor, Trametinib were tested in 18 CRC patients with promising efficacy and safety (34). Similarly, KRASG12C inhibitors in combination with CDK4/6 inhibitors such as Palbociclib demonstrated significant downregulation of KRAS pathway phosphorylation (35). Another emerging strategy includes simultaneously targeting other components of the KRAS pathway. One such target is the SHP2 which promotes KRAS signaling and CRC progression. SHP2 inhibition increases GDP-bound KRASG12C and shows synergism with KRAS inhibitors in vitro (36, 37).
Ongoing phase 1 trials are actively progressing on novel SHP2 inhibitors such as TN0155, BBP-398 and RMC-4630 with plans to test in combination with KRAS inhibitors (38–40). BI-3406, an SOS1 inhibitor demonstrates increased response in combination with Trametinib (20).
A phase 1b trial (NCT04449874) reported the activity of Divarasib, a covalent KRASG12C inhibitor (Table 2) that turns off oncogenic signaling by irreversibly locking the protein in an inactive state, and Cetuximab in 29 CRC patients with KRASG12C mutation. The ORR was 62.5% and median duration of response was 6.9 months. This encouraging anti-tumor activity of the combination therapy supports further investigation (41). Strikingly, Divarasib is also shown to be 5 to 20 times more potent and fifty times more selective than Sotorasib and Adagrasib (42).
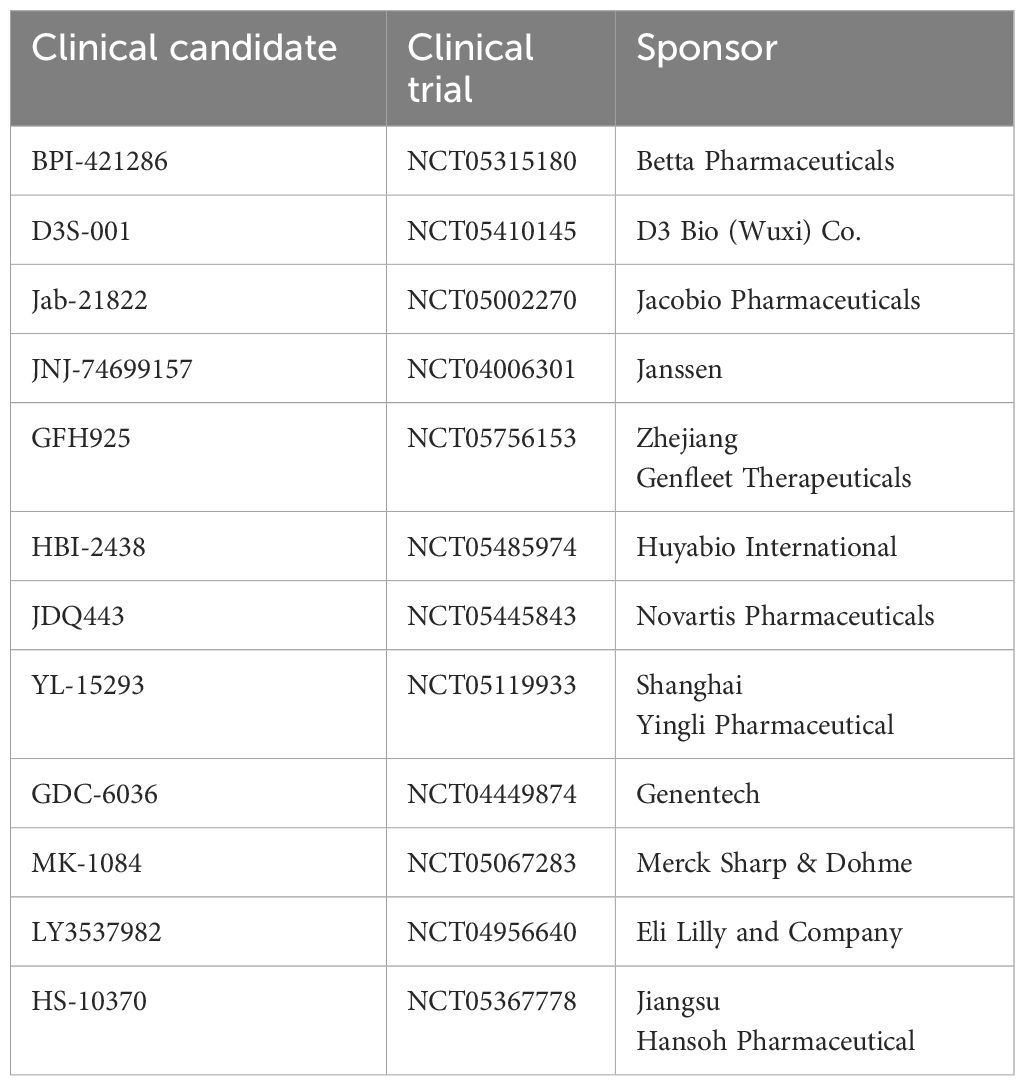
Table 2 A summary of registered trials listed on clinicaltrials.gov. as of March 24, 2024.
KRYSTAL-10 (NCT04793958) is a randomized phase 3 trial to test Adagrasib (600mg BID) and Cetuximab (500mg) in patients with KRASG12C metastatic CRC. This combination therapy is being compared with standard chemotherapy receiving FOLFIRI (leucovorin calcium (folinic acid), fluorouracil, and irinotecan hydrochloride). Another phase 3 randomized trial NCT05198934 is testing Sotorasib and Panitumumab and comparing with Tipiracil or Regorafenib is underway (Table 3) in previously treated metastatic KRASG12C mutant CRC patients (43).
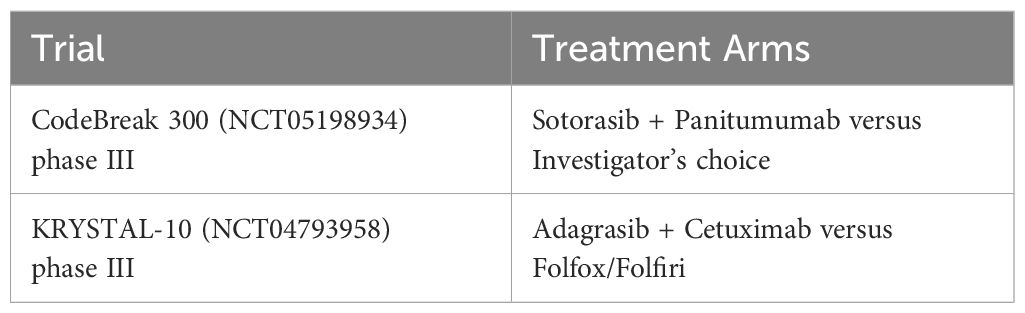
Table 3 A summary of current clinical trials in patients with KRASG12C mutant CRC listed on clinicaltrials.gov. as of March 24, 2024.
Other targets
BAY-293, an SOS1 inhibitor exhibits synergistic activity when combined with ARS-1620 in KRASG12C mutant CRC cancer cell lines proving that targeting the inactive GDP-bound form is a promising approach for generating anti-RAS therapeutics. Another novel SOS1 inhibitor BI-1701963 is currently under investigation as a single agent or in combination with Trametinib (NCT04111458) or Adagrasib (NCT04975256) (44, 45). Recently, BI-3406, another SOS1 inhibitor was demonstrated to be more potent and selective for inhibiting SOS1, decreasing KRAS-GTP levels and suppressing cancer cell proliferation (46). The tyrosine phosphorylation of SHP2 recruits GRB2-SOS complex promoting RAS nucleotide exchange acting as a scaffold protein. Currently, SHP2 inhibitors have gained attention and several of them are in the early phase of clinical trials. For example, RMC-4630 in combination with ERK inhibitors LY3214996 is in phase 1 clinical trial for KRASG12C CRC. (NCT04916236). Another inhibitor, TN0155 is in phase 1b/2 clinical trial (NCT0469918) in combination with KRASG12C inhibitor, JDQ443 in KRASG12C mutant CRC patients.
A phase 1 trial (NCT01085331) evaluated the effects of MEK inhibitor Pimasertib combined with FOLFIRI as a second line treatment of KRAS metastatic CRC. However, GI and skin toxicity were reported with Pimasertib (47). A novel RAF dimer inhibitor, Lifirefenib, demonstrated acceptable safety in phase 1 trials, but no activity was observed in KRAS mutant CRC patients (48). Three enzymes engage in post-translational modifications of KRAS which is the 1st step of membrane localization, FTase, RCE1 and (Isoprenylcysteine carboxymethyltransferase) ICMT. Inhibitors of ICMT such as cysmethynil and UCM-1336 showed potential to inhibit KRAS activity and impair the growth of KRAS mutant cell lines (49). However, clinical data are not yet available in patients with KRASG12C mutant CRC.
Current CRC treatment and limitations
The primary therapeutic strategy for resectable colorectal cancer is surgical removal and in non-resectable CRC, the strategies include chemotherapy, radiotherapy, and immunotherapy along with combination therapies. However, these approaches do not come without limitations such as relapse of acquired multi-drug resistance CRC. Recently, immune checkpoint inhibitors, T cell receptor alterations, cytokine therapy, RNA-based therapies such as siRNA and miRNA have yielded promising results (50).
Radiotherapy
Two of the adjuvant radiotherapies, a short course and long course are currently available, which are better options for treating stage II and stage III CRC. However, acute toxicity rates are high with long course radiotherapy. Decreased toxicity is observed with new delivery methods such as intensity-modulated radiotherapy (51, 52).
Chemotherapy
The commonly approved chemotherapy medications for stage III and IV CRC include fluoropyrimidines (capecitabine, Fluorouracil), topoisomerase I inhibitors (irinotecan, oxaliplatin) and tri-fluridine/tipiracil). After surgery for CRC, adjuvant fluoropyrimidine based chemotherapy is standard to reduce tumor recurrence and increases survival rate (53). Topoisomerase I inhibitor irinotecan and oxaliplatin are added to 5-flurouracil and folinic acid (leucovorin) as combination therapy regimens for metastatic CRC (known as FLOFOX and FOLFIRI) or to capecitabine (CAPOX). Regorafenib is an FDA approved tyrokinase inhibitor targeting VEGF, platelet derived growth factor, fibroblast growth factor in metastatic CRC (54).
Target specific treatment
Monoclonal antibodies such as Cetuximab, Panitumumab etc. are epidermal growth factor receptor inhibitors, while Bevacizumab and Ramucirumab target vascular epidermal growth factor and its receptor respectively (55). Cetuximab and Panitumumab are FDA approved first line treatment for CRC (56). CTLA-4 inhibition could inhibit tumor progression by upregulating effector T cell activity and suppressing regulatory T cells. FDA approved low dose Ipilimumab in combination with Nivolumab is used for previously treated microsatellite instability-high and deficient mismatch repair metastatic CRC. Pembrolizumab and Nivolumab (PD1 inhibitors) are also used in CRC (57).
Vaccines
Several clinical trials are conducted on introducing vaccines against CRC. The tumor associated antigens that are targeted include surviving, EGFR, VEGFR1 etc. These vaccines could activate local immune cells, releasing tumor antigens, increasing T cells and dendritic cell infiltration to the site of action (58).
Thus, in summary every patient has a unique tumor microenvironment, and individualized approaches to treating CRC are needed. Although conventional cytotoxic drugs are the first line of agents for CRC, their shortcomings include, toxicity and drug resistance leading to recurrent CRC. In addition to these, chemotherapy is associated with systemic toxicity, fever, stomatitis, mucositis, leukopenia, and thrombocytopenia. New approaches are emerging for treating CRC to overcome these drawbacks.
RAS PROTAC study
Despite the clinical success of KRASG12C inhibitors, acquired resistance is the major drawback with these agents. RAS was considered undruggable initially due to its insufficient binding pockets. The 1st half of the RAS protein is referred to as effector lobe (residues 1–85) while the second half of the G-domain (residues 86–166) is referred to as allosteric lobe. The exploration of high affinity macromolecular binders against the effector lobe potentially to inhibit RAS signaling is in the spotlight recently. Effective targets on the effector lobe include switch regions for which GDP and GTP specific binders have been identified (59). By genetically fusing E3-ligase subunits such as Von Hippel-Lindau tumor suppressor to monobodies NS1 and 12VC1, RAS degrader constructs were generated. These degraders have potent RAS signaling suppression and anti-proliferative activities (60).
These degraders emulate PROTAC (proteolysis targeting chimera) mode of action. Compared with competitive inhibitors, PROTACs instruct the degradation of protein by recruiting ubiquitin-proteasome system to target protein. They can therefore bind outside of an active protein site and after degradation abrogate any scaffolding functions of the target. This is attributed to their hybrid structure, containing one bonder (the warhead) for the target protein that is tethered via a linker to a moiety recuring the E3 ligase (61). PROTACs can be reused after reversible binding and degradation of target proteins. Current RAS targeting protacs (XX-4–88, LC-2, KP-14) are all built on covalent G12C inhibitor and cannot be beneficial as these inhibitors are consumed due to covalent cysteine engagement (62).
A remarkable development in this line is the reversible covalent inhibitor YF135 which employs cyanoacrylamide for cystine linkage (63). However, optimization of linker length requires further research and developmental efforts (64).
Given the spatial temporal distinct expression of E3 ligase in tissues and cells. PROTACs may provide a more controlled drug action and it remains to be seen if any of the RAS ligands can be converted to PROTACs.
Japanese guidelines for KRASG12C
The increase in targeted therapy for CRC based on genomic status has led to the clinical development of new agents which could be potentially used in patients with microsatellite instability and/or mismatch repair and metastatic CRC (mCRC) due to BRAFV600E mutations. In Japan, Trastuzumab combined with Pertuzumab was approved in March 2022, for ERBB2 (ErbB2 Receptor Tyrosine Kinase 2) positive mCRC. This development devised a better strategy for precision oncology for rare genomic alterations. The tumor genomic status in mCRC was determined for KRAS and NRAS, BRAFV600E mutations, ERBB2 and microsatellite instability (MSI)/mismatch repair (MMR) (65, 66).
The SCRUM-Japan GI-SCREEN was launched in Japan by the National cancer center hospital East in 2015. Approximately, 30,000 patients were screened using tissue and plasma assays in this nationwide screening project. In 2017, the regulatory graded registry platform (SCRUM-Japan-Registry) was established to collect efficacy data of standard therapy in patients with rare molecular alterations. Their treatment strategy was based on the 4 genomic status, along with the primary tumor location (67, 68).
In this SCRUM-Japan-GI-screen, the phase II TRIUMPH study, demonstrated the efficacy of Trastuzumab plus Pertuzumab in patients with ERBB2 amplification and this study showed ORR of 30% in 27 patients who were ERBB2 positive in tissues (69). Another interesting ongoing randomized multicenter phase II trial SWOG S1613 is recruiting patients with RAS and RAF wild type ERBB2 positive mCRC who received at least one prior line of therapy. The aim of this study is to compare the efficacy of trastuzumab plus Pertuzumab versus Cetuximab plus Irinotecan (NCT03365882) (65). A combination of chemotherapy plus anti-VEGF therapy along with immune oncology therapy could potentially be more effective in triggering immunogenic cell death and release of tumor antigens (70). In summary, for patients with MSI/MMR or BRAF V600E mCRC, Pembrolizumab is the first line therapy and Encoratinib plus Cetuximab with or without Binimetinib is considered as the second line therapy. New agents are proposed for rare molecular fractions such as ERBB2 amplification. While the efficacies of Trastuzumab plus Pertuzumab were indicated in single -arm trails, no anti-ERBB2 therapies are approved in the United States and European Union (65).
Conclusion
The understanding of KRAS signaling, structural biology, and biochemistry over the last several years has led to FDA approval of the first direct acting KRASG12C inhibitors. While KRASG12C inhibitors are efficacious for NSCLC, their use for CRC faces challenges due primarily to the development of resistance. Although the monotherapy response rates remain low in CRC patients, combination therapies are more promising. Another option recently explored are the pan-KRAS and pan-RAS inhibitors which inhibit RAS regardless of the mutated allele, or in the latter case, also independent of the RAS isozyme, which may compensate for effects of mutant specific KRAS inhibitors (71, 72).
The development of KRASG12C inhibitors for CRC is ongoing and larger randomized clinical trials may reveal more promising approaches. The recent development of KRASG12C targeted therapy in CRC has clearly ignited the field to develop new RAS inhibitors potentially with broader scope and reduced potential for resistance.
Author contributions
GP: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. PC: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. YM: Investigation, Validation, Writing – review & editing. KB: Investigation, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. NIH/NCI R01CA254197, R01CA238514.
Conflict of interest
Author GP is a co-founder of ADT Pharmaceuticals Inc. and consultant. Author KIB is a co-founder and CEO-Scientist of CerFlux.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic signaling pathways in the cancer genome atlas. Cell. (2018) 173:321–37.e10. doi: 10.1016/j.cell.2018.03.035
3. Ou SHI, Sokol ES, Madison R, Chung J, Ross JS, Miller VA, et al. 92PD - Comprehensive pan-cancer analysis of KRAS genomic alterations (GA) including potentially targetable subsets. Ann Oncol. (2019) 30:v26. doi: 10.1093/annonc/mdz239.003
4. Henry JT, Coker O, Chowdhury S, Shen JP, Morris VK, Dasari A, et al. Comprehensive clinical and molecular characterization of KRAS (G12C)-mutant colorectal cancer. JCO Precis Oncol. (2021) 5. doi: 10.1200/PO.20.00256
5. Muñoz-Maldonado C, Zimmer Y, Medová M. A comparative analysis of individual RAS mutations in cancer biology. Front Oncol. (2019) 9:1088. doi: 10.3389/fonc.2019.01088
6. Drosten M, Barbacid M. Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell. (2020) 37:543–50. doi: 10.1016/j.ccell.2020.03.013
7. Koulouridi A, Karagianni M, Messaritakis I, Sfakianaki M, Voutsina A, Trypaki M, et al. Prognostic value of KRAS mutations in colorectal cancer patients. Cancers (Basel). (2022) 14. doi: 10.3390/cancers14143320
8. Kwan AK, Piazza GA, Keeton AB, Leite CA. The path to the clinic: a comprehensive review on direct KRAS(G12C) inhibitors. J Exp Clin Cancer Res. (2022) 41:27. doi: 10.1186/s13046-021-02225-w
9. Zhu G, Pei L, Xia H, Tang Q, Bi F. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol Cancer. (2021) 20:143. doi: 10.1186/s12943-021-01441-4
10. Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. (2017) 170:17–33. doi: 10.1016/j.cell.2017.06.009
11. Nussinov R, Jang H, Tsai CJ. The structural basis for cancer treatment decisions. Oncotarget. (2014) 5:7285–302. doi: 10.18632/oncotarget.v5i17
12. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. (2003) 3:11–22. doi: 10.1038/nrc969
13. Papke B, Der CJ. Drugging RAS: know the enemy. Science. (2017) 355:1158–63. doi: 10.1126/science.aam7622
14. Fakih MG, Kopetz S, Kuboki Y, Kim TW, Munster PN, Krauss JC, et al. Sotorasib for previously treated colorectal cancers with KRAS(G12C) mutation (CodeBreaK100): a prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol. (2022) 23:115–24. doi: 10.1016/S1470-2045(21)00605-7
15. Yaeger R, Weiss J, Pelster MS, Spira AI, Barve M, Ou SI, et al. Adagrasib with or without cetuximab in colorectal cancer with mutated KRAS G12C. N Engl J Med. (2023) 388:44–54. doi: 10.1056/NEJMoa2212419
16. Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, et al. EGFR blockade reverts resistance to KRAS(G12C) inhibition in colorectal cancer. Cancer Discovery. (2020) 10:1129–39. doi: 10.1158/2159-8290.CD-20-0187
17. Fakih MG, Salvatore L, Esaki T, Modest DP, Lopez-Bravo DP, Taieb J, et al. Sotorasib plus panitumumab in refractory colorectal cancer with mutated KRAS G12C. N Engl J Med. (2023) 389:2125–39. doi: 10.1056/NEJMoa2308795
18. Desai J, Han S-W, Lee J-S, Shacham-Shmueli E, Massarelli E, Cervantes A, et al. Abstract CT029: Phase Ib study of GDC-6036 in combination with cetuximab in patients with colorectal cancer (CRC) with KRAS G12C mutation. Cancer Res. (2023) 83:CT029–CT. doi: 10.1158/1538-7445.AM2023-CT029
19. Désage AL, Léonce C, Swalduz A, Ortiz-Cuaran S. Targeting KRAS mutant in non-small cell lung cancer: novel insights into therapeutic strategies. Front Oncol. (2022) 12:796832. doi: 10.3389/fonc.2022.796832
20. Koga T, Suda K, Fujino T, Ohara S, Hamada A, Nishino M, et al. KRAS secondary mutations that confer acquired resistance to KRAS G12C inhibitors, sotorasib and adagrasib, and overcoming strategies: insights from in vitro experiments. J Thorac Oncol. (2021) 16:1321–32. doi: 10.1016/j.jtho.2021.04.015
21. Zhao Y, Murciano-Goroff YR, Xue JY, Ang A, Lucas J, Mai TT, et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature. (2021) 599:679–83. doi: 10.1038/s41586-021-04065-2
22. Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature. (2020) 577:421–5. doi: 10.1038/s41586-019-1884-x
23. Dunnett-Kane V, Nicola P, Blackhall F, Lindsay C. Mechanisms of resistance to KRAS(G12C) inhibitors. Cancers (Basel). (2021) 13. doi: 10.3390/cancers13010151
24. Lee SK, Jeong WJ, Cho YH, Cha PH, Yoon JS, Ro EJ, et al. β-Catenin-RAS interaction serves as a molecular switch for RAS degradation via GSK3β. EMBO Rep. (2018) 19. doi: 10.15252/embr.201846060
25. Akhave NS, Biter AB, Hong DS. The next generation of KRAS targeting: reasons for excitement and concern. Mol Cancer Ther. (2022) 21:1645–51. doi: 10.1158/1535-7163.MCT-22-0356
26. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med. (2020) 383:1207–17. doi: 10.1056/NEJMoa1917239
27. Lorthiois E, Gerspacher M, Beyer KS, Vaupel A, Leblanc C, Stringer R, et al. JDQ443, a structurally novel, pyrazole-based, covalent inhibitor of KRASG12C for the treatment of solid tumors. J Medicinal Chem. (2022) 65:16173–203. doi: 10.1021/acs.jmedchem.2c01438
28. Weiss A, Lorthiois E, Barys L, Beyer KS, Bomio-Confaglia C, Burks H, et al. Discovery, preclinical characterization, and early clinical activity of JDQ443, a structurally novel, potent, and selective covalent oral inhibitor of KRASG12C. Cancer Discovery. (2022) 12:1500–17. doi: 10.1158/2159-8290.CD-22-0158
29. Wang J, Martin-Romano P, Cassier P, Johnson M, Haura E, Lenox L, et al. Phase I study of JNJ-74699157 in patients with advanced solid tumors harboring the KRAS G12C mutation. Oncologist. (2022) 27:536–e53. doi: 10.1093/oncolo/oyab080
30. Murciano-Goroff YR, Heist RS, Kuboki Y, Koyama T, Ammakkanavar NR, Hollebecque A, et al. Abstract CT028: A first-in-human phase 1 study of LY3537982, a highly selective and potent KRAS G12C inhibitor in patients with KRAS G12C-mutant advanced solid tumors. Cancer Res. (2023) 83:CT028–CT. doi: 10.1158/1538-7445.AM2023-CT028
31. Ji J, Wang C, Fakih M. Targeting KRAS (G12C)-mutated advanced colorectal cancer: research and clinical developments. Onco Targets Ther. (2022) 15:747–56. doi: 10.2147/OTT.S340392
32. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. (2019) 575:217–23. doi: 10.1038/s41586-019-1694-1
33. Zhu C, Guan X, Zhang X, Luan X, Song Z, Cheng X, et al. Targeting KRAS mutant cancers: from druggable therapy to drug resistance. Mol Cancer. (2022) 21:159. doi: 10.1186/s12943-022-01629-2
34. Ramalingam S, Fakih M, Strickler J, Govindan R, Li BT, Goldberg S, et al. Abstract P05–01: A phase 1b study evaluating the safety and efficacy of sotorasib, a KRASG12C inhibitor, in combination with trametinib, a MEK inhibitor, in KRAS p.G12C-Mutated Solid Tumors. Mol Cancer Ther. (2021) 20:P05–1-P-1. doi: 10.1158/1535-7163.TARG-21-P05-01
35. Lou K, Steri V, Ge AY, Hwang YC, Yogodzinski CH, Shkedi AR, et al. KRAS(G12C) inhibition produces a driver-limited state revealing collateral dependencies. Sci Signal. (2019) 12. doi: 10.1126/scisignal.aaw9450
36. Huang Y, Wang J, Cao F, Jiang H, Li A, Li J, et al. SHP2 associates with nuclear localization of STAT3: significance in progression and prognosis of colorectal cancer. Sci Rep. (2017) 7:17597. doi: 10.1038/s41598-017-17604-7
37. Fedele C, Li S, Teng KW, Foster CJR, Peng D, Ran H, et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med. (2021) 218. doi: 10.1084/jem.20201414
38. Ou SI, Koczywas M, Ulahannan S, Janne P, Pacheco J, Burris H, et al. A12 the SHP2 inhibitor RMC-4630 in patients with KRAS-mutant non-small cell lung cancer: preliminary evaluation of a first-in-man phase 1 clinical trial. J Thorac Oncol. (2020) 15:S15–S6. doi: 10.1016/j.jtho.2019.12.041
39. Stice JP, Donovan S, Sun Y, Kohl N, Czako B, Mseeh F, et al. Abstract P207: BBP-398, a potent, small molecule inhibitor of SHP2, enhances the response of established NSCLC xenografts to KRASG12C and mutEGFR inhibitors. Mol Cancer Ther. (2021) 20:P207–P. doi: 10.1158/1535-7163.TARG-21-P207
40. Liu C, Lu H, Wang H, Loo A, Zhang X, Yang G, et al. Combinations with allosteric SHP2 inhibitor TNO155 to block receptor tyrosine kinase signaling. Clin Cancer Res. (2021) 27:342–54. doi: 10.1158/1078-0432.CCR-20-2718
41. Desai J, Alonso G, Kim SH, Cervantes A, Karasic T, Medina L, et al. Divarasib plus cetuximab in KRAS G12C-positive colorectal cancer: a phase 1b trial. Nat Med. (2024) 30:271–8. doi: 10.1038/s41591-023-02696-8
42. Purkey H. Abstract ND11: Discovery of GDC-6036, a clinical stage treatment for KRAS G12C-positive cancers. Cancer Res. (2022) 82:ND11–ND. doi: 10.1158/1538-7445.AM2022-ND11
43. Kuboki Y, Pietrantonio F, Salvatore L, Esaki T, Modest D, Paez D, et al. 91MO Sotorasib plus panitumumab versus standard-of-care for chemorefractory KRAS G12C-mutated metastatic colorectal cancer (mCRC): CodeBreak 300 phase III study. Ann Oncol. (2023) 34:S1504–S5. doi: 10.1016/j.annonc.2023.10.226
44. Hillig RC, Sautier B, Schroeder J, Moosmayer D, Hilpmann A, Stegmann CM, et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. Proc Natl Acad Sci U S A. (2019) 116:2551–60. doi: 10.1073/pnas.1812963116
45. Ross SJ, Revenko AS, Hanson LL, Ellston R, Staniszewska A, Whalley N, et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci Transl Med. (2017) 9. doi: 10.1126/scitranslmed.aal5253
46. Hofmann MH, Gmachl M, Ramharter J, Savarese F, Gerlach D, Marszalek JR, et al. BI-3406, a potent and selective SOS1-KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discovery. (2021) 11:142–57. doi: 10.1158/2159-8290.CD-20-0142
47. Macarulla T, Cervantes A, Tabernero J, Roselló S, Van Cutsem E, Tejpar S, et al. Phase I study of FOLFIRI plus pimasertib as second-line treatment for KRAS-mutated metastatic colorectal cancer. Br J Cancer. (2015) 112:1874–81. doi: 10.1038/bjc.2015.144
48. Desai J, Gan H, Barrow C, Jameson M, Atkinson V, Haydon A, et al. Open-label, dose-escalation/dose-expansion study of lifirafenib (BGB-283), an RAF family kinase inhibitor, in patients with solid tumors. J Clin Oncol. (2020) 38:2140–50. doi: 10.1200/JCO.19.02654
49. Marín-Ramos NI, Balabasquer M, Ortega-Nogales FJ, Torrecillas IR, Gil-Ordóñez A, Marcos-Ramiro B, et al. A potent isoprenylcysteine carboxylmethyltransferase (ICMT) inhibitor improves survival in Ras-driven acute myeloid leukemia. J Medicinal Chem. (2019) 62:6035–46. doi: 10.1021/acs.jmedchem.9b00145
50. Zhou J, Ji Q, Li Q. Resistance to anti-EGFR therapies in metastatic colorectal cancer: underlying mechanisms and reversal strategies. J Exp Clin Cancer Res. (2021) 40:328. doi: 10.1186/s13046-021-02130-2
51. Ngan SY, Burmeister B, Fisher RJ, Solomon M, Goldstein D, Joseph D, et al. Randomized trial of short-course radiotherapy versus long-course chemoradiation comparing rates of local recurrence in patients with T3 rectal cancer: Trans-Tasman Radiation Oncology Group trial 01.04. J Clin Oncol. (2012) 30:3827–33. doi: 10.1200/JCO.2012.42.9597
52. Hanna CR, Slevin F, Appelt A, Beavon M, Adams R, Arthur C, et al. Intensity-modulated radiotherapy for rectal cancer in the UK in 2020. Clin Oncol (R Coll Radiol). (2021) 33:214–23. doi: 10.1016/j.clon.2020.12.011
53. Gustavsson B, Carlsson G, Machover D, Petrelli N, Roth A, Schmoll HJ, et al. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Clin Colorectal Cancer. (2015) 14:1–10. doi: 10.1016/j.clcc.2014.11.002
54. Latchman J, Guastella A, Tofthagen C. 5-Fluorouracil toxicity and dihydropyrimidine dehydrogenase enzyme: implications for practice. Clin J Oncol Nurs. (2014) 18:581–5. doi: 10.1188/14.CJON.581-585
55. Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. (2013) 381:303–12. doi: 10.1016/S0140-6736(12)61900-X
56. Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. (2007) 357:2040–8. doi: 10.1056/NEJMoa071834
57. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. (2017) 18:1182–91. doi: 10.1016/S1470-2045(17)30422-9
58. Wagner S, Mullins CS, Linnebacher M. Colorectal cancer vaccines: Tumor-associated antigens vs neoantigens. World J Gastroenterol. (2018) 24:5418–32. doi: 10.3748/wjg.v24.i48.5418
59. Steffen CL, Kaya P, Schaffner-Reckinger E, Abankwa D. Eliminating oncogenic RAS: back to the future at the drawing board. Biochem Soc Trans. (2023) 51:447–56. doi: 10.1042/BST20221343
60. Wallon L, Khan I, Teng KW, Koide A, Zuberi M, Li J, et al. Inhibition of RAS-driven signaling and tumorigenesis with a pan-RAS monobody targeting the Switch I/II pocket. Proc Natl Acad Sci U S A. (2022) 119:e2204481119. doi: 10.1073/pnas.2204481119
61. Békés M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discovery. (2022) 21:181–200. doi: 10.1038/s41573-021-00371-6
62. Bond MJ, Chu L, Nalawansha DA, Li K, Crews CM. Targeted degradation of oncogenic KRAS(G12C) by VHL-recruiting PROTACs. ACS Cent Sci. (2020) 6:1367–75. doi: 10.1021/acscentsci.0c00411
63. Yang F, Wen Y, Wang C, Zhou Y, Zhou Y, Zhang ZM, et al. Efficient targeted oncogenic KRAS(G12C) degradation via first reversible-covalent PROTAC. Eur J Med Chem. (2022) 230:114088. doi: 10.1016/j.ejmech.2021.114088
64. Paiva SL, Crews CM. Targeted protein degradation: elements of PROTAC design. Curr Opin Chem Biol. (2019) 50:111–9. doi: 10.1016/j.cbpa.2019.02.022
65. Bando H, Ohtsu A, Yoshino T. Therapeutic landscape and future direction of metastatic colorectal cancer. Nat Rev Gastroenterol Hepato. (2023) 20:306–22. doi: 10.1038/s41575-022-00736-1
66. Benson AB, Venook AP, Al-Hawary MM, Arain MA, Chen YJ, Ciombor KK, et al. Colon cancer, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2021) 19:329–59. doi: 10.6004/jnccn.2021.0012
67. Sakamoto Y, Bando H, Nakamura Y, Hasegawa H, Kuwaki T, Okamoto W, et al. Trajectory for the regulatory approval of a combination of pertuzumab plus trastuzumab for pre-treated HER2-positive metastatic colorectal cancer using real-world data. Clin Colorectal Cancer. (2023) 22:45–52. doi: 10.1016/j.clcc.2022.10.003
68. Nakamura Y, Fujisawa T, Taniguchi H, Bando H, Okamoto W, Tsuchihara K, et al. SCRUM-Japan GI-SCREEN and MONSTAR-SCREEN: Path to the realization of biomarker-guided precision oncology in advanced solid tumors. Cancer Sci. (2021) 112:4425–32. doi: 10.1111/cas.15132
69. Nakamura Y, Okamoto W, Kato T, Esaki T, Kato K, Komatsu Y, et al. Circulating tumor DNA-guided treatment with pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer: a phase 2 trial. Nat Med. (2021) 27:1899–903. doi: 10.1038/s41591-021-01553-w
70. Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol. (2019) 16:361–75. doi: 10.1038/s41575-019-0126-x
71. Kim D, Herdeis L, Rudolph D, Zhao Y, Böttcher J, Vides A, et al. Pan-KRAS inhibitor disables oncogenic signalling and tumour growth. Nature. (2023) 619:160–6. doi: 10.1038/s41586-023-06123-3
Keywords: colorectal cancer, KRAS G12C inhibitors, resistance, clinical trials, KRAS mutation G12C
Citation: Piazza GA, Chandrasekaran P, Maxuitenko YY and Budhwani KI (2024) Assessment of KRASG12C inhibitors for colorectal cancer. Front. Oncol. 14:1412435. doi: 10.3389/fonc.2024.1412435
Received: 04 April 2024; Accepted: 06 June 2024;
Published: 24 June 2024.
Edited by:
Shiv K. Gupta, Mayo Clinic, United StatesReviewed by:
Ayad A. Al-Hamashi, University of Baghdad, IraqPeng-Chan Lin, National Cheng Kung University, Taiwan
Copyright © 2024 Piazza, Chandrasekaran, Maxuitenko and Budhwani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gary A. Piazza, Z2FwMDAzNEBhdWJ1cm4uZWR1